

Abstract
The primary function of cyclin‐dependent kinases (CDKs) in complex with their activating cyclin partners is to promote mitotic division in somatic cells. This canonical cell cycle‐associated activity is also crucial for fertility as it allows the proliferation and differentiation of stem cells within the reproductive organs to generate meiotically competent cells. Intriguingly, several CDKs exhibit meiosis‐specific functions and are essential for the completion of the two reductional meiotic divisions required to generate haploid gametes. These meiosis‐specific functions are mediated by both known CDK/cyclin complexes and meiosis‐specific CDK‐regulators and are important for a variety of processes during meiotic prophase. The majority of meiotic defects observed upon deletion of these proteins occur during the extended prophase I of the first meiotic division. Importantly a lack of redundancy is seen within the meiotic arrest phenotypes described for many of these proteins, suggesting intricate layers of cell cycle control are required for normal meiotic progression. Using the process of male germ cell development (spermatogenesis) as a reference, this review seeks to highlight the diverse roles of selected CDKs their activators, and their regulators during gametogenesis.
Keywords: cyclin, cyclin‐dependent kinase, meiosis, meiotic crossover, recombination, synapsis
Abbreviations
CDKs, cyclin‐dependent kinases
CKIs, CDK‐inhibitor proteins
DSBs, double‐strand breaks
FSH, follicle‐stimulating hormone
GST, gonocyte‐spermatogonia transition
LINC, linker of nucleoskeleton and cytoskeleton
LRN, late recombination nodule‐associated
RPM, rapid prophase movements
SSCs, spermatogonial stem cells
Cyclin‐dependent kinases: a diverse family with numerous activators and regulators
Cyclin‐dependent kinases (CDKs) and their activating partner proteins, the cyclins, are often compared to the molecular engine and gearbox, which is utilized to drive a cell through the different phases of the cell cycle 1. This simplistic analogy is representative of the cell cycle in eukaryotic organisms of lower complexity, such as yeast. These unicellular organisms possess a single ‘engine’ (CDK) and multiple cyclin ‘gears’. The sequential association of different cyclins with the CDK promotes the activation of distinct CDK/cyclin complexes to advance through each stage of the cell cycle. In more complex multicellular eukaryotes such as mammals, the CDK and cyclin gene families have become increasingly larger, as a result of multiple gene duplication events occurring throughout eukaryotic evolution. This has allowed some CDKs and cyclins to acquire specialized functions either in addition to, or independently of their roles in cell cycle progression 2, 3. There are currently 21 CDKs, and upwards of 30 cyclin proteins described in humans and mice. Only CDK1, 2, 3, 4, and 6 in complex with an A‐, B‐, C‐, D‐, or E‐type cyclins, form canonical CDK/cyclin complexes with direct roles in driving mitotic cell cycle progression. The remaining CDKs and cyclins exhibit diverse functions and have been shown to facilitate many important cell cycle‐independent roles including the modulation of transcription and RNA splicing 4. Although much study has been devoted to the understanding of mammalian cyclins, it is clear that there are still many intricacies yet to be properly described. Recently, several ‘atypical’ cyclin family members have been identified. These proteins share limited homology with the cell cycle‐associated cyclins. Many of these have yet‐unknown functions, and their ability to bind and activate CDKs is mostly unexplored 4, 5, 6, 7, 8, 9, 10.
In addition to the cyclins, several mammalian CDKs can also be bound and activated by noncyclin CDK interactors from the Speedy/RINGO family of proteins. Noncanonical CDK/Speedy complexes are not subject to typical cell cycle regulation. Whereas the majority of CDK/cyclin complexes are reliant upon a critical activating phosphorylation to achieve full catalytic activity 11, 12, 13, CDK/Speedy complexes display activity in the absence of this modification 14, 15, 16, 17, 18. Furthermore, CDK/Speedy complexes are also insensitive to inhibition by CDK‐inhibitor proteins (CKIs), which physically bind and inactivate CDK/cyclin complexes 4, 19, 20. These unique properties are thought to allow the formation of active CDK/Speedy complexes under circumstances, whereby CDK/cyclin complexes would usually be inactivated.
In regard to the role of CDK‐associated activity in maintaining normal reproductive health, several mammalian CDKs, cyclins, and at least one Speedy protein (Speedy A) have been shown to be essential for the development and maturation of the germ cells within the male and female reproductive organs (gametogenesis). This is similarly true of members of the pRB‐E2F signaling pathway, which represents a major downstream target of CDK activity and also specific CKI proteins. Many of these proteins show preferential or heightened expression in these tissues or, in some cases, exhibit differential expression of splice isoforms 21, 22, 23, 24. In accordance with these observations, knockout mouse models of many of these proteins result in infertility due to arrested gametogenesis. Interestingly, the stage at which germ cell development is affected in these models is highly varied depending upon the protein which is knocked out. This highlights a complex network of often nonredundant interactions between CDKs, their activators, and their regulatory proteins during gametogenesis and offers a unique insight into the requirement of CDK‐associated activity for normal fertility.
Spermatogenesis as a model system to study the importance of CDK activity during germ cell development
Much of the research performed to elucidate the roles of CDK‐associated activity during gametogenesis has utilized the process of male germ cell development (spermatogenesis) as a developmental model. For such studies, female germ cell development (oogenesis) is disadvantaged by both the timing and frequency of the meiotic divisions observed in this system. In mice, the first meiotic division of female meiocytes (oocytes) is initiated in the embryonic ovary (~embryonic day E13.5 in the mouse). Meiosis is subsequently arrested prior to the onset of the second meiotic division and oocytes enter a long‐term arrest state known as dictyate 25, 26. As in humans, the generation of mouse oocytes occurs only once. Consequently, the analysis of early oocyte development and its first meiotic division must be performed during embryonic development. In contrast, once spermatogenesis is initiated in pubertal male mice (~postnatal day 14), the development of germ cells occurs in continuous waves throughout the lifetime of a mouse. This is facilitated by the continual production and maturation of male meiocytes (spermatocytes) from a self‐renewing source of spermatogonial stem cells [SSCs] 27. For normal spermatogenesis, a small pool of ‘fully undifferentiated’ or ‘primitive’ (As) type SSCs must be maintained to supply a steady supply of cells competent to enter meiotic divisions. The division of these cells gives rise to either more As SSCs, which retain the capacity to self‐renew, or early differentiating spermatogonia. Differentiating spermatogonia divide successively via mitosis without cytokinesis to form first pairs (Apr), and then aligned chains (Aal) of spermatogonia linked by cytoplasmic bridges. These chains can attain lengths of 16 or rarely 32 connected cells. It is generally considered that chains of at least 8 (AAl8) must be formed to enable further differentiation 28. Committal of Aal spermatogonia cells to gametogenesis occurs in the absence of mitotic division and is characterized by morphological changes as well as changes in transcription 29, 30. The resultant A1‐type spermatogonia undergo sequential mitotic divisions to become A2, A3, and then A4‐type spermatogonia. A4 spermatogonia undergo further divisions to become intermediate‐type (In) and then B‐type spermatogonia. At this stage, B‐type spermatogonia are competent to differentiate into primary spermatocytes that can enter meiosis. The stepwise nature of spermatogenesis as depicted in Fig. 1 is particularly suited to knockout studies. Here, the stage of germ cell development affected by the deletion of a single protein can be easily investigated to determine functionality.
Figure 1.
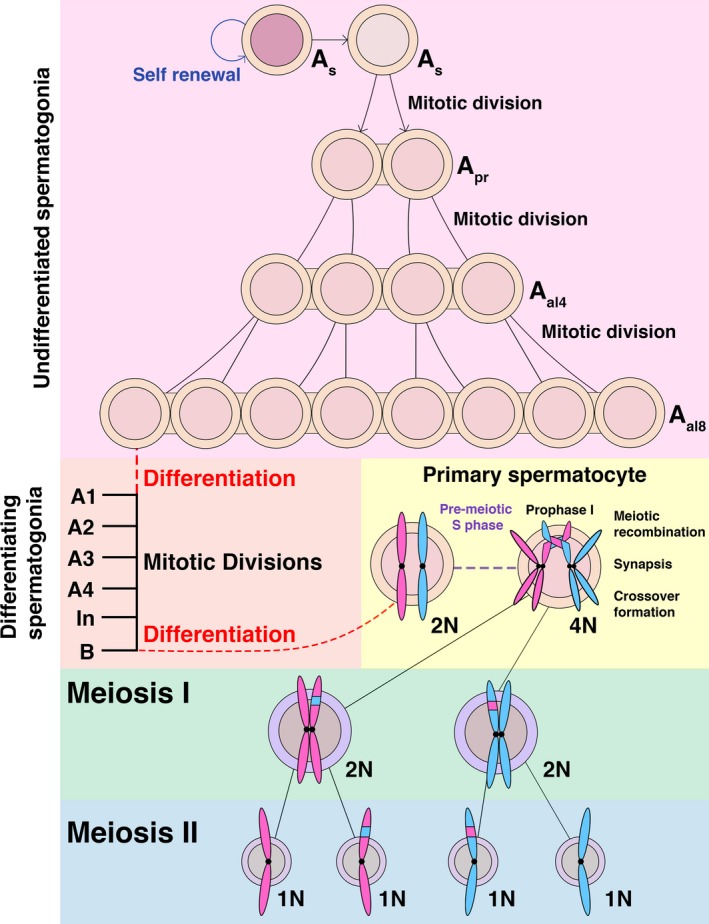
Schematic of adult spermatogenesis. Spermatogenesis is initiated by A single (As) type spermatogonial stem cells (SSCs). As type SSCs are thought to be able to produce both undifferentiated As type SSCs (self‐renewal) or separate populations of As SSCs capable of differentiation. As spermatogonia undergo consecutive rounds of mitotic division to produce pairs (Apr) or aligned chains (Aal) of SSCs. Aal SSCs represent clonal populations linked by shared cytoplasm and vary in the maximal chain length attained before further differentiation. Chains of 8 (Aal8) or more are considered proficient to enter further differentiation. Each Aal spermatogonia differentiates without division to form ‘differentiating’ A1 type spermatogonia. These undergo consecutive rounds of mitotic division to produce A2, A3, A3, Intermediate (In), and B‐type spermatogonia. B‐type spermatogonia subsequently differentiate without division to form primary spermatocytes capable of meiotic division. In premeiotic S phase, diploid primary spermatocytes duplicate their DNA to become tetraploid. During meiotic prophase I, chromosomal homologs initiate meiotic recombination and synapsis allowing the formation of meiotic crossovers. Upon resolution of crossovers, homologs are segregated into two diploid secondary spermatocytes (meiosis I). Each secondary spermatocyte divides rapidly to segregate sister chromatids into two haploid round spermatids (meiosis II). Thus, four haploid spermatids can be produced from two reductional divisions of a primary spermatocyte. Not shown here is the process of spermiogenesis whereby round spermatids undergo sequential steps of differentiation to form elongating spermatids and spermatozoa.
Meiotic prophase I: a hotbed for CDK‐associated functions
For the generation of haploid gametes, meiocytes must undergo two reductional meiotic divisions. The first meiotic division promotes the pairing and exchange of genetic material between maternal and paternal chromosomal pairs (homologs). This occurs during an extended prophase stage (prophase I) 31, 32 and is followed by the segregation of homologs of into two genetically heterogeneous daughter cells. This is followed by a second meiotic division, which is considered more comparable to mitotic divisions as it promotes the segregation of sister chromatids 33. The prolonged timeframe of meiotic prophase I, reflects the complexity of this developmental process and can be broken down into five further substages: Leptotene, Zygotene, Pachytene, Diplotene, and Diakinesis. In regard to the knockout mouse models discussed within this review, the majority of meiotic defects observed typically arise during one or more of these substages. Therefore, this section will introduce the events of meiotic prophase I in detail, in order that the meiotic functions of CDK activity described later in this review can be better understood.
During leptonema, the earliest stage of prophase I, paternal and maternal bivalent chromosomes locate and pair (synapse) with their homologous counterpart. An integral part of this process is the self‐imposed induction of double‐strand breaks (DSBs). This is mediated primarily by the DNA topoisomerase, SPO11 34, 35. SPO11‐mediated DSB formation occurs preferentially within so‐called ‘meiotic hotspots’ or ‘recombination hot regions’ of the genome. The biased nature of this process is enforced by epigenetic modifications placed proximal to the incipient break site 36, 37, 38, 39, 40, 41. These alterations are thought to create a preferential environment for the recombination machinery and are typically directed toward euchromatic regions depleted of nucleosomes 41, 42, 43, 44, 45, 46. Meiotic DSB formation results in the induction of approximately 150 and 200–300 DSBs during human and murine meiosis, respectively 34, 47, 48. Although this is a phenomenon, which occurs in all organisms that are capable of meiotic division, the number of break sites is highly variable between organisms. In mice, meiotic DSB formation is essential for proper chromosomal pairing 49, 50, 51 and is tightly integrated with the assembly of a tripartite scaffolding structure, known as the synaptonemal complex 52. The components comprising the axial/lateral elements of this structure (SYCP2/3) localize primarily to chromatin decorated with markers of meiotic DSBs, in a manner that remains poorly understood. The subsequent self‐assembly of axial filaments effectively anchors chromosome segments harboring meiotic break sites together within a single chromosomal core with the remaining chromatin organized in looped arrays tethered to these sites 53, 54, 55, 56.
Zygonema marks the onset of homolog pairing (synapsis). Here, the transverse (SYCP1) and central elements (SYCE1, SYCE2, SYCE2, TEX12, SIX6OS1) bind the axial element scaffold surrounding each homolog and bring them together in a zipper‐like fashion. This event brings meiotic DSBs close proximity with the near‐perfect DNA template of its chromosomal homolog within the context of the synaptonemal complex scaffold. Upon nucleolytic processing of DSB sites, single‐stranded 3′ DNA tails are generated and utilized to initiate homologous DNA damage repair in a process known as strand invasion 57. This effectively creates a large number of initial recombination sites (also known as early recombination nodules) 58.
An essential precondition for normal synapsis during zygonema is the tethering of telomeres to the inner nuclear envelope. Nuclear envelope tethering is stabilized by the interaction of proteins at the inner (SUN1, SUN2) and outer nuclear envelope (KASH5), in addition to additional ancillary interactors (TERB1, TERB2, MAJIN). Together, these proteins comprise the ‘LINC’ (Linker of Nucleoskeleton and Cytoskeleton) complex (59, 60, 61, 62, 63, 64, 65; for review, see 66). The LINC complex facilitates the creation of nucleocytoplasmic bridges connecting telomeres to microtubules within the cytoskeleton of the meiocyte allowing cytoskeletal forces to be transmitted via the nuclear envelope to drive rapid telomeric movements 67, 68, 69. These telomeric movements are crucial for establishing initial interactions between homologs and are thus essential for their eventual pairing. Much of the meiosis‐specific functions of the proteins discussed in this review seems to revolve around the formation and stabilization of the LINC complex and will be discussed at length later in this review.
In pachynema, chromosomal homologs achieve a state of full synapsis. At this time, numerous DNA repair complexes are recruited to chromosomal axes to stabilize and repair DNA‐intermediate structures created at recombination sites 70, 71, 72. In the majority of cases, these sites are repaired without the exchange of genetic material from its homologs (non‐crossovers). The remaining sites become stabilized in a manner which protects from this repair pathway. Instead, further processing results in the formation of crossover intermediate structures in so‐called late recombination nodules. These late recombination nodules represent the latest stages of homologous recombination repair, which occur prior to the formation of meiotic crossover sites 73. In mice, the formation meiotic crossovers resulting from the maturation of late recombination nodules occurs in a strictly regulated manner with 1–2 formed per homolog pair.
In diplonema, the synaptonemal complex lateral and central elements are disassembled, a process which is completed by diakinesis. Throughout these stages, sister chromatid arms remain closely connected via cohesins 74, but paired homologous chromosomes remain linked solely at meiotic crossover sites. These crossover sites act as anchor points, which allow bidirectional tension to be applied upon each homolog pair. This facilitates orderly alignment of homologs upon the meiotic spindle 75, 76, 77, 78, 79. Subsequent recognition and cleavage of crossover sites by structure‐specific endonucleases in anaphase I allows the release of tension on the spindle. This process is also known as crossover resolution 80, 81, 82. Successful crossover resolution allows the segregation of homologs into two daughter cells and completes the reciprocal exchange of maternal and paternal DNA at the resolved crossover site. Achieving this genetic exchange of information during meiotic prophase I is a hallmark of meiotic division and is the major determinant of heterogeneity within the genome of developing germ cells 55, 83, 84, 85, 86, 87.
Aims
Despite extensive research delineating the various roles of the CDKs, cyclins, and Speedy proteins during gametogenesis, pertinent questions remain even surrounding the best‐characterized members of these protein families, particularly CDK2, CDK4, the E‐ and D‐type cyclins, and Speedy A. This review aims to highlight the contribution of these selected CDKs in regard to their functions during gametogenesis. Specific effort is made in highlighting where gaps in the current literature can be addressed in future studies. Although also essential for gametogenesis, the A‐ and B‐type cyclins in conjunction with CDK1 and/or CDK2 are not discussed at length in this review. For a comprehensive analysis of these proteins in relation to gametogenesis, the reader is directed to the following reviews by Chotiner et al. and Risal et al. 24, 88.
Roles for canonical, cell cycle‐associated CDK activity during germ cell development
One of the major requirements for CDK‐associated activity during normal gametogenesis arises due to the need for mitotically dividing cells in the male and female reproductive organs. Typically, these mitotically active cell types consist of either precursor stem cells, which give rise to the meiocytes, or their respective supporting cell types, which promote meiocyte maturation. In this section, we will discuss the effects on fertility observed upon deletion of cell cycle‐associated cyclins, their associated CDKs, and the CKI proteins, which regulate the activity of these complexes. For added context, this section will also explore the effects occurring upon perturbation of downstream CDK/cyclin signaling. For simplicity, the order of discussion will follow the typical order of expression of these proteins throughout the cell cycle starting with the G1 stage D‐type cyclins and concluding with the S phase‐associated CDK2.
Proposed roles for the D‐type cyclins in regulating spermatogonial proliferation and differentiation
During mitotic cell division, D‐type cyclins act primarily as the activating partners of CDK4 and CDK6. Here, CDK4‐ and CDK6‐associated kinase activity serves an important role in driving entry into G1 from quiescence and also maintaining cellular proliferation in actively dividing cells 89, 90, 91, 92, 93. The proliferative role of CDK/cyclin proteins, in general, is mediated through the phosphorylation of the major CDK‐substrate, RB1 (Retinoblastoma) tumor suppressor, and its related proteins RBL1 (p107) and RBL2 (p130) [collectively referred to here as pRB]. In the absence of CDK‐driven phosphorylation, these pRB proteins act to inhibit cellular proliferation by sequestering E2F transcription factor family members; effectively holding them in an inactive state. When released from pRB repression, E2F transcription factors promote cell cycle progression through the downstream transcription of genes that drive cell cycle progression 94.
Based on their cell‐specific expression during spermatogenesis, cyclin D‐associated kinase activity is thought to play a primarily proliferative role in driving the mitotic expansion of differentiating spermatogonia. During the first wave of spermatogenesis in which spermatogenic cells differentiate in a synchronous manner 95, each of the three D‐type cyclin members (D1‐D3) can be detected in A1 type spermatogonia as they enter a differentiating state. Later, during adult spermatogenesis, cyclins D1 and D3 can be detected in dividing spermatogonia during many stages of spermatogenesis, suggesting specific roles for these cyclins in driving cellular proliferation 96, 97. Although deletion of cyclin D1 98, 99 or cyclin D3 in isolation does not affect fertility 100, the deletion of both cyclins D1 and D3 in combination leads to severe developmental defects resulting in early lethality 101. Since this early lethality precludes formal analysis of the relative requirement for cyclin D1 and/or D3 in spermatogonia, it is likely that as in many other cell types, that at least one of these proteins is required to promote cellular division in spermatogonia.
Unlike cyclins D1 and D3, cyclin D2 expression is required for normal fertility in both male and female mice. During spermatogenesis, the expression of cyclin D2 remains specifically restricted to differentiating A1‐type spermatogonia during adult spermatogenesis. It has been hypothesized that might reflect a role in the differentiation process of spermatogonia. Unfortunately, this is yet to be formally confirmed due to an incomplete analysis of the infertility phenotype in cyclin D2−/− testes.
In adult ovaries, which lack proliferating stem cells, cyclin D2 is expressed in the granulosa cells, which support the maturation of ovarian follicles. In this cell type, cyclin D2 expression is essential for cellular proliferation in response to the follicle‐stimulating hormone (FSH) 102. Interestingly, the proliferation of the corresponding testicular cell type, known as Sertoli cells, is similarly responsive to FSH signaling 103 and also seems to be influenced by cyclin D2 expression levels. This was best illustrated in studies of inhibin α−/− mice, which are unable to properly regulate FSH production. In these mice, additional deletion of cyclin D2 was shown to slow the growth of gonadotropin‐dependent gonadal tumors, which are comprised of the Sertoli or granulosa cell types in males and females, respectively 104. Together, these data suggest that cyclin D2 is a FSH‐responsive gene required for cellular proliferation in both testis and ovary. Future study is warranted to determine whether the spermatogenic defects observed in cyclin D2−/− mice arise from differentiation defects in spermatogonial stem cells or alternatively, the defective proliferation of Sertoli cells.
CDK4/CDK6
Mouse knockouts for the kinases partnering the D‐type cyclins, CDK4 105, 106, and CDK6 107 are viable. Expression of at least one of these proteins is required for the early development of hematopoietic precursors and their combined knockout results in embryonic lethality due to the development of severe anemia 107. This is also true of mice with the deletion of all D‐type cyclins 108. During spermatogenesis, the maximal expression of Cdk4 and Cdk6 is observed in immature testes at which time the testes consist primarily of spermatogonial stem cells 109, 110, 111, 112. Although Cdk6−/− mice show no overt defects in gametogenesis, Cdk4 deletion results in female infertility from birth and early‐onset infertility in male mice. Interestingly in regard to female fertility, the phenotype upon deletion of Cdk4, however, seems to be distinct from that of the cyclin D2 knockout as normal follicular maturation could be observed in these mice with no defect seen in the proliferation of granulosa cells. Instead, postovulatory progesterone secretion was markedly impaired and fertility in these mice could be rescued by progesterone treatment 113. In regard to male fertility, a low percentage (~20%) of Cdk4−/− males are initially fertile until around 2 months of age. The spermatogenic defects seen in Cdk4−/− testes increase in severity with age and fertility in these animals is invariably lost in older mice 105, 106. The importance of CDK4 for fertility remains poorly understood. One proposal was that early‐onset infertility in male Cdk4−/− mice might occur in a comorbid manner with the development of spontaneous nonobese diabetes mellitus 114, which is known to have a negative impact upon fertility 115, 116. Unfortunately, the analysis of the Cdk4−/− spermatogenic defect has not been extended further than the histological analysis of mutant testis sections. Potential spermatogonial stem cell proliferation/differentiation defects in this model are therefore yet to be investigated 114. Additional unexplored roles for CDK4 in meiotically dividing spermatocytes have also been proposed by several groups and will be discussed later in the latter sections of this review.
Regulation of CDK4/CDK6 activity by the INK4 class of CKI proteins during spermatogenesis
During mitotic division, control of CDK4 and CDK6‐associated kinase activity is exerted in part by the INK4 family of CKI proteins encoded by the genes of Cdkn2a, Cdkn2b, Cdkn2c, and Cdkn2d 117, 118. These proteins specifically inhibit CDK4 and CDK6 by inducing structural changes that prevent catalytic activation 119. Each of the INK4 proteins show distinct expression patterns during spermatogenesis and can be observed in either spermatogonia (p16INK4A, p19ARF), spermatocytes (p19INK4D), or both (p15INK4B, p18INK4C) 112, 120, 121, 122, 123. The Cdkn2a gene possesses distinct promoters upstream of alternate exons. This allows the expression of two distinct tumor suppressor proteins p16INK4A and p19ARF using different reading frames 124. In its capacity as a CDK4/CDK6 inhibitor, p16INK4A prevents the phosphorylation of pRB proteins by directly binding CDK4 or CDK6, effectively repressing downstream E2F signaling. p19ARF will not be discussed further here as it cannot bind to CDK4/CDK6 and is therefore not considered a CDK‐inhibitor 124. Deletion of Cdkn2a leading to the loss of both p16INK4A and p19ARF yields viable and fertile animals 125, as does specific deletion of p16INK4A 126. Similarly, singular, or combined deletion of either Cdkn2b (p15INK4B) or Cdkn2c (p18INK4C) does not result in overt defects in spermatogenesis suggesting nonessential roles for these proteins 127, 128. In contrast to the other INK4 family members, deletion of Cdkn2d (p19INK4D) results in the apoptosis of primary spermatocytes 129. The severity of this phenotype is worsened by the additional deletion of Cdkn2d (Cdkn2c−/−, Cdkn2d−/−), suggesting that these proteins might have overlapping functions. Further analysis suggested that Cdkn2c−/−, Cdkn2d−/− spermatogonia show defects entering meiotic divisions. Resultant spermatocytes also fail to correctly undergo normal meiotic division 111. The phenotypes seen in Cdkn2c−/− Cdkn2d−/− mice suggest that at least some inhibition of cyclin D‐dependent kinase activity is essential for the normal progression of spermatogenesis. Somewhat contradictory to these findings are observations that in knockin mice, whereby CDK4 (Cdk4R24C) or CDK6 (Cdk6R31C) are refractory to inhibition by the INK4 inhibitors 130, 131 are fertile, with no spermatogenic defects reported in either case 105, 132. Although compound Cdk4R24C Cdk6R31C mutant mice, which are fully insensitive to INK4 inhibitors have also been generated, it was not reported whether these mice exhibit any fertility defect. As such, it remains unknown to what extent the repression of CDK4/6/cyclin D activity might be required for normal spermatogenesis.
Regulation of the pRB/E2F pathway during spermatogenesis
During spermatogenesis, the prototypical CDK‐substrate RB1 has been demonstrated to be expressed in both spermatogonia and spermatocytes. The inhibitory CDK‐mediated phosphorylation at S795 has also been observed in subsets of spermatogonia. This indicates that active cellular proliferation is mediated by RB1 in these cells 121, 133. In contrast to a primarily anti‐proliferative role for RB1 during mitosis, RB1 in male germ cells seems to prevent differentiation. The conditional deletion of Rb1 expression in male germ cells promotes the differentiation of SSCs, preventing their continued self‐renewal. Despite initial fertility in these conditional Rb1 −/− animals, infertility occurs by 2 months of age due to a failure to replenish cells competent to enter meiosis 133. Interestingly, no overt meiotic defects during the first waves of spermatogenesis were observed in these animals, suggesting that RB1 is not essential for either of the two meiotic divisions. Constitutive deletion of Rb1 results in early embryonic lethality 134, 135, 136. In contrast, the RB‐related proteins p107 and p130 are not required for viability or fertility 137, 138, suggesting that these two proteins are not sufficient to compensate for the loss of Rb1 to ensure normal spermatogenesis. As RB1 is a repressor of E2F signaling, it is presumed that deletion of Rb1 results in unrestrained E2F transcriptional activity. Somewhat at odds with this interpretation is that overexpression of E2F1 in testes induces p53‐independent apoptosis in the testes of transgenic mice and results in the apoptosis of both spermatogonia and early primary spermatocytes 139, 140. Such apoptotic cell death was not observed in Rb1 conditional knockout germ cells.
Interestingly in the converse situation, mice with constitutive deletion of E2f1 exhibit testicular atrophy with aging. This results in the loss of germ cells and subfertility by 3 months of age. This phenotype was also described to progressively worsen with age suggesting that, E2F signaling, at least via E2F1 is required for normal spermatogenesis 141, 142, 143. Similarly to the conditional Rb1 knockout, E2f1−/− testis exhibit a depletion of self‐renewing SSCs 143. Additionally, during the first waves of spermatogenesis, the apoptotic removal of spermatogonia required for the correct establishment of germ/cell/Sertoli cell ratios 144, 145, 146 was noted to be perturbed and increased apoptosis was observed when these cells entered meiosis. E2F1 and several of the other E2F transcription factors have been described to exhibit specific localization during mouse spermatogenesis 143, 147, 148, 149, 150, both in spermatogonia and meiotically dividing spermatocytes. Despite this, the deletion of E2f2 151, E2f3 152, E2f4 153, or E2f5 154 does not cause overt spermatogenic defects when deleted in isolation. This, however, does not necessarily rule out the possibility of functional redundancy to allow compensation upon the deletion of certain E2F family members. The remaining E2F family members may modulate transcription independently of the pRB proteins as they lack a pocket protein binding domain and are, thus, less likely subject to CDK regulation 155, 156.
Roles for CDK2 in the maintenance of SSC homeostasis
In addition to appropriate pRB‐E2F signaling, the regulation of CDK2 activity is also important for normal SSC homeostasis. During spermatogenesis, CDK2 is primarily expressed at high levels in prophase I stage spermatocytes where it has essential functions in mediating homolog synapsis (for details, see below). Interestingly, CDK2 expression has also been noted in SSCs albeit at much lower levels 157, 158. The biological relevance of CDK2 activity in SSCs has only recently been addressed via the analysis of knockin mice with mutations which either partially (Cdk2Y15S/Y15S) or fully (Cdk2T14A,Y15F/T14A,Y15F) ablate the inhibitory phosphorylatable ‘TY’ motif within the glycine‐rich ‘inhibitory loop’ 159, 160. When phosphorylated, the conformation of the inhibitory loop renders the CDK unable to phosphorylate potential substrates due to multiple stable interactions occurring between the CDK catalytic site and bound ATP 161. As such, CDK2 activity in these models is expected to become uncoupled from its regulatory phosphorylation potentially increasing its activity under circumstances when it should usually be inactivated. Both Cdk2Y15S/Y15S and Cdk2T14A,Y15F/ T14A,Y15F were reported as autosomal semidominant male infertility alleles, which cause male infertility when homozygous and less severe spermatogenic defects when heterozygous 159, 160. Although the spermatogenic defects observed in Cdk2T14A,Y15F/T14A,Y15F mice have not been investigated in detail, male infertility in the case of Cdk2Y15S/Y15S arises due to a failure of SSC differentiation. Unlike spermatogenesis in adult animals, the first round of spermatogenesis is initiated by gonocytes 162. Subsequent waves of spermatogenesis are initiated by SSCs following their differentiation from gonocytes during process known as gonocyte‐spermatogonia transition (GST) 162, 163. Intriguingly, Cdk2Y15S/Y15S SSCs inappropriately exhibit properties of gonocytes including the cytoplasmic localization of FOXO1 164, 165, suggesting that GST is impaired in circumstances whereby CDK2 cannot be inhibited via its inhibitory loop phosphorylation. Although the initial wave of spermatogenesis is initiated as normal in Cdk2Y15S/Y15S mice and is able to generate spermatocytes proficient to enter meiosis I, subsequent rounds of spermatogenesis are not observed. Adult Cdk2Y15S/Y15S testes are severely atrophic and despite the presence of mitotically proficient SSC‐like cells positive for the self‐renewal marker GFRA1 166, 167, these cells lack the ability to mature into differentiating spermatogonia. Furthermore Aal chains of spermatogonia in these mice were observed to inappropriately retain GFRA1 expression, which is downregulated in wild‐type spermatogonia of the same stage. These results suggest that regulation of CDK2 activity is required to appropriately complete the developmental switch between gonocytes and spermatogonia, and the resultant differentiation status of abnormal SSCs is not sufficient to support their further differentiation 168. Here, it is hypothesized that premature CDK2 activity in Cdk2Y15S/Y15S gonocytes results in the unscheduled phosphorylation of a factor required for GST. FOXO1 has previously been identified as a CDK2 substrate 169, 170, making it an interesting candidate in mediating such a transitory role in the determination of germ cell fate 168. It may be of interest for future studies to investigate whether the repression of CDK‐associated kinase activity is a requirement for normal GST and whether this is only applicable to CDK2. Canonically, the ‘TY’ motif is phosphorylated and de‐phosphorylated by the WEE1 kinase and CDC25 family of phosphatases, respectively 1, 171, 172, 173. As such, pharmacological inhibition or conditional knockout of these regulatory proteins might be useful in addressing such questions.
Regulation of CDK activity by the CIP/KIP class of CKI proteins during spermatogenesis
In addition to the INK4 protein family of CKI proteins, knockout mice have also been generated for the CIP/KIP family of CKI proteins comprised of p21CIP1 (Cdkn1a), p27KIP1 (Cdkn1b), and p57KIP2 (Cdkn2c). This family is less specific than INK4 and is, thus, able to interact with a broader range of CDK/cyclin complexes including CDK1/CDK2 in addition to CDK4/CDK6.
The deletion of Cdkn1a, Cdkn1b, or both in combination is not associated with spermatogenic defects in male mice 174, 175, 176, 177. Although female infertility is seen in Cdkn1b −/− female animals, this results from hyperplasia of the supporting granulosa cells of the ovary which negatively impact the maturation of ovarian follicles 174, 175, 176. p21CIP1 and p27KIP1 also seem to play a role in restricting the proliferation of Sertoli cells in the testis in early development before these cells become mitotically quiescent. As such, testicular organomegaly is seen upon ablation of Cdkn1a, Cdkn1b, or both in combination 178, due to greater total numbers of Sertoli cells.
Interestingly, severe meiotic defects have been observed when endogenous levels of p27KIP1 are increased via deletion of the F‐box protein SKP2. Under normal circumstances, SKP2 is required for the ubiquitination of p27KIP1, and thus, in this mutant, p27KIP1 is degraded at a lower rate. Minimal fertility resulting from widespread apoptotic loss of spermatogenic cell types is seen in Skp2−/− males, and severe ovarian degeneration was also noted in female animals. Although a detailed analysis of the arrest stage of gametogenesis in these mice was not performed, apoptosis was noted to affect cells in most stages of spermatogenesis. Deletion of Cdkn1b in Skp2−/− mice restores fertility, in both male and female mice confirming that abnormally high levels of p27KIP1 were indeed the cause of defective gametogenesis in these mice 179, 180. As p27 KIP1 has the potential to inhibit both CDK2 and CDK4‐associated complexes and both Cdk2−/− and Cdk4−/− mice display fertility defects, it is likely that these defects arise due to a repression of either one of these kinases. Unlike p21CIP1 or p27KIP1, specific expression of p57KIP2 has been noted during spermatogenesis in both spermatogonia and early spermatocytes 181, 182. The deletion of Cdkn2c leads to neonatal death in the majority of mutant mice 183, 184. The small number of Cdkn2c−/− mice that survive until adulthood exhibits immaturity of germinal tissues including testis and ovaries, which suggests that this gene might be necessary for the normal differentiation of germ cells 185. However, this phenotype was not described in detail and is complicated by the many additional phenotypic defects caused by constitutive Cdkn2c deletion and would likely require the generation of a meiosis‐specific conditional knockout model for any further investigation.
Meiosis‐specific roles for CDK‐associated activity
Aside from the well‐characterized roles for the CDKs and their activating proteins during cell cycle progression, several of these proteins also exhibit meiosis‐specific functions. The standout example of this is arguably CDK2. In addition to the aforementioned role in the differentiation and fate decision process of SSCs, this kinase seems to be involved in several aspects of meiotic division, particularly during meiotic prophase I. One of the most best‐studied of these functions is promoting the normal synapsis of chromosomal homologs. In this context, CDK2 is reliant upon the expression of a non‐cyclin‐activating partner protein, Speedy A. To complicate matters, the canonical CDK2 activators, cyclin E1 and E2, are also essential for homolog synapsis. Accordingly, severe meiotic defects akin to that seen upon individual deletion of Cdk2 or Speedy A can also be observed upon the complete ablation of both E‐type cyclins. The lack of functional redundancy between the Speedy A and the E‐type cyclins, despite sharing CDK2 as their most likely kinase partner, is intriguing and requires further investigation to be properly understood. The following sections will primarily describe the known synaptic defects arising from deletion of CDK2 or its activating proteins. Additional topics discussed in this section are far more speculative and include poorly defined roles for the E‐type cyclins in promoting telomere stability in addition to potential roles for CDK2 and CDK4 in regard to various stages of meiotic recombination.
Potentiation of telomeric CDK2 binding by Speedy A during meiotic prophase I
A telomeric function for CDK2 during meiosis was first investigated due to the localization of this kinase to telomeres throughout the duration of meiotic prophase I alongside components of the multiprotein shelterin complex 157. In somatic cells, the shelterin complex forms a protective cap at telomeres, which prevents the inappropriate activation of the DNA damage response against telomeric DNA 186. During meiotic prophase I, the shelterin complex fulfill a secondary role in ‘tethering’ telomeres to the LINC complex during zygonema (see introduction for prior discussion of the LINC complex). One initial step in the formation of the LINC complex is the interaction between the core shelterin complex member TRF1 and the noncanonical CDK2 activator: Speedy A. Here, TRF1 is thought to act as a scaffold to promote the interaction of Speedy A and CDK2 at the telomeres 65. In accordance with this theory, telomeres of Trf1−/− spermatocytes show a complete failure to recruit both CDK2 and Speedy A 65. Additional evidence suggests that the loading of Speedy A to shelterin is likely to occur at the nuclear envelope, as Speedy A can be detected as multiple foci at this structure prior to the initial tethering of telomeres to the nuclear envelope. Importantly, this interaction is also a prerequisite step for the telomeric binding of CDK2, as telomeric CDK2 foci cannot be observed in Speedy A−/− spermatocytes 187. This CDK2‐recruiting role for Speedy A is supported by the observation that mutagenesis of key Speedy A‐interacting residues on CDK2 negatively impacts the ability of CDK2 to localize to telomeres 187. One key factor driving the interaction between CDK2 and Speedy A seems to be the existence of a longer meiocyte‐specific splice isoform of CDK2 (p39) 22. p39 CDK2 is a preferential partner of Speedy A in meiotic cells as opposed to the normal (≈ 33 kDa) isoform, which forms canonical complexes with cyclins 22, 187, 188. The relevance pertaining to the existence of two distinct isoforms of CDK2 during meiotic prophase is still not fully understood. However, this could potentially be utilized to compartmentalize the activities of both CDK2/Speedy A and CDK2/cyclin complexes for specific cellular processes.
The roles of CDK2 and Speedy A in promoting stable LINC complex formation
During the leptotene–zygotene transition, complex interactions occurring between components of the LINC complex and the cytoskeleton drive the polarization of membrane‐bound proteins and the tethered telomeres. This forces telomeres into close proximity driving their clustering within a small area of the nuclear envelope, which is responsible for the characteristic ‘bouquet’ pattern of chromosomes observed at this stage 189, 190. Bouquet formation is absolutely dependent upon the prior establishment of telomere–LINC complex interactions in the leptotene stage, as this does not occur in mutants where LINC complex components or their interacting proteins have been deleted 59, 61, 62, 65, 67, 187, 191, 192. Following initial telomeric clustering, cytoskeletal forces promote rapid prophase movements (RPM), which allow chromosomes to sample their surroundings for homology in a controlled manner 193, 194, 195. This is thought to be essential for homologs to locate each other and synapse without the occurrence of inappropriate pairing between nonhomologs (nonhomologous synapsis). The involvement of CDK2 and Speedy A during bouquet formation and synapsis is depicted in Fig. 2 with examples of meiotic defects observed upon a failure to establish normal telomere–nuclear envelope interactions.
Figure 2.
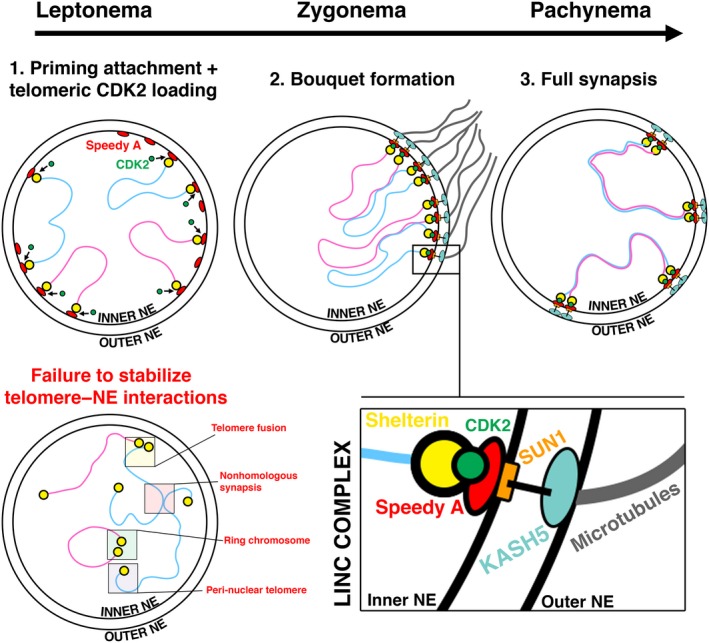
The roles of CDK2 and Speedy A in bouquet formation and synapsis. (1) During leptonema, Speedy A (red ovals) localizes to the inner nuclear envelope and interacts with the shelterin complex (yellow circles) on the telomeric ends of chromosomal homologs. This allows priming attachment or initial tethering of telomeres to the inner nuclear envelope. CDK2 (green circles) is then loaded onto telomeres in a manner dependent upon Speedy A. Here blue and pink lines are used to represent duplicated paternal and maternal chromosomal homologs, respectively; that is, each line contains two sister chromatids linked via their centromeres. (2) During zygonema, the interaction between shelterin, CDK2, and Speedy A is important in promoting the stable formation of the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex. A simplified representation of the main components of the LINC complex is shown as a blow up below. Formation of a stable LINC complex is likely mediated by interactions between CDK2 and the inner nuclear envelope protein SUN1 (orange rectangles). SUN1 in turn is linked to the outer nuclear envelope by interactions with KASH5 (teal ovals). Cytoskeletal forces mediated by microtubules (gray) act on the LINC complex via KASH5 to drive telomere movement along the inner nuclear envelope. This allows telomeres to be polarized on the inner nuclear envelope leading to the formation of a characteristic bouquet pattern of chromosomes. (3) Bouquet formation in pachynema brings chromosomal homologs into close proximity and facilitates homolog paring (synapsis), which is completed in pachynema. An example of meiotic defect occurring upon impaired formation and/or stability of the LINC complex is shown at the bottom left hand side. These defects are characteristic of Cdk2−/− and Speedy A−/− spermatocytes and also mutants whereby shelterin or LINC complex components have been deleted.
Upon deletion of CDK2 or Speedy A, stable telomere–nuclear envelope interactions are lost and telomeres are observed to become detached from the inner nuclear envelope despite initial binding. In both models, it was noted that detached telomeres remain associated with vesicles containing proteins associated with the inner nuclear membrane including SUN1, suggesting a disconnect between the telomeres and the components of the LINC complex at the inner nuclear envelope 61, 191. As a consequence, extensive nonhomologous synapsis occurs between chromosomes. In addition, telomeres engage in inappropriate inter‐ and intrachromosomal interactions resulting in telomeric fusions and the observance of ring chromosomes 61, 191. Upon failure of synapsis in Cdk2−/− and Speedy A−/− spermatocytes, chromosomes are no longer able to access their homologous counterparts to initiate the repair of meiotic DSBs by homologous recombination. Meiotically arrested spermatocytes subsequently undergo apoptosis, causing a complete block of spermatogenesis and infertility 61, 196, 197. The exact reason why CDK2 and Speedy A are required to maintain stable telomere–nuclear envelope interactions is uncertain. It is likely due to a failure to form proper interactions between the telomere and the inner nuclear envelope protein SUN1. In wild‐type spermatocytes, SUN1 localizes to telomeres tethered to the nuclear envelope during leptonema and remains bound whilst chromosomes adopt a bouquet arrangement 59, 62. In Cdk2−/− and Speedy A−/− spermatocytes, SUN1 fails to form a polarized cap along the nuclear envelope in association with its LINC complex partner KASH5 61, 62. Although interactions between CDK2, Speedy A, and SUN1 have not been confirmed in vivo, Ser48 within the N terminus of SUN1 has been shown to be phosphorylated by CDK2 61. Additionally, SUN1 was shown to be co‐immunoprecipitated with CDK2 in vitro, confirming at least that SUN1 is a worthwhile target for future studies 62. As the focal point linking the telomere and nuclear envelope, it is tempting to speculate that phosphorylation of SUN1 might stabilize its interaction with other LINC complex components, allowing telomeres to be grouped into their bouquet formation. The existence of a CDK2 substrate driving this process is supported by the fact that a knockin point mutant of Cdk2 where catalytic activity has been ablated (Cdk2D145N/D145N) shows essentially the same phenotype as both Cdk2−/− and Speedy A−/− spermatocytes 198. This is despite normal telomeric localization of both mutant CDK2D145N and Speedy A (unpublished data from the Kaldis laboratory).
This observed requirement for CDK2 catalytic activity is somewhat at odds with the observation that a truncated Speedy A protein, which is competent to bind telomeres and CDK2 but unable to activate CDK2, is able to rescue impaired telomere–nuclear envelope interactions (when electroporated into Speedy A−/− testis). This suggested that CDK2/Speedy A catalytic activity (against SUN1 or otherwise) is not required for this process 187. An alternate explanation is that catalytically inactive CDK2/Speedy A is able to maintain some degree of telomeric–nuclear envelope binding via noncatalytic interactions with SUN1 and/or its related protein SUN2. Relevant to this point is the observance that even in Sun1−/− mice, suboptimal levels of telomeric tethering and even bouquet formation can be observed although this is ultimately not sufficient for normal synapsis in pachynema 69. Further investigation into the candidacy of SUN1 as a biologically relevant CDK2/Speedy A substrate will likely require the generation of a nonphosphorylatable Sun1 mouse mutant.
The importance of E‐type cyclins for normal synapsis and telomere stability
Recent revelations obtained from knockout models of the LINC complex interactors TERB1, TERB2, and MAJIN have demonstrated the importance of a process termed telomere cap exchange for normal fertility. This event is thought to maintain the telomeric stability of membrane‐bound chromosomes during pachynema 64, 199. During cap exchange, telomeric DNA is transferred to a complex containing TERB1, TERB2, and MAJIN. This requires the dissociation of the shelterin complex proteins from the telomeric ends and results in the formation of a structure known as the telomere attachment plate, which integrates telomeric DNA within the inner nuclear envelope 64, 200, 201, 202, 203, 204, 205, 206, 207. The formation of telomere attachment plates can be determined via the appearance of conical thickenings at the ends of pachytene stage chromosomal axes. Interestingly, the typical formation of these structures is seemingly dependent upon the expression of the E‐type cyclins. Upon deletion of cyclin E2, the conical thickening of telomeric ends is compromised in a manner that can be progressively worsened upon additional deletion of one, or both copies of cyclin E1. In such cyclin E mutant spermatocytes, the failure to develop normal telomere attachment plates was associated with the observance of telomeres within the nuclear space. Furthermore, such abnormal telomeres showed reduced localization of shelterin complex components and positivity for the DNA damage marker, γ‐H2AX, suggesting inappropriate protection of telomeric DNA 208, 209. Despite such distinct telomeric defects, the E‐type cyclins do not exhibit telomeric localization during meiotic prophase, making their interaction with telomeric CDK2 uncertain 209. However, CDK2 has been shown to co‐immunoprecipitate both cyclin E1 and cyclin E2 in protein extracts from wild‐type pachytene spermatocytes. This suggests that a pool of likely nontelomeric CDK2/cyclin E is indeed present in meiotic cells 209. A functional relationship between CDK2 and the E‐type cyclins is also supported by the observation that the deletion of these proteins disrupts normal patterns of CDK2 localization during meiotic prophase. In cyclin E2−/− spermatocytes, for example, only 59% of telomeres were reported to show CDK2 binding at intensities similar to that of wild‐type controls. The additional deletion of one or both copies cyclin E1 subsequently resulted in complete loss of CDK2 binding to the telomeres. As both the severity of telomere defects and loss of CDK2 localization were similarly affected by progressive cyclin E loss 208, 209, it is likely that the action of these cyclins creates a stable environment at telomeres for CDK2 to bind.
In addition to the loss of shelterin integrity, another possibility could be that the cap exchange is defective in the absence of E‐type cyclins. Since the process of telomeric cap exchange requires the dissociation of the shelterin complex from telomeric ends, it is possible that if this process is perturbed, uncapped telomeric ends dislodged from the nuclear envelope might be exposed within the nuclear interior. This would trigger a DNA damage response directed against the uncapped telomeres, similar to that seen upon ablation of the E‐type cyclins. Fascinatingly, cap exchange has been shown to be influenced by CDK activity as the treatment of wild‐type spermatocytes with the unspecific CDK2‐inhibitor Roscovitine 210 leads to abolished cap exchange 64. In regard to a potential CDK‐substrate mediating this process, Thr647 of TERB1 was shown to be phosphorylated during cap exchange. However, electroporation of a nonphosphorylatable TERB1T647A protein into the testis of Terb1−/− mice was able to rescue otherwise defective cap exchange making the biological relevance of this phosphorylation uncertain 64.
Potential CDK2 functions in the formation and/or designation of meiotic crossover sites
In addition to promoting homolog synapsis via its function at meiotic telomeres, CDK2 is also known to localize to late recombination nodules during meiotic prophase. In this capacity, CDK2 foci can be transiently observed to localize to 1‐2 interstitial sites associated with the chromosomal axes of each homolog pair during midpachytene stage of prophase I 157, 159, 191, 209, 211, 212, 213, 214. Here, CDK2 co‐localizes with an E3 sumo‐ligase, RNF212, and a proposed sumo‐targeting ubiquitin ligase, CCNBIP1 (HEI10) 211, 213. These proteins, together with CDK2, have been described as pro‐crossover factors. This is due to their role in the formation and maturation of late recombination nodules, which is associated with the subsequent recruitment of MutLγ (MLH1, MLH3). This is one of the final steps required for the eventual repair of recombination intermediates to form type I crossover sites 215, 216, 217, 218, 219. Late recombination nodule‐associated CDK2 foci appear independently of Speedy A, which is solely observed at telomeres during meiotic prophase I 187, suggesting that any activity of CDK2 at these sites is likely mediated by other activating proteins.
During early meiotic prophase, abundant RNF212 foci can be observed to localize to sites of recombination. During pachynema, a subset of RNF212 foci are able to achieve a ‘stable’ state. This is associated with the subsequent localization of HEI10. Both the increase in RNF212 foci size and the presence of HEI10 foci are considered indicators of sites selected to become late recombination nodules. They are also termed as crossover designation 211. Prior to crossover designation, RNF212‐mediated SUMOylation is thought to stabilize the MutSγ (MSH5, MSH5) complex at recombination sites, promoting their progression toward crossover‐associated repair. At smaller ‘undesignated’ RNF212 foci, RNF212‐mediated SUMOylation also seems to act as a substrate for HEI10. This allows the ubiquitination and turnover of recombination proteins, including MutSγ and RNF212 itself, at recombination sites 220. Sites depleted of MutSγ and RNF212, fail to mature, and are subsequently repaired as non‐crossovers. Therefore, the action of HEI10 can be considered as a mechanism to deselect all but ‘designated’ RNF212 foci from maturing into late recombination nodules. This effectively prevents the formation of excess meiotic crossover sites. A model of crossover designation indicating the observance of CDK2 during this process is presented in Fig. 3. This is based upon the currently known functions of RNF212 and HEI10, as reported by the Hunter laboratory 211, 213. Figure 4 depicts the locations of CDK2 over the meiotic prophase.
Figure 3.
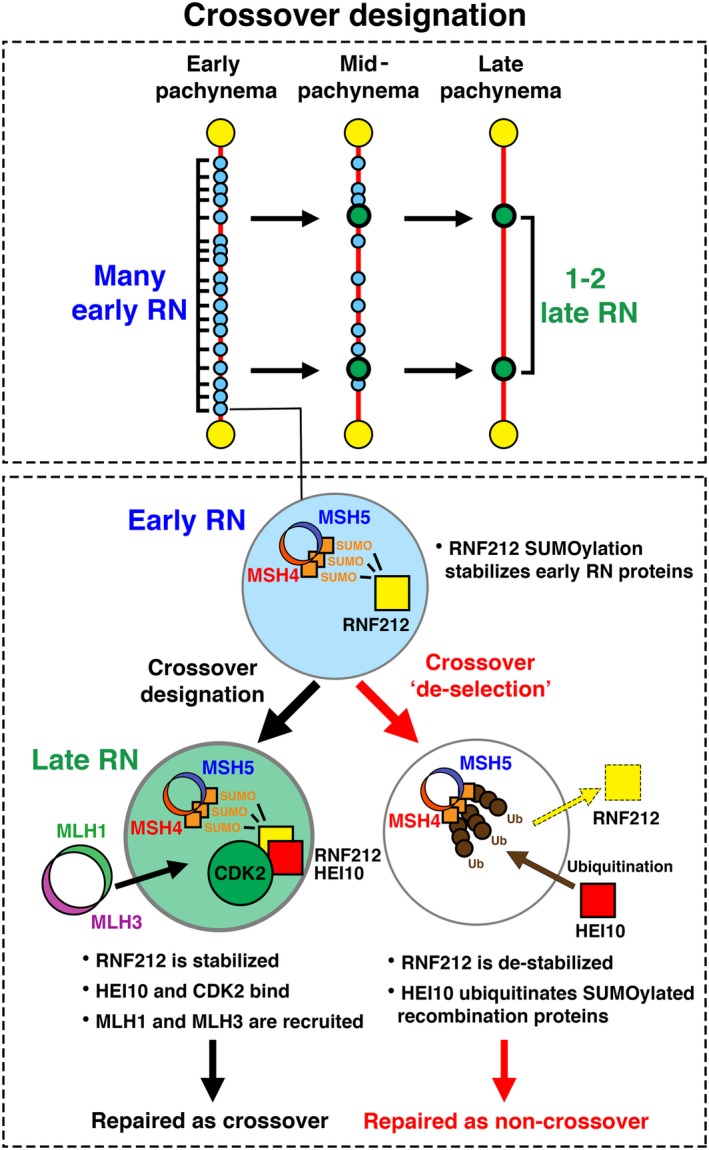
Schematic of the crossover designation process. Upper box: pattern of early and late recombination nodule formation during meiotic prophase I. During early pachynema, proteins associated with early recombination nodules (early RN) localize as many distinct foci along the chromosomal axes. Here, paired chromosomal axes are represented by a red line and early recombination nodules are represented by blue circles. By mid‐pachynema, crossover designation leads to the selection of 1 or maximally 2 early recombination nodules to mature into late recombination nodules (late RN) as shown by green circles. At the same time, nondesignated early recombination nodules are repaired and their associated proteins dissociate from the chromosomal axes. By late pachynema, only late RNs remain associated with the chromosomal axes. These mark the sites at which meiotic crossovers will form. Lower box: Events leading to crossover designation or crossover de‐selection at early recombination nodules. During early pachynema, early RNs exhibit specific localization of many proteins including the MutSγ complex comprised of MSH4 (blue ring) and MSH5 (red ring). MutSγ is thought to be stabilized by SUMOylation mediated by RNF212 (yellow square). During mid‐pachynema, one of two possibilities can occur at early RNs, crossover designation (left hand side) or crossover de‐selection (right hand side). Crossover de‐selection occurs when RNF212 becomes destabilized and dissociates from early RNs. This is thought to allow the ubiquitin ligase CCNBIP1 (HEI10) (red square) to target early RN proteins such as MutSγ for ubiquitination and degradation. These events are associated with the downstream repair of early RNs as non‐crossovers. Crossover designation occurs when RNF212 remains stabilized at early RNs. This is associated with the localization of HEI10 and CDK2. These events are characteristic of late RN formation and leads to the downstream recruitment of the MutLγ complex comprised of MLH1 (green ring) and MLH3 (purple ring). These events are required for the downstream repair of late RNs as crossovers.
Figure 4.
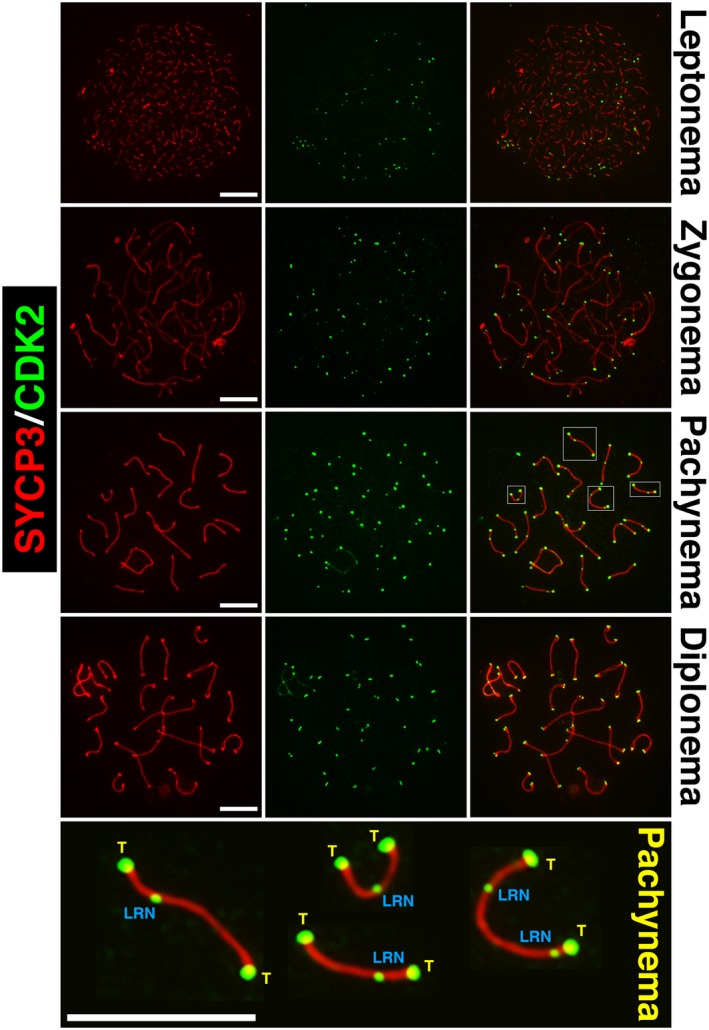
The pattern of CDK2 localization to chromosomal axes during meiotic prophase I. During leptonema, CDK2 foci (green) can be observed to localize to the telomeres of each chromosome. At this stage, chromosomal axes have not yet been formed as synaptonemal complex formation as determined by SYCP3 staining (red) is still at an early stage. During zygonema, homolog pairing is initiated and distinct axes of SYCP3 can be observed to associate with each other. At this stage, CDK2 localization is still specific to telomeres. During mid‐pachynema, intense singular CDK2 foci can be observed at paired telomeres of homologs. At this point, 1‐2 weaker but distinct interstitial foci of CDK2 can be observed marking the formation of late recombination nodules. In the lower most panel, several examples of paired homologs are shown at higher magnification to indicate both telomeric (T) and late recombination nodule‐associated (LRN) CDK2 foci. At this stage, staining can also be seen on the X‐Y chromosomes, but this is not addressed within the scope of this review. By diplonema, late recombination nodule‐associated CDK2 foci dissociate from the chromosomal axes but can still be observed as pairs of CDK2 foci at chromosomal ends. These represent the individual telomeres of each homolog, which separate upon the splitting of chromosomal axes. Scale bars for each image are shown in white and are equivalent to 5 µm in all pictures.
In addition to RNF212, HEI10, and CDK2, an atypical cyclin protein, ‘cyclin N‐terminal domain‐containing protein 1’ (CNTD1), has also been identified as a pro‐crossover factor. This protein seems to be essential for the de‐selection of undesignated crossover sites. In both Cntd1 −/− and Hei10 −/− spermatocytes, stable foci containing both MutSγ and RNF212 foci fail to undergo the de‐selection process in pachynema and crossover designation does not occur 213, 221. The relationship between CNTD1 and HEI10 is still not understood, but one possibility could be that CNTD1 is important for the destabilization of RNF212 at ‘nondesignated’ sites. This destabilization would then allow HEI10 to better target RNF212‐SUMOylated targets for degradation.
At present, the requirement for CDK2 localization at late recombination nodules remains unknown, but this is seemingly affected by the loss of other pro‐crossover factors. Upon singular deletion of Rnf212, Hei10, or Cntd1, CDK2 localization to recombination nodules is severely depleted whilst telomeric CDK2 binding remains seemingly unaffected 211, 214, 221. This indicates that these proteins are responsible or required for the localization of CDK2 to recombination nodules. As crossover designation fails to occur in each of these models, it is likely that the binding and/or action of CDK2 at these recombination nodules either occurs in parallel or directly after the observance of this process. In regard to the possible functions of CDK2 at these sites, it has previously been suggested that CDK2 might form a complex with CNTD1, which functions within the crossover designation process 221. However, at present there is no cytological evidence that CNTD1 is able to localize to recombination nodules or indeed that this protein might interact with CDK2 to form an active kinase complex. It should be noted here that the homolog of CNTD1 in C. elegans, COSA‐1, can localize to meiotic crossover sites 222, 223, 224. However, the comparison of COSA‐1 functions with mammalian CNTD1 is complicated by considerable differences in the process of crossover formation in C. elegans. For example, in this organism, MutSγ seems to function during the final stages of crossover formation and can be observed to localize at crossover sites in late meiotic prophase in a similar manner as described for mammalian MutLγ. Furthermore, although MutSγ foci are stabilized upon deletion of Cntd1, MutSγ becomes destabilized in Cosa‐1 mutants 222. One model addressing potential crossover‐associated CDK2 functions suggested that HEI10 might promote the destruction of CDK2‐bound cyclin allowing it to bind late recombination nodules, possibly in complex with another crossover‐specific interactor 214. As a matter of fact, the formal gene name for Hei10 is cyclin B1‐interacting protein 1 (Cnnb1ip1), which was given due to the discovery that HEI10 can interact with cyclin B1 in a yeast two‐hybrid screen 225. In addition, HEI10 is also a known substrate of cyclin B‐associated CDK activity in vitro 225 and contains several potential CDK consensus sites for phosphorylation within its C terminus. Of these sites, Ser242 (in mouse) is conserved in multiple eukaryotic species and harbors a putative cyclin binding motif upstream of this residue, making it a possible candidate substrate for CDK2 221. The importance of HEI10 for meiotic crossover formation was originally identified in an infertile, N‐ethyl‐N‐nitrosourea‐induced mutant mouse model (Hei10Mei4/Mei4) 214. This model resulted in a partial deletion of the N terminus of HEI10 within another predicted cyclin binding motif, suggesting that loss of HEI10 functions, in this case, might have been caused by the loss of cyclin interaction. To unravel the mysterious functions of CDK2 and its associated proteins at late recombination nodules, it may be a requirement to determine the network of proteins localized specifically to these sites. This might be achievable via immunoprecipitation of the crossover‐associated proteins HEI10 or RNF212 followed by mass spectrometry.
Unexplored roles for cyclin D‐dependent kinase activity during spermatogenesis
In addition to being readily detectable in spermatogonia, CDK4 alongside its partner proteins cyclin D2 and cyclin D3 can also be detected in meiotically dividing spermatocytes 97, 112, 157. In support of a functional role for CDK4 during meiotic division, at least two independent laboratories have indicated that CDK4 is able to localize to the chromosomal axes of spermatocytes in zygonema of meiotic prophase. Here, CDK4 appears as foci in areas where homologous chromosomes have synapsed. CDK4 foci persist until mid‐pachynema at which time they can no longer be observed 157, 221. Such localization has not been reported for CDK6, suggesting that these roles during meiotic prophase might be unique to CDK4. The localization pattern of CDK4 suggests that this kinase might be required for homolog synapsis sometime after meiotic recombination has been initiated. Based on this localization pattern, it is possible that CDK4 might be required to stabilize interactions between the axial and lateral/central elements of the synaptonemal complex to promote their interaction. Many such proteins including SYCP1, SYCE1, and TEX12 have already been noted to be phosphorylated during meiotic prophase I 226, 227. Another possibility is that CDK4 might help to destabilize proteins associated with asynapsed axes to promote their dissociation upon completion of synapsis. For example, the HORMA domain‐containing proteins, HORMAD1 and HORMAD2, are prevalent on asynapsed axes but are quickly removed upon synapsis 228, 229. One further possibility could be that CDK4 might interact with complexes such as MutSγ to stabilize early recombination nodules. In Cntd1−/− mice, CDK4 foci persist at high level throughout pachynema and can be observed in late pachytene spermatocytes. This was similar to the persistence of both RNF212 and the MutSγ complex, also observed in these mutant spermatocytes. This potentially suggests a link between stabilized recombination intermediates and CDK4. If such a stabilizing role is undertaken by CDK4, other possible CDK4 interactors could be the SPO16‐SHOC2 complex and/or TEX11. These proteins are required for stable MutSγ foci formation and also appear to localize to chromosomal axes during similar stages of meiotic prophase, as described for CDK4 230, 231. To date, interactions between CDK4 and meiotic interactors have not been investigated, and the role of this kinase is still uncertain. Elucidation of the possible function of axes associated CDK4 foci will likely require an in‐depth analysis of the spermatogenic defect of Cdk4−/− mice. In regard to this question, the preparation of meiotic surface spreads from Cdk4−/− might be informative as to whether CDK4 contributes toward meiotic processes such as synapsis or meiotic crossover formation.
Potential CDK2 functions related to the regulation of transcription in male germ cells
In addition to the known and potential meiotic roles of CDK2 discussed above, the Kaldis laboratory has recently identified a novel interaction between CDK2 and chromatin 232. This is mediated by interactions between CDK2 and the nuclear respiratory factor 1 transcription factor (NRF1) in male germ cells. Remarkably, through ChIPseq and ChIP‐reChIP approaches, we detected interactions of CDK2 with chromatin‐bound NRF1 at its target gene promoters. Despite the fact that this transcription factor is canonically associated with the regulation of mitochondrial respiration, many of the genes regulated by NRF1 in male germ cells were found to function within meiotic processes, a finding also noted by a prior study 232, 233. Although a thorough analysis of this relationship in meiotic cells was precluded by the early arrest stage of Cdk2−/− spermatocytes and the lethality associated with Nrf1 deletion 234, we were able to observe that CDK2 was a negative regulator of NRF1 transcriptional activity. In line with this theory, we were also able to demonstrate that the DNA binding domain of NRF1 is a substrate of CDK2 and that the phosphorylation by CDK2 decreased NRF1 binding activity to chromatin. In terms of the biological relevance of this interaction, we found that the expression of one of the many NRF1/CDK2 targets, Ehmt1, was elevated in germ cells upon conditional deletion of Cdk2. This was associated with the perturbation of EHMT1‐dependent placement of H3K9me2, during the zygotene–pachytene transition. Together, these findings led us to hypothesize that the expression of CDK2 might be important to modulate NRF1 transcriptional activity during meiotic prophase, which affects the transcription of many meiotic genes including Ehmt1, Msh4, Asz1, Syce1, and Tex19.1. This hypothesis might be best tested in future studies via the conditional deletion of Nrf1 in spermatocytes, utilizing a previously described Nrf1flox/flox model 233. This work further underlines that CDK2 has multiple functions in germ cell development in addition to the established function of tethering telomeres to the nuclear envelope. It will be interesting to determine the cyclin partners and the specific CDK2 substrates that regulate each of the diverse functions of CDK2.
Outstanding questions and outlook
Knockout mouse models have long been the method of choice when ascribing functions to the various cell cycle‐associated genes. An almost exhaustive list of CDK and cyclin knockout mouse models have now been described, offering a great deal of information about the activity of CDKs and their regulatory proteins during gametogenesis. Although many of these roles have been described in the sections above, this is by no means an exhaustive list. Notable exclusions excluded from this review are CDK1, as well as the A‐type and B‐type cyclins. Currently, it is not possible to investigate the effects of cyclin A2 or cyclin B1 deletion due to their requirement for normal development 235, 236. Although conditional deletion of Cdk1 in spermatocytes or constitutive deletion of cyclin A1 has been shown to cause male infertility, these proteins primarily function to drive the exit from meiotic prophase I. Accordingly, these proteins are not required for meiotic recombination, synapsis of homologs, or meiotic crossover formation 237, 238, 239, 240. Similarly, cyclins B2 and B3 are not required during meiotic prophase. Although cyclin B3 deletion results in female infertility, this arises due to its role in triggering anaphase I exit in oocytes 236, 241, 242. There is ample space for future investigations to determine the functions of these cell cycle regulators in germ cell development and meiosis.
In regard to future research, we perceive considerable scope to uncover novel information regarding CDK functions during germ cell development. For example, many of the so‐called atypical cyclins are known to exhibit biased or heightened expression in meiotic tissues 243. Although the function of these proteins is mostly unexplored, knockout models for several of these genes have been described to exhibit fertility defects 244, 245. In a similar vein, the Speedy/RINGO family members Speedy B1a, Speedy B1b, and Speedy B3 16, 246 also exhibit high levels of expression in meiotic tissues. At present, genetic knockouts for these proteins have not been described and it is, therefore, uncertain whether these proteins might be essential for meiotic division as has been described for Speedy A.
As illustrated by this review, normal reproductive health is dependent upon the proper expression and regulation of various CDK/complexes at distinct developmental stages of germ cell development. It is our view that important questions remain surrounding at least five major topics covered in this review:
What is the cause of spermatogenic arrest in Cdk4−/− mice? Does this arise from defects in spermatogonial proliferation/differentiation, or from a failure to complete meiotic division?
Why does CDK4 localize to early recombination nodules and what might its activating partner at such sites be?
Why do Speedy A and the E‐type cyclins have nonredundant roles in promoting chromosomal synapsis? Is this due to their actions at different stages of meiotic prophase? Do the E‐type cyclins form a complex with CDK2 during meiotic prophase or are their actions CDK2‐independent?
How does CDK2/Speedy A stabilize the LINC complex to ensure proper bouquet formation and synapsis? Could this be via the phosphorylation of SUN1 or other related proteins?
What is the purpose of CDK2 localization to late recombination nodules? Does CDK2 partner with a canonical cyclin at these sites or alternatively, an atypical interactor such as CNTD1? Does CDK2 have a specific substrate at these sites, such as HEI10, which it must phosphorylate to promote crossover designation/maturation?
To elucidate the molecular mechanisms behind, the functions of CDKs in germ cell development will not only uncover more details about meiosis but will also help to understand the prevalent fertility issues that has been observed in the human population.
Funding
The work in our laboratory is supported by the Biomedical Research Council, Agency for Science, Technology and Research (A*STAR) to PK, by SINGA (Singapore International Graduate Award) to NP, by the Biomedical Research Council—Joint Council Office Grant (1231AFG031 to PK); by the National Medical Research Council Singapore, NMRC (NMRC/CBRG/0091/2015) to PK, and by National Research Foundation Singapore grant NRF2016‐CRP001‐103 to PK.
Acknowledgments
We thank all present and past members of the Kaldis laboratory for discussions, input, and support, as well as Priti Singh and John Schimenti for productive collaboration. P.K. thanks Kui Liu for more than a decade of collaboration and friendship.
Philipp Kaldis is lead contact
Edited by Angel Nebreda
Contributor Information
Nathan Palmer, Email: npalmer91@live.co.uk.
Philipp Kaldis, Email: philipp.kaldis@med.lu.se.
References
- 1. Morgan DO (1997) Cyclin‐dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13, 261–291. [DOI] [PubMed] [Google Scholar]
- 2. Cao L, Chen F, Yang X, Xu W, Xie J and Yu L (2014) Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol Biol 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doonan JH and Kitsios G (2009) Functional evolution of cyclin‐dependent kinases. Mol Biotechnol 42, 14–29. [DOI] [PubMed] [Google Scholar]
- 4. Lim S and Kaldis P (2013) Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. [DOI] [PubMed] [Google Scholar]
- 5. Loyer P, Trembley JH, Katona R, Kidd VJ and Lahti JM (2005) Role of CDK/cyclin complexes in transcription and RNA splicing. Cell Signal 17, 1033–1051. [DOI] [PubMed] [Google Scholar]
- 6. Sherr CJ and Roberts JM (2004) Living with or without cyclins and cyclin‐dependent kinases. Genes Dev 18, 2699–2711. [DOI] [PubMed] [Google Scholar]
- 7. Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, Morgan DO, Tsai LH and Wolgemuth DJ (2009) Cyclin‐dependent kinases: a family portrait. Nat Cell Biol 11, 1275–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malumbres M (2014) Cyclin‐dependent kinases. Genome Biol 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murray AW and Marks D (2001) Can sequencing shed light on cell cycling? Nature 409, 844–846. [DOI] [PubMed] [Google Scholar]
- 10. Hydbring P, Malumbres M and Sicinski P (2016) Non‐canonical functions of cell cycle cyclins and cyclin‐dependent kinases. Nat Rev Mol Cell Biol 17, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stevenson LM, Deal MS, Hagopian JC and Lew J (2002) Activation mechanism of CDK2: role of cyclin binding versus phosphorylation. Biochemistry 41, 8528–8534. [DOI] [PubMed] [Google Scholar]
- 12. Morgan D (2012) The Cell Cycle Principles of Control. Oxford University Press, Oxford. [Google Scholar]
- 13. Solomon MJ (1993) Activation of the various cyclin/cdc2 protein kinases. Curr Opin Cell Biol 5, 180–186. [DOI] [PubMed] [Google Scholar]
- 14. Cheng A, Gerry S, Kaldis P and Solomon MJ (2005) Biochemical characterization of Cdk2‐Speedy/Ringo A2. BMC Biochem 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McGrath DA, Fifield BA, Marceau AH, Tripathi S, Porter LA and Rubin SM (2017) Structural basis of divergent cyclin‐dependent kinase activation by Spy1/RINGO proteins. EMBO J 36, 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Porter LA, Dellinger RW, Tynan JA, Barnes EA, Kong M, Lenormand JL and Donoghue DJ (2002) Human Speedy: a novel cell cycle regulator that enhances proliferation through activation of Cdk2. J Cell Biol 157, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karaiskou A, Perez LH, Ferby I, Ozon R, Jessus C and Nebreda AR (2001) Differential regulation of Cdc2 and Cdk2 by RINGO and cyclins. J Biol Chem 276, 36028–36034. [DOI] [PubMed] [Google Scholar]
- 18. Ferby I, Blazquez M, Palmer A, Eritja R and Nebreda AR (1999) A novel p34cdc2‐binding and activating protein that is necessary and sufficient to trigger G2/M progression in Xenopus oocytes. Genes Dev 13, 2177–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cheng A, Xiong W, Ferrell JE Jr and Solomon MJ (2005) Identification and comparative analysis of multiple mammalian Speedy/Ringo proteins. Cell Cycle 4, 155–165. [DOI] [PubMed] [Google Scholar]
- 20. Dinarina A, Perez LH, Davila A, Schwab M, Hunt T and Nebreda AR (2005) Characterization of a new family of cyclin‐dependent kinase activators. Biochem J 386, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang R, Morosetti R and Koeffler HP (1997) Characterization of a second human cyclin A that is highly expressed in testis and in several leukemic cell lines. Cancer Res 57, 913–920. [PubMed] [Google Scholar]
- 22. Ellenrieder C, Bartosch B, Lee GY, Murphy M, Sweeney C, Hergersberg M, Carrington M, Jaussi R and Hunt T (2001) The long form of CDK2 arises via alternative splicing and forms an active protein kinase with cyclins A and E. DNA Cell Biol 20, 413–423. [DOI] [PubMed] [Google Scholar]
- 23. Nguyen TB, Manova K, Capodieci P, Lindon C, Bottega S, Wang XY, Refik‐Rogers J, Pines J, Wolgemuth DJ and Koff A (2002) Characterization and expression of mammalian cyclin B3, a prepachytene meiotic cyclin. J Biol Chem 277, 41960–41969. [DOI] [PubMed] [Google Scholar]
- 24. Chotiner JY, Wolgemuth DJ and Wang PJ (2019) Functions of cyclins and CDKs in mammalian gametogenesis. Biol Reprod, 101, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Borum K (1961) Oogenesis in the mouse. A study of the meiotic prophase. Exp Cell Res 24, 495–507. [DOI] [PubMed] [Google Scholar]
- 26. Speed RM (1982) Meiosis in the foetal mouse ovary. I. An analysis at the light microscope level using surface‐spreading. Chromosoma 85, 427–437. [DOI] [PubMed] [Google Scholar]
- 27. Oakberg EF (1956) Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am J Anat 99, 507–516. [DOI] [PubMed] [Google Scholar]
- 28. de Rooij DG and Russell LD (2000) All you wanted to know about spermatogonia but were afraid to ask. J Androl 21, 776–798. [PubMed] [Google Scholar]
- 29. Manova K, Nocka K, Besmer P and Bachvarova RF (1990) Gonadal expression of c‐kit encoded at the W locus of the mouse. Development 110, 1057–1069. [DOI] [PubMed] [Google Scholar]
- 30. Yoshinaga K, Nishikawa S, Ogawa M, Hayashi S, Kunisada T and Fujimoto T (1991) Role of c‐kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c‐kit expression and function. Development 113, 689–699. [DOI] [PubMed] [Google Scholar]
- 31. Liskay RM (1977) Absence of a measurable G2 phase in two Chinese hamster cell lines. Proc Natl Acad Sci USA 74, 1622–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leblond CP and El‐Alfy M (1998) The eleven stages of the cell cycle, with emphasis on the changes in chromosomes and nucleoli during interphase and mitosis. Anat Rec 252, 426–443. [DOI] [PubMed] [Google Scholar]
- 33. Bennett MD (1977) The time and duration of meiosis. Philos Trans R Soc Lond B Biol Sci 277, 201–226. [DOI] [PubMed] [Google Scholar]
- 34. Keeney S, Giroux CN and Kleckner N (1997) Meiosis‐specific DNA double‐strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384. [DOI] [PubMed] [Google Scholar]
- 35. Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A and Forterre P (1997) An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386, 414–417. [DOI] [PubMed] [Google Scholar]
- 36. Baudat F, Imai Y and de Massy B (2013) Meiotic recombination in mammals: localization and regulation. Nat Rev Genet 14, 794–806. [DOI] [PubMed] [Google Scholar]
- 37. Brick K, Smagulova F, Khil P, Camerini‐Otero RD and Petukhova GV (2012) Genetic recombination is directed away from functional genomic elements in mice. Nature 485, 642–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grey C, Barthes P, Chauveau‐Le Friec G, Langa F, Baudat F and de Massy B (2011) Mouse PRDM9 DNA‐binding specificity determines sites of histone H3 lysine 4 trimethylation for initiation of meiotic recombination. PLoS Biol 9, e1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baudat F, Buard J, Grey C, Fledel‐Alon A, Ober C, Przeworski M, Coop G and de Massy B (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parvanov ED, Petkov PM and Paigen K (2010) Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walker M, Billings T, Baker CL, Powers N, Tian H, Saxl RL, Choi K, Hibbs MA, Carter GW, Handel MA et al (2015) Affinity‐seq detects genome‐wide PRDM9 binding sites and reveals the impact of prior chromatin modifications on mammalian recombination hotspot usage. Epigenetics Chromatin 8, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND et al. (2011) A hierarchical combination of factors shapes the genome‐wide topography of yeast meiotic recombination initiation. Cell 144, 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baudat F and Nicolas A (1997) Clustering of meiotic double‐strand breaks on yeast chromosome III. Proc Natl Acad Sci USA 94, 5213–5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO and Petes TD (2000) Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 97, 11383–11390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Buhler C, Borde V and Lichten M (2007) Mapping meiotic single‐strand DNA reveals a new landscape of DNA double‐strand breaks in Saccharomyces cerevisiae. PLoS Biol 5, e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Blitzblau HG, Bell GW, Rodriguez J, Bell SP and Hochwagen A (2007) Mapping of meiotic single‐stranded DNA reveals double‐stranded‐break hotspots near centromeres and telomeres. Curr Biol 17, 2003–2012. [DOI] [PubMed] [Google Scholar]
- 47. Barlow AL, Benson FE, West SC and Hulten MA (1997) Distribution of the Rad51 recombinase in human and mouse spermatocytes. EMBO J 16, 5207–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cole F, Kauppi L, Lange J, Roig I, Wang R, Keeney S and Jasin M (2012) Homeostatic control of recombination is implemented progressively in mouse meiosis. Nat Cell Biol 14, 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Loidl J, Klein F and Scherthan H (1994) Homologous pairing is reduced but not abolished in asynaptic mutants of yeast. J Cell Biol 125, 1191–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baudat F, Manova K, Yuen JP, Jasin M and Keeney S (2000) Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol Cell 6, 989–998. [DOI] [PubMed] [Google Scholar]
- 51. Romanienko PJ and Camerini‐Otero RD (2000) The mouse Spo11 gene is required for meiotic chromosome synapsis. Mol Cell 6, 975–987. [DOI] [PubMed] [Google Scholar]
- 52. Page SL and Hawley RS (2004) The genetics and molecular biology of the synaptonemal complex. Annu Rev Cell Dev Biol 20, 525–558. [DOI] [PubMed] [Google Scholar]
- 53. Kleckner N (2006) Chiasma formation: chromatin/axis interplay and the role(s) of the synaptonemal complex. Chromosoma 115, 175–194. [DOI] [PubMed] [Google Scholar]
- 54. Kleckner N (1996) Meiosis: how could it work? Proc Natl Acad Sci USA 93, 8167–8174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zickler D and Kleckner N (1999) Meiotic chromosomes: integrating structure and function. Annu Rev Genet 33, 603–754. [DOI] [PubMed] [Google Scholar]
- 56. Moens PB and Pearlman RE (1990) Telomere and centromere DNA are associated with the cores of meiotic prophase chromosomes. Chromosoma 100, 8–14. [DOI] [PubMed] [Google Scholar]
- 57. Szostak JW, Orr‐Weaver TL, Rothstein RJ and Stahl FW (1983) The double‐strand‐break repair model for recombination. Cell 33, 25–35. [DOI] [PubMed] [Google Scholar]
- 58. Carpenter AT (1975) Electron microscopy of meiosis in Drosophila melanogaster females: II. The recombination nodule–a recombination‐associated structure at pachytene? Proc Natl Acad Sci USA 72, 3186–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ding X, Xu R, Yu J, Xu T, Zhuang Y and Han M (2007) SUN1 is required for telomere attachment to nuclear envelope and gametogenesis in mice. Dev Cell 12, 863–872. [DOI] [PubMed] [Google Scholar]
- 60. Haque F, Mazzeo D, Patel JT, Smallwood DT, Ellis JA, Shanahan CM and Shackleton S (2010) Mammalian SUN protein interaction networks at the inner nuclear membrane and their role in laminopathy disease processes. J Biol Chem 285, 3487–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mikolcevic P, Isoda M, Shibuya H, del Barco Barrantes I, Igea A, Suja JA, Shackleton S, Watanabe Y and Nebreda AR (2016) Essential role of the Cdk2 activator RingoA in meiotic telomere tethering to the nuclear envelope. Nat Commun 7, 11084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Viera A, Alsheimer M, Gomez R, Berenguer I, Ortega S, Symonds CE, Santamaria D, Benavente R and Suja JA (2015) CDK2 regulates nuclear envelope protein dynamics and telomere attachment in mouse meiotic prophase. J Cell Sci 128, 88–99. [DOI] [PubMed] [Google Scholar]
- 63. Shibuya H, Ishiguro K and Watanabe Y (2014) The TRF1‐binding protein TERB1 promotes chromosome movement and telomere rigidity in meiosis. Nat Cell Biol 16, 145–156. [DOI] [PubMed] [Google Scholar]
- 64. Shibuya H, Hernandez‐Hernandez A, Morimoto A, Negishi L, Hoog C and Watanabe Y (2015) MAJIN links telomeric DNA to the nuclear membrane by exchanging telomere cap. Cell 163, 1252–1266. [DOI] [PubMed] [Google Scholar]
- 65. Wang L, Tu Z, Liu C, Liu H, Kaldis P, Chen Z and Li W (2018) Dual roles of TRF1 in tethering telomeres to the nuclear envelope and protecting them from fusion during meiosis. Cell Death Differ 25, 1174–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shibuya H and Watanabe Y (2014) The meiosis‐specific modification of mammalian telomeres. Cell Cycle 13, 2024–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Horn HF, Kim DI, Wright GD, Wong ES, Stewart CL, Burke B and Roux KJ (2013) A mammalian KASH domain protein coupling meiotic chromosomes to the cytoskeleton. J Cell Biol 202, 1023–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Morimoto A, Shibuya H, Zhu X, Kim J, Ishiguro K, Han M and Watanabe Y (2012) A conserved KASH domain protein associates with telomeres, SUN1, and dynactin during mammalian meiosis. J Cell Biol 198, 165–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Link J, Leubner M, Schmitt J, Gob E, Benavente R, Jeang KT, Xu R and Alsheimer M (2014) Analysis of meiosis in SUN1 deficient mice reveals a distinct role of SUN2 in mammalian meiotic LINC complex formation and function. PLoS Genet 10, e1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Moens PB, Kolas NK, Tarsounas M, Marcon E, Cohen PE and Spyropoulos B (2002) The time course and chromosomal localization of recombination‐related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA‐DNA interactions without reciprocal recombination. J Cell Sci 115, 1611–1622. [DOI] [PubMed] [Google Scholar]
- 71. Bishop DK (1994) RecA homologs Dmc1 and Rad51 interact to form multiple nuclear complexes prior to meiotic chromosome synapsis. Cell 79, 1081–1092. [DOI] [PubMed] [Google Scholar]
- 72. Tarsounas M, Morita T, Pearlman RE and Moens PB (1999) RAD51 and DMC1 form mixed complexes associated with mouse meiotic chromosome cores and synaptonemal complexes. J Cell Biol 147, 207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Carpenter AT (1979) Synaptonemal complex and recombination nodules in wild‐type Drosophila melanogaster females. Genetics 92, 511–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Buonomo SB, Clyne RK, Fuchs J, Loidl J, Uhlmann F and Nasmyth K (2000) Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 103, 387–398. [DOI] [PubMed] [Google Scholar]
- 75. Hassold T and Hunt P (2001) To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet 2, 280–291. [DOI] [PubMed] [Google Scholar]
- 76. Lacefield S and Murray AW (2007) The spindle checkpoint rescues the meiotic segregation of chromosomes whose crossovers are far from the centromere. Nat Genet 39, 1273–1277. [DOI] [PubMed] [Google Scholar]
- 77. Sakuno T, Tanaka K, Hauf S and Watanabe Y (2011) Repositioning of aurora B promoted by chiasmata ensures sister chromatid mono‐orientation in meiosis I. Dev Cell 21, 534–545. [DOI] [PubMed] [Google Scholar]
- 78. Watanabe Y (2012) Geometry and force behind kinetochore orientation: lessons from meiosis. Nat Rev Mol Cell Biol 13, 370–382. [DOI] [PubMed] [Google Scholar]
- 79. Klein F, Mahr P, Galova M, Buonomo SB, Michaelis C, Nairz K and Nasmyth K (1999) A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell 98, 91–103. [DOI] [PubMed] [Google Scholar]
- 80. Allers T and Lichten M (2001) Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106, 47–57. [DOI] [PubMed] [Google Scholar]
- 81. Wyatt HD and West SC (2014) Holliday junction resolvases. Cold Spring Harb Perspect Biol 6, a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Osman F, Dixon J, Doe CL and Whitby MC (2003) Generating crossovers by resolution of nicked Holliday junctions: a role for Mus81‐Eme1 in meiosis. Mol Cell 12, 761–774. [DOI] [PubMed] [Google Scholar]
- 83. Moens PB (1973) Mechanisms of chromosome synapsis at meiotic prophase. Int Rev Cytol 35, 117–134. [DOI] [PubMed] [Google Scholar]
- 84. Marcon E and Moens PB (2005) The evolution of meiosis: recruitment and modification of somatic DNA‐repair proteins. BioEssays 27, 795–808. [DOI] [PubMed] [Google Scholar]
- 85. Moens PB, Marcon E, Shore JS, Kochakpour N and Spyropoulos B (2007) Initiation and resolution of interhomolog connections: crossover and non‐crossover sites along mouse synaptonemal complexes. J Cell Sci 120, 1017–1027. [DOI] [PubMed] [Google Scholar]
- 86. Hunter N (2015) Meiotic recombination: the essence of heredity. Cold Spring Harb Perspect Biol 7, a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zickler D and Kleckner N (2015) Recombination, pairing, and synapsis of homologs during meiosis. Cold Spring Harb Perspect Biol 7, a016626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Risal S, Adhikari D and Liu K (2016) Animal models for studying the in vivo functions of cell cycle CDKs. Methods Mol Biol 1336, 155–166. [DOI] [PubMed] [Google Scholar]
- 89. Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM and Sherr CJ (1999) The p21Cip1 and p27Kip1 CDK 'inhibitors' are essential activators of cyclin D‐dependent kinases in murine fibroblasts. EMBO J 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cobrinik D, Dowdy SF, Hinds PW, Mittnacht S and Weinberg RA (1992) The retinoblastoma protein and the regulation of cell cycling. Trends Biochem Sci 17, 312–315. [DOI] [PubMed] [Google Scholar]
- 91. Dyson N (1998) The regulation of E2F by pRB‐family proteins. Genes Dev 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- 92. Nevins JR (1998) Toward an understanding of the functional complexity of the E2F and retinoblastoma families. Cell Growth Differ 9, 585–593. [PubMed] [Google Scholar]
- 93. Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI and Weinberg RA (1992) Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70, 993–1006. [DOI] [PubMed] [Google Scholar]
- 94. Dyson NJ (2016) RB1: a prototype tumor suppressor and an enigma. Genes Dev 30, 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bellve AR, Cavicchia JC, Millette CF, O'Brien DA, Bhatnagar YM and Dym M (1977) Spermatogenic cells of the prepuberal mouse. Isolation and morphological characterization. J Cell Biol 74, 68–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. de Rooij DG and Grootegoed JA (1998) Spermatogonial stem cells. Curr Opin Cell Biol 10, 694–701. [DOI] [PubMed] [Google Scholar]
- 97. Beumer TL, Roepers‐Gajadien HL, Gademan IS, Kal HB and de Rooij DG (2000) Involvement of the D‐type cyclins in germ cell proliferation and differentiation in the mouse. Biol Reprod 63, 1893–1898. [DOI] [PubMed] [Google Scholar]
- 98. Fantl V, Stamp G, Andrews A, Rosewell I and Dickson C (1995) Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev 9, 2364–2372. [DOI] [PubMed] [Google Scholar]
- 99. Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, Haslam SZ, Bronson RT, Elledge SJ and Weinberg RA (1995) Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82, 621–630. [DOI] [PubMed] [Google Scholar]
- 100. Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H et al. (2003) Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 4, 451–461. [DOI] [PubMed] [Google Scholar]
- 101. Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, Gardner H and Sicinski P (2002) Development of mice expressing a single D‐type cyclin. Genes Dev 16, 3277–3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD et al. (1996) Cyclin D2 is an FSH‐responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384, 470–474. [DOI] [PubMed] [Google Scholar]
- 103. Walker WH and Cheng J (2005) FSH and testosterone signaling in Sertoli cells. Reproduction 130, 15–28. [DOI] [PubMed] [Google Scholar]
- 104. Burns KH, Agno JE, Sicinski P and Matzuk MM (2003) Cyclin D2 and p27 are tissue‐specific regulators of tumorigenesis in inhibin alpha knockout mice. Mol Endocrinol 17, 2053–2069. [DOI] [PubMed] [Google Scholar]
- 105. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP and Barbacid M (1999) Loss of Cdk4 expression causes insulin‐deficient diabetes and Cdk4 activation results in beta‐islet cell hyperplasia. Nat Genet 22, 44–52. [DOI] [PubMed] [Google Scholar]
- 106. Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A and Kiyokawa H (1999) Targeted disruption of CDK4 delays cell cycle entry with enhanced p27Kip1 activity. Mol Cell Biol 19, 7011–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S, Dubus P and Barbacid M (2004) Mammalian cells cycle without the D‐type cyclin‐dependent kinases Cdk4 and Cdk6. Cell 118, 493–504. [DOI] [PubMed] [Google Scholar]
- 108. Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, Geng Y, Yu Q, Bhattacharya S, Bronson RT et al (2004) Mouse development and cell proliferation in the absence of D‐cyclins. Cell 118, 477–491. [DOI] [PubMed] [Google Scholar]
- 109. Rhee K and Wolgemuth DJ (1995) Cdk family genes are expressed not only in dividing but also in terminally differentiated mouse germ cells, suggesting their possible function during both cell division and differentiation. Dev Dyn 204, 406–420. [DOI] [PubMed] [Google Scholar]
- 110. Zhang Q, Wang X and Wolgemuth DJ (1999) Developmentally regulated expression of cyclin D3 and its potential in vivo interacting proteins during murine gametogenesis. Endocrinology 140, 2790–2800. [DOI] [PubMed] [Google Scholar]
- 111. Zindy F, den Besten W, Chen B, Rehg JE, Latres E, Barbacid M, Pollard JW, Sherr CJ, Cohen PE and Roussel MF (2001) Control of spermatogenesis in mice by the cyclin D‐dependent kinase inhibitors p18Ink4c and p19Ink4d . Mol Cell Biol 21, 3244–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bartkova J, Lukas C, Sorensen CS, Rajpert‐De Meyts E, Skakkebaek NE, Lukas J and Bartek J (2003) Deregulation of the RB pathway in human testicular germ cell tumours. J Pathol 200, 149–156. [DOI] [PubMed] [Google Scholar]
- 113. Moons DS, Jirawatnotai S, Tsutsui T, Franks R, Parlow AF, Hales DB, Gibori G, Fazleabas AT and Kiyokawa H (2002) Intact follicular maturation and defective luteal function in mice deficient for cyclin‐ dependent kinase‐4. Endocrinology 143, 647–654. [DOI] [PubMed] [Google Scholar]
- 114. Mettus RV and Rane SG (2003) Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene 22, 8413–8421. [DOI] [PubMed] [Google Scholar]
- 115. Cameron DF, Murray FT and Drylie DD (1985) Interstitial compartment pathology and spermatogenic disruption in testes from impotent diabetic men. Anat Rec 213, 53–62. [DOI] [PubMed] [Google Scholar]
- 116. Murray FT, Cameron DF and Orth JM (1983) Gonadal dysfunction in the spontaneously diabetic BB rat. Metabolism 32, 141–147. [DOI] [PubMed] [Google Scholar]
- 117. Sherr CJ and Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1‐phase progression. Genes Dev 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- 118. Malumbres M and Barbacid M (2005) Mammalian cyclin‐dependent kinases. Trends Biochem Sci 30, 630–641. [DOI] [PubMed] [Google Scholar]
- 119. Russo AA, Tong L, Lee JO, Jeffrey PD and Pavletich NP (1998) Structural basis for inhibition of the cyclin‐dependent kinase Cdk6 by the tumour suppressor p16INK4a . Nature 395, 237–243. [DOI] [PubMed] [Google Scholar]
- 120. Zindy F, Quelle DE, Roussel MF and Sherr CJ (1997) Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 15, 203–211. [DOI] [PubMed] [Google Scholar]
- 121. Bartkova J, Rajpert‐De Meyts E, Skakkebaek NE, Lukas J and Bartek J (2003) Deregulation of the G1/S‐phase control in human testicular germ cell tumours. APMIS 111, 252–265. [DOI] [PubMed] [Google Scholar]
- 122. Bartkova J, Thullberg M, Rajpert‐De Meyts E, Skakkebaek NE and Bartek J (2000) Lack of p19INK4d in human testicular germ‐cell tumours contrasts with high expression during normal spermatogenesis. Oncogene 19, 4146–4150. [DOI] [PubMed] [Google Scholar]
- 123. Gromley A, Churchman ML, Zindy F and Sherr CJ (2009) Transient expression of the Arf tumor suppressor during male germ cell and eye development in Arf‐Cre reporter mice. Proc Natl Acad Sci USA 106, 6285–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Quelle DE, Zindy F, Ashmun RA and Sherr CJ (1995) Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83, 993–1000. [DOI] [PubMed] [Google Scholar]
- 125. Serrano M, Lee H, Chin L, Cordon‐Cardo C, Beach D and DePinho RA (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell 85, 27–37. [DOI] [PubMed] [Google Scholar]
- 126. Churchman ML, Roig I, Jasin M, Keeney S and Sherr CJ (2011) Expression of arf tumor suppressor in spermatogonia facilitates meiotic progression in male germ cells. PLoS Genet 7, e1002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen‐Kiang S, Su L and Xiong Y (1998) CDK inhibitors p18INK4c and p27Kip1 mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev 12, 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Latres E, Malumbres M, Sotillo R, Martin J, Ortega S, Martin‐Caballero J, Flores JM, Cordon‐Cardo C and Barbacid M (2000) Limited overlapping roles of p15INK4b and p18INK4c cell cycle inhibitors in proliferation and tumorigenesis. EMBO J 19, 3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zindy F, van Deursen J, Grosveld G, Sherr CJ and Roussel MF (2000) INK4d‐deficient mice are fertile despite testicular atrophy. Mol Cell Biol 20, 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann‐Hieb E, De Plaen E, Hankeln T,Meyer zum Buschenfelde KH and Beach D (1995) A p16INK4a‐insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269, 1281–1284. [DOI] [PubMed] [Google Scholar]
- 131. Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, Hayward N and Dracopoli NC (1996) Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet 12, 97–99. [DOI] [PubMed] [Google Scholar]
- 132. Rodriguez‐Diez E, Quereda V, Bellutti F, Prchal‐Murphy M, Partida D, Eguren M, Kollmann K, Gomez de Cedron M, Dubus P, Canamero M et al. (2014) Cdk4 and Cdk6 cooperate in counteracting the INK4 family of inhibitors during murine leukemogenesis. Blood 124, 2380–2390. [DOI] [PubMed] [Google Scholar]
- 133. Hu YC, de Rooij DG and Page DC (2013) Tumor suppressor gene Rb is required for self‐renewal of spermatogonial stem cells in mice. Proc Natl Acad Sci USA 110, 12685–12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH and Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359, 288–294. [DOI] [PubMed] [Google Scholar]
- 135. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA and Weinberg RA (1992) Effects of an Rb mutation in the mouse. Nature 359, 295–300. [DOI] [PubMed] [Google Scholar]
- 136. Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A and te Riele H (1992) Requirement for a functional Rb‐1 gene in murine development. Nature 359, 328–330. [DOI] [PubMed] [Google Scholar]
- 137. Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, Harlow E, Beach D, Weinberg RA and Jacks T (1996) Shared role of the pRB‐related p130 and p107 proteins in limb development. Genes Dev 10, 1633–1644. [DOI] [PubMed] [Google Scholar]
- 138. Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, Harlow E and Jacks T (1996) Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev 10, 1621–1632. [DOI] [PubMed] [Google Scholar]
- 139. Holmberg C, Helin K, Sehested M and Karlstrom O (1998) E2F‐1‐induced p53‐independent apoptosis in transgenic mice. Oncogene 17, 143–155. [DOI] [PubMed] [Google Scholar]
- 140. Agger K, Santoni‐Rugiu E, Holmberg C, Karlstrom O and Helin K (2005) Conditional E2F1 activation in transgenic mice causes testicular atrophy and dysplasia mimicking human CIS. Oncogene 24, 780–789. [DOI] [PubMed] [Google Scholar]
- 141. Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E and Dyson NJ (1996) Tumor induction and tissue atrophy in mice lacking E2F–1. Cell 85, 537–548. [DOI] [PubMed] [Google Scholar]
- 142. Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM, Orkin SH and Greenberg ME (1996) E2F–1 functions in mice to promote apoptosis and suppress proliferation. Cell 85, 549–561. [DOI] [PubMed] [Google Scholar]
- 143. Rotgers E, Nurmio M, Pietila E, Cisneros‐Montalvo S and Toppari J (2015) E2F1 controls germ cell apoptosis during the first wave of spermatogenesis. Andrology 3, 1000–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Shaha C, Tripathi R and Mishra DP (2010) Male germ cell apoptosis: regulation and biology. Philos Trans R Soc Lond B Biol Sci 365, 1501–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rodriguez I, Ody C, Araki K, Garcia I and Vassalli P (1997) An early and massive wave of germinal cell apoptosis is required for the development of functional spermatogenesis. EMBO J 16, 2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. De Rooij DG and Lok D (1987) Regulation of the density of spermatogonia in the seminiferous epithelium of the Chinese hamster: II. Differentiating spermatogonia. Anat Rec 217, 131–136. [DOI] [PubMed] [Google Scholar]
- 147. Toppari J, Suominenf JS and Yan W (2003) The role of retinoblastoma protein family in the control of germ cell proliferation, differentiation and survival. APMIS 111, 245–251. [DOI] [PubMed] [Google Scholar]
- 148. Yan W, Kero J, Suominen J and Toppari J (2001) Differential expression and regulation of the retinoblastoma family of proteins during testicular development and spermatogenesis: roles in the control of germ cell proliferation, differentiation and apoptosis. Oncogene 20, 1343–1356. [DOI] [PubMed] [Google Scholar]
- 149. El‐Darwish KS, Parvinen M and Toppari J (2006) Differential expression of members of the E2F family of transcription factors in rodent testes. Reprod Biol Endocrinol 4, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hoja MR, Liu JG, Mohammadieh M, Kvist U and Yuan L (2004) E2F1 deficiency impairs murine spermatogenesis and augments testicular degeneration in SCP3‐nullizygous mice. Cell Death Differ 11, 354–356. [DOI] [PubMed] [Google Scholar]
- 151. Murga M, Fernandez‐Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, Balomenos D, Vicario A, Carrera AC, Orkin SH et al. (2001) Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 15, 959–970. [DOI] [PubMed] [Google Scholar]
- 152. Cloud JE, Rogers C, Reza TL, Ziebold U, Stone JR, Picard MH, Caron AM, Bronson RT and Lees JA (2002) Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Mol Cell Biol 22, 2663–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rempel RE, Saenz‐Robles MT, Storms R, Morham S, Ishida S, Engel A, Jakoi L, Melhem MF, Pipas JM, Smith C et al. (2000) Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol Cell 6, 293–306. [DOI] [PubMed] [Google Scholar]
- 154. Lindeman GJ, Dagnino L, Gaubatz S, Xu Y, Bronson RT, Warren HB and Livingston DM (1998) A specific, nonproliferative role for E2F–5 in choroid plexus function revealed by gene targeting. Genes Dev 12, 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Chen HZ, Tsai SY and Leone G (2009) Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer 9, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Morgunova E, Yin Y, Jolma A, Dave K, Schmierer B, Popov A, Eremina N, Nilsson L and Taipale J (2015) Structural insights into the DNA‐binding specificity of E2F family transcription factors. Nat Commun 6, 10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ashley T, Walpita D and de Rooij DG (2001) Localization of two mammalian cyclin dependent kinases during mammalian meiosis. J Cell Sci 114, 685–693. [DOI] [PubMed] [Google Scholar]
- 158. Ravnik SE and Wolgemuth DJ (1999) Regulation of meiosis during mammalian spermatogenesis: the A‐type cyclins and their associated cyclin‐dependent kinases are differentially expressed in the germ‐cell lineage. Dev Biol 207, 408–418. [DOI] [PubMed] [Google Scholar]
- 159. Singh P and Schimenti JC (2015) The genetics of human infertility by functional interrogation of SNPs in mice. Proc Natl Acad Sci USA 112, 10431–10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhao H, Chen X, Gurian‐West M and Roberts JM (2012) Loss of cyclin‐dependent kinase 2 (CDK2) inhibitory phosphorylation in a CDK2AF knock‐in mouse causes misregulation of DNA replication and centrosome duplication. Mol Cell Biol 32, 1421–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Welburn JP, Tucker JA, Johnson T, Lindert L, Morgan M, Willis A, Noble ME and Endicott JA (2007) How tyrosine 15 phosphorylation inhibits the activity of cyclin‐dependent kinase 2‐cyclin A. J Biol Chem 282, 3173–3181. [DOI] [PubMed] [Google Scholar]
- 162. Yoshida S, Sukeno M, Nakagawa T, Ohbo K, Nagamatsu G, Suda T and Nabeshima Y (2006) The first round of mouse spermatogenesis is a distinctive program that lacks the self‐renewing spermatogonia stage. Development 133, 1495–1505. [DOI] [PubMed] [Google Scholar]
- 163. de Rooij DG (1998) Stem cells in the testis. Int J Exp Pathol 79, 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Goertz MJ, Wu Z, Gallardo TD, Hamra FK and Castrillon DH (2011) Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest 121, 3456–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Pui HP and Saga Y (2017) Gonocytes‐to‐spermatogonia transition initiates prior to birth in murine testes and it requires FGF signaling. Mech Dev 144, 125–139. [DOI] [PubMed] [Google Scholar]
- 166. Meng X, Lindahl M, Hyvonen ME, Parvinen M, de Rooij DG, Hess MW, Raatikainen‐Ahokas A, Sainio K, Rauvala H, Lakso M et al. (2000) Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 287, 1489–1493. [DOI] [PubMed] [Google Scholar]
- 167. Grasso M, Fuso A, Dovere L, de Rooij DG, Stefanini M, Boitani C and Vicini E (2012) Distribution of GFRA1‐expressing spermatogonia in adult mouse testis. Reproduction 143, 325–332. [DOI] [PubMed] [Google Scholar]
- 168. Singh P, Patel RK, Palmer N, Grenier JK, Paduch D, Kaldis P, Grimson A and Schimenti JC (2019) CDK2 kinase activity is a regulator of male germ cell fate. Development, 10.1242/dev.180273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Huang H, Regan KM, Lou Z, Chen J and Tindall DJ (2006) CDK2‐dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 314, 294–297. [DOI] [PubMed] [Google Scholar]
- 170. Huang H and Tindall DJ (2007) CDK2 and FOXO1: a fork in the road for cell fate decisions. Cell Cycle 6, 902–906. [DOI] [PubMed] [Google Scholar]
- 171. Den Haese GJ, Walworth N, Carr AM and Gould KL (1995) The Wee1 protein kinase regulates T14 phosphorylation of fission yeast Cdc2. Mol Biol Cell 6, 371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Parker LL and Piwnica‐Worms H (1992) Inactivation of the p34cdc2‐cyclin B complex by the human WEE1 tyrosine kinase. Science 257, 1955–1957. [DOI] [PubMed] [Google Scholar]
- 173. Johnson LN and Lewis RJ (2001) Structural basis for control by phosphorylation. Chem Rev 101, 2209–2242. [DOI] [PubMed] [Google Scholar]
- 174. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY and Nakayama K (1996) Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85, 707–720. [DOI] [PubMed] [Google Scholar]
- 175. Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM et al. (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27Kip1‐deficient mice. Cell 85, 733–744. [DOI] [PubMed] [Google Scholar]
- 176. Kiyokawa H, Kineman RD, Manova‐Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA and Koff A (1996) Enhanced growth of mice lacking the cyclin‐dependent kinase inhibitor function of p27Kip1 . Cell 85, 721–732. [DOI] [PubMed] [Google Scholar]
- 177. Beumer TL, Kiyokawa H, Roepers‐Gajadien HL, van den Bos LA, Lock TM, Gademan IS, Rutgers DH, Koff A and de Rooij DG (1999) Regulatory role of p27kip1 in the mouse and human testis. Endocrinology 140, 1834–1840. [DOI] [PubMed] [Google Scholar]
- 178. Holsberger DR, Buchold GM, Leal MC, Kiesewetter SE, O'Brien DA, Hess RA, Franca LR, Kiyokawa H and Cooke PS (2005) Cell‐cycle inhibitors p27Kip1 and p21Cip1 regulate murine Sertoli cell proliferation. Biol Reprod 72, 1429–1436. [DOI] [PubMed] [Google Scholar]
- 179. Fotovati A, Nakayama K and Nakayama KI (2006) Impaired germ cell development due to compromised cell cycle progression in Skp2‐deficient mice. Cell Div 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Fotovati A, Abu‐Ali S, Nakayama K and Nakayama KI (2011) Impaired ovarian development and reduced fertility in female mice deficient in Skp2. J Anat 218, 668–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Kim ST, Park NC, Yi LS and Gye MC (2006) Expression of p57kip2 in germ cells and Leydig cells in human testis. Arch Androl 52, 463–469. [DOI] [PubMed] [Google Scholar]
- 182. Kim ST and Gye MC (2004) Expression of p57 in mouse and human testes. Dev Growth Differ 46, 495–502. [DOI] [PubMed] [Google Scholar]
- 183. Yan Y, Frisen J, Lee MH, Massague J and Barbacid M (1997) Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev 11, 973–983. [DOI] [PubMed] [Google Scholar]
- 184. Zhang P, Wong C, DePinho RA, Harper JW and Elledge SJ (1998) Cooperation between the Cdk inhibitors p27KIP1 and p57KIP2 in the control of tissue growth and development. Genes Dev 12, 3162–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Takahashi K, Nakayama K and Nakayama K (2000) Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J Biochem 127, 73–83. [DOI] [PubMed] [Google Scholar]
- 186. Palm W and de Lange T (2008) How shelterin protects mammalian telomeres. Annu Rev Genet 42, 301–334. [DOI] [PubMed] [Google Scholar]
- 187. Tu Z, Bayazit MB, Liu H, Zhang J, Busayavalasa K, Risal S, Shao J, Satyanarayana A, Coppola V, Tessarollo L et al. (2017) Speedy A‐Cdk2 binding mediates initial telomere‐nuclear envelope attachment during meiotic prophase I independent of Cdk2 activation. Proc Natl Acad Sci USA 114, 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tsai LH, Harlow E and Meyerson M (1991) Isolation of the human cdk2 gene that encodes the cyclin A‐ and adenovirus E1A‐associated p33 kinase. Nature 353, 174–177. [DOI] [PubMed] [Google Scholar]
- 189. Harper L, Golubovskaya I and Cande WZ (2004) A bouquet of chromosomes. J Cell Sci 117, 4025–4032. [DOI] [PubMed] [Google Scholar]
- 190. Siderakis M and Tarsounas M (2007) Telomere regulation and function during meiosis. Chromosome Res 15, 667–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Viera A, Rufas JS, Martinez I, Barbero JL, Ortega S and Suja JA (2009) CDK2 is required for proper homologous pairing, recombination and sex‐body formation during male mouse meiosis. J Cell Sci 122, 2149–2159. [DOI] [PubMed] [Google Scholar]
- 192. Luo Y, Lee IW, Jo YJ, Namgoong S and Kim NH (2016) Depletion of the LINC complex disrupts cytoskeleton dynamics and meiotic resumption in mouse oocytes. Sci Rep 6, 20408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Lee CY, Horn HF, Stewart CL, Burke B, Bolcun‐Filas E, Schimenti JC, Dresser ME and Pezza RJ (2015) Mechanism and regulation of rapid telomere prophase movements in mouse meiotic chromosomes. Cell Rep 11, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Koszul R and Kleckner N (2009) Dynamic chromosome movements during meiosis: a way to eliminate unwanted connections? Trends Cell Biol 19, 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hiraoka Y and Dernburg AF (2009) The SUN rises on meiotic chromosome dynamics. Dev Cell 17, 598–605. [DOI] [PubMed] [Google Scholar]
- 196. Berthet C, Aleem E, Coppola V, Tessarollo L and Kaldis P (2003) Cdk2 knockout mice are viable. Curr Biol 13, 1775–85. [DOI] [PubMed] [Google Scholar]
- 197. Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M and Barbacid M (2003) Cyclin‐dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35, 25–31. [DOI] [PubMed] [Google Scholar]
- 198. Chauhan S, Diril MK, Lee JH, Bisteau X, Manoharan V, Adhikari D, Ratnacaram CK, Janela B, Noffke J, Ginhoux F et al. (2016) Cdk2 catalytic activity is essential for meiotic cell division in vivo. Biochem J 473, 2783–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Zhang J, Tu Z, Watanabe Y and Shibuya H (2017) Distinct TERB1 domains regulate different protein interactions in meiotic telomere movement. Cell Rep 21, 1715–1726. [DOI] [PubMed] [Google Scholar]
- 200. Long J, Huang C, Chen Y, Zhang Y, Shi S, Wu L, Liu Y, Liu C, Wu J and Lei M (2017) Telomeric TERB1‐TRF1 interaction is crucial for male meiosis. Nat Struct Mol Biol 24, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Pendlebury DF, Fujiwara Y, Tesmer VM, Smith EM, Shibuya H, Watanabe Y and Nandakumar J (2017) Dissecting the telomere‐inner nuclear membrane interface formed in meiosis. Nat Struct Mol Biol 24, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Dunce JM, Milburn AE, Gurusaran M, da Cruz I, Sen LT, Benavente R and Davies OR (2018) Structural basis of meiotic telomere attachment to the nuclear envelope by MAJIN‐TERB2‐TERB1. Nat Commun 9, 5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Link J and Jantsch V (2019) Meiotic chromosomes in motion: a perspective from Mus musculus and Caenorhabditis elegans . Chromosoma, [Epub ahead of print], 10.1007/s00412-019-00698-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Alsheimer M, von Glasenapp E, Hock R and Benavente R (1999) Architecture of the nuclear periphery of rat pachytene spermatocytes: distribution of nuclear envelope proteins in relation to synaptonemal complex attachment sites. Mol Biol Cell 10, 1235–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Esponda P and Gimenez‐Martin G (1972) The attachment of the synaptonemal complex to the nuclear envelope. An ultrastructural and cytochemical analysis. Chromosoma 38, 405–417. [DOI] [PubMed] [Google Scholar]
- 206. Woollam DH, Millen JW and Ford EH (1967) Points of attachment of pachytene chromosomes to the nuclear membrane in mouse spermatocytes. Nature 213, 298–299. [DOI] [PubMed] [Google Scholar]
- 207. Liebe B, Alsheimer M, Hoog C, Benavente R and Scherthan H (2004) Telomere attachment, meiotic chromosome condensation, pairing, and bouquet stage duration are modified in spermatocytes lacking axial elements. Mol Biol Cell 15, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Manterola M, Sicinski P and Wolgemuth DJ (2016) E‐type cyclins modulate telomere integrity in mammalian male meiosis. Chromosoma 125, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Martinerie L, Manterola M, Chung SS, Panigrahi SK, Weisbach M, Vasileva A, Geng Y, Sicinski P and Wolgemuth DJ (2014) Mammalian E‐type cyclins control chromosome pairing, telomere stability and CDK2 localization in male meiosis. PLoS Genet 10, e1004165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M and Kim SH (1997) Inhibition of cyclin‐dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. Eur J Biochem 243, 518–526. [DOI] [PubMed] [Google Scholar]
- 211. Reynolds A, Qiao H, Yang Y, Chen JK, Jackson N, Biswas K, Holloway JK, Baudat F, de Massy B, Wang J et al (2013) RNF212 is a dosage‐sensitive regulator of crossing‐over during mammalian meiosis. Nat Genet 45, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Liu W, Wang L, Zhao W, Song G, Xu R, Wang G, Wang F, Li W, Lian J, Tian H et al (2014) Phosphorylation of CDK2 at threonine 160 regulates meiotic pachytene and diplotene progression in mice. Dev Biol 392, 108–116. [DOI] [PubMed] [Google Scholar]
- 213. Qiao H, Prasada Rao HB, Yang Y, Fong JH, Cloutier JM, Deacon DC, Nagel KE, Swartz RK, Strong E, Holloway JK et al. (2014) Antagonistic roles of ubiquitin ligase HEI10 and SUMO ligase RNF212 regulate meiotic recombination. Nat Genet 46, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ward JO, Reinholdt LG, Motley WW, Niswander LM, Deacon DC, Griffin LB, Langlais KK, Backus VL, Schimenti KJ, O'Brien MJ et al. (2007) Mutation in mouse hei10, an e3 ubiquitin ligase, disrupts meiotic crossing over. PLoS Genet 3, e139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Hunter N and Borts RH (1997) Mlh1 is unique among mismatch repair proteins in its ability to promote crossing‐over during meiosis. Genes Dev 11, 1573–1582. [DOI] [PubMed] [Google Scholar]
- 216. Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A et al. (1996) Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet 13, 336–342. [DOI] [PubMed] [Google Scholar]
- 217. Santucci‐Darmanin S, Neyton S, Lespinasse F, Saunieres A, Gaudray P and Paquis‐Flucklinger V (2002) The DNA mismatch‐repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum Mol Genet 11, 1697–706. [DOI] [PubMed] [Google Scholar]
- 218. Lipkin SM, Wang V, Jacoby R, Banerjee‐Basu S, Baxevanis AD, Lynch HT, Elliott RM and Collins FS (2000) MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat Genet 24, 27–35. [DOI] [PubMed] [Google Scholar]
- 219. Woods LM, Hodges CA, Baart E, Baker SM, Liskay M and Hunt PA (1999) Chromosomal influence on meiotic spindle assembly: abnormal meiosis I in female Mlh1 mutant mice. J Cell Biol 145, 1395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Rao HB, Qiao H, Bhatt SK, Bailey LR, Tran HD, Bourne SL, Qiu W, Deshpande A, Sharma AN, Beebout CJ et al. (2017) A SUMO‐ubiquitin relay recruits proteasomes to chromosome axes to regulate meiotic recombination. Science 355, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Holloway JK, Sun X, Yokoo R, Villeneuve AM and Cohen PE (2014) Mammalian CNTD1 is critical for meiotic crossover maturation and deselection of excess precrossover sites. J Cell Biol 205, 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Yokoo R, Zawadzki KA, Nabeshima K, Drake M, Arur S and Villeneuve AM (2012) COSA‐1 reveals robust homeostasis and separable licensing and reinforcement steps governing meiotic crossovers. Cell 149, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Zhang L, Kohler S, Rillo‐Bohn R and Dernburg AF (2018) A compartmentalized signaling network mediates crossover control in meiosis. eLife 7, e30789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Woglar A and Villeneuve AM (2018) Dynamic architecture of DNA repair complexes and the synaptonemal complex at sites of meiotic recombination. Cell 173, 1678–1691 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Toby GG, Gherraby W, Coleman TR and Golemis EA (2003) A novel RING finger protein, human enhancer of invasion 10, alters mitotic progression through regulation of cyclin B levels. Mol Cell Biol 23, 2109–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Gao J and Colaiacovo MP (2018) Zipping and unzipping: protein modifications regulating synaptonemal complex dynamics. Trends Genet 34, 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Jordan PW, Karppinen J and Handel MA (2012) Polo‐like kinase is required for synaptonemal complex disassembly and phosphorylation in mouse spermatocytes. J Cell Sci 125, 5061–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Shin YH, Choi Y, Erdin SU, Yatsenko SA, Kloc M, Yang F, Wang PJ, Meistrich ML and Rajkovic A (2010) Hormad1 mutation disrupts synaptonemal complex formation, recombination, and chromosome segregation in mammalian meiosis. PLoS Genet 6, e1001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Wojtasz L, Daniel K, Roig I, Bolcun‐Filas E, Xu H, Boonsanay V, Eckmann CR, Cooke HJ, Jasin M, Keeney S et al. (2009) Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA‐ATPase. PLoS Genet 5, e1000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Zhang Q, Ji SY, Busayavalasa K and Yu C (2019) SPO16 binds SHOC1 to promote homologous recombination and crossing‐over in meiotic prophase I. Sci Adv 5, eaau9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yang F, Gell K, van der Heijden GW, Eckardt S, Leu NA, Page DC, Benavente R, Her C, Hoog C, McLaughlin KJ et al. (2008) Meiotic failure in male mice lacking an X‐linked factor. Genes Dev 22, 682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Palmer N, Talib SZA, Ratnacaram CK, Low D, Bisteau X, Lee JHS, Pfeiffenberger E, Wollmann H, Tan JHL, Wee S et al. (2019) CDK2 regulates the NRF1/Ehmt1 axis during meiotic prophase I. J Cell Biol 218, 2896–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Wang J, Tang C, Wang Q, Su J, Ni T, Yang W, Wang Y, Chen W, Liu X, Wang S et al. (2017) NRF1 coordinates with DNA methylation to regulate spermatogenesis. FASEB J 31, 4959–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Huo L and Scarpulla RC (2001) Mitochondrial DNA instability and peri‐implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol 21, 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Murphy M, Stinnakre MG, Senamaud‐Beaufort C, Winston NJ, Sweeney C, Kubelka M, Carrington M, Brechot C and Sobczak‐Thepot J (1997) Delayed early embryonic lethality following disruption of the murine cyclin A2 gene. Nat Genet 15, 83–86. [DOI] [PubMed] [Google Scholar]
- 236. Brandeis M, Rosewell I, Carrington M, Crompton T, Jacobs MA, Kirk J, Gannon J and Hunt T (1998) Cyclin B2‐null mice develop normally and are fertile whereas cyclin B1‐null mice die in utero. Proc Natl Acad Sci USA 95, 4344–4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Gopinathan L, Szmyd R, Low D, Diril MK, Chang HY, Coppola V, Liu K, Tessarollo L, Guccione E, van Pelt AMM et al (2017) Emi2 is essential for mouse spermatogenesis. Cell Rep 20, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Clement TM, Inselman AL, Goulding EH, Willis WD and Eddy EM (2015) Disrupting cyclin dependent kinase 1 in spermatocytes causes late meiotic arrest and infertility in mice. Biol Reprod 93, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Liu D, Matzuk MM, Sung WK, Guo Q, Wang P and Wolgemuth DJ (1998) Cyclin A1 is required for meiosis in the male mouse. Nat Genet 20, 377–380. [DOI] [PubMed] [Google Scholar]
- 240. van der Meer T, Chan WY, Palazon LS, Nieduszynski C, Murphy M, Sobczak‐Thepot J, Carrington M and Colledge WH (2004) Cyclin A1 protein shows haplo‐insufficiency for normal fertility in male mice. Reproduction 127, 503–511. [DOI] [PubMed] [Google Scholar]
- 241. Li Y, Wang L, Zhang L, He Z, Feng G, Sun H, Wang J, Li Z, Liu C, Han J et al. (2019) Cyclin B3 is required for metaphase to anaphase transition in oocyte meiosis I. J Cell Biol 218, 1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Karasu ME, Bouftas N, Keeney S and Wassmann K (2019) Cyclin B3 promotes anaphase I onset in oocyte meiosis. J Cell Biol 218, 1265–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Diederichs S, Baumer N, Schultz N, Hamra FK, Schrader MG, Sandstede ML, Berdel WE, Serve H and Muller‐Tidow C (2005) Expression patterns of mitotic and meiotic cell cycle regulators in testicular cancer and development. Int J Cancer 116, 207–217. [DOI] [PubMed] [Google Scholar]
- 244. Terré B, Lewis M, Gil‐Gómez G, Han Z, Lu H, Aguilera M, Prats N, Roy S, Zhao H and Stracker TH (2019) Defects in efferent duct multiciliogenesis underlie male infertility in GEMC1‐, MCIDAS‐ or CCNO‐deficient mice. Development 146, dev162628 10.1242/dev.162628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Nunez‐Olle M, Jung C, Terre B, Balsiger NA, Plata C, Roset R, Pardo‐Pastor C, Garrido M, Rojas S, Alameda F et al. (2017) Constitutive Cyclin O deficiency results in penetrant hydrocephalus, impaired growth and infertility. Oncotarget 8, 99261–99273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Chauhan S, Zheng X, Tan YY, Tay BH, Lim S, Venkatesh B and Kaldis P (2012) Evolution of the Cdk‐activator Speedy/RINGO in vertebrates. Cell Mol Life Sci 69, 3835–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]