

Abstract
Background
Multiple sclerosis (MS) is a complex chronic inflammatory and degenerative disorder of the central nervous system. Accelerated brain volume loss, or also termed atrophy, is currently emerging as a popular imaging marker of neurodegeneration in affected patients, but, unfortunately, can only be reliably interpreted at the time when irreversible tissue damage likely has already occurred. Timing of treatment decisions based on brain atrophy may therefore be viewed as suboptimal.
Main body
This Narrative Review focuses on alternative techniques with the potential of detecting neurodegenerative events in the brain of subjects with MS prior to the atrophic stage. First, metabolic and molecular imaging provide the opportunity to identify early subcellular changes associated with energy dysfunction, which is an assumed core mechanism of axonal degeneration in MS. Second, cerebral hypoperfusion has been observed throughout the entire clinical spectrum of the disorder but it remains an open question whether this serves as an alternative marker of reduced metabolic activity, or exists as an independent contributing process, mediated by endothelin-1 hyperexpression. Third, both metabolic and perfusion alterations may lead to repercussions at the level of network performance and structural connectivity, respectively assessable by functional and diffusion tensor imaging. Fourth and finally, elevated body fluid levels of neurofilaments are gaining interest as a biochemical mirror of axonal damage in a wide range of neurological conditions, with early rises in patients with MS appearing to be predictive of future brain atrophy.
Conclusions
Recent findings from the fields of advanced neuroradiology and neurochemistry provide the promising prospect of demonstrating degenerative brain pathology in patients with MS before atrophy has installed. Although the overall level of evidence on the presented topic is still preliminary, this Review may pave the way for further longitudinal and multimodal studies exploring the relationships between the abovementioned measures, possibly leading to novel insights in early disease mechanisms and therapeutic intervention strategies.
Keywords: Brain atrophy, magnetic resonance imaging, multiple sclerosis, neurodegeneration, neurofilaments
Background
Multiple sclerosis (MS) is a chronic inflammatory demyelinating and degenerative disorder of the central nervous system (CNS) affecting over 2.5 million people worldwide. The first symptoms usually occur during young adulthood, after which patients may experience a heterogeneous relapsing or progressive disease course with a modulating role for age, sex and comorbidities [1–5]. Clinical outcomes are difficult to predict but cumulative axonal loss is generally seen as the primary determinant of long-term prognosis. Brain volume (BV) reduction, or atrophy, has recently emerged as a popular magnetic resonance imaging (MRI) marker of neurodegeneration in MS, pathologically corresponding with demyelination, decreased axonal count and neuronal cell death [6, 7]. In healthy individuals, the rate of BV decline, ranges between 0.2 and 0.5% per year, depending on the age of the subjects [8], whereas this process appears to advance approximately 3 to 5 times more rapid in individuals with MS. [9] Brain atrophy is present and clinically relevant in all subtypes, from subjects with a clinically isolated syndrome (CIS) to those with longstanding progressive disease [10, 11], being more pronounced in the later stages but seemingly proceeding at a similar pace throughout the entire clinical spectrum [12]. At the group level, development of brain atrophy, particularly in the grey matter, has become recognized as an independent predictor of current and future physical disability, mood disturbances and cognitive impairment; notably outperforming classical MRI markers of white matter inflammation [10, 13].
The therapeutic landscape of MS is changing dramatically since the past 10 to 15 years [14], (Table 1) and the arrival of more potent drugs raised the standards of optimal disease control along this process. It has been suggested that achieving a ‘no evidence of disease activity’ (NEDA) status; defined as the absence of clinical relapses, new or enlarging MRI lesions and sustained progression on the Expanded Disability Status Scale (EDSS) [15]; (Figure 1) 2 years after starting disease-modifying treatment is predictive of a favourable outcome over up to 7 years [16]. This concept has subsequently found its way into clinical practice and research. Most first-line drugs have initially showed only a modest impact on brain atrophy but after the first year or treatment, when the possible confounding effects of pseudo-atrophy - i.e. the volume decrease resulting from oedema reduction in response to the anti-inflammatory therapy - can be eliminated, various drugs do seem to slow down the rate of BV loss [17–21]. Moreover, this effect looks to be more pronounced for second-line agents and correlates for a substantial degree with the treatment effect on disability [22]. Brain atrophy measures now start to become increasingly incorporated as supplementary endpoints in MS treatment trials, expanding the goal from NEDA-3 to NEDA-4 (the latter can be defined as the combined absence of clinical relapses, new/enlarging MRI lesions, sustained EDSS progression and accelerated rate of BV loss – a threshold of 0.4% decline per year has been suggested for the brain atrophy parameter) [23].
Table 1.
Development timeline for disease-modifying therapy in MS
| FDA or EMA approval^ | Product | Main immunological mechanism of action |
|---|---|---|
| 1993/1996 | INF β-1b/-1a | Induces changes in cytokine balance |
| 1996 | Glatiramere acetate | Interferes with antigen presentation to T-lymphocytes |
| 2000 | Mitoxantrone | Reduces the number of circulating leukocytes |
| 2004 | Natalizumab | Blocks leukocyte migration across the BBB |
| 2010 | Fingolimod | Prevents lymphocyte egression from the lymph nodes |
| 2012 | Teriflunomide | Inhibits lymphocyte proliferation |
| 2013 | Dimethyl fumarate | Induces changes in cytokine balance |
| 2014 |
Alemtuzumab Pegylated INF β-1a |
Destructs circulating lymphocytes, followed by repopulation Induces changes in cytokine balance |
| 2016° | Daclizumab | Induces immune tolerance |
| 2017 |
Cladribine* Ocrelizumab |
Reduces the number of circulating lymphocytes Depletes B-lymphocyte population |
| 2019 | Siponimod | Prevents lymphocyte egression from the lymph nodes |
| Pipeline |
Ozanimod and ponesimod Ofatumumab Evobrutinib AHSCT |
Prevents lymphocyte egression from the lymph nodes Depletes circulating B-lymphocytes Inhibits B-lymphocyte signaling and maturation Immune system reconstitution |
FDA Food and Drug Administration, EMA European Medical Agency, INF interferon, BBB blood-brain barrier, AHSCT autologous stem cell transplantation, MS multiple sclerosis.
^ based on which approval came first
* No FDA approval at present
° Withdrawn in 2018
Fig. 1.

Cascade of events potentially linking inflammatory activity to slowly progressive neurodegeneration in MS. Chronic inflammatory demyelination leads to redistribution of sodium channels along the denudated axolemma, resulting in sodium influx. Elevated intracellular sodium levels increase the work-load of the energy-dependent Na/K pump. Mitochondrial function is impaired in multiple sclerosis (mainly resulting from the oxidative stress associated with inflammatory activity) which causes insufficient energy supply to compensate this imbalance. Intracellular sodium levels rise, leading to accumulation of calcium, e.g. by reversal of the transmembrane Na/Ca exchanger and release from intracellular sources. Acidosis contributes to sodium and calcium influx through opening of acid-sensing ion channels. Calcium stacking induces protease and lipase activity, eventually ending with cellular breakdown. ATP: adenosine triphosphate, ASIC: acid-sensing ion channel, MS: multiple sclerosis
On the other hand, it must be stated that the implementation of brain atrophy into clinical MS practice develops more difficult than originally expected, mainly due to several unresolved biological and technical issues [10], and its use as a marker of disease progression is still not recommended according to academic imaging guidelines [24, 25]. An additional and even more important problem might be the reasonable likelihood that a substantial degree of irreversible tissue damage is already present at the time when brain atrophy becomes apparent. Expert opinion suggests that BV changes should be interpreted over a period of at least 2 years in the individual patient trajectory [8], and none of the currently available MS drugs have so far been able to reverse this phenomenon. Consequently, even under optimal methodological circumstances, real-life treatment decisions based on the detection of accelerated BV loss risk to fall well beyond the window of opportunity. This Review focuses on the question which alternative techniques might be able to demonstrate early signs of neurodegenerative brain pathology in subjects with MS, in the absence of atrophic changes and hopefully before structural tissue damage has become definite.
Main text
Molecular and metabolic imaging
Linking inflammation to neurodegeneration
We believe that the most logical approach to our postulated question would be an attempt to outline the biological processes taking place inside degenerating neurons of individuals with MS, at the stage where the cellular homeostasis becomes threatened, yet still preceding the imminence of cell death. The precise causative mechanism remains unknown but in the traditional teaching MS is initiated by auto-reactive lymphocytes entering the CNS from the peripheral immune system to provoke hallmark demyelinating lesions, also termed plaques [26, 27]. Axonal transection can occur during the formation of these plaques and subsequently trigger anterograde (Wallerian) and retrograde degeneration [28, 29]. In general, however, most axons will survive such acute inflammatory activity [30], and various radiological and neuropathological studies have demonstrated only a modest correlation between total lesion burden and axonal loss [31–35]. Acute tissue damage due to inflammation therefore does not seem to be a major contributor to brain atrophy in MS. Nevertheless, longitudinal MRI observations have revealed a more slowly proceeding axonal decay within chronic inactive plaques [36], and post-mortem studies in highly disabled subjects with MS have reported a reduction in the corticospinal axonal count of up to 70% [37]. Sustained demyelination is believed to be the responsible element, by triggering a cascade of events that will eventually, when initial compensatory mechanisms have become exhausted, evolve into axonal degeneration. Prominent factors associated with this pathway include oxidative stress, mitochondrial injury, energy failure and ionic dysregulation [30].
Chronic demyelination leads to loss of trophic support and redistribution of sodium channels along the denuded axolemma which, under normal physiologic circumstances, would have remained concentrated at the nodes of Ranvier. Extracellular sodium is expected to migrate inwards through these channels, based on the transmembrane concentration difference, implying that the energy-consuming Na/K pump will have to work at an increased rate to maintain correct ionic gradients and polarization across the cell membrane [38–40]. Axons highly depend on mitochondrial activity for local energy production, whereas it is precisely mitochondrial integrity that can become compromised in subjects with MS. [41, 42] Inflammatory responses are associated with the production of ROS and NO by activated cells of the innate immune system (i.e. microglia and macrophages), and mitochondria appear to be extremely vulnerable to this kind of oxidative stress. For instance, significant reductions of respiratory chain cytochrome c-oxidase I and complex IV enzymes in active MS lesions have been attributed to the effects of ROS and NO. [30, 43, 44] As a consequence, demyelinated axons may experience a mismatch between higher energy needs and decreased mitochondrial metabolic functioning. The increased mitochondrial density and activity, as seen in chronically inactive plaques [45, 46], likely act as initial compensatory adaptations, but one can assume that an overt energy deficiency arises once the oxidative damage surpasses a certain threshold. The Na/K pump may start to slow down when energy supply is no longer sufficient, resulting in intracellular sodium accumulation which, in turn, can lead to a rise of intracellular calcium, due to reversal of the transmembrane Na/Ca exchanger and release from intracellular calcium sources such as the endoplasmic reticulum. Exorbitant intracellular calcium release is recognized as a common final pathway of cellular death, as calcium can activate various lipases and proteases (e.g. ubiquitin and calpain), which cause breakdown of structural components and the process hereby to reach a point-of-no-return [30, 43, 44]. In addition, a number of self-reinforcing mechanisms are hidden within this cascade. Energy dysfunction will, for instance, lead to intracellular acidosis, promoting both supplementary sodium and calcium entrance by activation of acid-sensing ion channel (ASIC) 1a [43, 47]. Intracellular calcium stacking can support further sodium influx by opening other types of ion channels in the cell membrane [43], and the whole process of mitochondrial and diffuse structural damage will generate even more oxidative stress, ultimately ending in a detrimental vicious circle (overview presented in Figure 1).
Molecular and metabolic imaging provide the exciting ability to reflect some of these proposed subcellular alterations throughout the otherwise normal-appearing white and grey matter of patients with MS. Molecular imaging compromises advanced MRI applications in which advanced contrast agents are used to label the biological target, and positron emission tomography (PET) which relies on radioligands selectively binding to the proteins of interest. Metabolic imaging techniques, in turn, exploit the magnetic properties of atomic nuclei, such as protons or sodium ions. The idea that early neurodegenerative MS pathology can be captured in the absence of atrophy is thus far best supported by magnetic resonance spectroscopy (MRS), sodium imaging, chemical exchange saturation transfer (CEST) and PET findings.
Mitochondrial failure
N-acetyl-aspartate (NAA) is almost exclusively produced by neuronal mitochondria in mature CNS and figures as a marker of mitochondrial function and/or integrity in both white and grey matter [48, 49]. The precise biological functions remain unknown, but during early postnatal development NAA is transported in large quantities to oligodendrocytes and subsequently metabolized into building blocks for myelin formation [49]. Levels in the brain and spinal cord can be measured with 1H-MRS and are usually expressed as NAA/creatine ratios for reasons of stability [43, 50, 51]. Because mitochondrial failure appears to be a crucial feature of neurodegeneration in patients with MS, regional estimates may provide valuable insights on local neuronal health. In acute MS lesions, NAA is decreased when compared to surrounding normal-appearing white matter and the white matter of controls subjects [52, 53]. In part this may still be explained by inflammatory oedema, but an analogue reduction has also been observed in chronic white matter plaques appearing hypointense on T1-weighted MRI [54]. MRS studies of both relapsing and progressive patients with MS have fairly consistently demonstrated reduced concentrations in the normal-appearing white and grey matter, as compared to controls, with a tendency of being more pronounced in progressive stages and related to clinical disability [54–57].
Definite proof that a chronic NAA reduction unequivocally precedes brain atrophy in a longitudinal neurodegenerative scenario affecting patients with MS, and which is detectable within this context by MRS, is still lacking, but at least a few interesting findings can be extracted from the current literature.
First, MRS follow-up studies of acute lesions in the brain and spinal cord have demonstrated that reduced NAA, albeit not necessarily a return to normal, partially recovered over time [52, 58, 59]. Interestingly, progressive atrophy occurred at the same time as the NAA restoration (the latter likely reflecting an adaptive change of the surviving mitochondria), thus creating a time line in which the NAA decrease did precede atrophy, and where a partial causal relation between both may not be excluded. Kirov and co-workers found that NAA concentrations in cerebral normal-appearing white matter of recently diagnosed and mildly disabled individuals with MS, which were all on disease-modifying treatment, were 6% lower than in control subjects, but significantly increased over the next 3 years [60]. These studies support the concept that reduced NAA levels may be reversible and subject to therapeutic intervention (at least when they reflect metabolic alterations), whereas this has not yet been the case with atrophy. Second, reduced levels of NAA have also been reported in the brain of patients with a clinically and even radiologically isolated syndrome (RIS) [61–63], suggesting that mitochondrial dysfunction may already occur at the earliest stages of the disease, where less atrophy would be expected. In contrast, one study found an isolated decrease in normalized global brain and cortical volumes of subjects with a RIS. However, a trend in NAA decrease was still observed in those which were at high risk for later MS conversion [64]. Third, lower NAA concentrations were measured in the spinal cord of 21 early primary progressive MS patients without extensive cord atrophy, and were associated with higher EDSS scores [65]. Fourth, MRS is also able to quantify cerebral glutamate, an excitatory neurotransmitter, which has been found to be increased in the normal-appearing white matter of all subtypes of MS, when compared with healthy controls [66, 67], potentially due to increased production by inflammatory cells in combination with reduced clearance by oligodendrocytes and astrocytes [48]. Later on, however, there are indications of a continuous glutamate decline [67], in contrast to NAA levels which remain relatively stable as a likely result of numeric mitochondrial adaptation. Glutamate itself may act as an inductor of neurodegeneration, by influencing transmembrane sodium and calcium channels in a mechanism termed excitotoxicity [68]. Higher concentrations in the normal-appearing white matter of a large cohort predominantly consisting of patients with a relapsing-remitting subtype, were associated with a more pronounced NAA decline (after 2 years) and BV reduction (after 3 years) over time [66]. When glutamate levels are normalized for NAA, they will probably provide more accurate information on its assumed neurodegenerative stimulus, because this ratio corrects for the amount of already existing neuronal loss. Longitudinal studies exploring the temporal relationship between glutamate/NAA, NAA/creatine and BV within this context would be very much of interest.
Sodium accumulation
Intraneuronal sodium accumulation is an early transduction of energy dysfunction in our proposed cascade of neurodegenerative events in MS. Under normal physiological circumstances, CNS sodium concentrations are actively kept low in the intracellular compartment (tissue fraction 80%), as compared to those in the smaller extracellular counterpart (20%) [48], in order to preserve a correct transmembrane electric gradient. In vivo, sodium can be quantified using 23Na MRI, based on which several groups have found increased concentrations within plaques, normal-appearing white matter and cortical and deep grey matter of subjects with MS, as compared to controls [69–71]. Higher values appear to be linked with progressive disease and greater disability [70]. In a combined 23Na MRI and MRS analysis, sodium accumulation was directly associated with neuronal metabolic dysfunction, as reflected by decreased NAA levels, in focal lesions, grey matter and normal-appearing white matter of 21 individuals with relapsing-remitting MS. [72]
The abovementioned studies are somewhat limited by the fact that they have assessed total sodium concentrations without distinguishing between the intra- and extracellular fraction, and theoretically, it cannot be excluded that an augmentation of total levels is predominantly driven by expansion of the extracellular space, in parallel with neuronal atrophy. However, abnormally high sodium concentrations were also found in grey matter zones which were spared by atrophy in progressive MS [73], as well as in isolated brain regions of patients with an early relapsing-remitting course [71], suggesting that sodium accumulation can be a marker of neuronal metabolic dysfunction rather than merely resulting from BV loss. More compelling evidence for this theory was provided by Petracca and co-workers who performed an ultra-high field (7 T) triple-quantum filtered 23Na MRI study enabling the quantification of the intracellular sodium concentration and extracellular sodium fraction, an indirect measure of the extracellular sodium concentration. They reported an increase of both total and intracellular sodium levels in the grey and normal-appearing white matter of 19 patients with relapsing-remitting MS, as compared to healthy controls, whereas extracellular sodium fractions in fact were reduced, arguing against the simultaneous presence of atrophy [74]. Interestingly, in a recent French publication cognitive impairment in relatively early relapsing-remitting MS was better explained by total sodium accumulation then volume loss in the grey matter [75], while a British group reported that increased total cortical levels were associated with multiple physical and cognitive outcome measures (e.g. EDSS and Symbol Digit Modalities Test scores) in patients with a relapsing onset, independent of grey matter atrophy [76].
Intracellular acidosis
CEST allows to image compounds at concentrations that are too low to be detected using standard MRI. The technique is based on the exchange of protons between specific tissues, that are first saturated, and water [77]. One of the most common applications is the assessment of amide proton transfer (APT) effects in proteins and peptides, which has been successful at demonstrating elevated protein concentrations in brain tumours, as well as at the detection of acute ischemic stroke, the latter due to the sensitivity of the APT effect to pH changes [78]. Within the field of MS, a 7 T study - including only 4 subjects - delivered preliminary evidence that the APT signal varied with lesion type and differed between plaques and healthy white matter [79]. A more recent study showed APT changes in the normal-appearing white matter of the cervical spinal cord of individuals with MS, as compared to controls, at 3 T [80]. Both acute inflammatory damage and chronic energy failure may result in intracellular acidosis, potentially accounting for the encountered alterations in these patients. Extreme or prolonged acidosis is toxic to CNS neurons in a process believed to be mediated by ASIC variants, and the positive effect of amiloride, which is an ASIC antagonist, on brain atrophy in subjects with primary progressive MS hints towards neuroprotective properties of blocking this pathway [81]. Although APT-CEST is a promising technique, its low specificity and dependence on acquisition parameters, manufacturers and coils, does currently not support a widespread clinical application. The lack of studies incorporating APT-CEST obstructs us to elaborate yet on whether this method is able to detect degenerative changes related to MS earlier than conventional imaging.
Positron emission tomography findings
MS is multifactorial disorder in which many different radioligands can be used for experimental PET imaging purposes, depending on whether the biological process of interest is inflammation, myelin breakdown, neuronal degeneration or astrocyte activation [48]. One of the best-known applications in human brain is fluorodeoxyglucose (FDG)-PET, which measures the rate of cerebral glucose consumption. Higher uptake, as compared to healthy controls, has been observed in patients with MS and was attributed to increased regional glucose utilization by activated inflammatory cells [82, 83], but it seems equally plausible for the global signal to become suppressed due to loss of neuronal metabolic integrity. In support of this hypothesis, FDG uptake was decreased in the spinal cord of 8 patients with MS, negatively correlating with autonomic and motor symptoms [84]. Based on FDG-PET imaging alone, however, the ability to discriminate structurally still uncomplicated metabolic dysfunction from overt atrophy in the brain seems rather unlikely.
11C-flumazenil is a different PET radioligand with selectivity for the benzodiazepine site of the class A gamma-aminobutyric acid (GABA) receptor complex in synapses of grey matter neurons. Freeman and co-workers found a reduced cortical 11C-flumazenil binding in a mixture of patients with relapsing-remitting and secondary progressive MS, as compared to healthy controls. Interestingly, this effect was already noticeable in the subset of relapsing-remitting subjects, who did not have a significant degree of grey matter atrophy, and was globally associated with reduced information processing speed [85]. Cao and co-workers described in a classical MRS study that GABA levels themselves were decreased in the posterior cingulate cortex and left hippocampus of patients with relapsing-remitting disease, which was correlated impaired verbal memory and executive function [86]. If and how dysfunctional GABA-minergic neurotransmission relates to neuronal metabolic dysfunction in MS remains unknown, but these findings do suggest that it can be detectable at a clinically relevant stage, in the absence of cortical volume loss.
Microglia are local macrophage residents that, together with infiltrating blood-derived monocytic cells, play a key role in regulating CNS inflammation. In MS, microglial activation may contribute to both tissue damage, mainly through the production of oxidative agents [87–89], and repair mechanisms, e.g. by scavenging for cell debris. Activation of microglia has been associated with increased PET signals in subjects with MS, as compared to heathy controls. The most investigated target is the translocator protein receptor (TSPO), which is normally expressed in small amounts by the outer mitochondrial membrane. TSPO studies using 11C-PK11195 have demonstrated increased uptake in active lesions, as well as in normal-appearing white and cortical grey matter, correlating with disability and brain atrophy measures [90–93]. Second generation TSPO tracers, such as 11C-PBR28, have been developed to improve affinity and specificity of the receptor binding and, interestingly, microglial activation was associated with brain volume loss at the cross-sectional and longitudinal level (correlation coefficients were -0.54 and 0.86, respectively) in two recent studies using this compound [94, 95].
Cerebral blood flow
Cerebral hypoperfusion in multiple sclerosis
Cerebral blood flow (CBF) is the most commonly used numerical expression of cerebral perfusion and represents the volume of blood flowing through a given amount of brain tissue per time unit [96]. As blood supply in the CNS is actively regulated by astrocytes to match regional metabolic activity [97], energy failure in MS would be expected to be accompanied by a reduction in CBF, which, theoretically, may serve as an alternative marker of the early neurodegenerative process. The first reports of cerebral hypoperfusion in patients with MS were published more than 3 decades ago, based on single-photon emission computed tomography and PET findings [98–101]. These studies, though, received little attention at the time due to technical limitations, such as low spatial resolution, and lack of conceptual understanding. The development of more accurate perfusion-weighted MRI methods has revived interest in this field during the past 15 years. Studies using dynamic susceptibility contrast-enhanced MRI have demonstrated a globally decreased CBF, as compared to healthy controls, in the normal-appearing white matter of patients with relapsing-remitting and primary progressive MS, as well as in those with a CIS [102–104]. Similar alterations have been found in the deep grey matter of patients with a CIS and relapsing-remitting MS. [105] Subsequently, global brain hypoperfusion, including in the cerebral cortex and cerebellum, was observed in mixed cohorts of MS subjects, by means of arterial spin labelling (ASL), a non-invasive technique which has the advantage of not requiring contrast administration [106, 107]; (see Figure 2 for an example from the authors’ own records, unpublished data). Overall, these findings suggest that reduced CBF is an early and integral feature of the disease, independent of its clinical course.
Fig. 2.

Brain perfusion is globally decreased in MS. Perfusion-weighted brain MRI (arterial spin labelling) from a 35-year old healthy male volunteer (left) versus a 34-year old man with relapsing-remitting MS (right), with CBF color-coded map overlay. Brain perfusion is globally reduced in the MS patients, as compared to the healthy volunteer. CBF: cerebral blood flow (expressed as mL/100 g/min), MS: multiple sclerosis, MRI: magnetic resonance imaging
The exact underlying mechanism and pathophysiological significance of reduced brain perfusion in MS remain poorly understood at present. Somewhat surprisingly, Saindane and co-workers concluded that perfusion and diffusion tensor MRI properties of the normal-appearing corpus callosum were compatible with primary ischemia in relapsing-remitting patients, rather than with hypoperfusion secondary to impaired axonal integrity [108]. This theory was later supported by a combined ASL perfusion and MRS study, in which both CBF and NAA levels were lower in the centrum semiovale of individuals with MS, as compared to control subjects. NAA/CBF ratios in the MS group were significantly higher than in the controls, suggesting that the reduced perfusion is greater than what would be expected from decreased axonal metabolism (or loss) alone [51]. These findings might be explained by overexpression of the vasospastic agent endothelin-1 by reactive astrocytes in focal lesions, possibly induced through inflammatory cytokines, and subsequent release in the cerebral circulation [107, 109]. In support of this theory, microcirculatory disturbances, characterized by impaired arteriolar vasoreactivity measured with hypercapnic perfusion MRI, have recently been associated with MS. [110, 111]
Active involvement in disease pathology?
The idea that brain hypoperfusion precedes or even induces neurodegenerative changes in MS, is currently still speculative yet intriguing. Some pathological data do support a theoretical framework for this type of ‘reverse causality’ between both. Enhanced expression of hypoxia-inducible factor (HIF)-1α and its downstream genes, a universal salvage pathway that becomes activated in response to tissue hypoxia, has been observed in post-mortem normal-appearing white matter of patients with MS. [112] Cerebral white matter myelin and axons tissue appear to be particularly susceptible to chronic hypoxia [113], and animal models have demonstrated that chronically impaired brain perfusion induces mitochondrial dysfunction and production of free radicals - both of which are assumed to be involved in the progressive neurodegenerative process of MS - eventually resulting in axonal injury [114].
Prospective longitudinal imaging data are missing but several cross-sectional analyses have already suggested the presence of a functionally relevant hypoperfusion in non-atrophic brain regions of subjects with MS. Hence, Debernard and co-workers have found a global perfusion reduction in the cerebral cortex and deep grey matter of patients with early relapsing-remitting MS, compared to controls, in the absence of corresponding volume loss. This CBF reduction was positively correlated with objective memory disturbances [115]. A similar atrophy-independent perfusion decrease has been described in cortical and frontal grey matter of cognitively impaired versus non-impaired patients [116, 117]. Doche and co-workers evaluated grey matter CBF of subjects with relapsing-remitting MS and matched controls, in combination with voxel-based-morphometry analysis in order to exclude atrophic regions. Significant hypoperfusion was measured in both thalami of the MS subjects, and positively correlated with Multiple Sclerosis Functional Composite evaluations (i.e. combined assessment of mobility, dexterity and cognition), nearly reaching an additional significant negative correlation with EDSS scores [118]. Poor overlap between grey matter atrophy and CBF was, finally, also observed in a recent combined conventional 3D-T1 and ASL study [119].
Connectivity assessment
Network structures
Some neurological functions are organized in a way that focal damage can cause very straightforward clinical impairment. For example, a single lesion in the middle temporal visual area, which is involved in the perception of motion, may lead to motion blindness [120]. Other functions, mainly concerning behaviour and cognition, appear to be distributed more diffusely within the brain, and rely on dynamic communication between regions operating in higher network structures. The pathological relevance of such network disruption is becoming increasingly recognized in various disorders, including MS, depression, Alzheimer’s disease and schizophrenia [121, 122]. Studies in these fields usually refer to 2 types of connectivity issues. The term structural connectivity embraces the direct anatomical linkage between different brain areas, while functional connectivity (FC) is based on the idea that remote regions expressing a similar temporal pattern of neuronal activation share information and are functionally connected [123]. One can assume that white matter tract integrity is an important prerequisite for correct interregional cross talk, whereas precisely myelin loss and axonal degeneration are hallmark features of MS pathology. In the hypothesis that energy failure, due to chronic demyelination and/or perfusion impairment, leads to early axonal dysfunction in subjects with MS, this might as well be reflected by changes at the network level.
Functional imaging
Functional MRI (fMRI) detects brain activity by measuring changes in CBF as a surrogate parameter, supported by the principle that regional perfusion and neuronal activation are coupled [97]. Blood oxygenation level dependent (BOLD) imaging is the standard technique for activity mapping in fMRI studies. Activation of brain tissue will lead to increased oxygen consumption in the neighbouring capillaries and venules, resulting in a decreased oxygenated/deoxygenated hemoglobin ratio. A compensatory augmentation in local CBF will take place over the next few seconds, delivering an immediate surplus of oxygenated hemoglobin. It is this pronounced rebound effect which will be visualized as the BOLD signal, because of the paramagnetic differences between both isoforms [124].
The most primary application of fMRI explores BOLD responses during specific tasks or stimuli. Increased zones of activation while performing motor or cognitive exercises have been consistently demonstrated in patients with MS, as compared to healthy controls [125, 126]. These observations possibly reflect the capacity of the brain to recruit supplementary cortical areas to maintain a certain level of performance. Already in the CIS stage, patients showed increased activations and connectivity during the Paced Auditory Serial Addition Test [127]. In recent years, the analysis of the ‘brain-at-rest’ has emerged as an alternative to task-based studies, driven by advantages such as the ease and replicability of the paradigm, the fact that encountered differences are less likely to result from variations in task-performance, and the observation that large neuronal networks are activated during rest. Resting-state FC has been found to be substantially different in patients with MS, as compared to controls. In general, subjects in the CIS or early relapsing-remitting stage (disease duration less than 2 years) display an increased FC of the default network [128–130], i.e. the network that typically becomes activated during wakeful rest (i.e. conscious state but not engaged in a distinct mental activity) [131], with subsequent decrease to normal levels and reduced FC in more advanced patients [132–134], all compared to normal controls. Remarkably, impaired cognition hereby seemed to be associated with early elevations as well as late decrements of FC. These findings suggest an ‘inverted U-shaped’-like evolution of resting-state FC along the disease trajectory, ending in overt clinical deterioration when initial compensatory mechanisms have become overturned (Figure 3). Whether decreased axonal functioning prior to irreversible damage is sufficient to induce detectable network changes in MS is unknown, but this hypothesis may be supported by findings of disturbed network efficiency in CIS patients with reduced FC in occipital, temporal and frontal cortices [135].
Fig. 3.
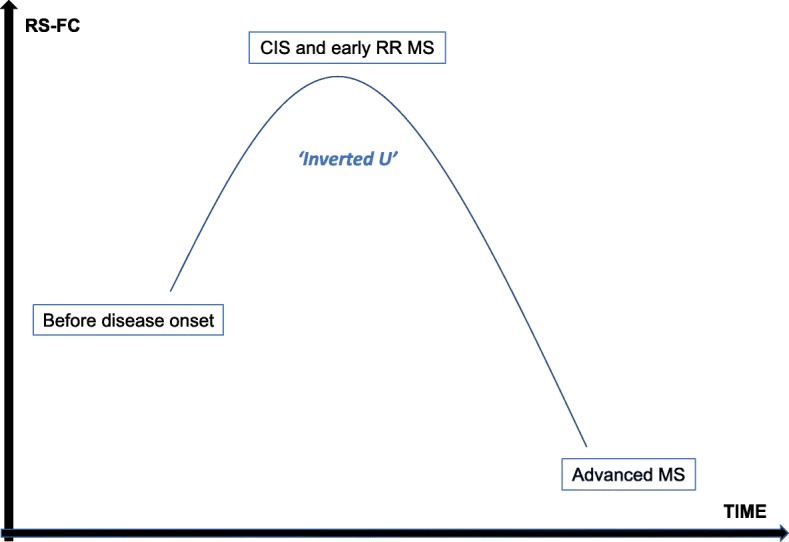
Longitudinal evolution of the default network’s RS-FC in MS. RS-CF is elevated early in the disease, as compared to healthy age-related controls, but subsequently drops down again, first to normal levels and then to reduced levels in more advanced patients. Impaired cognition seems to be associated with early increases and late decreases, most likely reflecting initial functional compensation followed by exhaustion of these mechanisms. RS-FC: resting-state functional connectivity, CIS: clinically isolated syndrome. RR: relapsing-remitting, MS: multiple sclerosis
Diffusion tensor imaging
Diffusion tensor imaging (DTI) is a specific form of diffusion-weighted MRI that allows tracking of water molecules through biological tissues [136]. In a medium free from barriers, these molecules are expected to disperse equally in all directions (i.e. isotropic diffusion). Axons, however, are organized in parallel bundles and have surrounding myelin, resulting in an internal diffusion pattern which is not random but heavily favoured to follow the main directional axis of the fibres. Profiting from its relatively limited scan time duration, DTI is the most commonly used method to study structural connectivity in the human brain. Computer-based reconstructions can generate visual impressions called tractography, (Figure 4) and from a mathematical perspective, the diffusion tensor is generally summarized in 2 parameters: mean diffusivity (MD) representing the rate of random molecular diffusion (lower values correspond to low diffusivity), and fractional anisotropy (FA) as a normalized measure of the isotropic character of the diffusion (0 = isotropic; 1 = anisotropic). Although the interpretation of these scalar values depends on the brain region, cellular basis and the specific disease process [137], normal-appearing white matter of patients with MS typically shows an increase in MD and a decrease in FA [129, 138–141], pointing towards a reduced fibral integrity. Pre-atrophic disruption of axonal architecture, whether this involves myelin breakdown, axolemma leakage or both, could be reflected by similar DTI alterations.
Fig. 4.

Examples of white matter tract orientation on processed DTI. Color denotes the direction of the first eigenvector at each voxel, weighted with the FA of that voxel. Red: left-right; blue: superior-inferior; green: anterior-posterior. Left: sagittal plane; middle: coronal plane; right: axial plane. DTI: diffusion tensor imaging, FA: fractional anisotropy
In a 2 year follow-up study of 37 CIS patients, Rocca and colleagues observed a distributed pattern of increased white matter diffusivity at baseline (2-60 days after diagnosis), which remained stable for one year but resulted in a further increase at 24 months. In the same study, an early increase in grey matter volume of the bilateral middle and inferior temporal gyri was noted at baseline, followed by a further increase in several grey matter structures at 3 months and a volume decrease of the deep nuclei, cerebellar cortex and several cortical areas from month 12 onwards [142]. Whereas the early increase in grey matter volume may be explained by either pseudo-atrophy or structural plasticity (or a combination of both), the early increase in MD may either point to early microstructural changes or an effect similar to pseudo-atrophy. Indeed, inflammation induced swelling of the brain may allow water molecules to diffuse more freely, but the further increase in MD at 24 months could then reflect true axonal pathology. Yet, it is important to note that conflicting results have been reported in the literature: while the Rocca group observed an increase in MD without changes in FA, researchers at the Buffalo Neuroimaging Analysis Centre have reported reduced FA in different cortical and subcortical brain structures of 45 CIS patients without differences in MD [143]. Interestingly, DTI alterations have been found in the volumetrically normal hippocampus of CIS patients, as compared to healthy controls, and these were negatively correlated with episodic verbal memory performance, thus arguing that clinically relevant microstructural brain damage can be detected prior to atrophy [144].
The growing presence of DTI in MS research can additionally be illustrated by studies demonstrating pharmaceutical effects on diffusion parameters. In a recent longitudinal observation, a relatively stable pattern in diffusion metrics was encountered in patients with relapsing-remitting MS which were administered dimethyl fumarate (DMF). In the same study, healthy controls showed a significantly greater rate of diffusion parameters increase in the thalamus and normal-appearing white matter, compared to the subjects with MS [145], cautiously (due to small sample sizes and lack of a placebo-controlled arm) suggesting a supplementary neuroprotective effect of DMF. Other studies reported a significant FA increase and radial diffusivity decrease in the corticospinal tract after initiation of fingolimod [146], or a stable pattern of diffusion parameters in the normal-appearing white matter for up to 48 months under natalizumab treatment [147]. In contrast, recent findings at the New York Mount Sinai School of Medicine demonstrated that white matter microstructural deterioration still occurred in relapsing-remitting MS patients, classified as having NEDA over an approximate 2 year evaluation period [148].
Body fluid markers
Although numerous contenders have been piloted over the past decades, only two body fluid markers are currently being used in routine MS practice: i.e. the presence of oligoclonal bands (OCBs) and an elevated IgG index, both in the cerebrospinal fluid (CSF). Both are helpful in establishing the diagnosis, and the former has showed potential as an independent predictor of conversion to MS in CIS and RIS subjects [149], but overall these measures do not show enough responsiveness to disease activity to be useful in clinical follow-up [150]. One of the most promising alternatives is neurofilament light chain (Nf-L). Neurofilaments are an abundant and cell-exclusive part of the cytoskeleton in neurons (where they play an important role in maintaining axonal calibre and functionality) which makes them excellent biomarker candidates of axonal damage [151]. Nf-L can be measured in CSF, and, following the development of highly sensitive single-molecule array assays [152], since recently also in serum, where concentrations are much lower than in the CSF [151]. Values in both compartments appear to be highly correlated to each other [153–155], but serum has the advantage of being much more easily accessible.
Neuro-axonal damage is the pathological substrate of disability in a large scale of neurological diseases. Elevated neurofilament concentrations have been found in the serum and CSF of patients with MS, but also in individuals with traumatic, vascular and other degenerative conditions of the CNS [151]. In MS, Nf-L levels in the CSF have been associated with clinical endpoints (e.g. degree of disability, disease activity and time since last relapse) for the first time in 1998 [156], findings that subsequently have been replicated and extended by several other and larger studies [151], even providing prognostic information over up to 15 years [157]. Baseline levels are significantly higher in patients with a CIS that will convert to MS [158], and predict disease activity over 2 years [159], but the predictive value for conversion is still considered weak at this time [158, 160]. At a group level, CSF concentrations appear to be responsive to treatment with natalizumab, fingolimod, mitoxantrone and rituximab in both relapsing and progressive MS. [151]
Returning to the crux question of this paper, we are not aware of studies demonstrating elevated body fluid neurofilament levels in patients with MS, as a result of neuronal homeostasis disruption and in the objectivated absence of brain atrophy. Nf-L values were found to be increased in the serum of subjects with a CIS [161], where less pronounced brain atrophy might be expected, but not in the CSF of those with a radiologically isolated equivalent [162]. Levels in both body fluids did correlate significantly with upcoming brain parenchymal fraction changes (coefficients for CSF were -0.564 over 3 years and -0.892 over 5 years) [158, 163, 164], but elevations at baseline are possibly be linked to a higher concurrent inflammatory activity [165], which may cause a pseudo-atrophy phenomenon in these subjects. On the other hand, with longer follow-up times, effects of pseudo-atrophy should decrease, and Petzold and co-workers reported a very high odds ratio (36, p < 0.01) for the development of pronounced BV loss after 15 years [166]. Some have reported a more modest predictive value [167–169], but, in general, these findings do support the idea that a neurofilament rise can precede the development of brain atrophy in MS.
Conclusions
Despite the growing availability of efficacious disease-controlling therapy, MS remains a significant burden for global health. Accelerated BV loss has recently emerged as a popular imaging biomarker of neurodegeneration, which is considered to be the main determinant of long-term prognosis in affected individuals. Unfortunately, irreversible tissue damage is already likely at the time when brain atrophy can be reliably measured. This Review highlights potential neurodegenerative markers, that may precede these atrophic changes and, (Figure 5) therefore, possibly provide a more beneficial window of opportunity for treatment adjustment. One candidate is the detection of mitochondrial dysfunction, by means of decreased NAA levels, in normal-appearing white matter, which has been described as early as in the CIS and RIS stage. Reduced metabolic activity, all the more together with increased needs due to chronic demyelination and ion channel redistribution, may induce a chronic energy deficiency, resulting in intracellular sodium accumulation. Recent high-field MRI has demonstrated raised intracellular sodium concentrations in patients with relapsing-remitting MS, combined with decreased extracellular fractions, arguing against the concomitant presence of atrophy. Axonal metabolic dysfunction may also be associated with a reduced energy consumption and, hence, a reduced CBF, which has been demonstrated to be independent from atrophy. Yet, cerebral hypoperfusion may as well represent a distinct pathological mechanism in MS, caused by overexpression of the vasospastic agent endothelin-1. Next, microstructural fibral changes, as assessed by DTI and interpreted as reduced axonal integrity, were observed in the volumetrically normal hippocampus of subjects with a CIS, where they were associated with clinically relevant cognitive changes. Finally, Nf-L concentrations in blood and CSF have recently developed into a marker of axonal damage in a variety of neurological disorders. Elevated levels have been described in early stages of MS and appear to be predictive at the group level for the degree of future BV loss over a sizeable time period.
Fig. 5.

Biological background of potential early neurodegenerative markers in MS. Blue: biological processes possibly connecting inflammation with neurodegeneration in MS. Orange: techniques to address the potential corresponding biomarker. DTI: diffusion tensor imaging, CEST: chemical exchange saturation transfer, fMRI: functional magnetic resonance imaging, PET: positron emission tomography, CBF: cerebral blood flow, CSF: cerebrospinal fluid, MS: multiple sclerosis.
The statements and assumptions throughout this paper are not without limitations. First, definite proof for the proposed model linking inflammation to neurodegeneration in MS is, although plausible and frequently put forth in the literature, still lacking and therefore our basis remains hypothetical. MS is an extremely complex disease in which many other pathways may lead to neurodegeneration and atrophy, including primary deficits in neurons, oligodendrocytes or astrocytes [170–172]. But if this pathophysiological basis is correct, halting early inflammatory disease activity will prevent neurodegeneration and result in a better clinical outcome for newly diagnosed subjects. BV assessment is becoming increasingly incorporated in treatment trials for MS, with positive data for both licensed and more experimental agents [22, 173, 174]. Interestingly, it has recently been suggested that teriflunomide may reduce disability worsening mainly through its effects on BV preservation [175]. We hope that insights from our paper will help the development of biomarkers that enable treatment decisions to anticipate on brain atrophy/irreversible tissue damage, hereby further favoring clinical prognosis. Second, this paper was not written as a systematic review based on a general literature search. The authors provided input from their respective areas of expertise, grafted on the subcellular model for neurodegeneration in MS, and complemented by specific individual database revisions (e.g. for the section on cerebral perfusion, the items ‘brain volume’ and ‘brain atrophy’ were used in PubMed, in combination with ‘cerebral blood flow’ and ‘cerebral hypoperfusion’) and bibliographies of key publications. Third, it remains unsure whether therapeutic decisions based on advanced MRI or biochemical findings suggestive of axonal suffering eventually will ensure better clinical outcomes. Studies in this field are non-existing, individual cut-off values of the abnormal are unknown, and it cannot be excluded that subcellular damage reaches a point-of-no-return before we are able to detect it. Fourth, BV loss has been granted the central role as marker of the neurodegenerative process in MS, while other potential and emerging candidates such as spinal cord and retinal atrophy have not been addressed [176, 177]. Reduced retinal nerve layer thickness (RNFL), as measured with optical coherence tomography, has been associated with both measures of brain atrophy and inflammatory disease activity in subjects with MS. [178, 179] Pisa and colleagues have recently demonstrated that the RNFL was relatively preserved over approximately 2 years in NEDA patients with a relapsing-remitting subtype while this was not the case in those with a progressive clinical subtype but which were stable according to their EDSS score. Based on these findings, the authors suggested that the anterior optic pathways share the same mechanisms of degeneration as the rest of the CNS, rather than predominantly resulting from transsynaptic degeneration secondary to lesional activity in the optic radiation [180]. Retinal degeneration seems to be an interesting reflection of analogue brain processes but it remains unsure whether detecting the former may eventually serve as a prior warning for the latter, as a correlating coexistence of both has already been demonstrated very early in the disease [181–183]. Preliminary data suggest that mitochondrial dysfunction might by reflected by alternative imaging and body fluid biomarkers (e.g. uric acid) as well [184, 185]. Fifth, the techniques described in this Review are not free from reliability and reproducibility issues. A very specific technical expertise is usually required and the impact of several potential biological confounders (e.g. normal aging, exercise, hydration status) on all presented measures still needs to be fully elucidated. Advanced acquisition schemes are more difficult to reproduce across different scanner vendors and clinical sites, which can be illustrated by a recent meta-analysis indicating a poor intraclass correlation coefficient (0.29) of fMRI connectivity estimates [186]. Blood and CSF neurofilament levels can increase with growing age [187, 188], and research has been mainly focusing on (younger) relapsing-remitting patients while there is a relative lack of data in more advanced disease stages. Sixth, it has been widely acknowledged that depression, vascular and autoimmune comorbidities are more prevalent in patients with MS than in age-matched controls [5]. These comorbid conditions may affect clinical outcomes and brain pathology, and confound clinical research. Zivadinov and colleagues assessed a large cohort of 815 subjects with MS, of which 29.6% had autoimmune comorbidities and demonstrated a reduction of whole-brain and cortical volumes. The main autoimmune diseases affecting brain volumes were psoriasis, thyroid disease, and type 1 diabetes mellitus [189]. Additionally, Lorefice and co-workers observed a reduced grey matter volume in individuals with type I diabetes, which correlated with diabetes disease duration [190]. Our selection of candidate biomarkers is informed by the different pathological processes and does not take comorbidities into account. Also, the potential coexistence of other neurodegenerative disorders such as Alzheimer’s disease needs to considered in an ageing population [191]. As illustrated by the findings from the Nun and Honolulu-Asia Aging studies, the total burden of comorbid neuropathology may actually be more relevant for the development of clinical symptoms than any single lesion [192]. In this context, combining several biomarkers that assess different pathways resulting in neurodegeneration could be valuable.
Studies exploring BV in combination with metabolic or molecular MRI, perfusion assessment, connectivity analysis and/or neurofilament measurement are scarce but should guide new work in this field. Thus far, only CBF and DTI alterations have been directly evaluated against brain volumetry, and provide the highest level of evidence. In order to disentangle the different possible mechanisms leading to atrophy and their timelines, longitudinal and multimodal studies are required. Studies of particular interest would be to explore the longitudinal relationship between glutamate, NAA and BV, to investigate the hypothesized U-shaped curve of functional MRI connectivity, and to investigate whether metabolic dysfunction induces a reduced cerebral blood flow or the other way around. These studies could provide important novel insights in disease mechanisms and provide new pathways to counteract the neurodegenerative effects of MS before irreparable damage has occurred. Finally, it is worth noting that with the advent of big medical data and artificial intelligence (AI) techniques, new models can be generated that may be able to predict atrophy before irreversible tissue loss has set in. Yet, AI requires a huge amount of data that is currently unavailable for most of the modalities presented in this paper. A specific (imaging) modality should first have some clinical value before large datasets can be collected. Based on the current literature, we only found one study that applied a deep learning strategy to uncover sustained EDSS progression [193], that reached a modest area under the receiver operator characteristic curve of 0.66. Other papers have explored the possibility of using non-linear models (like support vector machines and artificial neural networks) with standard brain volumetric measurements as input [194, 195]. Although these models may help to predict clinical deterioration (EDSS score change), they are already based on atrophy estimates and thus fall beyond the scope of this paper.
Acknowledgements
Not applicable.
Abbreviations
- AHSCT
Autologous stem cell transplantation
- AI
Artificial intelligence
- APT
Amide proton transfer
- ASIC
Acid-sensing ion channel
- ASL
Arterial spin labelling
- ATP
Adenosine triphosphate
- BBB
Blood-brain barrier
- BOLD
Blood oxygenation level dependent
- BV
Brain volume
- CBF
Cerebral blood flow
- CEST
Chemical exchange saturation transfer
- CIS
Clinically isolated syndrome
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- DMF
Dimethylfumarate
- DTI
Diffusion tensor imaging
- EDSS
Expanded Disability Status Scale
- EMA
European Medical Agency
- FA
Fractional anisotropy
- FC
Functional connectivity
- FDA
Food and Drug Administration
- FDG
Fluorodeoxyglucose
- fMRI
Functional magnetic resonance imaging
- GABA
Gamma-aminobutyric acid
- INF
Interferon
- MD
Mean diffusivity
- MRI
Magnetic resonance imaging
- MRS
Magnetic resonance spectroscopy
- MS
Multiple sclerosis
- NAA
N-acetyl-aspartate
- NEDA
No evidence of disease activity
- Nf-L
Neurofilament light chain
- NO
Nitric oxide
- OCBs
Oligoclonal bands
- PET
Positron emission tomography
- RIS
Radiologically isolated syndrome
- RNFL
Retinal nerve layer thickness
- ROS
Reactive oxygen species
- RR
Relapsing-remitting
- RS-FC
Resting-state functional connectivity
Authors’ contributions
MD conceptualized the paper. JVS, GN and MD wrote the first draft of the manuscript. All authors were involved in the critical reading process and approved the final version of the manuscript. JVS and MD created the figures and tables.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
GN is a shareholder of icometrix which is a software provider for BV measurements, and currently on secondment from his employer (i.e. Universitair Ziekenhuis) to icometrix for 10% of his time in the role of ‘Medical Director Neurology’. MBD, GN and MD have received consultancy fees from the manufacturers of the disease-modifying treatments mentioned in the paper.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Pugliatti M, Sotgiu S, Rosati G. The worldwide prevalence of multiple sclerosis. Clin Neurol Neurosurg. 2002;104(3):182–191. doi: 10.1016/S0303-8467(02)00036-7. [DOI] [PubMed] [Google Scholar]
- 3.Scalfari A, Neuhaus A, Daumer M, Ebers GC, Muraro PA. Age and disability accumulation in multiple sclerosis. Neurology. 2011;77(13):1246–1252. doi: 10.1212/WNL.0b013e318230a17d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leray E, Yaouanq J, Le Page E, Coustans M, Laplaud D, Oger J, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133(Pt 7):1900–1913. doi: 10.1093/brain/awq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marrie RA. Comorbidity in multiple sclerosis: Past, present and future. Clin Invest Med. 2019;42(1):E5–E12. doi: 10.25011/cim.v42i1.32383. [DOI] [PubMed] [Google Scholar]
- 6.Miller DH, Barkhof F, Frank JA, Parker GJ, Thompson AJ. Measurement of atrophy in multiple sclerosis: pathological basis, methodological aspects and clinical relevance. Brain. 2002;125(Pt 8):1676–1695. doi: 10.1093/brain/awf177. [DOI] [PubMed] [Google Scholar]
- 7.Minagar A, Barnett MH, Benedict RH, Pelletier D, Pirko I, Sahraian MA, et al. The thalamus and multiple sclerosis: modern views on pathologic, imaging, and clinical aspects. Neurology. 2013;80(2):210–219. doi: 10.1212/WNL.0b013e31827b910b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zivadinov R, Dwyer MG, Bergsland N. Brain atrophy measurements should be used to guide therapy monitoring in MS - YES. Mult Scler. 2016;22(12):1522–1524. doi: 10.1177/1352458516649253. [DOI] [PubMed] [Google Scholar]
- 9.Barkhof F. Brain atrophy measurements should be used to guide therapy monitoring in MS - NO. Mult Scler. 2016;22(12):1524–1526. doi: 10.1177/1352458516649452. [DOI] [PubMed] [Google Scholar]
- 10.Sastre-Garriga J, Pareto D, Rovira A. Brain Atrophy in Multiple Sclerosis: Clinical Relevance and Technical Aspects. Neuroimaging Clin N Am. 2017;27(2):289–300. doi: 10.1016/j.nic.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Zivadinov R, Jakimovski D, Gandhi S, Ahmed R, Dwyer MG, Horakova D, et al. Clinical relevance of brain atrophy assessment in multiple sclerosis. Implications for its use in a clinical routine. Expert Rev Neurother. 2016;16(7):777–793. doi: 10.1080/14737175.2016.1181543. [DOI] [PubMed] [Google Scholar]
- 12.De Stefano N, Giorgio A, Battaglini M, Rovaris M, Sormani MP, Barkhof F, et al. Assessing brain atrophy rates in a large population of untreated multiple sclerosis subtypes. Neurology. 2010;74(23):1868–1876. doi: 10.1212/WNL.0b013e3181e24136. [DOI] [PubMed] [Google Scholar]
- 13.Rocca MA, Battaglini M, Benedict RH, De Stefano N, Geurts JJ, Henry RG, et al. Brain MRI atrophy quantification in MS: From methods to clinical application. Neurology. 2017;88(4):403–413. doi: 10.1212/WNL.0000000000003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis - success from bench to bedside. Nat Rev Neurol. 2019;15(1):53–58. doi: 10.1038/s41582-018-0082-z. [DOI] [PubMed] [Google Scholar]
- 15.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33(11):1444–1452. doi: 10.1212/WNL.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 16.Rotstein DL, Healy BC, Malik MT, Chitnis T, Weiner HL. Evaluation of no evidence of disease activity in a 7-year longitudinal multiple sclerosis cohort. JAMA Neurol. 2015;72(2):152–158. doi: 10.1001/jamaneurol.2014.3537. [DOI] [PubMed] [Google Scholar]
- 17.Tsivgoulis G, Katsanos AH, Grigoriadis N, Hadjigeorgiou GM, Heliopoulos I, Papathanasopoulos P, et al. The effect of disease-modifying therapies on brain atrophy in patients with clinically isolated syndrome: a systematic review and meta-analysis. Ther Adv Neurol Disord. 2015;8(5):193–202. doi: 10.1177/1756285615600381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsivgoulis G, Katsanos AH, Grigoriadis N, Hadjigeorgiou GM, Heliopoulos I, Papathanasopoulos P, et al. The Effect of Disease Modifying Therapies on Disease Progression in Patients with Relapsing-Remitting Multiple Sclerosis: A Systematic Review and Meta-Analysis. PLoS One. 2015;10(12):e0144538. doi: 10.1371/journal.pone.0144538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branger P, Parienti JJ, Sormani MP, Defer G. The Effect of Disease-Modifying Drugs on Brain Atrophy in Relapsing-Remitting Multiple Sclerosis: A Meta-Analysis. PLoS One. 2016;11(3):e0149685. doi: 10.1371/journal.pone.0149685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vollmer T, Signorovitch J, Huynh L, Galebach P, Kelley C, DiBernardo A, et al. The natural history of brain volume loss among patients with multiple sclerosis: a systematic literature review and meta-analysis. J Neurol Sci. 2015;357(1-2):8–18. doi: 10.1016/j.jns.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Zivadinov R, Reder AT, Filippi M, Minagar A, Stuve O, Lassmann H, et al. Mechanisms of action of disease-modifying agents and brain volume changes in multiple sclerosis. Neurology. 2008;71(2):136–144. doi: 10.1212/01.wnl.0000316810.01120.05. [DOI] [PubMed] [Google Scholar]
- 22.Sormani MP, Arnold DL, De Stefano N. Treatment effect on brain atrophy correlates with treatment effect on disability in multiple sclerosis. Ann Neurol. 2014;75(1):43–49. doi: 10.1002/ana.24018. [DOI] [PubMed] [Google Scholar]
- 23.Kappos L, De Stefano N, Freedman MS, Cree BA, Radue EW, Sprenger T, et al. Inclusion of brain volume loss in a revised measure of 'no evidence of disease activity' (NEDA-4) in relapsing-remitting multiple sclerosis. Mult Scler. 2016;22(10):1297–1305. doi: 10.1177/1352458515616701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rovira A, Wattjes MP, Tintore M, Tur C, Yousry TA, Sormani MP, et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis-clinical implementation in the diagnostic process. Nat Rev Neurol. 2015;11(8):471–482. doi: 10.1038/nrneurol.2015.106. [DOI] [PubMed] [Google Scholar]
- 25.Wattjes MP, Rovira A, Miller D, Yousry TA, Sormani MP, de Stefano MP, et al. Evidence-based guidelines: MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis--establishing disease prognosis and monitoring patients. Nat Rev Neurol. 2015;11(10):597–606. doi: 10.1038/nrneurol.2015.157. [DOI] [PubMed] [Google Scholar]
- 26.Weiner HL. Multiple sclerosis is an inflammatory T-cell-mediated autoimmune disease. Arch Neurol. 2004;61(10):1613–1615. doi: 10.1001/archneur.61.10.1613. [DOI] [PubMed] [Google Scholar]
- 27.Frohman EM, Racke MK, Raine CS. Multiple sclerosis--the plaque and its pathogenesis. N Engl J Med. 2006;354(9):942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 28.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 29.Dziedzic T, Metz I, Dallenga T, Konig FB, Muller S, Stadelmann C, et al. Wallerian degeneration: a major component of early axonal pathology in multiple sclerosis. Brain Pathol. 2010;20(5):976–985. doi: 10.1111/j.1750-3639.2010.00401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–193. doi: 10.1016/S1474-4422(14)70256-X. [DOI] [PubMed] [Google Scholar]
- 31.Bielekova B, Kadom N, Fisher E, Jeffries N, Ohayon J, Richert N, et al. MRI as a marker for disease heterogeneity in multiple sclerosis. Neurology. 2005;65(7):1071–1076. doi: 10.1212/01.wnl.0000178984.30534.f9. [DOI] [PubMed] [Google Scholar]
- 32.Chard DT, Brex PA, Ciccarelli O, Griffin CM, Parker GJ, Dalton C, et al. The longitudinal relation between brain lesion load and atrophy in multiple sclerosis: a 14 year follow up study. J Neurol Neurosurg Psychiatry. 2003;74(11):1551–1554. doi: 10.1136/jnnp.74.11.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DeLuca GC, Williams K, Evangelou N, Ebers GC, Esiri MM. The contribution of demyelination to axonal loss in multiple sclerosis. Brain. 2006;129(Pt 6):1507–1516. doi: 10.1093/brain/awl074. [DOI] [PubMed] [Google Scholar]
- 34.Evangelou N, DeLuca GC, Owens T, Esiri MM. Pathological study of spinal cord atrophy in multiple sclerosis suggests limited role of local lesions. Brain. 2005;128(Pt 1):29–34. doi: 10.1093/brain/awh323. [DOI] [PubMed] [Google Scholar]
- 35.Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(Pt 5):1175–1189. doi: 10.1093/brain/awp070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klistorner A, Wang C, Yiannikas C, Parratt J, Dwyer M, Barton J, et al. Evidence of progressive tissue loss in the core of chronic MS lesions: A longitudinal DTI study. Neuroimage Clin. 2018;17:1028–1035. doi: 10.1016/j.nicl.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bjartmar C, Kidd G, Mork S, Rudick R, Trapp BD. Neurological disability correlates with spinal cord axonal loss and reduced N-acetyl aspartate in chronic multiple sclerosis patients. Ann Neurol. 2000;48(6):893–901. doi: 10.1002/1531-8249(200012)48:6<893::AID-ANA10>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 38.Black JA, Felts P, Smith KJ, Kocsis JD, Waxman SG. Distribution of sodium channels in chronically demyelinated spinal cord axons: immuno-ultrastructural localization and electrophysiological observations. Brain Res. 1991;544(1):59–70. doi: 10.1016/0006-8993(91)90885-Y. [DOI] [PubMed] [Google Scholar]
- 39.Lee JY, Taghian K, Petratos S. Axonal degeneration in multiple sclerosis: can we predict and prevent permanent disability? Acta Neuropathol Commun. 2014;2:97. doi: 10.1186/s40478-014-0097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009;8(3):280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- 41.Mahad D, Lassmann H, Turnbull D. Review: Mitochondria and disease progression in multiple sclerosis. Neuropathol Appl Neurobiol. 2008;34(6):577–589. doi: 10.1111/j.1365-2990.2008.00987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cambron M, D'Haeseleer M, Laureys G, Clinckers R, Debruyne J, De Keyser J. White-matter astrocytes, axonal energy metabolism, and axonal degeneration in multiple sclerosis. J Cereb Blood Flow Metab. 2012;32(3):413–424. doi: 10.1038/jcbfm.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ciccarelli O, Barkhof F, Bodini B, De Stefano N, Golay X, Nicolay K, et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol. 2014;13(8):807–822. doi: 10.1016/S1474-4422(14)70101-2. [DOI] [PubMed] [Google Scholar]
- 44.Kawachi I, Lassmann H. Neurodegeneration in multiple sclerosis and neuromyelitis optica. J Neurol Neurosurg Psychiatry. 2017;88(2):137–145. doi: 10.1136/jnnp-2016-313300. [DOI] [PubMed] [Google Scholar]
- 45.Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS, et al. Mitochondrial changes within axons in multiple sclerosis. Brain. 2009;132(Pt 5):1161–1174. doi: 10.1093/brain/awp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witte ME, Bo L, Rodenburg RJ, Belien JA, Musters R, Hazes T, et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol. 2009;219(2):193–204. doi: 10.1002/path.2582. [DOI] [PubMed] [Google Scholar]
- 47.Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13(12):1483–1489. doi: 10.1038/nm1668. [DOI] [PubMed] [Google Scholar]
- 48.Moccia M, Ciccarelli O. Molecular and Metabolic Imaging in Multiple Sclerosis. Neuroimaging Clin N Am. 2017;27(2):343–356. doi: 10.1016/j.nic.2016.12.005. [DOI] [PubMed] [Google Scholar]
- 49.Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol. 2007;81(2):89–131. doi: 10.1016/j.pneurobio.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steen C, Wilczak N, Hoogduin JM, Koch M, De Keyser J. Reduced creatine kinase B activity in multiple sclerosis normal appearing white matter. PLoS One. 2010;5(5):e10811. doi: 10.1371/journal.pone.0010811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steen C, D'Haeseleer M, Hoogduin JM, Fierens Y, Cambron M, Mostert JP, et al. Cerebral white matter blood flow and energy metabolism in multiple sclerosis. Mult Scler. 2013;19(10):1282–1289. doi: 10.1177/1352458513477228. [DOI] [PubMed] [Google Scholar]
- 52.Narayana PA, Doyle TJ, Lai D, Wolinsky JS. Serial proton magnetic resonance spectroscopic imaging, contrast-enhanced magnetic resonance imaging, and quantitative lesion volumetry in multiple sclerosis. Ann Neurol. 1998;43(1):56–71. doi: 10.1002/ana.410430112. [DOI] [PubMed] [Google Scholar]
- 53.Ciccarelli O, Wheeler-Kingshott CA, McLean MA, Cercignani M, Wimpey K, Miller DH, et al. Spinal cord spectroscopy and diffusion-based tractography to assess acute disability in multiple sclerosis. Brain. 2007;130(Pt 8):2220–2231. doi: 10.1093/brain/awm152. [DOI] [PubMed] [Google Scholar]
- 54.Caramanos Z, Narayanan S, Arnold DL. 1H-MRS quantification of tNA and tCr in patients with multiple sclerosis: a meta-analytic review. Brain. 2005;128(Pt 11):2483–2506. doi: 10.1093/brain/awh640. [DOI] [PubMed] [Google Scholar]
- 55.Chard DT, Griffin CM, McLean MA, Kapeller P, Kapoor R, Thompson AJ, et al. Brain metabolite changes in cortical grey and normal-appearing white matter in clinically early relapsing-remitting multiple sclerosis. Brain. 2002;125(Pt 10):2342–2352. doi: 10.1093/brain/awf240. [DOI] [PubMed] [Google Scholar]
- 56.Aboul-Enein F, Krssak M, Hoftberger R, Prayer D, Kristoferitsch W. Reduced NAA-levels in the NAWM of patients with MS is a feature of progression. A study with quantitative magnetic resonance spectroscopy at 3 Tesla. PLoS One. 2010;5(7):e11625. doi: 10.1371/journal.pone.0011625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adalsteinsson E, Langer-Gould A, Homer RJ, Rao A, Sullivan EV, Lima CA, et al. Gray matter N-acetyl aspartate deficits in secondary progressive but not relapsing-remitting multiple sclerosis. AJNR Am J Neuroradiol. 2003;24(10):1941–1945. [PMC free article] [PubMed] [Google Scholar]
- 58.Ciccarelli O, Altmann DR, McLean MA, Wheeler-Kingshott CA, Wimpey K, Miller DH, et al. Spinal cord repair in MS: does mitochondrial metabolism play a role? Neurology. 2010;74(9):721–727. doi: 10.1212/WNL.0b013e3181d26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Stefano N, Matthews PM, Antel JP, Preul M, Francis G, Arnold DL. Chemical pathology of acute demyelinating lesions and its correlation with disability. Ann Neurol. 1995;38(6):901–909. doi: 10.1002/ana.410380610. [DOI] [PubMed] [Google Scholar]
- 60.Kirov II, Tal A, Babb JS, Herbert J, Gonen O. Serial proton MR spectroscopy of gray and white matter in relapsing-remitting MS. Neurology. 2013;80(1):39–46. doi: 10.1212/WNL.0b013e31827b1a8c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wattjes MP, Harzheim M, Lutterbey GG, Bogdanow M, Schild HH, Traber F. High field MR imaging and 1H-MR spectroscopy in clinically isolated syndromes suggestive of multiple sclerosis: correlation between metabolic alterations and diagnostic MR imaging criteria. J Neurol. 2008;255(1):56–63. doi: 10.1007/s00415-007-0666-9. [DOI] [PubMed] [Google Scholar]
- 62.Rocca MA, Mezzapesa DM, Falini A, Ghezzi A, Martinelli V, Scotti G, et al. Evidence for axonal pathology and adaptive cortical reorganization in patients at presentation with clinically isolated syndromes suggestive of multiple sclerosis. Neuroimage. 2003;18(4):847–855. doi: 10.1016/S1053-8119(03)00043-0. [DOI] [PubMed] [Google Scholar]
- 63.Stromillo ML, Giorgio A, Rossi F, Battaglini M, Hakiki B, Malentacchi G, et al. Brain metabolic changes suggestive of axonal damage in radiologically isolated syndrome. Neurology. 2013;80(23):2090–2094. doi: 10.1212/WNL.0b013e318295d707. [DOI] [PubMed] [Google Scholar]
- 64.Labiano-Fontcuberta A, Martinez-Gines ML, Aladro Y, Ayuso L, Mitchell AJ, Puertas-Martin V, et al. A comparison study of cognitive deficits in radiologically and clinically isolated syndromes. Mult Scler. 2016;22(2):250–253. doi: 10.1177/1352458515591072. [DOI] [PubMed] [Google Scholar]
- 65.Abdel-Aziz K, Schneider T, Solanky BS, Yiannakas MC, Altmann DR, Wheeler-Kingshott CA, et al. Evidence for early neurodegeneration in the cervical cord of patients with primary progressive multiple sclerosis. Brain. 2015;138(Pt 6):1568–1582. doi: 10.1093/brain/awv086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Azevedo CJ, Kornak J, Chu P, Sampat M, Okuda DT, Cree BA, et al. In vivo evidence of glutamate toxicity in multiple sclerosis. Ann Neurol. 2014;76(2):269–278. doi: 10.1002/ana.24202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.MacMillan EL, Tam R, Zhao Y, Vavasour IM, Li DK, Oger J, et al. Progressive multiple sclerosis exhibits decreasing glutamate and glutamine over two years. Mult Scler. 2016;22(1):112–116. doi: 10.1177/1352458515586086. [DOI] [PubMed] [Google Scholar]
- 68.Kostic M, Zivkovic N, Stojanovic I. Multiple sclerosis and glutamate excitotoxicity. Rev Neurosci. 2013;24(1):71–88. doi: 10.1515/revneuro-2012-0062. [DOI] [PubMed] [Google Scholar]
- 69.Inglese M, Madelin G, Oesingmann N, Babb JS, Wu W, Stoeckel B, et al. Brain tissue sodium concentration in multiple sclerosis: a sodium imaging study at 3 tesla. Brain. 2010;133(Pt 3):847–857. doi: 10.1093/brain/awp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paling D, Solanky BS, Riemer F, Tozer DJ, Wheeler-Kingshott CA, Kapoor R, et al. Sodium accumulation is associated with disability and a progressive course in multiple sclerosis. Brain. 2013;136(Pt 7):2305–2317. doi: 10.1093/brain/awt149. [DOI] [PubMed] [Google Scholar]
- 71.Zaaraoui W, Konstandin S, Audoin B, Nagel AM, Rico A, Malikova I, et al. Distribution of brain sodium accumulation correlates with disability in multiple sclerosis: a cross-sectional 23Na MR imaging study. Radiology. 2012;264(3):859–867. doi: 10.1148/radiol.12112680. [DOI] [PubMed] [Google Scholar]
- 72.Donadieu M, Le Fur Y, Maarouf A, Gherib S, Ridley B, Pini L, et al. Metabolic counterparts of sodium accumulation in multiple sclerosis: A whole brain (23) Na-MRI and fast (1) H-MRSI study. Mult Scler. 2019;25(1):39–47. doi: 10.1177/1352458517736146. [DOI] [PubMed] [Google Scholar]
- 73.Maarouf A, Audoin B, Konstandin S, Rico A, Soulier E, Reuter F, et al. Topography of brain sodium accumulation in progressive multiple sclerosis. MAGMA. 2014;27(1):53–62. doi: 10.1007/s10334-013-0396-1. [DOI] [PubMed] [Google Scholar]
- 74.Petracca M, Vancea RO, Fleysher L, Jonkman LE, Oesingmann N, Inglese M. Brain intra- and extracellular sodium concentration in multiple sclerosis: a 7 T MRI study. Brain. 2016;139(Pt 3):795–806. doi: 10.1093/brain/awv386. [DOI] [PubMed] [Google Scholar]
- 75.Maarouf A, Audoin B, Pariollaud F, Gherib S, Rico A, Soulier E, et al. Increased total sodium concentration in gray matter better explains cognition than atrophy in MS. Neurology. 2017;88(3):289–295. doi: 10.1212/WNL.0000000000003511. [DOI] [PubMed] [Google Scholar]
- 76.Brownlee WJ, Solanky B, Prados F, Yiannakas M, Da Mota P, Riemer F, et al. Cortical grey matter sodium accumulation is associated with disability and secondary progressive disease course in relapse-onset multiple sclerosis. J Neurol Neurosurg Psychiatry. 2019;90(7):755–760. doi: 10.1136/jnnp-2018-319634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu B, Warnock G, Zaiss M, Lin C, Chen M, Zhou Z, et al. An overview of CEST MRI for non-MR physicists. EJNMMI Phys. 2016;3(1):19. doi: 10.1186/s40658-016-0155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang YZ, Xu TL. Acidosis, acid-sensing ion channels, and neuronal cell death. Mol Neurobiol. 2011;44(3):350–358. doi: 10.1007/s12035-011-8204-2. [DOI] [PubMed] [Google Scholar]
- 79.Dula AN, Asche EM, Landman BA, Welch EB, Pawate S, Sriram S, et al. Development of chemical exchange saturation transfer at 7 T. Magn Reson Med. 2011;66(3):831–838. doi: 10.1002/mrm.22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.By S, Barry RL, Smith AK, Lyttle BD, Box BA, Bagnato FR, et al. Amide proton transfer CEST of the cervical spinal cord in multiple sclerosis patients at 3T. Magn Reson Med. 2018;79(2):806–814. doi: 10.1002/mrm.26736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MI, et al. Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain. 2013;136(Pt 1):106–115. doi: 10.1093/brain/aws325. [DOI] [PubMed] [Google Scholar]
- 82.Matthews PM, Comley R. Advances in the molecular imaging of multiple sclerosis. Expert Rev Clin Immunol. 2009;5(6):765–777. doi: 10.1586/eci.09.66. [DOI] [PubMed] [Google Scholar]
- 83.Faria Dde P, Copray S, Buchpiguel C, Dierckx R, de Vries E. PET imaging in multiple sclerosis. J Neuroimmune Pharmacol. 2014;9(4):468–482. doi: 10.1007/s11481-014-9544-2. [DOI] [PubMed] [Google Scholar]
- 84.Kindred JH, Koo PJ, Rudroff T. Glucose uptake of the spinal cord in patients with multiple sclerosis detected by (1, 8) F-fluorodeoxyglucose PET/CT after walking. Spinal Cord. 2014;52(Suppl 3):S11–S13. doi: 10.1038/sc.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freeman L, Garcia-Lorenzo D, Bottin L, Leroy C, Louapre C, Bodini B, et al. The neuronal component of gray matter damage in multiple sclerosis: A [(11) C] flumazenil positron emission tomography study. Ann Neurol. 2015;78(4):554–567. doi: 10.1002/ana.24468. [DOI] [PubMed] [Google Scholar]
- 86.Cao G, Edden RAE, Gao F, Li H, Gong T, Chen W, et al. Reduced GABA levels correlate with cognitive impairment in patients with relapsing-remitting multiple sclerosis. Eur Radiol. 2018;28(3):1140–1148. doi: 10.1007/s00330-017-5064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019. [DOI] [PubMed]
- 88.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 89.van der Veen BS, de Winther MP, Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid Redox Signal. 2009;11(11):2899–2937. doi: 10.1089/ars.2009.2538. [DOI] [PubMed] [Google Scholar]
- 90.Rissanen E, Tuisku J, Rokka J, Paavilainen T, Parkkola R, Rinne JO, et al. In Vivo Detection of Diffuse Inflammation in Secondary Progressive Multiple Sclerosis Using PET Imaging and the Radioligand (1, 1) C-PK11195. J Nucl Med. 2014;55(6):939–944. doi: 10.2967/jnumed.113.131698. [DOI] [PubMed] [Google Scholar]
- 91.Politis M, Giannetti P, Su P, Turkheimer F, Keihaninejad S, Wu K, et al. Increased PK11195 PET binding in the cortex of patients with MS correlates with disability. Neurology. 2012;79(6):523–530. doi: 10.1212/WNL.0b013e3182635645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Debruyne JC, Versijpt J, Van Laere KJ, De Vos F, Keppens J, Strijckmans K, et al. PET visualization of microglia in multiple sclerosis patients using [11C]PK11195. Eur J Neurol. 2003;10(3):257–264. doi: 10.1046/j.1468-1331.2003.00571.x. [DOI] [PubMed] [Google Scholar]
- 93.Versijpt J, Debruyne JC, Van Laere KJ, De Vos F, Keppens J, Strijckmans K, et al. Microglial imaging with positron emission tomography and atrophy measurements with magnetic resonance imaging in multiple sclerosis: a correlative study. Mult Scler. 2005;11(2):127–134. doi: 10.1191/1352458505ms1140oa. [DOI] [PubMed] [Google Scholar]
- 94.Datta G, Violante IR, Scott G, Zimmerman K, Santos-Ribeiro A, Rabiner EA, et al. Translocator positron-emission tomography and magnetic resonance spectroscopic imaging of brain glial cell activation in multiple sclerosis. Mult Scler. 2017;23(11):1469–1478. doi: 10.1177/1352458516681504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Datta G, Colasanti A, Rabiner EA, Gunn RN, Malik O, Ciccarelli O, et al. Neuroinflammation and its relationship to changes in brain volume and white matter lesions in multiple sclerosis. Brain. 2017;140(11):2927–2938. doi: 10.1093/brain/awx228. [DOI] [PubMed] [Google Scholar]
- 96.Wuerfel J, Paul F, Zipp F. Cerebral blood perfusion changes in multiple sclerosis. J Neurol Sci. 2007;259(1-2):16–20. doi: 10.1016/j.jns.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 97.Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71(5):782–797. doi: 10.1016/j.neuron.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 98.Swank RL, Roth JG, Woody DC., Jr Cerebral blood flow and red cell delivery in normal subjects and in multiple sclerosis. Neurol Res. 1983;5(1):37–59. doi: 10.1080/01616412.1983.11739631. [DOI] [PubMed] [Google Scholar]
- 99.Brooks DJ, Leenders KL, Head G, Marshall J, Legg NJ, Jones T. Studies on regional cerebral oxygen utilisation and cognitive function in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1984;47(11):1182–1191. doi: 10.1136/jnnp.47.11.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lycke J, Wikkelso C, Bergh AC, Jacobsson L, Andersen O. Regional cerebral blood flow in multiple sclerosis measured by single photon emission tomography with technetium-99m hexamethylpropyleneamine oxime. Eur Neurol. 1993;33(2):163–167. doi: 10.1159/000116926. [DOI] [PubMed] [Google Scholar]
- 101.Sun X, Tanaka M, Kondo S, Okamoto K, Hirai S. Clinical significance of reduced cerebral metabolism in multiple sclerosis: a combined PET and MRI study. Ann Nucl Med. 1998;12(2):89–94. doi: 10.1007/BF03164835. [DOI] [PubMed] [Google Scholar]
- 102.Law M, Saindane AM, Ge Y, Babb JS, Johnson G, Mannon LJ, et al. Microvascular abnormality in relapsing-remitting multiple sclerosis: perfusion MR imaging findings in normal-appearing white matter. Radiology. 2004;231(3):645–652. doi: 10.1148/radiol.2313030996. [DOI] [PubMed] [Google Scholar]
- 103.Adhya S, Johnson G, Herbert J, Jaggi H, Babb JS, Grossman RI, et al. Pattern of hemodynamic impairment in multiple sclerosis: dynamic susceptibility contrast perfusion MR imaging at 3.0 T. Neuroimage. 2006;33(4):1029–1035. doi: 10.1016/j.neuroimage.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Varga AW, Johnson G, Babb JS, Herbert J, Grossman RI, Inglese M. White matter hemodynamic abnormalities precede sub-cortical gray matter changes in multiple sclerosis. J Neurol Sci. 2009;282(1-2):28–33. doi: 10.1016/j.jns.2008.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Papadaki EZ, Mastorodemos VC, Amanakis EZ, Tsekouras KC, Papadakis AE, Tsavalas ND, et al. White matter and deep gray matter hemodynamic changes in multiple sclerosis patients with clinically isolated syndrome. Magn Reson Med. 2012;68(6):1932–1942. doi: 10.1002/mrm.24194. [DOI] [PubMed] [Google Scholar]
- 106.D'Haeseleer M, Steen C, Hoogduin JM, van Osch MJ, Fierens Y, Cambron M, et al. Performance on Paced Auditory Serial Addition Test and cerebral blood flow in multiple sclerosis. Acta Neurol Scand. 2013;128(5):e26–e29. doi: 10.1111/ane.12129. [DOI] [PubMed] [Google Scholar]
- 107.D'Haeseleer M, Beelen R, Fierens Y, Cambron M, Vanbinst AM, Verborgh C, et al. Cerebral hypoperfusion in multiple sclerosis is reversible and mediated by endothelin-1. Proc Natl Acad Sci U S A. 2013;110(14):5654–5658. doi: 10.1073/pnas.1222560110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saindane AM, Law M, Ge Y, Johnson G, Babb JS, Grossman RI. Correlation of diffusion tensor and dynamic perfusion MR imaging metrics in normal-appearing corpus callosum: support for primary hypoperfusion in multiple sclerosis. AJNR Am J Neuroradiol. 2007;28(4):767–772. [PMC free article] [PubMed] [Google Scholar]
- 109.D'Haeseleer M, Hostenbach S, Peeters I, Sankari SE, Nagels G, De Keyser J, et al. Cerebral hypoperfusion: a new pathophysiologic concept in multiple sclerosis? J Cereb Blood Flow Metab. 2015;35(9):1406–1410. doi: 10.1038/jcbfm.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Marshall O, Lu H, Brisset JC, Xu F, Liu P, Herbert J, et al. Impaired cerebrovascular reactivity in multiple sclerosis. JAMA Neurol. 2014;71(10):1275–1281. doi: 10.1001/jamaneurol.2014.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Metzger A, Le Bars E, Deverdun J, Molino F, Marechal B, Picot MC, et al. Is impaired cerebral vasoreactivity an early marker of cognitive decline in multiple sclerosis patients? Eur Radiol. 2018;28(3):1204–1214. doi: 10.1007/s00330-017-5068-5. [DOI] [PubMed] [Google Scholar]
- 112.Graumann U, Reynolds R, Steck AJ, Schaeren-Wiemers N. Molecular changes in normal appearing white matter in multiple sclerosis are characteristic of neuroprotective mechanisms against hypoxic insult. Brain Pathol. 2003;13(4):554–573. doi: 10.1111/j.1750-3639.2003.tb00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wakita H, Tomimoto H, Akiguchi I, Matsuo A, Lin JX, Ihara M, et al. Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat. Brain Res. 2002;924(1):63–70. doi: 10.1016/S0006-8993(01)03223-1. [DOI] [PubMed] [Google Scholar]
- 114.Aliev G, Obrenovich ME, Smith MA, Perry G. Hypoperfusion, Mitochondria Failure, Oxidative Stress, and Alzheimer Disease. J Biomed Biotechnol. 2003;2003(3):162–163. doi: 10.1155/S1110724303305029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Debernard L, Melzer TR, Van Stockum S, Graham C, Wheeler-Kingshott CA, Dalrymple-Alford JC, et al. Reduced grey matter perfusion without volume loss in early relapsing-remitting multiple sclerosis. J Neurol Neurosurg Psychiatry. 2014;85(5):544–551. doi: 10.1136/jnnp-2013-305612. [DOI] [PubMed] [Google Scholar]
- 116.Aviv RI, Francis PL, Tenenbein R, O'Connor P, Zhang L, Eilaghi A, et al. Decreased frontal lobe gray matter perfusion in cognitively impaired patients with secondary-progressive multiple sclerosis detected by the bookend technique. AJNR Am J Neuroradiol. 2012;33(9):1779–1785. doi: 10.3174/ajnr.A3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hojjat SP, Cantrell CG, Vitorino R, Feinstein A, Shirzadi Z, MacIntosh BJ, et al. Regional reduction in cortical blood flow among cognitively impaired adults with relapsing-remitting multiple sclerosis patients. Mult Scler. 2016;22(11):1421–1428. doi: 10.1177/1352458515622696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Doche E, Lecocq A, Maarouf A, Duhamel G, Soulier E, Confort-Gouny S, et al. Hypoperfusion of the thalamus is associated with disability in relapsing remitting multiple sclerosis. J Neuroradiol. 2017;44(2):158–164. doi: 10.1016/j.neurad.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 119.Lagana MM, Mendozzi L, Pelizzari L, Bergsland NP, Pugnetti L, Cecconi P, et al. Are cerebral perfusion and atrophy linked in multiple sclerosis? Evidence for a multifactorial approach to assess neurodegeneration. Curr Neurovasc Res. 2018;15(4):282–291. doi: 10.2174/1567202616666181123164235. [DOI] [PubMed] [Google Scholar]
- 120.Zihl J, von Cramon D, Mai N. Selective disturbance of movement vision after bilateral brain damage. Brain. 1983;106(Pt 2):313–340. doi: 10.1093/brain/106.2.313. [DOI] [PubMed] [Google Scholar]
- 121.Hunt MJ, Kopell NJ, Traub RD, Whittington MA. Aberrant Network Activity in Schizophrenia. Trends Neurosci. 2017;40(6):371–382. doi: 10.1016/j.tins.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mohan A, Roberto AJ, Mohan A, Lorenzo A, Jones K, Carney MJ, et al. The Significance of the Default Mode Network (DMN) in Neurological and Neuropsychiatric Disorders: A Review. Yale J Biol Med. 2016;89(1):49–57. [PMC free article] [PubMed] [Google Scholar]
- 123.Tahedl M, Levine SM, Greenlee MW, Weissert R, Schwarzbach JV. Functional Connectivity in Multiple Sclerosis: Recent Findings and Future Directions. Front Neurol. 2018;9:828. doi: 10.3389/fneur.2018.00828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Matthews PM, Jezzard P. Functional magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2004;75(1):6–12. [PMC free article] [PubMed] [Google Scholar]
- 125.White AT, Lee JN, Light AR, Light KC. Brain activation in multiple sclerosis: a BOLD fMRI study of the effects of fatiguing hand exercise. Mult Scler. 2009;15(5):580–586. doi: 10.1177/1352458508100034. [DOI] [PubMed] [Google Scholar]
- 126.Colorado RA, Shukla K, Zhou Y, Wolinsky JS, Narayana PA. Multi-task functional MRI in multiple sclerosis patients without clinical disability. Neuroimage. 2012;59(1):573–581. doi: 10.1016/j.neuroimage.2011.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Forn C, Rocca MA, Valsasina P, Bosca I, Casanova B, Sanjuan A, et al. Functional magnetic resonance imaging correlates of cognitive performance in patients with a clinically isolated syndrome suggestive of multiple sclerosis at presentation: an activation and connectivity study. Mult Scler. 2012;18(2):153–163. doi: 10.1177/1352458511417744. [DOI] [PubMed] [Google Scholar]
- 128.Faivre A, Rico A, Zaaraoui W, Crespy L, Reuter F, Wybrecht D, et al. Assessing brain connectivity at rest is clinically relevant in early multiple sclerosis. Mult Scler. 2012;18(9):1251–1258. doi: 10.1177/1352458511435930. [DOI] [PubMed] [Google Scholar]
- 129.Hawellek DJ, Hipp JF, Lewis CM, Corbetta M, Engel AK. Increased functional connectivity indicates the severity of cognitive impairment in multiple sclerosis. Proc Natl Acad Sci U S A. 2011;108(47):19066–19071. doi: 10.1073/pnas.1110024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou F, Zhuang Y, Gong H, Wang B, Wang X, Chen Q, et al. Altered inter-subregion connectivity of the default mode network in relapsing remitting multiple sclerosis: a functional and structural connectivity study. PLoS One. 2014;9(7):e101198. doi: 10.1371/journal.pone.0101198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain's default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- 132.Bonavita S, Gallo A, Sacco R, Corte MD, Bisecco A, Docimo R, et al. Distributed changes in default-mode resting-state connectivity in multiple sclerosis. Mult Scler. 2011;17(4):411–422. doi: 10.1177/1352458510394609. [DOI] [PubMed] [Google Scholar]
- 133.Cruz-Gomez AJ, Ventura-Campos N, Belenguer A, Avila C, Forn C. The link between resting-state functional connectivity and cognition in MS patients. Mult Scler. 2014;20(3):338–348. doi: 10.1177/1352458513495584. [DOI] [PubMed] [Google Scholar]
- 134.Louapre C, Perlbarg V, Garcia-Lorenzo D, Urbanski M, Benali H, Assouad R, et al. Brain networks disconnection in early multiple sclerosis cognitive deficits: an anatomofunctional study. Hum Brain Mapp. 2014;35(9):4706–4717. doi: 10.1002/hbm.22505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu Y, Wang H, Duan Y, Huang J, Ren Z, Ye J, et al. Functional Brain Network Alterations in Clinically Isolated Syndrome and Multiple Sclerosis: A Graph-based Connectome Study. Radiology. 2017;282(2):534–541. doi: 10.1148/radiol.2016152843. [DOI] [PubMed] [Google Scholar]
- 136.Mori S, Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron. 2006;51(5):527–539. doi: 10.1016/j.neuron.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 137.Soares JM, Marques P, Alves V, Sousa N. A hitchhiker's guide to diffusion tensor imaging. Front Neurosci. 2013;7:31. doi: 10.3389/fnins.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Roosendaal SD, Geurts JJ, Vrenken H, Hulst HE, Cover KS, Castelijns JA, et al. Regional DTI differences in multiple sclerosis patients. Neuroimage. 2009;44(4):1397–1403. doi: 10.1016/j.neuroimage.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 139.Vrenken H, Geurts JJ, Knol DL, Polman CH, Castelijns JA, Pouwels PJ, et al. Normal-appearing white matter changes vary with distance to lesions in multiple sclerosis. AJNR Am J Neuroradiol. 2006;27(9):2005–2011. [PMC free article] [PubMed] [Google Scholar]
- 140.Moll NM, Rietsch AM, Thomas S, Ransohoff AJ, Lee JC, Fox R, et al. Multiple sclerosis normal-appearing white matter: pathology-imaging correlations. Ann Neurol. 2011;70(5):764–773. doi: 10.1002/ana.22521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.de Kouchkovsky I, Fieremans E, Fleysher L, Herbert J, Grossman RI, Inglese M. Quantification of normal-appearing white matter tract integrity in multiple sclerosis: a diffusion kurtosis imaging study. J Neurol. 2016;263(6):1146–1155. doi: 10.1007/s00415-016-8118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rocca MA, Preziosa P, Mesaros S, Pagani E, Dackovic J, Stosic-Opincal T, et al. Clinically Isolated Syndrome Suggestive of Multiple Sclerosis: Dynamic Patterns of Gray and White Matter Changes-A 2-year MR Imaging Study. Radiology. 2016;278(3):841–853. doi: 10.1148/radiol.2015150532. [DOI] [PubMed] [Google Scholar]
- 143.Cappellani R, Bergsland N, Weinstock-Guttman B, Kennedy C, Carl E, Ramasamy DP, et al. Diffusion tensor MRI alterations of subcortical deep gray matter in clinically isolated syndrome. J Neurol Sci. 2014;338(1-2):128–134. doi: 10.1016/j.jns.2013.12.031. [DOI] [PubMed] [Google Scholar]
- 144.Planche V, Ruet A, Coupe P, Lamargue-Hamel D, Deloire M, Pereira B, et al. Hippocampal microstructural damage correlates with memory impairment in clinically isolated syndrome suggestive of multiple sclerosis. Mult Scler. 2017;23(9):1214–1224. doi: 10.1177/1352458516675750. [DOI] [PubMed] [Google Scholar]
- 145.Zivadinov R, Hagemeier J, Bergsland N, Tavazzi E, Weinstock-Guttman B. Effect of dimethyl fumarate on gray and white matter pathology in subjects with relapsing multiple sclerosis: a longitudinal study. Eur J Neurol. 2018;25(3):584–e36. doi: 10.1111/ene.13562. [DOI] [PubMed] [Google Scholar]
- 146.Gurevich M, Waknin R, Stone E, Achiron A. Fingolimod-improved axonal and myelin integrity of white matter tracts associated with multiple sclerosis-related functional impairments. CNS Neurosci Ther. 2018;24(5):412–419. doi: 10.1111/cns.12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ontaneda D, Sakaie K, Lin J, Wang XF, Lowe MJ, Phillips MD, et al. Measuring Brain Tissue Integrity during 4 Years Using Diffusion Tensor Imaging. AJNR Am J Neuroradiol. 2017;38(1):31–38. doi: 10.3174/ajnr.A4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harel A, Sperling D, Petracca M, Ntranos A, Katz-Sand I, Krieger S, et al. Brain microstructural injury occurs in patients with RRMS despite 'no evidence of disease activity'. J Neurol Neurosurg Psychiatry. 2018;89(9):977–982. doi: 10.1136/jnnp-2017-317606. [DOI] [PubMed] [Google Scholar]
- 149.Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162–173. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 150.Barro C, Leocani L, Leppert D, Comi G, Kappos L, Kuhle J. Fluid biomarker and electrophysiological outcome measures for progressive MS trials. Mult Scler. 2017;23(12):1600–1613. doi: 10.1177/1352458517732844. [DOI] [PubMed] [Google Scholar]
- 151.Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577–589. doi: 10.1038/s41582-018-0058-z. [DOI] [PubMed] [Google Scholar]
- 152.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28(6):595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Disanto G, Barro C, Benkert P, Naegelin Y, Schadelin S, Giardiello A, et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann Neurol. 2017;81(6):857–870. doi: 10.1002/ana.24954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kuhle J, Barro C, Disanto G, Mathias A, Soneson C, Bonnier G, et al. Serum neurofilament light chain in early relapsing remitting MS is increased and correlates with CSF levels and with MRI measures of disease severity. Mult Scler. 2016;22(12):1550–1559. doi: 10.1177/1352458515623365. [DOI] [PubMed] [Google Scholar]
- 155.Novakova L, Zetterberg H, Sundstrom P, Axelsson M, Khademi M, Gunnarsson M, et al. Monitoring disease activity in multiple sclerosis using serum neurofilament light protein. Neurology. 2017;89(22):2230–2237. doi: 10.1212/WNL.0000000000004683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lycke JN, Karlsson JE, Andersen O, Rosengren LE. Neurofilament protein in cerebrospinal fluid: a potential marker of activity in multiple sclerosis. J Neurol Neurosurg Psychiatry. 1998;64(3):402–404. doi: 10.1136/jnnp.64.3.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Petzold A. The prognostic value of CSF neurofilaments in multiple sclerosis at 15-year follow-up. J Neurol Neurosurg Psychiatry. 2015;86(12):1388–1390. doi: 10.1136/jnnp-2014-309827. [DOI] [PubMed] [Google Scholar]
- 158.Arrambide G, Espejo C, Eixarch H, Villar LM, Alvarez-Cermeno JC, Picon C, et al. Neurofilament light chain level is a weak risk factor for the development of MS. Neurology. 2016;87(11):1076–1084. doi: 10.1212/WNL.0000000000003085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hakansson I, Tisell A, Cassel P, Blennow K, Zetterberg H, Lundberg P, et al. Neurofilament light chain in cerebrospinal fluid and prediction of disease activity in clinically isolated syndrome and relapsing-remitting multiple sclerosis. Eur J Neurol. 2017;24(5):703–712. doi: 10.1111/ene.13274. [DOI] [PubMed] [Google Scholar]
- 160.Tejeda-Velarde A, Costa-Frossard L. Sainz de la Maza S, Carrasco A, Espino M, Picon C, et al. Clinical usefulness of prognostic biomarkers in optic neuritis. Eur J Neurol. 2018;25(4):614–618. doi: 10.1111/ene.13553. [DOI] [PubMed] [Google Scholar]
- 161.Disanto G, Adiutori R, Dobson R, Martinelli V, Dalla Costa G, Runia T, et al. Serum neurofilament light chain levels are increased in patients with a clinically isolated syndrome. J Neurol Neurosurg Psychiatry. 2016;87(2):126–129. doi: 10.1136/jnnp-2014-309690. [DOI] [PubMed] [Google Scholar]
- 162.Pawlitzki M, Sweeney-Reed CM, Bittner D, Lux A, Vielhaber S, Schreiber S, et al. CSF-Progranulin and Neurofilament Light Chain Levels in Patients With Radiologically Isolated Syndrome-Sign of Inflammation. Front Neurol. 2018;9:1075. doi: 10.3389/fneur.2018.01075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mellergard J, Tisell A, Blystad I, Gronqvist A, Blennow K, Olsson B, et al. Cerebrospinal fluid levels of neurofilament and tau correlate with brain atrophy in natalizumab-treated multiple sclerosis. Eur J Neurol. 2017;24(1):112–121. doi: 10.1111/ene.13162. [DOI] [PubMed] [Google Scholar]
- 164.Barro C, Benkert P, Disanto G, Tsagkas C, Amann M, Naegelin Y, et al. Serum neurofilament as a predictor of disease worsening and brain and spinal cord atrophy in multiple sclerosis. Brain. 2018;141(8):2382–2391. doi: 10.1093/brain/awy154. [DOI] [PubMed] [Google Scholar]
- 165.Bodini B, Calabresi PA. From neurofilament research to multiple sclerosis clinical practice: Where do we stand? Neurology. 2017;88(9):816–817. doi: 10.1212/WNL.0000000000003676. [DOI] [PubMed] [Google Scholar]
- 166.Petzold A, Steenwijk MD, Eikelenboom JM, Wattjes MP, Uitdehaag BM. Elevated CSF neurofilament proteins predict brain atrophy: A 15-year follow-up study. Mult Scler. 2016;22(9):1154–1162. doi: 10.1177/1352458516645206. [DOI] [PubMed] [Google Scholar]
- 167.Canto E, Barro C, Zhao C, Caillier SJ, Michalak Z, Bove R, et al. Association Between Serum Neurofilament Light Chain Levels and Long-term Disease Course Among Patients With Multiple Sclerosis Followed up for 12 Years. JAMA Neurol. 2019. [DOI] [PMC free article] [PubMed]
- 168.Chitnis T, Gonzalez C, Healy BC, Saxena S, Rosso M, Barro C, et al. Neurofilament light chain serum levels correlate with 10-year MRI outcomes in multiple sclerosis. Ann Clin Transl Neurol. 2018;5(12):1478–1491. doi: 10.1002/acn3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jakimovski D, Kuhle J, Ramanathan M, Barro C, Tomic D, Hagemeier J, et al. Serum neurofilament light chain levels associations with gray matter pathology: a 5-year longitudinal study. Ann Clin Transl Neurol. 2019;6(9):1757–1770. doi: 10.1002/acn3.50872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chaudhuri A. Multiple sclerosis is primarily a neurodegenerative disease. J Neural Transm (Vienna). 2013;120(10):1463–1466. doi: 10.1007/s00702-013-1080-3. [DOI] [PubMed] [Google Scholar]
- 171.Akassoglou K, Bauer J, Kassiotis G, Pasparakis M, Lassmann H, Kollias G, et al. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol. 1998;153(3):801–813. doi: 10.1016/S0002-9440(10)65622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Laureys G, Clinckers R, Gerlo S, Spooren A, Wilczak N, Kooijman R, et al. Astrocytic beta (2)-adrenergic receptors: from physiology to pathology. Prog Neurobiol. 2010;91(3):189–199. doi: 10.1016/j.pneurobio.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 173.Fox RJ, Coffey CS, Conwit R, Cudkowicz ME, Gleason T, Goodman A, et al. Phase 2 Trial of Ibudilast in Progressive Multiple Sclerosis. N Engl J Med. 2018;379(9):846–855. doi: 10.1056/NEJMoa1803583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383(9936):2213–2221. doi: 10.1016/S0140-6736(13)62242-4. [DOI] [PubMed] [Google Scholar]
- 175.Sprenger T, Kappos L, Radue EW, Gaetano L, Mueller-Lenke N, Wuerfel J, et al. Association of brain volume loss and long-term disability outcomes in patients with multiple sclerosis treated with teriflunomide. Mult Scler. 2019:1352458519855722. [DOI] [PMC free article] [PubMed]
- 176.Schlaeger R, Papinutto N, Panara V, Bevan C, Lobach IV, Bucci M, et al. Spinal cord gray matter atrophy correlates with multiple sclerosis disability. Ann Neurol. 2014;76(4):568–580. doi: 10.1002/ana.24241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Petzold A, Balcer LJ, Calabresi PA, Costello F, Frohman TC, Frohman EM, et al. Retinal layer segmentation in multiple sclerosis: a systematic review and meta-analysis. Lancet Neurol. 2017;16(10):797–812. doi: 10.1016/S1474-4422(17)30278-8. [DOI] [PubMed] [Google Scholar]
- 178.Saidha S, Al-Louzi O, Ratchford JN, Bhargava P, Oh J, Newsome SD, et al. Optical coherence tomography reflects brain atrophy in multiple sclerosis: A four-year study. Ann Neurol. 2015;78(5):801–813. doi: 10.1002/ana.24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pisa M, Guerrieri S, Di Maggio G, Medaglini S, Moiola L, Martinelli V, et al. No evidence of disease activity is associated with reduced rate of axonal retinal atrophy in MS. Neurology. 2017;89(24):2469–2475. doi: 10.1212/WNL.0000000000004736. [DOI] [PubMed] [Google Scholar]
- 180.Pisa M, Ratti F, Vabanesi M, Radaelli M, Guerrieri S, Moiola L, et al. Subclinical neurodegeneration in multiple sclerosis and neuromyelitis optica spectrum disorder revealed by optical coherence tomography. Mult Scler. 2019:1352458519861603. [DOI] [PubMed]
- 181.Vural A, Okar S, Kurne A, Sayat-Gurel G, Acar NP, Karabulut E, et al. Retinal degeneration is associated with brain volume reduction and prognosis in radiologically isolated syndrome. Mult Scler. 2018:1352458518817987. [DOI] [PubMed]
- 182.Pietroboni AM, Dell'Arti L, Caprioli M, Scarioni M, Carandini T, Arighi A, et al. The loss of macular ganglion cells begins from the early stages of disease and correlates with brain atrophy in multiple sclerosis patients. Mult Scler. 2019;25(1):31–38. doi: 10.1177/1352458517740214. [DOI] [PubMed] [Google Scholar]
- 183.Maghzi AH, Graves J, Revirajan N, Spain R, Liu S, McCulloch CE, et al. Retinal axonal loss in very early stages of multiple sclerosis. Eur J Neurol. 2015;22(7):1138–1141. doi: 10.1111/ene.12722. [DOI] [PubMed] [Google Scholar]
- 184.Lazzarino G, Amorini AM, Petzold A, Gasperini C, Ruggieri S, Quartuccio ME, et al. Serum Compounds of Energy Metabolism Impairment Are Related to Disability, Disease Course and Neuroimaging in Multiple Sclerosis. Mol Neurobiol. 2017;54(9):7520–7533. doi: 10.1007/s12035-016-0257-9. [DOI] [PubMed] [Google Scholar]
- 185.Wood ET, Ercan E, Sati P, Cortese ICM, Ronen I, Reich DS. Longitudinal MR spectroscopy of neurodegeneration in multiple sclerosis with diffusion of the intra-axonal constituent N-acetylaspartate. Neuroimage Clin. 2017;15:780–788. doi: 10.1016/j.nicl.2017.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Noble S, Scheinost D, Constable RT. A decade of test-retest reliability of functional connectivity: A systematic review and meta-analysis. Neuroimage. 2019;203:116157. doi: 10.1016/j.neuroimage.2019.116157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yilmaz A, Blennow K, Hagberg L, Nilsson S, Price RW, Schouten J, et al. Neurofilament light chain protein as a marker of neuronal injury: review of its use in HIV-1 infection and reference values for HIV-negative controls. Expert Rev Mol Diagn. 2017;17(8):761–770. doi: 10.1080/14737159.2017.1341313. [DOI] [PubMed] [Google Scholar]
- 188.Mattsson N, Andreasson U, Zetterberg H, Blennow K. Alzheimer's Disease Neuroimaging I. Association of Plasma Neurofilament Light With Neurodegeneration in Patients With Alzheimer Disease. JAMA Neurol. 2017;74(5):557–566. doi: 10.1001/jamaneurol.2016.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zivadinov R, Raj B, Ramanathan M, Teter B, Durfee J, Dwyer MG, et al. Autoimmune Comorbidities Are Associated with Brain Injury in Multiple Sclerosis. AJNR Am J Neuroradiol. 2016;37(6):1010–1016. doi: 10.3174/ajnr.A4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lorefice L, Fenu G, Pitzalis R, Scalas G, Frau J, Coghe G, et al. Autoimmune comorbidities in multiple sclerosis: what is the influence on brain volumes? A case-control MRI study. J Neurol. 2018;265(5):1096–1101. doi: 10.1007/s00415-018-8811-1. [DOI] [PubMed] [Google Scholar]
- 191.Luczynski P, Laule C, Hsiung GR, Moore GRW, Tremlett H. Coexistence of Multiple Sclerosis and Alzheimer's disease: A review. Mult Scler Relat Disord. 2019;27:232–238. doi: 10.1016/j.msard.2018.10.109. [DOI] [PubMed] [Google Scholar]
- 192.White LR, Edland SD, Hemmy LS, Montine KS, Zarow C, Sonnen JA, et al. Neuropathologic comorbidity and cognitive impairment in the Nun and Honolulu-Asia Aging Studies. Neurology. 2016;86(11):1000–1008. doi: 10.1212/WNL.0000000000002480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tousignant A, Lemaître P, Precup D, Arnold DL, Arbel T. Prediction of disease progression in multiple sclerosis patients using deep learning analysis of MRI data. PMLR. 2019;102:483–492. [Google Scholar]
- 194.Filippi M, Preziosa P, Copetti M, Riccitelli G, Horsfield MA, Martinelli V, et al. Gray matter damage predicts the accumulation of disability 13 years later in MS. Neurology. 2013;81(20):1759–1767. doi: 10.1212/01.wnl.0000435551.90824.d0. [DOI] [PubMed] [Google Scholar]
- 195.Zhao Y, Healy BC, Rotstein D, Guttmann CR, Bakshi R, Weiner HL, et al. Exploration of machine learning techniques in predicting multiple sclerosis disease course. PLoS One. 2017;12(4):e0174866. doi: 10.1371/journal.pone.0174866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.