

Abstract
Many sensory and chemical signal inputs are transmitted by intracellular GTP-binding (G) proteins. G proteins make up two major subfamilies: “large” G proteins comprising three subunits and “small” G proteins, such as the proto-oncogene product RAS, which contains a single subunit. Members of both subfamilies are regulated by post-translational modifications, including lipidation, proteolysis, and carboxyl methylation. Emerging studies have shown that these proteins are also modified by ubiquitination. Much of our current understanding of this post-translational modification comes from investigations of the large G-protein α subunit from yeast (Gpa1) and the three RAS isotypes in humans, NRAS, KRAS, and HRAS. Gα undergoes both mono- and polyubiquitination, and these modifications have distinct consequences for determining the sites and mechanisms of its degradation. Genetic and biochemical reconstitution studies have revealed the enzymes and binding partners required for addition and removal of ubiquitin, as well as the delivery and destruction of both the mono- and polyubiquitinated forms of the G protein. Complementary studies of RAS have identified multiple ubiquitination sites, each having distinct consequences for binding to regulatory proteins, shuttling to and from the plasma membrane, and degradation. Here, we review what is currently known about these two well-studied examples, Gpa1 and the human RAS proteins, that have revealed additional mechanisms of signal regulation and dysregulation relevant to human physiology. We also compare and contrast the effects of G-protein ubiquitination with other post-translational modifications of these proteins.
Keywords: G protein, yeast, ubiquitin, Ras protein, E3 ubiquitin ligase, Cdc34, E3 ligase, OTUB1, Rabex-5, Ubp12
Introduction
A variety of sensory and chemical signals are detected by receptors at the cell surface. In many cases, these inputs are transmitted by intracellular GTP-binding proteins (Fig. 1). Generally speaking, large G proteins are activated by seven transmembrane segment receptors, also known as G-protein–coupled receptors (GPCRs).3 When these receptors bind to an agonist ligand, the G-protein α subunit releases GDP, binds to GTP, and dissociates from the Gβγ subunit dimer. GTP-bound Gα, Gβγ, or both go on to activate intracellular effectors that transduce the signal. Similarly, many small G proteins are activated by growth factor receptors and their associated guanine nucleotide–exchange factors (GEFs). As with GPCRs, GEFs promote the exchange of GTP for GDP and subsequent activation of downstream effectors. For both large and small G proteins, signaling persists until GTP is converted to GDP, and this inactivation step is accelerated by binding to GTPase-activating proteins (GAPs). Thus, GEFs and GAPs work in opposition to one another to regulate the activity of their cognate G proteins, both large and small. Here, we review newer mechanisms of G-protein regulation by ubiquitination, with a focus on a large G protein from yeast and three small G proteins from humans: NRAS, KRAS, and HRAS. We compare and contrast the effects of ubiquitination with that of other post-translational modifications, where such comparisons have proven to be instructive.
Figure 1.
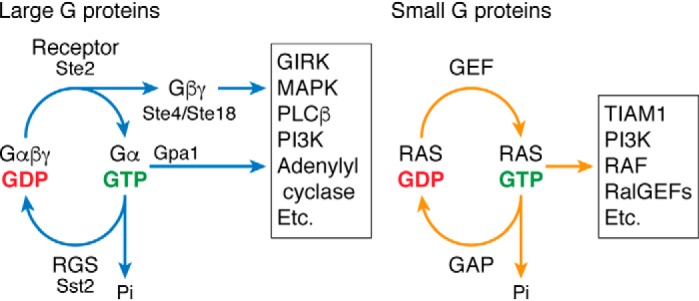
Large and small G proteins. G-protein signaling is initiated by GDP release and GTP binding, which are accelerated by an agonist-bound receptor (left) or guanine nucleotide-exchange factor (right). GTP-bound G proteins activate effector enzymes and ion channels within the cell (boxes). G-protein signaling is concluded by GTP hydrolysis to GDP and phosphate (Pi), which is accelerated by a regulator of G protein signaling (RGS, left) or GTPase-activating protein (GAP, right). Components of the large G-protein signaling complex from yeast are noted. GIRK, G protein–coupled inwardly-rectifying potassium channels; PLC, phospholipase C; PI3K, phosphatidylinositol 3-kinase; TIAM1, T-cell lymphoma invasion and metastasis-inducing protein, RAF, rapidly-accelerated fibrosarcoma.
Small (∼21 kDa) G proteins are part of a large superfamily of GTPases that contains over 170 members in humans. In addition to RAS, these include members of the RAB, RHO, ARF, and RAN subfamilies (reviewed in Ref. 1). The mammalian RAS proteins— composed of HRAS, NRAS, and two alternatively spliced forms of KRAS (KRAS4A and KRAS4B)—play key roles in regulating a host of cellular processes, including cell growth, differentiation, morphology, motility, and survival. These RAS proteins show high (∼90%) sequence identity within the core guanine–nucleotide-binding (G) domain but show substantial sequence divergence in the C-terminal hypervariable region. In addition, several post-translational modifications occur within the hypervariable region, including prenylation, proteolysis, and carboxyl methylation (reviewed in Ref. 2). These modifications facilitate membrane association needed for RAS protein function.
Prenylation is the transfer of either a farnesyl (C15) or geranyl–geranyl group (C20) to the C-terminal Cys of a target protein. Correspondingly, farnesylation is common to all three RAS proteins, and it is the first step required for membrane association. Following the prenylation step, three C-terminal amino acids are cleaved, and the new C terminus is carboxyl-methylated. HRAS, NRAS, and KRAS4A further undergo palmitoylation (2, 3), which refers to the reversible thioester linkage of palmitic acid (C16:0) to cysteine residues. Whereas lipidation is required for membrane association, sequence differences in the hypervariable region give rise to distinct and dynamic partitioning of the RAS isoforms between various endomembranes and the plasma membrane. This, in turn, contributes to isoform-specific localization and signaling (2).
Large G proteins undergo many of the post-translational modifications reported for RAS, and these processes play a particularly important role in the life cycle of yeast Saccharomyces cerevisiae. Yeast can exist as one of two haploid cell types (a and α) that secrete specific peptide pheromones (a factor and α factor), which bind to cell-surface receptors that activate a heterotrimeric G protein. This initiates events necessary for mating or the fusion of a and α cells to form an a/α diploid, including new gene transcription, morphological changes, cell-cycle arrest and cell–cell adhesion. In this instance the Gβγ subunit dimer (Ste4/18) binds to effector proteins, which include a MAPK scaffold (Ste5) and a GEF for the small G-protein Cdc42 (Cdc24) (reviewed in Ref. 4). In addition, the Gα subunit Gpa1 activates the phosphatidylinositol 3-kinase Vps34, located predominantly at endosomes (5).
Proper localization of the heterotrimeric G-protein complex depends on post-translational modification. Gpa1, like most other Gα proteins (6, 7), is myristoylated (8, 9) and palmitoylated (10, 11 and reviewed in Ref. 12). Myristoylation refers to the covalent attachment of myristic acid (C14:0) to the N-terminal glycine of proteins, a process that occurs cotranslationally and is considered irreversible. The Gβγ dimer is anchored to the plasma membrane through prenylation (introduced above) and palmitoylation of the γ subunit, Ste18 (11, 13, 14).
While past efforts have focused on protein lipidation, our focus here is on regulation by ubiquitination. Ubiquitination and lipidation share the ability to regulate protein–protein interactions and to direct modified proteins to specific compartments within the cell. Our discussion is centered on two of the best-characterized substrates for ubiquitination, RAS and Gpa1. In this context it is worth noting the important role of yeast in the identification of the enzymes that modify these G proteins. Yeast genetic screens coupled with biochemical analysis, using RAS as a test substrate, revealed the first enzymes responsible for protein palmitoylation (15–17) as well as farnesylation, proteolysis, and carboxymethylation (reviewed in Ref. 18). The first myristoyltransferase was isolated from yeast and initially characterized using Gpa1 as a substrate (19–22). The first farnesyltransferase inhibitors, used subsequently to inhibit RAS oncogenesis, were identified in yeast (18) and with Gβγ signaling as a functional readout of inhibitor activity (13). Below, we describe the identification of enzymes that ubiquitinate G proteins and the functional consequences of this modification for yeast Gpa1 and mammalian RAS.
Gα regulation by mono- and polyubiquitination: lessons from yeast
Given their role as transducers between cell-surface receptors and intracellular effectors, G proteins are well-positioned to serve as targets of regulation. Recent attention has focused on the regulation of G-protein signaling by ubiquitination (Fig. 2) (reviewed in Ref. 23). Although originally considered a tag for the irreversible destruction of protein substrates, ubiquitination is now recognized to serve as a reversible modification that affects a wide array of cellular processes, including enzyme activity, subcellular localization, and protein–protein interaction.
Figure 2.

Ubiquitination controls trafficking of a Gα in yeast. Ubiquitination is mediated by three distinct enzymes (E1, E2, and E3), the last of which defines substrate specificity and the timing of ubiquitination. A protein complex containing the E3 Rsp5 is necessary and sufficient for monoubiquitination of the G protein α-subunit Gpa1. The modified protein is internalized by a cascade of ubiquitin-binding domain proteins and is degraded in the vacuole. A protein complex containing the E3 Cdc53 and F-box adapter Cdc4 is necessary and sufficient for polyubiquitination of Gpa1, resulting in degradation by the proteasome protease complex. The ubiquitin protease Ubp12 is required for Gpa1 deubiquitination.
Ubiquitination is the process by which the ubiquitin polypeptide is covalently attached to a lysine residue in a given substrate. This three-step process is mediated by distinct enzymes (E1, E2, and E3), the last of which defines substrate specificity and the timing of ubiquitination. The E1-activating enzyme transfers ubiquitin to the E2-conjugating enzyme, which in conjunction with the E3 ligase transfers ubiquitin to lysine residues on the substrate. These are subdivided into two major families known as HECT (Homologous to E6AP C Terminus) family ligases and RING (Really Interesting New Gene) family ligases (Fig. 2). Ubiquitin can itself be ubiquitinated at any of seven internal lysine residues, resulting in various branch-chain forms of polyubiquitin. Generally speaking, polyubiquitinated substrates are recognized and degraded by the proteasome protease complex. More rarely, proteins are monoubiquitinated and subsequently transported to the lysosome (or vacuole in yeast) where they are degraded by resident proteases (reviewed in Refs. 24–26). In that regard, ubiquitination, like myristoylation and prenylation, has the ability to direct G proteins to specific compartments within the cell. Whereas myristoylation and prenylation are required for delivery to the plasma membrane, ubiquitination is required for delivery to the site of degradation. As detailed below, ubiquitination of RAS promotes degradation but also affects binding to activators, regulators, and effectors.
Although myristoylation and prenylation occur within well-defined consensus sequences, ubiquitination does not. This has created challenges to investigations of the biological role of ubiquitination. Although it is possible to systematically replace lysine residues and monitor the presence or absence of ubiquitin-modified protein, this approach may be impractical for proteins with a large number of these residues (there are 43 lysines in Gpa1). Moreover, such attempts are often futile because mutation of a ubiquitinated lysine can lead to the modification of a second nonphysiological site, possibly affecting the function of the protein (27, 28). Given that the mutagenesis approach can be misleading, an unbiased approach is needed to identify the native site of ubiquitination for each protein before it can be characterized functionally. A breakthrough in these efforts has been the implementation of MS methods to map ubiquitination sites, as first reported for Gpa1 (29). This advance required several technical refinements necessary to obtain sufficient quantities of material, particularly because the ubiquitinated species represents a very small proportion of the total protein and, once modified, is usually destined for rapid degradation.
The example of Gpa1 is illustrative of the approach, which was later used to map more than 19,000 unique sites of ubiquitination in human cells (30) and an additional 100,000 + sites currently entered in PhosphoSitePlus (www.phosphosite.org)4 (101). These efforts have revealed over 400 ubiquitination sites for all 16 subtypes of Gα in mammalian cells and >180 sites in RAS superfamily proteins. Furthermore, these advances have facilitated the development of powerful new bioinformatics tools designed to predict sites of modification (23, 31).
As originally described (29), His6-tagged Gpa1 was used because it will bind to metal affinity resins even in the presence of denaturing agents such as urea. The purified protein was analyzed by electrospray ionization tandem MS, and the resulting spectra scanned for peptides corresponding to the predicted tryptic cleavage products of ubiquitin ligated to Gpa1 (29). In particular, trypsin cleavage separates the C-terminal Gly–Gly dipeptide from the preceding Arg residue of ubiquitin. Alternatively, the di-Gly motif can be detected with specific antibodies (30).
The next objective was to determine the function(s) of ubiquitinated Gpa1. As noted above, Gpa1 is an unusual example of a protein that undergoes both mono- and polyubiquitination. Other prominent examples include the human tumor suppressor p53 (32) and the proliferating cell nuclear antigen (33). Such dually-modified proteins afford the opportunity to compare regulatory mechanisms and functional consequences for each type of ubiquitin linkage, for a given substrate. Gpa1 is particularly amenable to such analysis, given the large number of quantitative functional readouts of G-protein signaling and the sophisticated genetic tools available in yeast.
Initial efforts at functional characterization revealed that Gpa1 abundance is elevated in mutants deficient in either proteasomal (cim3) or vacuolar protease (pep4) function (34). Whereas the vacuolar protease mutants accumulate monoubiquitinated Gpa1, proteasome-defective mutants accumulate polyubiquitinated Gpa1. Whereas monoubiquitinated Gpa1 becomes concentrated within the lumen of the vacuole, polyubiquitinated Gpa1 exhibits a punctate expression pattern in the cytoplasm. In cells lacking the deubiquitinating protease Ubp12, Gpa1 accumulates in both mono- and polyubiquitinated forms and is enriched in both the cytoplasm and vacuole. Thus, mono- and polyubiquitination represent important targeting signals, both of which lead to the eventual degradation of Gpa1. The yeast Gβ (Ste4) and G-protein–coupled receptor (Ste2) are likewise monoubiquitinated (35, 36). In contrast to Gpa1, however, monoubiquitination of Ste4 does not lead to internalization and delivery to the vacuole. Rather monoubiquitination occurs in response to pheromone stimulation and promotes proper cell polarization and pheromone gradient detection (36).
After identifying the enzymes responsible for monoubiquitination of Gpa1, the next challenge was to establish the machinery used for delivery of modified protein to the vacuole. Critical to this process is recognition by ubiquitin-binding domain (UBD)-containing proteins (reviewed in Ref. 37). Numerous UBDs have been defined and are annotated in the protein descriptions found in the Saccharomyces Genome Database (www.yeastgenome.org).4 Although structurally diverse, UBDs share the ability to bind noncovalently to ubiquitin-conjugated substrates and to transport monoubiquitinated proteins through the various stages of endocytosis to their final destination (reviewed in Ref. 38). With 39 known UBD-containing proteins as a starting point, gene deletion mutants were systematically screened for any with defects in the vacuolar delivery of GFP-tagged Gpa1 (39). Because Gpa1 and Ste2 are targeted for internalization by the same modification, the two proteins were tracked side–by–side. This analysis revealed seven ubiquitin-binding domain–containing proteins required for constitutive internalization of Gpa1. Of these, four were required for Gpa1 but not Ste2, thereby demonstrating that endocytosis of the G protein and receptor are distinct processes regulated by distinct binding partners. The question remained: were mono- and polyubiquitinations mediated by the same or distinct ubiquitin ligases?
Given that the receptor Ste2 and Gα protein Gpa1 are both monoubiquitinated and, given that both are eventually delivered to the vacuole, it seemed likely that both proteins were modified by the same ligase. To the best of our knowledge, a single enzyme (Rsp5) is responsible for most, if not all, monoubiquitination in yeast. Indeed, early genetic analysis had shown that monoubiquitination of Ste2 requires Rsp5 (NEDD4 in humans) (40), and it was subsequently shown that monoubiquitination of Gpa1 likewise requires Rsp5 (41). To determine whether Rsp5 is sufficient as well as necessary for Gpa1 monoubiquitination, the proteins were purified and shown to reconstitute the monoubiquitination reaction in vitro (41).
Notably, although monoubiquitination was diminished in the absence of Rsp5, there was a concomitant increase in Gpa1 polyubiquitination as well as a marked redistribution of protein from the vacuolar compartment to puncta within the cytoplasm, possibly representing aggregates or association with the proteasome protease complex (41). Thus, although removal of Rsp5 could prevent monoubiquitination, it was evident that a second enzyme was responsible for Gpa1 polyubiquitination. The identity of that second enzyme was revealed as part of a separate effort to identify new regulators of the mating pathway, through a systematic analysis of yeast essential genes (42). In that endeavor, nearly every essential gene was placed under the control of a tetracycline-regulatable promoter (TetO7 promoter), which allows for precise control of gene expression with no change in protein sequence or function. A total of 870 TetO7 strains were transformed with a pathway-specific transcription reporter and exposed to a range of pheromone concentrations. Of 92 genes required for proper signaling, a disproportionate fraction was involved in protein degradation. These included gene products that either form (Cdc53 and Cdc34) or function with (Cdc4) the SCF (Skp1, Cullin, and F-box protein) ubiquitin ligase (43, 44). Knockdown of CDC4, CDC34, or CDC53 genes all resulted in diminished activation of the effector MAPK as well as the transcription response. The functional similarity of the mutants, and the fact that the proteins exist as a complex in cells, suggested that SCF/Cdc4 had a particularly important role in G-protein signal regulation. Because SCF is a negative regulator of protein stability, the reduction in signal output indicated that the likely target itself had an inhibitory function in signaling, most likely Gpa1. In support of the hypothesis, reconstitution of purified SCF and Cdc4 was sufficient for the ubiquitination of Gpa1. Conversely, ubiquitination was abrogated by knockdown of SCF or by removing the ubiquitination site within Gpa1 (42). Together, these results indicate that SCF is necessary and sufficient to polyubiquitinate Gpa1. Loss of SCF stabilizes the Gα protein and limits signaling, presumably by sequestering free Gβγ.
In a follow-up analysis, it was shown that while the F-box protein Cdc4 selectively targets Gpa1, other F-box proteins target downstream components of the pheromone-signaling pathway as follows: Dia2 selectively limits pheromone-induced vacuolar targeting; Ucc1 selectively limits MAPK induction; and Pfu1 is needed for proper mating morphogenesis and the disassembly of unproductive mating projections (45).
To summarize, our current understanding is that mono- and polyubiquitinations are catalyzed by different ubiquitin ligases with different functional consequences (Fig. 2). Whereas monoubiquitinated Gpa1 is targeted to the vacuole, polyubiquitinated Gpa1 is directed to the proteasome. There may be some important benefits to having two ubiquitinating pathways that can target the same protein. For example, degradation could be triggered in response to different signals and/or functional states of the protein. In support of this model, SCF (but not Rsp5) is thought to recognize only substrates that are phosphorylated (reviewed in Ref. 46). A subsequent search, done through a comprehensive screen of 109 kinase deletion mutants, revealed that Elm1 is sufficient and necessary to phosphorylate Gpa1, at least under nutrient-rich conditions (47). Elm1 is expressed predominantly during the G2–M phase of the cell cycle, and this expression pattern was reflected in a cell cycle–dependent pattern of Gpa1 phosphorylation and polyubiquitination. Elm1 was already known to be one of three protein kinases that phosphorylate and activate the ADP-activated protein kinase Snf1 under low glucose conditions, where ADP accumulates (48, 49). Phosphorylation of Snf1 is reversed by the protein phosphatase composed of Reg1 and Glc7 (50, 51). Gpa1 is likewise phosphorylated under low-glucose conditions and is regulated by the same kinases and phosphatase as those that act on Snf1. When all three kinases are deleted, pheromone signaling is amplified. Deletion of the phosphatase has the opposite effect (52).
The opposing functional effects of the kinase and SCF mutants are somewhat paradoxical given that phosphorylation is needed for ubiquitination by SCF. We speculate that phosphorylation has additional functional effects on Gpa1 activity apart from its role in directing ubiquitination and degradation. Regardless, current evidence indicates that cell-cycle progression and the glucose-sensing pathway act to directly modulate the pheromone-signaling pathway and do so through the combined effects of phosphorylation and polyubiquitination of the G protein. In this context it is important to consider the important role of G proteins and GPCRs in regulating cellular growth and metabolism. Classical studies, performed over the past half-century, revealed that G proteins and second messengers—most notably cAMP—lead to the mobilization of glucose from glycogen stores (53). More recent studies have implicated G proteins and GPCRs in tumorigenesis (reviewed in Refs. 54, 55). The results summarized above demonstrate how cross-pathway regulation can also occur in the opposite direction, wherein glucose availability and cell-cycle progression regulate G-protein signaling.
Ubiquitination regulates RAS localization, activity, and stability
RAS is the founding member of an expansive superfamily of small GTPases. Like Gα, RAS functions as a molecular switch, cycling between active GTP- and inactive GDP-bound states to regulate signaling pathways important for cellular growth control. The four mammalian RAS proteins (HRAS, NRAS, and KRAS 4A and 4B) contain a highly-conserved core guanine nucleotide–binding (G)-domain and a hypervariable C-terminal region. The core G-domain dictates high affinity and specificity for guanine nucleotides (GDP and GTP) as well as protein-binding partners. The hypervariable region specifies post-translational modifications that facilitate membrane association. Both of these regions contain sites of ubiquitination, as detailed below.
There are many parallels in the activation cycle for large and small G proteins. In an unstimulated cell, RAS is populated in the “inactive” GDP-bound conformation, despite the high ratio of intracellular GTP/GDP. Exchange factors bind and up-regulate RAS activity by promoting association with GTP, whereas RAS GAPs accelerate the intrinsic rate of GTP hydrolysis to down-regulate RAS activity. Binding of either GDP or GTP promotes distinct conformational changes in two key regions, termed switch I and II. The active GTP-bound conformation is recognized by downstream targets and culminates in downstream signaling. The best-characterized RAS effectors are RAF kinases, PI3K, and Ral exchange factors (RalGEFs). All three effectors contain a conserved RAS-binding domain, have well-characterized roles in RAS-mediated tumorigenesis, and have been extensively targeted for therapeutic drug treatment (reviewed in Ref. 56).
RAS mutations are commonly found in cancer and developmental disorders (e.g. RASopathies). Notably, RAS is the most frequently mutated oncogene, with ∼30% of human tumors containing an activating RAS mutation. Of the three isoforms, KRAS is the most highly mutated, particularly in pancreatic (∼90%), colon (∼50%), and lung (∼30%) cancers. These mutations chronically activate RAS and promote dysregulated cell proliferation. Point mutations in RAS can lead to chronic cellular activation in two ways, by increasing the rate of GDP dissociation or by inhibiting the rate of GTP hydrolysis; either of these mutational effects populates RAS in its biologically-active GTP-bound form. The subset of RAS mutations that accelerate guanine nucleotide exchange are located in two motifs (SAK and NKXD), which form key interactions with the guanine nucleotide. Mutations within these motifs perturb nucleotide binding and promote GTP binding, given the high GTP/GDP ratio in cells. However, most oncogenic KRAS mutations are found at glycine 12 and glycine 13 and less frequently glutamine 61. These mutations populate the active GTP-bound form of RAS by interfering with GTP hydrolysis (reviewed in Ref. 57).
In addition to mutations that permanently activate RAS, there is growing interest in protein modifications that suppress or mimic the effects of such mutations. Understanding the molecular basis of these modifications, and how they might be regulated pharmacologically, would complement wide-scale efforts to generate agents that antagonize aberrant RAS function. For example, early efforts to develop anti-RAS therapies relied on developing inhibitors of RAS farnesylation, but these drugs proved ineffective against KRAS as an alternative lipid modification, geranylgeranylation, can occur instead (58 and reviewed in Refs. 59–62). More recently, compounds have been developed that chemically modify a cysteine unique to the oncogenic KRAS G12C variant, which is particularly prevalent in lung cancer, resulting in inhibition of KRAS activity (63–67 and reviewed in Ref. 68). These covalent inhibitors are now in phase I clinical trials (Clinical Trial numbers: NCT03600883 and NCT03785249). Although these inhibitors represent an exciting breakthrough, there is a need to develop inhibitors that recognize other oncogenic RAS mutants. Understanding novel mechanisms of RAS regulation may prove helpful in identifying new therapeutic approaches for targeting RAS-driven tumors and developmental disorders.
RAS proteins are also substrates for ubiquitination. This modification targets multiple lysine residues and has been shown to alter RAS function in at least three distinct ways: (i) altering its subcellular localization; (ii) altering protein–protein interactions; and (iii) promoting its degradation (Fig. 3). Below, we consider these three mechanisms in turn. For each, we highlight important differences in the ubiquitin machinery responsible, the specific residues that are modified, and the RAS isoforms affected.
Figure 3.

Ubiquitination controls multiple interactions of RAS. A, RAS activity is regulated by ubiquitination. Primary sites of monoubiquitination occur at residues 147 in KRAS, whereas in HRAS it is at 117. Monoubiquitination of KRAS at 147 up-regulates RAS activity through a GAP defect leading to enhanced MAPK activation. In contrast, monoubiquitination of HRAS at 117 induces fast exchange and activates RAS in a GEF-independent manner. B, RAS localization is regulated by ubiquitination. Rabex-5 promotes mono- and diubiquitination of HRAS and NRAS resulting in endosome localization and reduced MAPK signaling. The deubiquitinase, OTUB1, removes ubiquitin from RAS and promotes plasma membrane localization and MAPK signaling. C, ubiquitination by LZTR1, β-TrCP1, and SMURF2 promotes RAS degradation through proteasome and autolysosomes resulting in reduced MAPK signaling.
The earliest studies examining the effects of ubiquitination on RAS were conducted in mammalian and Drosophila cells. In both studies, Rabex-5 was identified as a key regulator of RAS function (69, 70). Rabex-5 is a multidomain protein that contains an exchange factor domain that promotes activation of RAB5 GTPases during endocytosis. It also contains a ubiquitin-binding and E3 ubiquitin ligase domain that catalyzes mono- and di-ubiquitination of H- and NRAS but not KRAS. Similar to Gpa1, ubiquitination of either H- or KRAS protein results in their endosomal membrane localization and a reduction in MAPK activation. An HRAS variant with several lysine residues mutated was more efficient at recruiting RAF1 and activating the MAPK cascade. Although these observations provide a link between RAS ubiquitination, membrane localization, and down-regulation of RAS activity, several questions remain. First, what are the sites of ubiquitination that drive endosomal localization? The membrane-anchoring hypervariable domain of HRAS was postulated to contain modification sites; however, the exact sites of ubiquitination were not identified. Indeed, the mutant also contained several lysine substitutions (including the strictly conserved Lys-117) in the G-domain that could affect localization and activity independent of ubiquitination. It is also unclear whether mono- versus diubiquitination have distinct functional effects on H- and NRAS and whether signaling pathways other than the MAPK (e.g. PI3K and RALGEF) are modulated by endosome localization. Another question lies in the isoform specificity associated with Rabex-5–mediated ubiquitination. Is the ability of Rabex-5 to selectively ubiquitinate H- and NRAS but not KRAS in CHO-K cells due to specific colocalization of Rabex-5 complexes with these RAS isoforms or is it through another mechanism? Future studies directed at identifying the key sites and sites of modification, for example by MS, may aid in clarifying the role of ubiquitination in RAS endosome localization/retention and signaling.
While these pioneering studies were the first to demonstrate that RAS signaling is modulated by mono- and diubiquitination, other factors that may link these two processes have been identified. More recently, a deubiquitinase, OTUB1, was identified that facilitates removal of mono- and diubiquitin from RAS proteins, and it appears to promote retention of RAS proteins at the plasma membrane in a nucleotide-independent manner (71). Consistent with findings that OTUB1 may up-regulate RAS function, overexpression of OTUB1 promotes MAPK signaling and is commonly observed in nonsmall cell lung carcinomas that contain WT KRAS (71).
Although the studies described above reveal a role of ubiquitination in RAS localization and suppression of RAS function, it was recently shown that monoubiquitination of KRAS alters protein–protein interactions and up-regulates KRAS activity. In one landmark study, MS was used to identify sites of ubiquitination of K- and HRAS isolated from HEK293T cells, with lysine 147 identified as the most frequent site of KRAS monoubiquitination. Compared with unmodified RAS, the ubiquitinated subpopulation was predominantly in the activated GTP-bound state and showed increased association with the downstream effectors RAF, PI3K, and RalGEF. Consistent with these observations, mutation of KRAS to prevent ubiquitination at lysine 147 impaired tumor growth in a mouse xenograft model system (72). As lysine 147 is part of the conserved SAK motif important for coordinating the guanine nucleotide base of GTP or GDP, it was initially hypothesized that monoubiquitination up-regulates KRAS activity by promoting guanine nucleotide exchange. However, our subsequent studies, detailed below, showed that monoubiquitination at lysine 147 does not significantly alter nucleotide exchange, but rather impairs GAP-mediated down-regulation in vitro (73). These findings are significant because they indicate that ubiquitination can activate RAS-mediated signaling and tumorigenesis. The activation mechanism is similar in nature to GAP-defective oncogenic RAS mutations. Thus, monoubiquitination of KRAS at lysine 147 represents a third means of stimulating RAS signaling.
To investigate the mechanism of activation, large quantities of fully-ubiquitinated substrate are often required, which is difficult to obtain by enzymatic reactions. Accordingly, KRAS was monoubiquitinated at position 147 using a chemical biology approach (73). In our approach, we replaced the native ubiquitin linkage with a disulfide bond between a substituted cysteine at position 147 of RAS and another cysteine at the C terminus of ubiquitin (G77C). A surface-accessible cysteine (Cys-118) in RAS was replaced with serine to avoid unwanted modification. Ubiquitin modification of RAS at position 147 was driven to completion by the addition of a 10-fold excess of ubiquitin (G77C) at pH 8.0. In contrast to previously described methods (74–79), our chemical ligation method does not require complicated intermediate chemical or enzymatic steps but instead provides a simple, specific approach to ubiquitination. The disulfide ligation strategy was validated in previous studies, using a more complicated cysteamine intermediate in proliferating cell nuclear antigen (PCNA), where it was shown that chemically and enzymatically monoubiquitinated PCNAs exhibit identical catalytic properties (75).
Using this disulfide ligation strategy, we showed that KRAS147 monoubiquitination impedes GAP-mediated GTP hydrolysis and effector interactions in vitro and in cell lysates (73). This activation mechanism appears site-specific, as monoubiquitination at three other sites did not alter GAP regulation. Intriguingly, ubiquitin does not make specific interactions with RAS, but rather it dynamically samples conformational space so as to occlude a subset of RAS conformers, thus generating a distinct and more compact RAS conformational ensemble that impairs recognition by GAP proteins. Consistent with these observations, GAP activity depended on the size of the modification and linker length (80). These findings have broader impact beyond RAS regulation, as larger protein modifications like sumoylation could modulate a variety of systems via a similar mechanism.
Whereas lysine 147 was the predominant site of KRAS monoubiquitination identified in HEK293 cells, lysine 117 is the major ubiquitination site in HRAS (81). Notably, this lysine 117 is strictly conserved in the RAS superfamily of GTPases, and it makes key contacts with the bound nucleotide. Mutations at this residue cause fast exchange, enhanced GTP loading, and RAS activation. Consistent with these observations, ubiquitin modification at lysine 117 in HRAS promotes RAS activation due to faster nucleotide cycling, rather than by disrupting GAP interactions (81). Although the side chain makes contacts with the bound nucleotide, it is possible that ubiquitination occurs when RAS is in complex with factors that expose this site, such as GEFs that stabilize the nucleotide-free state of RAS. Another residue that is a minor site of KRAS ubiquitination is lysine 104 (72). However, unlike lysines 117 and 147, ubiquitin modification of lysine 104 does not appear to modulate nucleotide cycling, and thus it may primarily serve as a recognition site for ubiquitin-binding proteins. Notably, lysines 104 and 147 in KRAS have also been shown to undergo other post-translational modifications, e.g. acetylation (82–84). Although the role of acetylation in RAS function is controversial, these observations suggest that other lysine post-translational modifications may compete with ubiquitination. Moreover, the ubiquitin machinery may be cell-specific, as N- and HRAS mono- and diubiquitination was observed in CHO-K cells, yet KRAS ubiquitination was not. This is perhaps not surprising, as H-, N-, and KRAS isoforms undergo distinct spatial/temporal localization, which likely contributes to different mechanisms of ubiquitin regulation.
The studies described above support a role of mono- and diubiquitination in the regulation of RAS localization and protein–protein interactions. In addition, and more classically, polyubiquitination can alter RAS function through degradation. Proteasomal degradation of HRAS has been reported to occur in response to activation by the Wnt/β-catenin signaling pathway (85–87). This polyubiquitin-dependent degradation occurs upon recruitment of β-TrCP–E3 ligase after HRAS phosphorylation by glycogen synthase kinase 3β. Conversely, inhibition of this pathway by aberrant Wnt/β-catenin signaling enhances RAS expression levels and RAS-induced colorectal tumorigenesis. In an effort to develop anticancer drugs targeting RAS, small molecules were identified that induce the degradation of RAS and β-catenin through the Wnt/β-catenin pathway (86, 88). These compounds act by enhancing formation of the β-catenin destruction complex and subsequent recruitment of β-TrCP E3 ligase, leading to polyubiquitination-dependent proteasomal degradation of RAS. Subsequent studies identified more specific compounds, which degrade RAS without affecting the β-catenin levels. Results from these studies indicate that down-regulation of RAS signaling is mediated through degradation of RAS (89, 90). In a separate study, the Smad ubiquitination regulatory factor 2 (SMURF2) was shown to monoubiquitinate its E2 ubiquitin–conjugating enzyme (UBCH5) to form an active E3/E2 complex. This complex polyubiquitinates and degrades β-TrCP1 E3 ligase that modulates KRAS protein stability. Of note, oncogenic KRAS mutant levels appear more sensitive than WT KRAS to SMURF2-mediated protein stability regulation (91). Degradation appears dependent on lysosomal proteolysis, as a lysosome inhibitor protected KRAS from SMURF2, whereas proteasomal inhibition was ineffective. Moreover, degradation by this route is enhanced by treatment with the estrogen antagonist 4-hydroxytamoxifen (91). These findings are consistent with previous observations that KRAS can undergo lysosomal degradation (92). Thus, both proteasome- and lysosome-mediated degradation mechanisms appear to modulate KRAS levels and do so in a stimulus-dependent manner.
More recently, a new player that regulates RAS protein levels was identified. LZTR1, or leucine zipper-like transcription regulator 1, associates with the cullin 3 E3 ubiquitin ligase and facilitates ubiquitination of all RAS isoforms. Recent work by three groups showed that LZTR1 is mutated in human cancers and developmental diseases and attenuates RAS signaling (93–95). Correspondingly, reduced expression or loss of LZTR1, in mammalian cells or Drosophila, results in constitutive RAS signaling. Consistent with these observations, LZTR1 haploinsufficiency in mice result in a phenotype resembling Noonan syndrome, a development disorder that arises from enhanced RAS and MAPK signaling. Moreover, a loss of LZTR1 in Schwann cells promotes differentiation and proliferation. These disease-associated LZTR1 mutations disrupt either formation of the LZTR1/CUL3 complex or its interaction with RAS proteins (94).
Although ubiquitination by LZTR1 was initially proposed to modulate RAS function by a nondegradative mechanism (94), a more recent study showed LZTR1-dependent proteolysis of RAS was prevented by treatment with a proteasome inhibitor, suggesting that LZTR1 facilitates polyubiquitination and degradation of RAS via the ubiquitin–proteasome pathway (95). Although LZTR1 drives multiple sites of RAS ubiquitination, lysine 170 in the hypervariable region of HRAS appears to be key, as loss of ubiquitination at this site leads to dissociation of RAS from the plasma membrane (94). Conversely, the hypervariable region and farnesylation of the C-terminal cysteine 186 of KRAS4A is required for LZTR1-mediated ubiquitination (94). These findings, taken together, suggest a mechanism whereby LZTR1-mediated RAS ubiquitination down-regulates RAS function. Although these studies identify a new regulatory mechanism, several questions remain unresolved. Is LZTR1-mediated ubiquitination dependent on the activation state of RAS? As several lysines undergo ubiquitination, are there sites other than lysine 170 that contribute to down-regulation of RAS signaling?
The advances noted above have created new opportunities for potential therapies. In particular, efforts to promote association with ubiquitinating (E2 and E3) enzymes may provide a unique strategy to specifically destroy persistently activated KRAS mutants, and thus ablate RAS-driven tumorigenesis. In support of this concept, an engineered E3 ubiquitin ligase was generated to target the KRAS oncoprotein for ubiquitination and proteasomal degradation (96). In this study, a fusion was generated between a U-box–based chimeric E3 ligase, a RAS-binding domain, and a membrane localization domain (cysteine-rich domain (CRD)), to target activated KRAS for ubiquitination and degradation. Expression of this fusion in pancreatic cancer cells resulted in reduced KRAS expression, MAPK signaling, and pancreatic cancer cell growth in vitro and in vivo. These same investigators have also developed a method to target proteins for degradation without ubiquitin-driven degradation, one that relies on fusion of the substrate to ornithine decarboxylase, an enzyme that plays a key role in polyamine biosynthesis. Interaction with a natural cellular inhibitor of ornithine decarboxylase (antizyme) exposes the proteasome-binding site and promotes proteolytic degradation. A fusion that links ornithine decarboxylase to a domain in RAF-1, which recognizes activated RAS, leads to diminished expression and RAS-mediated growth in pancreatic cancer cells lines that have been cotransfected with the antizyme (97).
Finally, an emerging and particularly powerful strategy is to use bivalent ligands known as PROTACS (PROteolysis TArgeting Chimeras) for inducing target protein degradation. These bifunctional small molecules combine a small molecule that binds to the target (RAS in this case), a linker, and a small molecule that binds and recruits an E3 ligase, thus enabling selective target ubiquitination and subsequent protein degradation. This novel technology has led to the first “degrading” drug that is targeted to the androgen receptor for prostate cancer treatment, and it is now in phase 1 clinical trials (reviewed in Refs. 98, 99).
Unlike kinases that bind weakly to nucleotides, RAS binds GDP and GTP with picomolar affinity. Hence, it is very difficult to generate inhibitors that target the nucleotide binding site with selectivity and affinity in this range. As RAS proteins lack other druggable pockets, it has been challenging to identify high-affinity compounds that specifically recognize activated RAS or an oncogenic mutant. PROTACs that target RAS for degradation may be a particularly attractive anti-cancer strategy. However, this approach not only requires identification of bivalent ligands that engage both RAS and a specific E3 ligase, but also the formation of a complex between the E3 ligase and RAS in order to catalyze addition of ubiquitin molecules onto specific lysine residues. Moreover, the polyubiquitinated substrate must be recognized by the proteasome machinery. For RAS, optimization may also require generation of PROTACS with appropriate cellular distribution and activity in a cancer cell-type–dependent manner. There are now several reports of small molecules that bind to RAS proteins with high affinity, which should open up new avenues for design of PROTACs (reviewed in Ref. 100).
Concluding remarks
Our hope is that this review will be instructive to a broad group of readers, including those who may be knowledgeable about G proteins but not about their ubiquitination, as well as readers who know about ubiquitination but not about G proteins. The advances summarized here reveal how (i) a single protein (yeast Gpa1) is targeted for two different forms of ubiquitination, each having profoundly distinct consequences for targeting and degradation of the substrate (Fig. 2), and (ii) how a single protein (mammalian RAS) is targeted at different sites, each having unique consequences for protein–protein interactions and pathway activation (Fig. 3). A major technological breakthrough was the ability to map sites of ubiquitination directly, through MS. Perhaps the biggest conceptual advance is the realization that RAS proteins can be activated by ubiquitination as they are by growth factor stimulation and oncogenic mutations.
Gpa1 and RAS proteins share similar structures, mechanism of activation, and cellular effectors (Fig. 1). Moreover, investigations of Gpa1 in yeast led to similar studies of other ubiquitinated proteins, including RAS. Indeed, studies in yeast revealed many of the proteins and processes required for RAS activation, post-translational modifications, membrane anchoring, and inactivation.
If history is any guide, the application of the yeast model will continue to inform advances in cancer biology. Conversely, the complexity of mammalian cancer biology will continue to inspire new questions that may be addressed more easily in yeast. Given the ubiquity of G proteins as signal transducers and of ubiquitination as a mechanism of cell regulation, the extension of genetic, biochemical, and chemical biology approaches employed for the studies described here should prove useful in the study of other important targets of ubiquitination.
Acknowledgments
We express our appreciation to Professors Adrienne Cox (University of North Carolina), Matthew Torres (Georgia Tech), and Atsuo Sasaki (University of Cincinnati) for their insightful comments and suggestions. We acknowledge Brenda Temple for database search on G protein ubiquitination sites.
This work was supported by National Institutes of Health Grants R35 GM118105 (to H. G. D.) and R01 GM114130 and P01CA203657 (to S. L. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- GPCR
- G-protein–coupled receptor
- GAP
- GTPase-activating protein
- GEF
- guanine nucleotide-exchange factor
- PROTACS
- PROteolysis TArgeting Chimeras
- MAPK
- mitogen-activated protein kinase
- UBD
- ubiquitin-binding domain
- SCF
- Skp1, Cullin, and F-box protein
- PCNA
- proliferating cell nuclear antigen.
References
- 1. Wennerberg K., Rossman K. L., and Der C. J. (2005) The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 10.1242/jcs.01660 [DOI] [PubMed] [Google Scholar]
- 2. Ahearn I., Zhou M., and Philips M. R. (2018) Posttranslational modifications of RAS proteins. Cold Spring Harb. Perspect. Med. 8, a031484 10.1101/cshperspect.a031484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hancock J. F., Magee A. I., Childs J. E., and Marshall C. J. (1989) All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 10.1016/0092-8674(89)90054-8 [DOI] [PubMed] [Google Scholar]
- 4. Alvaro C. G., and Thorner J. (2016) Heterotrimeric G protein-coupled receptor signaling in yeast mating pheromone response. J. Biol. Chem. 291, 7788–7795 10.1074/jbc.R116.714980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Slessareva J. E., Routt S. M., Temple B., Bankaitis V. A., and Dohlman H. G. (2006) Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein α subunit at the endosome. Cell 126, 191–203 10.1016/j.cell.2006.04.045 [DOI] [PubMed] [Google Scholar]
- 6. Wedegaertner P. B., and Bourne H. R. (1994) Activation and depalmitoylation of Gsα. Cell 77, 1063–1070 10.1016/0092-8674(94)90445-6 [DOI] [PubMed] [Google Scholar]
- 7. Degtyarev M. Y., Spiegel A. M., and Jones T. L. (1994) Palmitoylation of a G protein αi subunit requires membrane localization not myristoylation. J. Biol. Chem. 269, 30898–30903 [PubMed] [Google Scholar]
- 8. Stone D. E., Cole G. M., de Barros Lopes M., Goebl M., and Reed S. I. (1991) N-Myristoylation is required for function of the pheromone-responsive Gα protein of yeast: conditional activation of the pheromone response by a temperature-sensitive N-myristoyl transferase. Genes Dev. 5, 1969–1981 10.1101/gad.5.11.1969 [DOI] [PubMed] [Google Scholar]
- 9. Song J., Hirschman J., Gunn K., and Dohlman H. G. (1996) Regulation of membrane and subunit interactions by N-myristoylation of a G protein a subunit in yeast. J. Biol. Chem. 271, 20273–20283 10.1074/jbc.271.34.20273 [DOI] [PubMed] [Google Scholar]
- 10. Song J., and Dohlman H. G. (1996) Partial constitutive activation of pheromone responses by a palmitoylation-site mutant of a G protein α subunit in yeast. Biochemistry 35, 14806–14817 10.1021/bi961846b [DOI] [PubMed] [Google Scholar]
- 11. Manahan C. L., Patnana M., Blumer K. J., and Linder M. E. (2000) Dual lipid modification motifs in G(α) and G(γ) subunits are required for full activity of the pheromone response pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 11, 957–968 10.1091/mbc.11.3.957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Escribá P. V., Wedegaertner P. B., Goñi F. M., and Vögler O. (2007) Lipid–protein interactions in GPCR-associated signaling. Biochim. Biophys. Acta 1768, 836–852 10.1016/j.bbamem.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 13. Finegold A. A., Schafer W. R., Rine J., Whiteway M., and Tamanoi F. (1990) Common modifications of trimeric G proteins and ras protein: involvement of polyisoprenylation. Science 249, 165–169 10.1126/science.1695391 [DOI] [PubMed] [Google Scholar]
- 14. Hirschman J. E., and Jenness D. D. (1999) Dual lipid modification of the yeast Gγ subunit Ste18p determines membrane localization of Gβγ. Mol. Cell. Biol. 19, 7705–7711 10.1128/MCB.19.11.7705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhattacharya S., Chen L., Broach J. R., and Powers S. (1995) Ras membrane targeting is essential for glucose signaling but not for viability in yeast. Proc. Natl. Acad. Sci. U.S.A. 92, 2984–2988 10.1073/pnas.92.7.2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bartels D. J., Mitchell D. A., Dong X., and Deschenes R. J. (1999) Erf2, a novel gene product that affects the localization and palmitoylation of Ras2 in Saccharomyces cerevisiae. Mol. Cell. Biol. 19, 6775–6787 10.1128/MCB.19.10.6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lobo S., Greentree W. K., Linder M. E., and Deschenes R. J. (2002) Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J. Biol. Chem. 277, 41268–41273 10.1074/jbc.M206573200 [DOI] [PubMed] [Google Scholar]
- 18. Omer C. A., and Gibbs J. B. (1994) Protein prenylation in eukaryotic microorganisms: genetics, biology and biochemistry. Mol. Microbiol. 11, 219–225 10.1111/j.1365-2958.1994.tb00302.x [DOI] [PubMed] [Google Scholar]
- 19. Towler D. A., Adams S. P., Eubanks S. R., Towery D. S., Jackson-Machelski E., Glaser L., and Gordon J. I. (1987) Purification and characterization of yeast myristoyl CoA:protein N-myristoyltransferase. Proc. Natl. Acad. Sci. U.S.A. 84, 2708–2712; Correction (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 7523 10.1073/pnas.84.9.2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Towler D. A., Eubanks S. R., Towery D. S., Adams S. P., and Glaser L. (1987) Amino-terminal processing of proteins by N-myristoylation. Substrate specificity of N-myristoyl transferase. J. Biol. Chem. 262, 1030–1036 [PubMed] [Google Scholar]
- 21. Duronio R. J., Towler D. A., Heuckeroth R. O., and Gordon J. I. (1989) Disruption of the yeast N-myristoyl transferase gene causes recessive lethality. Science 243, 796–800 10.1126/science.2644694 [DOI] [PubMed] [Google Scholar]
- 22. Johnson D. R., Duronio R. J., Langner C. A., Rudnick D. A., and Gordon J. I. (1993) Genetic and biochemical studies of a mutant Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase, nmt72pLeu99 → Pro, that produces temperature-sensitive myristic acid auxotrophy. J. Biol. Chem. 268, 483–494 [PubMed] [Google Scholar]
- 23. Torres M. (2016) Heterotrimeric G protein ubiquitination as a regulator of G protein signaling. Prog. Mol. Biol. Transl. Sci. 141, 57–83 10.1016/bs.pmbts.2016.03.001 [DOI] [PubMed] [Google Scholar]
- 24. Varshavsky A. (2017) The ubiquitin system, autophagy, and regulated protein degradation. Annu. Rev. Biochem. 86, 123–128 10.1146/annurev-biochem-061516-044859 [DOI] [PubMed] [Google Scholar]
- 25. Zheng N., and Shabek N. (2017) Ubiquitin ligases: structure, function, and regulation. Annu. Rev. Biochem. 86, 129–157 10.1146/annurev-biochem-060815-014922 [DOI] [PubMed] [Google Scholar]
- 26. Dikic I. (2017) Proteasomal and autophagic degradation systems. Annu. Rev. Biochem. 86, 193–224 10.1146/annurev-biochem-061516-044908 [DOI] [PubMed] [Google Scholar]
- 27. Bachmair A., and Varshavsky A. (1989) The degradation signal in a short-lived protein. Cell 56, 1019–1032 10.1016/0092-8674(89)90635-1 [DOI] [PubMed] [Google Scholar]
- 28. Hochstrasser M., Ellison M. J., Chau V., and Varshavsky A. (1991) The short-lived MATα2 transcriptional regulator is ubiquitinated in vivo. Proc. Natl. Acad. Sci. U.S.A. 88, 4606–4610 10.1073/pnas.88.11.4606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Marotti L. A. Jr, Newitt R., Wang Y., Aebersold R., and Dohlman H. G. (2002) Direct identification of a G protein ubiquitination site by mass spectrometry. Biochemistry 41, 5067–5074 10.1021/bi015940q [DOI] [PubMed] [Google Scholar]
- 30. Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., and Gygi S. P. (2011) Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 10.1016/j.molcel.2011.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dewhurst H. M., Choudhury S., and Torres M. P. (2015) Structural analysis of PTM hotspots (SAPH-ire)–a quantitative informatics method enabling the discovery of novel regulatory elements in protein families. Mol. Cell. Proteomics 14, 2285–2297 10.1074/mcp.M115.051177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li M., Brooks C. L., Wu-Baer F., Chen D., Baer R., and Gu W. (2003) Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302, 1972–1975 10.1126/science.1091362 [DOI] [PubMed] [Google Scholar]
- 33. Hoege C., Pfander B., Moldovan G. L., Pyrowolakis G., and Jentsch S. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 10.1038/nature00991 [DOI] [PubMed] [Google Scholar]
- 34. Wang Y., Marotti L. A. Jr, Lee M. J., and Dohlman H. G. (2005) Differential regulation of G protein α subunit trafficking by mono- and polyubiquitination. J. Biol. Chem. 280, 284–291 10.1074/jbc.M411624200 [DOI] [PubMed] [Google Scholar]
- 35. Hicke L., and Riezman H. (1996) Ubiquitination of a yeast plasma membrane receptor signals its ligand- stimulated endocytosis. Cell 84, 277–287 10.1016/S0092-8674(00)80982-4 [DOI] [PubMed] [Google Scholar]
- 36. Zhu M., Torres M. P., Kelley J. B., Dohlman H. G., and Wang Y. (2011) Pheromone- and Rsp5-dependent ubiquitination of the G protein β subunit Ste4 in yeast. J. Biol. Chem. 286, 27147–27155 10.1074/jbc.M111.254193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hurley J. H., and Stenmark H. (2011) Molecular mechanisms of ubiquitin-dependent membrane traffic. Annu. Rev. Biophys. 40, 119–142 10.1146/annurev-biophys-042910-155404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Husnjak K., and Dikic I. (2012) Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 81, 291–322 10.1146/annurev-biochem-051810-094654 [DOI] [PubMed] [Google Scholar]
- 39. Dixit G., Baker R., Sacks C. M., Torres M. P., and Dohlman H. G. (2014) Guanine nucleotide-binding protein (Gα) endocytosis by a cascade of ubiquitin binding domain proteins is required for sustained morphogenesis and proper mating in yeast. J. Biol. Chem. 289, 15052–15063 10.1074/jbc.M114.566117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dunn R., and Hicke L. (2001) Multiple roles for Rsp5p-dependent ubiquitination at the internalization step of endocytosis. J. Biol. Chem. 276, 25974–25981 10.1074/jbc.M104113200 [DOI] [PubMed] [Google Scholar]
- 41. Torres M. P., Lee M. J., Ding F., Purbeck C., Kuhlman B., Dokholyan N. V., and Dohlman H. G. (2009) G protein mono-ubiquitination by the Rsp5 ubiquitin ligase. J. Biol. Chem. 284, 8940–8950 10.1074/jbc.M809058200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cappell S. D., Baker R., Skowyra D., and Dohlman H. G. (2010) Systematic analysis of essential genes reveals important regulators of G protein signaling. Mol. Cell 38, 746–757 10.1016/j.molcel.2010.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Feldman R. M., Correll C. C., Kaplan K. B., and Deshaies R. J. (1997) A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 91, 221–230 10.1016/S0092-8674(00)80404-3 [DOI] [PubMed] [Google Scholar]
- 44. Skowyra D., Craig K. L., Tyers M., Elledge S. J., and Harper J. W. (1997) F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin–ligase complex. Cell 91, 209–219 10.1016/S0092-8674(00)80403-1 [DOI] [PubMed] [Google Scholar]
- 45. Rangarajan N., Gordy C. L., Askew L., Bevill S. M., Elston T. C., Errede B., Hurst J. H., Kelley J. B., Sheetz J. B., Suzuki S. K., Valentin N. H., Young E., and Dohlman H. G. (2019) Systematic analysis of F-box proteins reveals a new branch of the yeast mating pathway. J. Biol. Chem. 294, 14717–14731 10.1074/jbc.RA119.010063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ardley H. C., and Robinson P. A. (2005) E3 ubiquitin ligases. Essays Biochem. 41, 15–30 10.1042/EB0410015 [DOI] [PubMed] [Google Scholar]
- 47. Torres M. P., Clement S. T., Cappell S. D., and Dohlman H. G. (2011) Cell cycle-dependent phosphorylation and ubiquitination of a G protein α subunit. J. Biol. Chem. 286, 20208–20216 10.1074/jbc.M111.239343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sutherland C. M., Hawley S. A., McCartney R. R., Leech A., Stark M. J., Schmidt M. C., and Hardie D. G. (2003) Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr. Biol. 13, 1299–1305 10.1016/S0960-9822(03)00459-7 [DOI] [PubMed] [Google Scholar]
- 49. Hong S. P., Leiper F. C., Woods A., Carling D., and Carlson M. (2003) Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. U.S.A. 100, 8839–8843 10.1073/pnas.1533136100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tu J., and Carlson M. (1995) REG1 binds to protein phosphatase type 1 and regulates glucose repression in Saccharomyces cerevisiae. EMBO J. 14, 5939–5946 10.1002/j.1460-2075.1995.tb00282.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ludin K., Jiang R., and Carlson M. (1998) Glucose-regulated interaction of a regulatory subunit of protein phosphatase 1 with the Snf1 protein kinase in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 95, 6245–6250 10.1073/pnas.95.11.6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clement S. T., Dixit G., and Dohlman H. G. (2013) Regulation of yeast G protein signaling by the kinases that activate the AMPK homolog Snf1. Sci. Signal. 6, ra78 10.1126/scisignal.2004143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Berthet J., Rall T. W., and Sutherland E. W. (1957) The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J. Biol. Chem. 224, 463–475 [PubMed] [Google Scholar]
- 54. O'Hayre M., Degese M. S., and Gutkind J. S. (2014) Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 27, 126–135 10.1016/j.ceb.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wu V., Yeerna H., Nohata N., Chiou J., Harismendy O., Raimondi F., Inoue A., Russell R. B., Tamayo P., and Gutkind J. S. (2019) Illuminating the Onco-GPCRome: novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 294, 11062–11086 10.1074/jbc.REV119.005601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Khan A. Q., Kuttikrishnan S., Siveen K. S., Prabhu K. S., Shanmugakonar M., Al-Naemi H. A., Haris M., Dermime S., and Uddin S. (2019) RAS-mediated oncogenic signaling pathways in human malignancies. Semin. Cancer Biol. 54, 1–13 10.1016/j.semcancer.2018.03.001 [DOI] [PubMed] [Google Scholar]
- 57. Li S., Balmain A., and Counter C. M. (2018) A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer 18, 767–777 10.1038/s41568-018-0076-6 [DOI] [PubMed] [Google Scholar]
- 58. Welsch M. E., Kaplan A., Chambers J. M., Stokes M. E., Bos P. H., Zask A., Zhang Y., Sanchez-Martin M., Badgley M. A., Huang C. S., Tran T. H., Akkiraju H., Brown L. M., Nandakumar R., Cremers S., et al. (2017) Multivalent small-molecule Pan-RAS inhibitors. Cell 168, 878–889.e29 10.1016/j.cell.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gysin S., Salt M., Young A., and McCormick F. (2011) Therapeutic strategies for targeting ras proteins. Genes Cancer 2, 359–372 10.1177/1947601911412376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stephen A. G., Esposito D., Bagni R. K., and McCormick F. (2014) Dragging ras back in the ring. Cancer Cell 25, 272–281 10.1016/j.ccr.2014.02.017 [DOI] [PubMed] [Google Scholar]
- 61. Samatar A. A., and Poulikakos P. I. (2014) Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov. 13, 928–942 10.1038/nrd4281 [DOI] [PubMed] [Google Scholar]
- 62. Papke B., and Der C. J. (2017) Drugging RAS: know the enemy. Science 355, 1158–1163 10.1126/science.aam7622 [DOI] [PubMed] [Google Scholar]
- 63. Ostrem J. M., Peters U., Sos M. L., Wells J. A., and Shokat K. M. (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 10.1038/nature12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lim S. M., Westover K. D., Ficarro S. B., Harrison R. A., Choi H. G., Pacold M. E., Carrasco M., Hunter J., Kim N. D., Xie T., Sim T., Jänne P. A., Meyerson M., Marto J. A., Engen J. R., and Gray N. S. (2014) Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Engl. 53, 199–204 10.1002/anie.201307387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hunter J. C., Gurbani D., Ficarro S. B., Carrasco M. A., Lim S. M., Choi H. G., Xie T., Marto J. A., Chen Z., Gray N. S., and Westover K. D. (2014) In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. U.S.A. 111, 8895–8900 10.1073/pnas.1404639111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lito P., Solomon M., Li L. S., Hansen R., and Rosen N. (2016) Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608 10.1126/science.aad6204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Janes M. R., Zhang J., Li L. S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S. J., Darjania L., Feng J., Chen J. H., Li S., Li S., Long Y. O., et al. (2018) Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e17 10.1016/j.cell.2018.01.006 [DOI] [PubMed] [Google Scholar]
- 68. No authors. (2019) AMG 510 first to inhibit “undruggable” KRAS. Cancer Discov. 9, 988–989 10.1158/2159-8290.CD-NB2019-073 [DOI] [PubMed] [Google Scholar]
- 69. Xu L., Lubkov V., Taylor L. J., and Bar-Sagi D. (2010) Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr. Biol. 20, 1372–1377 10.1016/j.cub.2010.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yan H., Jahanshahi M., Horvath E. A., Liu H. Y., and Pfleger C. M. (2010) Rabex-5 ubiquitin ligase activity restricts Ras signaling to establish pathway homeostasis in Drosophila. Curr. Biol. 20, 1378–1382 10.1016/j.cub.2010.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baietti M. F., Simicek M., Abbasi Asbagh L., Radaelli E., Lievens S., Crowther J., Steklov M., Aushev V. N., Martínez García D., Tavernier J., and Sablina A. A. (2016) OTUB1 triggers lung cancer development by inhibiting RAS monoubiquitination. EMBO Mol. Med. 8, 288–303 10.15252/emmm.201505972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sasaki A. T., Carracedo A., Locasale J. W., Anastasiou D., Takeuchi K., Kahoud E. R., Haviv S., Asara J. M., Pandolfi P. P., and Cantley L. C. (2011) Ubiquitination of K-Ras enhances activation and facilitates binding to select downstream effectors. Sci. Signal. 4, ra13 10.1126/scisignal.2001518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Baker R., Lewis S. M., Sasaki A. T., Wilkerson E. M., Locasale J. W., Cantley L. C., Kuhlman B., Dohlman H. G., and Campbell S. L. (2013) Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat. Struct. Mol. Biol. 20, 46–52 10.1038/nsmb.2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Merkley N., Barber K. R., and Shaw G. S. (2005) Ubiquitin manipulation by an E2 conjugating enzyme using a novel covalent intermediate. J. Biol. Chem. 280, 31732–31738 10.1074/jbc.M505205200 [DOI] [PubMed] [Google Scholar]
- 75. Chen J., Ai Y., Wang J., Haracska L., and Zhuang Z. (2010) Chemically ubiquitylated PCNA as a probe for eukaryotic translesion DNA synthesis. Nat. Chem. Biol. 6, 270–272 10.1038/nchembio.316 [DOI] [PubMed] [Google Scholar]
- 76. Purbeck C., Eletr Z. M., and Kuhlman B. (2010) Kinetics of the transfer of ubiquitin from UbcH7 to E6AP. Biochemistry 49, 1361–1363 10.1021/bi9014693 [DOI] [PubMed] [Google Scholar]
- 77. Kumar K. S., Spasser L., Ohayon S., Erlich L. A., and Brik A. (2011) Expeditious chemical synthesis of ubiquitinated peptides employing orthogonal protection and native chemical ligation. Bioconjug. Chem. 22, 137–143 10.1021/bc1004735 [DOI] [PubMed] [Google Scholar]
- 78. Virdee S., Kapadnis P. B., Elliott T., Lang K., Madrzak J., Nguyen D. P., Riechmann L., and Chin J. W. (2011) Traceless and site-specific ubiquitination of recombinant proteins. J. Am. Chem. Soc. 133, 10708–10711 10.1021/ja202799r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Eger S., Castrec B., Hübscher U., Scheffner M., Rubini M., and Marx A. (2011) Generation of a mono-ubiquitinated PCNA mimic by click chemistry. Chembiochem 12, 2807–2812 10.1002/cbic.201100444 [DOI] [PubMed] [Google Scholar]
- 80. Hobbs G. A., Gunawardena H. P., Baker R., and Campbell S. L. (2013) Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Small GTPases 4, 186–192 10.4161/sgtp.26270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Baker R., Wilkerson E. M., Sumita K., Isom D. G., Sasaki A. T., Dohlman H. G., and Campbell S. L. (2013) Differences in the regulation of K-Ras and H-Ras isoforms by monoubiquitination. J. Biol. Chem. 288, 36856–36862 10.1074/jbc.C113.525691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang M. H., Nickerson S., Kim E. T., Liot C., Laurent G., Spang R., Philips M. R., Shan Y., Shaw D. E., Bar-Sagi D., Haigis M. C., and Haigis K. M. (2012) Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. U.S.A. 109, 10843–10848 10.1073/pnas.1201487109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yang M. H., Laurent G., Bause A. S., Spang R., German N., Haigis M. C., and Haigis K. M. (2013) HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 11, 1072–1077 10.1158/1541-7786.MCR-13-0040-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yin G., Kistler S., George S. D., Kuhlmann N., Garvey L., Huynh M., Bagni R. K., Lammers M., Der C. J., and Campbell S. L. (2017) A KRAS GTPase K104Q mutant retains downstream signaling by offsetting defects in regulation. J. Biol. Chem. 292, 4446–4456 10.1074/jbc.M116.762435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim S. E., Yoon J. Y., Jeong W. J., Jeon S. H., Park Y., Yoon J. B., Park Y. N., Kim H., and Choi K. Y. (2009) H-Ras is degraded by Wnt/β-catenin signaling via β-TrCP-mediated polyubiquitylation. J. Cell Sci. 122, 842–848 10.1242/jcs.040493 [DOI] [PubMed] [Google Scholar]
- 86. Cho Y. H., Cha P. H., Kaduwal S., Park J. C., Lee S. K., Yoon J. S., Shin W., Kim H., Ro E. J., Koo K. H., Park K. S., Han G., and Choi K. Y. (2016) KY1022, a small molecule destabilizing Ras via targeting the Wnt/β-catenin pathway, inhibits development of metastatic colorectal cancer. Oncotarget 7, 81727–81740 10.18632/oncotarget.13172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jeong W. J., Ro E. J., and Choi K. Y. (2018) Interaction between Wnt/β-catenin and RAS–ERK pathways and an anti-cancer strategy via degradations of β-catenin and RAS by targeting the Wnt/β-catenin pathway. NPJ Precis. Oncol. 2, 5 10.1038/s41698-018-0049-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cha P. H., Cho Y. H., Lee S. K., Lee J., Jeong W. J., Moon B. S., Yun J. H., Yang J. S., Choi S., Yoon J., Kim H. Y., Kim M. Y., Kaduwal S., Lee W., Min do S., et al. (2016) Small-molecule binding of the axin RGS domain promotes β-catenin and Ras degradation. Nat. Chem. Biol. 12, 593–600 10.1038/nchembio.2103 [DOI] [PubMed] [Google Scholar]
- 89. Park J., Cho Y. H., Shin W. J., Lee S. K., Lee J., Kim T., Cha P. H., Yang J. S., Cho J., Min D. S., Han G., Lee H. Y., and Choi K. Y. (2019) A Ras destabilizer KYA1797K overcomes the resistance of EGFR tyrosine kinase inhibitor in KRAS-mutated non-small cell lung cancer. Sci. Rep. 9, 648 10.1038/s41598-018-37059-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lee S. K., Cho Y. H., Cha P. H., Yoon J. S., Ro E. J., Jeong W. J., Park J., Kim H., Il Kim T., Min D. S., Han G., and Choi K. Y. (2018) A small molecule approach to degrade RAS with EGFR repression is a potential therapy for KRAS mutation-driven colorectal cancer resistance to cetuximab. Exp. Mol. Med. 50, 153 10.1038/s12276-018-0182-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shukla S., Allam U. S., Ahsan A., Chen G., Krishnamurthy P. M., Marsh K., Rumschlag M., Shankar S., Whitehead C., Schipper M., Basrur V., Southworth D. R., Chinnaiyan A. M., Rehemtulla A., Beer D. G., et al. (2014) KRAS protein stability is regulated through SMURF2: UBCH5 complex-mediated β-TrCP1 degradation. Neoplasia 16, 115–128 10.1593/neo.14184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lu A., Tebar F., Alvarez-Moya B., López-Alcalá C., Calvo M., Enrich C., Agell N., Nakamura T., Matsuda M., and Bachs O. (2009) A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 184, 863–879 10.1083/jcb.200807186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bigenzahn J. W., Collu G. M., Kartnig F., Pieraks M., Vladimer G. I., Heinz L. X., Sedlyarov V., Schischlik F., Fauster A., Rebsamen M., Parapatics K., Blomen V. A., Müller A. C., Winter G. E., Kralovics R., et al. (2018) LZTR1 is a regulator of RAS ubiquitination and signaling. Science 362, 1171–1177 10.1126/science.aap8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Steklov M., Pandolfi S., Baietti M. F., Batiuk A., Carai P., Najm P., Zhang M., Jang H., Renzi F., Cai Y., Abbasi Asbagh L., Pastor T., De Troyer M., Simicek M., Radaelli E., et al. (2018) Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362, 1177–1182 10.1126/science.aap7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Abe T., Umeki I., Kanno S. I., Inoue S. I., Niihori T., and Aoki Y. (2019) LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 2019 10.1038/s41418-019-0395-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ma Y., Gu Y., Zhang Q., Han Y., Yu S., Lu Z., and Chen J. (2013) Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 12, 286–294 10.1158/1535-7163.MCT-12-0650 [DOI] [PubMed] [Google Scholar]
- 97. Ma Y., Xu J., Huang P., Bai X., and Gao H. (2019) Ubiquitin-independent, proteasome-mediated targeted degradation of KRAS in pancreatic adenocarcinoma cells using an engineered ornithine decarboxylase/antizyme system. IUBMB Life 71, 57–65 10.1002/iub.1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mullard A. (2019) First targeted protein degrader hits the clinic. Nat. Rev. Drug Discov. 10.1038/d41573-019-00043-6 [DOI] [PubMed] [Google Scholar]
- 99. Flanagan J. J., and Neklesa T. K. (2019) Targeting nuclear receptors with PROTAC degraders. Mol. Cell. Endocrinol. 493, 110452 10.1016/j.mce.2019.110452 [DOI] [PubMed] [Google Scholar]
- 100. Xi M., Chen Y., Yang H., Xu H., Du K., Wu C., Xu Y., Deng L., Luo X., Yu L., Wu Y., Gao X., Cai T., Chen B., Shen R., and Sun H. (2019) Small molecule PROTACs in targeted therapy: an emerging strategy to induce protein degradation. Eur. J. Med. Chem. 174, 159–180 10.1016/j.ejmech.2019.04.036 [DOI] [PubMed] [Google Scholar]
- 101. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]