

Abstract
Inositol 1,4,5-trisphosphate receptors (InsP3Rs) are endoplasmic reticulum–localized channels that mediate Ca2+ release from the endoplasmic reticulum into the cytoplasm. We previously reported that an EF-hand Ca2+-binding protein, neuronal calcium sensor 1 (NCS1), binds to the InsP3R and thereby increases channel open probability, an event associated with chemotherapy-induced peripheral neuropathy. However, the exact NCS1-binding site on InsP3R remains unknown. Using protein docking, co-immunoprecipitation, and blocking peptides, we mapped the NCS1-binding site to residues 66–110 on the suppressor domain of InsP3R type 1 (InsP3R1). We also identified Leu-89, a residue in the hydrophobic pocket of NCS1, as being critical for facilitating the NCS1–InsP3R1 interaction. Overexpression of WT NCS1 in MDA-MB231 breast cancer cells increased Ca2+ signaling and survival, whereas overexpression of Leu-89 NCS1 variants decreased Ca2+ signaling and survival, further suggesting the importance of this residue in the NCS1–InsP3R1 interaction. In conclusion, we show that NCS1–InsP3R1 interaction enhances intracellular Ca2+ signaling in cells and can be modulated by altering or occluding the hydrophobic pocket of NCS1. This improved understanding of the NCS1–InsP3R1 interaction may facilitate the development of management strategies for diseases resulting from aberrant NCS1 expression.
Keywords: calcium intracellular release, calcium-binding protein, protein–protein interaction, breast cancer, docking, cell line MDA-MB231
Introduction
As a universal second messenger involved in many signaling pathways, the intracellular Ca2+ concentration is tightly regulated through a network of ion channels and pumps located on the plasma membrane and organellar membranes such as mitochondria and the endoplasmic reticulum (ER).3 Inositol 1,4,5-trisphosphate receptors (InsP3Rs) are ER-localized ion channels that regulate Ca2+ flow from the ER into the cytoplasm. InsP3Rs are activated by inositol 1,4,5-trisphosphate (InsP3), which is produced by a signaling cascade starting with ligands binding to various G protein–coupled receptors, resulting in the activation of phospholipase C, which then cleaves phosphatidylinositol 4,5-bisphosphate to produce InsP3 (1). The InsP3Rs are involved in numerous cellular processes including apoptosis (2), secretion (3), and memory and learning (4). Dysfunction of InsP3R-dependent signaling underlies a multitude of diseases ranging from cerebellar ataxia (5, 6) to bile duct obstruction (7). Similar to many other Ca2+ channels, the opening probability of the InsP3Rs is regulated by Ca2+-binding proteins such as calmodulin (CaM) (8), Ca2+-binding protein 1 (CaBP1) (9), and neuronal calcium sensor 1 (NCS1) (10, 11), which allow for the fine tuning of Ca2+-mediated Ca2+ signaling.
We previously showed that NCS1 binding to the InsP3R is a critical trigger for the development of chemotherapy-induced peripheral neuropathy (10, 12–14). NCS1 is a member of the neuronal calcium sensor (NCS) family, with high affinity and low capacity for Ca2+ binding (15). We showed that paclitaxel binds NCS1, which enhances NCS1 binding to the InsP3R, subsequently amplifying Ca2+ flux from the ER into the cytosol and causing pathological activation of calpain (10, 12–14). In another study, NCS1 was shown to physically and functionally interact with InsP3R1 and InsP3R2, and deletion of NCS1 decreased survival and contractile function in young mice (16). Similarly, NCS1 and InsP3R1 were shown to interact to regulate neurite outgrowth in chick dorsal ganglion cells (17). These studies suggest that the interaction between NCS1 and the InsP3Rs is important in many contexts and becomes dysregulated in pathological states. However, the exact binding site between NCS1 and the InsP3Rs has not yet been determined.
This study maps the exact interaction between NCS1 and InsP3R1, with the eventual aim of targeting this interaction for therapeutic treatments. We showed that NCS1 binds to residues 1–225 in the N terminus of InsP3R type 1 (InsP3R1), a region also regulated by other Ca2+ sensors (8, 9). We also demonstrated that NCS1–InsP3R1 interaction is enhanced by Ca2+ in the physiologically relevant submicromolar range. Using competing polypeptides and mutagenesis, the important residues for the NCS1–InsP3R1 interaction were further restricted to Leu-89 on NCS1 and His-93 and His-94 InsP3R1. Blocking peptides corresponding to the region where InsP3R1 binds the NCS1 region reduced the interaction between NCS1 and InsP3R1 (1–225), as well as full-length InsP3R1. The cell-penetrant version of one blocking peptide also reduced InsP3R-dependent Ca2+ signaling in MDA-MB231 cells. To further investigate the functional relevance of Leu-89 NCS1 variants, we found that overexpression of WT NCS1 increased InsP3R-dependent Ca2+ signaling, whereas overexpression of Leu-89 NCS1 variants reduced Ca2+ signaling. Similarly, overexpression of WT NCS1 increased, whereas overexpression of Leu-89 NCS1 variants decreased, cell survival, further supporting the importance of residue Leu-89 of NCS1. Overall, our findings expand the understanding of the biochemical components needed for regulation of Ca2+ homeostasis, as well as identify potential therapeutic targets for alleviating pathological conditions involving NCS1 and the InsP3Rs.
Results
NCS1 binds the suppressor domain of InsP3R1
We previously showed that NCS1 bound full-length InsP3R1, and the interaction is enhanced by the addition of Ca2+ or paclitaxel (10, 12). Because the addition of NCS1 to the cytoplasmic side of InsP3R1 increased channel activity (13), we hypothesized that NCS1 bound to a cytoplasmic region of InsP3R1. To narrow down the binding site, HA-tagged NCS1 and various fragments of InsP3R1 were co-expressed in HEK293 cells, followed by co-immunoprecipitation using NCS1 or HA antibodies (Fig. 1, A–C). NCS1 co-immunoprecipitated the InsP3R fragment comprised of residues 1–346, but not other fragments, suggesting that NCS1 only binds to the suppressor region of InsP3R1 (Fig. 1, B and C). Subsequently, we found that a GST-tagged fragment containing residues 1–225, which corresponded to the suppressor domain of InsP3R1, pulled-down NCS1 (Fig. 1D). Similar to full-length InsP3R1, the interaction between NCS1 and InsP3R1 (1–225) was enhanced by the addition of Ca2+ (Fig. 1C) or paclitaxel (Fig. S1). To determine the Ca2+ concentration relevant for NCS1–InsP3R1 interaction, we performed pulldown between GST-InsP3R1 (1–225) and NCS1 in Ca2+ concentrations ranging from 0 to 100 μm (Fig. 1D). The interaction was significantly enhanced by Ca2+ concentrations between 100 nm and 1 μm, which closely matches the Ca2+-binding curve of NCS1 (15). Using a log(agonist) versus response fit from four experiments, the EC50 of the interaction was estimated to be −6.8, which corresponded to a Ca2+ concentration of 160 nm (Fig. 1E).
Figure 1.

NCS1 binds the N terminus of InsP3R1 in a Ca2+-dependent manner. A, diagram showing the InsP3R1 fragments tested in co-immunoprecipitation experiments using NCS1 antibody and HEK293 cell lysates co-expressing fragments of InsP3R1 and HA-tagged NCS1. A check shows fragments that pulled down NCS1, a cross shows fragments that did not pull down NCS1. B, representative results with fragments F1, F2, F3, and F4. All experiments were repeated at least three times with similar results. C, F1 was also co-immunoprecipitated along with HA-tagged NCS1 using HA antibody. D, a representative plot showed that NCS1 binding to InsP3R1-(1–225) (F6) was enhanced by Ca2+ concentrations above 1 μm. E, quantification from four pulldown experiments at different free Ca2+ concentrations. An estimation using the EC50 value from a log(agonist) versus response fit gave a value of 160 nm. Error bars show S.E. Ctrl, control; IB, immunoblotting.
Leu-89 residue of NCS1 facilitates NCS1–InsP3R1 Ca2+-dependent interaction
To experimentally identify the predicted binding site, we performed pulldown experiments between GST-tagged InsP3R1 (1–225) and NCS1 variants. These variants of NCS1 have mutations in pairs of hydrophobic residues that span the hydrophobic cleft and are not located within the Ca2+-binding motifs (Fig. 2A). Binding between InsP3R1 (1–225) and NCS1 variants F48E/W103K, Y108E/M121K, or F169E/M156K was enhanced by Ca2+, similar to WT NCS1 (Fig. 1B). However, NCS1 mutant L89E/W103K showed an opposite trend, where the addition of Ca2+ almost completely inhibited its binding to InsP3R1 (1–225). Additional experiments demonstrated that the W103K mutant binding to InsP3R1 (1–225) was comparable with WT NCS1. In contrast, when residue Leu-89 alone was mutated to Ala, Glu, or Lys, binding to InsP3R1 (1–225) in Ca2+-containing solutions was reduced with the largest deficit observed for the L89K mutant (Fig. 2B). Pulldown experiments between GST-tagged InsP3R1 (1–225) and WT, L89A, or L89K NCS1 at increasing Ca2+ concentrations also demonstrated the opposite binding trend between WT and Leu-89 NCS1 variants to InsP3R1 (1–225) (Fig. 2, C and D). To rule out the possibility that the reduced binding efficiency was due to the Ca2+-binding property of the variants, isothermal titration calorimetry measurements were performed and showed that the L89K mutant bound Ca2+ similarly to WT NCS1 (Fig. S2 and Table S1).
Figure 2.

The Leu-89 residue on NCS1 is important for the Ca2+-dependent NCS1–InsP3R1 interaction. A, GST-InsP3R1 (1–225) pulldown of WT and mutant NCS1 in Ca2+-free and Ca2+-containing conditions. Only the L89E/W103K mutant showed an inverted trend where Ca2+ reduced instead of increased interaction. B, mutation of the Leu-89 residue to Ala, Glu, or Lys all decreased the NCS1–InsP3R1 interaction in the presence of Ca2+. C, interaction between the WT and Leu-89 NCS1 variants over a range of physiologically relevant Ca2+ concentrations from 0 to 10 μm. D, quantification of four experiments for WT NCS1 (same data as shown in Fig. 1E), four experiments for L89K, and three experiments for L89A. *, statistically significant difference between WT NCS1 and Leu-89 NCS1 variants at that specific Ca2+ concentration (Student's t test, adjusted for multiple comparison). Fit show was from log(agonist) versus response. Error bars show S.E. IB, immunoblotting.
Docking analysis
After identifying InsP3R1 (1–225) as the NCS1-binding region, we took advantage of the available crystal structures of full-length NCS1 and the N-terminal fragments of InsP3R1 to perform computational docking to predict the binding site. Docking was performed between full-length, Ca2+-bound NCS1 (PDB code 1G8I, chain A) and two fragments of InsP3R1, 1–246 (PDB code 1XZZ) and (1–604) (PDB code 3UJ4, chain A), using the programs Z-dock and Cluster Pro. Top docking results indicated that the helical arm of InsP3R1 (1–225), which encompasses residues 66–110, interacts with the hydrophobic pocket of NCS1 (Fig. 3A). Docking results also supported our observations that Leu-89 is important for binding to InsP3R1 (Fig. 3B). Leu-89 appears to make direct contact with residues His-93 and His-94 of InsP3R1 and forms additional hydrophobic contacts. We confirmed this by showing that the H93A/H94A InsP3R1 variant showed decreased binding to NCS1 in the presence of Ca2+ (Fig. 3C). Similar results were obtained using Cluster Pro, which utilized a different docking algorithm (Fig. 3D).
Figure 3.

A, prediction of the molecular interaction between Ca2+-bound NCS1 (white, PDB code 1G8I) and a fragment of InsP3R1 (blue) containing residues 1–604 (PDB code 3UJ0) using the ZDOCK server. B, Leu-89 (shown in pink) of NCS1 forms interactions with the arm motif of the suppressor domain of InsP3R1 (residues 66–110). Mutations L89A, L89E, and L89K would likely disrupt binding to this region of the InsP3R. C, mutation of His-93/His-94 to Ala reduced NCS1–InsP3R1 interaction in the presence of Ca2+ D, a nearly identical binding geometry for complex formation between NCS1 and InsP3R1 was predicted by the ClusPro server using an independent docking algorithm. E, visualization of NCS1 binding to full-length InsP3R. Molecular interactions between NCS1 (white, PDB code 1G8I) and a fragment of InsP3R1 (blue) containing residues 1–604 (PDB code 3UJ0) were predicted using the ZDOCK server and superimposed in PyMOL on the structure of full-length InsP3R1. The predicted binding site is in the cytoplasmic domain and appears accessible by NCS1. IB, immunoblotting.
InsP3R1 peptides corresponding to NCS1 binding region reduce NCS1–InsP3R1 interaction
Because our docking model predicted that the region spanning from residues 66–110 of InsP3R1 interacted with NCS1, we tested this prediction by adding in polypeptides spanning the region (peptides 68–80, 82–96, and 84–98; Fig. 4A). The peptides were similar regarding charge, hydrophobic content, and length. Both peptides (68–80) and (82–96) reduced the interaction between NCS1 and InsP3R1 (1–225) (Fig. 4B), whereas another peptide (84–98) did not (Fig. 4, B and C). To further validate this, we repeated co-immunoprecipitation experiments between NCS1 and full-length InsP3R1 from mouse cerebellum lysate. The peptide (82–96) reduced this interaction (Fig. 4, D and E), further supporting the finding that this region of InsP3R1 is important for the binding between NCS1 and InsP3R1.
Figure 4.

InsP3R1 peptides corresponding to NCS1 binding region reduced NCS1-InsP3R1 interaction. A, diagram showing the InsP3R1 peptides tested within the predicted binding region spanning residues 66–110, with the peptides used shown in boxes. B, representative blot of pulldown between GST-InsP3R1 (1–225) and NCS1 in the presence of various concentrations of peptides and 10 μm Ca2+. C, quantification of 3 independent experiments with peptides added at 20X molar excess (2 μm peptide, 100 nm NCS1). Addition of peptides (68–80) and (82–96) resulted in a 58% (p = .0007) and 16% reduction (p = 0.02) in the amount of NCS1 pulled down respectively, whereas addition of peptide (84–98) did not change the amount of NCS1 pulldown (p = 0.09) (two-tailed student t-test comparing the no peptide control with the specific peptide). D, representative blot of co-immunoprecipitation between NCS1 and full-length InsP3R1 with blocking peptides. E, peptide (82–96) also reduced the interaction between NCS1 and full-length InsP3R1 by 60% in mouse cerebellum lysate (n = 3, two-tailed student t-test, p = 0.002).
Cell-penetrant TAT-(68–80) peptide decreases InsP3R-dependent Ca2+ signaling
To test whether InsP3R1 blocking peptides can block the interaction in vivo and lead to a functional difference, we treated MDA-MB231 cells, an epithelial mammary breast cell line derived from a metastatic site, with 20 μm cell-penetrant TAT-(68–80) or the TAT-scrambled control 45 min prior to Ca2+ imaging. To rule out Ca2+ flux from the extracellular environment, imaging was carried out in Ca2+-free buffer and imaging with UTP, a specific P2Y G protein–coupled receptor agonist that leads to the production of InsP3 (18). Compared with cells treated with the scrambled control, cells treated with TAT-(68–80) showed a similar rate of response (Fig. 5B) but lower maximum amplitude (Fig. 5C), area under the curve (Fig. 5D), and rate of rise (Fig. 5E) of response. These observations suggested that TAT-(68–80) could block NCS1–InsP3R1 in vivo to decrease Ca2+ signaling.
Figure 5.

Cell-penetrant TAT-(68–80) decreased InsP3R-dependent Ca2+ signaling. A, combined traces of all coverslips (n = 12 for each treatment) for MB231 cells treated with either 20 μm TAT-(68–80) or TAT-scrambled control (TAT-SCRB). UTP was added from 10 to 40 s. B, the number of cells responding per coverslip were similar across different cell types (p = 0.68). C–E, compared with the scrambled control, cells treated with TAT-(68–80) showed decreased maximum amplitude (C, p = 0.014), area under the curve (D, p = 0.036), and rate of rise (E, p = 0.04). Statistical analysis was carried out using unpaired two-tailed Student's t test.
WT NCS1 increases, whereas Leu-89 NCS1 variants decrease, Ca2+ signaling
Because Leu-89 NCS1 variants had diminished binding to InsP3R1, we predicted that Leu-89 NCS1 variants would be loss-of-function variants. To test this hypothesis, we overexpressed WT NCS1 and NCS1 variants in MDA-MB231 cells, a human breast cancer cell line in which we previously showed that overexpression of WT NCS1 led to increased cell survival and metastasis (27, 28). Monoclonal cell populations were generated in which NCS1 levels in overexpressing cells were severalfold more than the native level (Fig. S3A). Previous studies reported that overexpression of WT NCS1 increases, whereas reducing NCS1 decreases, InsP3R1-dependent Ca2+ signaling in several cell types (10, 12, 16, 18). Because Leu-89 NCS1 variants do not bind InsP3R1 efficiently, we predicted that overexpressing these variants would not increase Ca2+ signals or even decrease signaling. As expected, cells overexpressing WT NCS1 showed increased Ca2+ signaling, whereas cells overexpressing L89A and L89K NCS1 showed unchanged or decreased Ca2+ signaling (Fig. 6A). The percentage of cells responding to 3 μm UTP was comparable across the different cells tested (Fig. 6B). Compared with the control cells, cells overexpressing WT NCS1 had an increased maximum amplitude (Fig. 6C), rate of rise (Fig. 6D), and area under the curve (Fig. 6E). In contrast, cells overexpressing Leu-89 NCS1 variants showed unchanged or decreased maximum amplitude, rate of rise, and area under the curve. Because both WT NCS1 and L89K NCS1 bind Ca2+, the reduction in Ca2+ signaling is likely not due to altered Ca2+ buffering but rather due to the reduction in the NCS1–InsP3R1 interaction. Ca2+ signals following the addition of 1 μm of the ionophore ionomycin were similar in the different cell types, suggesting that intracellular Ca2+ loading was similar among the different cells (Fig. S4).
Figure 6.

Leu-89 NCS1 variants did not change or decreased InsP3R1-dependent Ca2+ signaling. A, combined traces of all coverslips for different cell types (n = 21 coverslips for empty vector control, 15 for WT NCS1, 15 for L89A NCS1, and 11 for L89K NCS1). UTP was added from 10 to 30 s. B, the number of cells responding per coverslip were similar across different cell types (one-way ANOVA, p = 0.22). C–E, compared with control, cells overexpressing WT NCS1 showed increased maximum amplitude (C), area under the curve (D), and rate of rise (E). Cells overexpressing L89A or L89K NCS1 showed either similar or decreased maximum amplitude (C), rate of rise (D), and area under the curve (E). One-way ordinary ANOVA was carried out, followed by post hoc Dunnett's multiple comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.005. CTRL, control.
WT NCS1 increases, whereas Leu-89 variants decrease, survival
NCS1 was previously shown to increase survival of neuronal SH-SY5Y cells (20), cortical neurons (21), and MDA-MB231 (22, 23), partly through up-regulating the pAKT pathway. Because we observed that the Leu-89 NCS1 variants decreased Ca2+ signaling, we hypothesized that overexpression of L89A would also decrease cell survival. Using a colony formation assay to measure cell survival, we found a significant difference between cells expressing WT or mutant NCS1 as compared with control cells (Fig. 7). When 100 cells were plated initially, the cells that overexpressed WT NCS1 covered 4.5-fold more area than CTRL cells expressing the empty vector. As expected, cells that overexpressed L89A NCS1 showed a 75% reduction in the area covered as compared with the control (Fig. 7B). Similar results were observed when 500 cells were initially plated (Fig. 7C). There was no significant difference in the proliferation rate among CTRL, WT NCS1 overexpression, and L89K NCS1 overexpression cells (Fig. S5), suggesting that the difference observed in the colony formation assay was primarily caused by differences in survival rate.
Figure 7.
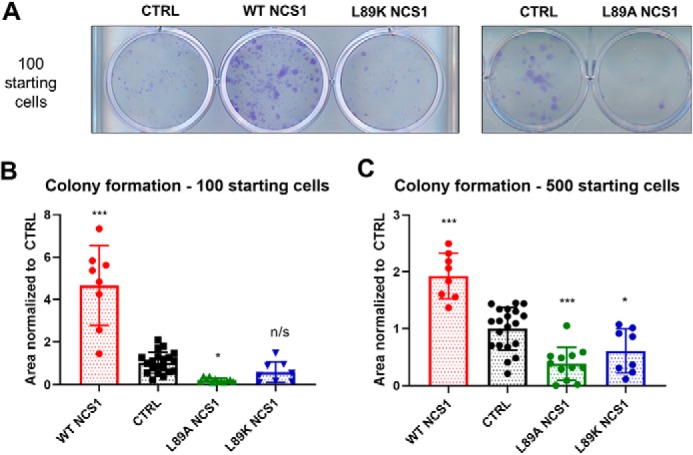
Leu-89 NCS1 variants decreased survival. A, representative image for colony formation assay. B, when 100 cells were initially plated, cells overexpressing WT NCS1 showed a significant increase in survival (p = 0.0001). In contrast, cells overexpressing L89A NCS1 showed a significant decrease in survival (p = 0.028). Cell overexpressing L89K NCS1 also showed a reduction in survival, although the reduction was not statistically significant (p = 0.44). C, similar results were obtained when 500 cells were initially plated, and the difference between CTRL and L89K cells was statistically significant (p = 0.035). Statistical analysis was performed on four independent experiments for each cell type, with duplicates or triplicates for each experiment. One-way ordinary ANOVA was carried out, followed by post hoc Dunnett's multiple comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.005. CTRL, control.
Discussion
Ca2+ is a universal second messenger that regulates diverse processes including neurotransmitter release, muscle contraction, and cell death. As such, cytosolic Ca2+ is tightly regulated, partly through the controlled release of Ca2+ from ER stores via InsP3Rs. One of the mechanisms used to regulate InsP3Rs is the binding of members of the EF-hand Ca2+-binding proteins, specifically, CaM, CaBP1, and NCS1. InsP3R1 contains three CaM-binding sites, of which two sites are in the suppressor domain (residues 1–225) and one site is in the modulatory domain (residues 1564–1585) (24). Binding of CaM to the suppressor domain decreases Ca2+ release (8), and binding of CaM to the modulatory domain does not affect Ca2+ release (24). There is less consensus concerning the effect of CaBP1 binding to the suppressor domain, where both inhibitory and activating effects have been reported (9, 25). We found that NCS1 binds only in the suppressor domain, and NCS1 binding is stimulatory (13). Each of these interactions between a Ca2+-binding protein and the InsP3R shows that regulating Ca2+ release through InsP3R1 channel is central to regulating cellular processes.
Physiological significance of NCS1–InsP3R interaction
NCS1 is a low-capacity, high-affinity Ca2+-binding protein. Although NCS1, CaM, and CaBP1 share <50% homology at the primary amino acid sequence, they are similar at the secondary and tertiary structure with four EF-hands. NCS1, CaM, and CaBP1 also bind to and regulate similar target proteins (26). We propose that regulation of the same protein by similar Ca2+-binding proteins may be essential in allowing the fine-tuning of a cell's response to fluctuation in Ca2+ levels. For example, NCS1 is sensitive to Ca2+ changes from 10 to 300 nm, whereas CaM is sensitive to Ca2+ changes from 100 nm to 100 μm (15). Our previous work shows that binding of NCS1 to InsP3R1 increases the open probability of InsP3R1, leading to a larger Ca2+ release (13). A further study may investigate possible interactions among NCS1, CaM, and CaBP1 for the suppressor domain of InsP3R1.
Although our study focuses on the interaction between NCS1 and InsP3R1, we propose that NCS1 also interacts with other subtypes of InsP3R, namely types 2 and 3. In fact, NCS1 was shown to interact with InsP3R2 in heart (16). Our preliminary docking results of NCS1 to the suppressor domain of InsP3R3 (Fig. S6) suggest that the binding is similar. The suppressor domains of the three subtypes of InsP3Rs are highly homologous, sharing more than 77% identity and 93% similarity. In comparison, the adjacent regulatory domains of InsP3Rs share 67% identity and 88% similarity. Residue His-94 is conserved across all three subtypes, whereas His-93 is changed to Gln in types 2 and 3 (Fig. S7). Whether this difference affects the binding affinity of NCS1 to types 2 and 3 remains to be investigated.
Identification of specific binding regions that can facilitate rational drug design
We showed that Leu-89 NCS1 variants displayed an opposite trend to WT NCS1 in which Ca2+ almost completely inhibited the binding to InsP3R1 (1–225). In our docking model, Leu-89 is close to the helical arm of the suppressor domain of the InsP3R1, specifically to His-93 (Fig. 3B). The importance of the Leu-89 NCS1 residue was further supported where Leu-89 NCS1 variants reduced instead of increased Ca2+ signaling. Previous work (27) also showed that human L89A NCS1 failed to rescue memory deficits in ncs1-null Caenorhabditis elegans, further suggesting the functional significance of this residue. The identification of the Leu-89 NCS1 residue as critical for NCS1–InsP3R1 binding can allow for drug screening focusing on the residue. A similar strategy was successful for blocking the interaction between NCS1 and the RIC8 guanine nucleotide exchange factor A (RIC8A), supporting this as a feasible approach (28).
Cell-penetrant peptides for modulating NCS1–InsP3R interaction
We showed that peptides consisting of residues 68–80 and 82–96 of the InsP3R could reduce the interaction between NCS1 and InsP3R1. Cell-penetrant TAT-(68–80) could also decrease InsP3R-dependent Ca2+ signaling 45 min after treatment. Interestingly, a D2R peptide used in a previous study (29) to reduce NCS1-D2R interaction increased NCS1–InsP3R1 interaction (Fig. S8). We hypothesize that the D2R peptide stabilizes the hydrophobic pocket of NCS1 to enhance NCS1–InsP3R1 interaction. Therefore, another approach would be to use these peptides in combination as scaffolds for producing small compounds that could moderate the interaction both up or down. For example, cell-penetrant peptides can be tested in animal models to test whether chemotherapy-induced peripheral neuropathy can be alleviated.
A limitation in our study is that we cannot decisively rule out whether other NCS1- or InsP3R1-protein partners also play a role in the functional differences observed. Because NCS1 is a multifunctional protein with multiple known interacting partners, Leu-89 NCS1 variants may also exhibit altered interaction with these partners. Similarly, the N terminus of InsP3R is regulated by various Ca2+ sensors, and a blocking peptide corresponding to this region may also block InsP3R interaction with these regulatory proteins. However, because modification of NCS1–InsP3R interaction through targeting either NCS1 or InsP3R1 resulted in similar changes, including decreased Ca2+ signaling, we are confident that NCS1–InsP3R1 interaction, at minimum, contributed to the differences observed.
Conclusion
Here, we show that NCS1 binds to the suppressor domain of InsP3R and that this binding is enhanced by the addition of Ca2+. We also identified one residue of NCS1 (Leu-89) and residues His-93 and His-94 of InsP3R1 as important for the interaction. Mutations to Leu-89 of NCS1 or peptides corresponding to the binding region of InsP3R1 reduced the interaction. To test whether NCS1–InsP3R1 binding was functionally relevant, we showed that changes in the interaction, either through a cell-penetrant peptide or through overexpressing Leu-89 NCS1 variants, modulated Ca2+ signaling. Further studies can examine how blocking the NCS1–InsP3R interaction, either by identification of novel drugs that inhibit binding or modifications of peptides centered on the relevant residues of NCS1 or InsP3R, can be used as therapeutic strategies.
Materials and methods
Mammalian IP3R1 fragments and NCS1 expression
All reagents were used per the manufacturers' instructions unless otherwise stated. Vectors for mammalian expression of full-length and fragments of InsP3R1 were gifts from Dr. David Yule (University of Rochester Medical Center). The vector for HA-tagged NCS1 was previously made in the laboratory. HEK293 cells (American Type Culture Collection, Manassas, VA) were co-transfected with 1 μg each of InsP3R1 and HA-tagged NCS1 using Lipofectamine 2000 (Thermo Fisher Scientific). The cells were harvested 48 h after transfection, lysed using M-PER mammalian protein extraction reagent (Thermo Fisher Scientific) supplemented with protease inhibitor (P2714; Sigma–Aldrich), and centrifuged at 13,000 rpm for 15 min at 4 °C. The pellet was discarded, and the lysate was collected. Protein concentration was determined using Pierce BCA protein assay (Thermo Fisher Scientific). Cell lysate was stored at −20 °C until further use.
Mammalian NCS1/IP3R1 co-immunoprecipitation
Transfected HEK293 cell lysate expressing InsP3R1 fragments and HA-tagged NCS1 were diluted to obtain 500 μl of 0.4 μg/μl per sample. Each sample was incubated with 10 μl of anti-NCS1 antibody (FL190; Santa Cruz Biotechnology) overnight at 4 °C, followed by incubation with 30 μl of protein A magnetic beads (PureProteome; Millipore Sigma) for 1 h at 4 °C. The beads were washed three times with cold PBS (American Bio, Canton, MA) and then eluted, and protein levels were quantified using Western blotting analysis.
Western blotting analysis
Western blots were performed using the NuPAGE system (Thermo Fisher Scientific) and polyvinylidene difluoride membrane with the Bio-Rad wet transfer system (Bio-Rad). Primary antibodies used were anti-NCS1 FL-190, anti-HA (Santa Cruz Biotechnology), anti-FLAG (Sigma–Aldrich), T1NH (1:1000, custom antibody that binds N-term of InsP3R1), and YU272 (1:1000, custom antibody that binds C-terminal of InsP3R1). Western blotting was performed, and the results were quantified as previously described (10).
Bacterial NCS1 and GST-IP3R1 (1–225) protein expression and purification
pGEX6p2 vector carrying the coding sequence for InsP3R1 (1–225) was a gift from Dr. Jan Parys (KU Leuven). pET-21 vector carrying the coding sequence for NCS1 was prepared in the lab previously. Each plasmid was transformed into BL21-CodonPlus (DE3)-RIPL competent cells (Agilent Technologies, Santa Clara, CA). Single clones were grown, and crude bacterial lysate was prepared as previously described (8, 30).
Preparing NCS1 mutant plasmid
Plasmids encoding WT NCS1 for protein expression (pET-21/NCS-) or in pcDNA3.1 for transfection were described previously (30). Plasmids encoding NCS1 variants were prepared by site-directed mutagenesis using a QuikChange site-directed mutagenesis kit (Stratagene). Primers were designed using the company's website. Plasmid sequences were confirmed by automated DNA sequencing. NCS1 expressed in bacteria was not tagged. For mammalian expression, a C-terminal tag was introduced using the reverse primer. WT, L89A, and L89K were tagged with HA, FLAG, and His6.
Monitoring calcium dependence
To monitor the calcium dependence of NCS1/InsP3R interactions, the EGTA concentration was fixed at 1 mm. To achieve different concentrations of free Ca2+, the amount of total CaCl2 needed was calculated using the Ca-EGTA calculator with T = 21.5 °C, pH 7.4, n = 0.163, and EGTA = 1 mm to obtain free Ca2+ concentration ranging from 10−8 to 10−3 m. Appropriate Ca2+ concentrations were maintained in all working solutions for each condition.
For GST pulldown, GSH-Sepharose 4B (GE Healthcare) was used according to the manufacturer's instructions. 30 μl of beads were washed three times with 1 ml of washing buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EGTA, and a predetermined concentration of Ca2+) and then incubated with 500 μg of crude GST-IP3R1 lysate for 1 h with gentle end-over-end rotation at room temperature. Subsequently, unbound proteins were removed, and the beads were washed three times with 1 ml of washing buffer. Next, the beads were incubated with 10 nm NCS1 in washing buffer for 2 h at room temperature. Unbound proteins were removed, and the beads were washed three times with 1 ml of washing buffer. Bound proteins were then eluted with 20 μl of 20 mm GSH (AmericanBio) in 50 mm Tris, pH 7.4.
Docking
The structural basis for interactions between NCS1 and InsP3R were independently predicted using the protein–protein docking programs ZDOCK (31, 32) and Cluster Pro 2.0 (33). In both ZDOCK and Cluster Pro, the structure of full-length calcium bound NCS1 (PDB code 1G8I) was input as the “ligand,” and the crystal structure of InsP3R containing residues 1–604 (PDB code 3UJ0) was input as the “receptor.” The structure of InsP3R used for docking contains both the suppressor domain (1–223) and the inositol 1,4,5-trisphosphate–binding core (224–579). The top-ranked complexes from ZDOCK (ZDOCK score, 1694) and Cluster Pro were chosen for analysis in the macromolecular visualization program PyMOL (version 0.99, Schrödinger, LLC).
Peptides
Synthetic peptides were purchased from Genscript and delivered with >95% purity, with TFA removal. Peptide sequences, solvents, and concentrations are detailed in Table 1.
Table 1.
Peptide information
| Label | Sequence | Stock concentration |
|---|---|---|
| (68–80) | AQKQFWKAAKPGA | 1 mm in PBS with 10% DMSO |
| (82–96) | TTDAVLLNKLHHAAD | |
| (84–98) | DAVLLNKLHHAADLE | |
| D2R | NIEFRKAFLKILHSR | |
| TAT-(68–80) | RKKRRQRRRGGAQKQFWKAAKPGA | 2 mm in PBS |
| TAT-SCRB | RKKRRQRRRGGFAKAQKPGQAAKW |
Cell culture and generation of stable cells
MDA-MB231 cells (American Type Culture Collection) were maintained at 37 °C, 5% CO2, in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% l-glutamine, and 1% penicillin/streptomycin. Stable MDA-MB231cells were generated through transfection with a control vector or vectors for overexpression of WT and mutant NCS1 using Lipofectamine. 2 days after selection, stable cells were selected with 2 mg/ml G418 (American Bio) for 2 weeks, and the overexpression was confirmed using Western blotting analysis. Single clones were selected and expanded for a homogeneous population. Stable cells were maintained in the same medium supplemented with 1 mg/ml G418.
Calcium imaging
22 × 22-mm glass coverslips were washed with 70% ethanol, rinsed three times with MilliQ water, left to dry, and sterilized under UV in a cell culture hood. 1 × 105 cells were plated on each coverslip 1 day before imaging. HEPES-buffered saline (140 mm NaCl, 1.13 mm MgCl2, 4.7 mm KCl, 2 mMCaCl2, 10 mm d-glucose, and 10 mm HEPES, adjusted to pH 7.4 with NaOH) was used for preparing Ca2+ dye solution and for imaging. For Ca2+-free HEPES buffer, CaCl2 was replaced with MgCl2 and 0.1 mm EGTA was added to chelate Ca2+. 4 μm Fluo-4-am dye was prepared by dissolving powder Fluo-4-am (Thermo Fisher Scientific) in Ca2+-containing HEPES buffer supplemented with 0.03% pluronic acid (Thermo Fisher Scientific). On the day of imaging, each coverslip was incubated in 4 μm Fluo-4-am for 45 min at 37 °C. The cells were then washed three times and bathed in HEPES-buffered saline solution for 5 min before Ca2+ measurements began. The signal for each cell was subtracted by the background signal and normalized to the first 10-s baseline. The cells that showed a UTP response of greater than or equal to 1.1-fold of baseline were considered to be responding and included in the data analysis. Rate of rise was estimated as the gradient between 25 and 75% maximum amplitude. All experiments were conducted at room temperature. All figures depicting Ca2+ imaging traces show the averages of all responding cells in 9–21 coverslips from at least three independent recordings.
Colony formation assay
For colony formation assays, once cells in a T75 flask reach confluence of 80–90%, they were detached by the addition of 2 ml of TrypLE (Thermo Fisher Scientific), followed by dilution in 5 ml of fresh medium. Cell density was determined using a hematocytometer. Subsequently, a total number of 100 or 500 cells was added to each well of a 12-well Corning plate (Thermo Fisher Scientific). The cells were then left undisturbed in the incubator over 14 days. After 14 days, the colonies were fixed and stained with 2.5% crystal violet solution, washed to remove excess dye, and scanned with a conventional scanner. The total area covered was determined with ImageJ using a published plugin (19). Data were obtained from three independent experiments and are represented as total area covered in every individual well normalized to controls.
Data analysis
Data management and calculations were performed using PRISM statistical software 7. For comparison between two groups, unpaired, two-tailed Student's t test was carried out. For comparison of more than two groups, one-way analysis of variance (ANOVA) followed by Dunnet's multiple comparisons test was carried out to determine whether the mean from one group is statistically significantly different from that of another group. A p value < 0.05 was considered to be statistically significant, and the following notations were used in all figures: *, p < 0.05; **, p < 0.01; ***, p < 0.005; and ****, p < 0.0001. All error bars show standard deviation unless otherwise stated.
Author contributions
L. D. N. and B. E. E. conceptualization; L. D. N. data curation; L. D. N., E. T. P., and L. K. H. formal analysis; L. D. N. validation; L. D. N., E. T. P., and L. K. H. investigation; L. D. N. methodology; L. D. N. and B. E. E. writing-original draft; L. D. N., E. T. P., L. K. H., and B. E. E. writing-review and editing; B. E. E. supervision; B. E. E. funding acquisition; B. E. E. project administration.
Supplementary Material
Acknowledgments
We acknowledge Dr. David Yule and Dr. Jan Parys for providing reagents and Allison Brill and Dr. Edward Kaftan for helpful discussion.
This work was supported by National Institutes of Health Grant 5P01DK057751 (to B. E. E.). B. E. E. is a founder of Osmol Therapeutics, a company that is targeting NCS1 for therapeutic purposes. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supporting text, Table S1, and Figs. S1-S8.
- ER
- endoplasmic reticulum
- InsP3R
- inositol 1,4,5-trisphosphate receptor
- NCS1
- neuronal calcium sensor 1
- CaM
- calmodulin
- CaBP1
- Ca2+-binding protein 1
- GST
- glutathione S-transferase
- PDB
- Protein Data Bank
- HA
- hemagglutinin
- ANOVA
- analysis of variance.
References
- 1. Berridge M. J. (2016) The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol. Rev. 96, 1261–1296 10.1152/physrev.00006.2016 [DOI] [PubMed] [Google Scholar]
- 2. Eckenrode E. F., Yang J., Velmurugan G. V., Foskett J. K., and White C. (2010) Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor–dependent Ca2+ signaling. J. Biol. Chem. 285, 13678–13684 10.1074/jbc.M109.096040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tengholm A., and Gylfe E. (2009) Oscillatory control of insulin secretion. Mol. Cell Endocrinol. 297, 58–72 10.1016/j.mce.2008.07.009 [DOI] [PubMed] [Google Scholar]
- 4. Fujii S., Matsumoto M., Igarashi K., Kato H., and Mikoshiba K. (2000) Synaptic plasticity in hippocampal CA1 neurons of mice lacking type 1 inositol-1,4,5-trisphosphate receptors. Learn. Mem. 7, 312–320 10.1101/lm.34100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schorge S., van de Leemput J., Singleton A., Houlden H., and Hardy J. (2010) Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 33, 211–219 10.1016/j.tins.2010.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matilla-Dueñas A., Ashizawa T., Brice A., Magri S., McFarland K. N., Pandolfo M., Pulst S. M., Riess O., Rubinsztein D. C., Schmidt J., Schmidt T., Scoles D. R., Stevanin G., Taroni F., Underwood B. R., et al. (2014) Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum 13, 269–302 10.1007/s12311-013-0539-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shibao K., Hirata K., Robert M. E., and Nathanson M. H. (2003) Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology 125, 1175–1187 10.1016/S0016-5085(03)01201-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kasri N. N., Bultynck G., Smyth J., Szlufcik K., Parys J. B., Callewaert G., Missiaen L., Fissore R. A., Mikoshiba K., and de Smedt H. (2004) The N-terminal Ca2+-independent calmodulin-binding site on the inositol 1,4,5-trisphosphate receptor is responsible for calmodulin inhibition, even though this inhibition requires Ca2+. Mol. Pharmacol. 66, 276–284 10.1124/mol.66.2.276 [DOI] [PubMed] [Google Scholar]
- 9. Li C., Chan J., Haeseleer F., Mikoshiba K., Palczewski K., Ikura M., and Ames J. B. (2009) Structural insights into Ca2+-dependent regulation of inositol 1,4,5-trisphosphate receptors by CaBP1. J. Biol. Chem. 284, 2472–2481 10.1074/jbc.M806513200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Boehmerle W., Splittgerber U., Lazarus M. B., McKenzie K. M., Johnston D. G., Austin D. J., and Ehrlich B. E. (2006) Paclitaxel induces calcium oscillations via an inositol 1,4,5-trisphosphate receptor and neuronal calcium sensor 1-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 103, 18356–18361 10.1073/pnas.0607240103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang K., Heidrich F. M., DeGray B., Boehmerle W., and Ehrlich B. E. (2010) Paclitaxel accelerates spontaneous calcium oscillations in cardiomyocytes by interacting with NCS-1 and the InsP3R. J. Mol. Cell Cardiol. 49, 829–835 10.1016/j.yjmcc.2010.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boehmerle W., Zhang K., Sivula M., Heidrich F. M., Lee Y., Jordt S. E., and Ehrlich B. E. (2007) Chronic exposure to paclitaxel diminishes phosphoinositide signaling by calpain-mediated neuronal calcium sensor-1 degradation. Proc. Natl. Acad. Sci. U.S.A. 104, 11103–11108 10.1073/pnas.0701546104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schlecker C., Boehmerle W., Jeromin A., DeGray B., Varshney A., Sharma Y., Szigeti-Buck K., and Ehrlich B. E. (2006) Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. Clin. J. Invest. 116, 1668–1674 10.1172/JCI22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mo M., Erdelyi I., Szigeti-Buck K., Benbow J. H., and Ehrlich B. E. (2012) Prevention of paclitaxel-induced peripheral neuropathy by lithium pretreatment. FASEB J. 26, 4696–4709 10.1096/fj.12-214643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burgoyne R. D. (2007) Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 8, 182–193 10.1038/nrn2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakamura T. Y., Jeromin A., Mikoshiba K., and Wakabayashi S. (2011) Neuronal calcium sensor-1 promotes immature heart function and hypertrophy by enhancing Ca2+ signals. Circ Res. 109, 512–523 10.1161/CIRCRESAHA.111.248864 [DOI] [PubMed] [Google Scholar]
- 17. Iketani M., Imaizumi C., Nakamura F., Jeromin A., Mikoshiba K., Goshima Y., and Takei K. (2009) Regulation of neurite outgrowth mediated by neuronal calcium sensor-1 and inositol 1,4,5-trisphosphate receptor in nerve growth cones. Neuroscience 161, 743–752 10.1016/j.neuroscience.2009.04.019 [DOI] [PubMed] [Google Scholar]
- 18. Koizumi S., Rosa P., Willars G. B., Challiss R. A., Taverna E., Francolini M., Bootman M. D., Lipp P., Inoue K., Roder J., and Jeromin A. (2002) Mechanisms underlying the neuronal calcium sensor-1-evoked enhancement of exocytosis in PC12 cells. J. Biol. Chem. 277, 30315–30324 10.1074/jbc.M201132200 [DOI] [PubMed] [Google Scholar]
- 19. Guzmán C., Bagga M., Kaur A., Westermarck J., and Abankwa D. (2014) ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One 9, e92444 10.1371/journal.pone.0092444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakamura T. Y., Jeromin A., Smith G., Kurushima H., Koga H., Nakabeppu Y., Wakabayashi S., and Nabekura J. (2006) Novel role of neuronal Ca2+ sensor-1 as a survival factor up-regulated in injured neurons. J. Cell Biol. 172, 1081–1091 10.1083/jcb.200508156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yip P. K., Wong L. F., Sears T. A., Yáñez-Muñoz R. J., and McMahon S. B. (2010) Cortical overexpression of neuronal calcium sensor-1 induces functional plasticity in spinal cord following unilateral pyramidal tract injury in rat. PLoS Biol. 8, e1000399 10.1371/journal.pbio.1000399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moore L. M., England A., Ehrlich B. E., and Rimm D. L. (2017) Calcium sensor NCS-1, promotes tumor aggressiveness and predicts patient survival. Mol. Cancer Res. 15, 942–952 10.1158/1541-7786.MCR-16-0408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Apasu J. E., Schuette D., LaRanger R., Steinle J. A., Nguyen L. D., Grosshans H. K., Zhang M., Cai W. L., Yan Q., Robert M. E., Mak M., and Ehrlich B. E. (2019) Neuronal calcium sensor 1 (NCS1) promotes motility and metastatic spread of breast cancer cells in vitro and in vivo. FASEB J. 33, 4802–4813 10.1096/fj.201802004R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamada M., Miyawaki A., Saito K., Nakajima T., Yamamoto-Hino M., Ryo Y., Furuichi T., and Mikoshiba K. (1995) The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. Biochem. J. 308, 83–88 10.1042/bj3080083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J., McBride S., Mak D. O., Vardi N., Palczewski K., Haeseleer F., and Foskett J. K. (2002) Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. USA 99, 7711–7716 10.1073/pnas.102006299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schaad N. C., De Castro E., Nef S., Hegi S., Hinrichsen R., Martone M. E., Ellisman M. H., Sikkink R., Rusnak F., Sygush J., and Nef P. (1996) Direct modulation of calmodulin targets by the neuronal calcium sensor NCS-1. Proc. Natl. Acad. Sci. U.S.A. 93, 9253–9258 10.1073/pnas.93.17.9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin V. M., Johnson J. R., Haynes L. P., Barclay J. W., and Burgoyne R. D. (2013) Identification of key structural elements for neuronal calcium sensor-1 function in the regulation of the temperature-dependency of locomotion in C. elegans. Mol. Brain 6, 39 10.1186/1756-6606-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mansilla A., Chaves-Sanjuan A., Campillo N. E., Semelidou O., Martínez-González L., Infantes L., González-Rubio J. M., Gil C., Conde S., Skoulakis E. M., Ferrús A., Martínez A., and Sánchez-Barrena M. J. (2017) Interference of the complex between NCS-1 and Ric8a with phenothiazines regulates synaptic function and is an approach for fragile X syndrome. Proc. Natl. Acad. Sci. U.S.A. 114, E999–E1008 10.1073/pnas.1611089114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saab B. J., Georgiou J., Nath A., Lee F. J., Wang M., Michalon A., Liu F., Mansuy I. M., and Roder J. C. (2009) NCS-1 in the dentate gyrus promotes exploration, synaptic plasticity, and rapid acquisition of spatial memory. Neuron 63, 643–656 10.1016/j.neuron.2009.08.014 [DOI] [PubMed] [Google Scholar]
- 30. Benbow J. H., DeGray B., and Ehrlich B. E., (2011) Protection of neuronal calcium sensor 1 protein in cells treated with paclitaxel. J. Biol. Chem. 286, 34575–34582 10.1074/jbc.M111.265751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pierce B. G., Hourai Y., and Weng Z. (2011) Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS One 6, e24657 10.1371/journal.pone.0024657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 10.1093/bioinformatics/btu097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kozakov D., Hall D. R., Xia B., Porter K. A., Padhorny D., Yueh C., Beglov D., and Vajda S. (2017) The ClusPro web server for protein–protein docking. Nat. Protoc. 12, 255–278 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.