

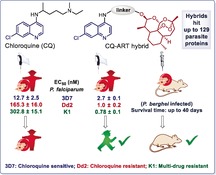
Abstract
A substantial challenge worldwide is emergent drug resistance in malaria parasites against approved drugs, such as chloroquine (CQ). To address these unsolved CQ resistance issues, only rare examples of artemisinin (ART)‐based hybrids have been reported. Moreover, protein targets of such hybrids have not been identified yet, and the reason for the superior efficacy of these hybrids is still not known. Herein, we report the synthesis of novel ART–isoquinoline and ART–quinoline hybrids showing highly improved potencies against CQ‐resistant and multidrug‐resistant P. falciparum strains (EC50 (Dd2) down to 1.0 nm; EC50 (K1) down to 0.78 nm) compared to CQ (EC50 (Dd2)=165.3 nm; EC50 (K1)=302.8 nm) and strongly suppressing parasitemia in experimental malaria. These new compounds are easily accessible by step‐economic C−H activation and copper(I)‐catalyzed azide–alkyne cycloaddition (CuAAC) click reactions. Through chemical proteomics, putatively hybrid‐binding protein targets of the ART‐quinolines were successfully identified in addition to known targets of quinoline and artemisinin alone, suggesting that the hybrids act through multiple modes of action to overcome resistance.
Keywords: antimalarial agents, artemisinin, drug resistance, hybridization, proteomics
ARTful hybrids: Artemisinin (ART)‐based hybrids that are highly potent against CQ/drug‐sensitive and multidrug‐resistant parasites and of strong efficacy in experimental malaria were synthesized. New putatively hybrid‐binding proteins, in addition to known target proteins of quinoline and artemisinin, were identified by chemical proteomics, suggesting that the hybrids act through multiple modes of action to overcome resistance.
Introduction
Every year malaria parasites cause approximately 200 million infections in humans and almost half a million deaths according to the World Health Organization.1 Plasmodium falciparum (P. falciparum), representing the most pathogenic species of this eukaryotic genus, is mostly responsible for lethal courses of infection. Currently, artemisinin (ART; Figure 1 A) and quinoline derivatives (e.g., chloroquine (CQ); Figure 1 B) are the two main classes of antimalarial drugs.2 ART is an enantiopure sesquiterpene lactone, which was first extracted from the Chinese medicinal plant Artemisia annua L. (sweet wormwood) in 1972 by Youyou Tu (Nobel Prize 2015).3 Isoquinolines and quinolines are further privileged scaffolds, used clinically as antimalarial and antiviral drugs, for example, through the different commercial drugs amodiaquine, paritaprevir, and tafenoquine (Figure 1 B). Inspired by these traditional and pharmacologically well‐established compounds, many derivatives of artemisinin and quinoline have been designed, synthesized, and biologically investigated.4
Figure 1.
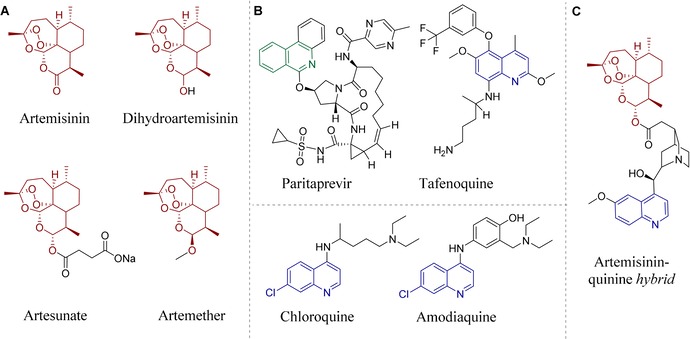
Selected artemisinin derivatives and pharmaceutically relevant isoquinolines/quinolines. A) Artemisinin (naturally occurring), dihydroartemisinin, artesunate, and artemether (semisynthetic derivatives). B) Clinically applied drugs with a quinoline or isoquinoline core: paritaprevir, tafenoquine, chloroquine, and amodiaquine. C) First example of an artemisinin–quinine hybrid.9a
The emergence of parasite resistance against approved drugs has been on the rise over the last decades, and medical concerns have thus increased dramatically. Two resistant strains Dd2 and K1 carry mutations and a number of copies for genes5 encoding for multidrug resistance transporters, resulting in resistances against currently available antimalarial drugs. Notably, Dd2 is resistant to CQ, mefloquine, and the antifolates sulfadoxine, pyrimethamine, and cycloguanil, whereas K1 is resistant to CQ and antifolates.5 In addition, they show different mutations, leading to an amino acid exchange of a target enzyme resulting in resistance against antifolates (sulfadoxine, pyrimethamine, cycloguanil). Compared with chloroquine/drug‐sensitive strains (3D7), the fitness and thus the reproduction rate of chloroquine/multidrug‐resistant (Dd2 and K1) parasites is slightly impaired by resistance‐mediating polymorphisms in putative drug transporters and/or in target enzymes,6 even without drug pressure.
CQ‐resistant P. falciparum has been reported from almost all malaria‐endemic countries. Moreover, the therapeutic achievements based on the natural product ART and its semi‐synthetic derivatives (dihydroartemisinin, artesunate, artemether; Figure 1 A) are also threatened by emerging drug resistance.7 Therefore, the WHO discourages mono‐therapy, which primarily promotes further development of resistance to ART in endemic areas, and recommends the use of artemisinin‐based combination therapies (ACTs) to reduce the risk of drug resistance. ACTs use ART or its derivatives together with one or more other antimalarial drugs that act through different mechanisms.8
Recently, structural fusions of artemisinins and quinolines, namely hybrid molecules (see Figure 1 C for an example), have been described as being even more efficient against different biological threats, such as malaria parasites and cancer cells.9 Hybridization appears as a powerful concept10 to increase the activity or pharmacological efficacy of known drugs or the bioactive constituents of the hybrid molecule11 and to potentially overcome drug resistance,12, 13 as hybrids can be less susceptible to drug resistance.
Whereas some examples of hybrid molecules of quinoline and ART exist, neither systematic SAR (structure–activity relationship) studies with different linkers nor identifications of their protein targets are known yet, while ART–isoquinoline hybrids have not even been reported thus far. To fill these gaps, we herein present the structural design, synthesis, and assessment of the in vitro antimalarial activities of 17 new ART–isoquinoline and ART–quinoline hybrid compounds able to combat multidrug‐resistant malaria. The antimalarial activities of the resulting hybrids were tested on the P. falciparum drug‐sensitive strain 3D7 and the two multidrug‐resistant strains Dd2 and K1.
The applied new isoquinoline precursors were accessible by versatile cobalt(III)‐catalyzed step‐economic C−H activation methods, while the linkers in the hybrids were generated using CuAAC (copper‐catalyzed azide–alkyne cycloaddition) click reactions and a novel rearrangement of the in situ formed tertiary amides to secondary amides. The SAR study revealed important aspects of the linker group, which led us to identify even more potent antimalarial hybrid compounds able to overcome drug resistance. One reason for the increased activity of the hybrids versus their individual constituents could be the simultaneous cellular uptake of both pharmacophores owing to their covalent linking, which is not possible for combination therapy. Besides, a possible synergistic effect of the applied pharmacophores might partially explain the pronounced bioactivity of the hybrids.
We further experimentally demonstrated the efficacy of selected hybrid compounds against malaria in P. berghei‐infected mice. Importantly, the proteomic characteristics of the hit hybrid compounds were assessed to identify putative targets in the Plasmodium proteome. We have shown that the hybridization of two individual pharmacophores into a new bioactive compound has high potential in addressing resistance issues because hybrids can simultaneously target proteins of both pharmacophores (artemisinin and quinoline), as well as new hybrid‐binding proteins. Overall, our findings are highly relevant towards the future development of effective multi‐action drugs against resistant malaria.
Results and Discussion
Chemistry
The synthesis of the new hybrids 1–17 (Figure 2) is discussed in two parts according to the chemical reactions that were applied to form the corresponding linker: 1) hybrid synthesis by CuI‐catalyzed click reactions; and 2) hybrid synthesis by esterification, amide bond formation, and a novel rearrangement of the in situ formed tertiary amide into a secondary amide.
Figure 2.
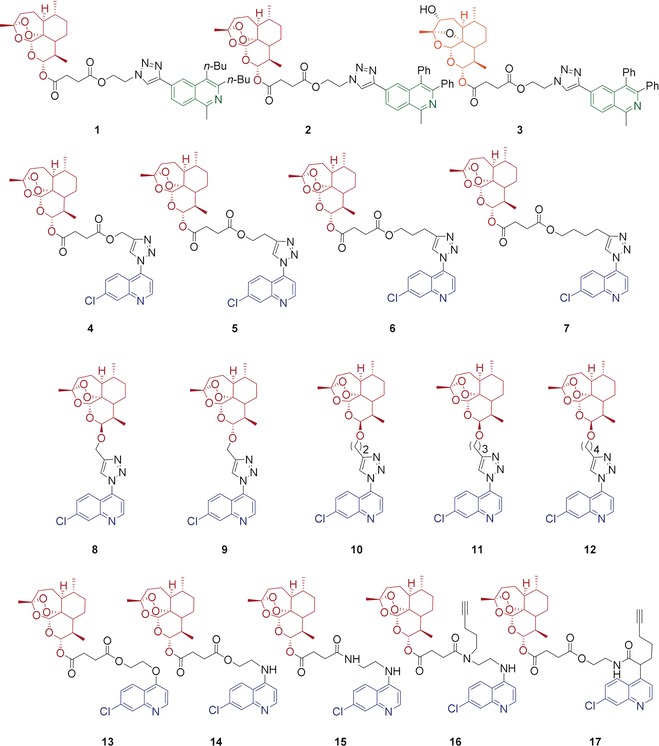
Novel hybrids 1–17 applied for activity examination against P. falciparum 3D7, Dd2, and K1 strains. Red, blue, and green indicate the parent pharmacophores of the molecules. The black moieties represent the linker groups of the molecules. The 3‐hydroxy‐desoxydihydroartemisinin unit of hybrid 3 is shown in orange.
Hybrid Synthesis by Copper(I)‐Catalyzed Click Reactions
We first planned to investigate the variation of bioactivity when the linker bears a triazole entity. The CuAAC click reaction has become one of the most important chemical reactions in the field of medicinal chemistry with its highly preferable features. The resulting products feature a five‐membered triazole ring as the linker group, formed by 1,3‐dipolar cycloaddition (click) reaction. This five‐membered ring is a building block very often employed in valuable pharmaceuticals.14 As illustrated in Schemes 1 A and 1 B, we thus obtained hybrids 1–12, bearing a triazole linker.
Scheme 1.
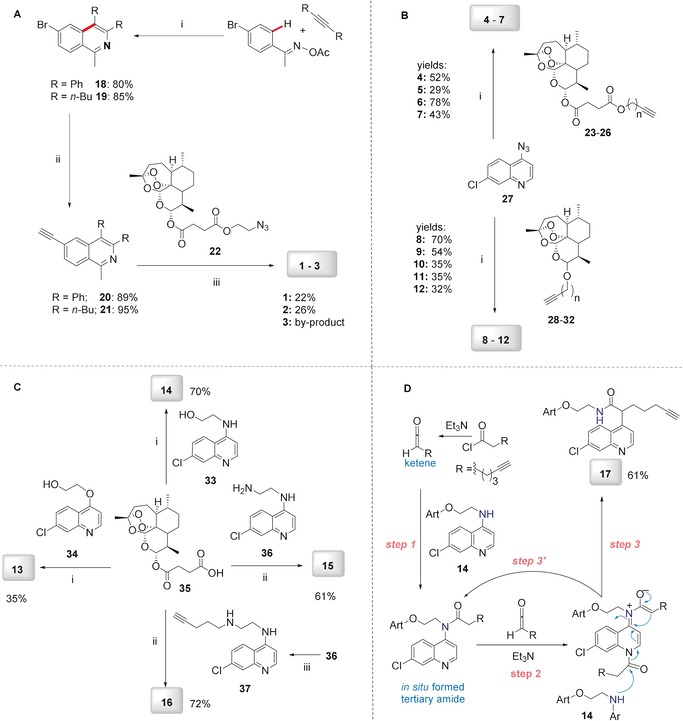
A) Synthesis of isoquinoline–artesunic acid hybrids 1–3. B) Synthesis of 7‐chloroquinoline–artesunic acid hybrids 4–7 and 7‐chloroquinoline–artemisinin hybrids 8–12. C) Synthesis of 7‐chloroquinoline–artesunic acid hybrids 13–16. D) Post‐modification of hybrid 14 to hybrid 17 in a novel domino process: in situ tertiary amide formation/intramolecular rearrangement reaction. Reagents and Conditions: A) (i) [Cp*Co(CO)I2] (10 mol %), AgSbF6 (20 mol %), NaOAc (20 mol %), 1,2‐dichloroethane, 120 °C, 1 h; (ii) 1) PdCl2(PPh3)2 (1.0 mol %), trimethylsilylacetylene, triethylamine, 50 °C, 2 h, 2) K2CO3, methanol, 25 °C, 4 h; (iii) CuSO4⋅5 H2O, sodium ascorbate, CH2Cl2/H2O, overnight. B) (i) CuSO4⋅5 H2O, sodium ascorbate, CH2Cl2/H2O, rt, overnight. 23: n=1; 24: n=2; 25: n=3; 26: n=4; 28: n=1 (C‐10β); 29: n=1 (C‐10α); 30: n=2 (C‐10β); 31: n=3 (C‐10β); 32: n=4 (C‐10β). C) (i) DCC, DMAP, CH2Cl2, 0 °C→rt, overnight, N2; (ii) EDCI, DMAP, CH2Cl2, 0 °C→rt, overnight, N2; (iii) K2CO3, acetonitrile, 130 °C, overnight. D) In CH2Cl2, 0 °C→rt, 18 h, N2.
The artemisinin–isoquinoline hybrid compounds 1–3 were prepared (Scheme 1 A) through a CuAAC click reaction of new artesunic acid based azide 22 with the isoquinoline‐derived alkynes 20 and 21 in the presence of CuSO4⋅5 H2O and sodium ascorbate. The corresponding isoquinoline precursors 20 and 21 were synthesized by facile C−H cobaltation of O‐acetyl oximes with internal alkynes,15 followed by Sonogashira coupling of the obtained 6‐bromo‐isoquinolines 18 and 19 with trimethylsilylacetylene (Scheme 1 A and the Supporting Information). We were also able to isolate compound 3, which is a side product of the formation of hybrid 2. We found that the artemisinin moiety is hydroxylated at the C3 position to a desoxydihydroartemisinin derivative in the presence of copper(I) species, as reported previously.16 This 3‐hydroxy‐desoxydihydroartemisinin derivative 3 was also subjected to bioactivity examination.
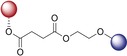
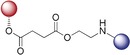
The required new artesunic acid alkyne derivatives 23–26 (Scheme 1 B) were prepared by esterification reaction using EDCI (1‐ethyl‐3‐(3‐dimethylaminopropyl)carbodiimide), DCC (N,N′‐dicyclohexylcarbodiimide), and DMAP (4‐dimethylaminopyridine) in CH2Cl2 at room temperature, starting from artesunic acid (35), and 2‐propyn‐1‐ol, 3‐butyn‐1‐ol, 4‐pentyn‐1‐ol, and 5‐hexyn‐1‐ol, respectively (see the Supporting Information). Using the same set of alcohols, etherification reactions were performed with dihydroartemisinin to obtain precursors 28–32. The subsequent 1,3‐dipolar cycloaddition reactions of artesunic acid based and dihydroartemisinin‐based alkyne derivatives 23–32 with 4‐azido‐7‐chloroquinoline (27) towards the corresponding hybrids 4–12 are presented in Scheme 1 B.
Hybrid Synthesis by Esterification, Amidation, and a Novel Rearrangement
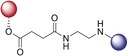
We next enlarged the scope of the hybrids by diversifying the linker to examine the SAR for further medicinal chemistry insight. The new hybrid molecules 13–16 were synthesized from artesunic acid 35 with the corresponding 7‐chloroquinoline derivatives 33, 34, 36, and 37 (Scheme 1 C and the Supporting Information). While the quinoline‐derived precursors 33, 34, and 36 were prepared based on previous reports,17 compound 37 was synthesized for the first time by a nucleophilic aliphatic substitution reaction of 5‐chloropentyne with quinoline‐based amine 36 (Scheme 1 C and the Supporting Information). The hybrid compounds 13 and 14 were obtained by esterification of artesunic acid 35 with the 7‐chloroquinoline‐derived primary alcohols 34 and 33, using the coupling reagents DCC and DMAP in CH2Cl2 under nitrogen atmosphere. Primary and secondary amine derivatives of 7‐chloroquinoline (36 and 37) were used for amide coupling reactions in the presence of EDCI and DMAP in CH2Cl2 to furnish hybrids 15 and 16 (Scheme 1 C).
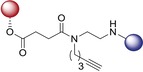
The alkyne‐tagged hybrid 16 was synthesized to identify its target proteins in P. falciparum by proteomics. To obtain another alkyne‐tagged hybrid compound for proteomics studies, we applied hybrid 14 for further modification through acylation of the secondary amine with hept‐6‐ynoyl chloride (Scheme 1 D). This experiment did not result in the expected tertiary amide product. Instead, we observed the product of a novel metal‐free intramolecular rearrangement of the in situ formed tertiary amide into the secondary amide hybrid 17, which we used for proteomics studies. A possible mechanism of this novel multistep domino reaction is shown in Scheme 1 D. Triethylamine promotes dehydrohalogenation of chloro acetyl chloride to generate the corresponding ketene,18 which subsequently reacts with 14 to a tertiary amide (step 1), which undergoes a second acylation reaction (step 2). The in situ formed C1‐ammonium enolate intermediate undergoes an intramolecular C−C bond formation/C−N bond cleavage sequence (step 3) towards the final product 17. Secondary amine 14 facilitates the rearomatization process via its acylation to a tertiary amide (step 3′).
Antimalarial Activity
Remarkably, all investigated artemisinin‐derived hybrid compounds in this study (Figure 2) exhibited potent antimalarial activity in the nanomolar to picomolar range against chloroquine/drug‐sensitive (3D7) and chloroquine/multidrug‐resistant (Dd2 and K1) parasite strains (Table 1).
Table 1.
EC50 values for hybrids 1–17, hybrid precursors 33–37, 18, chloroquine (CQ), artemisinin (ART), and artesunic acid tested against P. falciparum parasite strains 3D7, Dd2, and K1.
|
Compound |
EC50 [nm]±S.E.M. |
|||
|---|---|---|---|---|
|
|
3D7 |
Dd2 |
K1 |
|
|
1 |
|
9.2±0.7 |
10±0.8 |
4.2±0.4 |
|
2 |
|
5.3±0.2 |
4.8±0.1 |
3.3±0.7 |
|
3 [a] |
|
646.6±94.4 |
444.0±107.5 |
255.5±68.6 |
|
4 |
|
5.3±1.0 |
4.2±0.3 |
1.8±0.2 |
|
5 |
|
8.0±1.3 |
4.9±0.3 |
2.6±0.2 |
|
6 |
|
5.4±0.6 |
4.6±0.2 |
2.4±0.1 |
|
7 |
|
6.2±0.4 |
4.4±0.4 |
2.5±0.4 |
|
8 |
|
9.0±1.5 |
5.9±0.6 |
4.3±0.7 |
|
9 [b] |
|
27.5±2.9 |
10.6±1.3 |
5.9±1.4 |
|
10 |
|
8.8±1.1 |
4.5±0.3 |
2.0±0.2 |
|
11 |
|
9.4±1.1 |
5.1±0.2 |
3.8±0.4 |
|
12 |
|
8.4±1.6 |
6.1±0.2 |
3.7±0.5 |
|
13 |
|
14.8±0.4 |
10.6±0.4 |
5.5±0.4 |
|
14 |
|
4.5±0.5 |
2.3±0.6 |
1.7±0.3 |
|
15 |
|
2.7±0.1 |
1.0±0.2 |
0.78±0.1 |
|
16 |
|
3.5±0.3 |
1.6±0.3 |
1.3±0.3 |
|
17 |
|
6.0±0.4 |
4.8±0.9 |
2.2±0.2 |
|
18 |
|
16 400 |
18 400 |
22 100 |
|
27 |
|
1980 |
1890 |
2860 |
|
33 |
|
760 |
1600 |
2400 |
|
36 |
|
17.4±0.6 |
483.3±44.0 |
512.3±49.7 |
|
37 |
|
13.3±0.7 |
41.5±4.9 |
53.8±7.5 |
|
CQ |
|
12.7±2.5 |
165.3±16 |
302.8±15.1 |
|
ART |
|
26.8±2.4 |
11.3±1.8 |
5.4±0.5 |
|
artesunic acid (35) |
|
14.4±2.0 |
10.9±0.7 |
5.2±0.8 |
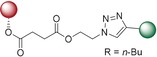
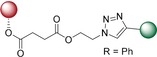
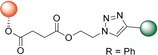
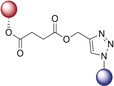
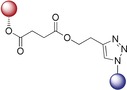
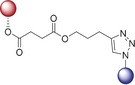
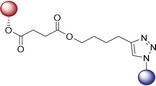
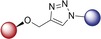
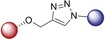
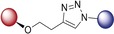
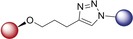
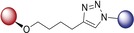
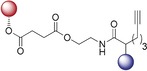


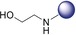
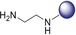
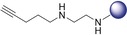

[a] Because of the limited available quantity, biological replicates were only done twice for the Dd2 and K1 strain. [b] Precipitates: The compound had to be heated at 55 °C for 15 min to dissolve, which resulted in an extreme variation in the EC50 values especially in the case of hybrid 11. EC50=half‐maximal effective concentration; S.E.M.=standard error of the mean.
While quinolines are well‐known for their antimalarial activity, much less is known about the antimalarial activity of isoquinolines and their structural determinants. In the following, the novel ART–isoquinolines hybrids 1–3 were initially investigated. The EC50 (half‐maximal effective concentration) values of hybrids 1 and 2 against 3D7, Dd2, and K1 strains of P. falciparum are 9.2, 10.0, and 4.2 nm and 5.3, 4.8, and 3.3 nm, respectively, indicating the potent activity of these novel hybrids compared to parent compounds artesunic acid 35 and isoquinoline precursor 18. Furthermore, we observed that the antimalarial activity of hybrid 3 is 100 times lower than that of hybrid 2. The drop in activity of 3‐hydroxy‐desoxydihydroartemisinin derivative 3 underlines the importance of the 1,2,4‐trioxane fragment for high activity.
Next, we examined the influence of the linker length on the activity of the artesunic acid–quinoline hybrid compounds 4–7 (Figure 2), where the linker length is elongated from one to four carbon atoms. However, among these hybrids 4–7, the elongation of the linker did not change the antimalarial activity of the compounds in a substantial manner (Table 1). These hybrids are highly potent antimalarial compounds, with EC50 values in the nanomolar range, and the shortest linker allowed us to achieve the highest activity observed against all P. falciparum strains used in this study. CQ gave an EC50 value of 12.7 nm against 3D7, whereas the hybrids showed EC50 values of 5.3, 8.0, 5.4, and 6.2 nm (short to long carbon chain linkers), being more active than CQ (Table 1). In the case of resistant strains, hybrids 4–7 are even more potent, showing 34 to 39 times higher potency against Dd2 (4.2–4.9 nm), and 116 to 168 times higher activity against K1 (1.8–2.6 nm) than CQ. Moreover, triazole‐linked artesunic acid–7‐chloroquinoline hybrids are highly potent against resistant P. falciparum strains. All hybrids were able to overcome CQ drug resistance and multidrug resistance; most importantly they also outperformed the reference drug artemisinin. Remarkably, the hybrids are more active than their parent compound artesunic acid (35) and 4‐azido‐7‐chloroquinoline (27; 14.4/10.9/5.2 nm and 1982/2523/2939 nm, respectively) in all cases (Table 1). Thus hybridization can be used to access more potent bioactive compounds than their parent precursors for this set of hybrids 4–7.
Similar to our previous results (see Table 1, hybrids 4–7), ART–quinoline hybrids 8–12 showed great activity against malaria parasites ranging from 2.0 nm to 27.5 nm, revealing that these hybrids are also more active against CQ/multidrug‐resistant parasites than against CQ/drug‐sensitive parasites. Hybrids 8, 10, 11, and 12 showed almost the same potency when they were tested against three different strains. The hybrids exhibited EC50 values of 9.0 nm, 8.8 nm, 9.4 nm, and 8.4 nm against 3D7; 5.9 nm, 4.5 nm, 5.1 nm, and 6.1 nm against Dd2; and 4.3 nm, 2.0 nm, 3.8 nm, and 3.7 nm against K1 strains, respectively (Table 1). Interestingly, we again did not observe a correlation between activity and linker length. Hybrids 8 and 9 are β‐ and α‐anomers because of the stereochemistry at the C10 position of the artemisinin unit. As previously reported for α‐ and β‐arteethers,19 we have also observed that the β‐isomer 8 is more active than the α‐isomer 9. Nevertheless, with EC50 values of 27.5 nm, 10.6 nm, and 5.9 nm, the α‐isomer of this hybrid is still highly active against P. falciparum strains 3D7, Dd2, and K1.
Next, we tested the new 7‐chloroquinoline–artesunic acid hybrids 13–15, containing ester and amide linkers, against P. falciparum parasites. Hybrid 14 is more active than hybrid 13. Replacing the H‐bond acceptor oxygen atom at the C4 position of the 7‐chloroquinoline unit of hybrid 13 with an H‐bond donor amine group resulted in the formation of 4‐aminoquinoline chemical space (hybrid 14).
As expected, this chemical modification substantially increased the potency against all P. falciparum strains, which is explained through the formation of the 4‐aminoquinoline, a known antimalarial core structure. The activities increased, as seen from decreases in the EC50 values from 14.8 to 4.5 nm for 3D7, from 10.6 to 2.3 nm for Dd2, and from 5.5 to 1.7 nm for K1 strains (Table 1). Based on these results, we assumed that the composition of the linker affects the activity. Furthermore, we decided to replace the second oxygen atom by a nitrogen atom. This designed compound 15 gave EC50 values of 2.7 nm, 1.0 nm, and 780 pm against 3D7, Dd2, and K1 strains, respectively (Table 1). Through this systematic replacement of heteroatoms, we examined requirements on the linker for high antimalarial activity, which led to EC50 values as low as 780 pm against the K1 strain with hybrid 15. These results are consistent with those obtained with previous hybrids (13 and 14) when comparing the activities of the individual constituents of each hybrid against chloroquine/drug‐sensitive (3D7) and chloroquine/multidrug‐resistant (Dd2 and K1) parasites, as higher efficiencies were always observed against resistant parasites. It is impressive that the generated compounds are able to counteract all resistances presented by the Dd2 and K1 strains. On the basis of the observed SAR, we hypothesized that these artesunic acid–7‐chloroquinoline based hybrids are more potent when they bear a nitrogen atom on the linker. Moreover, they show excellent antimalarial activity.
Next, compounds 16 and 17 were evaluated as alkyne‐tagged hybrids. They were designed for proteomics experiments, which require an alkyne unit to tag the molecule to a biotin affinity tag through a click reaction. The binding proteins of these hybrids were investigated to understand the mechanism of action of the most active antimalarial hybrid. To the best of our knowledge, none of the artemisinin–quinoline hybrids have been previously investigated based on their target profiling. Hybrid 16 gave EC50 values of 3.5, 1.6, and 1.3 nm against 3D7, Dd2, and K1 strains, respectively (Table 1). As an analogue, our hybrid 16 can be compared with hybrid 15, and it can be implied that the hydrogen atom on the secondary amide improves antimalarial activity. In contrast, hybrid 16 is a tertiary amide and does not provide an N−H bond, unlike hybrid 15, and it is thus less active. When the amide nitrogen atom is occupied by an alkyne chain, the activity is lower as it is the case for hybrid 14, which features an ester bond and a H‐bond acceptor. This finding might also be a confirmation that the H‐bond donor improves the activity. As expected, when we tested the antimalarial activity of the post‐modified hybrid 17, the activity dropped compared to its pre‐modified version, that is, hybrid 14. The decrease in activity is possible due to depletion of the 4‐aminoquinoline core, which is essential for heme binding.20 This initial set of compounds has shown that artemisinin and 7‐chloroquinoline are a promising pair to improve the activity against malaria parasites, and that the composition of the linker is a structural determinant for modulating activity.
Parasitemia Suppression in Experimental Malaria
We evaluated the suppressive activity of the hybrids in P. berghei‐infected mice over four consecutive days of treatment.21 Compared with the vehicle group, mice treated subcutaneously with 105 μmol kg−1 (40 mg kg−1) of artesunate (ARE) showed a >99 % reduction in blood parasitemia, and this dose also conferred 100 % animal survival (Table 2, entry 2). Based on these results, we chose a dose of 105 μmol kg−1 given by subcutaneous injection as a starting point for testing the hybrid compounds. To gain insight into the SARs of our compounds, we selected the artesunic acid–7‐chloroquinoline based hybrid 14, containing an ester linker group (the thermal, hydrolytic, and enzymatic stability of 14 is high; see the Supporting Information), as its potency is similar to that of alkyne‐tagged hybrid 16, prepared for target identification experiments. In addition, the artemisinin–7‐chloroquinoline based hybrid 12, containing a triazole linker, was selected. The use of hybrids 12 and 14 enabled us to investigate the importance of the linker group for antimalarial efficacy (Figures S1–S3 in the Supporting Information).
Table 2.
Summary of the activities of hybrid 14 and artesunate against blood‐stage P. berghei‐infected mice.
|
Entry |
Compound |
Dose [μmol/kg body weight] |
Parasitemia inhibition [%][a] |
Median survival [days] |
Number of alive animals (%)[b] |
|---|---|---|---|---|---|
|
subcutaneous administration (s.c.) | |||||
|
1 |
vehicle |
– |
– |
20 |
0/10 (0) |
|
2 |
ARE |
105 |
>99 |
>41 |
5/5 (100) |
|
3 |
hybrid 14 |
105 |
>99 |
>41 |
5/5 (100) |
|
4 |
ARE |
35 |
– |
– |
0/8 (0)[c] |
|
5 |
hybrid 14 |
35 |
95.2±4.9 |
>41 |
5/5 (80)[d] |
|
|
|
|
|
|
|
|
oral administration by gavage (P. Os.) | |||||
|
6 |
hybrid 14 |
105 |
78.6±10[e] |
35[e] |
6/10 (60)[e] |
[a] Values are mean±S.D. and were taken from 10 dpi. [b] Until 41 dpi. [c] Value taken from Ref. 21. [d] One of five mice were blood smear positive for parasites 41 dpi. [e] Median of two independent experiments, where error is S.E.M. dpi=days post‐infection. ARE=Artesunate. S.D.=standard deviation. S.E.M.=standard error of the mean.
At a dose of 105 μmol kg−1 (65 mg kg−1), hybrid 14 reduced parasitemia by >99 %, while at a dose of 35 μmol kg−1, it reduced parasitemia by 95.2±4.9 % (Table 2, entries 3 and 5). As a result of the strong parasitemia suppression, all mice treated with 105 μmol kg−1 of hybrid 14 remained free of parasites for up to 41 dpi (days post‐infection), and they were considered cured. When hybrid 14 was given at a dose of 35 μmol kg−1 (s.c.), 80 % of mice remained free of parasites up to 41 dpi, which is a cure rate higher than for artesunate treatment at the same dosage and with the experimental model.21 In contrast to hybrid compound 14, hybrid 12 featuring a triazole linker did not reduce parasitemia when administered at a dose of 105 μmol kg−1 (Figure S2).
Having ascertained the efficacy of hybrid 14 upon subcutaneous administration, we tested whether this compound is able to inhibit parasitemia upon oral administration (Table 2, entry 6). Compound 14 displayed strong antiparasitic activity, reducing parasitemia by 78.6±10 % and increasing the median animal survival time compared to the vehicle group. From two independent experiments, 60 % of mice remained free of parasites in the group treated orally with hybrid 14 (Table 2, entry 6). In contrast, infected mice orally treated with ARE at a dose of 105 μmol kg−1 were not cured. In fact, ARE is curative only when administered subcutaneously22 or in combination with mefloquine. The fact that hybrid 14 inhibited parasitemia and provided protection from mortality upon oral uptake indicates that this compound has oral efficacy, which is a highly desired property for a new antimalarial drug candidate.
ART and ARE are known to have a fast onset of antimalarial action, which causes a rapid decrease in parasitemia, but they also have a short half‐life.23 Unlike ARE, hybrid 14 presented a more long‐lasting action. This was inferred when instead of the standard treatment for four consecutive days, mice were treated at intercalated days at 3 and 48 h post‐infection (Figure S4 and Table S1). At day 10 post‐infection, both hybrid 14 and artesunate treatment suppressed parasitemia; however, at day 12 post‐infection, parasitemia had increased rapidly (i.e., recrudescence of parasitemia) in four out of five mice, and at day 16 post‐infection, all mice treated with ARE showed parasite recrudescence. In contrast, only two of five mice treated with hybrid 14 presented detectable parasites at day 12 post‐infection, showing that treatment with the hybrid compound is more efficient in delaying the recrudescence of parasites compared with ARE treatment.
Curative Potential in Experimental Malaria
The fact that hybrid 14 presented impressive parasitemia suppression led us to determine its curative potential (Thompson test). As expected, hybrid 14 cured 100 % of mice, while artesunate only cured 33 % of mice when administered at the same dose (Figure 3 and Table S2). Based on the cure ratio, we estimated that hybrid 14 is threefold more effective than ARE.
Figure 3.
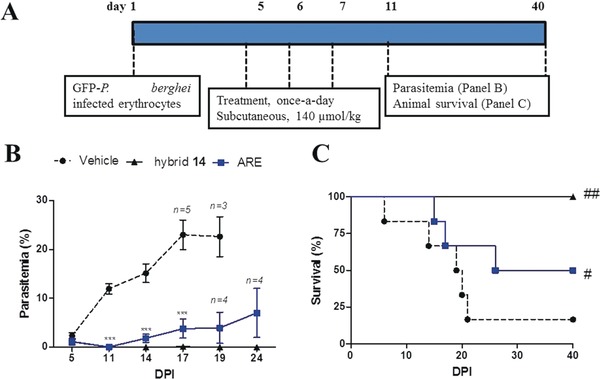
Experimental design of the curative Thompson test (A), follow‐up of parasitemia (B), and animal survival (C) in P. berghei‐infected mice. Parasitemia and animal survival were measured using an n=6 group, unless indicated otherwise. Error bars indicate S.D. ***p<0.01 (two‐way ANOVA) versus vehicle group. # p<0.01, ## p<0.005 (Log‐rank, Mantel‐Cox test) versus vehicle. dpi=days post‐infection. S.D.=standard deviation.
Apart from piperaquine, the required dose of antimalarials to cure mice in the curative model of Thompson is at the very least two times higher than the required dose to protect mice from mortality in the suppressive model of Peters.24 Strikingly, we observed that the sum of dosages of the hybrid compound employed in the suppressive test was enough to provide cure. This puts hybrid compound 14 in an efficacy range that is only comparable to those of piperaquine and ACTs.
The Underlying Mechanism of Potency Enhancement
To study the origin of potency enhancement, we analyzed the potential of hybrid 14 in impairing malarial pigment formation in in vitro and in vivo models. ARE lacks inhibitory properties in the in vitro β‐hematin formation while hybrid 14 inhibited β‐hematin formation with a potency as high as that of chloroquine (Figure 4 and Table S3). Assuming that only the 7‐chloroquinoline part of hybrid compound 14 is responsible for the β‐hematin inhibitory properties,20b, 20c the observation that compound 14 is as potent as chloroquine is explained by the fact that the 7‐chloroquinoline moiety in the hybrid compound can adopt different conformations and orientations, in part greatly influenced by the linker group. While this kind of potency enhancement against β‐hematin formation has been observed for quinoline derivatives,20c it has not been demonstrated in experimental malaria. Here, we introduce the notion that the potency enhancement of hybrid compound 14 against β‐hematin correlates well with its efficacy in impairing parasite hemozoin in experimental malaria. This was confirmed in a designed experiment with P. berghei‐infected mice by simultaneously monitoring parasitemia and heme species, including the cellular hemozoin (Figure 4).
Figure 4.
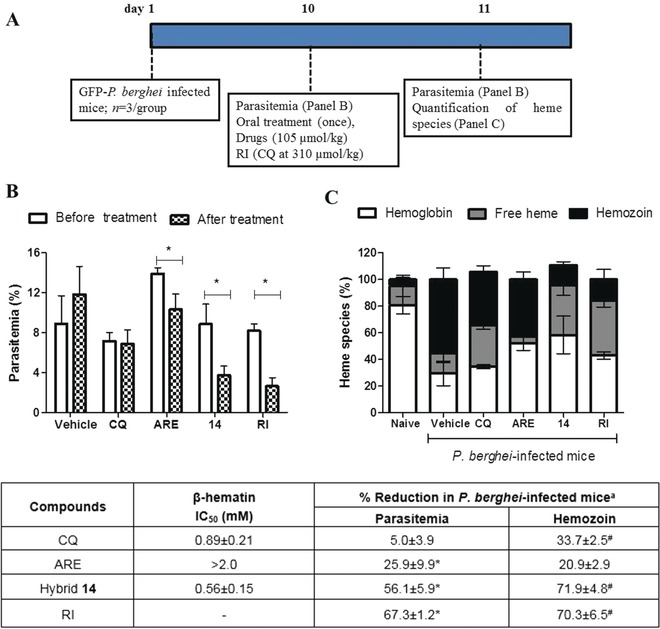
Experimental design (A), parasitemia (B), heme species (C), and summary of results (Table) from P. berghei‐infected mice within 24 h of oral treatment. In (B), parasitemia was determined by GFP signal using flow cytometry. In (C), heme species were quantified from the peripheral blood after mouse euthanasia. The results of two independent experiments with n=3 groups for each experiment are shown. Error bars are means and 95 % CI (95 % confidence interval). IC50 and % reduction values are mean±S.D. In the table, β‐hematin formation was determined 24 h after drug incubation. [a] Values relative to the infected untreated group (vehicle). *p<0.05 (one‐way ANOVA and Newman‐Keuls multiple comparison test) for before versus after treatment. # p<0.05 in comparison to vehicle. CQ=Chloroquine, ARE=Artesunate. RI=Reference inhibitor (CQ at 310 μmol kg−1).
These experiments revealed that 24 h after administration of a single oral dose (105 μmol kg−1), compound 14 had reduced parasitemia by 56.1 %. In addition to an impressive suppression of parasitemia, the hybrid was effective in impairing hemozoin formation by 71.9 % in comparison to untreated mice. At the same dose, ARE was only half as effective as compound 14 in reducing parasitemia but did not significantly reduce hemozoin. In contrast, CQ did not reduce parasitemia but attenuated hemozoin formation, albeit twice less than compound 14. Concomitant to a decrease in the hemozoin level, both CQ and compound 14 increased the levels of free heme, while ARE did not, clearly showing that CQ and compound 14 reduce parasite viability by interrupting the biosynthesis of hemozoin. In terms of efficacy, compound 14 behaved similarly to the group receiving a reference inhibitor of hemozoin (CQ‐treated mice at a high dose of 100 mg kg−1, 310 μmol kg−1). Our combination of in vitro and in vivo experiments, using pharmacologically relevant drug doses, in addition to a critical quantification of free heme and hemozoin levels, allowed us to recognize that hemozoin inhibition is due to a direct effect of hybrid drug treatment rather than a consequence of parasitemia reduction. This also supports the notion that the efficacy of hybrid 14 in P. berghei‐infected mice is not only due to the presence of the artemisinin moiety, but also highly dependent on the quinoline.
Chemical Proteomics Profiling of the Interactive Protein Targets of Hybrids 16 and 17 in P. falciparum
With the alkyne‐tagged artemisinin–chloroquinoline hybrids 16 and 17 in hand, we linked the alkyne‐tagged analogues to biotin azide through click chemistry (Figure 5 A). Then, the biotin‐bearing analogues were incubated with avidin beads to prepare the drug affinity beads. The enriched and purified P. falciparum parasite protein lysates were incubated with the drug‐bearing beads for 4 h. The drug binding targets were affinity‐purified by avidin beads and identified by tandem mass spectrometry. A total of 129 and 107 parasite proteins were identified as direct targets of hybrids 16 and 17 (see Table S5), while pull‐down of the dimethyl sulfoxide treated control beads did not identify any parasite proteins. The exponentially modified protein abundance index (emPAI) was used to provide a semi‐quantification of protein abundance.25
Figure 5.
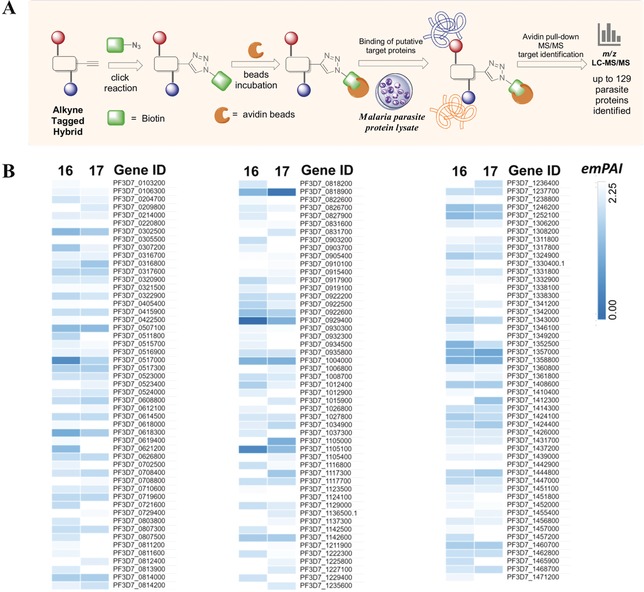
Target Identification. A) The alkyne‐tagged hybrids were reacted with biotin azide to generate the affinity probes for interactive target enrichment. Affinity pull‐down coupled mass spectrometry identification was carried out to characterize the hybrids targets. B) Heat map representation of the relative abundance of individual targets of hybrids 16 and 17. The emPAI scores of individual proteins were used to generate the heat map with morpheus software. Complete datasets of the drug targets are shown in Table S5.
Heat map analysis (Figure 5 B) showed that the target profiles of hybrids 16 and 17 are generally similar to each other as the hybrids possess a similar artemisinin moiety. Consistent with the higher antimalarial potency, the target labeling potential of hybrid 16 is also slightly higher than that of hybrid 17. Along with the target identification we found a number of proteins, such as heat shock protein 70, high‐molecular‐weight rhoptry protein 2, and 40S ribosomal protein S15, that were strongly enriched by both probes. Interestingly, we found that both artemisinin hybrid compounds 16 and 17 can interact with PfATP6, a well‐known artemisinin target.
Gene ontology pathway analysis was performed to map out the critical pathways of the drug targets involved in the antimalarial targets of the hybrid compounds (Figure 6). A number of metabolic processes are the most significant pathways affected by the two hybrids 16 and 17, including glycolytic and organonitrogen compound metabolic processes (Figure 7).
Figure 6.
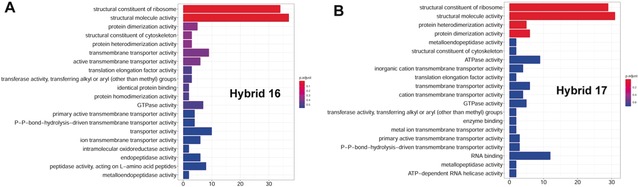
Gene ontology (GO) analysis of interactive proteins for A) hybrid 16 and B) hybrid 17.
Figure 7.
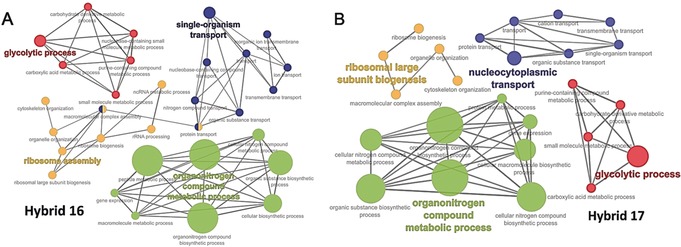
Hybrid 16 (A) and hybrid 17 (B) targets are involved in multiple biological processes essential for parasite survival.
The findings of the proteomic analysis support the notion that hybrid compounds 16 and 17 affect multiple pathways classically recognized for artemisinin action (such as the PfATP6 enzyme) and for quinolines (40S ribosomal protein machinery), which are important targets for development and survival during blood stage P. falciparum growth.26 These findings have important future implications for the development of efficient drug candidates based on ART‐derived hybrid compounds.
Conclusion
The burden of malaria, especially its most fatal species P. falciparum, and the emergence of resistances to known antimalarial drugs (e.g., CQ and ART) are two of the biggest concerns for global infectious disease control. Our work provides access by, among others, environmentally benign C−H activation to novel ART‐based hybrid compounds, which show outstanding activities against the drug‐sensitive 3D7 wild‐type strain and against two multidrug‐resistant strains (Dd2, K1) of P. falciparum parasites, with EC50 values ranging from 780 pm to 27.5 nm. The hybrids are significantly more potent towards resistant parasite strains. Thus these compounds illustrate the high potential of the hybridization concept as an alternative drug discovery approach for efficient treatment and to overcome drug resistance. The potency enhancement of hybrids in comparison to their corresponding parent compounds can be explained through the simultaneous cellular uptake of both pharmacophores covalently bound via a linker, and by possible synergistic effects. These concepts are supported by the observation that one of the most potent hybrid compounds against P. falciparum, the ART–quinoline hybrid 14, demonstrated superior efficacy in comparison to the antimalarial drug artesunate in P. berghei‐infected mice, being in its efficacy only comparable to ACTs. The outstanding efficacy of hybrid 14 might be due to the combination of the pharmacological features of quinolines (potent hemozoin inhibition) and artemisinins (fast parasite killing).
Finally, using a chemical proteomics approach, we identified a comprehensive set of P. falciparum parasite proteins as direct targets of the selected highly active hybrids 16 and 17. The results demonstrate that the hybrid compounds affect targets important for development and survival during blood‐stage P. falciparum development, such as the PfATP6 enzyme (responsible for artemisinin action) and the 40S ribosomal protein machinery (classically recognized for quinoline action).
In general, these findings should provide a valuable basis for further molecular investigations of putative target proteins of these artemisinin‐based compounds. This is an important implication for antimalarial drug design, in particular for hybrid drugs that can simultaneously target protein synthesis (as observed by proteomics) and hemozoin (Hz) formation (as observed by β‐hematin assay) as well as surmount multidrug resistance (inferred by EC50 values). Our results should also encourage further mechanistic studies and be a stepping stone towards overcoming multidrug resistance.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG) through grant TS 87/16‐3. Generous support by the DFG (Gottfried Wilhelm Leibniz award to L.A.), the CSC (fellowship to H.W.), and the Alexander von Humboldt foundation (fellowship to M.M.L.) is gratefully acknowledged. We also thank the Interdisciplinary Center for Molecular Materials (ICMM), the Graduate School Molecular Science (GSMS), as well as the Emerging Fields Initiative (EFI) “Chemistry in Live Cells” supported by Friedrich‐Alexander‐Universität Erlangen‐Nürnberg for funding. We also acknowledge support from the National Natural Science Foundation of China (81841001) and the Major National Science and Technology Program of China for Innovative Drug (2017ZX09101002‐001‐001‐05). We further acknowledge generous support by FAPESB (Brazil, grant number APP0088/2016) and Fiocruz/Inova (grant number 1642178247). A CNPq scholarship for M.C.B.S. and financial support by the German Academic Exchange Service (DAAD) through a doctoral research fellowship for A.Ç. is gratefully acknowledged.
A. Çapcı, M. M. Lorion, H. Wang, N. Simon, M. Leidenberger, M. C. Borges Silva, D. R. M. Moreira, Y. Zhu, Y. Meng, J. Y. Chen, Y. M. Lee, O. Friedrich, B. Kappes, J. Wang, L. Ackermann, S. B. Tsogoeva, Angew. Chem. Int. Ed. 2019, 58, 13066.
Dedicated to Professor Youyou Tu
The copyright line for this article was changed on 5 November 2019 after original online publication.
Contributor Information
Prof. Dr. Jigang Wang, Email: jgwang@icmm.ac.cn.
Prof. Dr. Lutz Ackermann, Email: lutz.ackermann@chemie.uni-goettingen.de.
Prof. Dr. Svetlana B. Tsogoeva, Email: svetlana.tsogoeva@fau.de.
References
- 1.World Health Organization (2018), World Malaria Report 2018; http://www.who.int/iris/handle/10665/275867. License: CC BY-NC-SA 3.0 IGO.
- 2. Bray P., Ward S., O'Neill P. in Malaria: drugs, disease and post-genomic biology, Springer, Berlin, 2005, pp. 3–38. [Google Scholar]
- 3.
- 3a. Tu Y., Nat. Med. 2011, 17, 1217–1220; [DOI] [PubMed] [Google Scholar]
- 3b. Tu Y., Angew. Chem. Int. Ed. 2016, 55, 10210–10226; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10366–10382. [Google Scholar]
- 4.For reviews, see:
- 4a. Jung M., Lee K., Kim H., Park M., Curr. Med. Chem. 2004, 11, 1265–1284; [DOI] [PubMed] [Google Scholar]
- 4b. O'Neill P. M., Ward S. A., Berry N. G., Jeyadevan J. P., Biagini G. A., Asadollaly E., Park B. K., Bray P. G., Curr. Top. Med. Chem. 2006, 6, 479–507. [DOI] [PubMed] [Google Scholar]
- 5. Ding X. C., Ubben D., Wells T. N., Malar. J. 2012, 11, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenthal P. J., Mol. Microbiol. 2013, 89, 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dondorp A. M., Yeung S., White L., Nguon C., Day N. P., Socheat D., von Seidlein L., Nat. Rev. Microbiol. 2010, 8, 272–280. [DOI] [PubMed] [Google Scholar]
- 8. Eastman R. T., Fidock D. A., Nat. Rev. Microbiol. 2009, 7, 864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Walsh J. J., Coughlan D., Heneghan N., Gaynor C., Bell A., Bioorg. Med. Chem. Lett. 2007, 17, 3599–3602; [DOI] [PubMed] [Google Scholar]
- 9b. Capela R., Cabal G. G., Rosenthal P. J., Gut J., Mota M. M., Moreira R., Lopes F., Prudêncio M., Antimicrob. Agents Chemother. 2011, 55, 4698–4706; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9c. Feng T.-S., Guantai E. M., Nell M., van Rensburg C. E., Ncokazi K., Egan T. J., Hoppe H. C., Chibale K., Biochem. Pharmacol. 2011, 82, 236–247; [DOI] [PubMed] [Google Scholar]
- 9d. Teixeira C., Vale N., Perez B., Gomes A., Gomes J. R., Gomes P. J. C. r., Chem. Rev. 2014, 114, 11164–11220. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Tietze L. F., Bell H. P., Chandrasekhar S., Angew. Chem. Int. Ed. 2003, 42, 3996–4028; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4128–4160; [Google Scholar]
- 10b. Gademann K., Chimia 2006, 60, 841–845; [Google Scholar]
- 10c. Meunier B., Acc. Chem. Res. 2008, 41, 69–77; [DOI] [PubMed] [Google Scholar]
- 10d. Tsogoeva S. B., Mini-Rev. Med. Chem. 2010, 10, 773–793; [DOI] [PubMed] [Google Scholar]
- 10e. Fröhlich T., Çapcı Karagöz A., Reiter C., Tsogoeva S. B., J. Med. Chem. 2016, 59, 7360–7388. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Posner G. H., Paik I.-H., Sur S., McRiner A. J., Borstnik K., Xie S., Shapiro T. A., J. Med. Chem. 2003, 46, 1060–1065; [DOI] [PubMed] [Google Scholar]
- 11b. Griesbeck A. G., Neudörfl J. r., Hörauf A., Specht S., Raabe A., J. Med. Chem. 2009, 52, 3420–3423; [DOI] [PubMed] [Google Scholar]
- 11c. Mott B. T., He R., Chen X., Fox J. M., Civin C. I., Arav-Boger R., Posner G. H., Bioorg. Med. Chem. 2013, 21, 3702–3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For pioneering examples of 4-aminoquinoline–trioxaquine hybrids, see:
- 12a. Dechy-Cabaret O., Benoit-Vical F., Robert A., Meunier B., ChemBioChem 2000, 1, 281–283; [DOI] [PubMed] [Google Scholar]
- 12b. Basco L. K., Dechy-Cabaret O., Ndounga M., Meche F. S., Robert A., Meunier B., Antimicrob. Agents Chemother. 2001, 45, 1886–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Reiter C., Herrmann A., Çapci A., Efferth T., Tsogoeva S. B., Bioorg. Med. Chem. 2012, 20, 5637–5641; [DOI] [PubMed] [Google Scholar]
- 13b. Reiter C., Çapcı Karagöz A., Fröhlich T., Klein V., Zeino M., Viertel K., Held J., Mordmüller B., Öztürk S. E., Anıl H., Efferth T., Tsogoeva S. B., Eur. J. Med. Chem. 2014, 75, 403–412; [DOI] [PubMed] [Google Scholar]
- 13c. Ackermann A., Çapcı Karagöz A., Ghoochani A., Buchfelder M., Eyüpoglu I., Tsogoeva S. B., Savaskan N., Oncotarget 2017, 8, 61457; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Held F. E., Guryev A. A., Frohlich T., Hampel F., Kahnt A., Hutterer C., Steingruber M., Bahsi H., von Bojnicic-Kninski C., Mattes D. S., Foertsch T. C., Nesterov-Mueller A., Marschall M., Tsogoeva S. B., Nat. Commun. 2017, 8, 15071; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13e. Fröhlich T., Hahn F., Belmudes L., Leidenberger M., Friedrich O., Kappes B., Coute Y., Marschall M., Tsogoeva S. B., Chem. Eur. J. 2018, 24, 8103–8113; [DOI] [PubMed] [Google Scholar]
- 13f. Çapcı Karagöz A., Reiter C., Seo E.-J., Gruber L., Hahn F., Leidenberger M., Klein V., Hampel F., Friedrich O., Marschall M., Bioorg. Med. Chem. 2018, 26, 3610–3618. [DOI] [PubMed] [Google Scholar]
- 14. Zhou C.-H., Wang Y., Curr. Med. Chem. 2012, 19, 239–280. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Ackermann L., Chem. Rev. 2011, 111, 1315–1345; [DOI] [PubMed] [Google Scholar]
- 15b. Wang H., Koeller J., Liu W., Ackermann L., Chem. Eur. J. 2015, 21, 15525–15528; [DOI] [PubMed] [Google Scholar]
- 15c. Gandeepan P., Muller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452. [DOI] [PubMed] [Google Scholar]
- 16. Kapkoti D. S., Singh S., Luqman S., Bhakuni R. S., New J. Chem. 2018, 42, 5978–5995. [Google Scholar]
- 17.
- 17a. Natarajan A., Guo Y., Harbinski F., Fan Y.-H., Chen H., Luus L., Diercks J., Aktas H., Chorev M., Halperin J. A., J. Med. Chem. 2004, 47, 4979–4982; [DOI] [PubMed] [Google Scholar]
- 17b. De Souza M. V., Pais K. C., Kaiser C. R., Peralta M. A., Ferreira M. d. L., Lourenço M. C., Bioorg. Med. Chem. 2009, 17, 1474–1480. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Paull D. H., Weatherwax A., Lectka T., Tetrahedron 2009, 65, 6771; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Getlik M. u., Wilson B. J., Morshed M. M., Watson I. D., Tang D., Subramanian P., Al-awar R., J. Org. Chem. 2013, 78, 5705–5710. [DOI] [PubMed] [Google Scholar]
- 19. Tripathi R., Dutta G. P., Vishwakarma R. A., Am. J. Trop. Med. Hyg. 1991, 44, 560–563. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Joshi M. C., Okombo J., Nsumiwa S., Ndove J., Taylor D., Wiesner L., Hunter R., Chibale K., Egan T. J., J. Med. Chem. 2017, 60, 10245–10256; [DOI] [PubMed] [Google Scholar]
- 20b. Fielding A. J., Lukinović V., Evans P. G., Alizadeh-Shekalgourabi S., Bisby R. H., Drew M. G., Male V., Del Casino A., Dunn J. F., Randle L. E., Chem. Eur. J. 2017, 23, 6811–6828; [DOI] [PubMed] [Google Scholar]
- 20c. Olafson K. N., Nguyen T. Q., Vekilov P. G., Rimer J. D., Chem. Eur. J. 2017, 23, 13638–13647. [DOI] [PubMed] [Google Scholar]
- 21. Janse C. J., Waters A. P., Kos J., Lugt C. B., Int. J. Parasitol. 1994, 24, 589–594. [DOI] [PubMed] [Google Scholar]
- 22. Conyers R. C., Mazzone J. R., Tripathi A. K., Sullivan D. J., Posner G. H., Bioorg. Med. Chem. Lett. 2015, 25, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klonis N., Crespo-Ortiz M. P., Bottova I., Abu-Bakar N., Kenny S., Rosenthal P. J., Tilley L., Proc. Natl. Acad. Sci. USA 2011, 108, 11405–11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Guo J., Guiguemde A. W., Bentura-Marciano A., Clark J., Haynes R. K., Chan W.-C., Wong H.-N., Hunt N. H., Guy R. K., Golenser J., Antimicrob. Agents Chemother. 2012, 56, 163–173; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Terzić N., Konstantinović J., Tot M., Burojević J., Djurković-Djaković O., Srbljanovic J., Štajner T., Verbić T., Zlatović M., Machado M., Albuquerque I. S., Prudêncio M., Sciotti R. J., Pecic S., D′Alessandro S., Taramelli D., Šolaja B. A., J. Med. Chem. 2016, 59, 264–281. [DOI] [PubMed] [Google Scholar]
- 25. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M., Mol. Cell. Proteomics 2005, 4, 1265–1272. [DOI] [PubMed] [Google Scholar]
- 26. Wong W., Bai X. C., Sleebs B. E., Triglia T., Brown A., Thompson J. K., Jackson K. E., Hanssen E., Marapana D. S., Fernandez I. S., Ralph S. A., Cowman A. F., Scheres S. H. W., Baum J., Nat. Microbiol. 2017, 2, 17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary