

Abstract
Dementias are prevalent brain disorders in the aged population. Dementias pose major socio‐medical burden, but currently there is no cure available. Novel proteomics approaches hold promise to identify alterations of the brain proteome that could provide clues on disease etiology, and identify candidate proteins to develop further as a biomarker. In this review, we focus on recent proteomics findings from brains affected with Alzheimer's Disease, Parkinson Disease Dementia, Frontotemporal Dementia, and Amyotrophic Lateral Sclerosis. These studies confirmed known cellular changes, and in addition identified novel proteins that may underlie distinct aspects of the diseases.
https://onlinelibrary.wiley.com/page/journal/14714159/homepage/virtual_issues.htm.
Keywords: Alzheimer's disease, brain, dementia, mass spectrometer, neuroproteomics
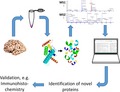
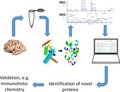
Mass spectrometry based proteomics is an ideal technology to elucidate aberrant protein expression in disease‐affected brain. In this review, we describe the recent proteomics findings from brains affected with Alzheimer's Disease, Parkinson Disease Dementia, Frontotemporal Dementia, and Amyotrophic Lateral Sclerosis. Brain is collected. Proteins are digested by trypsin, and the resulting peptides are analysed by LC‐MS/MS. Results are validated by alternative means, for example immunohistochemistry.
https://onlinelibrary.wiley.com/page/journal/14714159/homepage/virtual_issues.htm.
Abbreviations used
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- Aβ
amyloid beta protein
- CAA
cerebral amyloid angiopathy
- CV
median coefficient of variation
- DDA
data dependent acquisition
- DLB
dementia with Lewy bodies
- ECM
extracellular matrix proteins
- FTD
frontotemporal dementia
- LC‐MS/MS
liquid chromatography coupled to tandem mass spectrometry
- LMD
laser microdissection
- NFTs
neurofibrillary tangles
- PDD
Parkinson disease dementia
- rpAD
rapid AD
Dementias are prevalent brain disorders in the aged population. These collectively affect more than 50 million people worldwide, with an estimated economic cost of one trillion US$ in 2018 (World Alzheimer Report 2015). This major health problem will further escalate due to the increase in life expectancy. Dementia is the consequence of neurodegeneration in the brain, which develops slowly and gradually worsens over many years. Despite extensive research and clinical trials in the past decades, there is no cure for dementia, no large‐scale early diagnostic test is available for the disorders, and crucial mechanistic causes remain unclear or under debate.
There are hundreds of described neurodegenerative diseases (Przedborski et al. 2003), the most common form is sporadic Alzheimer's disease (AD). During the slow progression of AD, patients suffer from initial memory impairments, subsequent decline in multiple cognitive domains, and eventually their complete dependence on daily caretaking. In Western Europe 40% of people at the age of 80 and above suffer from AD. Aging is considered the major risk factor of AD, with few strong genetic risk factors, notably the APOE4 (Mishra et al. 2018) and TREM2 (Sims et al. 2017) genotypes. These genes have been implicated in neuroinflammation through dysfunctional microglia (Krasemann et al. 2017), thereby influencing AD progression. Other genes that have specifically been linked to microglia‐mediated innate immunity in AD are ABI3 and PLCG2 (Sims et al. 2017). Early onset familial AD (Bird 1999) is linked to mutations in genes for amyloid precursor protein (APP) and PSEN‐1 and ‐2. In addition, genome‐wide association studies implicated a number of genes in AD including SORL1, BIN1, CD33, ABCA7 (Seshadri et al. 2010; Wijsman et al. 2011; Shen and Jia 2016; Wang et al. 2016). In particular, distinct SORL1 variants have been shown to have strong influence on AD risk (Holstege et al. 2017).
AD is not a homogenous disease type. Apart from the typical pathological manifestations of AD, such as aggregation of Amyloid beta protein (Aβ) in plaques and hyperphosphorylated Tau (MAPT) in neurofibrillary tangles (NFTs), brain vascular dysfunction in the form of, for example, cerebral amyloid angiopathy (CAA) (Attems et al. 2011) in which Aβ accumulates in the walls of the brain vasculature, may occur in a spectrum ranging from none to high severity in different AD patients. In addition, some AD patients first show non‐amnestic AD symptoms, often with early onset, and are classified as atypical AD patients (Galton et al. 2000).
The second most common neurodegenerative disease is Parkinson disease dementia (PDD). It affects 2–3% of the population older than 65 (Poewe et al. 2017) whereas the related dementia with Lewy bodies (DLB) accounts for 15% to 20% of late‐onset dementias (Campbell et al. 2001). Patients often suffer from mild memory impairment, together with a high burden of motor symptoms. The disease is characterised by the deposition of α‐synuclein (SNCA) positive inclusions, called Lewy bodies, in the substantia nigra. The cause of the disease is incompletely understood, but it is thought to result from a complex interplay of genetic and environmental factors. Onset of the disease before age 50 is rare, but multiple mutations are known to cause early‐onset Parkinson disease (Poewe et al. 2017).
Amyotrophic lateral sclerosis (ALS) and Frontotemporal dementia (FTD) are two other commonly occurring forms of neurodegenerative disease. ALS patients develop muscle weakness, whereas FTD patients develop cognitive dysfunction, often including behavioural changes (Ferrari et al. 2011). Many patients have overlapping clinical and pathological phenotypes of both FTD and ALS, suggesting shared underlying pathological mechanisms of these two seemingly distinct disorders. Multiple gene mutations have been implicated in FTD and ALS, partly overlapping with other neurodegenerative disorders, such as mutations in the MAPT and Progranulin (PGRN) gene, which can also be seen in AD, progressive supranuclear palsy and corticobasal degeneration, and TDP‐43 (TARDBP) mutations, which occur both in ALS and FTD. Other mutations in ALS and FTD include SOD1, FUS and c9orf72 mutations and lead to specific disease subtypes (Ferrari et al. 2011).
A typical pathological hallmark of neurodegenerative diseases is the accumulation of one or more distinct proteins in aggregates, as well as their distribution and spread throughout the brain. These proteins may be modified post‐translationally in the affected brains, for example the hyperphosphorylated Tau that aggregates into NFTs in the cytoplasm of neurons, and the processing of APP via the β‐secretase (BACE1), that leads to the increased formation of Aβ plaques (Serrano‐Pozo et al. 2011). This suggests that proteinopathies play important roles in disease progression, where the capacity of proteins to aggregate is correlated with toxicity (Rubinsztein 2006). Misfolded proteins often possess prion‐like properties and propagate into connected healthy brain tissue (Goedert et al. 2017; Ruiz‐Riquelme et al. 2018).
This observation forms the basis of current disease staging schemes, which are based on the spatio‐temporal distribution of protein aggregates in different brain regions as revealed by (immuno‐) histochemical examination of the post‐mortem brain. In AD, the main staging schemes include the Braak stage for NFTs (Braak and Braak 1991), the CERAD score for neuritic plaques (Mirra et al. 1991) and the Thal phase for Aβ deposition (Thal et al. 2000). Since the presence and severity of each of these pathologies is thought to be important for the development and severity of AD, the National Institute on Aging‐Alzheimer's Association has introduced a staging system built up of all three pathologies (Hyman et al. 2012). NFTs first appear in the entorhinal cortex and hippocampal formation and later during the disease also appear in the neocortex. The spread of NFTs into the temporal cortex in Braak stage III to IV often coincides with the first appearance of symptoms, and Braak stage V or VI is observed in more severe stages of the disease (Braak and Braak 1991; Imhof et al. 2007). Aβ plaques first appear in the neocortex and only later affect the hippocampal formation and brain stem (Thal et al. 2000). Aβ accumulation preferentially starts in the precunes, medial orbitofrontal and posterior cingulate cortices, which may affect brain connectivity (Palmqvist et al. 2017). However, the distribution of Aβ plaques does not correlate with disease severity (Nelson et al. 2012). Recent studies provide clues to reconcile the spatio‐temporal segregation of Aβ and Tau in the progression of AD. The pathogenic Tau molecules initially deposited in the entorhinal cortex may spread in a prion‐like manner (Kfoury et al. 2012) via connecting tracks such as the hippocampal cingulum bundle to the medial parietal, lateral parietal and medial prefrontal cortex (Jacobs et al. 2018). These are regions with heavy and early deposition of Aβ. The convergence of toxic Aβ and Tau negatively affects these brain regions, and may account for the early episodic memory loss.
For PDD, the α‐synuclein inclusions initially appear in medulla oblongata and the olfactory bulb (stage 1–2), spread to pons, midbrain and basal forebrain (stage 3–4), and finally fill up the whole neocortex (stage 5–6). However, the association between stages and clinical severity of PDD are not apparent (Jellinger 2009). It should be noted that α‐synuclein accumulations can also be observed in around 40% of all AD cases, and parkinsonism can often be seen in advanced AD patients and cases with other forms of dementia (Spires‐Jones et al. 2017; Umoh et al. 2018). Especially in older patients, such overlap of pathologies as co‐morbidities is common. How these pathologies interact, aggravate each other, or have an additive effect on the observed symptoms, is still debated (Spires‐Jones et al. 2017).
The majority of dementias occur sporadically with unclear etiopathogenesis. As the underlying molecular changes of the diseases are believed to be polygenic and complex, the explorative nature of the –omics technologies, which give global views of the alterations at the levels of genome/transcriptome, proteome and metabolome, hold promise to unravel the molecular pathways underlying dementias (Redensek et al. 2018). Furthermore, they may give clues to uncover biomarkers for diagnosis. GWAS studies implicated several gene loci that may associate with AD. GWAS studies vary greatly in methodology and sample size, and consequently identify different, but also overlapping loci (Visscher et al. 2017). Some loci have repeatedly been reported and validated over several years, such as BIN1 and CLU (Seshadri et al. 2010; Wijsman et al. 2011; Wang et al. 2016), while new loci were reported recently, including epigenomic changes (Marioni et al. 2018) and certain variants of SORL1 (Holstege et al. 2017). How these gene products contribute to the development of AD is largely unknown. Transcriptomics is the method of choice to reveal differences in gene expression, and has been applied to examine differential expression of mRNA in AD patients. However, a recent large‐scale RNAseq analysis showed that the mRNA from AD patients was of generally poorer quality, which complicated their comparative analyses with the healthy controls (Miller et al. 2017). Furthermore, overlap of the RNA and protein dysregulation from AD studies showed only the modest similarity (Seyfried et al. 2017).
Proteomics technology
Many dementias are described as proteinopathies. As proteomics aims to reveal tissue‐ or cell‐specific proteomes both qualitatively and quantitatively, it can potentially detect post‐translational protein modifications. It is therefore thought to be an ideal technology to elucidate the aberrant protein expression in disease‐affected brain. However, proteomics analysis of dementias met limited success for years; it was hindered by the complexity of disease etiology in brain areas with specific spatio‐temporal protein expression patterns, the profound individual variation between human subjects, and the scarcity and limited availability of correctly diagnosed diseased brain tissues of high quality. The advancement of mass spectrometry technology today with fast acquisition rate, high mass accuracy, high resolution, and ultra‐sensitivity allows a proteomics workflow that approaches the whole tissue/cell proteome detection in a reasonable time frame using low amount of tissue/cells (Hosp and Mann 2017) obtained from clinically relevant human samples.
A potential confounding factor for the interpretation of the data is the effect of post‐mortem delay time on brain tissue. A recent study revealed that none to only a few genes in human cerebellum and cortex, respectively, showed a changed expression within a few hours of post‐mortem cold ischaemia interval (Ferreira et al. 2018). In addition, cytoskeletal and synaptic proteins in mouse brain were shown to be generally stable up to 6 h post‐mortem delay, whereas at 24 h post‐mortem delay many of these were partially degraded (ElHajj et al. 2016). It is expected that short post‐mortem interval of a few hours, which nowadays is becoming the desirable norm for the collection of brain autopsies, should generally improve the analysis of brain proteomes. Thus, the collaboration of neuroproteomics researchers, neuropathologists and national and international brain banks, aids in alleviating the problem in proper sample acquisition. Together, the global changes of proteins in specific brain regions in various human dementias are starting to become elucidated.
In the present review we focus on recent publications that generally quantified thousands of proteins from which the affected cellular pathways were revealed or implicated. The prior proteomics studies (e.g., Chen et al. 2012; Donovan et al. 2012; Zhou et al. 2013; Musunuri et al. 2014; Dammer et al. 2015; Zelaya et al. 2015), and studies involving animal and cellular models, have largely been covered by previous reviews (Fasano et al. 2016; Moya‐Alvarado et al. 2016; Monti et al. 2018) and are not included in this review.
Proteomics is a term referring to studies interrogating the proteome of interest from biological samples qualitatively or quantitatively. Currently, the bottom‐up proteomics approach is the method of choice. It makes use of liquid chromatography coupled to tandem mass spectrometry (LC‐MS/MS) for the identification and quantification of tryptic peptides derived from the solubilised and trypsin‐digested proteins. Recent studies reported the quantification of 2000 or more proteins, and in most cases provided sufficient details to reveal the major alteration of molecular pathways in the disease‐affected brain (e.g., Hondius et al. 2016, 2018a) and others, see below sections for detailed description).
Two main quantitative proteomics approaches are often used for the analysis of biological samples, the label‐free data dependent acquisition (DDA) and the isobaric multiplex labeling strategies for relative quantitative proteomics (using reagents iTRAQ or TMT). The workflow of neuroproteomics is shown in Fig. 1. Peptides of different hydrophobicity are partially separated by reverse phase LC. All peptides eluted at a given chromatographic time are electro‐sprayed and simultaneously detected by the mass spectrometer as distinct precursor ions in the MS1 scan. The intensity of each precursor ion (peak height or peak area) correlates linearly to peptide concentration, and is used for peptide quantitation by DDA. For peptide identification, several abundant precursor ions, which can range from 5 to 40 depending on the speed and sensitivity of the mass spectrometers, are sequentially selected within a time frame of usually 1–2 s for fragmentation. A longer acquisition time per MS/MS improves detection, however, reduces the number of interrogated peptides. Subsequent database search of the fragmentation pattern on the MS2 spectrum infers the amino acid sequence of the peptide. Today, many database search engines are available as commercial packages or freeware. MaxQuant (Tyanova et al. 2016) is a popular freeware for academics. Alternatively, Morpheus (Wenger and Coon 2013) performs equally well with several times faster analysis time, but it only works with MS spectra generated from high resolution mass spectrometers. After a database search, the output of a large number of identified and quantified proteins should be translated into meaningful biological findings by, for example, statistical testing and clustering of functional relevant proteins. Perseus is one of the useful tools for this analysis (Tyanova and Cox 2018).
Figure 1.
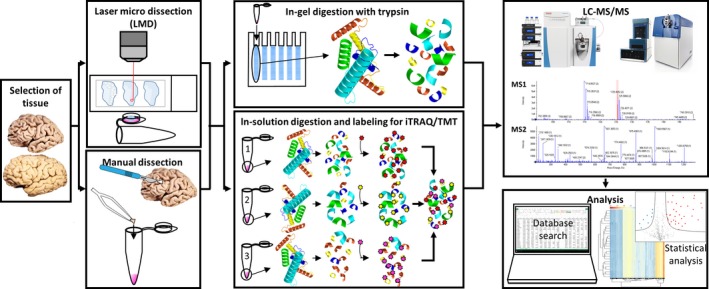
Quantitative neuroproteomics workflow. From left to right panels are the schematic representations of sample selection from Alzheimer's disease brains or heathy controls, the collection of brain areas by laser microdissection (LMD; upper panel) or manual dissection (lower panel), sample preparation for the downstream analysis by data dependent acquisition (upper panel) or isobaric multiplex labeling strategies for relative quantitative proteomics (lower panel), and the actual measurements of the peptides by LC‐MS/MS and the subsequent data analysis.
In iTRAQ/TMT experiments, all tryptic peptides from a certain sample are N‐terminally tagged by one of the eight sets of iTRAQ or the 10 sets of TMT reagents, respectively. Peptides from the next sample are tagged by the second set of iTRAQ or TMT reagents, etc. In total, peptides from a maximum of eight (iTRAQ) and 10 (TMT) samples can be separately tagged, and then pooled for LC‐MS/MS analysis. MS/MS of the peptide produces fragment ions for peptide identification. It also breaks the tags to release the signature reporter ions that are detected at low mass range and have no interference from known peptide fragment ions. The intensities of the reporter ions correlate to the peptide concentration and are used for comparative quantitation of each fragmented peptide tagged from the eight (iTRAQ) or 10 (TMT) samples.
In addition to the abundance analysis of proteomes in neurodegenerative diseases, LC‐MS/MS is also applicable for the studies of global protein post‐translational modifications including phosphorylation and glycosylation (Lassen et al. 2017; Wang et al. 2017; Ferrer et al. 2018) and the metabolomes (Toledo et al. 2017; Gill et al. 2018). Sample preparation and the analysis are generally more challenging than that of a typical proteomics experiment.
Proteome changes during AD progression
It is widely accepted that neurodegenerative diseases are caused by aberrant molecular and cellular alterations in the brain that worsen over decades after onset. Nevertheless, identity of the early disease‐related processes and their development into neurodegeneration are largely unknown. This pre‐symptomatic state of AD is of great interest for designing preventive or progression‐halting treatments.
At present the earliest molecular changes documented come from the study of Hondius et al. (2016), who examined the protein profile changes over all AD Braak stages. The distribution of NFTs correlates with the severity of the dementia, and the topographical distribution of NFTs matches the neuropsychological profiles of typical AD. As neurofibrillary degeneration starts in the allocortex of the medial temporal lobe, the hippocampal subregions CA1 and subiculum were targeted for analysis. This defined brain region was collected by laser microdissection (LMD) from 40 cases with 5–7 cases per Braak stage. Samples were selected to not exhibit comorbidities; for example AD patients with lewy body pathology in the hippocampus were excluded. Each sample was run on an sodium dodecyl sulfate‐acrylamide gel and the gel was split into 12 slices, digested by trypsin, and LC‐MS/MS analysed in an Orbitrap Discovery mass spectrometer. Database search was performed with MaxQuant. A linear regression analysis was applied across the Braak stages at a significance cutoff level of 10% false discovery rate. In total, about 3000 proteins were identified of which 166 proteins showed higher, and 206 showed lower levels. From these 372 proteins, 89 showed significant differences in the early phase of AD between Braak stages 0‐III. Cluster analysis showed the profiles of Braak stages 0‐III and IV–VI as two major groups, suggesting major changes occurring around Braak stages III–IV.
To validate the mass spectrometric data, immunoblotting and immunohistochemical staining of selected proteins were performed. As expected, the established markers for AD, MAPT, GFAP and CD44, all showed profound up‐regulation that followed the Braak stages. For other proteins including ANXA2 and 5, FLNA, VCL, TLN and TNC, the direction and extent of fold changes in AD were consistent among these three different approaches.
The clustering analysis grouped proteins with similar expression patterns over the Braak stages that showed (i) progressive decrease, (ii) early up, late down and (iii) progressive increase. The up‐regulated group was over‐represented by astrocytic genes. This is in agreement with the increasing astrogliosis during the progression of AD (Garwood et al. 2017). Astrogliosis is known to contribute to inflammation that can cause damage to the brain (Ferrer 2017). Indeed, the higher expressed proteins also included those involved in response to NRF2‐mediated oxidative stress, glycolysis and cytoskeletal rearrangement. In the lower expressed protein group, neuronal proteins were over‐represented. This correlates well to the loss of synapses and eventual neuronal death. The high‐low group is suggested to reflect a compensatory mechanism that includes proteins involved in axonal guidance and oxidative phosphorylation pathways.
An alternative analysis is to examine the expression pattern of proteins that belong to the same functional group to emphasise their concomitant changes in AD progression. The extracellular matrix proteins (ECM), such as TNC, VCAN, LAMB2 and LGALS1, and the adhesion molecules that interact with ECM, such as CD44, ITGB1 and ITGAV, showed global higher expression. The increase in ECM has also been detected in an AD mouse model and has been shown to affect neuronal plasticity (Vegh et al. 2014).
Microtubules and associated proteins are essential for intra‐cellular transport. These proteins either showed no change (MAPs) or decrease (TUBBs). The exception was MAPT which increased progressively. This reflects the abundant accumulation of aberrant forms of MAPT in the neuron as neurofibrillary tangles. It is known that the pathologic MAPTs are hyper‐phosphorylated. As this study was not designed to detect phosphorylation nor to differentiate MAPT isoforms, it remains unclear which MAPT isoforms and their phosphorylation patterns were altered during progression of AD. A previous study identified seven types of tau modifications at 63 sites in mice (Morris et al. 2015). The glutamate receptors AMPA receptor subunits GRIA 1–3 and NMDA receptor subunits GRIN1 and 2B) showed progressive decline. A recent study (Reinders et al. 2016) indicated that loss of GRIA3 may protect against Aβ‐associated memory impairment. Unlike α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionate and NMDA receptors, several presynaptic proteins (BSN, LIN7A, RIMS1 and RAB3C) and post‐synaptic proteins (DLG2‐4, DLGAP1, HOMER1 and SHANK3) showed an initial up‐regulation in Braak stages I–III, followed by a decline towards Braak stage VI. On the other hand, typical vesicle integral proteins, such as VAMP2 and SV2A, showed no change. Several endocytotic proteins such as CLTA and AP2M1 showed reduced expression, and SNAP91, AP2A1, AP2B1 and DNM1 showed early decrease, suggesting an early endocytic dysfunction. The global decline of all these pre‐ and post‐synaptic proteins from stage IV onwards signals the loss of synapses, through which cognitive decline might occur.
More recently, Lachen‐Montes and colleagues (Lachen‐Montes et al. 2017) examined proteome changes in the olfactory bulb from each 3–5 cases of healthy controls, as well as initial, intermediate and advanced AD stages. Samples were extracted in urea lysis buffer containing CHAPS and run very briefly in an sodium dodecyl sulfate acrylamide gel. The small gel piece containing all the proteins was cut and digested using trypsin. The peptides mixtures were fractionated with a 240 min gradient of LC run and analysed in a label‐free data dependent mode using a Sciex 5600 TripleTop MS. The relatively long LC run time should allow acquisition of a higher number of MS/MS events, which should lead to an increase in number of proteins identified. From the 1311 quantified proteins across all experimental groups, 110, 125 and 159 proteins from initial, intermediate and advanced AD cases, respectively, were differentially expressed when compared to the healthy controls. Significantly, more than 50% of these proteins tend to localise to the synaptic terminal, indicating a progressive synaptic degeneration during AD progression. When network analysis was performed, the initial stages showed dysregulation in the actin based cytoskeleton and the regulators of the interaction between components of the cell‐cell junction, the intermediate stages showed impaired mitochondrial functions and imbalanced redox signaling, and the advanced stages seemed to have impaired RNA stability and pre‐mRNA splicing processes.
Proteomes of asymptomatic and symptomatic AD
One major pathological hallmarks of AD is deposition of Aβ leading to Aβ plaques formation in the brain. However, the spatio‐temporal appearance pattern of Aβ plaques does not always correlate to the severity of the AD symptoms, especially at advanced ages (Sperling et al. 2011; Ganz et al. 2018).
To examine why individuals can have high amounts of Aβ without clinical signs of AD, Seyfried and coworkers compared 15 asymptomatic (AsymAD) and 20 symptomatic AD cases with 15 healthy controls from dorsolateral prefrontal cortex and precuneus (Seyfried et al. 2017). Precuneus is the first brain region with Aβ aggregation that spreads to orbitofrontal cortex and then neighbouring areas. This early Aβ accumulation is associated with hypoconnectivity within the default mode network and to the frontoparietal network (Palmqvist et al. 2017). At later stages the patients show glucose hypometabolism and eventually atrophy.
In the study of Seyfried, each piece of brain tissue was individually weighed (∼0.1 g) and homogenised in 500 μL of urea lysis buffer. After digestion with lysyl endopeptidase and trypsin, peptides were analysed on a Q‐Exactive plus mass spectrometer with a 2 h LC gradient acquired in DDA mode. About 5000 proteins were identified, from which 2735 protein groups which were present in 90% of samples were used for quantitation. When compared to the controls, 63 and 280 proteins from AsymAD and AD, respectively, showed significant differences. Thus, the much larger number of changes in AD correlated to the increased pathological burden.
Several proteins showed progressive changes from controls to AsymAD and AD. The ECM proteins CSPG, ACAN and PTN showed progressive increase, whereas proteins involved in synaptic function and synaptogenesis showed progressive decrease (SYNPO1, VGF, CAMKK2, CAMK4). This is in general agreement with the proteome changes observed in the hippocampus according to the Braak stages, in which synaptic proteins showed a decrease and ECM proteins increase progressively (Hondius et al. 2016). CLU and PEPD were altered exclusively in the symptomatic phase of AD.
Proteins involved in a distinct biological process could be co‐regulated in different disease stages. To reveal protein co‐expression, weighted gene co‐expression network analysis was applied and identified 16 modules ranging from the biggest module of 396 proteins to the smallest 28 proteins. Several modules of expression profiles showed significant decrease in AD. They were enriched with neuronal markers or mitochondrial proteins, consistent with the hypometabolism and synapse loss observed in AD patients (Mattson 2004). A module linked to inflammatory response was increased in AsymAD. This suggests the activation and proliferation of astrocytes and microglia. Another module enriched with microglia and astrocyte markers was increased only in AD. When the risk loci based on the GWAS data were used, the AD candidate genes were over‐represented in a module enriched in oligodendrocyte, and a module in astrocyte and microglia.
Together, the studies of Seyfried and Hondius (Hondius et al. 2016) reveal similarities in the alteration of functional protein groups in AD; both point to the increase of glial cell protein networks and the decrease in specific neuronal proteins as important factors that probably lead to cognitive and memory impairment.
Region specific protein expression in AD
It is clear that different brain regions develop different AD pathologies at different times. To reveal these spatial changes in the brain, Xu and colleagues (Xu et al. 2018) reported the proteomics analysis of six brain regions from controls and AD patients. The selected regions were grouped as heavily affected (hippocampus, entorhinal cortex and cingulate gyrus), lightly affected (sensory cortex and motor cortex), and the least affected cerebellum that lacked NFTs and showed only low numbers of Aβ Plaques. 8‐plex iTRAQ technology was used for quantitative proteomics analysis. A total of 4825 proteins were quantified in at least one brain region, and 1899 proteins were assigned to all six regions.
As expected, the heavily affected regions showed the highest number of protein level changes of around 30%, whereas the lightly affected regions have only 11–13% abundance changes. Unsupervised clustering analysis demonstrated a similar pattern of expression changes for these five regions, and that the lightly affected regions undergo molecular alterations equivalent to those in earlier stage of AD in the heavily affected regions. Interestingly, the supposedly spared cerebellum registered a relatively large change of 20%, with a profile distinct from the other five regions. The most consistent changes across all brain regions were the proteins and pathways involved in the immune response. Proteins involved in apoptosis and cell cycle regulation were dysregulated in severely affected regions. Metabolic impairments were also observed. Correlation network analysis identified four proteins with the most overall influence on AD pathogenesis (STXBP1, CRMP1, ACTR10 and AMPH). This points to the importance of neurotransmission processes. This study also recapitulated the molecular changes observed in hippocampus and precuneus.
The protein changes observed in the cerebellum were distinct from those in other regions. The cerebellum showed stronger reduction in the abundance of proteins involved in the electron transport chain and an increase in oxidative defense proteins. Together, they may provide a protective mechanism that decreases ROS‐production while increase ROS defenses. Furthermore, the Purine ribonucleosides degradation pathway was activated. Therefore, it is proposed that the cerebellum actively induces a unique pattern of up‐regulated neuronal survival pathways alongside protection against oxidative and inflammatory damage, which protects the cerebellum from degeneration.
Proteome from AD patients with cerebral amyloid angiopathy
CAA involves the deposition of amyloid in the brain vasculature. Patients with CAA have an increased risk of cerebral infarction, cerebral haemorrhage and micro‐bleeds. There are several types of CAA based on the amyloid protein involved (Yamada 2015). Among them, the sporadic Aβ type CAA is often detected in up to 80% of all AD patients. CAA occurs as two distinct types. CAA‐type 2 refers to cases where deposition of Aβ is present only in larger blood vessels, which include leptomeningeal vessels, cortical arteries and arterioles. In CAA‐type 1 cases, brain capillaries are also affected (Yamada 2015). In some AD cases, CAA type‐1 appeared to be the primary cause of rapid progressive dementia.
To examine the pathological differences of Aβ deposition in parenchyma and those related to the vasculature, Hondius and colleagues performed a discovery proteomics analysis on laser capture microdissected tissues of the occipital tissues from six cases of healthy controls without any Aβ or tau pathologies, seven cases of AD with severe plaque pathology but no vascular deposits, and seven cases of AD with severe CAA‐type 1 pathology and a negligible amount of plaque pathology (Hondius et al. 2018a). The occipital lobe was chosen because it is the brain region most heavily affected with CAA pathology. Grey matter enriched for Aβ aggregates was collected for analysis. The selected tissue areas have high plaque load in the AD cases, and contained many affected capillaries and larger vessels in the CAA cases. The same anatomical regions were isolated from control cases. More than 2000 proteins were identified in total. The authors took an exploratory approach with relaxed tests aimed to discover CAA‐type 1 specific proteins, while allowing a higher rate of false positives that can subsequently be falsified by independent focused studies. For example, a protein should be present in most CAA cases but at most once in AD and/or control cases, and vice versa. Nine proteins fulfilled this criterion (HLA‐DRA/DQA2, HTRA1, APCS, COL6A2, MOB2, POTE1, KIAA1468, TMF1 and SGIP1). For most other proteins, a simple t‐test with p < 0.05 was applied, which yielded 20 proteins (CLU, APOE, SUCLG2, PPP2R4, KTN1, ACTG1, TNR, COL6A3, NFASC, APP, UBLCP1, SR1, NDP, PNP, C1orf123, DHX15, SYNPO, TPM1, CADPS2 and SERPINA3).
Based on the fold change or specific expression in the CAA group compared to the AD and control groups, several proteins were selected for validation. APOE, NDP, APCS and COL6A2 all showed the same trend of an increased abundance in the CAA group. Immunohistochemistry subsequently confirmed the MS results. For example, NDP was abundantly present around the vasculature in the CAA cases. For cases that have dysphoric Aβ, diffuse staining of NDP was found in the parenchyma. NDP was not detectable in controls. When IHC was quantified, NDP and COL6A2 showed strongly increased immmunoreactivity specifically in CAA cases, and moderate increase for APCS, HTRA1 and APOE.
IHC revealed that the CAA specific proteins are also present in other brain vascular defects. In short, COL6A2 appeared as a general small vessel disease marker. NDP, APOE and APCS were abundant in CAA and disease with cotton wool plaque pathology, as well as prion CAA. They were present in lower level in cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy (CADASIL). In particular, the study suggested NDP as a useful marker to separate CAA from Aβ plaque pathology.
Two recent studies reported the proteomics of CAA‐type 2. As leptomeningeal and cortical vessels are large and easily distinguished from other structures, they were either removed from the brain with a micro‐scalpel or dissected via LMD. Inoue and coworkers performed proteomics on eight cases of severe CAA, 12 cases of mild CAA and 10 controls (Inoue et al. 2017), 60% of the CAA cases and 15% of control cases had Braak stages of three and above. It is stated that six proteins were significantly higher expressed in severe CAA, i.e., Aβ, APOE, GFAP, ENO 1/3, and SRPX1. IHC revealed the co‐accumulation of SRPX1 and Aβ in cerebral blood vessels. A subsequent functional study indicated that SRPX1 may increase Aβ‐induced cerebrovascular degeneration in CAA. Manousopoulou and coworkers collected five cases of young controls, seven cases of elderly controls and five cases of severe CAA (Manousopoulou et al. 2017). The iTRAQ technology was chosen for quantitative proteomics analysis. The caveat is that a maximum of eight samples can be processed in parallel per analysis. Therefore, two young and two elderly controls and four CAA cases were randomly selected for the proteomics analysis. With this low number of replications, data analysis to reveal differential expression of proteins is challenging. The authors reported a large number of proteins that show differences among the young and elderly controls and the CAA cases. IHC revealed the co‐localisation of CLU with Aβ within the walls of leptomeningeal arteries in the CAA cases. TIMP2 staining was found to be restricted to walls of arteries, and increased in CAA. In a separate study using a mouse model (Wojtas et al. 2017), CLU was suggested to function as a major Aβ chaperone to maintain Aβ solubility along interstitial fluid drainage pathways and prevent CAA formation.
Analysis of amyloid plaques distinguish rapidly progressive and sporadic AD
In a small population of AD patients the disorder progress rapidly, with a median survival time of 7–10 months (called rapid AD; rpAD). This is in contrast to the typical sporadic AD, which develops slowly for years. The rpAD patients do not have pathogenic mutations in APP or PSEN1/2. The pathological hallmarks of Aβ plaque and neurofibrillary tangle are comparable between the rapidly progressive AD and the sporadic patients.
To better understand rpAD, Drummond and colleagues compared the proteomes of amyloid plaques isolated from 22 cases each of rpAD and sporadic AD (Drummond et al. 2017). The immunohistochemically stained plaques from formalin‐fixed paraffin‐embedded tissue blocks were selected and microdissected via LMD and about 740 plaques per sample were collected. The tryptic peptides were analysed with a Q‐Exactive MS using a 2 h LC‐gradient in DDA mode. About 900 proteins were identified per sample, in which 279 proteins were consistently detected from every case, including the typical amyloid plaque proteins Aβ, APOE, tau, GFAP and CLU. When compared to sporadic AD cases, the rpAD cases have 85 proteins with increased expression (for example SNCA) and 56 with decreased (for example Aβ, GSN and GFAP) expression. Some other important plaque‐associated proteins (for example tau, ubiquitin and APOE) were similarly expressed. The rpAD plaques contained significantly higher level of neuronal proteins and lower levels of astrocyte proteins. Pathway analysis suggested an increase in neurotransmission related proteins. It was proposed that the altered process of synaptic vesicle release may play a role in plaque formation, which may then contribute to the rapid disease progression.
Proteomics analysis of other dementias
AD and Lewy body dementia are the most common forms of neurodegenerative diseases. Bereczki and colleagues compared the prefrontal cortex proteomes from 32 cases (eight controls, eight PDD, seven DLB and nine AD) (Bereczki et al. 2018). Peptides from each sample were labelled with TMT 10‐plex reagents. After pooling eight samples and two internal control samples, peptides were fractionated by isoelectric focusing on an IPG strip that separated peptides according to their charges. 72 fractions were recovered from the IPG strip and analysed by LC‐MS/MS separately. The data were acquired on a Q‐Exactive mass spectrometer with a 74 min LC gradient. In total 10 325 proteins were identified, of which 7033 proteins were common to all 32 samples. When compared to controls, there were 1010 differentially expressed proteins in DLB, 485 in PDD, and 593 in AD. Among the 851 proteins related to synaptic transmission, 25 were significantly altered in these dementias including presynaptic proteins (GRIK2, CAMK2A, BDNF, PDYN), synaptic vesicle priming proteins (SNAP47), synaptic vesicle proteins (SV2, SYT2), proteins found in both pre‐ and post‐synapse (GAP43, LRFN2) and post‐synaptic proteins (GRIA3, GRIA4, ARC, CNIH2, PVRL3, NRGN). As this study focused on the synaptic markers of neurodegenerative diseases, the bulk of other regulated proteins that may give clues to disease mechanisms remains to be interrogated in detail.
To differentiate the differences between PDD and DLB, Datta and coworkers performed an iTRAQ experiment on two replicates of pooled samples from BA9 area from patients of PDD, DLB and controls (Datta et al. 2017). The lysed tissues were trypsin digested, labelled with iTRAQ, fractionated by anion‐exchange chromatography into 20 fractions. Each fraction was analysed by LC‐MS/MS on a QStar mass spectrometer with a 90 min LC gradient. A total of 1914 proteins were identified, but none of the proteins showed a significant and opposite regulation between DLB and PDD when compared to controls. The authors contributed the similarity of DLB and PDD to the amyloid burden, which is a common co‐morbidity, especially in the elderly cases (Spires‐Jones et al. 2017).
ALS and FTD are clinically distinct diseases, yet many ALS patients also develop FTD, and vice versa, suggesting common disease mechanisms. Umoh and coworkers compared the frontal cortex proteome changes from patients of ALS, FTD, ALS/FTD, and healthy controls (Umoh et al. 2018). Protein co‐expression network analysis revealed 15 modules. The modules showed that proteins with increased expression in FTD were enriched for RNA splicing, response to biotic stimuli, zinc ion binding, homeostatic processes and blood microparticles and circulation immunoglobulin complexes. The latter process implicates damage of the blood‐brain barrier. The decreased modules include synaptic and neuronal proteins and mitochondrial proteins. The protein expression for ALS cases was similar to the controls. This is consistent with the fact that TDP‐43 pathology is not found in the frontal cortex, and that there is no cognitive decline. For the ALS/FTD group, the changes in the modules were the intermediate between ALS and FTD groups.
Proteomics studies of fluid biomarkers for neurodegenerative diseases
The current diagnostic tests for AD and FTD are dominated by the identification and quantification of Aβ, tau or phospho‐tau in the cerebrospinal fluid (CSF) (Blennow and Zetterberg 2018; Mattsson et al. 2018), which reflect the plaque and tangle pathologies in the brains of patients. However, co‐pathologies are common in dementia and cannot be distinguished by measuring Aβ/tau alone. Additional biomarkers should be included to improve specificity. In the past decade, studies on biomarkers in CSF focused mainly on one or a few proteins, and only few studies took an explorative proteomics approach (Portelius et al. 2017). While a few potential biomarkers have been proposed, none have reached clinical use as diagnostic tool.
A label free MS quantitation has been carried out on CSF of FTD patients with TDP‐43 (n = 12) or tau (n = 8) pathology, and cases with subjective memory complaints (n = 10) as controls (Teunissen et al. 2016). More than 1900 proteins were identified in total, of which 56 proteins were considered differentially regulated between groups. ELISA assays were performed on a small set of these proteins, namely FABP4, YKL‐40, complement factor D, IL1RAP and APOL1, where only YKL‐40 could be validated to exhibit higher levels in FTD cases than controls. YKL‐40 has been found to be increased in different neurodegenerative diseases (Alcolea et al. 2014), and may reflect the abnormality of astrocyte activity associated with inflammatory processes (Llorens et al. 2017).
The comparison of CSF proteomes of AD and controls was reported by Khoonsari et al. (Khoonsari et al. 2016). About 200–300 proteins were identified, many of which showed differences between the AD and control groups. Eight proteins (A2GL, APOM, C1QB, C1QC, C1s, FBLN3, PTPRZ and SEZ6) were further verified by an antibody‐based method to have lower levels in AD cases.
Future perspectives in neuroproteomics
A typical brain tissue sample contains thousands proteins, and an even larger number of isoforms (proteoforms) generated from alternative splicing of a gene or post‐translational modifications of the protein. A pre‐fractionation of tryptic peptides in conjunction with the use of a state‐of‐the‐art mass spectrometer, such as the Obitrap HF‐X with 40 Hz scanning speed (Kelstrup et al. 2018) or the ion mobility Q‐Top mass spectrometer (Tims TOF pro) equipped with PASEF technology (Meier et al. 2015), enable the identification of over 10 000 protein groups and tens of thousands of phosphopeptides by data dependent acquisition (Bekker‐Jensen et al. 2017). These most advanced proteomic pipelines rival the coverage generally achievable by transcriptomics. This degree of coverage, however, should be balanced by the analysis time, the reproducibility across multiple fractions from single sample, and the high cost and availability of the advanced mass spectrometers. Alternatively, data independent acquisition (SWATH) holds promise to interrogate several thousand proteins in 1–2 h of analysis time from commonly used research grade mass spectrometers (Kelstrup et al. 2018; Koopmans et al. 2018). This technique has the potential to develop into a high throughput methodology and is particularly useful for experiments of large sample sizes, such as the analysis of multiple brain regions across different neurodegenerative stages from various dementias.
In addition to the mass spectrometry itself, also the auxiliary technology, such as LMD as a mean to get to single cell type resolution in proteomics analysis, are being developed. Specific mass spectrometry compatible staining protocols (Hondius et al. 2018b) are contributing tremendously to the use of mass spectrometry in neurodegenerative disease.
Conclusion
LC‐MS/MS is the driving force of proteomics studies. With the current advancement of mass spectrometers in sensitivity and speed, it is now possible to interrogate thousands of proteins from whole tissue lysates. The application of quantitative bottom‐up proteomics in the past few years is starting to reveal the alteration of proteins and their networks during progression of neurodegeneration, and suggest candidate proteins as potential biomarkers for specific neurodegenerative diseases.
Acknowledgments and conflict of interest disclosure
Ganz, A.B.is funded by Stichting Dioraphte (VSM 14 04 14 02). The authors acknowledge no potential conflicts of interest.
References
- Alcolea D., Carmona‐Iragui M., Suarez‐Calvet M. et al (2014) Relationship between beta‐Secretase, inflammation and core cerebrospinal fluid biomarkers for Alzheimer's disease. J. Alzheimers Dis. 42, 157–167. [DOI] [PubMed] [Google Scholar]
- Attems J., Jellinger K., Thal D. R. and Van Nostrand W. (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 37, 75–93. [DOI] [PubMed] [Google Scholar]
- Bekker‐Jensen D. B., Kelstrup C. D., Batth T. S. et al (2017) An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 4, 587–599.e584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereczki E., Branca R. M., Francis P. T. et al (2018) Synaptic markers of cognitive decline in neurodegenerative diseases: a proteomic approach. Brain 141, 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird T. D. (1999) Sep 24 [Updated 2012 Oct 18], Early‐Onset Familial Alzheimer Disease, In: Adam M. P., Ardinger H. H., Pagon R. A.et al., editors. GeneReviews® [Internet]. pp. 1993–2018. Seattle (WA): University of Washington, Seattle: Available from: https://www.ncbi.nlm.nih.gov/books/NBK1236/ [Google Scholar]
- Blennow K. and Zetterberg H. (2018) The past and the future of Alzheimer's disease fluid biomarkers. J. Alzheimers Dis. 62, 1125–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H. and Braak E. (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Campbell S., Stephens S. and Ballard C. (2001) Dementia with Lewy bodies: clinical features and treatment. Drugs Aging 18, 397–407. [DOI] [PubMed] [Google Scholar]
- Chen S., Lu F. F., Seeman P. and Liu F. (2012) Quantitative proteomic analysis of human substantia nigra in Alzheimer's disease, Huntington's disease and Multiple sclerosis. Neurochem. Res. 37, 2805–2813. [DOI] [PubMed] [Google Scholar]
- Dammer E. B., Lee A. K., Duong D. M., Gearing M., Lah J. J., Levey A. I. and Seyfried N. T. (2015) Quantitative phosphoproteomics of Alzheimer's disease reveals cross‐talk between kinases and small heat shock proteins. Proteomics 15, 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A., Chai Y. L., Tan J. M., Lee J. H., Francis P. T., Chen C. P., Sze S. K. and Lai M. K. P. (2017) An iTRAQ‐based proteomic analysis reveals dysregulation of neocortical synaptopodin in Lewy body dementias. Mol. Brain 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan L. E., Higginbotham L., Dammer E. B. et al (2012) Analysis of a membrane‐enriched proteome from postmortem human brain tissue in Alzheimer's disease. Proteomics Clin. Appl. 6, 201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E., Nayak S., Faustin A. et al (2017) Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer's disease. Acta Neuropathol. 133, 933–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ElHajj Z., Cachot A., Muller T., Riederer I. M. and Riederer B. M. (2016) Effects of postmortem delays on protein composition and oxidation. Brain Res. Bull. 121, 98–104. [DOI] [PubMed] [Google Scholar]
- Fasano M., Monti C. and Alberio T. (2016) A systems biology‐led insight into the role of the proteome in neurodegenerative diseases. Expert Rev. Proteomics 13, 845–855. [DOI] [PubMed] [Google Scholar]
- Ferrari R., Kapogiannis D., Huey E. D. and Momeni P. (2011) FTD and ALS: a tale of two diseases. Curr. Alzheimer Res. 8, 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira P. G., Munoz‐Aguirre M., Reverter F. et al (2018) The effects of death and post‐mortem cold ischemia on human tissue transcriptomes. Nat. Commun. 9, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I. (2017) Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol. 27, 645–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I., Garcia M. A., Gonzalez I. L. et al (2018) Aging‐related tau astrogliopathy (ARTAG): not only tau phosphorylation in astrocytes. Brain Pathol. 10.1111/bpa.12593. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galton C. J., Patterson K., Xuereb J. H. and Hodges J. R. (2000) Atypical and typical presentations of Alzheimer's disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain 123(Pt 3), 484–498. [DOI] [PubMed] [Google Scholar]
- Ganz A. B., Beker N., Hulsman M. et al (2018) Neuropathology and cognitive performance in self‐reported cognitively healthy centenarians. Acta Neuropathol. Commun. 6, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garwood C. J., Ratcliffe L. E., Simpson J. E., Heath P. R., Ince P. G. and Wharton S. B. (2017) Review: astrocytes in Alzheimer's disease and other age‐associated dementias: a supporting player with a central role. Neuropathol. Appl. Neurobiol. 43, 281–298. [DOI] [PubMed] [Google Scholar]
- Gill E. L., Koelmel J. P., Yost R. A., Okun M. S., Vedam‐Mai V. and Garrett T. J. (2018) Mass spectrometric methodologies for investigating the metabolic signatures of Parkinson's disease: current progress and future perspectives. Anal. Chem. 90, 2979–2986. [DOI] [PubMed] [Google Scholar]
- Goedert M., Masuda‐Suzukake M. and Falcon B. (2017) Like prions: the propagation of aggregated tau and alpha‐synuclein in neurodegeneration. Brain 140, 266–278. [DOI] [PubMed] [Google Scholar]
- Holstege H., van der Lee S. J., Hulsman M. et al (2017) Characterization of pathogenic SORL1 genetic variants for association with Alzheimer's disease: a clinical interpretation strategy. Eur. J. Hum. Genet. 25, 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondius D. C., van Nierop P., Li K. W., Hoozemans J. J., van der Schors R. C., van Haastert E. S., van der Vies S. M., Rozemuller A. J. and Smit A. B. (2016) Profiling the human hippocampal proteome at all pathologic stages of Alzheimer's disease. Alzheimers Dement. 12, 654–668. [DOI] [PubMed] [Google Scholar]
- Hondius D. C., Eigenhuis K. N., Morrema T. H. J. et al (2018a) Proteomics analysis identifies new markers associated with capillary cerebral amyloid angiopathy in Alzheimer's disease. Acta Neuropathol. Commun. 6, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondius D. C., Hoozemans J. J. M., Rozemuller A. J. M., Li K. W. and Smit A. B. (2018b) A laser microdissection‐liquid chromatography‐tandem mass spectrometry workflow for post‐mortem analysis of brain tissue. Methods Mol. Biol. 1723, 371–383. [DOI] [PubMed] [Google Scholar]
- Hosp F. and Mann M. (2017) A primer on concepts and applications of proteomics in neuroscience. Neuron 96, 558–571. [DOI] [PubMed] [Google Scholar]
- Hyman B. T., Phelps C. H., Beach T. G. et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof A., Kovari E., von Gunten A., Gold G., Rivara C. B., Herrmann F. R., Hof P. R., Bouras C. and Giannakopoulos P. (2007) Morphological substrates of cognitive decline in nonagenarians and centenarians: a new paradigm? J. Neurol. Sci. 257, 72–79. [DOI] [PubMed] [Google Scholar]
- Inoue Y., Ueda M., Tasaki M. et al (2017) Sushi repeat‐containing protein 1: a novel disease‐associated molecule in cerebral amyloid angiopathy. Acta Neuropathol. 134, 605–617. [DOI] [PubMed] [Google Scholar]
- Jacobs H. I. L., Hedden T., Schultz A. P. et al (2018) Structural tract alterations predict downstream tau accumulation in amyloid‐positive older individuals. Nat. Neurosci. 21, 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger K. A. (2009) A critical evaluation of current staging of alpha‐synuclein pathology in Lewy body disorders. Biochim. Biophys. Acta 1792, 730–740. [DOI] [PubMed] [Google Scholar]
- Kelstrup C. D., Bekker‐Jensen D. B., Arrey T. N., Hogrebe A., Harder A. and Olsen J. V. (2018) Performance evaluation of the Q exactive HF‐X for shotgun proteomics. J. Proteome Res. 17, 727–738. [DOI] [PubMed] [Google Scholar]
- Kfoury N., Holmes B. B., Jiang H., Holtzman D. M. and Diamond M. I. (2012) Trans‐cellular propagation of Tau aggregation by fibrillar species. J. Biol. Chem. 287, 19440–19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoonsari P. E., Haggmark A., Lonnberg M. et al (2016) Analysis of the cerebrospinal fluid proteome in Alzheimer's disease. PLoS ONE 11, e0150672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans F., Ho J. T. C., Smit A. B. and Li K. W. (2018) Comparative analyses of data independent acquisition mass spectrometric approaches: DIA, WiSIM‐DIA, and untargeted DIA. Proteomics 18, 1700304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S., Madore C., Cialic R. et al (2017) The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachen‐Montes M., Gonzalez‐Morales A., Zelaya M. V., Perez‐Valderrama E., Ausin K., Ferrer I., Fernandez‐Irigoyen J. and Santamaria E. (2017) Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of prohibitin complex during Alzheimer's disease progression. Sci. Rep. 7, 9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassen P. S., Thygesen C., Larsen M. R. and Kempf S. J. (2017) Understanding Alzheimer's disease by global quantification of protein phosphorylation and sialylated N‐linked glycosylation profiles: a chance for new biomarkers in neuroproteomics? J. Proteomics. 161, 11–25. [DOI] [PubMed] [Google Scholar]
- Llorens F., Thune K., Tahir W. et al (2017) YKL‐40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 12, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manousopoulou A., Gatherer M., Smith C. et al (2017) Systems proteomic analysis reveals that clusterin and tissue inhibitor of metalloproteinases 3 increase in leptomeningeal arteries affected by cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 43, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni R. E., McRae A. F., Bressler J., et al. (2018) Meta‐analysis of epigenome‐wide association studies of cognitive abilities. Mol Psychiatry. doi: http://10.1038/s41380-017-0008-y. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. (2004) Pathways towards and away from Alzheimer's disease. Nature 430, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N., Grigoriou S. and Zetterberg H. (2018) Flud biomarkers in Alzheimer's disease and frontotemporal dementia, in Neurodegenerative Diseases (Galimberti D., Scarpini E., ed.), pp. 221–252. Springer, Cham. [Google Scholar]
- Meier F., Beck S., Grassl N., Lubeck M., Park M. A., Raether O. and Mann M. (2015) Parallel Accumulation‐Serial Fragmentation (PASEF): multiplying sequencing speed and sensitivity by synchronized scans in a trapped ion mobility device. J. Proteome Res. 14, 5378–5387. [DOI] [PubMed] [Google Scholar]
- Miller J. A., Guillozet‐Bongaarts A., Gibbons L. E. et al (2017) Neuropathological and transcriptomic characteristics of the aged brain. Elife 6, e31126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra S. S., Heyman A., McKeel D. et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Mishra S., Blazey T. M., Holtzman D. M., Cruchaga C., Su Y., Morris J. C., Benzinger T. L. S. and Gordon B. A. (2018) Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE varepsilon4 genotype. Brain 141, 1828–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti C., Colugnat I., Lopiano L., Chio A. and Alberio T. (2018) Network analysis identifies disease‐specific pathways for Parkinson's disease. Mol. Neurobiol. 55, 370–381. [DOI] [PubMed] [Google Scholar]
- Morris M., Knudsen G. M., Maeda S., Trinidad J. C., Ioanoviciu A., Burlingame A. L. and Mucke L. (2015) Tau post‐translational modifications in wild‐type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya‐Alvarado G., Gershoni‐Emek N., Perlson E. and Bronfman F. C. (2016) Neurodegeneration and Alzheimer's disease (AD). What can proteomics tell us about the Alzheimer's brain? Mol. Cell Proteomics 15, 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuri S., Wetterhall M., Ingelsson M., Lannfelt L., Artemenko K., Bergquist J., Kultima K. and Shevchenko G. (2014) Quantification of the brain proteome in Alzheimer's disease using multiplexed mass spectrometry. J. Proteome Res. 13, 2056–2068. [DOI] [PubMed] [Google Scholar]
- Nelson P. T., Alafuzoff I., Bigio E. H. et al (2012) Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist S., Scholl M., Strandberg O. et al (2017) Earliest accumulation of beta‐amyloid occurs within the default‐mode network and concurrently affects brain connectivity. Nat. Commun. 8, 1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W., Seppi K., Tanner C. M., Halliday G. M., Brundin P., Volkmann J., Schrag A. E. and Lang A. E. (2017) Parkinson disease. Nat. Rev. Dis. Primers 3, 17013. [DOI] [PubMed] [Google Scholar]
- Portelius E., Brinkmalm G., Pannee J., Zetterberg H., Blennow K., Dahlen R., Brinkmalm A. and Gobom J. (2017) Proteomic studies of cerebrospinal fluid biomarkers of Alzheimer's disease: an update. Expert Rev. Proteomics 14, 1007–1020. [DOI] [PubMed] [Google Scholar]
- Przedborski S., Vila M. and Jackson‐Lewis V. (2003) Neurodegeneration: what is it and where are we? J. Clin. Invest. 111, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redensek S., Dolzan V. and Kunej T. (2018) From genomics to omics landscapes of Parkinson's disease: revealing the molecular mechanisms. OMICS 22, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders N. R., Pao Y., Renner M. C., da Silva‐Matos C. M., Lodder T. R., Malinow R. and Kessels H. W. (2016) Amyloid‐beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc. Natl Acad. Sci. USA 113, E6526–E6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein D. C. (2006) The roles of intracellular protein‐degradation pathways in neurodegeneration. Nature 443, 780–786. [DOI] [PubMed] [Google Scholar]
- Ruiz‐Riquelme A., Lau H. H. C., Stuart E., Goczi A. N., Wang Z., Schmitt‐Ulms G. and Watts J. C. (2018) Prion‐like propagation of beta‐amyloid aggregates in the absence of APP overexpression. Acta Neuropathol. Commun. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano‐Pozo A., Frosch M. P., Masliah E. and Hyman B. T. (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect Med. 1, a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S., Fitzpatrick A. L., Ikram M. A. et al (2010) Genome‐wide analysis of genetic loci associated with Alzheimer disease. JAMA 303, 1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried N. T., Dammer E. B., Swarup V. et al (2017) A multi‐network approach identifies protein‐specific co‐expression in asymptomatic and symptomatic Alzheimer's disease. Cell Syst. 4, 60–72.e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L. and Jia J. (2016) An overview of genome‐wide association studies in Alzheimer's disease. Neurosci. Bull. 32, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R., van der Lee S. J., Naj A. C. et al (2017) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat. Genet. 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R. A., Aisen P. S., Beckett L. A. et al (2011) Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires‐Jones T. L., Attems J. and Thal D. R. (2017) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol. 134, 187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teunissen C. E., Elias N., Koel‐Simmelink M. J. et al (2016) Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement. (Amst) 2, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal D. R., Rub U., Schultz C., Sassin I., Ghebremedhin E., Del Tredici K., Braak E. and Braak H. (2000) Sequence of Abeta‐protein deposition in the human medial temporal lobe. J. Neuropathol. Exp. Neurol. 59, 733–748. [DOI] [PubMed] [Google Scholar]
- Toledo J. B., Arnold M., Kastenmuller G. et al (2017) Metabolic network failures in Alzheimer's disease: a biochemical road map. Alzheimers Dement. 13, 965–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S. and Cox J. (2018) Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer research. Methods Mol. Biol. 1711, 133–148. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T. and Cox J. (2016) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat. Protoc. 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- Umoh M. E., Dammer E. B., Dai J., Duong D. M., Lah J. J., Levey A. I., Gearing M., Glass J. D. and Seyfried N. T. (2018) A proteomic network approach across the ALS‐FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med. 10, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegh M. J., Heldring C. M., Kamphuis W. et al (2014) Reducing hippocampal extracellular matrix reverses early memory deficits in a mouse model of Alzheimer's disease. Acta Neuropathol. Commun. 2, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher P. M., Wray N. R., Zhang Q., Sklar P., McCarthy M. I., Brown M. A. and Yang J. (2017) 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Z., Bi R., Hu Q. X. et al (2016) Validating GWAS‐identified risk loci for Alzheimer's disease in Han Chinese populations. Mol. Neurobiol. 53, 379–390. [DOI] [PubMed] [Google Scholar]
- Wang S., Yang F., Petyuk V. A. et al (2017) Quantitative proteomics identifies altered O‐GlcNAcylation of structural, synaptic and memory‐associated proteins in Alzheimer's disease. J. Pathol. 243, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger C. D. and Coon J. J. (2013) A proteomics search algorithm specifically designed for high‐resolution tandem mass spectra. J. Proteome Res. 12, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijsman E. M., Pankratz N. D., Choi Y. et al (2011) Genome‐wide association of familial late‐onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet. 7, e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtas A. M., Kang S. S., Olley B. M. et al (2017) Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways. Proc. Natl Acad. Sci. USA 114, E6962–E6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Patassini S., Rustogi N. et al (2018) Regional protein expression in human Alzheimer's brain correlates with disease severity. bioRxiv doi: 10.1101/283705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M. (2015) Cerebral amyloid angiopathy: emerging concepts. J. Stroke 17, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelaya M. V., Perez‐Valderrama E., de Morentin X. M., Tunon T., Ferrer I., Luquin M. R., Fernandez‐Irigoyen J. and Santamaria E. (2015) Olfactory bulb proteome dynamics during the progression of sporadic Alzheimer's disease: identification of common and distinct olfactory targets across Alzheimer‐related co‐pathologies. Oncotarget 6, 39437–39456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J., Jones D. R., Duong D. M., Levey A. I., Lah J. J. and Peng J. (2013) Proteomic analysis of postsynaptic density in Alzheimer's disease. Clin. Chim. Acta 420, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]