

Abstract
Inositol-requiring enzyme 1 (IRE1) is an endoplasmic reticulum (ER)-resident transmembrane protein that senses ER stress and is evolutionarily conserved from yeast to humans. IRE1 possesses both Ser/Thr protein kinase and endoribonuclease (RNase) activities within its cytoplasmic domain and is activated through autophosphorylation and dimerization/oligomerization. It mediates a critical arm of the unfolded protein response to manage ER stress provoked by lumenal overload of unfolded/misfolded proteins. Emerging lines of evidence have revealed that in mammals, IRE1α functions as a multifunctional signal transducer that responds to metabolic cues and nutrient stress conditions, exerting profound and broad effects on metabolic homeostasis. In this review, we cover recent advances in our understanding of how IRE1α integrates a variety of metabolic and stress signals and highlight its tissue-specific or context-dependent metabolic activities. We also discuss how dysregulation of this metabolic stress sensor during handling of excessive nutrients in cells contributes to the progression of obesity and metabolic disorders.
Keywords: endoplasmic reticulum stress (ER stress), unfolded protein response (UPR), X-box–binding protein 1 (XBP1), ER-associated degradation, signal transduction, endoplasmic reticulum to nucleus signaling 1 (Ern1), IRE1α, metabolic inflammation, nutrient sensing, regulated IRE1-dependent decay (RIDD)
Introduction
In eukaryotic cells, the endoplasmic reticulum (ER)2 is the largest membrane-bound organelle that forms a contiguous network with interconnected sheets and tubules (1). Spreading throughout the cell, it is typically classified into two types, smooth ER and rough ER. The ER performs a diversity of essential cellular functions, including calcium (Ca2+) storage and release, protein synthesis and processing, lipid biosynthesis and trafficking, and membrane biogenesis. Dynamically interacting with many other organelles (e.g. mitochondria, endosomes, Golgi, lysosomes, and lipid droplets) through membrane contact sites (2–4), the ER is generally thought to be implicated not only in exchanging ions, metabolites, lipids, and proteins, but also in conveying crucial cellular signals. Thus, adaptive regulatory mechanisms for maintaining the dynamic ER integrity are vitally important for cell homeostasis and survival in response to changes in nutritional states and a variety of other environmental challenges.
The ER is the primary site for the synthesis, folding, modification, and trafficking of nearly one-third of the cellular proteome, particularly transmembrane or secreted proteins. An overload of unfolded proteins or excess accumulation of misfolded proteins within the ER lumen instigates a state of the so-called “ER stress,” leading to activation of a highly-conserved adaptive response referred to as the unfolded protein response (UPR) (5–10). The notion of UPR was initially proposed over 30 years ago by Kozutsumi et al. (11) based on the observation that malfolded protein-elicited signals from the ER could trigger the induction of two glucose-regulated proteins, which are later known to be the ER protein chaperones. This was followed by elaborate characterizations, through yeast genetics approaches, of the delicate intracellular UPR signaling pathways (6, 12, 13). The ER-resident transmembrane proteins, PKR-like endoplasmic reticulum kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1), are the essential signal transducers in mediating the three classical arms of the UPR. Coordinated activation of these UPR sensors serves as the quality control mechanism to govern ER proteostasis and cope with ER stress. This is achieved through shutting down the cellular protein translation machinery, while transcriptionally activating gene expression programs to enhance the ER's protein-folding capacity and to promote the elimination of terminally unfolded/misfolded proteins by ER-associated degradation (ERAD) (14). Under prolonged or severe ER stress conditions, however, homeostasis cannot be restored at the ER, and overactivation of the UPR leads to activation of cell death programs (15). A plethora of studies over the last 30 years or so have uncovered the central roles of the UPR signaling arms in the control of many aspects of cell physiology, particularly cell fate decisions (16, 17), as well as their implications in a wide range of pathologies, including cancer, neurodegeneration, and metabolic diseases (18–25).
IRE1, encoded by the Ern1 (Endoplasmic reticulum to nucleus signaling 1) gene, is the most ancient and evolutionarily-conserved UPR sensor that was originally identified in yeast Saccharomyces cerevisiae (26, 27). As an ER-localized type I transmembrane protein (i.e. a membrane-spanning protein with the extracellular/lumenal portion at its N terminus), IRE1 harbors an N-terminal lumenal domain (LD) that can sense the protein-folding status within the ER, and a C-terminal cytoplasmic effector domain that possesses both protein serine/threonine kinase and endoribonuclease (RNase) activities (Fig. 1) (5, 6, 28). Two IRE1 isoforms, designated IRE1α and IRE1β, are encoded by the mammalian genome, with IRE1α found to be the most abundantly and ubiquitously expressed (29). Upon ER stress, IRE1α is activated through dimerization/oligomerization and trans-autophosphorylation, resulting in allosteric activation of its C-terminal RNase domain (30, 31). Activation of IRE1α's RNase activity in turn catalyzes the unconventional splicing of the mRNA encoding X-box–binding protein 1 (XBP1), removing an intron (26-nucleotide in mouse and human) that contains a double hairpin structure (6, 7). Subsequent ligation by an RNA ligase (RtcB in mammals (32)) leads to generation of a spliced form of Xbp1 mRNA that encodes a more stable and transcriptionally-active XBP1s protein, initiating a major UPR gene expression program through up-regulation of protein chaperones, ERAD components, and molecules involved in ER biogenesis (6, 7). IRE1α is also known to exert its signal-transducing actions through two additional mechanisms (Fig. 1). Activated IRE1α RNase can also catalyze the degradation of a number of mRNAs as well as certain pre-miRNAs in a process referred to as regulated IRE1-dependent decay (RIDD) (33), which was initially thought to eliminate select ER-localized mRNA species (34). Moreover, IRE1 is known to form high-order clusters at the ER membrane (35, 36), and IRE1α can interact with many functionally important protein partners such as the proapoptotic BCL-2 family members BAX and BAK, the scaffold proteins TRAF2, TRAF6, and RACK1, and the transcription factor STAT3, potentially acting as a signaling platform to regulate apoptosis, inflammation, and proliferation (37–44). Thus, IRE1 is a genuine multitasked stress-sensing machinery that performs a myriad of cellular functions via a complex signaling network.
Figure 1.

Activation of IRE1α as a metabolic stress sensor. In addition to protein folding overload within the ER lumen, the mammalian ER-localized transmembrane stress sensor IRE1α can also sense the indicated nutrient or metabolic signals that activate its downstream effector activities. As a UPR signal transducer with an N-terminal LD for detecting unfolded/misfolded proteins, IRE1α possesses a cytoplasmic domain with dual kinase/RNase activities. Upon typical ER stress, IRE1α activation involves dimerization/oligomerization and autophosphorylation. Activated IRE1α RNase can catalyze the unconventional splicing of Xbp1 mRNA, removing a 26-nucleotide intron from the unspliced Xbp1 (Xbp1u) mRNA. Subsequently, RNA ligase RtcB-mediated ligation yields the spliced form of Xbp1 (Xbp1s) mRNA and generates a transcriptionally active transcription factor XBP1s to initiate a key UPR gene expression program to cope with ER stress and maintain the homeostatic control of cell proliferation and metabolism. IRE1α can also degrade select sets of mRNAs or pre-miRNAs through a process termed RIDD. In addition, IRE1α can interact with an increasing number of protein partners (indicated by X, Y, and Z) and presumably serves as a signaling platform composed of regulatory factors as well as signaling effectors. Under metabolic stress conditions, IRE1α may act as a multitasked protein machinery that is involved in regulating many aspects of metabolism.
Numerous studies have implicated IRE1α signaling, through the three aforementioned mechanisms, in a diverse range of biological processes in mammals, including cell survival/death determination, immunity, and metabolism, which have been previously reviewed (16, 22, 45–47). In particular, ER stress is highly associated with obesity and insulin resistance (22, 48, 49), which arises from chronic imbalance of food intake and energy expenditure. As indicated by the implication of the IRE1α–XBP1 pathway in obesity-related metabolic disorders (50), molecular elucidation of the complex mechanisms linking the IRE1α signaling network during handling of an excessive load of nutrients to chronic inflammation and metabolic dysfunctions has been a very active and vigorous research field. Currently, it is increasingly recognized how important it is to dissect the metabolic roles of IRE1α as well as its mechanisms of action in the context of different cell types and various nutrient stress conditions. Therefore, we intend to summarize herein the recent progress with regard to the physiological roles of IRE1α in metabolic organs based on studies of various animal models, with a focus upon its tissue- or cell type-specific regulatory actions in energy balance and glucose/lipid metabolism. We will also discuss how dysregulation of IRE1α under overnutrition-elicited metabolic ER stress may mediate the pathological progression of obesity and related metabolic diseases, including type 2 diabetes, nonalcoholic fatty liver disease (NAFLD), and atherosclerosis.
IRE1α integrates nutrient, hormonal, and metabolic stress signals
Given the unique ER structure spreading throughout the cell to handle both the protein folding task and the intracellular flow of ions, nutrients, and metabolites, it is naturally not surprising that the ER undergoes dynamic remodeling in response to nutritional changes and metabolic challenges. As the key signal transducer in the regulatory machinery that governs proteostasis and ER's functional integrity, IRE1α can respond not only to an increased load of protein folding within the ER lumen, but also to other intracellular signals derived from metabolite fluxes such as lipid composition alterations that lead to disruption of ER Ca2+ storage (51). It has been well-documented how the lumenal domain of IRE1α senses unfolded/misfolded proteins during ER stress, either through directly binding to unfolded proteins or via its dissociation from the ER chaperone protein BiP/GRP78 (binding-immunoglobulin protein/glucose-regulated protein 78) (8). It remains largely unclear, however, whether there exist alternative modes of IRE1α-activating mechanisms that do not stem from within the ER lumen.
Animal model studies, particularly in the setting of obesity or other metabolic stress conditions, have led to an increasing recognition that IRE1α in metabolic organs can respond to changes in the body's nutritional and metabolic states (Fig. 1). The activation of IRE1α signaling, which can be typically evaluated by measuring its phosphorylation state or Xbp1 mRNA splicing activity, has been frequently observed in multiple tissues and cell types from dietary and genetic obesity mouse models. This state of so-called “metabolic ER stress” is presumably provoked by a multitude of stimuli arising from both systemic and tissue microenvironmental changes in the face of energy surplus. From studies of metabolically-active organs or cell types in response to metabolic stress-inducing factors, emerging lines of evidence suggest that IRE1α can virtually integrate three major types of signals: nutrients, hormones, and immunological stimuli. Although an altered load of protein folding, i.e. increased accumulation of lumenal unfolded/misfolded proteins, can be viewed as a major stressor for activating IRE1α signaling under most circumstances, additional factors are also very likely involved in triggering its activation, through either cytosolic signaling cross-talk or perturbations in the ER's structural integrity, e.g. lipid composition changes that cause ER membrane remodeling (51).
Glucose-sensing of IRE1α
The nutrient-sensing property of IRE1α can be best-exemplified by its ability to respond to high-glucose stimulation in professional secretory pancreatic islet β-cells (42, 52, 53). Several groups have reported that high glucose is able to stimulate the phosphorylation of IRE1α at Ser724 within its kinase activation domain in cultured insulinoma β-cell lines as well as in primary pancreatic islets, resulting in elevated Xbp1 mRNA splicing (53) without inducing the activation of the PERK–eIF2α branch of the UPR (42). Although the molecular signaling mechanism by which glucose stimulates the selective activation of the IRE1α arm is largely obscure, this glucose-responsive phosphorylation of IRE1α, mostly likely reflecting its activation, is coupled to insulin biosynthesis (42, 52). Furthermore, research from our group revealed that glucose-stimulated IRE1α phosphorylation can be negatively regulated through glucose-induced IRE1α association with the adaptor protein receptor for activated C kinase 1 (RACK1), which in turn recruits protein phosphatase PP2A for its dephosphorylation (42). Interestingly, prolonged high-glucose exposure was found to cause dissociation of PP2A from RACK1 along with a higher degree of IRE1α phosphorylation, mimicking that under the chemically-induced ER stress state (42). This suggests that IRE1α's glucose-sensing activation is dynamically controlled.
Lipid-sensing of IRE1α
Lipid species can also stimulate the activation of the IRE1α-signaling pathway in different types of cells such as β-cells, macrophages, and endothelial cells. Free fatty acid (FFA) has been documented as a typical ER stressor, which is thought to have a key role in lipotoxicity-induced β-cell death (54). In macrophages, it was demonstrated that saturated fatty acid (SFA) can activate IRE1α in a manner that does not rely on its unfolded protein-sensing ability. This, in turn, promotes the activation of the NOD-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome in the “metabolically-activated” macrophages that drive high-fat diet (HFD)-induced tissue inflammation (55). Interestingly, the ability of SFAs to stimulate IRE1α activation is thought to be associated with its flux into phosphatidylcholine (PC), which may contribute to the perturbation of ER lipid composition. In addition, cholesterol trafficking to the ER membrane has been proposed to cause depletion of ER Ca2+ storage, activating the UPR, including the IRE1α–XBP1 pathway (56). However, it remains unknown whether IRE1α directly mediates cholesterol-induced cytotoxicity or exerts cytoprotecting actions during ER stress–associated apoptosis of cholesterol-loaded macrophages, which has a key role in the progression of atherosclerosis (56). In addition, oxidized low-density lipoproteins, as well as the lipid oxidation products 7-ketocholesterol and 4-hydroxynonenal, were also shown to evoke the activation of the IRE1α–XBP1 pathway in human endothelial cells (57). How is IRE1α sensing these lipid molecules? Recent studies have shown that IRE1α can be activated by SFA through an unexpected unique mechanism without involving its lumenal sensing of protein folding load (58). The transmembrane (TM) domain of IRE1α was found to contain intrinsic structural elements for detecting ER membrane lipid saturation, allowing the IRE1α's TM domain to dimerize for its activation in response to membrane aberrancy (59–61). It will be interesting to interrogate further whether such lipid sensing of IRE1α occurs in localized regions/subdomains of the ER membrane undergoing lipid bilayer stress and through a similar mechanism as suggested for yeast Ire1 protein (62).
IRE1α activation by metabolic hormones
Investigations of how IRE1α is metabolically regulated, mostly carried out in the liver, have revealed that IRE1α can be activated by both anabolic and catabolic hormones. Due to the onset of insulin resistance in mice following HFD feeding, hyperinsulinemia was found to accompany elevations in phosphorylation at Ser724 of hepatic IRE1α and Xbp1 mRNA splicing (63). Furthermore, prolonged insulin exposure could activate the IRE1α–XBP1 pathway in nonobese diabetic livers as well as in primary hepatocytes, which was linked to up-regulation of the de novo lipogenic program through XBP1s-mediated control of sterol regulatory element-binding protein 1c (SREBP1c) (63). Notably, this could be suppressed by inhibition of the mammalian target of rapamycin–dependent protein synthesis. Acute activation of the IRE1α–XBP1 pathway was also documented in the liver of mice upon refeeding, presumably attributable, at least in part, to the increased postprandial blood insulin level (64). Insulin is an anabolic hormone known to stimulate both protein synthesis and lipogenesis in the liver, and insulin stimulation can lead to an increased load of protein folding within the ER lumen while altering the lipid composition of the ER membrane as well. Paradoxically, animal studies from our group uncovered that, upon short-term starvation, phosphorylation at Ser724 of IRE1α was stimulated without significant activation of its Xbp1 mRNA–splicing activity in the liver when compared with that under the ad libitum–fed state; furthermore, glucagon secreted from pancreatic islet α-cells during fasting was able to induce IRE1α phosphorylation in primary hepatocytes (65). More surprisingly, we found that glucagon or epinephrine signaling-activated PKA could directly phosphorylate IRE1α at Ser724 in a fashion independent of its autophosphorylation ability (65). This catabolic activation of IRE1α was implicated in promoting glucagon stimulation of the hepatic gluconeogenic program (65), but the precise mechanism by which IRE1α regulates hepatic glucose production remains unclear. It is tempting to speculate the possibility that hepatic IRE1α phosphorylation at Ser724 in response to the fasting/feeding cycle is coupled to the circadian control of its activation (66), and IRE1α is likely to sense the circadian input and participate in the rhythmic control of hepatic lipid metabolism (66) as well as gluconeogenesis (67).
IRE1α activation by inflammatory stimuli
Early studies have indicated that IRE1α is connected to JNK and IKKβ–NF-κB inflammatory pathways through interacting with TNF-receptor–associated factor 2 (TRAF2) (41, 68). More recent immunometabolic research has pointed to the critical involvement of IRE1α in regulating or mediating immune responses that can profoundly influence metabolic homeostasis. In this context, it has been shown that IRE1α in primary macrophages could be selectively activated by Toll-like receptor agonists such as lipopolysaccharide (LPS), which did not appear to elicit typical ER stress response; and this, in turn, could drive M1-polarized activation of macrophages (also termed classically activated macrophages) and the inflammatory gene expression program (69–71). Moreover, our research on IRE1α signaling in adipose tissue macrophages (ATMs) in relation to obesity-associated metabolic inflammation showed that IL-4, a cytokine known to promote M2-polarized activation of macrophages (also termed alternatively-activated macrophages) (72, 73), was also able to induce IRE1α phosphorylation and activate its Xbp1 mRNA-splicing activity (70). Interestingly, two innate immune sensors, nucleotide-binding oligomerization domain 1 (NOD1) and NOD2, were found to be associated with the IRE1α–TRAF2 axis, and pharmacological inhibition analyses suggested that IRE1α might serve as a key component in sensing cytosolic danger signals to trigger the pro-inflammatory responses (e.g. IL-6 production) during tissue damage or microbial infection (74). These findings suggest a crucial role of IRE1α in coupling ER stress to inflammation, which may have particular relevance in obesity-associated metabolic inflammation.
Taken together, it is most likely that under metabolic ER stress, IRE1α may respond to a cluster of metabolic conditions, including hyperglycemia, hyperinsulinemia, hyperlipidemia, abnormal levels of blood glucagon, and SFAs, as well as various immune response stimuli (e.g. higher LPS levels as a result of increased intestinal permeability during diet-induced obesity) or pro-inflammatory cytokines. It is also worth noting that the IRE1α–XBP1 pathway can be activated in the liver of mice following long-term starvation or when fed a ketogenic (75) or high-fructose diet (76), as well as in cancer cells when mice are fed a low-protein isocaloric diet but not a low-carbohydrate diet (77). Thus, this may reflect IRE1α's unique metabolic sensing capacity for responding to various diet-induced changes in nutrients (e.g. carbohydrates, fatty acids, or amino acids) and hormones. In addition to metabolic hormones, a vast diversity of metabolites, such as short-chain fatty acids and branched-chain amino acids, is known to be involved in regulation of many aspects of energy metabolism (78). It can be anticipated that more extensive research will offer new insights into IRE1α's nutrient- or metabolite-sensing mechanisms and its important roles in metabolic functions and dysfunctions, as in the case of G-protein–coupled receptor (GPCR)-mediated signaling of various metabolites (79).
Regulatory actions of IRE1α in metabolic tissues
Thus far, we have summarized recent results revealing IRE1α as a nutrient stress sensor that can be activated in response to changes of metabolic cues. Now, we will focus on its regulatory functions in metabolic homeostasis. As mentioned previously, a great number of animal model investigations, especially in the settings of obesity or other stress conditions, have demonstrated critical roles of IRE1α signaling in various metabolic tissues, including hypothalamus, pancreatic islets, liver, and adipose tissue (Fig. 2). In particular, the IRE1α–XBP1 pathway has been most extensively interrogated using loss–of–function and gain–of–function strategies (23, 50). These studies have revealed the tissue- or cell type-specific, and in most cases context-dependent, features of IRE1α regulation of metabolism, unveiling the complex network of IRE1α signaling that interconnects with metabolic and inflammatory pathways.
Figure 2.
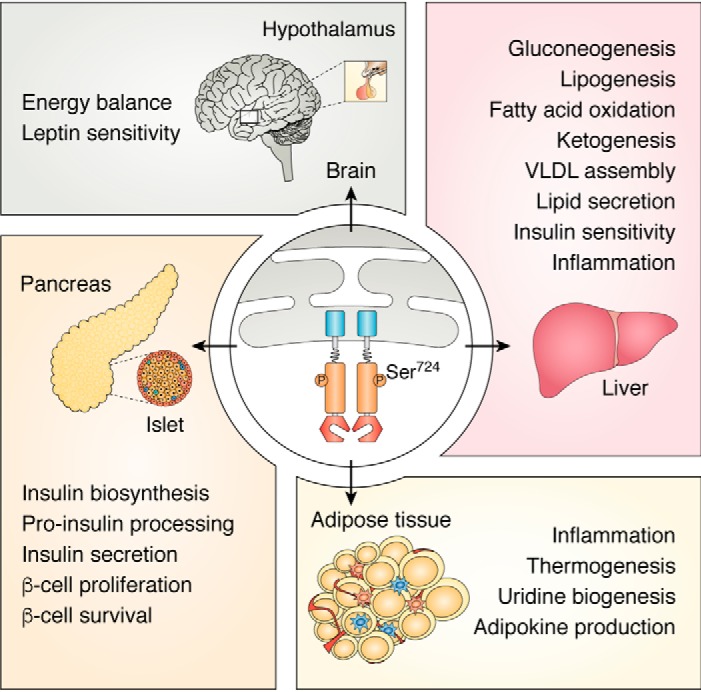
IRE1α exerts a broad range of tissue- or cell type-specific functions in metabolic organs. IRE1α responds to metabolic cues and nutrient stress signals, exerting broad and profound metabolic effects. In the hypothalamus, IRE1α is implicated in energy balance control and regulation of leptin sensitivity. In pancreatic islets, IRE1α responds to glucose stimulation and acts to control insulin biosynthesis, processing, and secretion, as well as the compensatory proliferation and growth of β-cells in the setting of metabolic ER stress and insulin resistance. In the liver, IRE1α responds to both anabolic and catabolic stimulatory signals and regulates a variety of metabolic pathways in glucose and lipid metabolism as indicated. In adipose tissues, IRE1α performs regulatory functions in adipogenesis, thermogenesis, and metabolic inflammation.
IRE1α regulation of energy balance in hypothalamus
The hypothalamus of the central nervous system serves as the headquarters of energy balance control via a complex neuroendocrine circuitry to regulate food intake and energy expenditure. Located in the arcuate nucleus of hypothalamus, orexigenic agouti-related peptide neurons, and anorexigenic pro-opiomelanocortin (POMC) neurons are the best-characterized leptin-responsive neurons with opposing regulatory functions, which act to integrate the nutritional states of the body and orchestrate the central control of energy balance (80). Documented studies in mouse models have shown that ER stress is implicated in hypothalamic inflammation-associated insulin and leptin resistance during diet-induced obesity (81, 82). Interestingly, Williams et al. (83) reported a mouse model with inducible expression of XBP1s specifically in POMC neurons, which exhibited protection against HFD-induced obesity with improved leptin and hepatic insulin sensitivity. XBP1s expression in POMC neurons also led to an up-regulated thermogenic program in adipose tissues, along with higher Xbp1 mRNA splicing in the liver that is thought to represent a cell-nonautonomous UPR signaling mechanism between the brain and liver (84). In addition, Yao et al. (85) reported another mouse model in which exons 16 and 17 encoding a portion of the kinase domain within the Ern1 gene was deleted in POMC neurons, and in this model, POMC neuron-specific IRE1α deficiency resulted in marginal acceleration of HFD-induced obesity with considerable impairments in food intake and energy expenditure. Loss of IRE1α in POMC neurons was thought to aggravate ER stress-induced leptin and insulin resistance (85). In contrast, Xiao et al. (86) reported a different IRE1α-deficient mouse model in which exon 2 of the Ern1 gene was specifically deleted in POMC neurons, which exhibited significant resistance to HFD-induced obesity and improvement of insulin resistance. It was also shown that POMC neuron-specific ablation of IRE1α led to increased energy expenditure and leptin sensitivity with higher production of α-melanocyte–stimulating hormone in the hypothalamus (86). This apparent discrepancy in the phenotypes of these two mouse models likely resulted from the different regions of IRE1α protein that were targeted for genetic deletion. Whereas removal of exon 2 was shown to result in a complete abrogation of IRE1α protein expression (75), deletion of exons 16 and 17 could lead to generation of a mutant IRE1 protein with deletion within its kinase domain, namely a 115-kDa ΔIRE1α protein as recently reported in B-cells (87). Although it remains unclear whether such a mutant IRE1α protein could act in POMC neurons (e.g. through forming signaling clusters with its interacting partners) to affect leptin sensitivity, it is reasonable to speculate that the findings in these animal models largely reflect the multifaceted functions of IRE1α protein.
IRE1α modulation of pancreatic islet cells
Studies of several conditional IRE1α or XBP1 knockout mouse models have established that IRE1α can regulate the entire process of insulin production from pancreatic islet β-cells, ranging from insulin biosynthesis, processing, and secretion through both its Xbp1 mRNA splicing and RIDD activities. Despite that sustained production of XBP1s was shown to induce dysfunction and apoptosis in primary β-cells (88), genetic ablation of XBP1 in mice resulted in the loss of β-cells, decreased insulin content, and impaired insulin maturation, largely ascribable to hyperactivation of IRE1α whose increased RIDD activity could act to degrade mRNAs encoding proinsulin-processing enzymes (89). Furthermore, in two mouse models with genetic deletion of IRE1α in pancreatic β-cells, the IRE1α–XBP1 pathway was shown to not only regulate oxidative proinsulin folding, insulin processing, glucose-stimulated secretion of insulin, and protection against oxidative stress (53, 90), but also to promote pancreatic islet growth and β-cell proliferation in the face of obesity and insulin resistance (91). Therefore, properly-balanced control of IRE1α's Xbp1 mRNA splicing versus its RIDD activity is essential for the maintenance of β-cell homeostasis and insulin-producing capacity. Notably, a mouse model with pancreatic islet α-cell–specific XBP1 knockout was reported to display glucose intolerance, with an inability to suppress glucagon secretion upon glucose stimulation (92). Similar to β-cells, loss of XBP1 also caused hyperactivation of IRE1α, leading to increased JNK activation and impaired insulin signaling in α-cells. It remains unknown, however, whether IRE1α's RNase activity is directly involved in regulating glucagon secretion in α-cells.
IRE1α regulation of liver glucose and lipid metabolism
The liver is an essential metabolic organ in which various metabolic pathways in glucose and lipid metabolism are interconnected and converged. A series of animal model studies have demonstrated that IRE1α serves as a key component of the ER's nutrient-sensing network (22), acting to regulate many aspects of hepatic metabolism. There exists an intricate network connecting the IRE1α–XBP1 branch to metabolic and inflammatory pathways (21). Conceivably, the regulatory roles of IRE1α signaling revealed by these investigations largely rely upon the feeding conditions and dietary compositions, unveiling the dynamic nature of IRE1α signaling actions in response to different types of metabolic stress.
The IRE1α–XBP1 pathway in the liver can be activated upon refeeding and after insulin stimulation (63, 64), indicating that it can mediate the anabolic actions of insulin. This notion is supported by XBP1s promotion of hepatic lipogenesis directly through XBP1s-dependent up-regulation of SREBP1c and fatty-acid synthase (63). Accordingly, studies of mice with liver-specific XBP1 knockouts also revealed its role in regulating the lipogenic program when fed high-carbohydrate diets (93). Moreover, following feeding of high-fructose chow, mice with liver-specific XBP1 deficiency showed reduced hepatic steatosis and improved hepatic insulin sensitivity in parallel with reduced hepatic diacylglycerol (DAG) content and protein kinase Cϵ (PKCϵ) activity, despite increased ER stress and IRE1α activation-associated JNK (76). Remarkably, the nuclear translocation of XBP1s protein has been shown to be facilitated by associating with the regulatory subunits of phosphoinositide 3-kinase, p85α, and p85β, the critical components of the insulin-signaling network (94, 95). Moreover, this nuclear translocation process was reported to be enhanced by interacting with bromodomain-containing protein 7 (BRD7), which could be further promoted by insulin but was found to be suppressed in the state of obesity and insulin resistance (96).
Additional evidence further suggests that XBP1s may serve as an effector in contributing to the metabolic actions of insulin. XBP1s was demonstrated to interact with FoxO1, the transcription factor that is negatively controlled by insulin signaling, acting to direct its proteosomal degradation and suppress hepatic gluconeogenesis (97). Given that XBP1u protein was also known to interact with FoxO1 for its proteosomal degradation (98), it is of particular interest to unveil the molecular details regarding how endogenous XBP1u/s proteins can cooperate in the control of FoxO1 protein turnover during metabolic stress. Moreover, both the stress-activated protein kinase p38 mitogen-activated protein kinase (MAPK) and IκB kinase β (IKKβ) of the inflammatory pathway were shown to phosphorylate XBP1s (99, 100), thereby enhancing its nuclear translocation (99) or protein stability (100). Depending upon the genetic or HFD-induced obesity mouse models, nuclear XBP1s localization was arguably found to be impaired (50) along with abnormal hepatic p38 MAPK and IKKβ activity (99, 100), and restoration of nuclear XBP1s expression in the liver could ameliorate hyperglycemia. It remains enigmatic, however, whether phosphorylation of endogenous XBP1s by p38 MAPK or IKKβ in vivo can influence its selection of targeted gene sets for transcriptional activation or its ability and selectivity for interacting with other protein factors, such as the orphan nuclear receptor small heterodimer partner or the Cullin3-SPOP (speckle-type POZ protein) E3 ligase complex (101), under different metabolic stress conditions. Highlighting again the relevance of differing animal models and experimental contexts, adenovirus-mediated XBP1s expression in the liver exerted an unexpected anti-lipogenic effect in both HFD-induced and genetically-obese mice with insulin resistance, leading to suppressed lipogenesis and alleviated hepatic steatosis with lower PKCϵ activity and enhanced macrolipophagy (102).
Research from our group has uncovered that hepatic IRE1α can also be activated under catabolic states. We found in mice that glucagon-activated PKA kinase could stimulate IRE1α phosphorylation at Ser724 without activating its Xbp1 mRNA splicing in the liver following a short-term fast, and IRE1α phosphorylation at Ser724 was markedly elevated owing to higher hepatic PKA activation in mice with genetic obesity (65). Moreover, knockdown of hepatic IRE1α expression reduced the gluconeogenic program and normalized glucose intolerance in the face of obesity-associated metabolic stress (65). However, we found that prolonged starvation or feeding of a ketogenic diet resulted in activation of the IRE1α–XBP1 pathway in the liver (75), presumably arising from higher levels of metabolic stress signals (e.g. glucagon as well as FFAs). Further interrogations using hepatocyte-specific IRE1α knockout mice revealed that IRE1α could promote fatty acid oxidation and ketogenesis as well as lipid secretion in the liver, and it exerted these catabolic effects largely through XBP1s-dependent transcriptional up-regulation of peroxisome proliferator-activated receptor α (PPARα), the master starvation regulator (75). Interestingly, the IRE1α–XBP1 pathway was also documented to directly activate the transcriptional expression of starvation hormones fibroblast growth factor 21 (FGF21) (103) and growth differentiation factor 15 (GDF15) (104), which might also contribute to promoting hepatic fatty acid β-oxidation and ketogenesis, thereby counteracting ER stress-related liver steatosis.
In line with its role in hepatic lipid homeostasis, Wang et al. (105) employed hepatocyte-specific IRE1α knockout mice and showed that the IRE1α–XBP1 pathway could regulate the assembly and secretion of triglyceride (TG)-rich very-low-density lipoproteins (VLDLs); and loss of IRE1α in the liver exerted a hypolipidemic effect, which could be partially attributed to XBP1s up-regulation of protein-disulfide isomerase, a key enzyme in the control of VLDL assembly by affecting microsomal triglyceride transfer protein activity (105). Interestingly, So et al. (106) took advantage of the fact that XBP1 deficiency in mice led to a feedback hyperactivation of IRE1α and identified the RIDD activity of such highly-activated IRE1αs in down-regulating a number of mRNAs encoding proteins involved in lipid and lipoprotein biogenesis such as angiopoietin-like protein 3, which is also an XBP1s target gene. As anticipated, hepatic XBP1 ablation showed a plasma lipid-lowering effect in dyslipidemic ApoE-deficient mice (106). Further supporting its regulatory role in hepatic lipid secretion, the IRE1α–XBP1 pathway was recently found to orchestrate the transcriptional up-regulation of the COPII vesicle secretory program in another mouse model with hepatocyte-specific IRE1α abrogation (107), thus affecting ApoB-associated lipid secretion in response to nutritional changes. Putting these findings together, it can be postulated that bidirectional modulating strategies of targeting IRE1α RNase for inhibiting its Xbp1 mRNA splicing while boosting its RIDD activity would bring about beneficial effects upon lowering atherogenic blood lipids.
From these extensive studies, it can be appreciated that the IRE1α–XBP1 pathway actively participates in the control of a wide range of metabolic pathways under either anabolic or catabolic conditions in the liver. It is worth further dissecting the molecular modifications of IRE1α or XBP1s under these opposing metabolic states, which may govern its functional outputs. Nonetheless, the findings reported thus far have illustrated the highly-complex mechanisms by which hepatic IRE1α responds to metabolic cues and operates to regulate the metabolic switches and pathways in fuel metabolism. It should be reiterated that, most likely due to differences in genetic manipulations or experimental conditions, different phenotypic effects have been observed. For instance, XBP1 was shown to have a lipogenic role in hepatocyte-specific XBP1 knockout mice when fed a fructose chow diet (76), whereas an anti-lipogenic effect of adenoviral XBP1s expression was reported in obese mouse models with elevated plasma triglyceride levels observed (102). In the former model, reduced lipid secretion due to hyperactivation of IRE1α's RIDD activity might also contribute to the decreased plasma TG levels, while the abolished XBP1s-dependent lipogenesis showed its prominent effect when fed a high-fructose diet (76); whereas in the latter, XBP1s promotion of VLDL secretion could be a predominant factor for alleviating hepatic steatosis under the reported experimental settings. For another example, mice with liver-specific knockouts of IRE1α (through deletion of exons 16 and 17 within the kinase domain that presumably could result in production of the 115-kDa ΔIRE1α protein (87)) showed an elevated degree of hepatic steatosis upon chemical ER stress or following HFD (45% fat) feeding (108, 109). However, we observed that when fed a HFD (60% fat), mice with hepatocyte ablation of IRE1α (through removal of exon 2 of the Ern1 gene) exhibited a slight alleviation of hepatic steatosis in the obesity-accelerated, diethylnitrosamine-induced hepatocellular carcinoma (HCC) model (110). These results point to potential previously unrecognized functional outputs of IRE1α that may be independent of its kinase or RNase activities.
IRE1α regulation of adipose tissue function
Adipose tissue plays a pivotal role in the control of whole-body energy balance and glucose metabolism, functioning not only as the energy storage depot but also as an active endocrine organ that secretes many adipokines such as leptin, adiponectin, and inflammatory cytokines (111, 112). Three types of adipocytes have been characterized, including unilocular, energy-storing white adipocytes, and energy-dissipating, mitochondrial uncoupling protein 1 (UCP1)-expressing brown adipocytes and beige/brite adipocytes with thermogenic capacity (113). Adipose tissue is known to regulate many physiological aspects of metabolic homeostasis, ranging from glucose uptake, lipid storage, to energy expenditure. Adipose tissue inflammation is a hallmark of obesity (114), and adipose-resident immune cells, particularly polarized activation of macrophages, are believed to exert profound effects upon adipocyte function and insulin sensitivity (71–73, 115). Increased ER stress signaling in white adipose tissue has been described in both animal models and human patients with obesity and diabetes (22, 48, 116), and emerging evidence from studies of XBP1 in adipocytes and IRE1α in ATMs suggests that IRE1α signaling is critically involved in the regulation of adipose tissue inflammation, metabolism, and remodeling.
The IRE1α–XBP1 pathway has been documented to be required for adipogenesis, with defective adipocyte differentiation observed in XBP1-deficient mouse embryonic fibroblasts and 3T3-L1 cells with knockdown of XBP1 or IRE1α (117). Because the adipogenic defect in XBP1-deficient cells could be rescued by restored XBP1s expression, IRE1α-mediated Xbp1 mRNA splicing was thought to be a crucial mechanism in adipogenic control (117). Interestingly, PKA-dependent activation of the IRE1α–XBP1 pathway was shown to up-regulate the expression of UCP1 in primary brown adipocytes (118), the essential mitochondrial protein that dissipates chemical energy as heat during thermogenic respiration. However, a mouse model study showed that deletion of adipocyte XBP1 had no effect on adipocyte formation or on systemic metabolism when fed a regular or high-fat diet (119). By contrast, inducible XBP1s overexpression in adipocytes resulted in reduced adiposity in genetic or diet-induced obese mouse models (120), and this anti-obesity effect of adipocyte XBP1s was thought to result from stimulated uridine biosynthesis in adipocytes, which has been linked to maintaining whole-body metabolic balance, including thermogenesis (121). Moreover, XBP1s overexpression in adipocytes was also reported in lean and obese mouse models that exhibited improvement in insulin resistance and glucose intolerance (122), largely because of the enhancement of ER chaperone-mediated maturation of high-molecular-weight adiponectin, the multifunctional adipokine that exerts a broad range of beneficial effects upon metabolic health (123). These studies thus suggest that adipocyte IRE1α plays regulatory roles in adipose tissue metabolism through an XBP1-dependent mechanism under certain circumstances, although its XBP1-independent actions have yet to be explored.
In addition, given that macrophages are the primary effector cells in orchestrating tissue-immune and inflammatory responses (124), we studied metabolic ER stress and IRE1α's function in ATMs in response to nutrient overload. Utilizing mice in which IRE1α was specifically ablated, by removal of exon 2 within the Ern1 gene, in myeloid cells, we found that IRE1α deficiency led to balanced M1/M2 polarization of ATMs, in parallel with marked reduction of adipose tissue inflammation, prominent enhancement of brown adipose tissue activation, and white adipose tissue browning (70). Mice with myeloid IRE1α deletions were completely protected against HFD-induced obesity, insulin resistance, hyperglycemia, dyslipidemia, and hepatic steatosis (70), indicating a powerful role for IRE1α in governing the activation states of ATM to promote energy expenditure through boosting the thermogenic ability of brown and beige adipocytes. These findings suggest that macrophage IRE1α can sense nutrient overload and promote pro-inflammatory M1 polarization while suppressing M2 polarization, thus linking metabolic ER stress to adipose inflammation, energy imbalance, and metabolic disorders. However, the following remains to be further deciphered: the molecular nature of the signals derived from nutrient overload that IRE1α senses, and how IRE1α-deficient macrophages communicate with adipocytes to affect their metabolic properties (125).
Mechanisms of IRE1α dysregulation linking ER stress to metabolic dysfunction
Now that we have summarized the broad metabolic roles of IRE1α signaling in different metabolic tissues, we will discuss the potential mechanisms that may lead to its dysregulation during metabolic ER stress. Overnutrition, i.e. chronic energy surplus, is a major factor driving the pathogenic progression of obesity, which is highly associated with a chronic, subacute state of inflammation along with dramatic structural remodeling of the gut microbiome (126–128). Since the early reports that implicated ER stress in obesity, insulin resistance, and type 2 diabetes (129, 130), a great deal has been learned about the potential mechanisms by which the IRE1α signaling branch of the UPR links metabolic ER stress to obesity-associated tissue inflammation and metabolic disorders, including type 2 diabetes, NAFLD, and atherosclerosis. IRE1α is believed to be subjected to multiple layers of regulatory surveillance (50), and very complex mechanisms have been indicated to cause its dysregulation under various stress conditions. It can be hypothesized that under chronic metabolic stress, nutrient overload may provoke a number of intrinsic alterations to IRE1α, such as phosphorylation status and other post-translational modifications, which may underlie its conversion from an adaptive activation state in physiological settings to a maladaptive, aberrant activation state under pathological conditions (Fig. 3). Such conversion is supposed to profoundly affect IRE1α's functional effector outputs, leading to abnormal enzyme activities (e.g. Xbp1 mRNA splicing versus RIDD), substrate specificities, or signaling efficiencies. Mechanistically, IRE1α dysregulation may stem from altered sensing of metabolic stress signals, disrupted actions of its regulatory partners, and/or aberrant assembly of the multiprotein signaling platform.
Figure 3.
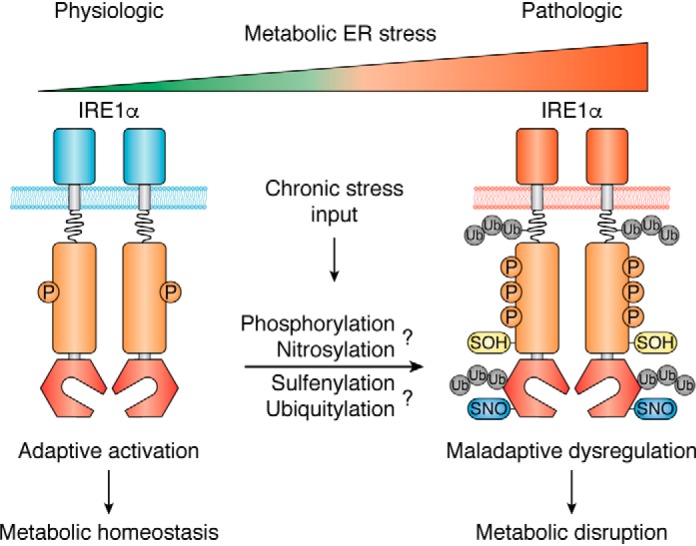
IRE1α dysregulation links metabolic ER stress to metabolic disorders. Under overnutrition-induced chronic ER stress, IRE1α can hypothetically undergo dysregulated activation arising from intrinsic alterations in its phosphorylation and/or other indicated modifications that may cause aberrant protein complex assembly. IRE1α's conversion from an adaptive activation state to a dysregulated form can result in maladaptive effector outputs, thereby exerting its disruptive actions upon metabolic homeostasis.
Aberrant IRE1α phosphorylation
Hyperactivation of IRE1α has been extensively observed and documented in the state of obesity based upon its increased phosphorylation level, mostly as determined by phosphorylation at Ser724 within the activation loop of the kinase domain, usually in parallel with elevated Xbp1 mRNA splicing. As a typical ER stress sensor, activation of IRE1α involves its dimerization/oligomerization and auto-phosphorylation at multiple sites (35, 131), but it is unclear whether IRE1α contains preferentially phosphorylated residues upon activation in response to different stress signals. Surprisingly, we found that hepatic IRE1α could be phosphorylated by glucagon-activated PKA kinase at Ser724, and it had much fewer phosphorylation sites in the livers of genetically-obese mice when compared with that induced by experimental ER stress or upon its overexpression (65). It has yet to be determined whether this PKA-dependent metabolic activation of IRE1α occurs in other metabolic tissues, or how it may affect IRE1α's autophosphorylation profile and regulate its downstream effector actions in metabolic control. Furthermore, IRE1α phosphorylation at particular residues may occur only selectively in response to certain types of stimuli, as exemplified by the bacterial subtilase cytotoxin-induced phosphorylation at Ser729 of IRE1α, which is also located within the activation loop of the kinase domain, in B-cells (87). Therefore, it requires more in-depth investigations to dissect whether and how the extent of IRE1α phosphorylation at critical sites within different functional domains can govern its activation state and dictate its RNase enzyme activity or specificity, e.g. its Xbp1 mRNA splicing versus RIDD actions. During the conversion from an adaptive to a maladaptive activation state, abnormal IRE1α phosphorylation states may ensue as a result of disruption of mechanisms that control its phosphorylation. For example, it is currently unclear whether disruption of the RACK1–PP2A feedback regulatory loop identified in pancreatic β-cells (42) occurs in the liver during obesity-induced metabolic ER stress, thus causing IRE1α hyperphosphorylation at Ser724 and subsequently the dysregulated gluconeogenic program and hyperglycemia (65). Interestingly, it was shown that the RACK1–PP2A regulatory loop was suppressed by TRAF6 association with IRE1α, resulting in LPS-induced hyperactivation of IRE1α RNase activity and enhancement of the inflammatory gene expression program in macrophages (44). It is worth further dissecting whether this mechanism is also involved in promoting M1 polarization of ATMs during HFD-induced adipose inflammation (70).
Post-translational IRE1α modifications
With respect to the possible alterations in IRE1α protein that may disturb its functional outputs, many other types of post-translational modifications, in addition to phosphorylation, of IRE1α are also likely to play a role. Remarkably, Yang et al. (132) reported that in the face of obesity, inflammation-associated increase of inducible nitric-oxide synthase (iNOS) activity resulted in S-nitrosylation of hepatic IRE1α within its RNase domain, leading to a progressive decline in its Xbp1 mRNA splicing activity in both genetic ob/ob and HFD-induced obesity mouse models. Moreover, restored expression of a nitrosylation-resistant version of IRE1α in obese mice with liver-specific IRE1α deficiency normalized Xbp1 mRNA splicing and improved glucose homeostasis. This suggests that inflammation-induced S-nitrosylation impairs IRE1α activity, which may represent one molecular type of obesity-associated IRE1α dysregulation (132). Interestingly, this iNOS-directed dysregulation of the IRE1α–XBP1 pathway was found to be a crucial mechanism in cardiomyocyte dysfunction in a mouse model of heart failure with preserved ejection fraction (133). Moreover, such S-nitrosylation was also shown to impair hepatic IRE1α's RIDD activity, resulting in derepression of miR-200 and miR-34, two miRNAs that target the 3′UTRs of PPARα and SIRT1 mRNAs to attenuate their translation (109). This in turn was thought to promote hepatic steatosis in mice following HFD feeding (109). In addition, it has to be pointed out that IRE1α protein can be subjected to additional types of modification that may affect its functional outputs. For instance, in response to oxidative stress, cytoplasmic reactive oxygen species (ROS) was shown to induce sulfenylation at a conserved cysteine within the kinase activation loop of IRE1α, inhibiting its phosphorylation and Xbp1 mRNA–splicing activity while initiating an antioxidant response (134). Given the important role of mitochondria in ROS generation and cellular metabolism, it is of particular interest to elucidate whether such sulfenylation can link mitochondrial dysfunction to metabolic ER stress in obesity and metabolic diseases. Moreover, although the Sel1L–Hrd1 ERAD complex was shown to regulate IRE1α protein stability (135), several E3 ubiquitin ligases have also been documented to interact with and catalyze IRE1α ubiquitination, including synoviolin (136), TRAF6 (44), C terminus of HSC70-interacting protein (CHIP) (137), and mitochondrial ubiquitin ligase (MITOL/MARCH5) (138). Although synoviolin-mediated ubiquitination was shown to promote its degradation, TRAF6- and CHIP-dependent ubiquitination of IRE1α was found to affect, respectively, its phosphorylation by interfering with the RACK1–PP2A regulatory loop or its activation of the TRAF2–JNK pathway. Intriguingly, MITOL-directed Lys-63–linked chain ubiquitination of IRE1α at lysine 481 was reported to occur at the mitochondria-associated ER membrane (MAM), preventing IRE1α hyper-oligomerization and reducing its RIDD activity. Therefore, it warrants more extensive, in-depth investigations in order to pinpoint the distinct types of modification of IRE1α in the context of how they may bring about maladaptive effector functions of the IRE1α signaling platform in metabolic control.
Alterations in regulatory proteins
Given the increasing number of proteins that have been identified as IRE1α interactors (38), it is conceivable that IRE1α most likely operates within a multiprotein complex machinery containing both regulatory partners and signaling effectors. Many regulatory proteins have been recently reported to either suppress or enhance IRE1α signaling, which include Bax inhibitor-1 (BI-1), an evolutionarily conserved ER-resident protein that interacts through its C-terminal cytosolic domain with IRE1α and negatively controls IRE1α's RNase activity (140–142); ERdj4/DNAJB9, an ER luminal co-chaperone acting as a selective repressor of IRE1α via promoting a complex between BiP and IRE1α (143); heat-shock protein 72 (Hsp72), a stress-inducible cytosolic molecular chaperone that forms a stable complex with the cytosolic domain of IRE1α to enhance its activation (144); protein kinase C substrate 80K-H (PRKCSH/hepatocystin), an ER lumenal noncatalytic subunit of β-subunit of glucosidase II that interacts with and boosts the autophosphorylation and oligomerization of IRE1α upon ER stress (145); and Hsp47, an evolutionarily conserved ER lumenal chaperone that directly binds to IRE1α with high affinity and displaces its negative regulator BiP to facilitate IRE1α oligomerization (146). Although the interplays between these potential IRE1α regulators under metabolic stress conditions remain to be determined, it will be intriguing to decipher how changes in their regulatory modes may contribute to the dysregulation of IRE1α during chronic metabolic ER stress, consequently affecting the homeostatic control of metabolism. Indeed, hepatic BI-1 protein has been found to be down-regulated in the state of obesity (147), along with hyperactivation of the IRE1α–XBP1 pathway (103). Adenoviral restoration of BI-1 protein expression suppressed hepatic IRE1α activation to decrease XBP1s protein production and gluconeogenesis, protecting mice from obesity-associated insulin resistance and glucose intolerance but resulting in higher hepatic steatosis with hypolipidemia (147). This again reflects the multifaceted effector functions of IRE1α signaling that could lead to bidirectional outputs, i.e. reduced hepatic lipid secretion due to deficient XBP1s production and improved insulin sensitivity and suppressed gluconeogenic programming due to inhibition of the activation degree of IRE1α. Supporting this notion, genetic ablation of BI-1 in mice was found to markedly aggravate ER stress–induced or chronic HFD feeding–induced nonalcoholic steatohepatitis (NASH), which was accompanied by unrestrained hepatic IRE1α signaling as indicated by higher XBP1s protein level and NLRP3 inflammasome activation (148). Curiously, pharmacological inhibition showed that the metabolic effects of this BI-1 deficiency-evoked dysregulation of IRE1α could be largely attributable to its elevated RNase activity (148), which in a certain extent contradicted the findings reported for the liver-specific IRE1α knockout mouse model (109). For yet another example, despite that metabolic ER stress may not arise solely from protein folding overload, alterations in ER lumenal chaperone proteins may affect the capacity of IRE1α for sensing or detecting abnormal protein folding, and this can also contribute to IRE1α dysregulation. In this regard, adenoviral expression of the ER chaperone BiP/glucose-regulated protein 78 (GRP78) in the livers of obese ob/ob mice reduced liver XBP1s protein and inhibited SREBP-1c production, leading to prominent alleviation of hepatic steatosis (149). Thus, these findings suggest the important roles of the regulatory molecules in provoking IRE1α dysregulation during chronic ER stress, but the predominant downstream effectors linking aberrant IRE1α signaling to its metabolic disrupting actions remain to be further defined.
Maladaptive outputs of IRE1α effector actions
As a multiprotein signaling platform, it has yet to be clearly illustrated how maladaptive activation of IRE1α through differing states of phosphorylation and/or modification affects its downstream effector outputs, whether by altering its kinase/RNase activities or disrupting its signaling cascades. Most of IRE1α's cellular functions are believed to be mainly performed through activation of its RNase activities. Whereas alterations in both Xbp1 mRNA splicing and RIDD activities of IRE1α have been frequently reported in animal models with obesity and metabolic dysfunctions, it remains obscure whether IRE1α can be selectively activated to dictate its RNase output in terms of Xbp1 mRNA splicing versus RIDD of select mRNA substrates.
In the state of obesity and metabolic ER stress, hyperactivation of IRE1α leads to elevated Xbp1 mRNA splicing, and XBP1s protein produced from this unconventional splicing may exert a broad range of metabolic effects in different metabolic tissues as mentioned previously. In most cases, XBP1s can be viewed as an adaptive effector in the homeostatic control of metabolism, and its defective nuclear translocation or stability has been linked to disruption of glucose metabolism (23, 50, 97, 99, 100). However, maladaptive consequences of higher Xbp1 mRNA splicing were also reported. In hepatocytes during palmitate-induced lipotoxic ER stress, XBP1s could promote the release of pro-inflammatory extracellular vesicles for recruiting macrophages to the liver (150), which is considered to be a potential NASH-promoting factor (151). In obese mouse models, elevated plasma LPS concentration due to increased intestinal permeability is thought to promote insulin resistance, and LPS-activated production of XBP1s was shown to induce the acetyltransferase P300, which in turn could acetylate insulin receptor substrate 1/2 (IRS1/2) to diminish insulin signaling (152). Therefore, these findings suggest the context-dependent metabolic impacts of IRE1α's Xbp1 mRNA splicing in response to different stress signals.
It has been increasingly recognized that hyperactivation of IRE1α's RIDD activity can also mediate its maladaptive, pathological effects during obesity-induced metabolic stress. For instance, overactivation of IRE1α in ATMs was found to down-regulate Irf4 and Klf4 mRNA levels through its RIDD activity, thereby suppressing M2-polarized macrophage activation and connecting metabolic ER stress to adipose inflammation and insulin resistance during HFD-induced obesity (70). In pancreatic β-cells, severe experimental ER stress or IRE1α overexpression was shown to cause RIDD of insulin mRNA as well as β-cell apoptosis (89, 153, 154), although it is unclear whether stress-induced disruption of the RACK1–PP2A control of IRE1α activity can contribute to the hyperactivation of its RIDD activity under chronic metabolic ER stress. Interestingly, hyperactivated IRE1α was also reported to increase the stability of mRNA encoding thioredoxin-interacting protein (TXNIP), likely by its RIDD control of a TXNIP-destabilizing microRNA, miR17, which in turn couples ER stress to activation of the NLRP3 inflammasome and pancreatic β-cell death (155, 156). Notably, mammalian IRE1α may also employ distinct catalytic mechanisms for Xbp1 mRNA splicing and RIDD as found in yeast (157), and opposing Xbp1 mRNA splicing and RIDD outputs have been documented in glioblastoma aggressiveness (158). In this sense, it requires more exquisite dissections to determine which of the RNase outputs may play predominant roles in metabolic dysfunctions in a given tissue or cell type under a certain metabolic stress condition with clinical relevance.
Aberrant IRE1α signaling represents another maladaptive mechanism stemming from chronic activation of IRE1α during metabolic stress. Mice with liver XBP1 haplodeficiency were shown to exhibit insulin resistance with increased JNK activation, which might be largely attributable to IRE1α hyperactivation in the context of XBP1 deficiency (129). Similarly, hepatocyte-specific Xbp1-deficient mice, when chronically fed a high-fat/sugar diet, developed enhanced liver injury and fibrosis but less steatosis in parallel with higher JNK activation and inflammatory signatures (159), which might also result from the IRE1α–TRAF2 signaling axis. Moreover, we found that in the face of HFD-induced obesity, hepatic IRE1α could promote activation of the IKKβ–NF-κB cascade, likely through its interaction with TRAF2, thereby enhancing, in a feed-forward fashion, the sustained activation of the IRE1α–STAT3 pathway during obesity acceleration of HCC progression (110). It can be anticipated that more signaling molecules from the IRE1α platform will be uncovered in mediating metabolic ER stress–related derangement of metabolic control.
All these studies highlight a critical role for IRE1α in linking metabolic ER stress to obesity and metabolic diseases, and it is essential to further decipher the molecular and cellular features of maladaptive dysregulation of IRE1α in different tissues or cell types in response to a diversity of factors from the tissue microenvironment at differing stages during metabolic disease progression (160). In addition to its phosphorylation, modification, enzyme activity, and downstream signaling effector, it should also be borne in mind that most likely IRE1α does not operate alone at the ER membrane in vivo as a homodimeric or homooligomeric machinery, as indicated by its in vitro structures (161), to perform its regulatory functions or exert its deteriorating effects upon metabolic homeostasis. Given IRE1α's clustering property in cells under ER stress or when overexpressed, it has yet to be pinpointed how the maladaptive dysregulation of IRE1α involves its interacting protein partners. In this regard, it is worth noting that cytosolic ABL kinases were recently documented to localize to the ER membrane and promote the clustering and hyperactivation of IRE1α, thereby driving ER stress-associated pancreatic β-cell death (162). Moreover, imatinib, the anti-cancer tyrosine kinase inhibitor that could disrupt the ABL–IRE1α interaction, was shown to blunt IRE1α hyperactivity and reduce β-cell apoptosis, resulting in the reversal of autoimmune diabetes in the mouse model (162). This underscores the translational importance of elucidating the mechanistic details of IRE1α dysregulation that may underpin the pathogenesis of metabolic disorders.
Concluding remarks
Over the last decade, remarkable progress has been made toward our molecular and physiological understanding of IRE1α's metabolic actions as well as its mechanistic connections to obesity-associated metabolic inflammation and metabolic disorders. However, it is only proper to conclude that what we have learned thus far can be largely regarded as the “tip of the iceberg” owing to the complex, multifaceted mechanisms by which IRE1α operates as a multitasked protein machinery in adaptively coping with metabolic stress, and how IRE1α regulation may go awry during the pathological progression of metabolic disorders. A plethora of outstanding and fascinating questions remain to be carefully addressed before we can translate our discoveries into developing IRE1α-targeting therapeutics to combat the devastating epidemic of metabolic diseases. For instance, functioning as a bona fide ER-localized “signalome,” what are the molecular components and cellular signatures of IRE1α machinery during the sensing of different metabolic stress signals? How is this multiprotein machinery temporally and spatially regulated in cells, or does it perform context-dependent functions in compartmentalized locations, i.e. at particular ER subdomains making contact sites with other organelles? Within the assembly of the IRE1α-signaling platform, are the interacting protein partners serving as regulatory auxiliary factors that are subjected to metabolic surveillance control, and how do they govern IRE1α's distinct modes of action or effector outputs? As recently reported, IRE1α might serve as a scaffold to regulate the distribution of inositol 1,4,5-trisphosphate receptors at MAMs, thus facilitating the Ca2+ transfer from the ER to mitochondria regardless of the ER stress state (163). It is likely that IRE1α localization at certain ER subdomains or membrane contact sites can respond to local metabolic changes or stress signals from within other organelles in the cell. Investigations aiming to answer these questions with respect to the subcellular context-dependent functions of IRE1α will provide new knowledge on the molecular determinants of IRE1α dysregulation that can be rather dynamic.
Among all the potential mechanisms that may give rise to maladaptive outputs from dysregulated IRE1α signaling, it is imperative to pinpoint the precise and distinct features in the control of IRE1α's dynamic clustering, RNase activity and substrate selectivity, and signaling effectors. This will allow us to gain more insight into how IRE1α dysregulation in metabolic tissues underlies its crippling actions on metabolic control. Toward this end, it is particularly worth exploring the key factors that drive the conversion of IRE1α from an adaptive to maladaptive activation state, leading to its aberrant effector outputs. Thus far, we have witnessed encouraging progress with therapeutic translation based on molecular dissection of the connection between macrophage IRE1α hyperactivation and metabolic inflammation in obesity, insulin resistance, and atherosclerosis. In this scenario, IRE1α represents a highly-attractive therapeutic target among the three UPR pathways (164). For instance, given that IRE1α was found to be highly activated in lipid-laden macrophages infiltrating the atherosclerotic lesions during metabolic ER stress (165), small molecule inhibitors of IRE1α RNase have shown promising efficacy in counteracting the progression of atherosclerosis in animal models (166). Employing novel single-cell methodologies as in the case for unraveling the pathogenesis of NASH (139), future research will lead to more thorough understanding of the complex cell type- and context-dependent mechanisms by which the IRE1α machinery operates in metabolic control, as well as the pathomechanisms by which its dysregulation promotes the pathological progression of metabolic disorders. This will pave the way for selective targeting with precision of IRE1α's distinct maladaptive outputs, offering new avenues for development of therapeutic agents against obesity and metabolic diseases.
Acknowledgments
We thank Drs. Jianmiao Liu from Huazhong University of Science and Technology, Ling Qi from University of Michigan, and Qiong A. Wang from City of Hope. We also thank all the laboratory members for their insightful discussions and suggestions. We are grateful to Bowen Lv from University of Chinese Academy of Sciences for the artistic preparation of the figures. We apologize to all the colleagues whose work could not be cited because of the scope and space limitations.
This work was supported by National Natural Science Foundation of China Grants 31690102, 91857204, and 81420108006 and Ministry of Science and Technology of China National Key R&D Program of China Grants 2018YFA0800700 and 2016YFA0500100 (to Y. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- ER
- endoplasmic reticulum
- UPR
- unfolded protein response
- PERK
- PKR-like endoplasmic reticulum kinase
- ERAD
- ER-associated degradation
- RIDD
- regulated IRE1-dependent decay
- STAT3
- signal transducer and activator of transcription 3
- PP2A
- protein phosphatase 2A
- FFA
- free fatty acid
- SFA
- saturated fatty acid
- JNK
- c-Jun N-terminal kinase
- IKK
- IκB kinase
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cells
- PKCϵ
- protein kinase Cϵ
- p38 MAPK
- p38 mitogen-activated protein kinase
- PKA
- protein kinase A
- PPARα
- peroxisome proliferator-activated receptor α
- ATM
- adipose tissue macrophage
- iNOS
- inducible nitric-oxide synthase
- CHIP
- C terminus of HSC70-interacting protein
- MITOL
- mitochondrial ubiquitin ligase
- Hsp72
- heat-shock protein 72
- PC
- phosphatidylcholine
- LPS
- lipopolysaccharide
- POMC
- pro-opiomelanocortin
- VLDL
- very-low-density lipoprotein
- TG
- triglyceride
- ApoB/E
- apolipoprotein B/E
- NASH
- nonalcoholic steatohepatitis
- NAFLD
- nonalcoholic fatty liver disease
- HCC
- hepatocellular carcinoma
- ROS
- reactive oxygen species
- MAM
- mitochondria-associated membrane
- LD
- lumenal domain
- HFD
- high-fat diet
- TM
- transmembrane
- TXNIP
- thioredoxin-interacting protein.
References
- 1. Chen S., Novick P., and Ferro-Novick S. (2013) ER structure and function. Curr. Opin. Cell Biol. 25, 428–433 10.1016/j.ceb.2013.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Phillips M. J., and Voeltz G. K. (2016) Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 17, 69–82 10.1038/nrm.2015.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen S., Valm A. M., and Lippincott-Schwartz J. (2018) Interacting organelles. Curr. Opin. Cell Biol. 53, 84–91 10.1016/j.ceb.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu H., Carvalho P., and Voeltz G. K. (2018) Here, there, and everywhere: the importance of ER membrane contact sites. Science 361, eaan5835 10.1126/science.aan5835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schröder M., and Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 10.1146/annurev.biochem.73.011303.074134 [DOI] [PubMed] [Google Scholar]
- 6. Ron D., and Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 7. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- 8. Kimata Y., and Kohno K. (2011) Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 23, 135–142 10.1016/j.ceb.2010.10.008 [DOI] [PubMed] [Google Scholar]
- 9. Hollien J. (2013) Evolution of the unfolded protein response. Biochim. Biophys. Acta 1833, 2458–2463 10.1016/j.bbamcr.2013.01.016 [DOI] [PubMed] [Google Scholar]
- 10. Hetz C., Chevet E., and Oakes S. A. (2015) Proteostasis control by the unfolded protein response. Nat. Cell Biol. 17, 829–838 10.1038/ncb3184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kozutsumi Y., Segal M., Normington K., Gething M.-J., and Sambrook J. (1988) The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464 10.1038/332462a0 [DOI] [PubMed] [Google Scholar]
- 12. Sidrauski C., Chapman R., and Walter P. (1998) The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol. 8, 245–249 10.1016/S0962-8924(98)01267-7 [DOI] [PubMed] [Google Scholar]
- 13. Mori K. (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem. 146, 743–750 10.1093/jb/mvp166 [DOI] [PubMed] [Google Scholar]
- 14. Vembar S. S., and L Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 10.1038/nrm2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 10.1038/nrm3270 [DOI] [PubMed] [Google Scholar]
- 16. Hetz C., and Papa F. R. (2018) The unfolded protein response and cell fate control. Mol. Cell 69, 169–181 10.1016/j.molcel.2017.06.017 [DOI] [PubMed] [Google Scholar]
- 17. Hetz C., and Glimcher L. H. (2011) Protein homeostasis networks in physiology and disease. Curr. Opin. Cell Biol. 23, 123–125 10.1016/j.ceb.2011.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cubillos-Ruiz J. R., Bettigole S. E., and Glimcher L. H. (2017) Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell 168, 692–706 10.1016/j.cell.2016.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matus S., Glimcher L. H., and Hetz C. (2011) Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr. Opin. Cell Biol. 23, 239–252 10.1016/j.ceb.2011.01.003 [DOI] [PubMed] [Google Scholar]
- 20. Hetz C., and Saxena S. (2017) ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 13, 477–491 10.1038/nrneurol.2017.99 [DOI] [PubMed] [Google Scholar]
- 21. Fu S., Watkins S. M., and Hotamisligil G. S. (2012) The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 15, 623–634 10.1016/j.cmet.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 22. Hotamisligil G. S. (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 10.1016/j.cell.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee J., and Ozcan U. (2014) Unfolded protein response signaling and metabolic diseases. J. Biol. Chem. 289, 1203–1211 10.1074/jbc.R113.534743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang M., and Kaufman R. J. (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529, 326–335 10.1038/nature17041 [DOI] [PubMed] [Google Scholar]
- 25. Cnop M., Foufelle F., and Velloso L. A. (2012) Endoplasmic reticulum stress, obesity and diabetes. Trends Mol. Med. 18, 59–68 10.1016/j.molmed.2011.07.010 [DOI] [PubMed] [Google Scholar]
- 26. Cox J. S., Shamu C. E., and Walter P. (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206 10.1016/0092-8674(93)90648-A [DOI] [PubMed] [Google Scholar]
- 27. Mori K., Ma W., Gething M.-J., and Sambrook J. (1993) A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 74, 743–756 10.1016/0092-8674(93)90521-Q [DOI] [PubMed] [Google Scholar]
- 28. Sidrauski C., and Walter P. (1997) The transmembrane kinase ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039 10.1016/S0092-8674(00)80369-4 [DOI] [PubMed] [Google Scholar]
- 29. Tirasophon W., Welihinda A. A., and Kaufman R. J. (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 12, 1812–1824 10.1101/gad.12.12.1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shamu C. E., and Walter P. (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 15, 3028–3039 10.1002/j.1460-2075.1996.tb00666.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Welihinda A. A., and Kaufman R. J. (1996) The unfolded protein response pathway in Saccharomyces cerevisiae: oligomerization and trans-phosphorylation of Ire1p (Ern1p) are required for kinase activation. J. Biol. Chem. 271, 18181–18187 10.1074/jbc.271.30.18181 [DOI] [PubMed] [Google Scholar]
- 32. Guo L., Giasson B. I., Glavis-Bloom A., Brewer M. D., Shorter J., Gitler A. D., and Yang X. (2014) A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 55, 15–30 10.1016/j.molcel.2014.04.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maurel M., Chevet E., Tavernier J., and Gerlo S. (2014) Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 39, 245–254 10.1016/j.tibs.2014.02.008 [DOI] [PubMed] [Google Scholar]
- 34. Hollien J., and Weissman J. S. (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313, 104–107 10.1126/science.1129631 [DOI] [PubMed] [Google Scholar]
- 35. Korennykh A. V., Egea P. F., Korostelev A. A., Finer-Moore J., Zhang C., Shokat K. M., Stroud R. M., and Walter P. (2009) The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 10.1038/nature07661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li H., Korennykh A. V., Behrman S. L., and Walter P. (2010) Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. U.S.A. 107, 16113–16118 10.1073/pnas.1010580107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hetz C., and Glimcher L. H. (2009) Fine-tuning of the unfolded protein response: assembling the IRE1α interactome. Mol. Cell 35, 551–561 10.1016/j.molcel.2009.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Woehlbier U., and Hetz C. (2011) Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem. Sci. 36, 329–337 10.1016/j.tibs.2011.03.001 [DOI] [PubMed] [Google Scholar]
- 39. Chen Y., and Brandizzi F. (2013) IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 23, 547–555 10.1016/j.tcb.2013.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hetz C., Bernasconi P., Fisher J., Lee A.-H., Bassik M. C., Antonsson B., Brandt G. S., Iwakoshi N. N., Schinzel A., Glimcher L. H., and Korsmeyer S. J. (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 312, 572–576 10.1126/science.1123480 [DOI] [PubMed] [Google Scholar]
- 41. Urano F., Wang X., Bertolotti A., Zhang Y., Chung P., Harding H. P., and Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 10.1126/science.287.5453.664 [DOI] [PubMed] [Google Scholar]
- 42. Qiu Y., Mao T., Zhang Y., Shao M., You J., Ding Q., Chen Y., Wu D., Xie D., Lin X., Gao X., Kaufman R. J., Li W., and Liu Y. (2010) A crucial role for RACK1 in the regulation of glucose-stimulated IRE1α activation in pancreatic β cells. Sci. Signal. 3, ra7 10.1126/scisignal.2000514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y., Shao M., Wu Y., Yan C., Jiang S., Liu J., Dai J., Yang L., Li J., Jia W., Rui L., and Liu Y. (2015) Role for the endoplasmic reticulum stress sensor IRE1α in liver regenerative responses. J. Hepatol. 62, 590–598 10.1016/j.jhep.2014.10.022 [DOI] [PubMed] [Google Scholar]
- 44. Qiu Q., Zheng Z., Chang L., Zhao Y. S., Tan C., Dandekar A., Zhang Z., Lin Z., Gui M., Li X., Zhang T., Kong Q., Li H., Chen S., Chen A., et al. (2013) Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. EMBO J. 32, 2477–2490 10.1038/emboj.2013.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hetz C., Martinon F., Rodriguez D., and Glimcher L. H. (2011) The unfolded protein response: integrating stress signals through the stress sensor IRE1α. Physiol. Rev. 91, 1219–1243 10.1152/physrev.00001.2011 [DOI] [PubMed] [Google Scholar]
- 46. Bettigole S. E., and Glimcher L. H. (2015) Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 33, 107–138 10.1146/annurev-immunol-032414-112116 [DOI] [PubMed] [Google Scholar]
- 47. Grootjans J., Kaser A., Kaufman R. J., and Blumberg R. S. (2016) The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 16, 469–484 10.1038/nri.2016.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boden G., Duan X., Homko C., Molina E. J., Song W., Perez O., Cheung P., and Merali S. (2008) Increase in endoplasmic reticulum stress–related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 57, 2438–2444 10.2337/db08-0604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boden G. (2009) Endoplasmic reticulum stress: another link between obesity and insulin resistance/inflammation? Diabetes 58, 518–519 10.2337/db08-1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sha H., He Y., Yang L., and Qi L. (2011) Stressed out about obesity: IRE1α–XBP1 in metabolic disorders. Trends Endocrinol. Metab. 22, 374–381 10.1016/j.tem.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fu S., Yang L., Li P., Hofmann O., Dicker L., Hide W., Lin X., Watkins S. M., Ivanov A. R., and Hotamisligil G. S. (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531 10.1038/nature09968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lipson K. L., Fonseca S. G., Ishigaki S., Nguyen L. X., Foss E., Bortell R., Rossini A. A., and Urano F. (2006) Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 4, 245–254 10.1016/j.cmet.2006.07.007 [DOI] [PubMed] [Google Scholar]
- 53. Hassler J. R., Scheuner D. L., Wang S., Han J., Kodali V. K., Li P., Nguyen J., George J. S., Davis C., Wu S. P., Bai Y., Sartor M., Cavalcoli J., Malhi H., Baudouin G., et al. (2015) The IRE1α/XBP1s pathway is essential for the glucose response and protection of beta cells. PLoS Biol. 13, e1002277 10.1371/journal.pbio.1002277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cunha D. A., Hekerman P., Ladrière L., Bazarra-Castro A., Ortis F., Wakeham M. C., Moore F., Rasschaert J., Cardozo A. K., Bellomo E., Overbergh L., Mathieu C., Lupi R., Hai T., Herchuelz A., et al. (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 121, 2308–2318 10.1242/jcs.026062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Robblee M. M., Kim C. C., Porter Abate J., Valdearcos M., Sandlund K. L., Shenoy M. K., Volmer R., Iwawaki T., and Koliwad S. K. (2016) Saturated fatty acids engage an IRE1α-dependent pathway to activate the NLRP3 inflammasome in myeloid cells. Cell Rep. 14, 2611–2623 10.1016/j.celrep.2016.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feng B., Yao P. M., Li Y., Devlin C. M., Zhang D., Harding H. P., Sweeney M., Rong J. X., Kuriakose G., Fisher E. A., Marks A. R., Ron D., and Tabas I. (2003) The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781–792 10.1038/ncb1035 [DOI] [PubMed] [Google Scholar]
- 57. Sanson M., Augé N., Vindis C., Muller C., Bando Y., Thiers J.-C., Marachet M.-A., Zarkovic K., Sawa Y., Salvayre R., and Nègre-Salvayre A. (2009) Oxidized low-density lipoproteins trigger endoplasmic reticulum stress in vascular cells. Circ. Res. 104, 328–336 10.1161/CIRCRESAHA.108.183749 [DOI] [PubMed] [Google Scholar]
- 58. Volmer R., and Ron D. (2015) Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell biol. 33, 67–73 10.1016/j.ceb.2014.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Volmer R., van der Ploeg K., and Ron D. (2013) Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. U.S.A. 110, 4628–4633 10.1073/pnas.1217611110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kono N., Amin-Wetzel N., and Ron D. (2017) Generic membrane-spanning features endow IRE1α with responsiveness to membrane aberrancy. Mol. Biol. Cell 28, 2318–2332 10.1091/mbc.e17-03-0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cho H., Stanzione F., Oak A., Kim G. H., Yerneni S., Qi L., Sum A. K., and Chan C. (2019) Intrinsic structural features of the human IRE1α transmembrane domain sense membrane lipid saturation. Cell Rep. 27, 307–320.e5 10.1016/j.celrep.2019.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Halbleib K., Pesek K., Covino R., Hofbauer H. F., Wunnicke D., Hänelt I., Hummer G., and Ernst R. (2017) Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 67, 673–684.e8 10.1016/j.molcel.2017.06.012 [DOI] [PubMed] [Google Scholar]
- 63. Ning J., Hong T., Ward A., Pi J., Liu Z., Liu H. Y., and Cao W. (2011) Constitutive role for IRE1α–XBP1 signaling pathway in the insulin-mediated hepatic lipogenic program. Endocrinology 152, 2247–2255 10.1210/en.2010-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deng Y., Wang Z. V., Tao C., Gao N., Holland W. L., Ferdous A., Repa J. J., Liang G., Ye J., Lehrman M. A., Hill J. A., Horton J. D., and Scherer P. E. (2013) The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. J. Clin. Invest. 123, 455–468 10.1172/JCI62819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mao T., Shao M., Qiu Y., Huang J., Zhang Y., Song B., Wang Q., Jiang L., Liu Y., Han J. D., Cao P., Li J., Gao X., Rui L., Qi L., et al. (2011) PKA phosphorylation couples hepatic inositol-requiring enzyme 1α to glucagon signaling in glucose metabolism. Proc. Natl. Acad. Sci. U.S.A. 108, 15852–15857 10.1073/pnas.1107394108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cretenet G., Le Clech M., and Gachon F. (2010) Circadian clock-coordinated 12 hr period rhythmic activation of the IRE1α pathway controls lipid metabolism in mouse liver. Cell Metab. 11, 47–57 10.1016/j.cmet.2009.11.002 [DOI] [PubMed] [Google Scholar]
- 67. Zhang E. E., Liu Y., Dentin R., Pongsawakul P. Y., Liu A. C., Hirota T., Nusinow D. A., Sun X., Landais S., Kodama Y., Brenner D. A., Montminy M., and Kay S. A. (2010) Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 16, 1152–1156 10.1038/nm.2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hu P., Han Z., Couvillon A. D., Kaufman R. J., and Exton J. H. (2006) Autocrine tumor necrosis factor α links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 26, 3071–3084 10.1128/MCB.26.8.3071-3084.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martinon F., Chen X., Lee A. H., and Glimcher L. H. (2010) TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 11, 411–418 10.1038/ni.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Shan B., Wang X., Wu Y., Xu C., Xia Z., Dai J., Shao M., Zhao F., He S., Yang L., Zhang M., Nan F., Li J., Liu J., Liu J., et al. (2017) The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 18, 519–529 10.1038/ni.3709 [DOI] [PubMed] [Google Scholar]
- 71. Chawla A., Nguyen K. D., and Goh Y. P. (2011) Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 11, 738–749 10.1038/nri3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Biswas S. K., and Mantovani A. (2012) Orchestration of metabolism by macrophages. Cell Metab. 15, 432–437 10.1016/j.cmet.2011.11.013 [DOI] [PubMed] [Google Scholar]
- 73. McNelis J. C., and Olefsky J. M. (2014) Macrophages, immunity, and metabolic disease. Immunity 41, 36–48 10.1016/j.immuni.2014.05.010 [DOI] [PubMed] [Google Scholar]
- 74. Keestra-Gounder A. M., Byndloss M. X., Seyffert N., Young B. M., Chávez-Arroyo A., Tsai A. Y., Cevallos S. A., Winter M. G., Pham O. H., Tiffany C. R., de Jong M. F., Kerrinnes T., Ravindran R., Luciw P. A., McSorley S. J., et al. (2016) NOD1 and NOD2 signalling links ER stress with inflammation. Nature 532, 394–397 10.1038/nature17631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shao M., Shan B., Liu Y., Deng Y., Yan C., Wu Y., Mao T., Qiu Y., Zhou Y., Jiang S., Jia W., Li J., Li J., Rui L., Yang L., and Liu Y. (2014) Hepatic IRE1α regulates fasting-induced metabolic adaptive programs through the XBP1s-PPARα axis signalling. Nat. Commun. 5, 3528 10.1038/ncomms4528 [DOI] [PubMed] [Google Scholar]
- 76. Jurczak M. J., Lee A. H., Jornayvaz F. R., Lee H. Y., Birkenfeld A. L., Guigni B. A., Kahn M., Samuel V. T., Glimcher L. H., and Shulman G. I. (2012) Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J. Biol. Chem. 287, 2558–2567 10.1074/jbc.M111.316760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rubio-Patiño C., Bossowski J. P., De Donatis G. M., Mondragon L., Villa E., Aira L. E., Chiche J., Mhaidly R., Lebeaupin C., Marchetti S., Voutetakis K., Chatziioannou A., Castelli F. A., Lamourette P., Chu-Van E., et al. (2018) Low-protein diet induces IRE1α-dependent anticancer immunosurveillance. Cell Metab. 27, 828–842.e7 10.1016/j.cmet.2018.02.009 [DOI] [PubMed] [Google Scholar]
- 78. Yang Q., Vijayakumar A., and Kahn B. B. (2018) Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 19, 654–672 10.1038/s41580-018-0044-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Husted A. S., Trauelsen M., Rudenko O., Hjorth S. A., and Schwartz T. W. (2017) GPCR-mediated signaling of metabolites. Cell Metab. 25, 777–796 10.1016/j.cmet.2017.03.008 [DOI] [PubMed] [Google Scholar]
- 80. Varela L., and Horvath T. L. (2012) Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 13, 1079–1086 10.1038/embor.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang X., Zhang G., Zhang H., Karin M., Bai H., and Cai D. (2008) Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73 10.1016/j.cell.2008.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ozcan L., Ergin A. S., Lu A., Chung J., Sarkar S., Nie D., Myers M. G. Jr., and Ozcan U. (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 9, 35–51 10.1016/j.cmet.2008.12.004 [DOI] [PubMed] [Google Scholar]
- 83. Williams K. W., Liu T., Kong X., Fukuda M., Deng Y., Berglund E. D., Deng Z., Gao Y., Liu T., Sohn J.-W., Jia L., Fujikawa T., Kohno D., Scott M. M., Lee S., et al. (2014) Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab. 20, 471–482 10.1016/j.cmet.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Mardones P., Dillin A., and Hetz C. (2014) Cell-nonautonomous control of the UPR: mastering energy homeostasis. Cell Metab. 20, 385–387 10.1016/j.cmet.2014.07.009 [DOI] [PubMed] [Google Scholar]
- 85. Yao T., Deng Z., Gao Y., Sun J., Kong X., Huang Y., He Z., Xu Y., Chang Y., Yu K.-J., Findley B. G., Berglund E. D., Wang R.-T., Guo H., Chen H., et al. (2017) Ire1α in Pomc neurons is required for thermogenesis and glycemia. Diabetes 66, 663–673 10.2337/db16-0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xiao Y., Xia T., Yu J., Deng Y., Liu H., Liu B., Chen S., Liu Y., and Guo F. (2016) Knockout of inositol-requiring enzyme 1α in pro-opiomelanocortin neurons decreases fat mass via increasing energy expenditure. Open Biol. 6, 160131 10.1098/rsob.160131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tang C.-H., Chang S., Paton A. W., Paton J. C., Gabrilovich D. I., Ploegh H. L., Del Valle J. R., and Hu C.-C. (2018) Phosphorylation of IRE1 at S729 regulates RIDD in B cells and antibody production after immunization. J. Cell Biol. 217, 1739–1755 10.1083/jcb.201709137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Allagnat F., Christulia F., Ortis F., Pirot P., Lortz S., Lenzen S., Eizirik D. L., and Cardozo A. K. (2010) Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 53, 1120–1130 10.1007/s00125-010-1699-7 [DOI] [PubMed] [Google Scholar]
- 89. Lee A. H., Heidtman K., Hotamisligil G. S., and Glimcher L. H. (2011) Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. U.S.A. 108, 8885–8890 10.1073/pnas.1105564108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Tsuchiya Y., Saito M., Kadokura H., Miyazaki J.-I. Tashiro F., Imagawa Y., Iwawaki T., and Kohno K. (2018) IRE1–XBP1 pathway regulates oxidative proinsulin folding in pancreatic β cells. J. Cell Biol. 217, 1287–1301 10.1083/jcb.201707143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Xu T., Yang L., Yan C., Wang X., Huang P., Zhao F., Zhao L., Zhang M., Jia W., Wang X., and Liu Y. (2014) The IRE1α–XBP1 pathway regulates metabolic stress-induced compensatory proliferation of pancreatic beta-cells. Cell Res. 24, 1137–1140 10.1038/cr.2014.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Akiyama M., Liew C. W., Lu S., Hu J., Martinez R., Hambro B., Kennedy R. T., and Kulkarni R. N. (2013) X-Box binding protein 1 is essential for insulin regulation of pancreatic α-cell function. Diabetes 62, 2439–2449 10.2337/db12-1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lee A. H., Scapa E. F., Cohen D. E., and Glimcher L. H. (2008) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 320, 1492–1496 10.1126/science.1158042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Park S. W., Zhou Y., Lee J., Lu A., Sun C., Chung J., Ueki K., and Ozcan U. (2010) The regulatory subunits of PI3K, p85α and p85β, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 16, 429–437 10.1038/nm.2099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Winnay J. N., Boucher J., Mori M. A., Ueki K., and Kahn C. R. (2010) A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box–binding protein-1 to modulate the unfolded protein response. Nat. Med. 16, 438–445 10.1038/nm.2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Park S. W., Herrema H., Salazar M., Cakir I., Cabi S., Basibuyuk Sahin F., Chiu Y. H., Cantley L. C., and Ozcan U. (2014) BRD7 regulates XBP1s' activity and glucose homeostasis through its interaction with the regulatory subunits of PI3K. Cell Metab. 20, 73–84 10.1016/j.cmet.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zhou Y., Lee J., Reno C. M., Sun C., Park S. W., Chung J., Lee J., Fisher S. J., White M. F., Biddinger S. B., and Ozcan U. (2011) Regulation of glucose homeostasis through a XBP-1–oxO1 interaction. Nat. Med. 17, 356–365 10.1038/nm.2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Zhao Y., Li X., Cai M.-Y., Ma K., Yang J., Zhou J., Fu W., Wei F.-Z., Wang L., Xie D., and Zhu W.-G. (2013) XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res. 23, 491–507 10.1038/cr.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lee J., Sun C., Zhou Y., Lee J., Gokalp D., Herrema H., Park S. W., Davis R. J., and Ozcan U. (2011) p38 MAPK-mediated regulation of Xbp1s is crucial for glucose homeostasis. Nat. Med. 17, 1251–1260 10.1038/nm.2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Liu J., Ibi D., Taniguchi K., Lee J., Herrema H., Akosman B., Mucka P., Salazar Hernandez M. A., Uyar M. F., Park S. W., Karin M., and Ozcan U. (2016) Inflammation improves glucose homeostasis through IKKβ-XBP1s interaction. Cell 167, 1052–1066.e18 10.1016/j.cell.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sun S., Kelekar S., Kliewer S. A., and Mangelsdorf D. J. (2019) The orphan nuclear receptor SHP regulates ER stress response by inhibiting XBP1s degradation. Genes Dev. 33, 1083–1094 10.1101/gad.326868.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Herrema H., Zhou Y., Zhang D., Lee J., Salazar Hernandez M. A., Shulman G. I., and Ozcan U. (2016) XBP1s is an anti-lipogenic protein. J. Biol. Chem. 291, 17394–17404 10.1074/jbc.M116.728949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jiang S., Yan C., Fang Q. C., Shao M. L., Zhang Y. L., Liu Y., Deng Y. P., Shan B., Liu J. Q., Li H. T., Yang L., Zhou J., Dai Z., Liu Y., and Jia W. P. (2014) Fibroblast growth factor 21 is regulated by the IRE1α–XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J. Biol. Chem. 289, 29751–29765 10.1074/jbc.M114.565960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhang M., Sun W., Qian J., and Tang Y. (2018) Fasting exacerbates hepatic growth differentiation factor 15 to promote fatty acid β-oxidation and ketogenesis via activating XBP1 signaling in liver. Redox Biol. 16, 87–96 10.1016/j.redox.2018.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang S., Chen Z., Lam V., Han J., Hassler J., Finck B. N., Davidson N. O., and Kaufman R. J. (2012) IRE1α–XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 16, 473–486 10.1016/j.cmet.2012.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. So J. S., Hur K. Y., Tarrio M., Ruda V., Frank-Kamenetsky M., Fitzgerald K., Koteliansky V., Lichtman A. H., Iwawaki T., Glimcher L. H., and Lee A. H. (2012) Silencing of lipid metabolism genes through IRE1α-mediated mRNA decay lowers plasma lipids in mice. Cell Metab. 16, 487–499 10.1016/j.cmet.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Liu L., Cai J., Wang H., Liang X., Zhou Q., Ding C., Zhu Y., Fu T., Guo Q., Xu Z., Xiao L., Liu J., Yin Y., Fang L., Xue B., et al. (2019) Coupling of COPII vesicle trafficking to nutrient availability by the IRE1α–XBP1s axis. Proc. Natl. Acad. Sci. U.S.A. 116, 11776–11785 10.1073/pnas.1814480116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhang K., Wang S., Malhotra J., Hassler J. R., Back S. H., Wang G., Chang L., Xu W., Miao H., Leonardi R., Chen Y. E., Jackowski S., and Kaufman R. J. (2011) The unfolded protein response transducer IRE1α prevents ER stress-induced hepatic steatosis. EMBO J. 30, 1357–1375 10.1038/emboj.2011.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang J.-M., Qiu Y., Yang Z., Kim H., Qian Q., Sun Q., Zhang C., Yin L., Fang D., Back S. H., Kaufman R. J., Yang L., and Zhang K. (2018) IRE1α prevents hepatic steatosis by processing and promoting the degradation of select microRNAs. Sci. Signal. 11, eaao4617 10.1126/scisignal.aao4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wu Y., Shan B., Dai J., Xia Z., Cai J., Chen T., Lv S., Feng Y., Zheng L., Wang Y., Liu J., Fang J., Xie D., Rui L., Liu J., and Liu Y. (2018) Dual role for inositol-requiring enzyme 1α in promoting the development of hepatocellular carcinoma during diet-induced obesity in mice. Hepatology 68, 533–546 10.1002/hep.29871 [DOI] [PubMed] [Google Scholar]
- 111. Ahima R. S., and Flier J. S. (2000) Adipose tissue as an endocrine organ. Trends Endocrinol. Metab. 11, 327–332 10.1016/S1043-2760(00)00301-5 [DOI] [PubMed] [Google Scholar]
- 112. Spiegelman B. M., and Flier J. S. (2001) Obesity and the regulation of energy balance. Cell 104, 531–543 10.1016/S0092-8674(01)00240-9 [DOI] [PubMed] [Google Scholar]
- 113. Rosen E. D., and Spiegelman B. M. (2014) What we talk about when we talk about fat. Cell 156, 20–44 10.1016/j.cell.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lumeng C. N., Bodzin J. L., and Saltiel A. R. (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 117, 175–184 10.1172/JCI29881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Brestoff J. R., and Artis D. (2015) Immune regulation of metabolic homeostasis in health and disease. Cell 161, 146–160 10.1016/j.cell.2015.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gregor M. F., Yang L., Fabbrini E., Mohammed B. S., Eagon J. C., Hotamisligil G. S., and Klein S. (2009) Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 58, 693–700 10.2337/db08-1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sha H., He Y., Chen H., Wang C., Zenno A., Shi H., Yang X., Zhang X., and Qi L. (2009) The IRE1α–XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 9, 556–564 10.1016/j.cmet.2009.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Asada R., Kanemoto S., Matsuhisa K., Hino K., Cui M., Cui X., Kaneko M., and Imaizumi K. (2015) IRE1α–XBP1 is a novel branch in the transcriptional regulation of Ucp1 in brown adipocytes. Sci. Rep. 5, 16580 10.1038/srep16580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gregor M. F., Misch E. S., Yang L., Hummasti S., Inouye K. E., Lee A. H., Bierie B., and Hotamisligil G. S. (2013) The role of adipocyte XBP1 in metabolic regulation during lactation. Cell Rep. 3, 1430–1439 10.1016/j.celrep.2013.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Deng Y., Wang Z. V., Gordillo R., Zhu Y., Ali A., Zhang C., Wang X., Shao M., Zhang Z., Iyengar P., Gupta R. K., Horton J. D., Hill J. A., and Scherer P. E. (2018) Adipocyte Xbp1s overexpression drives uridine production and reduces obesity. Mol. Metab. 11, 1–17 10.1016/j.molmet.2018.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Deng Y., Wang Z. V., Gordillo R., An Y., Zhang C., Liang Q., Yoshino J., Cautivo K. M., De Brabander J., Elmquist J. K., Horton J. D., Hill J. A., Klein S., and Scherer P. E. (2017) An adipo-biliary-uridine axis that regulates energy homeostasis. Science 355, eaaf5375 10.1126/science.aaf5375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sha H., Yang L., Liu M., Xia S., Liu Y., Liu F., Kersten S., and Qi L. (2014) Adipocyte spliced form of X-box-binding protein 1 promotes adiponectin multimerization and systemic glucose homeostasis. Diabetes 63, 867–879 10.2337/db13-1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Turer A. T., and Scherer P. E. (2012) Adiponectin: mechanistic insights and clinical implications. Diabetologia 55, 2319–2326 10.1007/s00125-012-2598-x [DOI] [PubMed] [Google Scholar]
- 124. Qiu Y., Shan B., Yang L., and Liu Y. (2016) Adipose tissue macrophage in immune regulation of metabolism. Sci. China Life Sci. 59, 1232–1240 10.1007/s11427-016-0155-1 [DOI] [PubMed] [Google Scholar]
- 125. Bujisic B., and Martinon F. (2017) IRE1 gives weight to obesity-associated inflammation. Nat. Immunol. 18, 479–480 10.1038/ni.3725 [DOI] [PubMed] [Google Scholar]
- 126. Hotamisligil G. S. (2017) Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 10.1038/nature21363 [DOI] [PubMed] [Google Scholar]
- 127. Ley R. E., Turnbaugh P. J., Klein S., and Gordon J. I. (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 10.1038/4441022a [DOI] [PubMed] [Google Scholar]
- 128. Zhang C., Li S., Yang L., Huang P., Li W., Wang S., Zhao G., Zhang M., Pang X., Yan Z., Liu Y., and Zhao L. (2013) Structural modulation of gut microbiota in life-long calorie-restricted mice. Nat. Commun. 4, 2163 10.1038/ncomms3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 10.1126/science.1103160 [DOI] [PubMed] [Google Scholar]
- 130. Ozcan U., Yilmaz E., Ozcan L., Furuhashi M., Vaillancourt E., Smith R. O., Görgün C. Z., and Hotamisligil G. S. (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313, 1137–1140 10.1126/science.1128294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Prischi F., Nowak P. R., Carrara M., and Ali M. M. (2014) Phosphoregulation of Ire1 RNase splicing activity. Nat. Commun. 5, 3554 10.1038/ncomms4554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Yang L., Calay E. S., Fan J., Arduini A., Kunz R. C., Gygi S. P., Yalcin A., Fu S., and Hotamisligil G. S. (2015) S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349, 500–506 10.1126/science.aaa0079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Schiattarella G. G., Altamirano F., Tong D., French K. M., Villalobos E., Kim S. Y., Luo X., Jiang N., May H. I., Wang Z. V., Hill T. M., Mammen P. P., Huang J., Lee D. I., Hahn V. S., et al. (2019) Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356 10.1038/s41586-019-1100-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hourihan J. M., Moronetti Mazzeo L. E., Fernández-Cárdenas L. P., and Blackwell T. K. (2016) Cysteine sulfenylation directs IRE-1 to activate the SKN-1/Nrf2 antioxidant response. Mol. Cell 63, 553–566 10.1016/j.molcel.2016.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Sun S., Shi G., Sha H., Ji Y., Han X., Shu X., Ma H., Inoue T., Gao B., Kim H., Bu P., Guber R. D., Shen X., Lee A. H., Iwawaki T., et al. (2015) IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 17, 1546–1555 10.1038/ncb3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gao B., Lee S. M., Chen A., Zhang J., Zhang D. D., Kannan K., Ortmann R. A., and Fang D. (2008) Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO Rep. 9, 480–485 10.1038/embor.2008.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Zhu X., Zhang J., Sun H., Jiang C., Dong Y., Shan Q., Su S., Xie Y., Xu N., Lou X., and Liu S. (2014) Ubiquitination of inositol-requiring enzyme 1 (IRE1) by the E3 ligase CHIP mediates the IRE1/TRAF2/JNK pathway. J. Biol. Chem. 289, 30567–30577 10.1074/jbc.M114.562868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Takeda K., Nagashima S., Shiiba I., Uda A., Tokuyama T., Ito N., Fukuda T., Matsushita N., Ishido S., Iwawaki T., Uehara T., Inatome R., and Yanagi S. (2019) MITOL prevents ER stress-induced apoptosis by IRE1α ubiquitylation at ER-mitochondria contact sites. EMBO J. 38, e100999 10.15252/embj.2018100999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Xiong X., Kuang H., Ansari S., Liu T., Gong J., Wang S., Zhao X. Y., Ji Y., Li C., Guo L., Zhou L., Chen Z., Leon-Mimila P., Chung M. T., Kurabayashi K., et al. (2019) Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol. Cell 75, 644–660.e5 10.1016/j.molcel.2019.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Chae H. J., Kim H. R., Xu C., Bailly-Maitre B., Krajewska M., Krajewski S., Banares S., Cui J., Digicaylioglu M., Ke N., Kitada S., Monosov E., Thomas M., Kress C. L., Babendure J. R., et al. (2004) BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol. Cell 15, 355–366 10.1016/j.molcel.2004.06.038 [DOI] [PubMed] [Google Scholar]
- 141. Bailly-Maitre B., Fondevila C., Kaldas F., Droin N., Luciano F., Ricci J. E., Croxton R., Krajewska M., Zapata J. M., Kupiec-Weglinski J. W., Farmer D., and Reed J. C. (2006) Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 103, 2809–2814 10.1073/pnas.0506854103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Lisbona F., Rojas-Rivera D., Thielen P., Zamorano S., Todd D., Martinon F., Glavic A., Kress C., Lin J. H., Walter P., Reed J. C., Glimcher L. H., and Hetz C. (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1α. Mol. Cell 33, 679–691 10.1016/j.molcel.2009.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Amin-Wetzel N., Saunders R. A., Kamphuis M. J., Rato C., Preissler S., Harding H. P., and Ron D. (2017) A J-protein co-chaperone recruits BiP to monomerize IRE1 and repress the unfolded protein response. Cell 171, 1625–1637.e13 10.1016/j.cell.2017.10.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Gupta S., Deepti A., Deegan S., Lisbona F., Hetz C., and Samali A. (2010) HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α–XBP1 signaling through a physical interaction. PLoS Biol. 8, e1000410 10.1371/journal.pbio.1000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Shin G. C., Moon S. U., Kang H. S., Choi H. S., Han H. D., and Kim K. H. (2019) PRKCSH contributes to tumorigenesis by selective boosting of IRE1 signaling pathway. Nat. Commun. 10, 3185 10.1038/s41467-019-11019-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sepulveda D., Rojas-Rivera D., Rodríguez D. A., Groenendyk J., Köhler A., Lebeaupin C., Ito S., Urra H., Carreras-Sureda A., Hazari Y., Vasseur-Cognet M., Ali M. M. U., Chevet E., Campos G., Godoy P., et al. (2018) Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1α. Mol. Cell 69, 238–252.e7 10.1016/j.molcel.2017.12.028 [DOI] [PubMed] [Google Scholar]
- 147. Bailly-Maitre B., Belgardt B. F., Jordan S. D., Coornaert B., von Freyend M. J., Kleinridders A., Mauer J., Cuddy M., Kress C. L., Willmes D., Essig M., Hampel B., Protzer U., Reed J. C., and Brüning J. C. (2010) Hepatic Bax inhibitor-1 inhibits IRE1α and protects from obesity-associated insulin resistance and glucose intolerance. J. Biol. Chem. 285, 6198–6207 10.1074/jbc.M109.056648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Lebeaupin C., Vallée D., Rousseau D., Patouraux S., Bonnafous S., Adam G., Luciano F., Luci C., Anty R., Iannelli A., Marchetti S., Kroemer G., Lacas-Gervais S., Tran A., Gual P., and Bailly-Maitre B. (2018) Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1α signaling in mice. Hepatology 68, 515–532 10.1002/hep.29847 [DOI] [PubMed] [Google Scholar]
- 149. Kammoun H. L., Chabanon H., Hainault I., Luquet S., Magnan C., Koike T., Ferré P., and Foufelle F. (2009) GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 119, 1201–1215 10.1172/JCI37007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kakazu E., Mauer A. S., Yin M., and Malhi H. (2016) Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1α-dependent manner. J. Lipid Res. 57, 233–245 10.1194/jlr.M063412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hirsova P., Ibrahim S. H., Krishnan A., Verma V. K., Bronk S. F., Werneburg N. W., Charlton M. R., Shah V. H., Malhi H., and Gores G. J. (2016) Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 150, 956–967 10.1053/j.gastro.2015.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Cao J., Peng J., An H., He Q., Boronina T., Guo S., White M. F., Cole P. A., and He L. (2017) Endotoxemia-mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat. Commun. 8, 131 10.1038/s41467-017-00163-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Han D., Lerner A. G., Vande Walle L., Upton J. P., Xu W., Hagen A., Backes B. J., Oakes S. A., and Papa F. R. (2009) IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138, 562–575 10.1016/j.cell.2009.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lipson K. L., Ghosh R., and Urano F. (2008) The role of IRE1α in the degradation of insulin mRNA in pancreatic beta-cells. PLoS One 3, e1648 10.1371/journal.pone.0001648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Lerner A. G., Upton J. P., Praveen P. V., Ghosh R., Nakagawa Y., Igbaria A., Shen S., Nguyen V., Backes B. J., Heiman M., Heintz N., Greengard P., Hui S., Tang Q., Trusina A., et al. (2012) IRE1α induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 16, 250–264 10.1016/j.cmet.2012.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Oslowski C. M., Hara T., O'Sullivan-Murphy B., Kanekura K., Lu S., Hara M., Ishigaki S., Zhu L. J., Hayashi E., Hui S. T., Greiner D., Kaufman R. J., Bortell R., and Urano F. (2012) Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 16, 265–273 10.1016/j.cmet.2012.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Tam A. B., Koong A. C., and Niwa M. (2014) Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 9, 850–858 10.1016/j.celrep.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lhomond S., Avril T., Dejeans N., Voutetakis K., Doultsinos D., McMahon M., Pineau R., Obacz J., Papadodima O., Jouan F., Bourien H., Logotheti M., Jegou G., Pallares-Lupon N., Schmit K., et al. (2018) Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 10, e7929 10.15252/emmm.201707929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Liu X., Henkel A. S., LeCuyer B. E., Schipma M. J., Anderson K. A., and Green R. M. (2015) Hepatocyte X-box binding protein 1 deficiency increases liver injury in mice fed a high-fat/sugar diet. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G965–G974 10.1152/ajpgi.00132.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Garfinkel B. P., and Hotamisligil G. S. (2017) ER stress promotes inflammation through Re-wIREd macrophages in obesity. Mol. Cell 66, 731–733 10.1016/j.molcel.2017.05.037 [DOI] [PubMed] [Google Scholar]
- 161. Korennykh A., and Walter P. (2012) Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 28, 251–277 10.1146/annurev-cellbio-101011-155826 [DOI] [PubMed] [Google Scholar]
- 162. Morita S., Villalta S. A., Feldman H. C., Register A. C., Rosenthal W., Hoffmann-Petersen I. T., Mehdizadeh M., Ghosh R., Wang L., Colon-Negron K., Meza-Acevedo R., Backes B. J., Maly D. J., Bluestone J. A., and Papa F. R. (2017) Targeting ABL-IRE1α signaling spares ER-stressed pancreatic beta cells to reverse autoimmune diabetes. Cell Metab. 25, 883–897.e8 10.1016/j.cmet.2017.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Carreras-Sureda A., Jaña F., Urra H., Durand S., Mortenson D. E., Sagredo A., Bustos G., Hazari Y., Ramos-Fernández E., Sassano M. L., Pihán P., van Vliet A. R., González-Quiroz M., Torres A. K., Tapia-Rojas C., et al. (2019) Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 21, 755–767 10.1038/s41556-019-0329-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hetz C., Chevet E., and Harding H. P. (2013) Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 12, 703–719 10.1038/nrd3976 [DOI] [PubMed] [Google Scholar]
- 165. Tabas I. (2010) The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 107, 839–850 10.1161/CIRCRESAHA.110.224766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Tufanli O., Telkoparan Akillilar P., Acosta-Alvear D., Kocaturk B., Onat U. I., Hamid S. M., Çimen I., Walter P., Weber C., and Erbay E. (2017) Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 114, E1395–E1404 10.1073/pnas.1621188114 [DOI] [PMC free article] [PubMed] [Google Scholar]