

Sudden cardiac death (SCD) is frequently the initial manifestation of a cardiac arrhythmia, resulting in about 350 000 deaths annually in the United States.1 Devices such as the implantable cardioverter‐defibrillator (ICD) seek to restore normal rhythm and may abort SCD.2 However, given the complex spatiotemporal dynamics of cardiac electrophysiology, predicting the onset of an arrhythmia and preventing the transition from a stable to an unstable rhythm is highly challenging. Deciphering the mechanisms that lead to an unstable heart rhythm and developing therapies to prevent unstable rhythms is an urgent clinical need.
In 1908, Heinrich Hering first described ECG alternans, a pattern of beat‐to‐beat oscillation in the ECG waveform.3 Subsequently, repolarization alternans (RA), or alternans that manifests during ventricular repolarization, has been associated with an increased risk for ventricular tachyarrhythmic events (VTEs) and SCD under a wide range of pathophysiologic substrates including ischemic and nonischemic cardiomyopathy and recent acute coronary syndromes.4, 5 RA may also be seen in structurally normal hearts under conditions of significant metabolic stress6 and chronotropic stimulation.7, 8 Early pioneering work has shown that different regions of the heart may alternate out of phase to form spatially discordant RA, and that phenomenon alone was a key factor promoting arrhythmogenesis by predisposing the heart to reentrant wave propagation.9, 10 Furthermore, in in silico studies, it was demonstrated that spatially discordant alternans led to markedly increased dispersion in repolarization (DR) that formed an ideal substrate for an ectopic trigger beat to instigate spiral‐wave breakups leading to the onset of lethal arrhythmias, such as ventricular tachycardia (VT) and ventricular fibrillation (VF).11
This review provides a contemporary perspective of the subcellular and cellular mechanisms that give rise to cardiac alternans and potential therapeutic approaches based on this mechanistic understanding.
Mechanisms of Cardiac Alternans
Two prevailing hypotheses have been put forth to explain the pathogenesis of cardiac alternans. The first posits that alternans is a membrane voltage or action potential (AP)–driven phenomenon. Under this hypothesis, alternation in cellular sarcolemmal currents, AP duration (APD) and AP amplitude drive alternation in intracellular Ca2+ concentration on an every‐other‐beat basis. In silico and in vitro studies support this hypothesis by demonstrating that the stability of Ca2+ homeostatic processes and the transition to stable alternans is driven by modulation of sarcolemmal Ca2+ 12 and K+ 13, 14 currents, driven primarily by fluctuation in AP morphology.15, 16, 17
The second hypothesis postulates that intracellular Ca2+ concentration ([Ca2+]i) alternans is the primary driver, which then results in membrane voltage (AP morphology) alternation.6, 15, 18, 19, 20, 21, 22, 23 Under the second hypothesis, stress‐induced8, 18 disruptions in Ca2+ transport processes can initiate [Ca2+]i alternans, which then results in AP alternans. Perturbations in intracellular Ca2+ transport can impact Ca2+ entry into the cytoplasm,14 sarcoplasmic reticulum (SR) Ca2+ uptake,24 intra‐SR Ca2+ redistribution,25, 26 SR Ca2+ release,6, 19 recovery of inactivated ryanodine receptors (RyRs) and coupling between intracellular Ca2+ cycling and surface membrane voltage.15, 17
Figure 1 exhibits Ca2+ cycling via calcium‐induced calcium release, the impact of SR Ca2+ content, and the role of mitochondria on a proposed model for the subcellular and cellular pathogenesis of alternans. The solid line indicates the SR Ca2+ baseline and the dashed line represents the threshold of SR Ca2+ content at which Ca2+ release occurs. In addition to providing ATP for excitation/contraction, mitochondria are centrally involved in Ca2+ signaling by serving as a Ca2+ buffer by taking up Ca2+ via the mitochondrial Ca2+ uniporter.27 Because of the spatial proximity of mitochondria to the RyR, mitochondria have been directly implicated in excitation‐contraction coupling. Whether mitochondrial Ca2+ uptake occurs on a beat‐to‐beat basis28, 29 or occurs in a more slowly integrated fashion30, 31 remains unclear.
Figure 1.
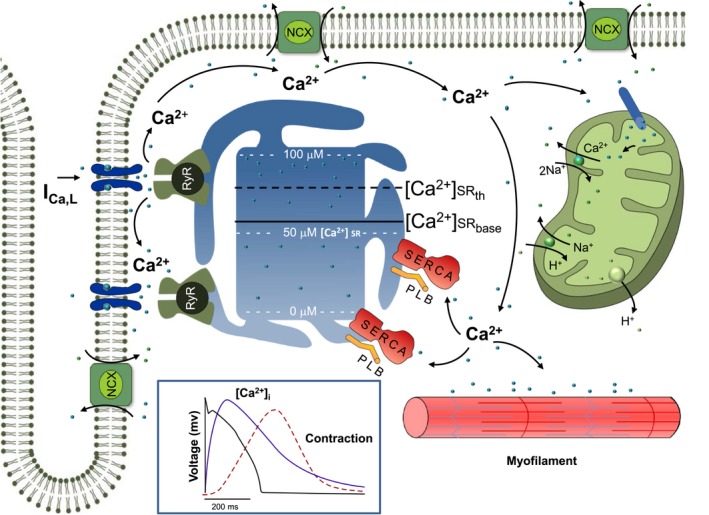
Schematic diagram of Ca2+ cycling includes the L‐type Ca2+ channel, the ryanodine receptor (RyR) channel, the phospholamban (PLB), the sarcoplasmic reticulum (SR) Ca2+ ATPase pump (SERCA2a), the Na+/Ca2+ exchanger (NCX), and the mitochondria. The effect of the SR Ca2+ content on a proposed model for cellular alternans is also demonstrated: the solid/dashed lines indicate the SR Ca2+ baseline ([Ca2+]SR‐base) and the SR Ca2+ content threshold ([Ca2+]SR‐th) at which Ca2+ release occurs, respectively. Calcium‐induced calcium release is manifested by an operational baseline of [Ca2+]SR in the diseased heart that is lower than the threshold to trigger spontaneous Ca2+ release in the normal heart. Inset: Representative traces of voltage (black) and intracellular calcium (purple) transients from a single cell along with the resultant mechanical contraction (red).
Metabolic Mechanisms of Alternans in Isolated Cardiac Myocytes
A preponderance of data have emerged that support the second hypothesis and invokes perturbations in Ca2+ handling as the primary driver of subcellular and cellular alternans. The effect of mitochondrial dysfunction on sarcoplasmic Ca2+ content during alternans has been studied by our group32 and others.33 These studies have provided insight on the changes that occur in Ca2+ handling in myocytes in diseased hearts and may open the door to novel therapeutic interventions. Our study used a customized photometry system in which Ca2+ dyes were excited at 2 discrete wavelengths to simultaneously excite 2 different dyes. Figure 2A presents examples of simultaneously measured cytosolic Ca2+ (Fluo‐4, AM; Thermo Fisher Scientific) and mitochondrial Ca2+ (x‐Rhod‐1, AM; Thermo Fisher Scientific) alternans. Figure 2B presents simultaneously measured cytosolic Ca2+ (Rhod‐2, AM; Thermo Fisher Scientific) and sarcoplasmic Ca2+ (Fluo‐5N, AM; Thermo Fisher Scientific) alternans.
Figure 2.
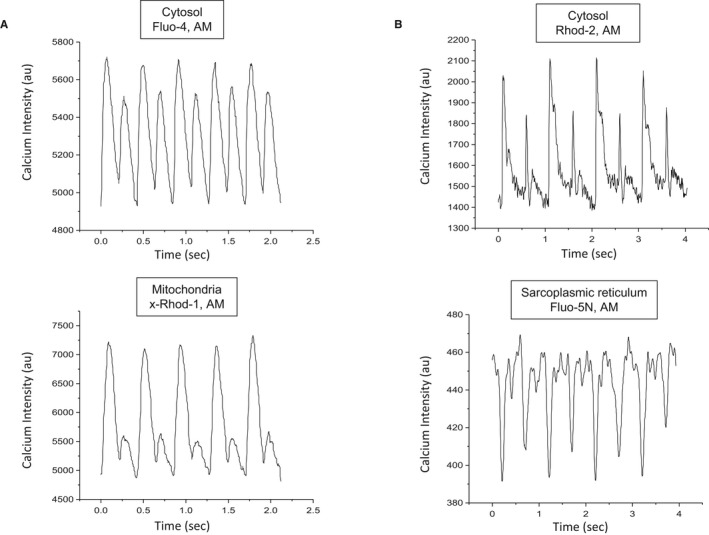
Representative examples of simultaneously measured (A) cytosolic Ca2+ (Fluo‐4, AM) and mitochondrial Ca2+ (x‐Rhod‐1, AM) alternans, and (B) cytosolic Ca2+ (Rhod‐2, AM) and sarcoplasmic Ca2+ (Fluo‐5N) alternans.
In the same study, we demonstrated that blocking cytochrome c oxidase, F0F1‐synthase, complex I and II, and α‐ketoglutarate dehydrogenase of the electron transport chain increased alternans in both control and SERCA2a upregulated mice. The increase in alternans in SERCA2a upregulated mice was significantly less than in control mice under 7 of 9 conditions tested (P<0.04). However, N‐Acetyl‐L‐cysteine reduced alternans in myocytes previously exposed to an oxidizing agent and CGP (an antagonist of mitochondrial sodium calcium exchanger). Blocking the mitochondrial permeability transition pore with cyclosporin A reduced CGP‐induced alternans.
In summary, our work32 demonstrates that mitochondrial Ca2+ handling impairments and energy production deficiencies increase alternans. This effect is lessened in SERCA2a upregulated mice, suggesting that these mice are better able to maintain electrical stability under conditions of stress. The data support a functional relationship between mitochondrial dysfunction, sarcoplasmic Ca2+ content, and the genesis of alternans and may help explain perturbations in Ca2+ signaling in myocytes from patients with heart failure.
While mitochondrial Ca2+ buffering can lead to alternans, studies have also shown that increased reactive oxygen species, especially following myocardial infarction (MI), may reduce SERCA2a function and lead to enhanced alternans.34 Furthermore, redox modulation of RyRs has been shown to promote Ca2+ alternans and create a proarrhythmic substrate following MI.35
Interplay of [Ca2+]i and AP Alternans
To further delineate the relationship between ionic currents and the SR Ca2+ uptake and release fluxes with the sarcolemmal membrane potential, we used a novel reverse engineering approach of a simultaneous AP and [Ca2+]i clamp of experimentally obtained data to a previously described left ventricular (LV) canine myocyte model.36 This hybrid (experimental and computational) approach was used to investigate whether model‐derived APs correlate with the APs obtained experimentally and to elucidate the subcellular (Figure 3C) and cellular mechanisms underlying cardiac alternans.17
Figure 3.
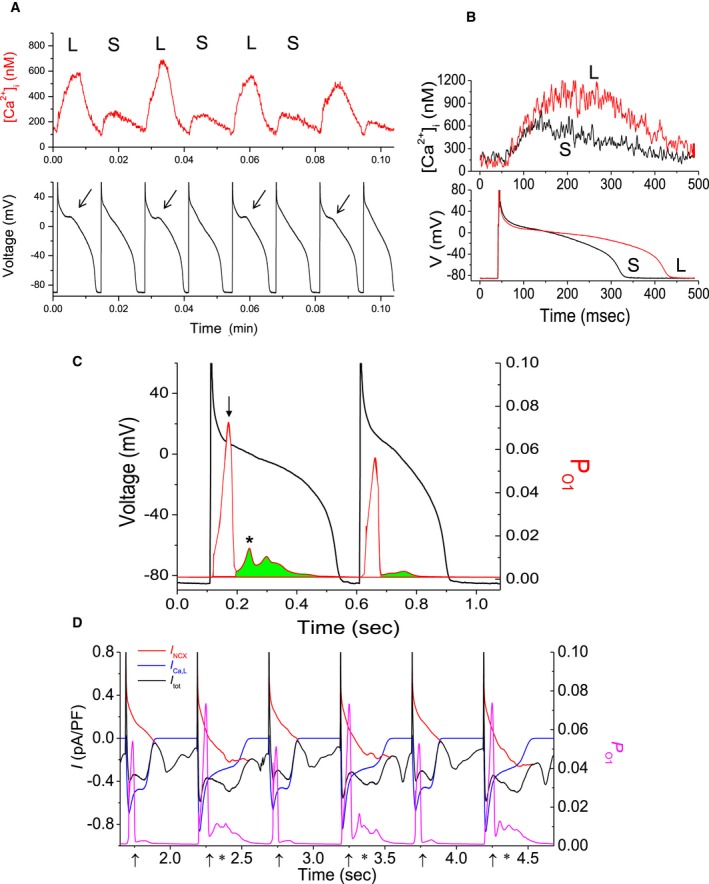
A, Examples of concordant calcium transients ([Ca2+]) and action potential (AP) alternans recorded in a left ventricular canine myocyte (at 0.8 seconds). Arrows indicate location of subthreshold early afterdepolarizations. L and S indicate Large/Long and Small/Short [Ca2+] AP, respectively. B, Superposition of intracellular Ca2+ concentration ([Ca2+]i) and APs from 2 consecutive beats during alternans. C, Left ordinate indicates the AP during alternans, while the right ordinate (in red) indicates open probability of state 1, PO 1 of the ryanodine receptor (RyR). The green area under the PO1V curve indicates the limits and the magnitude of the secondary RyR, while the same figure also indicates the timing of the peak of that secondary release with respect to the AP upstroke. D, Left ordinate presents the sum (black line) of the IC a,L (blue line) and INCX (red line), and the right ordinate the RyR state 1 open probability PO 1 (magenta line). The primary and secondary RyR releases are indicated by “↑” and an “*”, respectively.
Our work has demonstrated that APD prolongation is associated with a large [Ca2+]i and coincides with a secondary, much smaller, SR Ca2+ release (Figure 3B) manifested in the RyR state‐1 open probability P O1 on an every‐other‐beat basis (where primary/secondary SR Ca2+ releases are indicated by an “↑” and an “*”, respectively). This, in turn, triggers a larger inward depolarizing current attributed to both the L‐type Ca2+ channel (LTCC) and the sodium calcium exchanger (NCX), as shown in Figure 3D. This depolarizing current results from a large [Ca2+]i coincident with a smaller secondary SR Ca2+ release and a smaller RyR state‐1 open probability, as shown in Figure 3C and 3D. Also, careful inspection of Figure 3A reveals a small deflection on the AP, a subthreshold early afterdepolarization, which is associated with this depolarizing current. Importantly, this every‐other‐beat secondary Ca2+ release (Figure 3B) does not occur in recordings that do not exhibit alternans. The shape of the AP waveform is dependent on balance between the NCX and the LTCC. The NCX contributes either a depolarizing or repolarizing current during the AP, while the LTCC contributes a depolarizing current. Both the NCX and LTCC are directly mediated by [Ca2+]i. A large calcium transient causes the NCX to reverse earlier, thus contributing a smaller repolarizing current and leading to AP prolongation. It also causes acceleration of the LTCC Ca2+‐mediated inactivation, leading to AP shortening. A small calcium transient would expectedly cause opposite effects. Thus, the net balance of NCX and LTCC defines the effect of [Ca2+]i on the AP.37 Therefore, Ca2+ induced inactivation of the LTCC and Ca2+ transport across the sarcolemma through the NCX, which are both dependent on [Ca2+]i, mediate the relationship between [Ca2+]i and APD during alternans.17 Furthermore, under varying pathological conditions, this intricate balance between the NCX and LTCC could be altered leading to concordant or discordant alternans.
This observation further suggests that AP alternans is closely associated with the incidence of a spontaneous secondary SR Ca2+ release on alternate beats that occurs early during the AP plateau and provokes a subthreshold early afterdepolarization, which ultimately results in RA at the whole heart level. In prior studies it has been shown that elevation in SR lumenal Ca2+ is more likely to cause RyRs to be triggered by cytosolic Ca2+.38, 39, 40 In addition, spontaneous SR Ca2+ release41 and delayed afterdepolarization amplitude can ascend closer to AP trigger threshold42, 43 with an increase in SR Ca2+ content. Delayed afterdepolarization have, in turn, been shown to trigger abnormal electrical activity in response to catecholamines or high stimulation rates44 in normal ventricular myocytes,45 heart failure preparations,46 and cardiomyopathic human hearts.47 These studies support the hypothesis that SR Ca2+ stabilization can abolish alternans as evidenced by studies in which ryanodine and thapsigargin markedly reduced [Ca2+]i and eliminated AP alternans.18 Ryanodine has also been shown to eliminate both AP and tension alternans in papillary muscles.25, 26 Xie and Weiss48 provided further evidence for the relationship between SR Ca2+ content and alternans by demonstrating that rapid stimulation rates make myocytes more susceptible to Ca2+ overload and interactions between spontaneously occurring Ca2+ waves and AP‐triggered [Ca2+]i result in subcellular spatially discordant alternans. Therefore, the combination of increased likelihood of cytosolic Ca2+ to activate neighboring RyR clusters and increased sensitization of RyR luminal Ca2+ to cytosolic Ca2+ may reset local [Ca2+]SR and trigger subcellular alternans.48 These results also align well with the unified theory of Ca2+‐mediated alternans recently proposed by Qu et al,49 wherein alternans was shown to arise as a result of an instability in 3 properties of the Ca2+ release units, namely, randomness of Ca2+ sparks, recruitment of a Ca2+ spark by neighboring Ca2+ sparks, and refractoriness of the Ca2+ release units. In addition, they have successfully demonstrated that SR Ca2+, RyR sensitivity, and SR Ca2+ uptake rate all play an important role in Ca2+‐mediated alternans.
To test the hypothesis that secondary RyR openings are involved in AP alternans, we used isolated ventricular myocytes from mice hearts that were whole‐cell (in current‐clamp) patch‐clamped (37°C) at progressively faster rates until AP alternans was elicited (Figure 4A). Following the onset of alternans, pulses of −3.78 pA/pF and 5 ms duration were delivered 10 ms after the AP upstroke, on every other beat, which resulted in elimination of APD alternans (Figure 4B). Upon termination of stimulation (Figure 4C), APD alternans reappeared.
Figure 4.
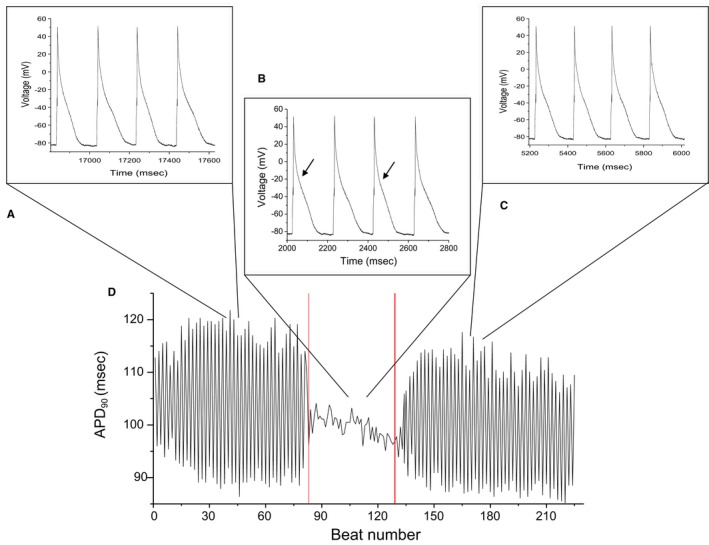
Representative example of the effect of pacing during the absolute refractory period during alternans in a mouse myocyte. A, Stable alternans resulting from pacing at increasingly higher stimulation rates. B, Pacing during the absolute refractory period eliminates alternans. C, Upon termination of pacing, alternans resumes. D, Beat‐to‐beat action potential duration (APD) for each of the interventions (A through C).
Mechanisms of Alternans in the Whole Heart
Early work demonstrated that electrical restitution, an intrinsic property of cardiac myocytes, can cause APD alternans at high heart rates. It has been shown in both in silico and in vitro studies that steep APD restitution slope (the relationship between the APD and the previous diastolic interval) and abnormal [Ca2+]i handling are the reasons for [Ca2+]i and APD alternans.11, 50, 51 In addition, tissue level studies demonstrate that ectopic beats and conduction velocity restitution promote spatially discordant alternans.52 However, despite evidence suggesting that sustained APD alternans occurs when the slope of the APD restitution at a given pacing cycle length is >1, the restitution hypothesis has not been validated in experimental studies.53 In both isolated ventricular myocytes and intact tissue, the onset of APD alternans occurs when APD restitution slope is considerably <1 and interventions that suppress [Ca2+]SR cycling eliminate AP alternans irrespective of the APD restitution.18, 54 While it was proposed that in certain cases, the complex interaction between the transient outward current I to and the I Ca,L can lead to alternans at slower heart rates,55 there is compelling evidence that in the intact heart, the onset of APD alternans is primarily attributable to an instability in [Ca2+]i handling rather than steep APD restitution.11, 56 Furthermore, in simultaneous voltage‐calcium optical mapping studies in isolated whole rabbit hearts, it has been demonstrated that the local onset of Ca2+ amplitude alternans precedes and triggers APD alternans.57 In aggregate, studies in isolated myocytes, intact tissue, and isolated hearts provide evidence that perturbations in Ca2+ cycling processes are the principal factors underlying APD alternans in the whole heart.
Localized alternans may lead to increased DR and susceptibility to VT/VF,22 and increased DR has been linked to concordant or discordant alternans (in which DR is found to be greater at sites of discordant compared with concordant alternans) and to VT/VF,58, 59 while many other studies have shown that APD alternans may become the substrate for reentry.22, 23, 60, 61, 62, 63, 64 Furthermore, it has been demonstrated that under conditions of reduced repolarization reserve such as long QT syndrome and bradycardia, RA can cause increased DR, which can trigger premature ventricular complexes and lead to reentrant ventricular arrhythmias.65
It should be noted, however, that the mechanisms that give rise to APD alternans may differ under different pathophysiological conditions and it remains unclear whether the presence of alternans always reflects a proarrhythmic substrate. It has been suggested that chronotropically induced alternans is nonspecific and does not necessarily reflect a proarrhythmic substrate. In contrast, discordant alternans resulting from acute ischemia or heart failure appears to be caused by subcellular Ca2+ handling perturbations, which are reflective of a proarrhythmic substrate.66, 67, 68 It seems likely that the more advanced the underlying heart disease, the higher the probability of inducing alternans with progressively smaller trigger events resulting in greater arrhythmia susceptibility.69
In addition to the voltage‐ and calcium‐mediated alternans hypotheses, over the past few years, some alternative theories pertinent to the formation of cardiac alternans have emerged. The presence of dynamic instabilities in the substrate that cause EADs were shown to lead to APD restitution discontinuities causing APD alternans.55, 70, 71 Sato et al70 demonstrated using in silico experiments that, while in smaller tissue sizes, EADs were able to synchronize globally, in larger tissues the spatial heterogeneity of EADs could lead to complex rhythms like APD alternans. In addition, EADs created a substrate conducive to the formation of premature ventricular complexes that then degraded into lethal arrhythmias such as VT or VF.
Furthermore, it has been shown that fibroblasts, which can electrotonically couple to myocytes via gap junctions, can affect APD and Ca2+ cycling dynamics. Xie et al72 demonstrated that modulation of fibroblast‐myocyte coupling can alter repolarization and Ca2+ cycling alternans at both the cellular and tissue levels, hence playing an important role in arrhythmogenesis, especially in diseased hearts with fibrosis. Both the conduction velocity and pacing frequency at which the alternans onset occurred have been shown to increase with increased gap junction coupling between cells.73 While lower gap junctional coupling enhanced Ca2+ alternans,73, 74 intermediate coupling enabled maximum spatial spread of alternans.73 Additionally, it has been proposed that tissue heterogeneity and the presence of structural barriers accentuate the presence and magnitude of alternans.75 The increased DR caused by the presence of tissue heterogeneities is conducive to the onset of spatially discordant alternans and potentiates the transition to VT/VF. And the DR affecting the presence of spatially discordant alternans is in effect not only caused by spatial dispersion of APD restitution, but likely also caused by dispersion of conduction velocity restitution.63, 76
RA and Short‐Term Arrhythmia Susceptibility
It is believed that RA is a marker of long‐term cardiac electrical instability.4, 5 In addition to risk stratifying patients for ICD therapy, recent clinical observations have also suggested that heightened RA may be an important predictor of short‐term arrhythmia susceptibility. Previous studies have established a plausible link between RA and susceptibility to VTEs and suggest that suppression of RA may prevent VTEs and SCD.10, 77, 78 This idea is supported by observations that increases in RA magnitude occur within minutes before spontaneous VTEs in patients with a history of cardiac arrest.79 In this study, compared with baseline, a 25% higher T‐wave alternans (TWA) magnitude was seen 10 minutes before the onset of a VTE. Similarly, increased TWA has also been demonstrated using ECG analysis (leads V1, V5, and aVF) in patients hospitalized for acute heart failure80 where an upsurge in TWA was observed 15 to 30 minutes before the onset of VTEs.
These data provide proof of concept that measuring changes in TWA from body surface ECGs may be capable of predicting acute arrhythmia susceptibility. Compared with body surface ECGs, intracardiac electrograms (EGMs) have significantly larger RA magnitude and may provide even more robust assessment of the link between surges in RA and short‐term arrhythmia susceptibility. Studies from our group81 and others82 have shown close correlation between simultaneous measurements of RA from body surface ECGs and intracardiac EGMs, suggesting that these methods are measuring the same phenomena.
The magnitude of RA measured from intracardiac EGMs from ICDs has been shown to rise sharply before spontaneous VTEs.83, 84 However, similar surges have not been noted before inappropriate ICD shocks or induced VTEs.83 In a prospective multicenter study,85 it was noted that the amplitude of alternans and nonalternans T‐wave variability (TWA/V) is significantly greater before spontaneous VTEs compared with during baseline rhythm, time‐matched ambulatory EGMs (from the same time of day at which spontaneous VTEs occurred), rapid pacing (at 105 bpm), and EGMs before the onset of supraventricular tachycardia. Each μV increase in TWA/V was associated with a >2‐fold increase in the odd ratio for experiencing a VTE.
These data suggest a close temporal relationship between surges in RA and spontaneous ventricular arrhythmias. To further these observations, in a tachypacing–induced heart failure model,86 we have noted a transition profile from concordant to discordant alternans in unipolar (as well as near‐field bipolar and far‐field bipolar [not shown here]) EGMs recorded from multiple sites across the left ventricle (base to apex) (Figure 5). The representation demonstrates that discordant alternans observed in both QRS and T waves originates in the region of the heart spanned by leads LV5 to LV10 with spatiotemporal propagation to neighboring regions. The presence of spatially discordant alternans furthers the DR, which, in turn, enables the triggering of VTEs.
Figure 5.
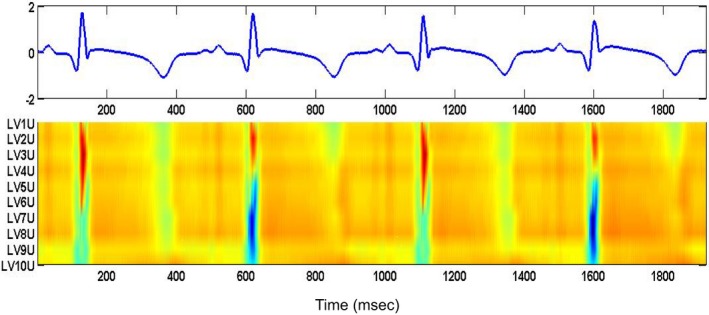
Transition from concordant alternans to discordant alternans during 4 consecutive beats for unipolar, near‐field bipolar, and far‐field bipolar signals from left ventricular (LV) catheter in the failing heart model. Amplitudes are normalized with red corresponding to higher amplitudes and blue pointing to lower amplitudes. The representation demonstrates that the discordant alternans observed in both QRS and T waves are originating in the region of the heart spanned by leads LV5 to LV10 with similar spatiotemporal propagation effect on the neighboring leads.
In summary, a robust body of evidence supports the hypothesis that there is a close relationship between surges in RA and short‐term susceptibility to VTEs. Elevated RA occurs either in conjunction with the development of a VTE or the heart passes through a state of elevated RA en route to a VTE. In either case, these observations suggest that detection of elevated levels of RA may be an important short‐term predictor of an impending VTE and raise the possibility that upstream therapies may be able to suppress RA and prevent the onset of VTEs.
Use of RA in Guiding Antiarrhythmic Therapy
RA is known to exhibit spatiotemporal heterogeneity.81 Therefore, attempts to deliver upstream RA suppressive therapy depend on the ability to detect RA regardless of where it originates in the heart. We have developed a novel intracardiac electrode configuration to detect RA in a highly reproducible manner despite spatiotemporal heterogeneity (using pairs of electrodes from catheters in the right ventricle and the coronary sinus [CS]).81 In an acute ischemia animal model, our data demonstrate that if significant RA is present, the right ventricular (RV) CS lead configuration will detect it >85% of the time.81
Using the same animal model, in Figure 6A we demonstrate the use of body surface as well as intracardiac leads comprised of catheters in the right ventricle, the left ventricle, and the CS to monitor RA immediately before the onset of a VTE. Figure 6B shows results (N=17) of alternans voltage and Kscore from body surface and LV CS leads before the onset of VT/VF, where Kscore reflects the statistical significance of the alternans voltage, relative to the background noise.86 Significant RA (ST segment and T wave) are present before VT/VF, at least in 1 lead in which the alternans voltage and Kscore are significant (alternans voltage >0.55 and 2.2 μV for body surface and LV CS leads, respectively; Kscore >3.0), indicating that RA can be accurately measured from intracardiac electrograms immediately before the onset of a VTE, and therefore may be used as an index to initiate upstream antiarrhythmic therapy.
Figure 6.
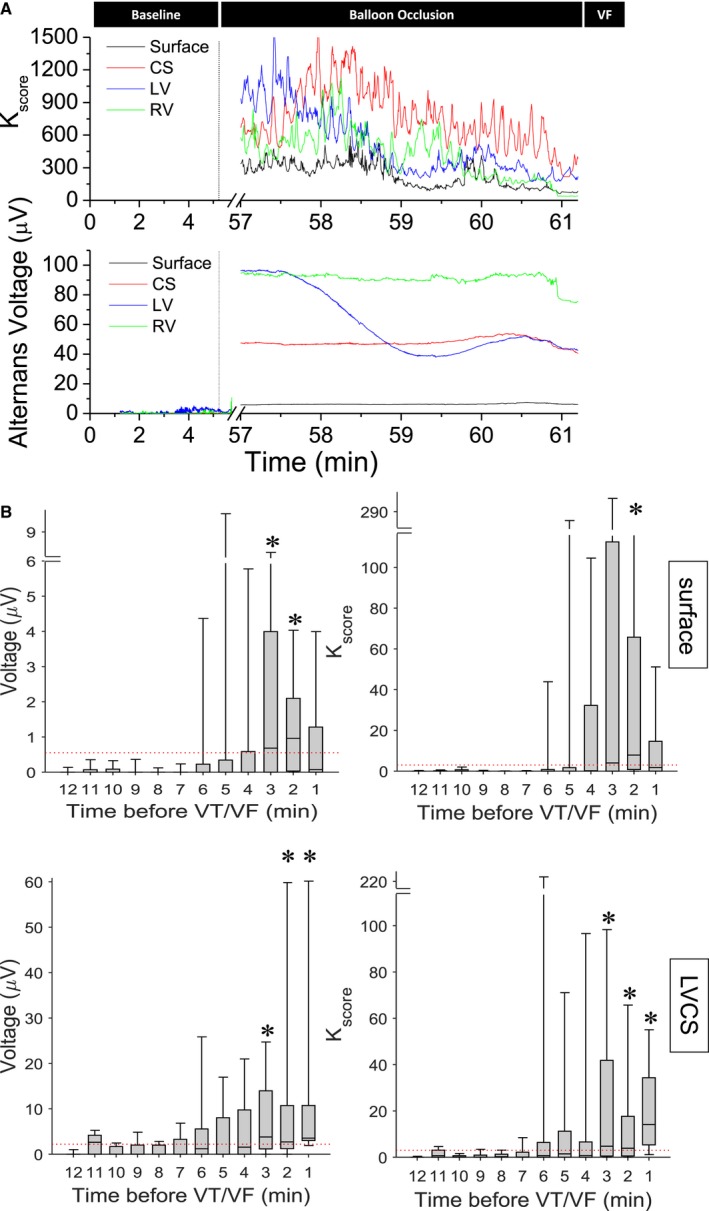
A, Circumflex coronary artery occlusion induced repolarization alternans, preceding ventricular fibrillation (VF). Alternans voltage and Kscore estimated from body surface as well intracardiac right ventricular (RV), left ventricular (LV), and coronary sinus (CS) unipolar leads. The beginning of the occlusion is marked by a vertical line. B, One observes a significant increase of alternans voltage and Kscore, for several minutes, preceding the onset of VF in both surface and LVCS leads. Red line indicates alternans voltage and Kscore thresholds for the respective leads (N=17). VT/VF indicates ventricular tachycardia/ventricular fibrillation. * denotes presence of significant repolarization alternans, defined as at least 1 min of data with both alternans voltage and Kscore greater than their respective thresholds, in at least one lead, in a 5 min window before the event.
Overall, despite the substantial spatiotemporal heterogeneity of RA, the RV CS lead configuration system can detect RA with a high degree of sensitivity. It also has clinical applicability because many implantable devices already utilize RV and CS leads (ie, in cardiac resynchronization therapy). The ability to detect heightened levels of RA from implantable devices opens the door to delivering upstream therapy from the device with the aim of suppressing RA and potentially preventing the development of a proarrhythmic substrate. Such upstream therapy may prevent the need for ICD shocks and mitigate the adverse impact of ICD shocks on quality of life. Upstream therapy may take the form of adaptive pacing protocols, which could be incorporated into an implantable device such that upon detection of an unstable substrate, as evidenced by detection of a surge in RA, the adaptive pacing protocol would be triggered to restabilize the unstable substrate and could be terminated when the RA magnitude drops below a predefined threshold. Homogenization of the electrical substrate, via adaptive pacing to suppress RA, would render the substrate less vulnerable to a trigger, such as a premature beat, which might have initiated a VTE under other circumstances.
Suppression of RA
Control of cardiac alternans has been the focus of several studies in in silico, in vitro, and in vivo models.77, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96 These studies have demonstrated control of alternans at the single cell level, as well as in in vitro preparations that have employed an adaptive negative feedback loop algorithm to adjust the pacing cycle length based on the alternans magnitude.77, 91, 92 This approach has demonstrated control of alternans in a tissue region of ≈2 to 2.5 cm. However, alternans control in a more complex spatiotemporal setting has been difficult to demonstrate.93, 95 Recently, Kulkarni et al97 demonstrated prevention of chronotropically induced alternans through real‐time control of the diastolic interval in optical mapping studies in healthy, isolated whole rabbit hearts. In an elegant in vivo demonstration of dynamic pacing therapy to control alternans, Christini et al98 demonstrated the ability to control AV nodal conduction alternans in humans undergoing electrophysiologic evaluation. However, AV nodal conduction alternans is a spatially constrained phenomenon that differs from the complex spatial‐temporal nature of ventricular alternans.
We have recently explored the utility of adaptive pacing86, 99 to suppress RA in vivo in an acute coronary artery occlusion swine model. In Figure 7A and 7B, we plotted the alternans voltage and Kscore, respectively, of an alternative, clinically relevant triangular intracardiac lead configuration comprising catheters in the CS and left ventricle.81 Upon detection of significant spontaneous RA at baseline, the phase of RA is estimated in real time and in phase, with positive polarity R‐wave triggered pacing delivered from a lead in the RV apex (intervention “pacing”)100 resulting in a significant decrease of RA (alternans voltage: ≈4‐fold decrease compared with baseline in panel a, P<0.0001; Kscore: ≈12‐fold decrease compared with baseline in panel a, P<0.0001). In panel c, RV12 pacing is stopped, resulting in a rise of the alternans voltage and Kscore (alternans voltage: ≈3‐fold rise compared with pacing in panel b, P<0.0001; Kscore: ≈7‐fold rise compared with pacing in panel b, P<0.0001).
Figure 7.
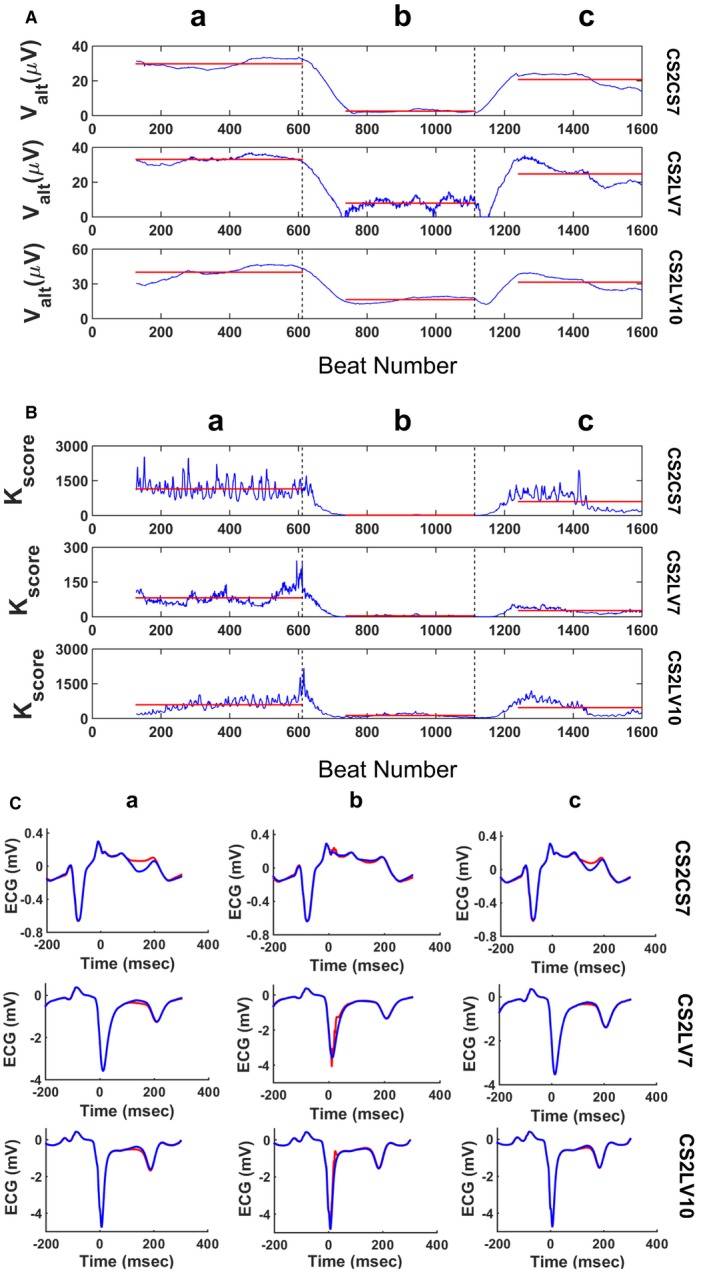
Example of the use of R‐wave triggered pacing during the absolute refractory period to suppress spontaneous repolarization alternans (RA) during acute ischemia. Alternans voltage (A) and Kscore (B) are plotted for intracardiac leads CS2CS7, CS2LV7, and CS2LV10. R‐wave triggered pacing (amplitude: +4 mA, pulse width: 10 ms, R‐wave coupling: 10 ms) is delivered from the right ventricular apex (RV12). a, spontaneous visible RA at baseline; (b) pacing delivered on every even beat results in RA suppression; (c) termination of pacing results in rising of the alternans voltage and Kscore to the baseline level. Transitions between interventions are indicated by dashed vertical lines, while red horizontal lines indicate the mean values of the alternans voltage and Kscore during each intervention. C, ECG morphology changes during the above‐described interventions. a, visible RA at baseline, (b) pacing decreases the RA level, (c) pacing is interrupted and RA becomes again visible. Panels show the median odd (red)/even (blue) beats of a 128‐beat sequence during each intervention.
Furthermore, Figure 8A and 8B present summary results across all experiments (N=7 animals; n=11 records) where R‐wave triggered pacing during the absolute refractory period was used to suppress spontaneously occurring RA. The figure demonstrates the alternans voltage (top row) and Kscore (bottom row) measured from RV CS leads depicting the suppression of RA when pacing is on. During balloon occlusion (baseline) in the presence of acute ischemia, markedly elevated levels of RA are observed. When triggered pacing is initiated, using the customized parameters (amplitude, pulse width, coupling interval, and phase), a significant decrease in alternans voltage and Kscore is observed. With cessation of pacing, alternans magnitude again returns to the elevated levels seen during baseline recordings in the presence of acute ischemia.
Figure 8.
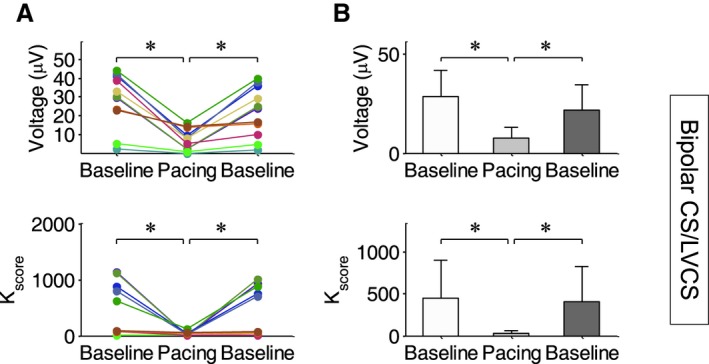
Demonstration of the utility of pacing during the absolute refractory period to suppress spontaneous repolarization alternans (RA) during acute ischemia (N=7 animals). (A) Alternans voltage and Kscore for each animal at baseline during balloon occlusion, during triggered pacing and during baseline again after cessation of pacing. (B) Summary results for alternans voltage and Kscore across all animals, during the three interventions. At baseline, during balloon occlusion, markedly elevated levels of RA are observed. When triggered pacing is initiated, a significant decrease in alternans voltage and Kscore is observed. With cessation of pacing, the RA magnitude again returns to the elevated levels seen at baseline. CS indicates coronary sinus; LV, left ventricular; RV, right ventricular. *denotes statistical significance of p<0.05.
In summary, these studies provide a proof of concept that RA can be suppressed using an adaptive (RA‐triggered and in real time) pacing protocol, and thus potentially preventing the formation of a proarrhythmic substrate. The concept of pacing during the absolute refractory period to suppress RA is supported by prior studies investigating its use in modulating cardiac contractility,101, 102, 103 where it has been shown in experimental studies and computer simulations that stimulation during the absolute refractory period may control the APD. This observation is in agreement with studies that have shown that pacing stimuli applied early during the absolute refractory period result in modulation of the transient outward current I to (see also Figure 4), which, in turn, may result in activation of the LTCC and [Ca2+]i modulation, suggesting that this form of stimulation aiming to control APD and RA and the pathogenesis of alternans at the myocyte level may share the same mechanisms.
Atrial Alternans
The majority of the mechanisms and understanding pertaining to cardiac alternans today has been derived from experimental and theoretical investigation of ventricular myocytes. More recently, however, there has been strong evidence supporting the role of alternans in promoting arrhythmogenic substrates in the atria as well.104, 105 In fact, it has been proposed that similar to RA in the ventricles, atrial alternans can serve as a precursor to severe atrial arrhythmias such as atrial fibrillation (AF).106, 107 Not only has the presence of atrial alternans been reported preceding episodes of AF,105, 107 clinically, P‐wave alternans have also been observed before and during atrial flutter preceding the transition to AF.108, 109 Furthermore, in a recent study, P‐wave abnormalities were shown to predict recurrence of AF in patients after electrical cardioversion.110
Despite the differences in atrial and ventricular physiology and AP morphologies, many similarities have been noted in terms of mechanisms of alternans origin. It was recently shown that similar to ventricular alternans, intracellular Ca2+ cycling abnormalities played a major role in initiating atrial alternans and blocking [Ca2+]i abolished alternans in isolated rabbit atrial myocytes.111 In addition, both in silico and in vitro experiments have demonstrated that recovery of RyR also plays a key role in initiation and maintenance of atrial Ca2+ alternans. However, Kanaporis and Blatter112 highlighted some important differences between atrial and ventricular alternans that could affect control and treatment strategies. Using isolated rabbit myocytes, they demonstrated that atrial alternans had a higher pacing frequency threshold for induction of alternans compared with ventricular alternans. Since there is higher SERCA activity in the atria,113 end‐diastolic [Ca2+]SR did not alternate during Ca2+ alternans in atrial myocytes,114 whereas imbalances in [Ca2+]SR load have been shown to promote ventricular alternans. In addition, they also showed that atrial myocytes have a higher density of the Ca2+‐activated Cl− channels, which play a key role in maintaining APD alternans in the atria.115 Finally, the structural complexity of the atria and increased spatiotemporal heterogeneity can affect the maintenance and propagation of atrial arrhythmias in a unique way, compared with the ventricles.
Effect of Autonomic Modulation on Cardiac Alternans
It is well established that both the parasympathetic and sympathetic branches of the autonomic nervous system innervate the heart and control normal cardiac function.116, 117, 118 A balance between these branches is essential for regulating cardiac function. Under pathophysiological conditions such as heart failure, hypertension, and MI, an offset in the autonomic regulation characterized by an increased sympathetic drive and decreased parasympathetic activity has been observed.119, 120 Extensive research in the past decade has focused on neuromodulation techniques to restore this autonomic imbalance by stimulating the parasympathetic nervous system as a potential therapy for the treatment of cardiovascular diseases. Several studies have demonstrated beneficial cardiovascular effects of vagus nerve stimulation (VNS), which is already a Food and Drug Administration–approved therapy for epilepsy and depression.120, 121, 122, 123, 124, 125
Initially, it was believed that while vagal fibers densely innervated the atria, sinoatrial node, and atrioventricular junction, there was little or no parasympathetic innervation of the ventricles.116 Hence, although the effect of parasympathetic activation on slowing the heart rate was well documented, a mechanistic understanding of its influence on ventricular function and electrophysiology was missing. Since then, studies have demonstrated the presence of parasympathetic innervation in the ventricles and highlighted its role in regulating ventricular electrophysiological properties.116, 126, 127 Several preclinical studies using heart failure animal models have reported significant improvements in ventricular hemodynamics with decreased mortality,122 significantly improved LV ejection fraction,128 and attenuated LV remodeling.129 In addition, VNS has been shown to exhibit anti‐inflammatory128, 130, 131 effects and inhibit SCD132 while markedly suppressing arrhythmias.133, 134, 135
The antiarrhythmic effects of VNS have been widely studied over the past decade, with promising results reported regarding suppression of both atrial and ventricular arrhythmias. Recently, in a randomized patient study, low‐level VNS was shown to suppress postoperative AF.136 Paroxysmal AF was also shown to be suppressed in patients using transcutaneous low‐level tragus stimulation along with a decrease in inflammatory cytokines.137 Similarly, VNS was shown to decrease VT inducibility in MI rats by preserving connexin43,138 reduce the occurrence of spontaneous ventricular arrhythmias and VT after coronary artery occlusion in dogs,139 decrease the maximum slope of restitution and electrical alternans, and increase VF threshold in isolated innervated rabbit hearts140 and reduce the levels of TWA in patients with drug‐refractory partial‐onset.141
The effect of sympathetic nervous system stimulation on the heart is complex and governed by the state of the myocardium. In the normal ventricle, sympathetic stimulation shortens the APD and reduces the DR, both of which have been associated with a decrease in the arrhythmogenic tendency.142 However, in pathological states, sympathetic stimulation is a potent stimulus for the generation of arrhythmias, perhaps by enhancing the DR, which may be why β‐blocker therapy reduces SCD in patients with heart failure.143, 144 In that context, interventions that reduce cardiac sympathetic activity have been shown to protect against arrhythmias,145, 146 whereas those that enhance sympathetic activity provoke them.145, 146, 147, 148
An upsurge in the magnitude of RA has been reported during periods of elevated sympathetic activity in humans149, 150 and in an end‐stage heart failure animal model.151 On the other hand, β‐blockers152, 153, 154, 155 have been reported to reduce the amplitude of RA. In patients with documented or suspected ventricular tachyarrhythmias who underwent RA testing, acute administration of the β‐blockers metoprolol and dl‐sotalol reduced overall RA amplitude by 35% and 38%, respectively,153 indicating that RA can be modulated, at least in some patients, by sympathetic activity. The possible effect of β‐blockers on the clinical utility of RA is mediated by at least 2 factors: blunting the chronotropic response to exercise, which may prevent some patients from reaching the specific threshold heart rate to develop RA,156 and reducing the magnitude of alternans.153
In basic science studies,140 alternans occurred at significantly longer cycle lengths and the peak alternans level was greater with sympathetic stimulation, compared with baseline. In all hearts,140 alternans level increased at progressively shorter pacing cycle length until VF occurred, albeit the cycle length at which VF occurred was not altered with sympathetic stimulation. VNS has caused a decrease in alternans level (as a result of a small decrease in the cycle length at which alternans occurred) and a small increase in cycle length at which VF occurred.140
The promising preclinical studies led to several clinical trials testing the efficacy of VNS to treat cardiovascular diseases in patients, which unfortunately showed mixed results. While the ANTHEM‐HF (Autonomic Neural Regulation Therapy to Enhance Myocardial Function in Heart Failure) study125 demonstrated significant improvements in LV function and decreased TWA in patients with heart failure, both the INOVATE‐HF (Increase of Vagal Tone in Heart Failure) and NECTAR‐HF (Neural Cardiac Therapy for Heart Failure) trials failed to demonstrate significant improvements with VNS therapy.125, 157, 158, 159 A major factor for the conflicting results was the difference in stimulation parameters used in the different studies. Ardell et al160 recently demonstrated that the effects of VNS depend on the balance between the efferent and afferent vagal fiber responses, which, in turn, is dependent on the stimulation parameters used. They proposed the existence of an optimal frequency–amplitude–pulse width–based operating point, also called neural fulcrum, when the efferent and afferent stimulation effects are balanced, producing a null heart rate response. They showed that it is possible to achieve cardiac control and beneficial cardiovascular effects of VNS by appropriate selection of stimulation parameters within the neural fulcrum.160 While many groups continue to decipher the underlying mechanisms behind the cardiovascular effects of VNS, it continues to be an active area of both preclinical and clinical research, offering a potentially promising nonpharmacological treatment for cardiac arrhythmias and cardiovascular diseases such as heart failure, hypertension, and MI.
Conclusions
While many hypotheses have been proposed to explain the genesis of cardiac alternans, the prevailing one proposes that subcellular disruptions of intracellular Ca2+ homeostatic mechanisms occurring dynamically on a beat‐to‐beat basis give rise to [Ca2+]i alternans, which, in turn, results in APD and ECG alternans. The manifestation of discordant APD alternans at the whole heart level is associated with increased spatial dispersion of refractoriness, wavefront fractionation, and the onset of reentrant VTEs. Thus, this conceptual framework regarding the pathophysiology of cardiac alternans suggests that an RA surge, beyond being linked to medium‐ and long‐term risk of VTEs and SCD, is likely to play a more central role in creating the necessary conditions for short‐term arrhythmia susceptibility. The temporal relationship between RA and short‐term arrhythmogenesis has significant clinical implications for triggering and guiding upstream antiarrhythmic therapy.
Sources of Funding
This work was supported by a Grand‐in‐Aid (#15GRNT23070001) from the American Heart Association (AHA), 2 Founders Affiliate Post‐doctoral Fellowships (#15POST22690003 and #12POST9310001) from the AHA, the Kenneth M. Rosen Fellowship in Cardiac Pacing and Electrophysiology (#13‐FA‐32‐HRS) from the Heart Rhythm Society, and the RICBAC Foundation, National Institutes of Health [NIH] 1 R01 HL135335‐01, NIH 1 R21 HL137870‐01 and NIH 1 R21EB026164‐01. This work was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (NIH Award #UL1 RR 025758 and financial contributions from Harvard University and its affiliated academic healthcare centers). The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University, and its affiliated academic healthcare centers, the National Center for Research Resources, or the NIH.
Disclosures
None.
(J Am Heart Assoc. 2019;8:e013750 DOI: 10.1161/JAHA.119.013750.)
References
- 1. Chugh SS. Sudden cardiac death in 2017: spotlight on prediction and prevention. Int J Cardiol. 2017;237:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greenspon AJ, Patel JD, Lau E, Ochoa JA, Frisch DR, Ho RT, Pavri BB, Kurtz SM. Trends in permanent pacemaker implantation in the United States from 1993 to 2009: increasing complexity of patients and procedures. J Am Coll Cardiol. 2012;60:1540–1545. [DOI] [PubMed] [Google Scholar]
- 3. Hering H. Das wesen des herzalternans. Munch Med Wochenschr. 1908;4:1417–1421. [Google Scholar]
- 4. Armoundas AA, Tomaselli GF, Esperer HD. Pathophysiological basis and clinical application of T‐wave alternans. J Am Coll Cardiol. 2002;40:207–217. [DOI] [PubMed] [Google Scholar]
- 5. Armoundas AA, Hohnloser SH, Ikeda T, Cohen RJ. Can microvolt T‐wave alternans testing reduce unnecessary defibrillator implantation? Nat Rev Cardiol. 2005;2:522. [DOI] [PubMed] [Google Scholar]
- 6. Hüser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation—contraction coupling underlies alternans in cat heart cells. J Physiol. 2000;524:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Turitto G, Caref EB, El‐Attar G, Helal M, Mohamed A, Pedalino RP, El‐Sherif N. Optimal target heart rate for exercise‐induced T‐wave alternans. Ann Noninvasive Electrocardiol. 2001;6:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laurita KR, Rosenbaum DS. Restitution, repolarization and alternans as arrhythmogenic substrates, in Cardiac Electrophysiology: From Cell to Bedside Zipes D.P., Jalife J., Ed. Cardiac Electrophysiology. 4th ed, Philadelphia, PA: Saunders; 2004;ch 26, 232–241. [Google Scholar]
- 9. Chialvo DR, Gilmour RF Jr, Jalife J. Low dimensional chaos in cardiac tissue. Nature. 1990;343:653. [DOI] [PubMed] [Google Scholar]
- 10. Karma A. Electrical alternans and spiral wave breakup in cardiac tissue. Chaos. 1994;4:461–472. [DOI] [PubMed] [Google Scholar]
- 11. Weiss JN, Karma A, Shiferaw Y, Chen P‐S, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98:1244–1253. [DOI] [PubMed] [Google Scholar]
- 12. Mahajan A, Sato D, Shiferaw Y, Baher A, Xie L‐H, Peralta R, Olcese R, Garfinkel A, Qu Z, Weiss JN. Modifying L‐type calcium current kinetics: consequences for cardiac excitation and arrhythmia dynamics. Biophys J. 2008;94:411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hua F, Johns DC, Gilmour RF Jr. Suppression of electrical alternans by overexpression of HERG in canine ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2342–H2351. [DOI] [PubMed] [Google Scholar]
- 14. Fox JJ, McHarg JL, Gilmour RF Jr. Ionic mechanism of electrical alternans. Am J Physiol Heart Circ Physiol. 2002;282:H516–H530. [DOI] [PubMed] [Google Scholar]
- 15. Jordan PN, Christini DJ. Action potential morphology influences intracellular calcium handling stability and the occurrence of alternans. Biophys J. 2006;90:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Allen D, Orchard C. Myocardial contractile function during ischemia and hypoxia. Circ Res. 1987;60:153–168. [DOI] [PubMed] [Google Scholar]
- 17. Armoundas AA. Mechanism of abnormal sarcoplasmic reticulum calcium release in canine left‐ventricular myocytes results in cellular alternans. IEEE Trans Biomed Eng. 2009;56:220–228. [DOI] [PubMed] [Google Scholar]
- 18. Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. [DOI] [PubMed] [Google Scholar]
- 19. Diaz ME, Eisner DA, O'Neill SC. Depressed ryanodine receptor activity increases variability and duration of the systolic Ca2+ transient in rat ventricular myocytes. Circ Res. 2002;91:585–593. [DOI] [PubMed] [Google Scholar]
- 20. Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J. 1999;77:2930–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kockskämper J, Zima AV, Blatter LA. Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J Physiol. 2005;564:697–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T‐wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. [DOI] [PubMed] [Google Scholar]
- 23. Pastore JM, Rosenbaum DS. Role of structural barriers in the mechanism of alternans‐induced reentry. Circ Res. 2000;87:1157–1163. [DOI] [PubMed] [Google Scholar]
- 24. Kameyama M, Hirayama Y, Saitoh H, Maruyama M, Atarashi H, Takano T. Possible contribution of the sarcoplasmic reticulum Ca2+ pump function to electrical and mechanical alternans. J Electrocardiol. 2003;36:125–135. [DOI] [PubMed] [Google Scholar]
- 25. Kihara Y, Morgan JP. Abnormal Cai2+ handling is the primary cause of mechanical alternans: study in ferret ventricular muscles. Am J Physiol Heart Circ Physiol. 1991;261:H1746–H1755. [DOI] [PubMed] [Google Scholar]
- 26. Lee JA. Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ Res. 1990;66:585–595. [DOI] [PubMed] [Google Scholar]
- 27. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566. [DOI] [PubMed] [Google Scholar]
- 28. O'Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46:767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Isenberg G, Han S, Schiefer A, Wendt‐Gallitelli MF. Changes in mitochondrial calcium concentration during the cardiac contraction cycle. Cardiovasc Res. 1993;27:1800–1809. [DOI] [PubMed] [Google Scholar]
- 30. Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 1991;261:H1123–H1134. [DOI] [PubMed] [Google Scholar]
- 31. Sedova M, Klishin A, Hüser J, Blatter LA. Capacitative Ca2+ entry is graded with degree of intracellular Ca2+ store depletion in bovine vascular endothelial cells. J Physiol. 2000;523:549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stary V, Puppala D, Scherrer‐Crosbie M, Dillmann WH, Armoundas AA. SERCA2a upregulation ameliorates cellular alternans induced by metabolic inhibition. J Appl Physiol. 2016;120:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tomek J, Tomková M, Zhou X, Bub G, Rodriguez B. Modulation of cardiac alternans by altered sarcoplasmic reticulum calcium release: a simulation study. Front Physiol. 2018;9:1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Plummer BN, Liu H, Wan X, Deschênes I, Laurita KR. Targeted antioxidant treatment decreases cardiac alternans associated with chronic myocardial infarction. Circ Arrhythm Electrophysiol. 2015;8:165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Belevych AE, Terentyev D, Viatchenko‐Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O'Rourke B. Role of sodium‐calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Armoundas AA. Discordant calcium transient and action potential alternans in a canine left‐ventricular myocyte. IEEE Trans Biomed Eng. 2009;56:2340–2344. [DOI] [PubMed] [Google Scholar]
- 38. Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase a phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–316. [DOI] [PubMed] [Google Scholar]
- 39. Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ikemoto N, Ronjat M, Meszaros LG, Koshita M. Postulated role of calsequestrin in the regulation of calcium release from sarcoplasmic reticulum. Biochemistry. 1989;28:6764–6771. [DOI] [PubMed] [Google Scholar]
- 41. Satoh H, Blatter LA, Bers DM. Effects of [Ca2+] i, SR Ca2+ load, and rest on Ca2+ spark frequency in ventricular myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H657–H668. [DOI] [PubMed] [Google Scholar]
- 42. Orchard C, Eisner D, Allen D. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 1983;304:735. [DOI] [PubMed] [Google Scholar]
- 43. Wier WG, Kort AA, Stern MD, Lakatta EG, Marban E. Cellular calcium fluctuations in mammalian heart: direct evidence from noise analysis of aequorin signals in Purkinje fibers. Proc Natl Acad Sci USA. 1983;80:7367–7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14:61–66. [DOI] [PubMed] [Google Scholar]
- 45. Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol Heart Circ Physiol. 1990;258:H1796–H1805. [DOI] [PubMed] [Google Scholar]
- 46. Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium‐calcium exchange, inward rectifier potassium current, and residual β‐adrenergic responsiveness. Circ Res. 2001;88:1159–1167. [DOI] [PubMed] [Google Scholar]
- 47. Gilmour RF, Heger JJ, Prystowsky EN, Zipes DP. Cellular electrophysiologic abnormalities of diseased human ventricular myocardium. Am J Cardiol. 1983;51:137–144. [DOI] [PubMed] [Google Scholar]
- 48. Xie LH, Weiss JN. Arrhythmogenic consequences of intracellular calcium waves. Am J Physiol Heart Circ Physiol. 2009;297:H997–H1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Qu Z, Liu MB, Nivala M. A unified theory of calcium alternans in ventricular myocytes. Sci Rep. 2016;6:35625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tolkacheva EG, Zhao X. Nonlinear dynamics of periodically paced cardiac tissue. Nonlinear Dyn. 2012;68:347–363. [Google Scholar]
- 51. Koller ML, Riccio ML, Gilmour RF Jr. Dynamic restitution of action potential duration during electrical alternans and ventricular fibrillation. Am J Physiol Heart Circ Physiol. 1998;275:H1635–H1642. [DOI] [PubMed] [Google Scholar]
- 52. Mironov S, Jalife J, Tolkacheva EG. Role of conduction velocity restitution and short‐term memory in the development of action potential duration alternans in isolated rabbit hearts. Circulation. 2008;118:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gilmour RF. Electrical restitution and ventricular fibrillation: negotiating a slippery slope. J Cardiovasc Electrophysiol. 2002;13:1150–1151. [DOI] [PubMed] [Google Scholar]
- 54. Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. [DOI] [PubMed] [Google Scholar]
- 55. Qu Z, Xie Y, Garfinkel A, Weiss JN. T‐wave alternans and arrhythmogenesis in cardiac diseases. Front Physiol. 2010;1:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Narayan SM, Bayer JD, Lalani G, Trayanova NA. Action potential dynamics explain arrhythmic vulnerability in human heart failure: a clinical and modeling study implicating abnormal calcium handling. J Am Coll Cardiol. 2008;52:1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Visweswaran R, McIntyre SD, Ramkrishnan K, Zhao X, Tolkacheva EG. Spatiotemporal evolution and prediction of [Ca2+] i and APD alternans in isolated rabbit hearts. J Cardiovasc Electrophysiol. 2013;24:1287–1295. [DOI] [PubMed] [Google Scholar]
- 58. Chinushi M, Restivo M, Caref EB, El‐Sherif N. Electrophysiological basis of arrhythmogenicity of QT/T alternans in the long‐QT syndrome: tridimensional analysis of the kinetics of cardiac repolarization. Circ Res. 1998;83:614–628. [DOI] [PubMed] [Google Scholar]
- 59. Chinushi M, Kozhevnikov D, Caref EB, Restivo M, El‐Sherif N. Mechanism of discordant T wave alternans in the in vivo heart. J Cardiovasc Electrophysiol. 2003;14:632–638. [DOI] [PubMed] [Google Scholar]
- 60. Tachibana H, Kubota I, Yamaki M, Watanabe T, Tomoike H. Discordant S‐T alternans contributes to formation of reentry: a possible mechanism of reperfusion arrhythmia. Am J Physiol. 1998;275:H116–H121. [DOI] [PubMed] [Google Scholar]
- 61. Shimizu W, Antzelevitch C. Cellular and ionic basis for T‐wave alternans under long‐QT conditions. Circulation. 1999;99:1499–1507. [DOI] [PubMed] [Google Scholar]
- 62. Fox JJ, Riccio ML, Hua F, Bodenschatz E, Gilmour RF Jr. Spatiotemporal transition to conduction block in canine ventricle. Circ Res. 2002;90:289–296. [DOI] [PubMed] [Google Scholar]
- 63. Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation. 2000;102:1664–1670. [DOI] [PubMed] [Google Scholar]
- 64. Watanabe MA, Fenton FH, Evans SJ, Hastings HM, Karma A. Mechanisms for discordant alternans. J Cardiovasc Electrophysiol. 2001;12:196–206. [DOI] [PubMed] [Google Scholar]
- 65. Liu W, Kim TY, Huang X, Liu MB, Koren G, Choi BR, Qu Z. Mechanisms linking T‐wave alternans to spontaneous initiation of ventricular arrhythmias in rabbit models of long QT syndrome. J Physiol. 2018;596:1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea‐pig hearts: mechanisms underlying concordant alternans. J Physiol. 2000;529:171–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wu Y, Clusin WT. Calcium transient alternans in blood‐perfused ischemic hearts: observations with fluorescent indicator fura red. Am J Physiol Heart Circ Physiol. 1997;273:H2161–H2169. [DOI] [PubMed] [Google Scholar]
- 68. Bayer JD, Narayan SM, Lalani GG, Trayanova NA. Rate‐dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm. 2010;7:1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Merchant FM, Armoundas AA. Role of substrate and triggers in the genesis of cardiac alternans, from the myocyte to the whole heart: implications for therapy. Circulation. 2012;125:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, Karagueuzian H, Garfinkel A, Weiss JN, Qu Z. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci USA. 2009;106:2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tran DX, Sato D, Yochelis A, Weiss JN, Garfinkel A, Qu Z. Bifurcation and chaos in a model of cardiac early afterdepolarizations. Phys Rev Lett. 2009;102:258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xie Y, Garfinkel A, Weiss JN, Qu Z. Cardiac alternans induced by fibroblast‐myocyte coupling: mechanistic insights from computational models. Am J Physiol Heart Circ Physiol. 2009;297:H775–H784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jia Z, Bien H, Shiferaw Y, Entcheva E. Cardiac cellular coupling and the spread of early instabilities in intracellular Ca2+ . Biophys J. 2012;102:1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hammer KP, Ljubojevic S, Ripplinger CM, Pieske BM, Bers DM. Cardiac myocyte alternans in intact heart: influence of cell–cell coupling and β‐adrenergic stimulation. J Mol Cell Cardiol. 2015;84:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Krogh‐Madsen T, Christini DJ. Action potential duration dispersion and alternans in simulated heterogeneous cardiac tissue with a structural barrier. Biophys J. 2007;92:1138–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Banville I, Gray RA. Effect of action potential duration and conduction velocity restitution and their spatial dispersion on alternans and the stability of arrhythmias. J Cardiovasc Electrophysiol. 2002;13:1141–1149. [DOI] [PubMed] [Google Scholar]
- 77. Rappel WJ, Fenton F, Karma A. Spatiotemporal control of wave instabilities in cardiac tissue. Phys Rev Lett. 1999;83:456. [Google Scholar]
- 78. Tolkacheva EG, Romeo MM, Guerraty M, Gauthier DJ. Condition for alternans and its control in a two‐dimensional mapping model of paced cardiac dynamics. Phys Rev E. 2004;69:031904. [DOI] [PubMed] [Google Scholar]
- 79. Shusterman V, Goldberg A, London B. Upsurge in T‐wave alternans and nonalternating repolarization instability precedes spontaneous initiation of ventricular tachyarrhythmias in humans. Circulation. 2006;113:2880–2887. [DOI] [PubMed] [Google Scholar]
- 80. Nearing BD, Wellenius GA, Mittleman MA, Josephson ME, Burger AJ, Verrier RL. Crescendo in depolarization and repolarization heterogeneity heralds development of ventricular tachycardia in hospitalized patients with decompensated heart failure. Circ Arrhythm Electrophysiol. 2012;5:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weiss EH, Merchant FM, d'Avila A, Foley L, Reddy VY, Singh JP, Mela T, Ruskin JN, Armoundas AA. A novel lead configuration for optimal spatio‐temporal detection of intracardiac repolarization alternans. Circ Arrhythm Electrophysiol. 2011;4:407–417. DOI: 10.1161/CIRCEP.109.934208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Paz O, Zhou X, Gillberg J, Tseng HJ, Gang E, Swerdlow C. Detection of T‐wave alternans using an implantable cardioverter‐defibrillator. Heart Rhythm. 2006;3:791–797. [DOI] [PubMed] [Google Scholar]
- 83. Kim JW, Pak HN, Park JH, Nam GB, Kim SK, Lee HS, Jang JK, Choi JI, Kim YH. Defibillator electrogram T wave alternans as a predictor of spontaneous ventricular tachyarrhythmias in defibrillator recipients. Circ J. 2009;73:55–62. [DOI] [PubMed] [Google Scholar]
- 84. Armoundas AA, Albert CM, Cohen RJ, Mela T; TOVA investigators . Utility of implantable cardioverter defibrillator electrograms to estimate repolarization alternans preceding a tachyarrhythmic event. J Cardiovasc Electrophysiol. 2004;15:594–597. [DOI] [PubMed] [Google Scholar]
- 85. Swerdlow C, Chow T, Das M, Gillis AM, Zhou X, Abeyratne A, Ghanem RN. Intracardiac electrogram t‐wave alternans/variability increases before spontaneous ventricular tachyarrhythmias in implantable cardioverter‐defibrillator patients: a prospective, multi‐center study. Circulation. 2011;123:1052–1060. [DOI] [PubMed] [Google Scholar]
- 86. Armoundas AA, Weiss EH, Sayadi O, Laferriere S, Sajja N, Mela T, Singh JP, Barrett CD, Heist EK, Merchant FM. A novel pacing method to suppress repolarization alternans in vivo: implications for arrhythmia prevention. Heart Rhythm. 2013;10:564–572. [DOI] [PubMed] [Google Scholar]
- 87. McIntyre SD, Kakade V, Mori Y, Tolkacheva EG. Heart rate variability and alternans formation in the heart: the role of feedback in cardiac dynamics. J Theor Biol. 2014;350:90–97. [DOI] [PubMed] [Google Scholar]
- 88. Wu R, Patwardhan A. Mechanism of repolarization alternans has restitution of action potential duration dependent and independent components. J Cardiovasc Electrophysiol. 2006;17:87–93. [DOI] [PubMed] [Google Scholar]
- 89. Zlochiver S, Johnson C, Tolkacheva E. Constant DI pacing suppresses cardiac alternans formation in numerical cable models. Chaos. 2017;27:093903. [DOI] [PubMed] [Google Scholar]
- 90. Christini DJ, Collins JJ. Using chaos control and tracking to suppress a pathological nonchaotic rhythm in a cardiac model. Phys Rev E. 1996;53:R49. [DOI] [PubMed] [Google Scholar]
- 91. Hall K, Christini DJ, Tremblay M, Collins JJ, Glass L, Billette J. Dynamic control of cardiac alternans. Phys Rev Lett. 1997;78:4518. [Google Scholar]
- 92. Hall GM, Gauthier DJ. Experimental control of cardiac muscle alternans. Phys Rev Lett. 2002;88:198102. [DOI] [PubMed] [Google Scholar]
- 93. Echebarria B, Karma A. Spatiotemporal control of cardiac alternans. Chaos. 2002;12:923–930. [DOI] [PubMed] [Google Scholar]
- 94. Jordan PN, Christini DJ. Adaptive diastolic interval control of cardiac action potential duration alternans. J Cardiovasc Electrophysiol. 2004;15:1177–1185. [DOI] [PubMed] [Google Scholar]
- 95. Christini DJ, Riccio ML, Culianu CA, Fox JJ, Karma A, Gilmour RF Jr. Control of electrical alternans in canine cardiac Purkinje fibers. Phys Rev Lett. 2006;96:104101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kanu UB, Iravanian S, Gilmour RF, Christini DJ. Control of action potential duration alternans in canine cardiac ventricular tissue. IEEE Trans Biomed Eng. 2011;58:894–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kulkarni K, Lee SW, Kluck R, Tolkacheva EG. Real‐time closed loop diastolic interval control prevents cardiac alternans in isolated whole rabbit hearts. Ann Biomed Eng. 2018;46:555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Christini DJ, Stein KM, Markowitz SM, Mittal S, Slotwiner DJ, Scheiner MA, Iwai S, Lerman BB. Nonlinear‐dynamical arrhythmia control in humans. Proc Natl Acad Sci USA. 2001;98:5827–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Merchant FM, Sayadi O, Sohn K, Weiss EH, Puppala D, Doddamani R, Singh JP, Heist EK, Owen C, Kulkarni K, Armoundas AA. Real‐time closed‐loop suppression of repolarization alternans reduces arrhythmia susceptibility in vivo. Sci Rep. 2019. In Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Sayadi O, Merchant FM, Puppala D, Mela T, Singh JP, Heist EK, Owen C, Armoundas AA. A novel method for determining the phase of T‐wave alternans: diagnostic and therapeutic implications. Circ Arrhythm Electrophysiol. 2013;6:818–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Brunckhorst CB, Shemer I, Mika Y, Ben‐Haim SA, Burkhoff D. Cardiac contractility modulation by non‐excitatory currents: studies in isolated cardiac muscle. Eur J Heart Fail. 2006;8:7–15. [DOI] [PubMed] [Google Scholar]
- 102. Winter J, Brack KE, Ng GA. The acute inotropic effects of cardiac contractility modulation (CCM) are associated with action potential duration shortening and mediated by β1‐adrenoceptor signalling. J Mol Cell Cardiol. 2011;51:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Borggrefe MM, Lawo T, Butter C, Schmidinger H, Lunati M, Pieske B, Misier AR, Curnis A, Böcker D, Remppis A. Randomized, double blind study of non‐excitatory, cardiac contractility modulation electrical impulses for symptomatic heart failure. Eur Heart J. 2008;29:1019–1028. [DOI] [PubMed] [Google Scholar]
- 104. Comtois P, Nattel S. Atrial repolarization alternans as a path to atrial fibrillation. J Cardiovasc Electrophysiol. 2012;23:1013–1015. [DOI] [PubMed] [Google Scholar]
- 105. Franz MR, Jamal SM, Narayan SM. The role of action potential alternans in the initiation of atrial fibrillation in humans: a review and future directions. Europace. 2012;14:v58–v64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hiromoto K, Shimizu H, Furukawa Y, Kanemori T, Mine T, Masuyama T, Ohyanagi M. Discordant repolarization alternans‐induced atrial fibrillation is suppressed by verapamil. Circ J. 2005;69:1368–1373. [DOI] [PubMed] [Google Scholar]
- 107. Narayan SM, Franz MR, Clopton P, Pruvot EJ, Krummen DE. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation. 2011;123:2922–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Narayan SM, Bode F, Karasik PL, Franz MR. Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation. 2002;106:1968–1973. [DOI] [PubMed] [Google Scholar]
- 109. Siniorakis E, Arvanitakis S, Tzevelekos P, Giannakopoulos N, Limberi S. P‐wave alternans predicting imminent atrial flutter. Cardiol J. 2017;24:706–707. [DOI] [PubMed] [Google Scholar]
- 110. Gonna H, Gallagher MM, Guo XH, Yap YG, Hnatkova K, Camm AJ. P‐wave abnormality predicts recurrence of atrial fibrillation after electrical cardioversion: a prospective study. Ann Noninvasive Electrocardiol. 2014;19:57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kanaporis G, Blatter LA. The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ Res. 2015;116:846–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kanaporis G, Blatter L. Alternans in atria: mechanisms and clinical relevance. Medicina (Kaunas). 2017;53:139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Lüss I, Boknik P, Jones LR, Kirchhefer U, Knapp J, Linck B, Lüss H, Meissner A, Müller FU, Schmitz W. Expression of cardiac calcium regulatory proteins in atrium v ventricle in different species. J Mol Cell Cardiol. 1999;31:1299–1314. [DOI] [PubMed] [Google Scholar]
- 114. Shkryl VM, Maxwell JT, Domeier TL, Blatter LA. Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am J Physiol Heart Circ Physiol. 2012;302:H2310–H2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kanaporis G, Blatter LA. Calcium‐activated chloride current determines action potential morphology during calcium alternans in atrial myocytes. J Physiol. 2016;594:699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Coote J. Myths and realities of the cardiac vagus. J Physiol. 2013;591:4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Kapa S, DeSimone CV, Asirvatham SJ. Innervation of the heart: an invisible grid within a black box. Trends Cardiovasc Med. 2016;26:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. 2014;114:1004–1021. [DOI] [PubMed] [Google Scholar]
- 119. Bibevski S, Dunlap ME. Evidence for impaired vagus nerve activity in heart failure. Heart Fail Rev. 2011;16:129–135. [DOI] [PubMed] [Google Scholar]
- 120. Schwartz PJ, De Ferrari GM. Sympathetic–parasympathetic interaction in health and disease: abnormalities and relevance in heart failure. Heart Fail Rev. 2011;16:101–107. [DOI] [PubMed] [Google Scholar]
- 121. Beaumont E, Southerland EM, Hardwick JC, Wright GL, Ryan S, Li Y, KenKnight BH, Armour JA, Ardell JL. Vagus nerve stimulation mitigates intrinsic cardiac neuronal and adverse myocyte remodeling post myocardial infarction. Am J Physiol Heart Circ Physiol. 2015;309:H1198–H1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Li M, Zheng C, Sato T, Kawada T, Sugimachi M, Sunagawa K. Vagal nerve stimulation markedly improves long‐term survival after chronic heart failure in rats. Circulation. 2004;109:120–124. [DOI] [PubMed] [Google Scholar]
- 123. Sabbah HN. Electrical vagus nerve stimulation for the treatment of chronic heart failure. Cleve Clin J Med. 2011;78(suppl 1):S24–S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Schwartz PJ, De Ferrari GM. Vagal stimulation for heart failure: background and first in‐man study. Heart Rhythm. 2009;6:S76–S81. [DOI] [PubMed] [Google Scholar]
- 125. Premchand RK, Sharma K, Mittal S, Monteiro R, Dixit S, Libbus I, DiCarlo LA, Ardell JL, Rector TS, Amurthur B. Extended follow‐up of patients with heart failure receiving autonomic regulation therapy in the ANTHEM‐HF study. J Card Fail. 2016;22:639–642. [DOI] [PubMed] [Google Scholar]
- 126. Armour JA, Murphy DA, Yuan BX, MacDonald S, Hopkins DA. Gross and microscopic anatomy of the human intrinsic cardiac nervous system. Anat Rec. 1997;247:289–298. [DOI] [PubMed] [Google Scholar]
- 127. Ulphani JS, Cain JH, Inderyas F, Gordon D, Gikas PV, Shade G, Mayor D, Arora R, Kadish AH, Goldberger JJ. Quantitative analysis of parasympathetic innervation of the porcine heart. Heart Rhythm. 2010;7:1113–1119. [DOI] [PubMed] [Google Scholar]
- 128. Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, Mazgalev TN. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high‐rate pacing model. Circ Heart Fail. 2009;2:692–699. [DOI] [PubMed] [Google Scholar]
- 129. Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin‐converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest. 1997;99:1926–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458. [DOI] [PubMed] [Google Scholar]
- 131. Yamakawa K, Matsumoto N, Imamura Y, Muroya T, Yamada T, Nakagawa J, Shimazaki J, Ogura H, Kuwagata Y, Shimazu T. Electrical vagus nerve stimulation attenuates systemic inflammation and improves survival in a rat heatstroke model. PLoS One. 2013;8:e56728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Vanoli E, De Ferrari GM, Stramba‐Badiale M, Hull SS Jr, Foreman RD, Schwartz PJ. Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ Res. 1991;68:1471–1481. [DOI] [PubMed] [Google Scholar]
- 133. Zheng C, Li M, Inagaki M, Kawada T, Sunagawa K, Sugimachi M. Vagal stimulation markedly suppresses arrhythmias in conscious rats with chronic heart failure after myocardial infarction. Conf Proc IEEE Eng Med Biol Soc. 2005;7:7072–7075. [DOI] [PubMed] [Google Scholar]
- 134. Brack KE, Coote JH, Ng GA. Vagus nerve stimulation protects against ventricular fibrillation independent of muscarinic receptor activation. Cardiovasc Res. 2011;91:437–446. [DOI] [PubMed] [Google Scholar]
- 135. Wu W, Lu Z. Loss of anti‐arrhythmic effect of vagal nerve stimulation on ischemia‐induced ventricular tachyarrhythmia in aged rats. Tohoku J Exp Med. 2011;223:27–33. [DOI] [PubMed] [Google Scholar]
- 136. Stavrakis S, Humphrey MB, Scherlag B, Iftikhar O, Parwani P, Abbas M, Filiberti A, Fleming C, Hu Y, Garabelli P. Low‐level vagus nerve stimulation suppresses post‐operative atrial fibrillation and inflammation: a randomized study. JACC Clin Electrophysiol. 2017;3:929–938. [DOI] [PubMed] [Google Scholar]
- 137. Stavrakis S, Humphrey MB, Scherlag BJ, Hu Y, Jackman WM, Nakagawa H, Lockwood D, Lazzara R, Po SS. Low‐level transcutaneous electrical vagus nerve stimulation suppresses atrial fibrillation. J Am Coll Cardiol. 2015;65:867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ando M, Katare RG, Kakinuma Y, Zhang D, Yamasaki F, Muramoto K, Sato T. Efferent vagal nerve stimulation protects heart against ischemia‐induced arrhythmias by preserving connexin43 protein. Circulation. 2005;112:164–170. [DOI] [PubMed] [Google Scholar]
- 139. Nasi‐Er BG, Wenhui Z, HuaXin S, Xianhui Z, Yaodong L, Yanmei L, Hongli W, TuEr‐Hong ZL, Qina Z, BaoPeng T. Vagus nerve stimulation reduces ventricular arrhythmias and increases ventricular electrical stability. Pacing Clin Electrophysiol. 2019;42:247–256. [DOI] [PubMed] [Google Scholar]
- 140. Ng GA, Brack KE, Patel VH, Coote JH. Autonomic modulation of electrical restitution, alternans and ventricular fibrillation initiation in the isolated heart. Cardiovasc Res. 2007;73:750–760. [DOI] [PubMed] [Google Scholar]
- 141. Schomer AC, Nearing BD, Schachter SC, Verrier RL. Vagus nerve stimulation reduces cardiac electrical instability assessed by quantitative T‐wave alternans analysis in patients with drug‐resistant focal epilepsy. Epilepsia. 2014;55:1996–2002. [DOI] [PubMed] [Google Scholar]
- 142. Takei M, Sasaki Y, Yonezawa T, Lakhe M, Aruga M, Kiyosawa K. The autonomic control of the transmural dispersion of ventricular repolarization in anesthetized dogs. J Cardiovasc Electrophysiol. 1999;10:981–989. [DOI] [PubMed] [Google Scholar]
- 143. Hjalmarson A, Goldstein S, Fagerberg B, Wedel H, Waagstein F, Kjekshus J, Wikstrand J, Westergren G, Thimell M, El Allaf D. Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomised intervention trial in congestive heart failure (MERIT‐HF). Lancet. 1999;353:2001–2007.10376614 [Google Scholar]
- 144. Poole‐Wilson PA, Swedberg K, Cleland JG, Di Lenarda A, Hanrath P, Komajda M, Lubsen J, Lutiger B, Metra M, Remme WJ. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the carvedilol or metoprolol European trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. [DOI] [PubMed] [Google Scholar]
- 145. Corr P, Yamada K, Witkowski F. The heart and cardiovascular system. 1986.
- 146. Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Janse MJ, Schwartz PJ, Wilms‐Schopman F, Peters R, Durrer D. Effects of unilateral stellate ganglion stimulation and ablation on electrophysiologic changes induced by acute myocardial ischemia in dogs. Circulation. 1985;72:585–595. [DOI] [PubMed] [Google Scholar]
- 148. Billman GE. A comprehensive review and analysis of 25 years of data from an in vivo canine model of sudden cardiac death: implications for future anti‐arrhythmic drug development. Pharmacol Ther. 2006;111:808–835. [DOI] [PubMed] [Google Scholar]
- 149. Lampert R, Shusterman V, Burg MM, Lee FA, Earley C, Goldberg A, Mcpherson CA, Batsford WP, Soufer R. Effects of psychologic stress on repolarization and relationship to autonomic and hemodynamic factors. J Cardiovasc Electrophysiol. 2005;16:372–377. [DOI] [PubMed] [Google Scholar]
- 150. Verrier RL, Antzelevitch C. Autonomic aspects of arrhythmogenesis: the enduring and the new. Curr Opin Cardiol. 2004;19:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Shusterman V, McTiernan CF, Goldberg A, Saba S, Salama G, London B. Adrenergic stimulation promotes T‐wave alternans and arrhythmia inducibility in a TNF‐α genetic mouse model of congestive heart failure. Am J Physiol Heart Circ Physiol. 2009;298:H440–H450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Kirk M. Beta adrenergic blockade decreases T wave alternans. J Am Coll Cardiol. 1999;33:108A (abstract). [Google Scholar]
- 153. Klingenheben T, Grönefeld G, Li YG, Hohnloser SH. Effect of metoprolol and d, l‐sotalol on microvolt‐level T‐wave alternans: results of a prospective, double‐blind, randomized study. J Am Coll Cardiol. 2001;38:2013–2019. [DOI] [PubMed] [Google Scholar]
- 154. Rashba EJ, Cooklin M, MacMurdy K, Kavesh N, Kirk M, Sarang S, Peters RW, Shorofsky SR, Gold MR. Effects of selective autonomic blockade on T‐wave alternans in humans. Circulation. 2002;105:837–842. [DOI] [PubMed] [Google Scholar]
- 155. Komiya N, Seto S, Nakao K, Yano K. The influence of β‐adrenergic agonists and antagonists on T‐wave alternans in patients with and without ventricular tachyarrhythmia. Pacing Clin Electrophysiol. 2005;28:680–684. [DOI] [PubMed] [Google Scholar]
- 156. Tapanainen JM, Still AM, Airaksinen KJ, Huikuri HV. Prognostic significance of risk stratifiers of mortality, including T wave alternans, after acute myocardial infarction: results of a prospective follow‐up study. J Cardiovasc Electrophysiol. 2001;12:645–652. [DOI] [PubMed] [Google Scholar]
- 157. Gold MR, Van Veldhuisen DJ, Hauptman PJ, Borggrefe M, Kubo SH, Lieberman RA, Milasinovic G, Berman BJ, Djordjevic S, Neelagaru S. Vagus nerve stimulation for the treatment of heart failure: the INOVATE‐HF trial. J Am Coll Cardiol. 2016;68:149–158. [DOI] [PubMed] [Google Scholar]
- 158. Libbus I, Nearing BD, Amurthur B, KenKnight BH, Verrier RL. Autonomic regulation therapy suppresses quantitative T‐wave alternans and improves baroreflex sensitivity in patients with heart failure enrolled in the ANTHEM‐HF study. Heart Rhythm. 2016;13:721–728. [DOI] [PubMed] [Google Scholar]
- 159. Zannad F, De Ferrari GM, Tuinenburg AE, Wright D, Brugada J, Butter C, Klein H, Stolen C, Meyer S, Stein KM. Chronic vagal stimulation for the treatment of low ejection fraction heart failure: results of the neural cardiac therapy for heart failure (NECTAR‐HF) randomized controlled trial. Eur Heart J. 2014;36:425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Ardell JL, Nier H, Hammer M, Southerland EM, Ardell CL, Beaumont E, KenKnight BH, Armour JA. Defining the neural fulcrum for chronic vagus nerve stimulation: implications for integrated cardiac control. J Physiol. 2017;595:6887–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]