

Abstract
During the late phase of the HIV-1 replication cycle, the viral Gag polyproteins are targeted to the plasma membrane for assembly. The Gag-membrane interaction is mediated by binding of Gag's N-terminal myristoylated matrix (MA) domain to phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2). The viral envelope (Env) glycoprotein is then recruited to the assembly sites and incorporated into budding particles. Evidence suggests that Env incorporation is mediated by interactions between Gag's MA domain and the cytoplasmic tail of the gp41 subunit of Env (gp41CT). MA trimerization appears to be an obligatory step for this interaction. Insufficient production of a recombinant MA trimer and unavailability of a biologically relevant membrane system have been barriers to detailed structural and biophysical characterization of the putative MA-gp41CT-membrane interactions. Here, we engineered a stable recombinant HIV-1 MA trimer construct by fusing a foldon domain (FD) of phage T4 fibritin to the MA C terminus. Results from NMR experiments confirmed that the FD attachment does not adversely alter the MA structure. Employing hydrogen–deuterium exchange MS, we identified an MA-MA interface in the MA trimer that is implicated in Gag assembly and Env incorporation. Utilizing lipid nanodiscs as a membrane mimetic, we show that the MA trimer binds to membranes 30-fold tighter than does the MA monomer and that incorporation of PI(4,5)P2 and phosphatidylserine enhances the binding of MA to nanodiscs. These findings advance our understanding of a fundamental mechanism in HIV-1 assembly and provide a template for investigating the interaction of MA with gp41CT.
Keywords: human immunodeficiency virus (HIV); plasma membrane; nuclear magnetic resonance (NMR); isothermal titration calorimetry (ITC); mass spectrometry (MS); Fab; Gag protein; myristoylated matrix (MA); phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2); phosphatidylserine (PS); nanodisc (ND)
Introduction
During the late stage of HIV-1 replication cycle, the Gag polyproteins are transported to the plasma membrane (PM)3 for assembly, virus budding, and release (1–7). During or shortly after virus budding, the viral protease cleaves the Gag polyprotein into matrix (MA), capsid (CA), nucleocapsid (NC), two spacer peptides (SP1 and SP2), and P6 to form mature virions (reviewed in Refs. 8–10). It is demonstrated that Gag binding to the PM is mediated by the MA domain, which for most retroviruses contains a bipartite signal consisting of a highly basic region (HBR) and an N-terminal myristoyl (myr) group. The affinity of Gag binding to membranes is regulated by electrostatic and hydrophobic interactions, protein multimerization, cellular and viral RNA, and recognition of specific phospholipids (7, 11–16). Pioneering studies from the Freed laboratory established that binding of HIV-1 Gag to the PM depends on phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) (7). Efficient Gag binding to membranes is also dependent on phosphatidylserine (PS), an abundant phospholipid in the inner leaflet of the PM (15, 17–19).
Analysis of the molecular arrangement of Gag in the immature HIV-1 particle and its cleaved domains in the mature particle has often relied on cryo-EM data. Although the molecular details of the hexameric CA lattice are well-understood (20–24), cryo-EM studies were unable to provide details of the MA domain due to lack of periodicity on the inner leaflet of the membrane (21). Structural studies using NMR or X-ray crystallography approaches indicated that the structures of myristoylated and unmyristoylated MA proteins are nearly identical (25–29). X-ray crystallography studies of the unmyristoylated MA protein (myr(−)MA) of HIV-1 (25) and simian immunodeficiency virus (30) revealed a trimer arrangement of MA molecules. However, NMR studies indicate that myr(−)MA is monomeric in solution (26, 28, 29). On the other hand, NMR studies of the MA protein confirmed that the protein is in a monomer-trimer equilibrium (29). That study also revealed that the myr group can adopt sequestered and exposed conformations and that myr exposure is coupled with protein trimerization (29). Structural details of the MA trimer in solution are not defined because of the lack of a stable recombinant construct that recapitulates the functional trimer protein (29). For example, it is not yet known whether the MA-MA interface in the MA trimer is similar to that in the X-ray structure of myr(−)MA (25).
Over the past decade, we and others have employed structural and biophysical approaches to characterize binding of retroviral monomeric MA proteins to various phospholipids and membrane mimetics, such as bicelles, micelles, and liposomes (28, 31–41). These studies provided invaluable insight into how the MA proteins interact with membranes, which enabled identification of key molecular determinants of MA-mediated assembly. However, structural studies of proteins in micelles and bicelles are sometimes complicated by their unfavorable effect on protein stability and folding. Liposomes, on the other hand, are large, with diameters on the order of micrometers and contain thousands to millions of phospholipids, which does not enable utilization in structural studies either by NMR spectroscopy or X-ray crystallography. Additionally, the convex curvature of the liposome surface may also be unfavorable for protein-binding studies. Over the last decade, lipid nanodiscs (NDs) have been increasingly used as membrane mimetics to characterize protein-membrane interactions (42–49). A lipid ND is a noncovalent assembly of phospholipids and a genetically engineered membrane scaffold protein (MSP) (43–48). Phospholipids (120–160 molecules) associate as a bilayer domain stabilized by two MSP molecules wrapped around the edges of the discoidal structure in a beltlike configuration. One advantage of using membrane NDs is that they can be modified in size and lipid composition to accommodate a variety of membrane proteins. Another major advantage of using NDs is the ability to obtain quantitative measurements of binding to proteins by calculating the ND concentration based on the absorbance of the MSP at 280 nm.
Genetic, in vivo, and biochemical studies suggested that incorporation of the Env protein into virus particles is mediated by interactions between the MA domain of Gag and the cytoplasmic tail of the transmembrane subunit of Env (gp41CT) (50–54). Higher-order organization of MA on the membrane appears to be essential for Env incorporation (55–58). The HIV-1 MA and MACA proteins have been shown to assemble as hexagonal cage lattices on PS/cholesterol membrane monolayers and as hexamers of trimers in the presence of PI(4,5)P2 (57, 59, 60). Of note, those trimer and hexamer arrangements form the basis of the models proposed to explain Env incorporation (55, 57, 58). A recent study provided biochemical evidence that MA trimerization is an obligatory step in the assembly of infectious HIV-1 by demonstrating a correlation between loss of MA trimerization and loss of Env incorporation (55). Therefore, structural and biophysical characterization of the MA trimer and how it interacts with membranes is required for elucidating the mechanisms of virus assembly and Env incorporation. Toward this end, we engineered a stable MA timer construct and identified the MA-MA interface within the trimer by hydrogen–deuterium exchange MS (HDX-MS). Furthermore, we developed methods to examine interactions of MA with lipid NDs by NMR, biochemical, and biophysical techniques.
Results
NMR studies of MA binding to lipid NDs
Reduction in signal intensity upon binding of proteins to large molecules such as NDs is often caused by slow tumbling due to an increase in the molecular weight of the complex and/or by an intermediate to slow exchange process on the NMR time scale between the free and bound states (61). NDs were prepared with various lipids, such as 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine (POPS), and PI(4,5)P2. We employed 2D 1H-15N HSQC NMR methods to determine how MA interacts with NDs. First, we assessed binding of myr(−)MA and MA to NDs made of 100% POPC. No chemical shift perturbations (CSPs) were observed upon the addition of POPC NDs into myr(−)MA, indicating no detectable binding. However, titration of POPC NDs into MA led to substantial loss in intensity for a subset of 1H-15N signals corresponding to residues 2–20, Val35, Trp36, Arg39, Gly49, Glu52, His89, Qln90, and Arg91 (Fig. S1).4 Those distinctive CSPs are indicative of exposure of the myr group (29, 32, 62). This result is in agreement with the previous finding that MA interactions with membrane mimetics such as micelles and bicelles triggered myr exposure (32).
Next, we examined whether PS enhances binding of MA to NDs. As observed in Fig. 1A and Fig. S2, titration of POPC:POPS (80:20) NDs into MA led to substantial loss in intensity for numerous signals across the NMR spectrum. At a 0.02:1 ND:MA molar ratio, the 1H-15N signals corresponding to residues in the N-terminal domain were severely broadened. A steady decrease in intensity for most of the 1H-15N signals was clearly observed as a function of increased ND concentration. At 0.16:1 ND:MA, the majority of 1H-15N signals were broadened except for those corresponding to residues located in the flexible C-terminal region of MA (Fig. 1A and Fig. S2). An increase of the PS concentration in the POPC:POPS (50:50) NDs led to an accelerated loss of signal intensity for numerous signals, indicating tighter binding than that observed for POPC:POPS (80:20) NDs (Fig. S2). This result suggests that the uniform intensity reduction of signals reflects the increase in molecular weight and that a more rapid reduction in intensity is caused by the exchange-broadening effect due to chemical shift changes upon binding.
Figure 1.
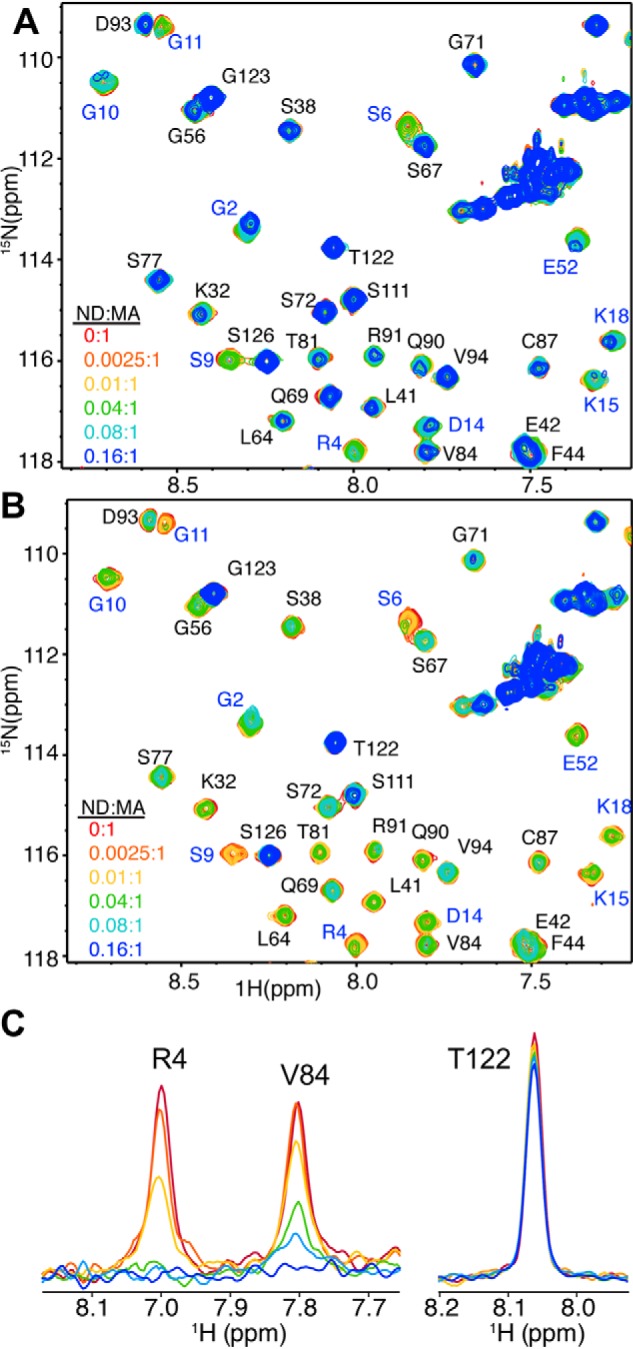
NMR spectra of MA upon titration with NDs. A and B, selected regions of 2D 1H-15N HSQC spectra obtained for 15N-labeled samples of MA at 100 μm in the presence of 0 (red), 0.0025 (orange), 0.01 (yellow), 0.04 (green), 0.08 (cyan), and 0.16 (blue) molar equivalents of POPC:POPS (80:20) NDs (A) and POPC:POPS:PI(4,5)P2 (72:20:8) NDs (B). C, 1D slices of representative cross-peaks from B showing the decrease in signal intensity upon the addition of POPC:POPS:PI(4,5)P2 (72:20:8) NDs.
To examine how incorporation of PI(4,5)P2 impacts binding of MA to NDs, we performed titrations of MA with NDs made of POPC:POPS:PI(4,5)P2 (72:20:8) followed by acquisition of 1H-15N 2D HSQC spectra. Considering the lipid composition and the fact that a single ND contains ∼150 lipid molecules (44), on average, six molecules of PI(4,5)P2 are incorporated within each leaflet of the ND. As shown in Fig. 1B and Fig. S2, the addition of POPC:POPS:PI(4,5)P2 (72:20:8) NDs to MA led to substantial signal dampening across the spectrum. Loss of signal intensity is larger than that observed for POPC:POPS at 80:20 or 50:50 ratios, indicating that MA binding to NDs is enhanced significantly upon inclusion of PI(4,5)P2. Of note, signals of residues in the HBR of MA (Lys26, Lys27, Lys30, and Lys32; Fig. 2) exhibited significant loss in intensity at low concentration of POPC:POPS:PI(4,5)P2 NDs (Fig. 1B). The further addition of POPC:POPS:PI(4,5)P2 NDs led to a steady decrease in signal intensity for the vast majority of signals except for those corresponding to the flexible C-terminal region (amino acids ∼115–132) (Fig. 1 (B and C) and Fig. S2). Altogether, our data indicated that the myr group is readily exposed and anchored to the membrane bilayer, that the affinity of MA binding to NDs is enhanced upon incorporation of PS and PI(4,5)P2, and that the interaction interface in MA is located in the HBR. These findings demonstrate that NDs are a relevant membrane mimetic to study binding of retroviral MA proteins.
Figure 2.
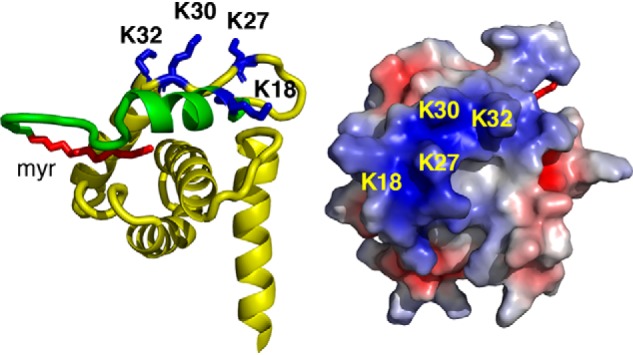
MA-ND interaction interface. Shown are a cartoon representation (left) and electrostatic map (right) of the HIV-1 MA structure (PDB code 2H3I) highlighting basic residues in the HBR (blue) and the N-terminal helix (green) that exhibited substantial loss in signal intensity and/or CSPs upon binding to POPC:POPS:PI(4,5)P2 NDs. The myr group is shown in red. Residues 116–132 are not shown for clarity.
Thermodynamics of MA binding to NDs
We employed ITC methods to obtain the thermodynamic parameters for MA binding to NDs. ITC provides values for the dissociation constant (Kd), stoichiometry of binding (n), enthalpy change (ΔH°) and the entropic term (TΔS°). ITC data were collected on MA titrations into NDs made of varying proportions of POPC, POPS, and PI(4,5)P2. Whereas titration of MA into 100% POPC NDs produced no detectable heat of binding, titration of MA into POPC:POPS (80:20) NDs yielded a thermogram with low heat values, which precluded accurate fitting of the data (not shown). These results are consistent with weak binding of MA to membranes containing POPC and low amounts of POPS (38). However, similar ITC titrations of MA into POPC:POPS:PI(4,5)P2 (75:20:5) NDs yielded a thermogram characteristic of an exothermic binding process (Fig. 3). Data were best fit with a model for a single set of identical binding sites and yielded an apparent Kd of 10.3 ± 1.9 μm and n = 10.3 ± 0.2 (Fig. 3). Of note, the n value indicates that 10 molecules of MA are capable of binding to one ND (two membrane surfaces). Remarkably, the apparent Kd of MA to POPC:POPS:PI(4,5)P2 (75:20:5) NDs is virtually identical to that obtained for MA binding to PI(4,5)P2 embedded in liposomes (38). Taken together, we have shown that MA binds to lipid NDs in a PI(4,5)P2- and PS-dependent manner, that up to 10 molecules of MA are capable of binding to one NDs, and that MA binding to NDs is mediated by the HBR implicated in Gag assembly on the PM.
Figure 3.
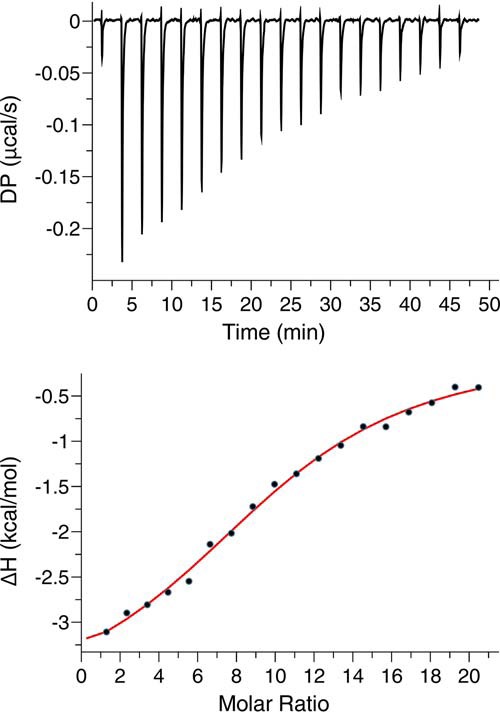
ITC analysis of MA binding to NDs. ITC data were obtained for MA (350 μm) upon titration into POPC:POPS:PI(4,5)P2 (75:20:5) NDs (3.3 μm). The top panel shows the baseline-adjusted thermogram, and the bottom panel depicts the binding isotherm and fit to the data (red line). DP, differential power between the reference and sample cells.
Production of HIV-1 MA trimer
As mentioned above, previous studies indicated that the HIV-1 MA protein resides in a monomer-trimer equilibrium (29). Although it has been shown that the monomer-trimer distribution is regulated by protein concentration and pH (29, 62), a stable and homogeneous trimer form of MA has not been produced or characterized. Previous studies have used the C-terminal domain of T4 fibritin (foldon; FD) as an artificial trimerization domain in the generation of HIV-1 gp120 and influenza hemagglutinin trimers (63–65). The FD domain, a 30-amino acid segment that forms a β-propeller consisting of monomeric β-hairpin segments, is obligatory for the formation of the fibritin trimer structure (66, 67). Here, we devised a strategy to produce a stable MA trimer by engineering a molecular clone encoding for the FD domain fused to the flexible C-terminal domain of MA (Fig. 4A). The matrix-foldon (MAFD) gene has been inserted into a vector harboring the yeast N-myristoyl transferase to produce a myristoylated MAFD protein. Previous structural studies indicated that residues 115–132 of MA are unstructured (26–29). We hypothesized that the flexibility in the C-terminal tail may render the MA trimer flexible. To decrease flexibility within the MAFD trimer without compromising the structure of the globular domain, we deleted residues 123–132 of MA. The resulting construct is referred to as MA122FD (Fig. 4A). Both myr(−)MA122FD and MA122FD have been prepared and characterized. The identity of proteins and efficiency of myristoylation were confirmed by mass spectrometry (MS). Electrospray ionization MS analysis of MA122FD (Fig. S3) identified a component with molecular mass of 18,212.0 Da corresponding to a myristoylated protein (theoretical mass = 18,212.6 Da).
Figure 4.
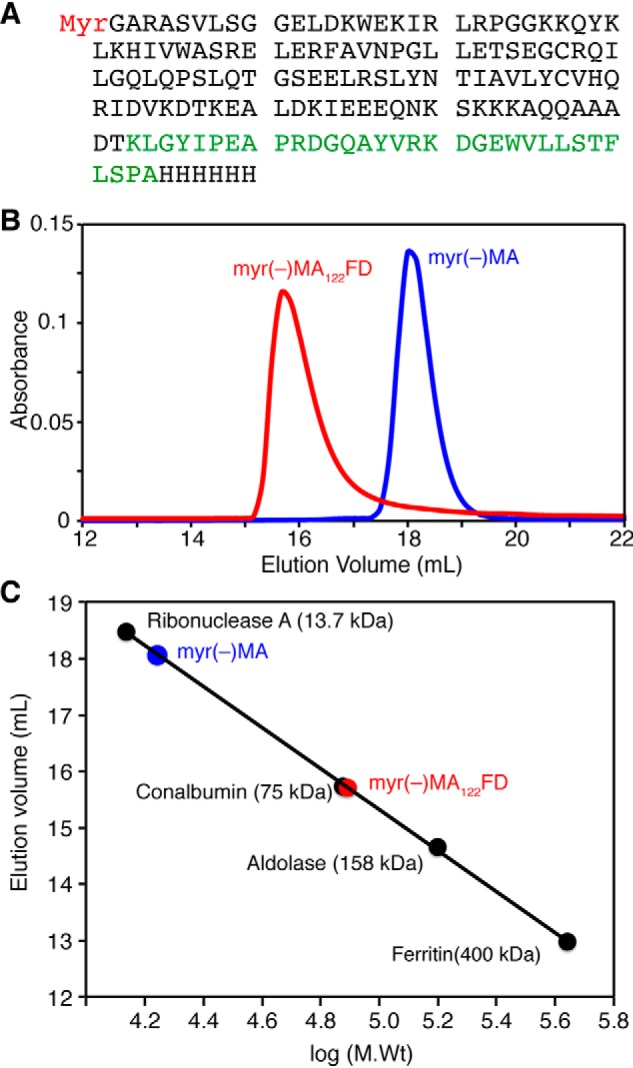
Gel filtration assay of MA proteins. A, protein sequence of MA122FD. Amino acids of FD are highlighted in green. B, elution profiles of myr(−)MA and myr(−)MA122FD on a HiLoad Superdex 200 (10/300 GL) column. C, a molecular weight calibration curve used to determine the approximate molecular weight of proteins.
To examine whether the FD domain promoted the formation of MA trimer, we ran a gel filtration assay for myr(−)MA122FD and compared it with the monomeric myr(−)MA protein. As shown in Fig. 4B, the myr(−)MA122FD protein eluted as a single peak at 15.8 ml, whereas that of the myr(−)MA protein eluted at 18 ml. A gel filtration mobility assay with known protein standards (Fig. 4C) indicated that the apparent molecular mass of myr(−)MA122FD was ∼75 kDa, which is slightly higher than the calculated mass of a trimer (∼60 kDa). The migration behavior of myr(−)MA122FD can be attributed to its shape, as shown previously with other proteins (68–70). In comparison, the elution profile of myr(−)MA is consistent with a monomeric ∼17-kDa species (Fig. 4, B and C). Similar experiments were conducted for the MA122FD protein and yielded similar results (see Fig. S6). Next, we performed sedimentation velocity experiments for the MA122FD and MA proteins. To ensure sample homogeneity, samples were run on a gel filtration column prior to sedimentation. As shown in Fig. 5, the sedimentation velocity profiles for both proteins exhibit a single sedimentation boundary distribution. Analysis of the data using SEDFIT yielded peaks at 1.7 and 4.9 S for MA and MA122FD, respectively. Molecular weight distribution analysis gave ∼17 and ∼70 kDa for the MA and MA122FD proteins, respectively. These results are consistent with the trimeric nature of the MA122FD protein. In summary, for the first time, we were able to produce a soluble, homogeneous, and myristoylated MA trimer.
Figure 5.
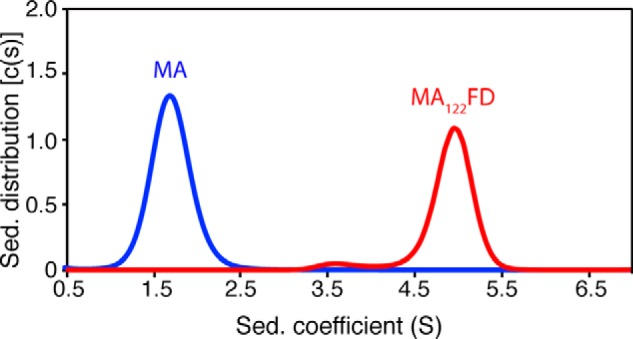
Sedimentation velocity of MA proteins. Shown are sedimentation coefficient distributions (c(s)) versus sedimentation coefficient (s) obtained from the sedimentation profiles of MA and MA122FD. Molecular weight distribution analysis indicates a largely monomeric MA and trimeric MA122FD species. Proteins were run using a buffer containing 50 mm sodium phosphate (pH 6.0) and 50 mm NaCl.
MA structure is not altered by trimerization
To examine whether the attachment of the FD fragment to the C terminus of MA and deletion of residues 123–132 altered the structure of MA, we obtained 2D HSQC data on 15N-labeled samples of both myr(−)MA122FD and MA122FD and compared them with those of the myr(−)MA and MA proteins, respectively (Fig. 6 and Fig. S4). As shown, the chemical shifts corresponding to the MA residues are very similar, demonstrating that the global structure of MA is not altered upon fusion of the FD fragment or deletion of residues 123–132. Interestingly, the spectrum for the MA122FD protein is significantly broader than that of the myr(−)MA122FD protein (Fig. 6 and Fig. S4). Moreover, for MA122FD, the 1H-15N signals corresponding to residues 2–20, Val35, Trp36, Arg39, Gly49, Glu52, His89, Qln90, and Arg91 are significantly shifted or severely broadened. These characteristic CSPs are indicative of conformational changes involving exposure of the myr group (29).
Figure 6.
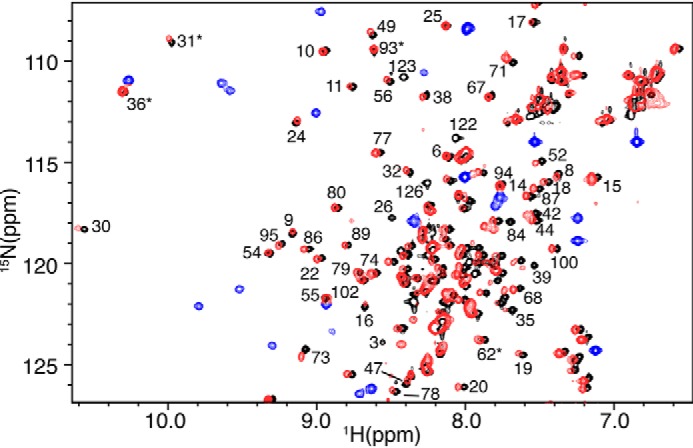
NMR data of myr(−)MA and myr(−)MA122FD. Shown is an overlay of 2D 1H-15N NMR spectra for myr(−)MA (black) and myr(−)MA122FD (red and blue). The great similarity of the spectra indicates that the global structure of MA is not altered upon fusion of the FD domain to the C terminus of MA. Signals shown in blue are those of the FD domain. Signals denoted by asterisks are aliased by 20 ppm.
Identification of the MA-MA interface by HDX-MS
HDX-MS is rapidly becoming a key technique for exploring protein structure and dynamics and protein-protein interactions (reviewed in Ref. 71). This approach exploits the fact that exposure of a protein to D2O induces amide hydrogen-to-deuterium exchange. Typically, the rate of the exchange depends on pH, temperature, participation in hydrogen bonding, and general solvent accessibility. This means that, at constant temperature and pH, tightly folded elements and regions involved in binding to other factors are much more protected from HDX. After incubation in D2O for various time periods, proteins are digested, and the deuterium uptake for individual peptides is analyzed by MS, allowing for structural characterization of individual regions within the protein. We applied this approach to determine whether introducing the FD domain promoted trimerization of the globular MA region and, if so, which regions of MA are involved in the trimer interface. HDX-MS experiments were conducted on the MA and MA122FD proteins. The deuterium incorporation profiles of peptide fragments obtained from both proteins were analyzed. By comparing the percentage of deuteration for peptides identified in both MA and MA122FD, we observed nearly identical profiles at all time points throughout most of the protein (Fig. S5). However, several overlapping peptides (residues 43–52, 45–50, 45–51, and 45–52) exhibited a significant increase in protection for MA122FD when compared with the deuteration profile of MA (Fig. 7A and Fig. S5). The disparity in protection is observed at shorter time points and converges as the exchange periods are increased. Small differences in protection evident at early times have previously been interpreted as due to time-dependent dissociation of a dynamic oligomer (72). Thus, this pattern likely indicates protection from exchange for this region upon induction of MA trimerization, leading us to conclude that this region is most likely located in the trimer interface. Remarkably, this region corresponds to one side of the interface displayed in the crystal structure of the MA trimer (Fig. 7B). Our data are not only in agreement with the currently accepted models for MA trimerization, but also provide the first biophysical evidence for the location of the trimerization interface in solution.
Figure 7.
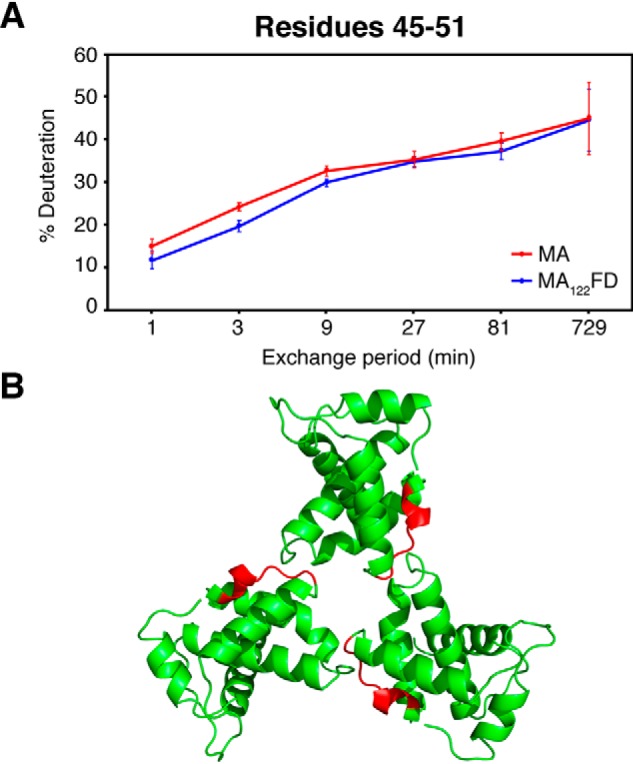
HDX-MS data of MA and MA122FD. A, deuterium exchange profile for residues 45–51 of MA and MA122FD at various time points. Points and error bars represent the average values and 95% confidence intervals calculated from six independent HDXMS experiments. B, cartoon representation of the HIV-1 MA crystal structure (PDB code 1HIW) with residues 45–51 highlighted in red located at the MA-MA interface.
Anti-FD Fab binding to the MA122FD protein
One of the major aims of this study was to characterize how the MA trimer interacts with lipid bilayers. Our initial attempts to characterize the interactions of MA122FD with NDs were unsuccessful due to heavy precipitation of the resulting complex. We hypothesized that precipitation of the MA122FD-ND complex is caused by overcrowding of the ND-binding surface, creating large insoluble aggregates. To overcome this challenge, we devised a novel approach in which the MA122FD protein was complexed with anti-FD Fab prior to characterization of its binding to NDs. We hypothesized that complexation of MA122FD with the anti-FD Fab may create steric hindrance and limit the number of MA122FD molecules bound to each layer of the ND.
First, we employed a gel filtration assay to assess whether the anti-FD Fab binds to the MA122FD protein. A sample of MA122FD-Fab complex prepared at a 1:1 molar ratio eluted as a single peak at 15.6 ml (Fig. S6). In comparison, the free MA122FD and anti-FD Fab proteins eluted at 17.5 and 18.2 ml, respectively. Fractions of the complex were analyzed via SDS-PAGE (Fig. S6). As shown, bands for the light and heavy chains of Fab as well as the MA122FD protein are observed with relatively equal intensities, suggesting the formation of a 1:1 MA122FD:Fab complex (three Fabs bound to an MA122FD trimer). A gel filtration mobility assay with known protein standards (Fig. S6) revealed that the free Fab migrates as a ∼50-kDa species, consistent with a complex of heavy and light chains cross-linked via a disulfide bond. Based on the mobility assay, the elution profile for the MA122FD-Fab complex corresponds to a molecular mass of ∼250 kDa, consistent with a MA122FD trimer bound to three Fab molecules.
Next, we employed ITC methods to obtain the thermodynamic parameters of Fab interaction with MA122FD. Titration of anti-FD Fab into MA122FD yielded a thermogram characteristic of an exothermic binding process. Data were best fit to a single set of identical sites (Fig. S7) and yielded a Kd of 60 ± 5 nm. As indicated by the thermodynamic parameters (Table 1), the interaction is enthalpically driven. Of note, the n value indicates that one Fab molecule binds to one MA122FD (i.e. 3 Fab molecules per MA122FD trimer). Altogether, our data demonstrate that anti-FD Fab forms a tight complex with MA122FD. Finally, we assessed whether binding of anti-FD Fab to the FD fragment in MA122FD had any effect on the structure of the MA globular domain. To do so, we collected 2D 1H-15N HSQC data on free and Fab-bound 15N-labeled myr(−)MA122FD. As shown in Fig. S7, the 1H-15N signals corresponding to FD residues shifted or became undetectable due to significant signal broadening. However, the amide signals corresponding to residues within the globular region of MA were still detectable and are unperturbed. This result indicates that the anti-FD Fab binds to FD without altering the structure of the globular domain of MA. Altogether, we presented a strategy in which anti-FD Fab is used to form a stable complex with the MA trimer to enable a quantitative assessment of binding to lipid bilayers.
Table 1.
ITC-binding constants and thermodynamic parameters for MA protein interactions with NDs and Fab
Titrations were conducted in a buffer containing 50 mm sodium phosphate (pH 6.0) and 50 mm NaCl.
| Titrations | n | Kd | ΔH° | −TΔS° | ΔG° |
|---|---|---|---|---|---|
| μm | kcal/mol | kcal/mol | kcal/mol | ||
| MA into POPC:POPS:PI(4,5)P2 (75:20:5) NDs | 10.3 ± 0.2 | 10.3 ± 1.9 | −4.3 ± 0.3 | −2.5 ± 0.4 | −6.8 ± 0.1 |
| MA122FD into Fab | 1.00 ± 0.03 | 0.060 ± 0.005 | −3.2 ± 0.2 | −6.8 ± 0.2 | −10.0 ± 0.1 |
| MA122FD-Fab into POPC:POPS:PI(4,5)P2 (75:20:5) NDs | 6.0 ± 0.2 | 0.32 ± 0.03 | −5.7 ± 1.1 | −3.3 ± 1.1 | −9.0 ± 0.1 |
Interaction of MA122FD-Fab with NDs
Formation of a stable and homogeneous MA122FD-Fab complex with a well-defined stoichiometry afforded an opportunity to study how the MA trimer interacts with lipid NDs. First, we employed ITC methods to obtain thermodynamic parameters of the MA122FD trimer interaction with NDs. To do so, we prepared a MA122FD-Fab complex and passed it through a gel filtration column as described above. Fractions were pooled from the center of the gel filtration elution peak of the MA122FD-Fab complex. Titration of the MA122FD-Fab complex into POPC:POPS:PI(4,5)P2 (75:20:5) NDs yielded a thermogram characteristic of an exothermic binding process (Fig. 8). Fitting the data to a one-site binding model provided a Kd of 320 ± 32 nm. Intriguingly, the affinity of MA122FD trimer binding to NDs is 30-fold tighter than that of the monomeric MA protein (Table 1). As indicated by the n value, two MA122FD trimers bind to one ND. The stoichiometry of binding is smaller compared with that observed for binding of monomeric MA to NDs (n = ∼10), likely due to steric hindrance caused by the presence of three Fab molecules. No binding was detected when Fab was titrated into POPC:POPS:PI(4,5)P2 (75:20:5) NDs (not shown), indicating direct binding between MA122FD and NDs.
Figure 8.
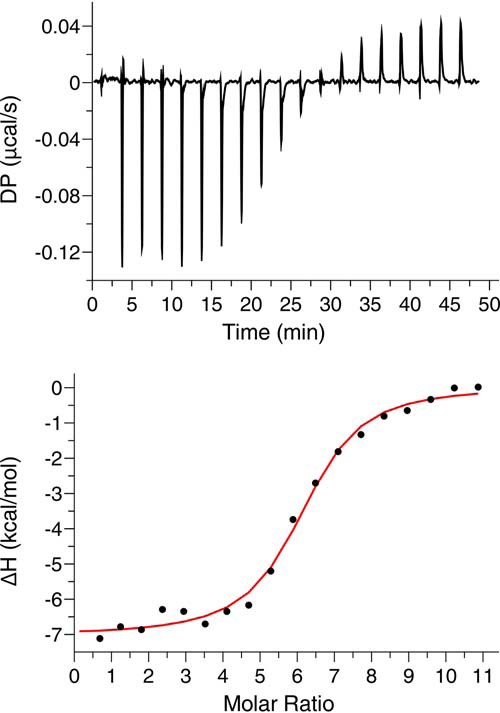
ITC analysis of MA trimer binding to NDs. Shown are ITC data obtained for titration of MA122FD-Fab (152 μm) into POPC:POPS:PI(4,5)P2 (75:20:5) NDs (2.7 μm). The top panel shows the baseline-adjusted thermogram, and the bottom panel depicts the binding isotherm and fit to the data (red line).
Next, we used a gel filtration assay to further assess binding of the MA122FD-Fab complex to POPC:POPS:PI(4,5)P2 (75:20:5) NDs. The MA122FD-Fab-ND complex was prepared by mixing a pure and homogeneous MA122FD-Fab complex with NDs at a molar ratio of 6:1, respectively. In contrast to mixing MA122FD with NDs, no precipitation was observed here. As shown in Fig. 9 (top), a peak with an elution volume of 12.8 ml was distinct from those of the free NDs (16.2 ml) and MA122FD-Fab complex (15.5 ml), indicating formation of a stable MA122FD-Fab-ND complex. All components of the MA122FD-Fab-ND complex were clearly observed in the SDS-PAGE (Fig. 9, bottom). The apparent molecular mass of the MA122FD-Fab-ND complex is estimated at 450 kDa. The elution profiles of free NDs and MA122FD-Fab complex are consistent with 150- and 170-kDa species, respectively. Based on the estimated molecular weight of the complex, two MA122FD-Fab complexes appear to bind to one ND (i.e. 2 trimers per one ND), which is consistent with the ITC data. These results indicate that a stable complex is formed between the MA122FD-Fab complex and lipid NDs enriched in PI(4,5)P2, demonstrating that Fab binding to the FD fragment does not affect binding of MA122FD to NDs.
Figure 9.
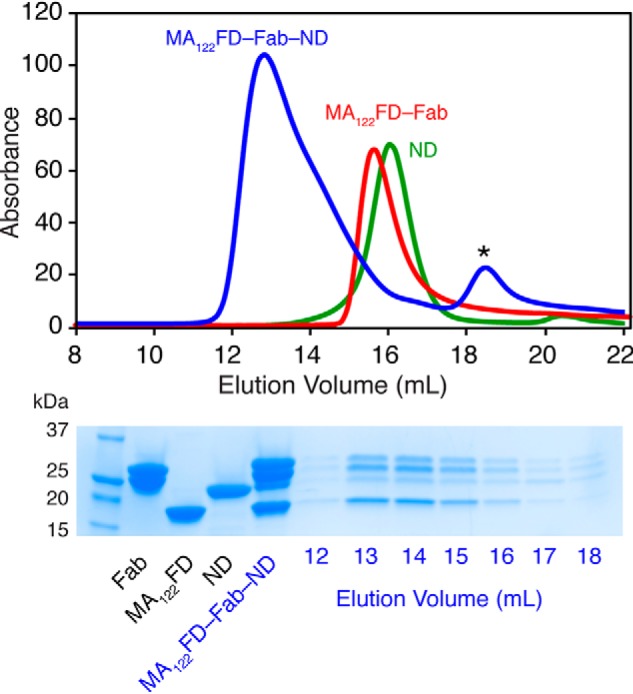
Gel filtration assay of the MA122FD-Fab-ND complex. Top, elution profiles of MA122FD-Fab, ND, and their complex on a Superose 6 (10/300 GL) column. Bottom, SDS-PAGE of MA122FD, Fab, ND, and their complexes. All components of the complex are clearly observed in the elution peak of the MA122FD-Fab-ND complex. Asterisk denotes a minor impurity.
Taken together, our data show that trimerization of the MA protein greatly enhances association with lipid bilayer, which supports the hypothesis that MA organizes as trimers on the membrane surface. These results also validate the utility of lipid NDs as membrane mimetics to study retroviral MA-membrane interactions.
Discussion
Biochemical and genetic data demonstrated that higher-order organization of MA is an important element for HIV-1 Gag assembly on the membrane and Env incorporation (55, 57–60, 73). Previous studies have shown that the HIV-1 MA and MACA proteins assembled as hexamers of trimers on membrane monolayers enriched with PI(4,5)P2 (59). This arrangement is widely used as a model to explain how the Env protein is incorporated into virus particles (55–57, 73). Cryo-EM studies of both mature and immature virus particles have provided detailed structures of the hexameric capsid lattice but were unable to resolve any well-ordered density for the membrane-bound MA domain (20–24). Structural investigation of the MA trimer and how it interacts with membranes is key to understanding Gag assembly and Env incorporation.
In this study, we engineered a stable MA trimer construct and characterized its interactions with lipid NDs by NMR, biochemical and biophysical tools. The rationale behind designing this construct was that tethering the three MAs together at the flexible C terminus would drive the natural equilibrium toward the trimeric state. NMR and HDX-MS data demonstrated that the global structure of MA was unperturbed by the attachment of FD. Intriguingly, relative to the monomeric MA protein, the HDX-MS data revealed an increased protection in multiple overlapping peptides in the MA122FD protein. This region (residues 43–52) corresponds to the MA-MA interface in the crystal structure of the myr(−)MA trimer (25). Of note, one of the mutants reported to inhibit trimerization and Env incorporation is located in this region (A45E) (55). Our data clearly established the overall structural similarities between the MA122FD and myr(−)MA trimer models and provided the first biophysical evidence for the trimer interface in solution. This is significant because the crystal structure of the trimer remains the basis for most models describing the organization of MA on the PM (55, 59, 60). The trimer structure of myr(−)MA was docked into the low-resolution density map of the 2D crystal lattice of MA on membrane (59). Our results indicated that a downstream oligomerizing domain such as the CA and NC domains of Gag, or FD as in our case, is essential for formation of an MA-MA interface that otherwise is undetectable in weakly associating MA molecules. Previous studies have shown that the trimeric, myristate-exposed species is enhanced upon inclusion of the capsid domain, indicating that exposure is enhanced by subdomains that promote self-association (29). Here, we have shown that attachment of the FD domain to the C terminus of MA may have also promoted myr exposure.
The second most important aspect of this study is the utilization of lipid NDs as a membrane bilayer mimetic, which enabled quantitative characterization of the interactions with MA monomer and trimer proteins. NDs are routinely used as a model lipid bilayer system for studying the structure and function of integral membrane proteins (reviewed in Ref. 42). NDs are flat lipid bilayers that are more analogous to the inner leaflet of cellular membrane than other lipid analogues such as micelles or liposomes (42). The ability to calculate the molar concentration of NDs based on the absorbance of the MSP protein at 280 nm (two copies per ND) allows for calculation of the binding parameters, including stoichiometry. Despite these advantages, NDs have been rarely used in studies investigating the binding properties of peripherally associated membrane proteins such as MA (48, 74, 75). Considering that the use of NDs in binding experiments is a relatively novel concept, we first conducted our studies with the MA monomer before expanding them to include the MA122FD trimer. We have shown that MA binding to NDs is significantly enhanced upon incorporation of PS and PI(4,5)P2 and that the interaction is mediated by the HBR located in the N terminus. Our data also indicated that the myr group is readily exposed for anchoring into the ND even in the absence of PS and PI(4,5)P2. These results demonstrated that NDs are a viable mimetic of biological membranes that can be reliably used to study membrane-associated proteins. Altogether, these findings allowed for construction of a model of MA trimer bound to a membrane bilayer (Fig. 10).
Figure 10.
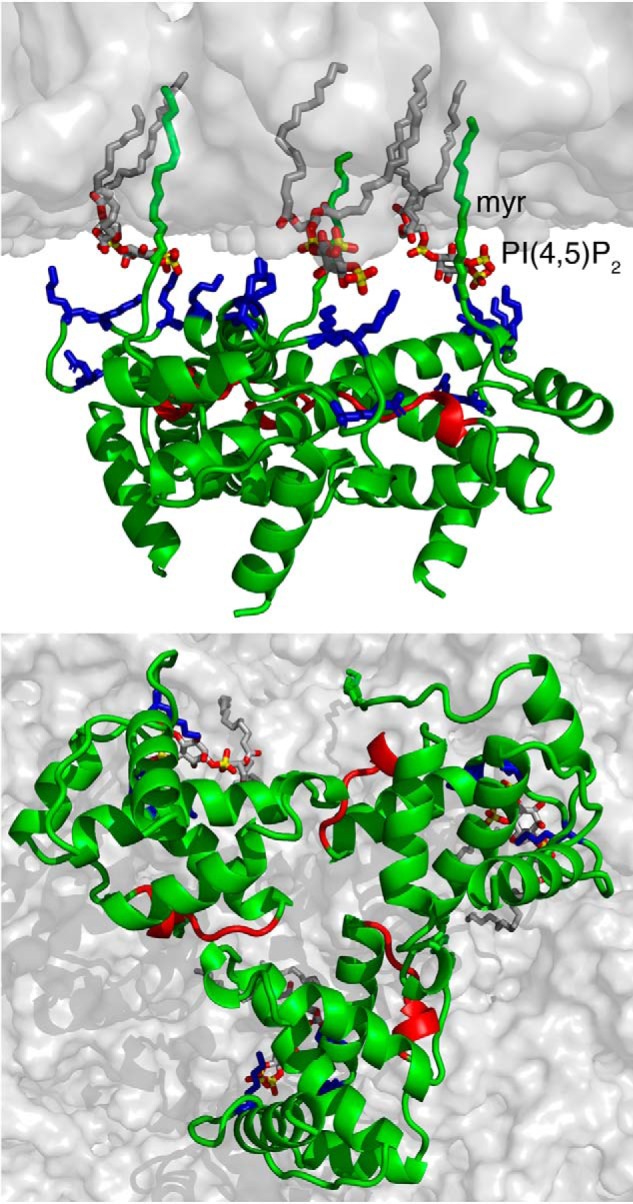
A model of HIV-1 MA trimer bound to membrane. Shown are top and bottom views of the MA trimer bound to a membrane bilayer. The interaction is mediated by the myr group, the acidic polar head of PI(4,5)P2, and basic residues (Arg22, Lys26, Lys27, Lys30, and Lys32) in the HBR (blue). Residues colored in red are located in the trimeric interface, as revealed by the HDX-MS data. Membrane bilayer was generated in the VMD membrane builder plug-in (92). PI(4,5)P2 was generated in Avogadro (93). The MA trimer was constructed by superimposition of the structure of myr-exposed MA and the X-ray structure of myr(−)MA.
Reported affinities of HIV-1 MA binding to membranes tend to vary, depending on the experimental technique. For example, in one study (15) using a liposome flotation assay with a lipid composition of POPC:POPS (2:1), Kd was found to be extremely weak (10 mm). In another study, by using surface plasmon resonance methods to study MA interaction with planar membrane models of similar lipid composition, the affinity was reported to be ∼2000-fold tighter (Kd ∼5 μm). The surface plasmon resonance study also provided a Kd of 1.4 μm for membranes composed of dioleoyl-phosphatidylcholine:dioleoyl-PS:PI(4,5)P2 (80:15:5) (76). More recently, by employing an NMR-detected liposome-binding assay, the apparent Kd of MA binding to PI(4,5)P2 embedded in liposomes was found to be ∼10 μm (38). Here, our ITC data of MA binding to POPC:POPS:PI(4,5)P2 (75:20:5) NDs afforded an apparent Kd of 10 μm, a result that is comparable with that obtained by the NMR-detected liposome binding assay. As mentioned above, one major advantage of using lipid NDs is the ability to obtain an accurate stoichiometry of binding. We have shown that 10 molecules of MA are capable of binding to one ND. Estimated from the NMR structure of HIV-1 MA (PDB entry 1UPH), one molecule should occupy about 7.5 nm2 of the membrane surface. Based on the inner diameter of the MSP1 ND (∼7.6 nm) (44) the total surface area of ND is ∼45 nm2. Therefore, each ND surface is capable of accommodating five MA molecules.
We initially attempted to perform binding studies of MA122FD to NDs in the same manner in which we conducted the experiments with the MA protein. However, titration of MA122FD into NDs led to the formation of insoluble aggregates. We circumvented this problem by forming a complex between MA122FD and an anti-FD Fab prior to titrations into NDs. Our ITC data indicated that the MA122FD-Fab complex binds to NDs at a stoichiometry of two trimers per ND. The addition of Fab into the complex most likely created enough steric hindrance so as to only allow a single trimer to bind each side of the ND. Perhaps this is an important consideration going forward with these types of binding experiments. If peripherally bound proteins contain multiple binding sites, then care must be taken to ensure that the binding surface only allows for a discrete number of binding partners. Of note, Fabs have emerged as a promising solution in structural studies of membrane proteins to yield a homogeneous population of the protein and to obtain diffraction quality crystals (77). They are often used to provide contrast and/or stability in EM and crystallographic studies (78, 79). As shown here, Fabs may also serve as a useful tool to prevent formation of insoluble aggregates in membrane binding experiments. In summary, we present the first successful utilization of NDs to assess retroviral MA-membrane interactions, which therefore can be expanded to examine binding of other retroviral MA and Gag proteins.
Previous studies have used MA fusion constructs to study the role of oligomerization in membrane interactions. In one study (80), two constructs were examined in which dimerization or hexamerization domains were fused to the C terminus of MA. These multimerization domains were chosen to mimic two well-characterized oligomeric states of CA. By utilizing liposome floatation assays, these studies revealed only modest increases in affinity for the dimeric and hexameric MA constructs relative to the MA monomer (∼1.5- and 2-fold, respectively) (80). Our data clearly indicate that the MA122FD trimer imparts a greater increase in affinity to membranes relative to the monomeric MA. One possible explanation for this disparity in the affinity of MA multimers binding to membranes is the different techniques and methods used to measure the affinity.
Recent studies from our laboratory revealed that the structure of gp41CT contains three α-helical domains that are tightly associated with the membrane (81). More recently, pulldown studies provided evidence for a direct MA-gp41CT interaction and suggested a role for MA trimerization (58). If a direct MA-gp41CT interaction occurs during Gag assembly, it is reasonable to suggest that a tight association of MA with membrane is required to facilitate this interaction. However, if the alternative model (55) based on steric exclusion of gp41CT is accurate, then tighter membrane association could be an unrelated consequence of the trimer formation. In either case, detailed structural studies of the MA trimer and gp41CT in a membrane context are warranted. We hope that the tools and techniques described in this report could facilitate such studies.
In summary, we developed new methods that allowed for the generation of a stable MA trimer and enabled characterization of its interaction with membranes using lipid NDs. These methods can be applied to study the binding properties of other membrane-interacting proteins, including those of other retroviruses, and can be used as a template for future investigation of the MA-gp41CT-membrane complex. Elucidation of the structural basis for MA-gp41CT-membrane interactions is key to understanding the mechanisms of virus assembly and Env incorporation.
Experimental procedures
Sample preparation
Plasmid construction
The DNA gene encoding for HIV-1 MA (amino acids 2–132) was amplified from the pNL4-3 isolate (GenBankTM number AF324493.2) with NcoI and HindIII restriction sites on the 5′- and 3′-end, respectively. The DNA gene encoding for the FD domain of phage T4 fibritin (GenBankTM number AF158101.6) plus a His6 tag at the 3′-end was amplified from the pWAC plasmid provided by Dr. Alasdair Steven (National Institutes of Health) (82). The amplified FD DNA fragment contained HindIII and XhoI restriction sites on its 5′- and 3′-end, respectively. The MA and FD DNA fragments were ligated via the HindIII site. The resulting MA-FD fragment was ligated into a co-expression plasmid encoding for yeast N-terminal myristoyltransferase provided by Dr. Michael Summers (Howard Hughes Medical Institute, University of Maryland Baltimore County). The DNA region encoding for residues 123–132 of MA was deleted using a QuikChange Lightning multi site-directed mutagenesis kit (Agilent Technologies).
Protein expression and purification
The HIV-1 MA and myr(−)MA proteins were expressed and purified as described (29). The MA122FD and myr(−)MA122FD proteins were overexpressed in Escherichia coli BL21-CodonPlus-RIL cells (Agilent Technologies). Cells were grown at 37 °C in Luria–Bertani broth medium containing 100 mg/liter ampicillin. When A600 reached ∼0.2, medium was supplemented with 60 μm myristic acid to produce MA122FD. When A600 reached 0.7–0.8, cells were induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (Gold Biotechnology). Cells were then grown at 16 °C for 18 h, spun down, and stored at −80 °C. The cell pellet was resuspended in 60 ml of lysis buffer (for 1 liter of culture) containing 50 mm sodium phosphate (pH 8.0), 1 m NaCl, 10% glycerol, 5 mm CHAPS (Fisher), 20 mm imidazole, 0.1 mm phenylmethanesulfonyl fluoride (Sigma–Aldrich), and 2 mm benzamidine (Sigma–Aldrich). Cells were then sonicated, and lysate was spun down at 35,000 × g for 40 min. The supernatant was filtered with a 0.45-μm syringe filter (Millex) and loaded into a cobalt affinity resin column equilibrated in lysis buffer (excluding protease inhibitors). Resin was then washed with lysis buffer (excluding protease inhibitors) followed by washing with a buffer containing 50 mm sodium phosphate (pH 8), 500 mm NaCl, 5% glycerol, and 20 mm imidazole. The MA122FD proteins were eluted via a gradient using a buffer containing 50 mm sodium phosphate (pH 8), 500 mm NaCl, 5% glycerol, and 500 mm imidazole. Protein purity was verified by SDS-PAGE. Fractions containing the protein were pooled and stored at −80 °C. Uniformly 15N-labeled MA samples were prepared by growing cells in M9 minimal medium containing 15NH4Cl. Protein purification was performed as described above. Identity of the proteins and efficiency of myristoylation were confirmed by MS (see below).
Production of anti-FD Fab
Plasmids encoding for the anti-FD IgG heavy and light chains were generously provided by Dr. Jason McLellan (University of Texas, Austin, TX). DH5α competent cells (Invitrogen) were transformed by introducing the plasmids, and the QIAfilter maxi kit (Qiagen) was used to purify plasmids for transfection. Plasmids were transfected in Freestyle 293-F cells (Gibco) grown in suspension culture with Freestyle 293 expression medium (Gibco). Cell culture was prepared for transfection according to the manufacturer's instructions with minor modifications. Transfection was performed by diluting 125 μg of each of the heavy and light chain plasmids into 25 ml of Opti-MEM reduced-serum medium (Gibco). A stock solution (1 mg/ml) of linear, transfection grade polymer polyethyleneimine (PEI 25K; Polysciences) was made in a buffer containing 25 mm HEPES (pH 7.5) and 150 mm NaCl. PEI stock solution (750 μl) was diluted in 25 ml of Opti-MEM. The plasmid solution was then rapidly combined with the 25-ml PEI solution and incubated for 20 min at room temperature before being added to ∼1.1 × 106 cells/ml 293F cells in 450 ml of Freestyle 293 expression medium. Cells were then placed in an incubator at 37 °C with a humidified atmosphere of 8% CO2 on an orbital shaker platform rotating at 125 rpm. Supernatant was harvested 6 days after transfection. Antibodies were purified using Pierce Protein A Plus agarose resin (Thermo Scientific) and digested by human rhinovirus 3C protease to obtain Fab. The FC domain was removed by passage over the Protein A resin and washing with PBS. Fab purity was assessed by SDS-PAGE under both reducing and nonreducing conditions, concentrated to ∼100 μm, and stored at −80 °C.
Preparation of lipid NDs
Membrane scaffold protein 1 (MSP1) plasmid (Addgene catalogue no. 20060) was expressed and purified as described (44, 46, 74). The MSP1 protein was stored at ∼200–300 μm in ND assembly buffer (20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 0.5 mm EDTA, and 0.01% NaN3). POPC, POPS, and PI(4,5)P2 (Avanti Polar Lipids) were stored as stock chloroform solutions at −20 °C. Chloroform stocks were mixed at the appropriate ratios (depending on the desired lipid composition in NDs), and the chloroform was evaporated under gentle airflow until visibly dry followed by 30 min under vacuum. Lipids were redissolved in ND assembly buffer supplemented with 100 mm sodium cholate by multiple cycles of freeze/thaw. A MSP1 stock sample was diluted to 120 μm in the assembly buffer before being added to lipid solution at an MSP1 to lipid molar ratio of 1:65. Sample was rotated on an orbital shaker at room temperature for 1 h. Sodium cholate was removed from solution by incubation with Bio-beads SM adsorbents (Bio-Rad) overnight at 4 °C on an orbital shaker. ND solution was then passed through a 0.2-μm filter to remove any precipitant or Bio-beads. Properly reconstituted lipid NDs were then run on a Superdex 200 Increase 10/300 GL column (GE Healthcare). ND fractions were pooled and used immediately or stored at −80 °C.
Gel filtration assay
The mobility of MA122FD, Fab, ND, and their complexes were analyzed by a gel filtration assay. Briefly, 0.5 ml of ∼30–100 μm protein samples were loaded on a Superdex S200 (10/300) column (GE Healthcare) in a buffer containing 50 mm sodium phosphate (pH 8) and 500 mm NaCl or on a Superose 6 (10/300) column (GE Healthcare) in a buffer containing 50 mm sodium phosphate (pH 7.4) and 150 mm NaCl. Protein fractions were analyzed by SDS-PAGE and stained by Coomassie Brilliant Blue. The approximate molecular weights of the loaded proteins were determined by molecular weight calibration kits (GE Healthcare).
NMR spectroscopy
NMR data were collected at 35 °C on a Bruker Avance II (700-MHz 1H) or Avance III (600- or 850-MHz 1H) spectrometers equipped with cryogenic triple-resonance probes, processed with NMRPIPE (83), and analyzed with NMRVIEW (84) or CCPN Analysis (85). 15N-Labeled myr(−)MA, MA, myr(−)MA122FD, and MA122FD protein samples were prepared at ∼100 μm in 50 mm sodium phosphate (pH 5.5 or 6.5) and 50 or 150 mm NaCl.
2D 1H-15N HSQC NMR titrations were conducted with 100 μm samples of 15N-labeled MA in 50 mm sodium phosphate (pH 6.5), 50 mm NaCl, and 2 mm DTT. Stock solutions of POPC:POPS and POPC:POPS:PI(4,5)P2 NDs were at 220 and 280 μm, respectively. Aliquots of NDs were added to 100 μm 15N-labeled MA to obtain molar ratios (ND:MA) of 0.0025:1, 0.01:1, 0.02:1, 0.04:1, 0.08:1, and 0.16:1. Each titration was followed by acquisition of 1D 1H and 2D 1H-15N HSQC spectra. Changes in signal intensities upon ND additions were expressed as relative reductions according to the formula, RRI = 1 − I/I0, where RRI is relative reduction of peak intensity (0 meaning no reduction, 1 meaning signal disappearance), I is peak integral in the presence of NDs, and I0 is peak integral in the absence of NDs. The RRIs were plotted as residue-by-residue histograms.
Analytical ultracentrifugation
Sedimentation velocity measurements were performed on a Beckman XL-I Optima system equipped with a 4-hole An-60-rotor (Beckman Coulter). The MA and MA122FD protein samples were at 20 μm in a buffer containing 50 mm phosphate (pH 6.0) and 50 mm NaCl. Rotor speed was set at 40,000 rpm, and scans were acquired at 280 nm and 20 °C. Partial specific volumes (v̄) and molar extinction coefficients were calculated using the program SENDTERP, and buffer densities were measured pycnometrically. Sedimentation velocity data were analyzed using SEDFIT (86–89).
Mass spectrometry
MS experiments were carried out on a Synapt G2-Si (Waters Corp.) and a LEAP HD/X-PAL (Trajan) fluidics system. The molecular mass measurement of MA122FD was conducted with 27 pmol of protein. Proteins were prepared in a buffer containing 20 mm sodium phosphate (pH 6) and 50 mm NaCl. Protein was partially separated based on hydrophobicity using a SecurityGuard ULTRA Widepore C4 trap column (Phenomenex). Liquid chromatography (LC) was carried out using a Shimadzu SPD-20 series pump system. Protein loading was carried out using a 0.1% formic acid solution at 0.1 ml/min. Samples for the HDX experiments contained 36 pmol of MA122FD or MA. Proteins were prepared in a buffer containing 20 mm sodium phosphate (pH 6) and 50 mm NaCl. Samples were diluted 10-fold in an equivalent buffer made with 99.8% D2O. Following dilution, proteins were incubated at 20 °C for an incubation period of 0, 1, 3, 9, 27, 81, and 729 min. The exchange reactions were quenched at 2 °C by a 1:1 dilution with a buffer consisting of 200 mm sodium phosphate (pH 2.1) and 4 m guanidine HCl. The quenched reaction was then digested by passage over an Enzymate pepsin column at 0.1 ml/min (Waters Corp., pore size 300 Å, particle size 5 μm, column size 2.1 × 30 mm). Enzymate pepsin column peptides were partially separated based on their hydrophobicity using an Acclaim PepMap C18 trap column (Thermo Fisher Scientific) and an ACQUITY BEH C18 reverse-phase column (Waters Corp., pore size 130 Å, particle size 1.7 μm, column size 1 × 50 mm). Liquid chromatography was carried out using a Shimadzu SPD-20 series pump system. Peptide loading was carried out using a 0.1% formic acid solution at 0.1 ml/min. Peptide separation was performed using an acetonitrile gradient in the presence of formic acid at 0.07–0.1 ml/min. Peptides were selected in PLGS (Waters) with a quality score of 6 or greater and a charge state of +1 to +4. The level of deuterium exchange was measured using HDExaminer software (Sierra Analytics).
Isothermal titration calorimetry
A sample of MA122FD-Fab complex was prepared and passed through a HiPrep 16/60 Sephacryl S-200 HR column (GE Healthcare) pre-equilibrated with a buffer containing 50 mm sodium phosphate (pH 7.4) and 150 mm NaCl. Fractions of the complex were taken from the center of the elution peak, dialyzed overnight in a buffer containing 50 mm sodium phosphate (pH 6.0) and 50 mm NaCl, and concentrated as needed. Thermodynamic parameters of Fab binding to MA122FD and those of MA and MA122FD-Fab binding to NDs were determined using a MicroCal PEAQ-ITC (Malvern Instruments). ITC data were obtained in a buffer containing 50 mm sodium phosphate (pH 6.0) and 50 mm NaCl. The heat of reaction was measured at 30 or 25 °C for 19 injections. The following concentrations were used: MA122FD at 300 μm into Fab at 35 μm; MA122FD-Fab at 160 μm into NDs at 2.7, 4.0, and 4.0 μm for POPC:POPS:PI(4,5)P2 (75:20:5), POPC:POPS (80:20), and POPC, respectively. MA at 350 μm into NDs at 3.5, 4.0, and 4.0 μm for POPC:POPS:PI(4,5)P2 (75:20:5), POPC:POPS (80:20), and POPC, respectively. The heat of dilution was measured by titrating MA, MA122FD, and MA122FD-Fab complex into buffer and was subtracted from the heat of binding. ITC experiments were collected in triplicates and yielded similar values. Mean values and S.D. values of the thermodynamic parameters were calculated based on the three independent experiments. Data analysis was performed using PEAQ analysis software. The thermodynamic parameters were determined by fitting baseline-corrected data to a binding model for a single set of identical sites.
Structure visualization
Visualization of structures was performed using PyMOL (PyMOL Molecular Graphics System, version 2.3.1 Schrödinger, LLC). Electrostatic potential maps were generated using PDB2PQR and APBS software (90, 91).
Author contributions
R. E. M., A. B. S., J. V., V. M., P. E. P., and J. S. S. data curation; R. E. M., A. B. S., J. V., P. E. P., and J. S. S. software; R. E. M., A. B. S., J. V., V. M., P. E. P., and J. S. S. formal analysis; R. E. M., A. B. S., J. V., V. M., P. E. P., and J. S. S. validation; R. E. M., A. B. S., J. V., V. M., P. E. P., and J. S. S. visualization; R. E. M., A. B. S., J. V., V. M., P. E. P., and J. S. S. methodology; R. E. M. and J. S. S. writing-original draft; R. E. M., J. V., P. E. P., and J. S. S. writing-review and editing; A. B. S., J. V., V. M., P. E. P., and J. S. S. investigation; P. E. P. and J. S. S. conceptualization; P. E. P. and J. S. S. resources; J. S. S. supervision; J. S. S. funding acquisition; J. S. S. project administration.
Supplementary Material
Acknowledgments
We thank members of our laboratory for fruitful discussion of the manuscript. We thank Dr. Jason S. McLellan and Daniel Wrapp at the University of Texas (Austin, TX) for help with the protocol of Fab production. We also thank Dr. John Kappes (University of Alabama, Birmingham, AL) for assistance with the logistics of Fab production. The high-field NMR facility at the University of Alabama at Birmingham was established through National Institutes of Health Grant 1S10RR026478 and is currently supported by NCI, National Institutes of Health, Comprehensive Cancer Center Grant P30 CA013148.
This work was supported by National Institutes of Health Grants 5R01GM117837 and 9R01AI150901 (to J. S. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S7.
The N-terminal Met, which is absent in the myristoylated protein, is designated as residue 1. In contrast, other studies considered the N-terminal Gly of the myristoylated protein as residue 1.
- PM
- plasma membrane
- MA
- myristoylated matrix
- myr
- myristoyl
- myr(−)MA
- unmyristoylated matrix
- FD
- foldon
- ND
- nanodisc
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- MSP
- membrane scaffold protein
- HSQC
- heteronuclear single quantum coherence
- CSP
- chemical shift perturbation
- ITC
- isothermal titration calorimetry
- HDX
- hydrogen–deuterium exchange
- CA
- capsid
- NC
- nucleocapsid
- SP1 and SP2
- spacer peptides 1 and 2, respectively
- HBR
- highly basic region
- PS
- phosphatidylserine
- 1D and 2D
- one- and two-dimensional, respectively
- MAFD
- matrix-foldon
- PDB
- Protein Data Bank
- PEI
- polyethyleneimine.
References
- 1. Mücksch F., Laketa V., Müller B., Schultz C., and Kräusslich H. G. (2017) Synchronized HIV assembly by tunable PIP2 changes reveals PIP2 requirement for stable Gag anchoring. Elife 6, e25287 10.7554/eLife.25287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hendrix J., Baumgärtel V., Schrimpf W., Ivanchenko S., Digman M. A., Gratton E., Kräusslich H. G., Müller B., and Lamb D. C. (2015) Live-cell observation of cytosolic HIV-1 assembly onset reveals RNA-interacting Gag oligomers. J. Cell Biol. 210, 629–646 10.1083/jcb.201504006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gousset K., Ablan S. D., Coren L. V., Ono A., Soheilian F., Nagashima K., Ott D. E., and Freed E. O. (2008) Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 4, e1000015 10.1371/journal.ppat.1000015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jouvenet N., Neil S. J. D., Bess C., Johnson M. C., Virgen C. A., Simon S. M., and Bieniasz P. D. (2006) Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 4, e435 10.1371/journal.pbio.0040435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Welsch S., Keppler O. T., Habermann A., Allespach I., Krijnse-Locker J., and Kräusslich H.-G. (2007) HIV-1 buds predominantly at the plasma membrane of primary human macrophages. PLoS Pathog. 3, e36 10.1371/journal.ppat.0030036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chukkapalli V., Hogue I. B., Boyko V., Hu W.-S., and Ono A. (2008) Interaction between HIV-1 Gag matrix domain and phosphatidylinositol-(4,5)-bisphosphate is essential for efficient Gag-membrane binding. J. Virol. 82, 2405–2417 10.1128/JVI.01614-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ono A., Ablan S. D., Lockett S. J., Nagashima K., and Freed E. O. (2004) Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. U.S.A. 101, 14889–14894 10.1073/pnas.0405596101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Freed E. O. (2015) HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 13, 484–496 10.1038/nrmicro3490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghanam R. H., Samal A. B., Fernandez T. F., and Saad J. S. (2012) Role of the HIV-1 matrix protein in Gag intracellular trafficking and targeting to the plasma membrane for virus assembly. Front. Microbiol. 3, 55 10.2165/00023210-200418060-00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ganser-Pornillos B. K., Yeager M., and Sundquist W. I. (2008) The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18, 203–217 10.1016/j.sbi.2008.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chukkapalli V., Inlora J., Todd G. C., and Ono A. (2013) Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J. Virol. 87, 7155–7159 10.1128/JVI.00075-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chukkapalli V., and Ono A. (2011) Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J. Mol. Biol. 410, 512–524 10.1016/j.jmb.2011.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Purohit P., Dupont S., Stevenson M., and Green M. R. (2001) Sequence-specific interaction between HIV-1 matrix protein and viral genomic RNA revealed by in vitro genetic selection. RNA 7, 576–584 10.1017/S1355838201002023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H., Dou J., Ding L., and Spearman P. (2007) Myristoylation is required for human immunodeficiency virus type 1 Gag-Gag multimerization in mammalian cells. J. Virol. 81, 12899–12910 10.1128/JVI.01280-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dalton A. K., Ako-Adjei D., Murray P. S., Murray D., and Vogt V. M. (2007) Electrostatic interactions drive membrane association of the human immunodeficiency virus type 1 Gag MA domain. J. Virol. 81, 6434–6445 10.1128/JVI.02757-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dick R. A., Goh S. L., Feigenson G. W., and Vogt V. M. (2012) HIV-1 Gag protein can sense the cholesterol and acyl chain environment in model membranes. Proc. Natl. Acad. Sci. U.S.A. 109, 18761–18766 10.1073/pnas.1209408109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chukkapalli V., Oh S. J., and Ono A. (2010) Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc. Natl. Acad. Sci. U.S.A. 107, 1600–1605 10.1073/pnas.0908661107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alfadhli A., Still A., and Barklis E. (2009) Analysis of human immunodeficiency virus type 1 matrix binding to membranes and nucleic acids. J. Virol. 83, 12196–12203 10.1128/JVI.01197-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ehrlich L. S., Fong S., Scarlata S., Zybarth G., and Carter C. (1996) Partitioning of HIV-1 gag and gag-related proteins to membranes. Biochemistry 35, 3933–3943 10.1021/bi952337x [DOI] [PubMed] [Google Scholar]
- 20. Briggs J. A., Riches J. D., Glass B., Bartonova V., Zanetti G., and Kräusslich H. G. (2009) Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. U.S.A. 106, 11090–11095 10.1073/pnas.0903535106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wright E. R., Schooler J. B., Ding H. J., Kieffer C., Fillmore C., Sundquist W. I., and Jensen G. J. (2007) Electron crytomography of immature HIV-1 virions reveals the structure of the CA and SP1 Gag shells. EMBO J. 26, 2218–2226 10.1038/sj.emboj.7601664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Briggs J. A., Johnson M. C., Simon M. N., Fuller S. D., and Vogt V. M. (2006) Cryo-electron microscopy reveals conserved and divergent features of gag packing in immature particles of Rous sarcoma virus and human immunodeficiency virus. J. Mol. Biol. 355, 157–168 10.1016/j.jmb.2005.10.025 [DOI] [PubMed] [Google Scholar]
- 23. Carlson L. A., Briggs J. A., Glass B., Riches J. D., Simon M. N., Johnson M. C., Müller B., Grunewald K., and Kräusslich H. G. (2008) Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host Microbe 4, 592–599 10.1016/j.chom.2008.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schur F. K., Hagen W. J., Rumlová M., Ruml T., Müller B., Kräusslich H. G., and Briggs J. A. (2015) Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 517, 505–508 10.1038/nature13838 [DOI] [PubMed] [Google Scholar]
- 25. Hill C. P., Worthylake D., Bancroft D. P., Christensen A. M., and Sundquist W. I. (1996) Crystal structures of the trimeric HIV-1 matrix protein: implications for membrane association. Proc. Natl. Acad. Sci. U.S.A. 93, 3099–3104 10.1073/pnas.93.7.3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Massiah M. A., Starich M. R., Paschall C., Summers M. F., Christensen A. M., and Sundquist W. I. (1994) Three dimensional structure of the human immunodeficiency virus type 1 matrix protein. J. Mol. Biol. 244, 198–223 10.1006/jmbi.1994.1719 [DOI] [PubMed] [Google Scholar]
- 27. Massiah M. A., Worthylake D., Christensen A. M., Sundquist W. I., Hill C. P., and Summers M. F. (1996) Comparison of the NMR and X-ray structures of the HIV-1 matrix protein: evidence for conformational changes during viral assembly. Protein Sci. 5, 2391–2398 10.1002/pro.5560051202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saad J. S., Miller J., Tai J., Kim A., Ghanam R. H., and Summers M. F. (2006) Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. U.S.A. 103, 11364–11369 10.1073/pnas.0602818103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang C., Loeliger E., Luncsford P., Kinde I., Beckett D., and Summers M. F. (2004) Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc. Natl. Acad. Sci. U.S.A. 101, 517–522 10.1073/pnas.0305665101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rao Z., Belyaev A. S., Fry E., Roy P., Jones I. M., and Stuart D. I. (1995) Crystal structure of SIV matrix antigen and implications for virus assembly. Nature 378, 743–747 10.1038/378743a0 [DOI] [PubMed] [Google Scholar]
- 31. Saad J. S., Loeliger E., Luncsford P., Liriano M., Tai J., Kim A., Miller J., Joshi A., Freed E. O., and Summers M. F. (2007) Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J. Mol. Biol. 366, 574–585 10.1016/j.jmb.2006.11.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vlach J., and Saad J. S. (2013) Trio engagement via plasma membrane phospholipids and the myristoyl moiety governs HIV-1 matrix binding to bilayers. Proc. Natl. Acad. Sci. U.S.A. 110, 3525–3530 10.1073/pnas.1216655110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saad J. S., Ablan S. D., Ghanam R. H., Kim A., Andrews K., Nagashima K., Soheilian F., Freed E. O., and Summers M. F. (2008) Structure of the myristylated HIV-2 MA protein and the role of phosphatidylinositol-(4,5)-bisphosphate in membrane targeting. J. Mol. Biol. 382, 434–447 10.1016/j.jmb.2008.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vlach J., Eastep G. N., Ghanam R. H., Watanabe S. M., Carter C. A., and Saad J. S. (2018) Structural basis for targeting avian sarcoma virus Gag polyprotein to the plasma membrane for virus assembly. J. Biol. Chem. 293, 18828–18840 10.1074/jbc.RA118.003944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamard-Peron E., Juillard F., Saad J. S., Roy C., Roingeard P., Summers M. F., Darlix J. L., Picart C., and Muriaux D. (2010) Targeting of murine leukemia virus gag to the plasma membrane is mediated by PI(4,5)P2/PS and a polybasic region in the matrix. J. Virol. 84, 503–515 10.1128/JVI.01134-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prchal J., Srb P., Hunter E., Ruml T., and Hrabal R. (2012) The structure of myristoylated Mason-Pfizer monkey virus matrix protein and the role of phosphatidylinositol-(4,5)-bisphosphate in its membrane binding. J. Mol. Biol. 423, 427–438 10.1016/j.jmb.2012.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brown L. A., Cox C., Baptiste J., Summers H., Button R., Bahlow K., Spurrier V., Kyser J., Luttge B. G., Kuo L., Freed E. O., and Summers M. F. (2015) NMR structure of the myristylated feline immunodeficiency virus matrix protein. Viruses 7, 2210–2229 10.3390/v7052210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mercredi P. Y., Bucca N., Loeliger B., Gaines C. R., Mehta M., Bhargava P., Tedbury P. R., Charlier L., Floquet N., Muriaux D., Favard C., Sanders C. R., Freed E. O., Marchant J., and Summers M. F. (2016) Structural and molecular determinants of membrane binding by the HIV-1 matrix protein. J. Mol. Biol. 428, 1637–1655 10.1016/j.jmb.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anraku K., Fukuda R., Takamune N., Misumi S., Okamoto Y., Otsuka M., and Fujita M. (2010) Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 49, 5109–5116 10.1021/bi9019274 [DOI] [PubMed] [Google Scholar]
- 40. Shkriabai N., Datta S. A., Zhao Z., Hess S., Rein A., and Kvaratskhelia M. (2006) Interactions of HIV-1 Gag with assembly cofactors. Biochemistry 45, 4077–4083 10.1021/bi052308e [DOI] [PubMed] [Google Scholar]
- 41. Fernandes F., Chen K., Ehrlich L. S., Jin J., Chen M. H., Medina G. N., Symons M., Montelaro R., Donaldson J., Tjandra N., and Carter C. A. (2011) Phosphoinositides direct equine infectious anemia virus gag trafficking and release. Traffic 12, 438–451 10.1111/j.1600-0854.2010.01153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Denisov I. G., and Sligar S. G. (2016) Nanodiscs for structural and functional studies of membrane proteins. Nat. Struct. Mol. Biol. 23, 481–486 10.1038/nsmb.3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borch J., and Hamann T. (2009) The nanodisc: a novel tool for membrane protein studies. Biol. Chem. 390, 805–814 10.1515/BC.2009.091 [DOI] [PubMed] [Google Scholar]
- 44. Bayburt T. H., and Sligar S. G. (2010) Membrane protein assembly into Nanodiscs. FEBS Lett. 584, 1721–1727 10.1016/j.febslet.2009.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hagn F., Etzkorn M., Raschle T., and Wagner G. (2013) Optimized phospholipid bilayer nanodiscs facilitate high-resolution structure determination of membrane proteins. J. Am. Chem. Soc. 135, 1919–1925 10.1021/ja310901f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kobashigawa Y., Harada K., Yoshida N., Ogura K., and Inagaki F. (2011) Phosphoinositide-incorporated lipid-protein nanodiscs: a tool for studying protein-lipid interactions. Anal. Biochem. 410, 77–83 10.1016/j.ab.2010.11.021 [DOI] [PubMed] [Google Scholar]
- 47. Ritchie T. K., Grinkova Y. V., Bayburt T. H., Denisov I. G., Zolnerciks J. K., Atkins W. M., and Sligar S. G. (2009) Chapter 11—Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 464, 211–231 10.1016/S0076-6879(09)64011-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yokogawa M., Kobashigawa Y., Yoshida N., Ogura K., Harada K., and Inagaki F. (2012) NMR analyses of the interaction between the FYVE domain of early endosome antigen 1 (EEA1) and phosphoinositide embedded in a lipid bilayer. J. Biol. Chem. 287, 34936–34945 10.1074/jbc.M112.398255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hagn F., Nasr M. L., and Wagner G. (2018) Assembly of phospholipid nanodiscs of controlled size for structural studies of membrane proteins by NMR. Nat. Protoc. 13, 79–98 10.1038/nprot.2017.094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dorfman T., Mammano F., Haseltine W. A., and Göttlinger H. G. (1994) Role of the matrix protein in the virion association of the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 68, 1689–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Freed E. O., and Martin M. A. (1996) Domains of the human immonodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J. Virol. 70, 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Freed E. O., and Martin M. A. (1995) Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J. Virol. 69, 1984–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu X., Yuan X., Matsuda Z., Lee T.-H., and Essex M. (1992) The matrix protein of human immunodeficiency virus type I is required for incorporation of viral envelope protein into mature virions. J. Virol. 66, 4966–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cosson P. (1996) Direct interaction between the envelope and matrix proteins of HIV-1. EMBO J. 15, 5783–5788 10.1002/j.1460-2075.1996.tb00964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tedbury P. R., Novikova M., Ablan S. D., and Freed E. O. (2016) Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc. Natl. Acad. Sci. U.S.A. 113, E182–E190 10.1073/pnas.1516618113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tedbury P. R., Ablan S. D., and Freed E. O. (2013) Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog. 9, e1003739 10.1371/journal.ppat.1003739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alfadhli A., Mack A., Ritchie C., Cylinder I., Harper L., Tedbury P. R., Freed E. O., and Barklis E. (2016) Trimer enhancement mutation effects on HIV-1 matrix protein binding activities. J. Virol. 90, 5657–5664 10.1128/JVI.00509-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Alfadhli A., Staubus A. O., Tedbury P. R., Novikova M., Freed E. O., and Barklis E. (2019) Analysis of HIV-1 matrix-envelope cytoplasmic tail interactions. J. Virol. 93, e01079–19 10.1128/JVI.01079-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alfadhli A., Barklis R. L., and Barklis E. (2009) HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology 387, 466–472 10.1016/j.virol.2009.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alfadhli A., Huseby D., Kapit E., Colman D., and Barklis E. (2007) Human immunodeficiency virus type 1 matrix protein assembles on membranes as a hexamer. J. Virol. 81, 1472–1478 10.1128/JVI.02122-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Matsuo H., Walters K. J., Teruya K., Tanaka T., Gassner G. T., Lippard S. J., Kyogoku Y., and Wagner G. (1999) Identification by NMR spectroscopy of residues at contact surfaces in large, slowly exchanging macromolecular complexes. J. Am. Chem. Soc. 121, 9903–9904 10.1021/ja991881g [DOI] [Google Scholar]
- 62. Fledderman E. L., Fujii K., Ghanam R. H., Waki K., Prevelige P. E., Freed E. O., and Saad J. S. (2010) Myristate exposure in the human immunodeficiency virus type 1 matrix protein is modulated by pH. Biochemistry 49, 9551–9562 10.1021/bi101245j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang X., Lee J., Mahony E. M., Kwong P. D., Wyatt R., and Sodroski J. (2002) Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J. Virol. 76, 4634–4642 10.1128/JVI.76.9.4634-4642.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lu Y., Welsh J. P., and Swartz J. R. (2014) Production and stabilization of the trimeric influenza hemagglutinin stem domain for potentially broadly protective influenza vaccines. Proc. Natl. Acad. Sci. U.S.A. 111, 125–130 10.1073/pnas.1308701110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Seok J. H., Kim J., Lee D. B., Cho K. J., Lee J. H., Bae G., Chung M. S., and Kim K. H. (2017) Conformational modulation of influenza virus hemagglutinin: characterization and in vivo efficacy of monomeric form. Sci. Rep. 7, 7540 10.1038/s41598-017-08021-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Güthe S., Kapinos L., Möglich A., Meier S., Grzesiek S., and Kiefhaber T. (2004) Very fast folding and association of a trimerization domain from bacteriophage T4 fibritin. J. Mol. Biol. 337, 905–915 10.1016/j.jmb.2004.02.020 [DOI] [PubMed] [Google Scholar]
- 67. Meier S., Güthe S., Kiefhaber T., and Grzesiek S. (2004) Foldon, the natural trimerization domain of T4 fibritin, dissociates into a monomeric A-state form containing a stable beta-hairpin: atomic details of trimer dissociation and local β-hairpin stability from residual dipolar couplings. J. Mol. Biol. 344, 1051–1069 10.1016/j.jmb.2004.09.079 [DOI] [PubMed] [Google Scholar]
- 68. Majava V., and Kursula P. (2009) Domain swapping and different oligomeric states for the complex between calmodulin and the calmodulin-binding domain of calcineurin A. PLoS One 4, e5402 10.1371/journal.pone.0005402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Agamasu C., Ghanam R. H., and Saad J. S. (2015) Structural and biophysical characterization of the interactions between calmodulin and the pleckstrin homology domain of Akt. J. Biol. Chem. 290, 27403–27413 10.1074/jbc.M115.673939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Samal A. B., Ghanam R. H., Fernandez T. F., Monroe E. B., and Saad J. S. (2011) NMR, biophysical and biochemical studies reveal the minimal calmodulin-binding domain of the HIV-1 matrix protein. J. Biol. Chem. 286, 33533–33543 10.1074/jbc.M111.273623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Konermann L., Pan J., and Liu Y. H. (2011) Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 40, 1224–1234 10.1039/C0CS00113A [DOI] [PubMed] [Google Scholar]
- 72. Huang R. Y., Garai K., Frieden C., and Gross M. L. (2011) Hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry determine the interface and dynamics of apolipoprotein E oligomerization. Biochemistry 50, 9273–9282 10.1021/bi2010027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tedbury P. R., and Freed E. O. (2014) The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol. 22, 372–378 10.1016/j.tim.2014.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Agamasu C., Ghanam R. H., Xu F., Sun Y., Chen Y., and Saad J. S. (2017) The interplay between calmodulin and membrane interactions with the pleckstrin homology domain of Akt. J. Biol. Chem. 292, 251–263 10.1074/jbc.M116.752816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wan C., Wu B., Song Z., Zhang J., Chu H., Wang A., Liu Q., Shi Y., Li G., and Wang J. (2015) Insights into the molecular recognition of the granuphilin C2A domain with PI(4,5)P2. Chem. Phys. Lipids 186, 61–67 10.1016/j.chemphyslip.2015.01.003 [DOI] [PubMed] [Google Scholar]
- 76. Barros M., Heinrich F., Datta S. A. K., Rein A., Karageorgos I., Nanda H., and Lösche M. (2016) Membrane binding of HIV-1 matrix protein: dependence on bilayer composition and protein lipidation. J. Virol. 90, 4544–4555 10.1128/JVI.02820-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kim J., Stroud R. M., and Craik C. S. (2011) Rapid identification of recombinant Fabs that bind to membrane proteins. Methods 55, 303–309 10.1016/j.ymeth.2011.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Griffin L., and Lawson A. (2011) Antibody fragments as tools in crystallography. Clin. Exp. Immunol. 165, 285–291 10.1111/j.1365-2249.2011.04427.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yu G., Li K., Huang P., Jiang X., and Jiang W. (2016) Antibody-based affinity cryoelectron microscopy at 2.6-Å resolution. Structure 24, 1984–1990 10.1016/j.str.2016.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dick R. A., Kamynina E., and Vogt V. M. (2013) Effect of multimerization on membrane association of Rous sarcoma virus and HIV-1 matrix domain proteins. J. Virol. 87, 13598–13608 10.1128/JVI.01659-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Murphy R. E., Samal A. B., Vlach J., and Saad J. S. (2017) Solution structure and membrane interaction of the cytoplasmic tail of HIV-1 gp41 protein. Structure 25, 1708–1718.e5 10.1016/j.str.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Miroshnikov K. A., Marusich E. I., Cerritelli M. E., Cheng N., Hyde C. C., Steven A. C., and Mesyanzhinov V. V. (1998) Engineering trimeric fibrous proteins based on bacteriophage T4 adhesins. Protein Eng. 11, 329–332 10.1093/protein/11.4.329 [DOI] [PubMed] [Google Scholar]
- 83. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 10.1007/bf00197809 [DOI] [PubMed] [Google Scholar]
- 84. Johnson B. A., and Blevins R. A. (1994) NMRview: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 10.1007/BF00404272 [DOI] [PubMed] [Google Scholar]
- 85. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., and Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 10.1002/prot.20449 [DOI] [PubMed] [Google Scholar]
- 86. Lebowitz J., Lewis M. S., and Schuck P. (2002) Modern analytical ultracentrifugation in protein science—a tutorial review. Protein Sci. 11, 2067–2079 10.1110/ps.0207702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Schuck P. (2000) Size distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schuck P. (2003) On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 320, 104–124 10.1016/S0003-2697(03)00289-6 [DOI] [PubMed] [Google Scholar]
- 89. Schuck P., Perugini M. A., Gonzales N. R., Howlett G. J., and Schubert D. (2002) Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys. J. 82, 1096–1111 10.1016/S0006-3495(02)75469-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 10.1093/nar/gkh381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 10.1073/pnas.181342398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 93. Hanwell M. D., Curtis D. E., Lonie D. C., Vandermeersch T., Zurek E., and Hutchison G. R. (2012) Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 17 10.1186/1758-2946-4-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.