

Abstract
The gut microbiome has emerged as an important factor affecting human health and disease. The recent development of –omics approaches, including phylogenetic marker-based microbiome profiling, shotgun metagenomics, metatranscriptomics, metaproteomics, and metabolomics, has enabled efficient characterization of microbial communities. These techniques can provide strain-level taxonomic resolution of the taxa present in microbiomes, assess the potential functions encoded by the microbial community and quantify the metabolic activities occurring within a complex microbiome. The application of these meta-omics approaches to clinical samples has identified microbial species, metabolic pathways, and metabolites that are associated with the development and treatment of human diseases. These findings have further facilitated microbiome-targeted drug discovery and efforts to improve human health management. Recent in vitro and in vivo investigations have uncovered the presence of extensive drug-microbiome interactions. These interactions have also been shown to be important contributors to the disparate patient responses to treatment that are often observed during disease therapy. Therefore, developing techniques or frameworks that enable rapid screening, detailed evaluation, and accurate prediction of drug/host-microbiome interactions is critically important in the modern era of microbiome research and precision medicine. Here we review the current status of meta-omics techniques, including integrative multi-omics approaches, for characterizing the microbiome’s functionality in the context of health and disease. We also summarize and discuss new frameworks for applying meta-omics approaches and microbiome assays to study drug-microbiome interactions. Lastly, we discuss and exemplify strategies for implementing microbiome-based precision medicines using these meta-omics approaches and high throughput microbiome assays.
Keywords: Drug-microbiome interactions, Host-microbiome interactions, Meta-omics, Microbiome, Microbiome assay, Multi-omics, Personalized medicine
Introduction
The human gut harbors trillions of microbial cells and thousands of different species from diverse phylogenetic backgrounds, including bacteria, archaea, and various microbial eukaryotes [1]. Altogether, this community of microorganisms, termed the gut microbiota, has a similar cell number to that of human cells [2] and 450-fold more genes than the human genome [3]. These gut microbiota genomes, namely the metagenome, encode functions and metabolic pathways that participate in various host biological processes, including metabolism, nutrition, and immunity [4–6]. Given the high complexity of the human gut microbiota and the challenges in culturing a high proportion of gut microbial species [7], most microbiome studies employ “meta-omics” approaches, including 16S rRNA gene sequencing, metagenomics, metatranscriptomics, metaproteomics, and metabolomics, which directly examine the phylogenetic markers, genes, transcripts, proteins, or metabolites from the samples [8].
In the past two decades, meta-omics based research has revealed significant associations between the gut microbiome and human diseases, including obesity, diabetes, inflammatory bowel disease (IBD), cardiovascular disease, and various cancers [4, 5]. Several studies have also demonstrated causative roles for the gut microbiome in inducing or alleviating the development of disease following variants of Koch’s postulates [9–11]. Given that the composition of the human microbiota is highly dynamic and can be altered with drugs or dietary interventions [12], the microbiome has been proposed as a druggable target in humans [13]. Accumulating evidence also supports the idea that many drugs, such as metformin [14, 15], may alleviate disease, at least in part, through modulating the gut microbiome. Recent large-scale screening of > 1000 drugs on the growth of selected gut bacterial species also highlighted the wide impacts of various drugs on individual microbes [16]. In addition, the existence of bidirectional drug-microbiome interactions for many clinically prescribed drugs has been demonstrated to impact drug efficacy and/or toxicity [17, 18]. As medicine is currently pursuing more precise disease treatment and health management, it is vital that the microbiome is fully integrated into future therapeutic strategies [19].
Nevertheless, our understanding on the mechanisms underlying host-microbiome and drug-microbiome interactions is still very limited. Several databases linking specific microbial species/strains or microbial metabolic pathways to specific diseases have been published [20–22]; however, these databases remain incomplete and most clinically prescribed drugs have not been assessed for their impact on the composition and function of human microbiomes. In addition, the composition of the human microbiome differs between individuals and is affected by various factors such as diet, lifestyle, and host genetics [23–26]. Thus, each patient’s microbiome will respond differently to therapeutic treatments, and we currently cannot accurately predict these responses in advance. Fortunately, recent microbiome studies have expanded beyond simply profiling microbiota compositions and are increasingly characterizing microbial functions by using functional meta-omics approaches such as metatranscriptomics and metaproteomics [27–29]. The development and optimization of various in vitro microbiome culturing models, such as HuMiX [30], SHIME [31], and RapidAIM [32], opens the door to rapidly screen drugs against individual microbiomes. Herein we summarize the current development of various functional meta-omics approaches, highlighting efforts to integrate findings across meta-omic platforms and discuss their applications in host-microbiome, drug-microbiome, and microbe-microbe interaction studies at the interface of precision medicine.
Functional meta-omics approaches for studying the microbiome
The human gut consists of host and microbial cells, as well as secreted proteins, metabolites, and microvesicles, all of which may interact with each other to impact human health. Different meta-omic approaches each examine different aspects of this intestinal ecosystem at different levels with their own advantages (detailed in this section) and disadvantages (or challenges discussed in Table 1) (Fig. 1). Technical details on these meta-omics techniques and their associated bioinformatic data processing tools have been reviewed elsewhere [43–47]. Here we focus on the key information that can be obtained from each –omic approach, with a particular focus on those that characterize functional and metabolic activities; namely metatranscriptomics, metaproteomics, and metabolomics.
Table 1.
Challenges for metatranscriptomics, metaproteomics, and metabolomics in microbiome studies
| Metatranscriptomics, metaproteomics, and metabolomics each have their own shortcomings. Metatranscriptomic experiments rely on obtaining sufficient high-quality RNA from the sample source; something which can be quite challenging due to the ubiquitous presence of RNases in host-derived samples. In addition, metatranscriptomic sequencing can often become saturated with reads from less-informative, but highly expressed transcripts (i.e., ribosomal proteins, translation factors, major outer membrane proteins) from the most abundant microbes present, obscuring the detection of functionally important, but less abundant transcripts/proteins. Therefore, the quality of RNA as well as the depth of measurement is important aspects that need to be evaluated or considered in metatranscriptomics. | |
| Compared to metagenomics and metatranscriptomics, metaproteomics has a lower depth of measurement and can only capture 10–20% of expressed proteins in human gut microbiomes [27, 33, 34]. MS spectra can also be saturated with the highly abundant proteins from dominant species, and this issue is unlikely to be resolved by increasing the speed or time of MS scanning. However, applying off-line protein/peptide separation (such as using sodium dodecyl sulfate polyacrylamide gel electrophoresis) or targeted enrichment strategies (such as using activity-based probes [35]) may to some extent address this limitation. In addition, as metaproteomics is still in its infancy for the study of microbiomes, there is still a lack of universal guidelines and protocols for properly performing metaproteomic experiments and interpreting metaproteomic results. Therefore, careful considerations should be made for sample preparation, MS measurement, bioinformatic workflows, and data reporting (readers are directed to this perspective article for more details [36]). | |
| The major challenge for metabolomics in microbiome studies is the difficulty to distinguish host- and microbiome-origin metabolites and directly link metabolites to specific taxa [37]. One feasible approach to address this issue is to identify co-variations between metabolites and microbial species, which is indicative for species-specific metabolite production, through integrative analysis of microbiota compositions with metabolite profiles [38–41]. Other approaches, such as protein stable-isotope probing (protein-SIP) [42], can also link the metabolism of a specific substrate to phylogenetic information by monitoring the isotopes in microbial protein sequences with mass spectrometers and may eventually aid in microbiome metabolic reconstructions. |
Fig. 1.
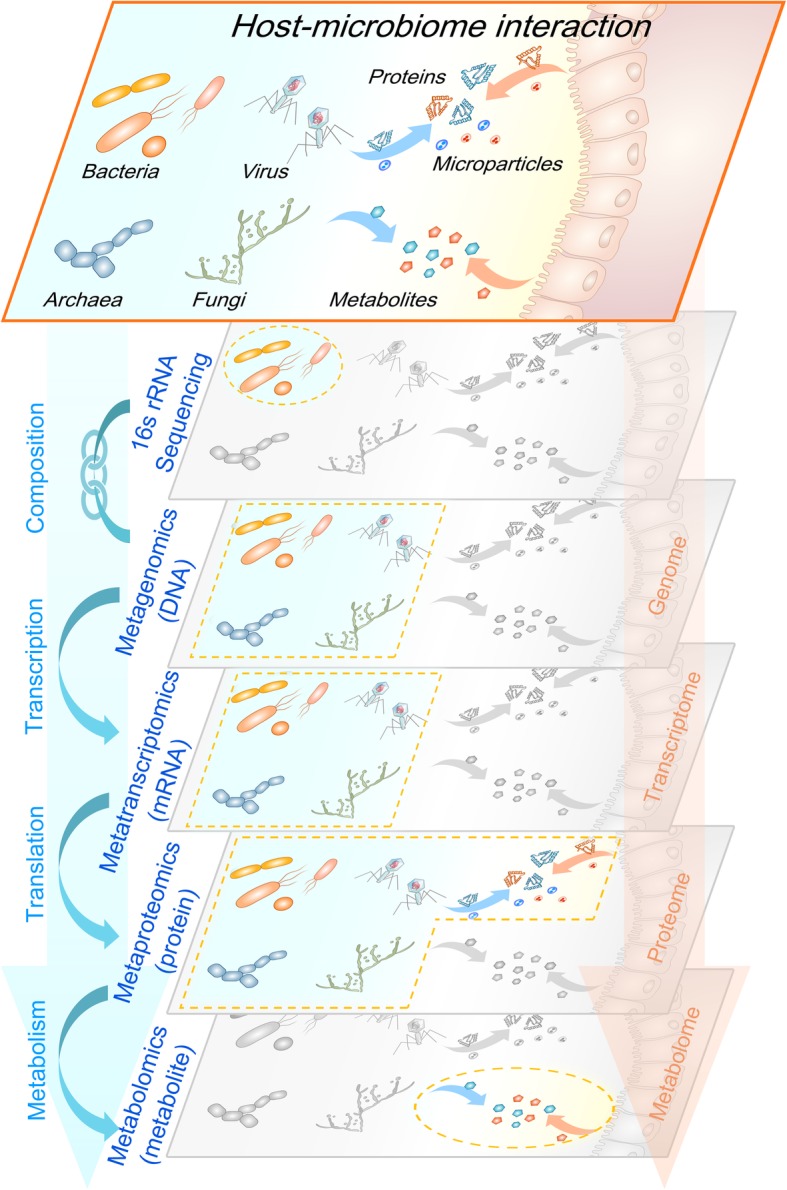
Meta-omics approaches for the study of host-associated microbiomes. Each meta-omics approach reveals different layers of information in the intestinal eco-systems
The composition and functional capacity of human microbiomes have been well characterized using next-generation sequencing techniques, such as amplicon sequencing (e.g., 16S rRNA gene) and shotgun metagenomics. In particular, shotgun metagenomics is now widely applied in microbiome studies, providing valuable functional information down to the strain level and for all types of microorganisms (including archaea, fungi, and viruses) [48–52]. More recently, metagenomic sequencing of hundreds to tens of thousands of samples was carried out in large scale projects studying the role of the microbiome in human disease, including studies on early-onset type 1 diabetes (T1D) [53, 54], IBD [55], pre-diabetes [56], and colorectal cancer [57, 58]. In particular, these studies employed longitudinal and/or multi-omic experimental designs, which enabled better characterization of the dynamic changes and functional activities of the microbiome during disease progression. Despite their costs and technical challenges, longitudinal and multi-omic experimental designs are becoming indispensable for unravelling host-microbiome interactions during disease and for assessing causality in clinical microbiome investigations. A beneficial spin-off from these massive metagenomic sequencing projects has been their deposition into easily accessible databases. This has allowed researchers to leverage these datasets to create reference databases for future studies. Examples include a database with > 150,000 microbial reference genomes [59] and a human gut microbial gene catalog database consisting of > 9,800,000 genes [3]. These are valuable resources for functional studies of the human microbiome using metatranscriptomic and metaproteomic approaches.
The presence of a gene does not necessarily mean the gene is expressed. Thus the direct measurement of transcripts or proteins using metatranscriptomics or metaproteomics, respectively, is becoming an important complementary approach for metagenomics. Metatranscriptomics employs similar analytical approaches (e.g., nucleic acid sequencing) as metagenomics. Accordingly, the software tools employed for metagenomics are often adapted for metatranscriptomic data processing [29]. Using the same tools for both metatranscriptomics and metagenomics provides a straightforward route for their integration in microbiome studies [29, 33, 60]. Their combination not only improves microbial genome assembly and gene prediction [33], but can also enable the identification of genes which are induced/repressed under specific conditions. In addition, identifying genomes with active transcription can distinguish metabolically active microbes from inert or dead microbes [33]. In contrast with metagenomics and metatranscriptomics, metaproteomics measures expressed proteins, the basic functional unit for most cellular biological processes, using high-resolution mass spectrometry (MS). Metaproteomics should in principle provide superior insight into gut microbial functionality as compared with metatranscriptomics, since not all transcripts are subsequently translated into proteins. In the past, metaproteomics was rarely employed in gut microbiome studies, at least in part, due to the lack of efficient bioinformatic tools and low protein measurement depth [61]. Fortunately, the recent development of metaproteomic data processing tools, such as MetaLab [62], MetaProteomeAnalyzer [63], and Galaxy-P [64], has greatly advanced our ability to analyze metaproteomic data (readers are directed to extensive reviews for more information [61, 65]). This has enabled deep characterization of microbiome protein compositions, with some reports quantifying > 50,000 unique microbial protein groups in a single study [38, 66]. It is noteworthy that metaproteomics identifies and quantifies proteins from all organisms present within the microbiome, regardless of their phylogenetic origin, and can quantify host proteins as well [27, 33, 67]. This feature is of particular importance when studying host-associated microbiomes in vivo and can uncover important players (e.g., extracellular vesicles [27]) mediating host-microbiome interactions.
In addition to the microbiome’s functional activity, a further goal of microbiome research is to measure metabolic outcomes. Metabolomics directly measures the metabolites present in the intestine using analytical techniques such as nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS). Given the higher sensitivity of MS compared to NMR [68], the field of metabolomics has increasingly shifted to MS-based approaches. Readers are directed to previous reviews for more details on methodologies for fecal metabolomics [69–71]. Fecal water is among the most commonly used sample types for fecal metabolomic studies, although there are increasing numbers of studies examining intracellular microbial metabolites as well [38, 72]. The fecal metabolome is often regarded as an endpoint readout of biological processes originating from the gut microbiome [73]. Identified metabolites in fecal metabolomics can include those derived from the microbiota (e.g., lipopolysaccharide and butyrate) or the host (e.g., anti-microbial peptides). These metabolites can often act as signalling markers that allow for communication between the host and microbiome. In fact, many metabolites in the intestine are produced by co-metabolisms of the host and their microbiome, and intestinal metabolic imbalances have been commonly implicated in disease development [37, 74, 75]. Profiling metabolomes in fecal samples or targeted analysis of drug metabolites during drug treatment can provide valuable information on bi-directional drug-microbiome interactions that may contribute to drug pharmacodynamics, pharmacokinetics, or toxicity.
Integrative multi-omics for studying the host-microbiome interactions
Integrating the data from multi-omic approaches provides additional insight into microbiome functions. For example, integrating metagenomics and metatranscriptomics enables the calculation of transcript/gene ratios, which is indicative of gene transcriptional activation or repression. Metaproteomics is also frequently integrated with metagenomics for either facilitating protein identification from MS spectra using a matched metagenome database search strategy or for calculating microbiome protein expression [34, 76, 77]. Metabolomics is increasingly integrated with metagenomics for identifying co-variation patterns between metabolites and microbiota composition/function and for characterizing phylogenetic specific contributions to metabolite production [39, 40, 78–81]. An excellent example of an integrative multi-omics study was carried out by Heintz-Buschart et al. [33], who characterized microbiome functions in patients with type 1 diabetes (T1D) using metagenomics, metatranscriptomics, and metaproteomics. Their study identified various differentially abundant microbial transcripts encoded by microbes whose abundance was unresponsive to T1D. In addition, the metaproteomic profiling identified several fecal human proteins that correlated with microbial functional profiles. These findings highlight the usefulness of integrating functional meta-omics approaches for host-microbiome interaction studies.
Unfortunately, integrating multi-omic datasets is not a trivial task due to the increased complexity and diversity of the collected data (e.g., data structure, measurement depth, potential errors, etc.). This integration is increasingly reliant on efficient bioinformatic tools, advanced statistical methods, such as multivariate statistics and machine-learning approaches (readers are directed to the following representative reviews for more details [45–47, 82–84]). Correlation analysis, such as Pearson’s or Spearman’s rank correlation, and correlation-based network analysis are the most straightforward and commonly used approaches for multi-omics data integration. Multivariate statistical methods, such as partial least squares regression, orthogonal partial least squares and nonmetric multidimensional scaling [39–41], have also been applied to identify key features that contribute to the association of two or more –omics data sets. The similarity/correlation between multi-omic datasets can be evaluated using statistical approaches such as Procrustes analysis and multiple co-inertia analysis [40, 78, 80]. A further goal of multi-omics data integration is to generate and validate microbiome metabolic networks/models. Although this is still challenging, promising steps forward have been made, including the generation of > 700 genome-scale metabolic reconstructions [85], the development of tools for microbiome metabolic modeling/prediction [86, 87], and the establishment of inter-species metabolic network databases [88]. Recently, several microbiome studies have also taken advantage of machine learning methods, such as random forest algorithms, to either differentiate between health and disease states or identify features that predict clinical outcomes [89–92]. The application of advanced machine-learning approaches will likely revolutionize our ability to integrate and interpret multi-omics data [93, 94]. These future integrations may include the generation of microbiome-scale metabolic reconstructions, which would further push the frontiers of translational microbiome research.
In summary, although multi-omics data integration is still challenging, the integration of multiple meta-omics datasets provides a promising approach to comprehensively characterize the composition, functional, and metabolic activity of microbiomes. This is of particular importance for microbiome research to be translated into clinical applications. The chronic human diseases, such as T1D, diabetes, or IBD, that are often associated with microbiome alterations, are unlikely to be caused by a single bacterium or a single protein/metabolite. Therefore, we anticipate that meta-omics approaches, along with their decreased costs and increased throughput, will become a first-choice analytical method for microbiome-based clinical or pharmaceutical practice.
Meta-omics in the study of drug-microbiome interactions
The responses of microbiome to external treatments, such as diet and drugs, are usually dependent on the initial microbiome composition, which is highly variable between individuals. A holistic understanding of the interactions between drugs and microbiomes using meta-omics approaches would be helpful in predicting the outcomes of drug treatment or guiding the usage of drugs. Many clinically prescribed drugs can be metabolized by the gut microbiome and/or modulate gut microbiome composition; these drug-microbiome interactions can thus affect drug efficacy and/or toxicity [17, 95, 96]. Examples of these include antibiotics (which would be expected to modulate the gut microbiome) [97], as well as host-targeting drugs, such as metformin and nonsteroidal anti-inflammatory drugs [14, 96]. A recent study by Maier et al. screened > 1000 marketed drugs against 40 human gut microbial strains and found that 24% of the non-antibiotic drugs could inhibit specific gut bacterial species [16]. Zimmermann et al. also reported that around two-thirds of their selected 271 oral drugs were metabolized by at least one of their 76 cultured human gut bacterial strains [98]. These findings provide further evidence for the widespread existence of drug-microbiome interactions in marketed drugs and the importance of evaluating their effects on entire microbiomes. Unfortunately, the detailed interactions between human gut microbiomes and these drugs are still largely unknown, and fewer than 100 drugs have been recorded in drug-microbiome interaction databases [99]. In addition, the few interactions that are recorded often provide little insight as to whether the drug-microbiome interactions may lead to positive, negligible or even negative outcomes for the host. Therefore, the development of high-throughput platforms to rapidly characterize drug-microbiome interactions is urgently needed. Most previous drug-microbiome interaction studies have been performed using animal models, which are time consuming, expensive and not always representative of what will occur in humans. Ex vivo culturing of entire human microbiomes when combined with meta-omics analysis provides a promising way to develop microbiome assays for rapidly screening drug-microbiome interactions against individual microbiomes.
The technology for high-throughput microbiome assays is often adapted from current cell culture-based, host-targeting drug screening platforms. However, there are several challenges inherent to microbiome assays and include (1) the representability of the cultured microbiome, (2) the throughput of microbiome culturing, and (3) the throughput of data generation and processing. Over the past few years, new developments have improved our ability to culture entire human gut microbiota. Lagier et al. reported the culture of > 1000 species from human gut microbiome samples and identified a set of 70 best culture conditions for growing gut microbiota [7]. Fenn et al. utilized a co-culture technique to culture human gut microbiota and identified an essential nutrient (menaquinone), which may help better maintain microbiomes in vitro [100]. Li et al. recently proposed an orthogonal experimental design to rapidly determine key factors in culture media that impact microbiome composition/function and thereby optimize in vitro culture media for specific microbiomes [101]. In addition to static batch culturing systems, microfluidic devices for continuous culturing, such as HuMiX and SHIME [30, 31], have also been developed. Continuous flow devices enable better simulation of in vivo intestinal conditions for the growth of microbiome; however, they are more expensive and cannot be easily adapted for high throughput culturing of many different microbiomes/conditions in a short timeframe. As such, most high throughput screening microbiome assays use batch culturing approaches. Rapid generation of microbiome data using a single –omics approach is now also feasible as technologies and bioinformatic tools for meta-omics analysis are available and being continuously optimized (see above). Multiple –omics approaches can be simultaneously applied to drug-microbiome screening; however, the throughput will be reduced, and costs will be greatly increased. As such, a two-stage approach consisting of an initial rapid screening with a single –omics approach and a second stage consisting of multi-omics characterization for the selected hits is currently more practical to enable high throughput screening and characterization (Fig. 2). Good examples of first step screening tools are 16S rRNA gene sequencing, due to its lower cost, or single-shot metaproteomics given that it provides information on microbiome biomass, composition, and function.
Fig. 2.

Framework of an ex vivo assay for screening drug-microbiome interactions. The individual’s microbiome is cultured and treated with drugs in anaerobic conditions simulating the in vivo environment. The cultured samples can then be analysed by 16S rRNA gene sequencing or single-shot metaproteomics to rapidly identify hit compounds taking advantage of well-established bioinformatic platforms. Detailed bidirectional drug-microbiome interactions for hit compounds can then be further evaluated with integrative multi-omics approaches
We have recently reported a proof-of-concept high throughput ex vivo microbiome assay, termed rapid assay of individual’s microbiome (RapidAIM) that is based on culturing an individual’s entire microbiome followed by metaproteomic measurement [32]. We showed that RapidAIM maintained microbiome structure and functional profiles for up to 48 hours and recapitulated known in vivo drug effects on microbiomes. We evaluated the responses of individual microbiomes against 43 compounds and found that 27 compounds had significant effects on microbiome composition and function. Chankhamjon et al. adopted a similar microbiome batch culture platform for the rapid screening and detailed characterization of microbiome-derived drug metabolism [102]. Briefly, a healthy microbiome was co-cultured with a library of drugs and the drug metabolites were analysed using HPLC-MS. Among the > 500 oral drugs tested, they discovered that 13% could be metabolized by the microbiome [102]. These studies demonstrate the feasibility of applying high throughput microbiome assays for assessing bi-directional interactions between microbiome and clinically used drugs. The extensive screening of drug-microbiome interactions may also represent an economic way to discover currently approved drugs which have impacts on the microbiome and potentially repurpose these drugs for microbiome-targeted disease therapy.
Meta-omics at the interface of microbiome and precision medicine
Precision medicine is an emerging concept for health management given that responses to therapeutic interventions usually vary between individuals. In the past, these variations were assumed to be simply caused by subtle differences between patient genetic backgrounds or due to epigenetic factors controlling host gene expression. For example, genomics-based precision medicine has often been applied in cancer therapy [103–106]. However, it should be noted that many cancer therapeutics could also alter the gut microbiome [16, 107, 108]. More recently, variations in patient responses to cancer immuno- and chemo-therapies were linked to inter-individual differences in gut microbiomes [74, 109–114]. These findings suggest an opportunity to further optimize disease therapies through microbiome-informed patient stratification, through personalized treatment decisions and/or through direct manipulation of patient microbiomes (Fig. 3). They also highlight the importance of including microbiomes into the framework of precision medicine [19].
Fig. 3.

Introducing microbiomes into clinical practice for precision medicine. The profiles of individual patient microbiomes are analyzed with meta-omics, which allow for patients to be classified into sub-groups, i.e., responders vs. non-responders to treatments (a). The in vivo response of an individual’s microbiome to drugs can also be predicted with ex vivo microbiome assays, allowing the selection of the best drugs or adjuvant treatments for different patients (b). Finally, health and disease management could be carried out by precisely manipulating of the microbiome through supplementing commensal bacteria, engineered bacteria, microbiome-targeted drugs or bacteriophages (c)
Patient stratification based on microbiome profiling
One important goal of precision medicine is to identify biomarkers for stratifying patients into subgroups that are likely to be responsive (or unresponsive) to a given treatment [115]. As mentioned above, heterogeneous responses of patients to treatment may be due in part to differences in their gut microbiomes. Therefore, a prior understanding of an individual’s microbiome may help predict treatment outcomes and/or suggest optimal therapeutic strategies (Fig. 3a). Gu et al. demonstrated that the gut microbiome of new-onset type 2 diabetes (T2D) patients could be classified into two clusters, namely cluster P (dominated by Prevotella) and B (dominated by Bacteroides), and found that cluster P patients had greater metabolic improvement after 3-month acarbose treatment as compared to cluster B patients [116]. In a prospective wellness study, Price et al. [117] illustrated the use of dense and dynamic personal data clouds, including host genetic traits, clinical analytes, metabolites, proteomes, and microbiomes, to identify candidate markers for predicting the transition from health to disease. In the disease-prone subgroup, life style interventions stemming from these personalized-data biomarkers successfully improved their health status [117]. In cancer therapy, immune checkpoint inhibitors targeting the programmed death 1 (PD-1) protein are important therapeutics but are only effective in a subset of patients. Recent studies have shown that patient’s failure to respond to anti-PD-1 therapy could be attributed to the absence or under-representation of certain immune-regulating bacterial species in gut, namely Akkermansia muciniphila, Faecalibacterium, and Bifidobacterium longum [109–111]. These findings suggest that quantifying these commensals in patient fecal samples may help predict therapeutic outcomes and stratify patients into potential responders or non-responders to PD-1 blockade.
Ex vivo microbiome assays for guiding treatment decisions
The gut microbiota is a highly diverse microbial community with high inter-individual variability, making in vivo drug-microbiome interactions complex and an individual’s response to drug treatment difficult to predict. In addition, as mentioned above, there is no knowledge on drug-microbiome interactions for the majority of clinically prescribed drugs. A prior understanding or prediction of drug-microbiome interactions in a patient through microbiome assays would be invaluable for optimizing the therapeutic outcomes in diseases that are known to be associated with gut microbial alterations. This would allow for patients to be prescribed the most effective drug for treating their disease (Fig. 3b). For example, digoxin is a commonly used cardiac drug and can be converted into its inactive form, dihydrodigoxin, by specific strains of the intestinal bacterium Eggerthella lenta, and this has been suggested to contribute to digoxin’s varied bioavailability among individuals [118, 119]. However, the extent of digoxin inactivation is also dependent on the presence of other gut microbes [118], indicating that a single biomarker using the presence of E. lenta species may not be sufficient for patient stratification. Instead, culture of digoxin with an ex vivo microbiome followed by metabolite measurement can more accurately predict the extent of digoxin inactivation and thereby guide the decision on whether adjuvant intervention, such as arginine supplements or antibiotics, is needed [118, 120]. In addition, for diseases with multiple drug candidates, such as IBD [121], culturing a set of candidate drugs with an individual’s ex vivo microbiome may help select the most likely effective drug candidate for treating each patient’s disease.
Targeted manipulation of microbiome for precision disease treatment
Although the gut microbiome has long been considered as a potential target for disease treatment [13] and an effective microbiome-targeted dietary intervention approach has been demonstrated [9], commercially available targeted therapeutics for precise modulation of microbiomes are still lacking. However, our understanding of the mechanisms underlying host-microbiome interactions is growing rapidly, and new potential targets (e.g., specific microbial species or metabolic pathways) in the microbiome are being identified. It may soon be feasible to precisely manipulate the microbiome through either supplementation of beneficial species (such as A. muciniphila) [111], engineered probiotics/commensals [122], prebiotics [9], bacteriophages [123], or highly selective drugs [124] (Fig. 3c). For example, Zhu et al. recently reported that tungstate can specifically inhibit molybdenum-cofactor-dependent gut microbial respiratory pathways under inflammatory conditions, which ameliorates intestinal colitis and restores gut microbial homeostasis in a mouse model of colitis [124]. More recently, Ho et al. reported that a genetically modified E. coli strain, which has selective affinity to cancer cells and secrets myrosinase for converting vegetable derived glucosinolate into anti-cancer compounds, effectively prevented the development of cancer in mice receiving a cruciferous vegetable diet [125]. Dietary intervention is another safe and promising approach for manipulating the microbiome. Zhao et al. utilized a specialized diet to promote the growth of a group of short-chain fatty acid-producing bacteria in the gut of T2D patients, which was proposed to have contributed to improved glucose homeostasis in these patients [9]. Along with the development of sophisticated tools for manipulating microbial genetics [122, 126], it is becoming feasible for targeted modulation of specific microbial metabolic pathways or species in microbiome, which further lays the foundation for future microbiome-targeted therapies.
Conclusions
The ultimate goal of human microbiome research is to facilitate health and disease management. Gut microbiome alterations have been associated with an increasing list of diseases, and selectively modifying the gut microbiota has been shown to alleviate the development of disease, including diabetes and colitis. These achievements highlight the importance of introducing the microbiome into the precision medicine framework, through either microbiome-guided patient stratification or interventions that specifically target microbial species/pathways. However, it is still a challenge to rapidly identify specific, actionable targets within microbiomes. Fortunately, the addition of metatranscriptomics, metaproteomics, and metabolomics to metagenomics is enhancing our functional understanding of the microbiome. Although more powerful and convenient bioinformatic tools are still needed, integrative functional meta-omics is becoming one of the most important approaches for dissecting microbial metabolic pathways in microbiomes. In addition, the development of microbiome-targeted drugs is also challenging. Therefore, efforts are underway to develop new ex vivo assays targeting panels of individual bacteria, simple microbial communities, or entire microbiomes. These are likely to rapidly increase our understanding of how microbiomes interact with drugs, food components, and natural compounds. Ex vivo microbiome assays will likely be useful in precision medicine by allowing individual microbiomes to be screened against panels of drugs/compounds to select the most efficient treatment.
Acknowledgements
DF acknowledges a Distinguished Research Chair at the University of Ottawa.
Abbreviation
- IBD
Inflammatory bowel disease
- MS
Mass spectrometry
- NMR
Nuclear magnetic resonance
- T1D
Type 1 diabetes
- T2D
Type 2 diabetes
- PD-1
Programmed death 1
Author’s contributions
XZ and DF drafted the initial manuscript, with further review and editing done by JB and AS. LL and XZ generated the figures. All authors read, edited, and approved the final manuscript.
Funding
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Government of Canada through Genome Canada and the Ontario Genomics Institute (OGI-114, OGI-149 and OGI-156), CIHR grant number GPH-129340, MOP-114872 and ECD-144627, and the Ontario Ministry of Economic Development and Innovation (REG1-4450 and ORF-LSARP-13440).
Availability of data and materials
Not applicable
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
DF and AS are co-founders of Biotagenics and MedBiome, clinical microbiomics companies. The remaining authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xu Zhang, Email: xzhan7@uottawa.ca.
Leyuan Li, Email: lli8@uottawa.ca.
James Butcher, Email: jbutcher@uottawa.ca.
Alain Stintzi, Email: astintzi@uottawa.ca.
Daniel Figeys, Email: dfigeys@uottawa.ca.
References
- 1.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010:464, 59–5. [DOI] [PMC free article] [PubMed]
- 2.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533. doi: 10.1371/journal.pbio.1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 4.Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012;13:260–270. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lagier JC, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P, et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol. 2016;1:16203. doi: 10.1038/nmicrobiol.2016.203. [DOI] [PubMed] [Google Scholar]
- 8.Marchesi JR, Ravel J. The vocabulary of microbiome research: a proposal. Microbiome. 2015;3:31. doi: 10.1186/s40168-015-0094-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359:1151–1156. doi: 10.1126/science.aao5774. [DOI] [PubMed] [Google Scholar]
- 10.Fei N, Zhao L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2013;7:880–884. doi: 10.1038/ismej.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neville BA, Forster SC, Lawley TD. Commensal Koch’s postulates: establishing causation in human microbiota research. Curr Opin Microbiol. 2018;42:47–52. doi: 10.1016/j.mib.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017;550:61–66. doi: 10.1038/nature23889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia W, Li H, Zhao L, Nicholson JK. Gut microbiota: a potential new territory for drug targeting. Nat Rev Drug Discov. 2008;7:123–129. doi: 10.1038/nrd2505. [DOI] [PubMed] [Google Scholar]
- 14.Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Manneras-Holm L, et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med. 2017;23:850–858. doi: 10.1038/nm.4345. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Zhao Y, Xu J, Xue Z, Zhang M, Pang X, et al. Modulation of gut microbiota by berberine and metformin during the treatment of high-fat diet-induced obesity in rats. Sci Rep. 2015;5:14405. doi: 10.1038/srep14405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018. [DOI] [PMC free article] [PubMed]
- 17.Wilson ID, Nicholson JK. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 2017;179:204–222. doi: 10.1016/j.trsl.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spanogiannopoulos P, Bess EN, Carmody RN, Turnbaugh PJ. The microbial pharmacists within us: a metagenomic view of xenobiotic metabolism. Nat Rev Microbiol. 2016;14:273–287. doi: 10.1038/nrmicro.2016.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuntz TM, Gilbert JA. Introducing the microbiome into precision medicine. Trends Pharmacol Sci. 2017;38:81–91. doi: 10.1016/j.tips.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Janssens Y, Nielandt J, Bronselaer A, Debunne N, Verbeke F, Wynendaele E, et al. Disbiome database: linking the microbiome to disease. BMC Microbiol. 2018;18:50. doi: 10.1186/s12866-018-1197-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma W, Zhang L, Zeng P, Huang C, Li J, Geng B, et al. An analysis of human microbe-disease associations. Brief Bioinform. 2017;18:85–97. doi: 10.1093/bib/bbw005. [DOI] [PubMed] [Google Scholar]
- 22.Noronha A, Modamio J, Jarosz Y, Guerard E, Sompairac N, Preciat G, et al. The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019;47:D614–D624. doi: 10.1093/nar/gkz323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Human Microbiome Project C Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, Ober C, et al. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe. 2016;19:731–743. doi: 10.1016/j.chom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolde R, Franzosa EA, Rahnavard G, Hall AB, Vlamakis H, Stevens C, et al. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med. 2018;10:6. doi: 10.1186/s13073-018-0515-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Deeke SA, Ning Z, Starr AE, Butcher J, Li J, et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat Commun. 2018;9:2873. doi: 10.1038/s41467-018-05357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schirmer M, Franzosa EA, Lloyd-Price J, Mclver LJ, Xavier R, et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol. 2018;3:337. doi: 10.1038/s41564-017-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abu-Ali GS, Mehta RS, Lloyd-Price J, Mallick H, Branck T, Ivey KL, et al. Metatranscriptome of human faecal microbial communities in a cohort of adult men. Nat Microbiol. 2018;3:356–366. doi: 10.1038/s41564-017-0084-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah P, Fritz JV, Glaab E, Desai MS, Greenhalgh K, Frachet A, et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat Commun. 2016;7:11535. doi: 10.1038/ncomms11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molly K, Vande Woestyne M, Verstraete W. Development of a 5-step multi-chamber reactor as a simulation of the human intestinal microbial ecosystem. Appl Microbiol Biotechnol. 1993;39:254–258. doi: 10.1007/BF00228615. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Ning Z, Zhang X, Mayne J, Cheng K, Stintzi A, et al. RapidAIM: A culture- and metaproteomics-based rapid assay of individual microbiome responses to drugs. bioRxiv 2019:543256. [DOI] [PMC free article] [PubMed]
- 33.Heintz-Buschart A, May P, Laczny CC, Lebrun LA, Bellora C, Krishna A, et al. Integrated multi-omics of the human gut microbiome in a case study of familial type 1 diabetes. Nat Microbiol. 2016;2:16180. doi: 10.1038/nmicrobiol.2016.180. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, Ning Z, Mayne J, Moore JI, Li J, Butcher J, et al. MetaPro-IQ: a universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome. 2016;4:31. doi: 10.1186/s40168-016-0176-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayers MD, Moon C, Stupp GS, Su AI, Wolan DW. Quantitative metaproteomics and activity-based probe enrichment reveals significant alterations in protein expression from a mouse model of inflammatory bowel disease. J Proteome Res. 2017;16:1014–1026. doi: 10.1021/acs.jproteome.6b00938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Figeys D. Perspective and guidelines for metaproteomics in microbiome studies. J Proteome Res. 2019;18:2370–2380. doi: 10.1021/acs.jproteome.9b00054. [DOI] [PubMed] [Google Scholar]
- 37.Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- 38.Maier TV, Lucio M, Lee LH, VerBerkmoes NC, Brislawn CJ, Bernhardt J, et al. Impact of dietary resistant starch on the human gut microbiome, metaproteome, and metabolome. MBio. 2017;8. [DOI] [PMC free article] [PubMed]
- 39.Jansson J, Willing B, Lucio M, Fekete A, Dicksved J, Halfvarson J, et al. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS One. 2009;4:e6386. doi: 10.1371/journal.pone.0006386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C, Yin A, Li H, Wang R, Wu G, Shen J, et al. Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine. 2015;2:968–984. doi: 10.1016/j.ebiom.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antharam VC, McEwen DC, Garrett TJ, Dossey AT, Li EC, Kozlov AN, et al. An integrated metabolomic and microbiome analysis identified specific gut microbiota associated with fecal cholesterol and coprostanol in clostridium difficile infection. PLoS One. 2016;11:e0148824. doi: 10.1371/journal.pone.0148824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jehmlich N, Schmidt F, Taubert M, Seifert J, Bastida F, von Bergen M, et al. Protein-based stable isotope probing. Nat Protoc. 2010;5:1957–1966. doi: 10.1038/nprot.2010.166. [DOI] [PubMed] [Google Scholar]
- 43.Morgan XC, Huttenhower C. Meta'omic analytic techniques for studying the intestinal microbiome. Gastroenterology. 2014;146:1437–1448. doi: 10.1053/j.gastro.2014.01.049. [DOI] [PubMed] [Google Scholar]
- 44.Segata N, Boernigen D, Tickle TL, Morgan XC, Garrett WS, Huttenhower C. Computational meta'omics for microbial community studies. Mol Syst Biol. 2013;9:666. doi: 10.1038/msb.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valles-Colomer M, Darzi Y, Vieira-Silva S, Falony G, Raes J, Joossens M. Meta-omics in inflammatory bowel disease research: applications, challenges, and guidelines. J Crohns Colitis. 2016;10:735–746. doi: 10.1093/ecco-jcc/jjw024. [DOI] [PubMed] [Google Scholar]
- 46.Mallick H, Ma S, Franzosa EA, Vatanen T, Morgan XC, Huttenhower C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017;18:228. doi: 10.1186/s13059-017-1359-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, et al. Best practices for analysing microbiomes. Nat Rev Microbiol. 2018;16:410–422. doi: 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- 48.Costea PI, Coelho LP, Sunagawa S, Munch R, Huerta-Cepas J, Forslund K, et al. Subspecies in the global human gut microbiome. Mol Syst Biol. 2017;13:960. doi: 10.15252/msb.20177589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 50.Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, et al. Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe. 2015;18:489–500. doi: 10.1016/j.chom.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart CJ, Ajami NJ, O'Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2018;562:583–588. doi: 10.1038/s41586-018-0617-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature. 2018;562:589–594. doi: 10.1038/s41586-018-0620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou W, Sailani MR, Contrepois K, Zhou Y, Ahadi S, Leopold SR, et al. Longitudinal multi-omics of host–microbe dynamics in prediabetes. Nature. 2019;569:663–671. doi: 10.1038/s41586-019-1236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat Med. 2019;25:667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat Med. 2019;25:679–689. doi: 10.1038/s41591-019-0406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell. 2019;176:649–662. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Franzosa EA, Morgan XC, Segata N, Waldron L, Reyes J, Earl AM, et al. Relating the metatranscriptome and metagenome of the human gut. Proc Natl Acad Sci U S A. 2014;111:E2329–E2338. doi: 10.1073/pnas.1319284111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heyer R, Schallert K, Zoun R, Becher B, Saake G, Benndorf D. Challenges and perspectives of metaproteomic data analysis. J Biotechnol. 2017;261:24–36. doi: 10.1016/j.jbiotec.2017.06.1201. [DOI] [PubMed] [Google Scholar]
- 62.Cheng K, Ning Z, Zhang X, Li L, Liao B, Mayne J, et al. MetaLab: an automated pipeline for metaproteomic data analysis. Microbiome. 2017;5:157. doi: 10.1186/s40168-017-0375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muth T, Kohrs F, Heyer R, Benndorf D, Rapp E, Reichl U, et al. MPA portable: a stand-alone software package for analyzing metaproteome samples on the go. Anal Chem. 2018;90:685–689. doi: 10.1021/acs.analchem.7b03544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jagtap PD, Blakely A, Murray K, Stewart S, Kooren J, Johnson JE, et al. Metaproteomic analysis using the Galaxy framework. Proteomics. 2015;15:3553–3565. doi: 10.1002/pmic.201500074. [DOI] [PubMed] [Google Scholar]
- 65.Starr AE, Deeke SA, Li L, Zhang X, Daoud R, Ryan J, et al. Proteomic and metaproteomic approaches to understand host-microbe interactions. Anal Chem. 2018;90:86–109. doi: 10.1021/acs.analchem.7b04340. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Chen W, Ning Z, Mayne J, Mack D, Stintzi A, et al. Deep metaproteomics approach for the study of human microbiomes. Anal Chem. 2017;89:9407–9415. doi: 10.1021/acs.analchem.7b02224. [DOI] [PubMed] [Google Scholar]
- 67.Gavin PG, Mullaney JA, Loo D, Cao KL, Gottlieb PA, Hill MM, et al. Intestinal metaproteomics reveals host-microbiota interactions in subjects at risk for type 1 diabetes. Diabetes Care. 2018;41:2178–2186. doi: 10.2337/dc18-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emwas AH. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol Biol. 2015;1277:161–193. doi: 10.1007/978-1-4939-2377-9_13. [DOI] [PubMed] [Google Scholar]
- 69.Karu N, Deng L, Slae M, Guo AC, Sajed T, Huynh H, et al. A review on human fecal metabolomics: methods, applications and the human fecal metabolome database. Anal Chim Acta. 2018;1030:1–24. doi: 10.1016/j.aca.2018.05.031. [DOI] [PubMed] [Google Scholar]
- 70.Lamichhane S, Sen P, Dickens AM, Oresic M, Bertram HC. Gut metabolome meets microbiome: a methodological perspective to understand the relationship between host and microbe. Methods. 2018;149:3–12. doi: 10.1016/j.ymeth.2018.04.029. [DOI] [PubMed] [Google Scholar]
- 71.Xu J, Zhang Q-F, Zheng J, Yuan B-F, Feng Y-Q. Mass spectrometry-based fecal metabolome analysis. TrAC Trends in Analytical Chemistry 2019.
- 72.Daniel H, Gholami AM, Berry D, Desmarchelier C, Hahne H, Loh G, et al. High-fat diet alters gut microbiota physiology in mice. ISME J. 2014;8:295–308. doi: 10.1038/ismej.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zierer J, Jackson MA, Kastenmuller G, Mangino M, Long T, Telenti A, et al. The fecal metabolome as a functional readout of the gut microbiome. Nat Genet. 2018;50:790–795. doi: 10.1038/s41588-018-0135-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scott TA, Quintaneiro LM, Norvaisas P, Lui PP, Wilson MP, Leung KY, et al. Host-microbe co-metabolism dictates cancer drug efficacy in C. elegans. Cell. 2017;169:442–456. doi: 10.1016/j.cell.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360. [DOI] [PMC free article] [PubMed]
- 76.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tanca A, Abbondio M, Palomba A, Fraumene C, Manghina V, Cucca F, et al. Potential and active functions in the gut microbiota of a healthy human cohort. Microbiome. 2017;5:79. doi: 10.1186/s40168-017-0293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McHardy IH, Goudarzi M, Tong M, Ruegger PM, Schwager E, Weger JR, et al. Integrative analysis of the microbiome and metabolome of the human intestinal mucosal surface reveals exquisite inter-relationships. Microbiome. 2013;1:17. doi: 10.1186/2049-2618-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blanco-Míguez Aitor, Fdez-Riverola Florentino, Sánchez Borja, Lourenço Anália. Resources and tools for the high-throughput, multi-omic study of intestinal microbiota. Briefings in Bioinformatics. 2017;20(3):1032–1056. doi: 10.1093/bib/bbx156. [DOI] [PubMed] [Google Scholar]
- 80.Ishii Chiharu, Nakanishi Yumiko, Murakami Shinnosuke, Nozu Ryoko, Ueno Masami, Hioki Kyoji, Aw Wanping, Hirayama Akiyoshi, Soga Tomoyoshi, Ito Mamoru, Tomita Masaru, Fukuda Shinji. A Metabologenomic Approach Reveals Changes in the Intestinal Environment of Mice Fed on American Diet. International Journal of Molecular Sciences. 2018;19(12):4079. doi: 10.3390/ijms19124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zarrinpar A, Chaix A, Yooseph S, Panda S. Diet and feeding pattern affect the diurnal dynamics of the gut microbiome. Cell Metab. 2014;20:1006–1017. doi: 10.1016/j.cmet.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Franzosa EA, Hsu T, Sirota-Madi A, Shafquat A, Abu-Ali G, Morgan XC, et al. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat Rev Microbiol. 2015;13:360–372. doi: 10.1038/nrmicro3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blanco-Miguez A, Fdez-Riverola F, Sanchez B, Lourenco A. Resources and tools for the high-throughput, multi-omic study of intestinal microbiota. Brief Bioinform. 2017. [DOI] [PubMed]
- 84.Huang S, Chaudhary K, Garmire LX. More is better: recent progress in multi-omics data integration methods. Front Genet. 2017;8:84. doi: 10.3389/fgene.2017.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Magnusdottir S, Heinken A, Kutt L, Ravcheev DA, Bauer E, Noronha A, et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat Biotechnol. 2017;35:81–89. doi: 10.1038/nbt.3703. [DOI] [PubMed] [Google Scholar]
- 86.Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, Jansson JK, et al. Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems 2016;1. [DOI] [PMC free article] [PubMed]
- 87.Shoaie S, Ghaffari P, Kovatcheva-Datchary P, Mardinoglu A, Sen P, Pujos-Guillot E, et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015;22:320–331. doi: 10.1016/j.cmet.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 88.Sung J, Kim S, Cabatbat JJT, Jang S, Jin YS, Jung GY, et al. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat Commun. 2017;8:15393. doi: 10.1038/ncomms15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Douglas GM, Hansen R, Jones CMA, Dunn KA, Comeau AM, Bielawski JP, et al. Multi-omics differentially classify disease state and treatment outcome in pediatric Crohn's disease. Microbiome. 2018;6:13. doi: 10.1186/s40168-018-0398-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rahman SF, Olm MR, Morowitz MJ, Banfield JF. Machine learning leveraging genomes from metagenomes identifies influential antibiotic resistance genes in the infant gut microbiome. mSystems.2018;3. [DOI] [PMC free article] [PubMed]
- 91.Subramanian S, Huq S, Yatsunenko T, Haque R, Mahfuz M, Alam MA, et al. Persistent gut microbiota immaturity in malnourished Bangladeshi children. Nature. 2014;510:417–421. doi: 10.1038/nature13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tripathi A, Xu ZZ, Xue J, Poulsen O, Gonzalez A, Humphrey G, et al. Intermittent hypoxia and hypercapnia reproducibly change the gut microbiome and metabolome across Rodent Model Systems. mSystems 2019;4. [DOI] [PMC free article] [PubMed]
- 93.Zhang L, Lv C, Jin Y, Cheng G, Fu Y, Yuan D, et al. Deep learning-based multi-omics data integration reveals two prognostic subtypes in high-risk neuroblastoma. Front Genet. 2018;9:477. doi: 10.3389/fgene.2018.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res. 2018;24:1248–1259. doi: 10.1158/1078-0432.CCR-17-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carmody RN, Turnbaugh PJ. Host-microbial interactions in the metabolism of therapeutic and diet-derived xenobiotics. J Clin Invest. 2014;124:4173–4181. doi: 10.1172/JCI72335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liang X, Bittinger K, Li X, Abernethy DR, Bushman FD, FitzGerald GA. Bidirectional interactions between indomethacin and the murine intestinal microbiota. Elife. 2015;4:e08973. doi: 10.7554/eLife.08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50. doi: 10.1016/j.cell.2012.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature.2019. [DOI] [PMC free article] [PubMed]
- 99.Saad R, Rizkallah MR, Aziz RK. Gut Pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog. 2012;4:16. doi: 10.1186/1757-4749-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fenn K, Strandwitz P, Stewart EJ, Dimise E, Rubin S, Gurubacharya S, et al. Quinones are growth factors for the human gut microbiota. Microbiome. 2017;5:161. doi: 10.1186/s40168-017-0380-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li L, Zhang X, Ning Z, Mayne J, Moore JI, Butcher J, et al. Evaluating in vitro culture medium of gut microbiome with orthogonal experimental design and a metaproteomics approach. J Proteome Res. 2018;17:154–163. doi: 10.1021/acs.jproteome.7b00461. [DOI] [PubMed] [Google Scholar]
- 102.Chankhamjon P, Javdan B, Lopez J, Hull R, Chatterjee S, Donia MS. Systematic mapping of drug metabolism by the human gut microbiome. bioRxiv 2019:538215. [DOI] [PMC free article] [PubMed]
- 103.Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nat Med. 2011;17:297–303. doi: 10.1038/nm.2323. [DOI] [PubMed] [Google Scholar]
- 104.Aung KL, Fischer SE, Denroche RE, Jang GH, Dodd A, Creighton S, et al. Genomics-driven precision medicine for advanced pancreatic cancer: early results from the COMPASS trial. Clin Cancer Res. 2018;24:1344–1354. doi: 10.1158/1078-0432.CCR-17-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Aguirre AJ, Nowak JA, Camarda ND, Moffitt RA, Ghazani AA, Hazar-Rethinam M, et al. Real-time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 2018;8:1096–1111. doi: 10.1158/2159-8290.CD-18-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Montassier E, Gastinne T, Vangay P, Al-Ghalith GA. Bruley des Varannes S, Massart S, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther. 2015;42:515–528. doi: 10.1111/apt.13302. [DOI] [PubMed] [Google Scholar]
- 108.Montassier E, Batard E, Massart S, Gastinne T, Carton T, Caillon J, et al. 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb Ecol. 2014;67:690–699. doi: 10.1007/s00248-013-0355-4. [DOI] [PubMed] [Google Scholar]
- 109.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 112.Garcia-Gonzalez AP, Ritter AD, Shrestha S, Andersen EC, Yilmaz LS, Walhout AJM. Bacterial metabolism affects the C. elegans response to cancer chemotherapeutics. Cell. 2017;169:431–441. doi: 10.1016/j.cell.2017.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–970. doi: 10.1126/science.1240527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trusheim MR, Berndt ER, Douglas FL. Stratified medicine: strategic and economic implications of combining drugs and clinical biomarkers. Nat Rev Drug Discov. 2007;6:287–293. doi: 10.1038/nrd2251. [DOI] [PubMed] [Google Scholar]
- 116.Gu Y, Wang X, Li J, Zhang Y, Zhong H, Liu R, et al. Analyses of gut microbiota and plasma bile acids enable stratification of patients for antidiabetic treatment. Nat Commun. 2017;8:1785. doi: 10.1038/s41467-017-01682-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Price ND, Magis AT, Earls JC, Glusman G, Levy R, Lausted C, et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat Biotechnol. 2017;35:747–756. doi: 10.1038/nbt.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ. Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science. 2013;341:295–298. doi: 10.1126/science.1235872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Saha JR, Butler VP, Jr, Neu HC, Lindenbaum J. Digoxin-inactivating bacteria: identification in human gut flora. Science. 1983;220:325–327. doi: 10.1126/science.6836275. [DOI] [PubMed] [Google Scholar]
- 120.Lindenbaum J, Rund DG, Butler VP, Jr, Tse-Eng D, Saha JR. Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med. 1981;305:789–794. doi: 10.1056/NEJM198110013051403. [DOI] [PubMed] [Google Scholar]
- 121.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. doi: 10.1016/S0140-6736(07)60751-X. [DOI] [PubMed] [Google Scholar]
- 122.Hwang IY, Koh E, Wong A, March JC, Bentley WE, Lee YS, et al. Engineered probiotic Escherichia coli can eliminate and prevent Pseudomonas aeruginosa gut infection in animal models. Nat Commun. 2017;8:15028. doi: 10.1038/ncomms15028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reyes A, Wu M, McNulty NP, Rohwer FL, Gordon JI. Gnotobiotic mouse model of phage-bacterial host dynamics in the human gut. Proc Natl Acad Sci U S A. 2013;110:20236–20241. doi: 10.1073/pnas.1319470110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhu Wenhan, Winter Maria G., Byndloss Mariana X., Spiga Luisella, Duerkop Breck A., Hughes Elizabeth R., Büttner Lisa, de Lima Romão Everton, Behrendt Cassie L., Lopez Christopher A., Sifuentes-Dominguez Luis, Huff-Hardy Kayci, Wilson R. Paul, Gillis Caroline C., Tükel Çagla, Koh Andrew Y., Burstein Ezra, Hooper Lora V., Bäumler Andreas J., Winter Sebastian E. Precision editing of the gut microbiota ameliorates colitis. Nature. 2018;553(7687):208–211. doi: 10.1038/nature25172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ho CL, Tan HQ, Chua KJ, Kang A, Lim KH, Ling KL, et al. Engineered commensal microbes for diet-mediated colorectal-cancer chemoprevention. Nature Biomed Eng. 2018;2:27. doi: 10.1038/s41551-017-0181-y. [DOI] [PubMed] [Google Scholar]
- 126.Lim B, Zimmermann M, Barry NA, Goodman AL. Engineered regulatory systems modulate gene expression of human commensals in the gut. Cell. 2017;169:547–558. doi: 10.1016/j.cell.2017.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable