

Abstract
Histone deacetylase (HDAC) enzymes that catalyze removal of acetyl-lysine post translational modifications are frequently post-translationally modified. HDAC8 is phosphorylated within the deacetylase domain at conserved residue serine 39 which leads to decreased catalytic activity. HDAC8 phosphorylation at S39 is unique in its location and function and may represent a novel mode of deacetylation regulation. To better understand the impact of phosphorylation of HDAC8 on enzyme structure and function, we performed crystallographic, kinetic, and molecular dynamics studies of the S39E HDAC8 phosphomimetic mutant. This mutation decreases deacetylation of peptides derived from acetylated nuclear and cytoplasmic proteins. However, the magnitude of the effect depends on the peptide sequence and the identity of the active site metal ion (Zn(II) vs Fe(II)) with the value of kcat/KM for the mutant decreasing 9- to >200-fold compared to wild-type HDAC8. Furthermore, the dissociation rate constant of the active site metal ion increases by ~15-fold. S39E HDAC8 was crystallized in complex with the inhibitor Droxinostat revealing that phosphorylation of S39, as mimicked by the glutamate side chain, perturbs local structure through distortion of the L1 loop. Molecular dynamics simulations of both S39E and phosphorylated S39 HDAC8 demonstrate that the perturbation of the L1 loop likely occurs because of the lost hydrogen bond between D29 and S39. Furthermore, the S39 perturbation causes structural changes that propagate through the protein scaffolding to influence function in the active site. These data demonstrate that phosphorylation plays an important regulatory role for HDAC8 by affecting ligand binding, catalytic efficiency and substrate selectivity.
Keywords: phosphorylation, crystallography, molecular dynamics, substrate recognition, structure, metal-dependent
Graphical Abstract
INTRODUCTION
Post translational modifications (PTMs) are a well-known mechanism used by cells to reversibly regulate protein structure and function. The family of histone deacetylases (HDACs), enzymes responsible for catalyzing the removal of acetylation PTMs on proteins, are frequently modified themselves by various PTMs, including phosphorylation. Metal-dependent HDACs 1–9 are phosphorylated at one or more sites, and the PTM regulates protein-protein interactions, protein complex formation, and subcellular localization.1–2 Of these, only HDAC5, HDAC6, and HDAC8 have a phosphorylation site within the deacetylase domain of their protein structure, which is more likely to impact enzyme activity.2
The phosphorylation site of HDAC8 within the deacetylase domain is unique among HDACs. HDAC8 is phosphorylated by protein kinase A at serine 39 (S39), a residue not conserved among the closely related class I HDACs (arginine in HDAC1 and HDAC2, alanine in HDAC3).3–4 HDAC4, 5, 7, and 9 have a nearby serine based on sequence alignment, although the local sequence environment is different.5 S39 is conserved in homologues from many species including Mus musculus (Mouse), Bos taurus (Bovine), Xenopus laevis (African clawed frog) and Danio rerio (Zebrafish),6 suggesting that the residue is important for HDAC8 function. However, S39 is not conserved in the orthologue of Schistosoma mansoni. This blood fluke is the most common parasite infecting humans and is responsible for intestinal schistosomiasis, a neglected tropical disease inflicting over 200 million people whose main treatment option is at risk for development of resistance. Since S. mansoni HDAC8 is targeted for antiparasitic drug development, clarification of S39 phosphorylation function could illuminate novel targeting approaches.7–9 HDAC8 phosphorylation is also unique among HDACs in function. While phosphorylation has been suggested to activate the catalytic activity of HDAC1 and HDAC2,3, 10–13 phosphorylation decreases HDAC8 activity.4 Moreover, despite conflicting results, phosphorylation appears to play an important role in HDAC1 and HDAC2 complex formation.3, 10–11 While it is unclear whether phosphorylation of HDAC8 impacts complex formation in cells, HDAC8 activity in vitro is not dependent on complex formation, as is the case for HDAC1 and HDAC2.
The purpose of HDAC8 S39 phosphorylation remains a mystery. However, S39 phosphorylation clearly impacts HDAC8 structure and function by means of several proposed mechanisms. First, HDAC8 phosphorylation appears to alter subcellular localization in certain cell types.14 Unlike other class I isozymes, HDAC8 has been observed in both the nucleus and the cytoplasm.14–16 In myometrial cells, unmodified HDAC8 is observed in the cytoplasmic, cytoskeletal, and nuclear fractions.14 In contrast, phosphorylated HDAC8 co-localizes almost entirely with the cytoskeleton.14 Intriguingly, phosphorylated HDAC8 levels are increased in the myometrial cells of pregnant women while overall HDAC8 expression remains unchanged.14 Phosphorylation may be used as a way to direct all or a portion of HDAC8 to specific compartments within the cell, potentially dependent on cell status, type, or location. Phosphorylation-dependent mechanisms of translocation have been observed in other HDACs as well. Providing a precedent for this behavior, phosphorylation regulates class II HDACs movement between nucleus and cytosol.17–22
In addition to regulating subcellular localization, HDAC8 phosphorylation also impacts acetylation levels in cells. Although within the deacetylase domain, S39 is located over 20 Å from the catalytic metal ion.23–29 Despite this distance, S39 phosphorylation leads to inhibition of catalytic activity by an unknown mechanism.4 Increased acetylation was observed on histones isolated from PKA-activated HeLa cells expressing Flag-tagged HDAC8 and treated with the adenylyl cyclase PKA activator forskolin, compared with untreated, non-overexpressed, and S39A-HDAC8 expressed control cells, indicative of PKA-mediated HDAC8 inhibition.4 Moreover, phosphorylated Flag-HDAC8, immunopurified from the PKA-activated cells, exhibited slower rates of deacetylation of core histones H3 and H4 compared to the non-phosphorylated HDAC8 control.4
One hypothesis suggests that S39 phosphorylation impacts catalytic activity through altering protein-protein interactions. Phosphorylation increases HDAC8 association with human Est1p-like protein B (hEST1B) and Hsp70 in co-immunoprecipitation experiments, demonstrating selectivity for binding phosphorylated HDAC8.30 Alternatively, S39 phosphorylation may have a direct impact on the active site and surrounding areas by leading to structural changes. R37 (Figure 1) is a highly conserved residue critical for catalytic activity that serves as the gatekeeper to the internal channel of HDAC8 by regulating hydrogen bond interactions between conserved ‘gate’ residues G303 and G305.31 Presumably, a conformational change of R37 due to phosphorylation of nearby S39 could lead to a similar outcome. Moreover, as shown in crystal structures of HDAC8, S39 is located at the base of loop L1 (Figure 1) and is poised to modulate interactions with binding partners, substrates, and/or inhibitors distal to the active site.23–24, 28–29 The L1 and L2 loops flank the active site of HDAC8 and their high flexibility accommodate binding of different ligands, as shown by crystallographic and molecular dynamics studies.23–24, 28–29, 32–33 Indeed, the L1 and L2 loops are critical for ligand recognition and binding in part through the interaction between K33 (L1 loop) and D101 (L2 loop),32–33 and in comparison with other isozymes, such as HDAC1 whose loops are more rigid, likely govern binding selectivity.34 Perturbing the size and charge of S39 would lead to distortions in the α2 helix, which could impact K33 positioning and possibly the K33-D101 interaction and thereby substrate binding affinity and selectivity.
Figure 1. Structure of HDAC8.

Crystal structure of human, wild-type HDAC8 (grey, PDB ID 2V5W) bound to a peptide substrate derived from p53 (cyan).26 Loops L1 and L2 are shown in green and the active site zinc(II) ion is shown in red. Indicated are helices α1 and α2 and residues S39, R37, K33, D101, G303 and G305.
Since HDAC8 function is implicated in a variety of diseases, this isozyme is a target for drug development.8 However, to date, no HDAC8-specific inhibitor has been approved for drug use. HDAC8 phosphorylation at S39 presents a unique mode of HDAC, and specifically HDAC8 regulation, and therefore is an important subject for further investigation to identify and develop isozyme-specific drug targeting routes. Here we combine structural and biochemical data of the S39E phosphomimetic mutant to demonstrate that phosphorylation modulates conformational changes, which alter the reactivity and substrate selectivity of HDAC8.
MATERIALS AND METHODS
Reagents
The HDAC inhibitor 4-(4-chloro-2-methylphenoxy)-N-hydroxybutanamide (Droxinostat) was obtained from Sigma Aldrich and was used without further purification. Tris(2-carboxyethyl)phosphine (TCEP) was purchased from GoldBio. All other reagents were purchased from Fisher unless otherwise specified.
Expression and purification of S39E HDAC8 for crystallization
The S39E mutation was introduced into the HDAC8-His6-pET20b construct24 using Quickchange site directed-mutagenesis kit protocols (Agilent Technologies, Inc.). Primers used for PCR mutagenesis are as follows: forward, 5’-GCT AAA ATC CCG AAA CGT GCA gag ATG GTG CAT TCT TTG ATT GAA G-3’; and reverse, 5’-C TTC AAT CAA AGA ATG CAC CAT ctc TGC ACG TTT CGG GAT TTT AGC-3’. Incorporation of the desired mutations was confirmed by DNA sequencing at the University of Pennsylvania Perelman School of Medicine. Recombinant S39E HDAC8 was expressed in BL21(DE3) Escherichia coli cells transformed with S39E HDAC8-His6-pET20b plasmid and purified according to a previously published procedure.35
Expression and purification of wild-type and S39E HDAC8 for assays
The S39E mutation was introduced into the pHD4 (codon-optimized HDAC8-TEV-His6) plasmid36 using custom primers (Integrated DNA Technologies) and Quickchange site directed-mutagenesis kit protocols (Agilent Technologies, Inc.). Primers used for PCR mutagenesis are as follows: forward, 5’-CCC GAA ACG TGC Aga GAT GGT GCA TTC TTT GAT TGA AGC ATA TG-3’; and reverse, 5’-CAT ATG CTT CAA TCA AAG AAT GCA CCA TCt cTG CAC GTT TCG GG-3’. Incorporation of desired mutations was confirmed by DNA sequencing performed by the University of Michigan DNA Sequencing Core, and the corresponding mass of the purified S39E HDAC8 was confirmed by Q-TOF HPLC-MS (Agilent) analysis. Wild-type (WT) and S39E HDAC8 were expressed in BL21 (DE3) Escherichia coli cells transformed with the appropriate plasmid and purified according to previously published procedures.36–37 Apo enzyme was prepared by dialyzing concentrated enzyme against 200-fold metal-chelation buffer containing 1 mM ethylenediaminetetraacetic acid (EDTA) followed by dialysis against 200-fold metal-free/EDTA-free buffer. Protein purity was assessed using SDS-PAGE, and apo enzyme was verified to contain less than 10% metal contamination using inductively coupled plasma mass spectrometry (ICP-MS). Metal-free enzyme was aliquoted, flash frozen with liquid N2, and stored at −80°C. The concentration of active sites in the enzyme stocks was determined by an active site titration where activity was measured using the Fluor-de-Lys assay upon titration with SAHA at enzyme concentrations well above the Ki.
Fluor-de-Lys assay
The Fluor-de-Lys assay38–39 (Enzo Life Sciences) was performed as described.36 Metal-free HDAC8 was reconstituted for 1 h on ice with stoichiometric Zn(II) (Fluka Zinc Atomic Spectroscopy standard #96457) or Fe(II) (iron(II)chloride, Sigma). Reactions were performed at 30°C using the Fluor-de-Lys HDAC8 or Sirt1 p53-based commercial fluorophore-conjugated peptide substrate. HDAC8 was reconstituted with stoichiometric Zn(II) or Fe(II) for activity assays. For Fe(II) assays, solid iron(II)chloride, solid ascorbic acid (Fluka), and HDAC8 assay buffer were equilibrated overnight in an anaerobic chamber (Coy Laboratory Products). The enzyme and substrate were separately equilibrated in the anaerobic chamber for 1 h prior to use. Fe(II)-HDAC8 was reconstituted in the anaerobic chamber in the presence of ascorbic acid. A stock of 500 μM Fe(II) in 5 mM ascorbic acid was prepared and used for a final enzyme reconstitution of 100 μM Fe(II)-S39E-HDAC8 in 1 mM ascorbic acid and HDAC8 assay buffer (25 mM HEPES, pH 8.0, 137 mM NaCl, 3 mM KCl). The reconstitution was incubated for at least one hour on ice or coolbox in the anaerobic chamber. Iron assays were performed outside the anaerobic chamber and were completed within 2 h, the effective working timespan for 1 mM ascorbic acid to maintain Fe(II) (data not shown). Reactions were initiated by addition of HDAC8 (0.5 – 1 μM) to HDAC8 assay buffer preincubated with substrate (10 – 1000 μM) for at least 5 min at 30°C. Time points were quenched using a combination of trypsin developer and trichostatin A (TSA) solution as previously described.36 After at least a 15 min incubation at room temperature, the fluorescence of product (ex. 340 nm, em. 450 nm) and substrate (ex. 340 nm, em. 380 nm) was measured using a PolarStar fluorescence plate reader. The ratio of product to substrate fluorescence of the linear portion of each reaction was used to calculate HDAC8 activity compared to a standard curve derived from the completed reaction of HDAC8 with known amounts of substrate. kcat/KM values were generated by fitting the Michaelis-Menten equation (Equation 1) to the ν0/[E] data at varying [S]. Standard errors for kcat/KM, kcat, and KM values were calculated using GraphPad Prism analysis and either non-linear or linear regression. Error for kcat/KM ratios were calculated using the propagation of uncertainty equation.40
| Equation 1. |
Enzyme-coupled deacetylation assay
Non-fluorophore conjugated peptides were assayed by coupling deacetylation of acetyl-lysine residues to the formation of NADH as described.41–42 Briefly, hydrolysis of acetyl-lysine forms acetate, which is converted to NADH via acetyl-CoA synthetase, citrate synthase, and malate dehydrogenase, and NADH is measured by fluorescence. Peptides (Peptide 2.0 or Synthetic Biomolecules) were N-terminally acetylated (N-terminal acetylation is not hydrolyzable by HDAC8) with C-terminal amides. The enzyme-coupled acetate-detection assay referred to simply as the ‘acetate assay’, was performed as described.41 As in the Fluor de Lys assay described above, metal-free HDAC8 was reconstituted for 1 h on ice with stoichiometric Zn(II) (Fluka Zinc Atomic Spectroscopy standard #96457). HDAC8 concentration ranged from 1–5 μM and substrate concentration varied from 10–1200 μM. Reaction time points (60 μL) were quenched into 5 μL 10% hydrochloric acid. Coupled enzyme solution reagents were purchased from Sigma, with the exception of acetyl-CoA synthetase, which was expressed and purified as described.41 Time points were neutralized with 15 μL 6% sodium bicarbonate (60 μL), loaded into wells of a black 96-well plate containing coupled enzyme solution (10 μL) and allowed to equilibrate protected from light. The fluorescence (ex. = 340 nm, em. = 460 nm) of the resulting NADH was measured and converted to product concentration using an acetate standard curve. An NADH standard curve was prepared to verify coupled solution activity. The linear portion of each reaction was used to determine the initial velocity, ν0. kcat/KM values were generated by fitting the Michaelis-Menten equation (Equation 1) to the ν0/[E] data at varying [S]. Standard errors for kcat/KM, kcat, and KM values were calculated using GraphPad Prism analysis and either non-linear or linear regression. Error for kcat/K ratios were calculated using the propagation of uncertainty equation.40
Determination of metal ion dissociation rate constants (koff)
The divalent metal off rate constants for HDAC8 were determined by measuring the time-dependent decrease in activity upon incubation with EDTA as described previously.43 As described above, HDAC8 was reconstituted with stoichiometric Zn(II) or Fe(II) for activity assays. Fe(II)-HDAC8 was reconstituted in the anaerobic chamber in the presence of 1 mM ascorbic acid. After at least one hour on ice or coolbox in the anaerobic chamber, the enzyme was diluted into assay buffer containing 1 mM EDTA at 30°C. At each time point (0–60 min), the enzyme in EDTA was diluted 4-fold into HDAC8 assay buffer containing 250 μM Fluor-de-Lys HDAC8 substrate, and initial rates of deacetylation were measured using the Fluor-de-Lys assay.38–39 Reaction time points were quenched by adding them to TSA and trypsin developer as described above. The linear, initial rate for product formation was determined for reactions at each EDTA incubation time. Control reactions without EDTA were incubated for the same times at 30°C and the initial rate of the EDTA reaction was divided by the non-EDTA reaction for each incubation time to yield the activity retained. The koff was determined by fitting the normalized activity retained (fraction activity/y-intercept of single exponential) to a single exponential (Equation 2) using GraphPad Prism. In the equation, ΔA is the normalized fraction activity, A is the normalized fraction activity at time zero, t is time, and koff is the dissociation rate constant. Standard errors for koff values were calculated using GraphPad Prism analysis. Error for koff ratios were calculated using the propagation of uncertainty equation.40
| Equation 2. |
Molecular dynamics (MD) simulations
The coordinates for the HDAC8-substrate complex for the simulations were taken from PDB ID: 2V5W.26 The spatial positions of K+ and Zn2+ ions were retained in the simulation from the original PDB. Three sets of simulations were completed: wild-type HDAC8, HDAC8 with phosphorylated S39 and HDAC8 with S39E mutation.
The modified phosphorylated protein simulation was made using the Forcefield PTM server (www.selene.princeton.edu/FFPTM). The AMBER forcefield parameters for post-translational modifications were taken from Khoury et al.44–45 The S39E mutant structure was obtained from the crystal structure reported in this manuscript. Disordered loops were reconstructed using 2V5W structure as the template. The substrate was introduced in the HDAC8 enzyme after superimposition with 2V5W structure. A total of six simulations were carried out, with and without substrate.
The parameters for substrate were generated via the Antechamber module of the AMBER software using Generalized AMBER force field.46–47 The charges were assigned to the substrate using the AM1-BCC method.48 The systems were set up using xleap module of AMBER14.49 K+ ions were used for neutralization and TIP3P water molecules were used for solvation. AMBER-adapted Joung and Cheatham parameters specific for TIP3P waters and K+ ions (radius 1.593 Å and well depth 0.4297054 kcal mol−1) were used.50 The system was solvated in a periodic box whose boundaries extended at least 10 Å from any solute atom. The periodic boundary conditions were defined by the PME algorithm and non-bonded cut-off was set to 10 Å.51 All chemical bonds involving hydrogen atoms were restrained using SHAKE, allowing for stable simulations with a 2 fs time step.52 Simulations were carried out using an NPT ensemble, using the Berendsen algorithm to control temperature and pressure.53 Standard equilibration protocols were used for initial minimization of the structure.54 The final MD simulations were carried out for 400 ns using ACEMD and the frames were collected every 10 ps using a timestep of 4 fs.55 Analyses of the trajectory were performed using the GROMACS 4.5 tools.56–57 The programs ICM, VMD, and PyMOL were used for visualization.58–60
Crystallization and data collection
Crystals of the S39E HDAC8-Droxinostat complex were grown in sitting drops at 21°C using the vapor diffusion method. A 500 nL drop containing 5.0 mg/mL S39E HDAC8, 50 mM Tris (pH 8.0), 150 mM KCl, 5% glycerol, 1 mM dithiothreitol, 2 mM Droxinostat, and 0.03 M glycylglycylglycine was added to a 500 nL drop of precipitant solution and equilibrated against a 100 μL reservoir of precipitant solution. The precipitant solution consisted of 100 mM BisTris (pH 6.5), 6% (w/v) PEG 8,000 (Hampton Research), and 4 mM TCEP.
Crystals appeared within 1 day. Crystals were flash-cooled in liquid nitrogen after transfer to a cryoprotectant solution consisting of precipitant solution supplemented with 25% glycerol. X-ray diffraction data were collected on beamline X29 at the National Synchrotron Light Source (NSLS, Brookhaven National Laboratory, New York). Data collection statistics are recorded in Table 1. Data were indexed, integrated and scaled using HKL2000.61
Table 1.
Data collection and refinement statistics for the S39E-HDAC8-Droxinostat Complex
| Unit cell | |
| Space group symmetry | P21 |
| a, b, c (Å) | 53.4, 84.4, 94.6 |
| α, β, γ (deg) | 90, 99.4, 90 |
| Data collection | |
| Wavelength (Å) | 1.075 |
| Resolution limits (Å) | 43.0–1.59 |
| Total/unique reflections | 819616/110604 |
| Rmerge1,2 | 0.080 (0.605) |
| I/σ(I)a | 19.3 (4.7) |
| Redundancy a | 7.4 (7.1) |
| Completeness (%)a | 100 (100) |
| Refinement | |
| Reflections used in refinement/test set | 110567/5539 |
| Rcryst3 | 0.142 |
| Rfree4 | 0.160 |
| Protein atoms5 | 5648 |
| Water moleculese | 777 |
| Ligand moleculese | 2 |
| Zn2+ ionse | 2 |
| K+ ionse | 4 |
| Glycerol moleculese | 2 |
| Average B-factors (Å2) | |
| Protein | 24 |
| Metal ions | 15 |
| Solvent | 38 |
| Inhibitor | 43 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.3 |
| Dihedral angles (°) | 12 |
| Ramachandran plot (%)6 | |
| allowed | 91.1 |
| additionally allowed | 8.9 |
| PDB accession code | 5BWZ |
Values in parentheses refer to the highest shell of data.
Rmerge = Σ|Ih - 〈I〉h|ΣIh, where 〈I〉h is the average intensity for reflection h calculated from replicate reflections.
Rcryst = Σ||Fo| - |Fc||/Σ|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
Rfree = Σ||Fo| - |Fc||/Σ|Fo| for reflections contained in the test set held aside during refinement.
Per asymmetric unit. f Calculated with PROCHECK version 3.4.4
Calculated with PROCHECK version 3.4.4.
Phasing, model building, and structure refinement
Crystals belonged to space group P21, with 2 molecules in the asymmetric unit. The crystal structure was solved by molecular replacement using PHENIX62 with the atomic coordinates of the H143A HDAC8–tetrapeptide substrate complex (PDB accession code 3EWF)24 less substrate, metal ions, and solvent molecules used as a search probe. The model was refined with iterative cycles of refinement in PHENIX62 and manual model rebuilding in COOT.63 Solvent molecules and inhibitors were added after initial rounds of refinement. Translation Libration Screw (TLS) refinement was performed in the late stages of refinement. TLS groups were automatically determined using PHENIX. Final refinement statistics are recorded in Table 1.
Portions of the N-terminus, the C-terminus, and the L1 and L2 loops were characterized by missing or broken electron density. These segments appeared to be disordered and were excluded from the final model as follows: M1-S13 (monomers A and B), A32-I34 (monomer B), G86-D89 (monomer A) G86-E95 (monomer B), I378-H389 (monomer B), and E379-H389 (monomer A). Likewise, side chains of residues that were partially or completely disordered were excluded from the model as follows: L14 (monomers A and B), K33 (monomer A), I34 (monomer A), K52 (monomer B), K58 (monomer B), K60 (monomers A and B), K81 (monomers A and B), Q84 (monomer A), E85 (monomer A), I94 (monomer A), E95 (monomer A), Y100 (monomer B), E106 (monomer B), K132 (monomers A and B), K221 (monomer B), E238 (monomer B), Q253 (monomers A and B), E358 (monomer B), K370 (monomer A), V377 (monomer A), and I378 (monomer A).
Occasional ambiguous electron density peaks were observed in the structure. These peaks were usually elongated and potentially correspond to disordered PEG fragments or other molecules present in the crystallization buffer. However, since these electron density peaks were not confidently interpretable, they were left unmodeled. Similarly, ambiguous electron density was observed around W141 in monomer A, possibly corresponding to alternative conformations. However, since such conformations were not confidently interpretable, the W141 side chain was modeled in only one primary conformation.
RESULTS
Crystal structure of S39E HDAC8
Because the phosphorylated enzyme is difficult to obtain in the large quantities and purity needed for crystallization, we used the S39E-HDAC8 phosphomimetic mutant, which mimics phosphorylation at this site. The mutant was crystallized in an inhibitor-bound state. This is the first crystal structure of an HDAC isozyme complexed with Droxinostat, a selective HDAC3, HDAC6, and HDAC8 inhibitor.64 The IC50 values for inhibition of cobalt (II)-substituted wild-type and S39E HDAC8 by Droxinostat were measured as 33 ± 11 nM and 119 ± 14 nM, respectively (Figure S1).
The structure of the S39E HDAC8-Droxinostat complex illustrates how phosphorylation of S39, as mimicked by the glutamate substitution, might influence inhibitor binding in the enzyme active site. Residue S39 is located in the α2 helix, approximately 20 Å away from the catalytic Zn2+ ion. The structure of the S39E HDAC8–Droxinostat complex is similar to that of the wild-type HDAC8–M344 complex (PDB accession code 1T67)23 with a root mean square (r.m.s.) deviation of 0.49 Å for 356 Cα atoms and 0.47 Å for 350 Cα atoms, for monomers A and B, respectively.
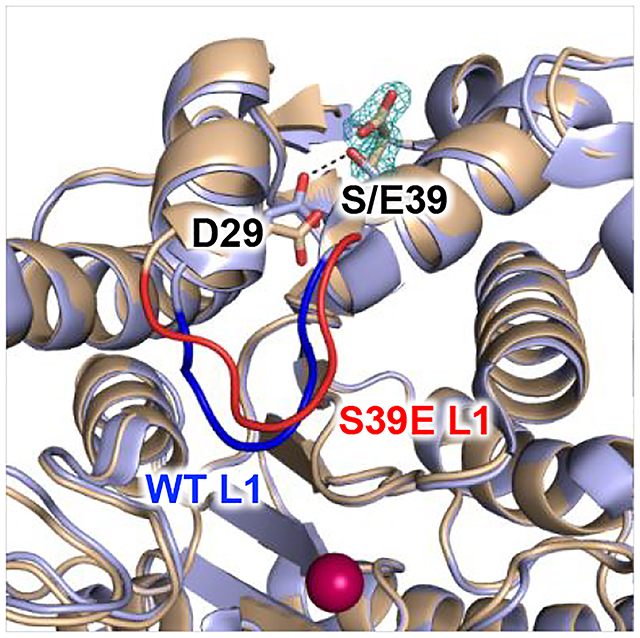
Although the S39E substitution does not cause large-scale changes in the HDAC8 structure (Figure 2A), local structural changes are observed in the vicinity of E39 that propagate through the protein scaffold to influence the conformation of loop L1 that is important for interactions with substrates and inhibitors.33 Presumably, these structural changes would be similarly triggered by phosphorylation of S39 in the wild-type enzyme.
Figure 2. Structural comparison of S39E and wild type HDAC8.

A. Stereoview superimposition of the S39E HDAC8-Droxinostat complex (monomer A: C = wheat, N = blue, O = red, Zn2+ = magenta sphere) and the wild-type HDAC8-M344 complex (PDB 1T67, color-coded as above except C = light blue). In the wild-type structure, S39 donates a hydrogen bond (black dashed line) to D29. Upon substitution to a glutamate (simulated omit map contoured at 4.0σ showing the E39 side chain), this interaction is not conserved and causes local rearrangement. The L1 loop adopts a different conformation as highlighted in red and blue for the S39E HDAC8-Droxinostat complex and the wild-type HDAC8-M344 complex, respectively. B. Stereoview simulated annealing omit map of Droxinostat bound in the active site of S39E HDAC8 (monomer A, contoured at 3.0σ). Atomic color codes are as follows: C = wheat (protein, monomer A), or green (inhibitor), N = blue, O = red, Zn2+ = magenta sphere. Metal coordination and selected hydrogen bond interactions are shown as solid black or dashed black lines, respectively. As in A, the L1 loop of S39E HDAC8 is highlighted in red.
In the wild-type HDAC8 structure, the hydroxyl group of S39 donates a hydrogen bond to the carboxylate group of D29, which is located on the adjacent α1 helix. In S39E HDAC8, the E39 side chain is oriented away from D29. However, the S39E substitution induces a slight shift and conformational change in D29 away from E39, allowing for the insertion of a hydrogen bonded water molecule between both residues (Figure 2A, Figure S2). This conformational change causes a slight twist in helix α1 containing D29 (the Cα atom shifts 0.6 Å), which leads to conformational changes in the L1 loop (L31–P35) connecting helices α1 and α2. The L1 loop is adjacent to the active site and can be flexible as it interacts with substrates and inhibitors.33 Here, this loop is similarly flexible as indicated by higher thermal B factors, along with missing electron density for the side chains of K33 and I34 in monomer A, and weak electron density in monomer B that did not allow modeling of the A32-I34 segment.
The 4-chloro-2-methylphenoxyl capping group of Droxinostat is characterized by unambiguous yet somewhat weaker electron density (Figure 2B) and higher thermal B factors compared with surrounding protein atoms (Table 1). The hydroxamate moiety of Droxinostat coordinates to the active site Zn2+ ion, forming a five-membered ring chelate, as typically observed in HDAC8-hydroxamate crystal structures.23–26, 29, 35 The coordination distances to the Zn2+ ion are 2.0 Å and 2.2 Å for the hydroxamate hydroxyl and carbonyl groups, respectively. The Zn2+-bound hydroxamate is also stabilized by hydrogen bond interactions with Y306, H142, and H143 (Figure 2B). The capping group of Droxinostat does not make hydrogen bond interactions with residues at the mouth of the active site. A contact is made between the chlorine atom of Droxinostat and the hydroxyl group of Y100 in the L2 loop (the Cl--O distance in monomer A is 3.2 Å; the side chain of Y100 is disordered in monomer B). However, Y100 is poorly oriented to characterize this interaction as a hydrogen bond. This interaction instead may be a halogen bond.
The conformational change of D29 resulting from the S39E substitution is not observed in S39D HDAC8; D29 remains in its wild-type conformation in this mutant (Figure S2).26 The D29---S39 hydrogen bond is lost in S39D HDAC8, and the D39 side chain is oriented away from D29. Since there is no conformational change of D29 in S39D HDAC8, there are no significant changes in the conformations of helix α1 and the L1 loop. Thus, in contrast with S39E HDAC8, S39D HDAC8 is more like wild-type HDAC8 in terms of the protein structure around residue 39.
S39E mutation decreases HDAC8 activity and changes substrate selectivity with peptides in vitro
While inhibition of HDAC8 activity by phosphorylation had been observed for deacetylation of core histones in cell-based experiments,4 detailed kinetic parameters for the phosphorylated enzyme or the phosphomimetic S39E mutant had not been determined. We hypothesized that perturbation of the L1 loop by S39 phosphorylation or mutation to glutamate could affect both the reactivity and the substrate recognition of the enzyme. To investigate whether S39 alteration affects substrate selectivity or simply dampens activity globally towards all substrates, we measured kcat/KM values, the catalytic efficiency parameter that is most important for determining substrate selectivity, for Zn(II)-constituted S39E HDAC8 and wild-type HDAC8 towards deacetylation of a library of peptide substrates in vitro. Since the methylcoumarin fluorophore on the widely used Fluor-de-Lys peptide substrates enhances activity and substrate affinity with HDAC8,41 we selected additional unlabeled peptide substrates to test using the acetate assay, an assay that couples the formation of acetate to a fluorescent NADH readout.41, 65 This selection of peptide substrates is taken from acetylated nuclear and cytoplasmic proteins that encompass a diverse range of cellular functions (Table 2). Additionally, the list includes peptides from putative and novel HDAC8 substrates determined from computational modeling and cell-based proteomics screens including a peptide from SMC3, the best validated putative in vivo HDAC8 substrate to date.66,67–69 Consistent with previous reports, S39E HDAC8 activity was decreased compared to wild-type HDAC8 in all cases tested (Table 2).
Table 2.
Kinetics of deacetylation of acetylated peptides catalyzed by S39E and wild-type HDAC8a
| Substrate | Function | Sequence | WT: kcat/KM (M−1s−1) | S39E: kcat/KM (M−1s−1) | Ratio (WT/S39E) |
|---|---|---|---|---|---|
| H3K9 | Chromatin | TKQTARK(ac)STGGKA | 51 ± 3b | 1.7 ± 0.2 | 30 ± 4 |
| SMC3 | Cell cycle | RVIGAKK(ac)DQY | 58 ± 2 | <0.5 | >120 |
| CSRP2BP | Acetyltransferase | STPVK(ac)FISR | 160 ± 27b | 11.5 ± 0.6 | 14 ± 2 |
| LARP1 | Translation | LGK(ac)FRR | 653 ± 99 | 70 ± 10 | 9 ± 2 |
| CAD Protein | Pyrimidine biosynthesis | LSK(ac)FLR | 660 ± 27 | 11 ± 1 | 60 ± 6 |
| MYH1 (CAD_LARP) | Muscle contraction | LSK(ac)FRR | 2400 ± 100 | 10.6 ± 0.3 | 226 ± 11 |
| LARP_CAD | Synthetic | LGK(ac)FLR | 2900 ± 700 | 19 ± 4 | 153 ± 49 |
| p53 | Cell cycle | RHK(ac)K(ac)-AMCc | 950 ± 96 | 26 ± 3 | 37 ± 6 |
| p53 | Cell cycle | RHKK(ac)-AMCc | 1030 ± 200 | 8 ± 3 | 129 ± 54 |
Values for kcat/KM were obtained by fitting the Michaelis-Menten equation (Equation 1) to the dependence of the initial rates of deacetylation on the substrate concentration catalyzed by Zn(II)-constituted S39E and wild-type HDAC8. The p53 fluorogenic peptide substrates were measured using the Fluor de Lys assay (FdL) and all other peptides were measured using the acetate assay. Enzyme concentration was 1 μM and substrate concentration was varied from 10–1200 μM. Standard error for kcat/KM values was calculated using GraphPad Prism analysis and either non-linear or linear regression, depending on best fit. Error for kcat/KM ratios were calculated using the propagation of uncertainty equation.40
Values reported previously.70 Control reactions were performed with 1 μM wild-type HDAC8 and 100 μM H3K9 and CSR2BP peptides parallel with the S39E reactions to verify that wild-type activity with peptide agreed with previously reported values.
AMC: 7-amino-4-methylcoumarin
Notably, the deacetylase activity of S39E HDAC8 was reduced to differing extents depending on the substrate (Table 2, Figure 3–4A) with values ranging from a 9-fold to 220-fold decrease in activity for the S39E mutant compared to wild-type HDAC8 (Figure 4B). For the peptide corresponding to SMC3, the mutation nearly abolished activity. However, for the peptide corresponding to LARP1, the mutation caused a modest 9-fold reduction in activity compared to wild-type (Figure 3). Furthermore, while the wild-type catalytic efficiencies for the CAD and LARP1 peptides (660 ± 27 versus 653 ± 99 M−1s−1, respectively) were comparable, the S39E phosphomimetic displayed a much larger (60-fold) decrease in activity for the CAD peptide. Similarly, the value of kcat/KM for wild-type HDAC8-catalyzed deacetylation of the fluorogenic p53 HDAC8 peptide is comparable to the values for CAD and LARP1, yet S39E HDAC8 exhibited a 37-fold decreased catalytic efficiency for this peptide. These data indicate that the S39E mutation not only decreases activity but also has an impact on peptide substrate selectivity. These results suggest an alternate set of substrate recognition patterns triggered by S39 phosphorylation. Since the sequences for the CAD and LARP1 peptides are similar, we wondered whether swapping the C- and N-terminal sequences would impact selectivity. Unexpectedly, these sequence changes increased the reactivity of wild-type HDAC8 with both peptides by 4–5-fold while decreasing the reactivity with S39E HDAC8 by 4–7-fold. Therefore, the WT/S39E selectivity ratio increased significantly (Table 2). This surprising result suggests the formation of alternate contacts between the peptide and the binding interface of the two enzymes caused by the shift in loop L1 due to the S39E mutation.
Figure 3. Deacetylation of LARP1 peptide by S39E and wild-type HDAC8.

Representative peptide assay data. Dependence of the initial rates of deacetylation of the LARP1 peptide (LGK(ac)FRR) on the substrate concentration catalyzed by Zn(II)-constituted S39E (closed blue circles, right y-axis) and wild-type HDAC8 (open blue circles, left y-axis) measured using the acetate assay.41 Enzyme concentration was 1 μM and substrate concentration was 10–800 μM. The data are a combination of two experiments (Zn(II)-S39E), or one experiment (Zn(II)-WT), and the Michaelis-Menten equation (Equation 1) was fit to the data using global regression analysis (GraphPad Prism). Standard errors were determined from the fits.
Figure 4. Comparison of S39E and wild-type HDAC8 deacetylation of peptides.

A. Catalytic efficiencies, kcat/KM, of wild-type and S39E HDAC8-catalyzed deacetyation of peptides listed in Table 2 as measured by the Fluor de Lys assay (FdL-HDAC8 and FdL-Sirt1 peptides) and the acetate assay (remaining peptides). B. fold change in catalytic efficiency of S39E HDAC8 compared to wild type for peptides. For all three graphs, peptides are ordered from most to least active with wild-type HDAC8. Error bars are shown in same colors as columns. Substrate names on X-axis correspond to peptides listed in Table 2.
S39E mutation increases metal dissociation rates and decreases metal-activation for iron(II) and zinc(II)
Additionally, we investigated the impact of phosphorylation on metal-selectivity. HDAC8 is activated by different metals, most notably iron(II) and the canonical catalytic ion zinc(II), and substrate selectivity changes based on the identity of the active site metal.71 S39E HDAC8 activity was measured using the Fluor-de-Lys assay with Zn(II) and Fe(II) bound at the active site and the catalytic efficiencies were compared to those of Zn(II)- and Fe(II)-constituted wild-type HDAC8 (Table 3, Figure 5). Fe(II)- and Zn(II)-dependent catalytic efficiency (kcat/KM) was 6-fold and 34-fold lower, respectively, for the S39E mutant compared to wild type. The differential effects on reactivity of the two metal-substituted enzymes provide further evidence for S39E-induced alterations at the active site. Comparing the KM and kcat values for the Fe(II)-constituted WT and S39E enzymes reveals that the decrease in kcat/KM is due to both a 2-fold increase in KM and a 3-fold reduction in kcat. This suggests that the serine to glutamate mutation may affect both substrate recognition and catalysis. To further evaluate effects on the metal site, the kinetics of Zn(II) and Fe(II) metal dissociation (koff) was measured for the phosphomimetic mutant and compared to that of wild-type HDAC8 (data not shown). For both Zn(II) and Fe(II), the rate of metal-dissociation from S39E increased approximately 15-fold compared to wild type (Figure 6) providing further evidence of structural changes near the metal site. Taken together, the S39E mutation enhances metal dissociation, decreases catalytic activity, and alters substrate selectivity. These results indicate that HDAC8 phosphorylation may be an important modulator of HDAC8 activity.
Table 3.
Kinetics of Fe(II)- and Zn(II)-S39E and wild-type HDAC8a
| HDAC8 | kcat/KM (M−1s−1) | KM (μM) | kcat (s−1) | kcat/KM Ratio (WT/S39E) | koff (min−1) | koff Ratio (S39E/WT) |
|---|---|---|---|---|---|---|
| Fe(II)-S39E | 440 ± 60 | 170 ± 30 | 0.077 ± 0.005 | 6 ± 2 | 0.48 ± 0.05 | 9 ± 1 |
| Fe(II)-WT | 2800 ± 700 | 90 ± 30 | 0.25 ± 0.02 | 0.055 ± 0.005 | ||
| Zn(II)-S39E | 26 ± 3 | > 400b | > 0.05b | 37 ± 6 | 0.57 ± 0.07 | 14 ± 3 |
| Zn(II)-WT | 950 ± 96 | 1200 ± 300 | 1.1 ± 0.3 | 0.040 ± 0.006 |
Values for kcat/KM, KM, and kcat were obtained by fitting the Michaelis-Menten equation (Equation 1) to the dependence of the initial rates of deacetylation on the substrate concentration catalyzed by Zn(II)- and Fe(II)-constituted S39E and wild-type HDAC8 as measured using the FdL assay. Enzyme concentration was 0.5–1 μM and substrate concentration was varied from 10–1000 μM. Values for koff were determined by fitting a single exponential (Equation 2) to the time-dependent decrease in activity upon incubation with 1 mM EDTA. Standard errors for kcat/KM, KM, kcat, and koff values were calculated using GraphPad Prism analysis. Error for kcat/KM and koff ratios were calculated using the propagation of uncertainty equation.40
Little curvature was observed in the dependence of activity on substrate concentration so that KM and kcat values are poorly defined by this data set. The necessity for excessively high enzyme and substrate concentrations preclude the accurate determination of these parameters, individually.
Figure 5. Zinc(II)- and iron(II)-constituted S39E and wild-type HDAC8 catalyzed deacetylation of fluorescently-labeled Fluor de Lys HDAC8 test substrate.

Dependence of the initial rates of deacetylation of the Fluor-de-Lys HDAC8 peptide substrate on substrate concentration catalyzed by A. Zn(II)-constituted S39E HDAC8 (closed blue circles) and Zn(II)-constituted WT HDAC8 (open blue circles) and B. Fe(II)-constituted S39E HDAC8 (closed red circles) and Fe(II)-constituted WT HDAC8 (open red circles). Enzyme concentration was 0.5–1 μM and substrate concentration was 10–1000 μM. The data are a combination of four experiments (Zn(II)-S39E), or one experiment (Zn(II)-WT, Fe(II)-S39E, Fe(II)-WT), and the Michaelis-Menten equation (Equation 1) was fit to the data using global regression analysis (GraphPad Prism).
Figure 6. Metal ion dissociation rates for zinc(II) and iron(II)-constituted S39E HDAC8.

Initial rates for Zn(II)-constituted S39E HDAC8 (filled blue circles) and Fe(II)-constituted S39E HDAC8 (filled red circles) deacetylation activity as a function of time after addition of 1 mM EDTA, as measured using the Fluor-de-Lys assay. The fraction activity is determined by dividing the activity in EDTA by the activity of HDAC8 incubated in the absence of EDTA. The koff values were calculated by fitting an exponential decay equation to data from replicates on different days using global regression analysis (GraphPad Prism).
Molecular dynamics simulations suggest phospho-HDAC8 structural changes
The crystal structure provides a snapshot of inhibitor-bound S39E HDAC8. To investigate the effects of phosphorylation on substrate-HDAC8 interactions, molecular dynamics simulations were performed, starting with an HDAC8-peptide substrate complex crystal structure (PDB ID: 2V5W).26 We compared the substrate binding dynamics of phosphorylated HDAC8 (pS39-HDAC8, modeled), S39E HDAC8 (with substrate, modeled), and wild-type HDAC8. By simulating phosphorylation on the wild-type HDAC8 structure, we were able to predict the structure and dynamics of residues in the L1 loop that were not resolved in the S39E crystal structure and validate the S39E mutation as a mimic of phosphorylated HDAC8. While the mutant was crystalized in complex with an inhibitor, the simulations predict the structure of pS39-HDAC8 with and without peptide substrate bound. First, wild-type HDAC8 was modeled with and without the Fluor-de-Lys p53-based peptide substrate, to identify key residues in substrate binding. Relevant residue conformations surrounding S39 are highlighted in Figure 7, with the most important being those of the substrate, Y306, D29 and K33. Figure 7 shows a comparison of wild-type, pS39- and S39E HDAC8 snapshots during substrate binding. The simulations demonstrate that the pS39-HDAC8 structural behavior is comparable to that of the S39E-HDAC8 mutant, which validates use of the S39E-HDAC8 mutant for prediction of pS39-HDAC8 behavior in vitro. Importantly, the simulations also illustrate differences between wild-type and pS39/S39E-HDAC8 substrate binding conformations. Based on the simulations, modification of S39 leads to a disruption of the interaction between Y306 and K33, which perturbs substrate binding. In wild-type HDAC8, the interaction between S39-D29-K36 tethers the L1 loop and maintains the orientation of K36. This is distinct from the S39E-HDAC8 mutant dynamic simulation, in which there is no interaction between E39 and D29 (Figure 8). The interaction between S39 and D29 is disrupted by the addition of negative charge at position 39. Instead, D29 interacts directly with K36 (Figure 8). The strength of these interactions, and the disruption of interaction between residue 39 and D29 in the S39E mutant, is evident in the distance versus time plots in Figure 8B. The K36-D29 interaction and altered Y306-K33 interaction preclude binding of substrate at the wild-type position. Instead, the peptide binds in a channel between K33 and Y306 where it is not optimally positioned for deacetylation by the active site metal-water nucleophile. These simulations provide insight into the basis for decreased pS39 and S39E HDAC8 activity and altered substrate selectivity, and although the D29-K36 contact is not noticeable in the S39E crystal structure, the altered orientation of D29 is consistent with the 0.6 Å shift for D29 that was observed in the crystal structure.
Figure 7. Simulations of wild-type HDAC8 binding to substrate.

Top panel (red box): The orientation of key residues in wild-type HDAC8 at A. the start, B. 51 ns, and C. 70 ns of the substrate binding simulation. S39 is solvent-exposed. Y306 bends at 90° toward K33, and the Hε of Y306 forms a hydrogen bond with the carbonyl of K33 (B). This opens the tunnel for substrate interaction with the active site, which is otherwise blocked by Y306. Yellow and purple spheres represent Zn2+ and K+ ions respectively. Center and bottom panels: Simulations of pS39 (blue box) and S39E (green box) bound to substrate. In the center panel (pS39 modeled on wild-type HDAC8), two snapshots (D. start and E. 400 ns) during the simulation demonstrate that the substrate is shifted in the active site between K33 and Y306 compared to wild-type HDAC8 (Figure 7A–C). Y306 interacts with substrate but does not interact with K33. The bottom panel (S39E HDAC8 with modeled substrate) is a representation at F. the start and G. 400 ns of the simulation of key residues in S39E HDAC8 and demonstrates that the enzyme behaves similarly to pS39-HDAC8. The L1 loop is altered, Y306 and K33 do not interact (unlike in wild-type HDAC8 (Figure 7B) where Y306 forms a hydrogen bond with the carbonyl oxygen of K33), and Y306 does not interact with substrate in this simulation. Substrate access to the active site is altered. Yellow and purple spheres represent Zn2+ and K+ ions respectively.
Figure 8. Interaction between K36-D29-S39.

A. The wild-type (blue) and mutant HDAC8 (yellow) is illustrated. The S39-D29 interaction tethers loop 1 and maintains the orientation of K33 in the wild-type protein. S39E can interact with K36 but not with D29 and therefore K33 orientation is not maintained in the mutant. B-C. Distance plot showing the interaction between D29, K36 and S39 in (B) wild-type and (C) S39E mutant HDAC8. In wild type, S39 can interact with D29 directly while in S39E, mutant residue S39E and D29 are beyond interacting distance (green).
DISCUSSION
The mechanistic and functional roles of phosphorylation on HDAC8 are important facets of HDAC8 regulation that have not been well studied up to this point. To examine the effect of addition of a bulky negative charge at S39, S39E HDAC8 was used as a phosphomimetic. The validity of the mimic was bolstered by the fact that S39E HDAC8 and phospho-HDAC8 behave similarly in assays where immunoprecipitated S39E HDAC8 showed equivalent enzymatic activity to phosphorylated wild-type HDAC8 immunoprecipitated from HeLa cells treated with PKA-activating agent forskolin.4 Moreover, a S39A mutation was slightly activating compared to the wild-type enzyme.4 The molecular simulations reported here provide further evidence to support the use of S39E as a viable phosphomimetic for pHDAC8. Previously, crystal structures of the S39D HDAC8 have been solved, and the structure of this mutant is reported to be essentially the same as the wild type.26, 33 The S39E mutant, however, is a more appropriate mimic of phosphorylation4 and the S39E-HDAC8-Droxinostat structure exhibits noticeable differences from the structure of the wild-type enzyme. The structure of the S39E HDAC8-Droxinostat complex reveals that the L1 loop undergoes a conformational change and the interaction between S39 and D29 is disrupted by the glutamate substitution. Loop L1 is important for HDAC8 substrate and inhibitor interactions, so this structural perturbation likely contributes to decreased catalytic efficiency and altered substrate selectivity.33 It is important to note that a structure of wild-type HDAC8 complexed with Droxinostat has not been solved and comparing S39E HDAC8-Droxinostat directly to wild-type-HDAC8-Droxinostat would be useful to eliminate the possibility of structural changes induced by the identity of the inhibitor.
Interestingly, contrary to previous hypotheses, the position of R37 is not altered in either the inhibitor-bound crystal structure or the phosphorylation simulation. As mentioned earlier, since R37 is in close proximity to S39E, a conformational change of this important residue would not have been surprising.31 Additionally, an electrostatic interaction between K36 and E39 had been anticipated but was not evident in the structure. Instead, the relevant structural changes observed in the crystal structure and/or simulation were: 1) perturbed interactions between Y306, substrate, and K33, 2) the loss of a hydrogen bond between the hydroxyl of S39 and the carboxylate of D29 (which bridges the α1 and α2 helices), and 3) the gain of an interaction between D29 and K36. Regarding the lost D29-S39 hydrogen bond, the inhibitor-bound structure shows that this change is correlated with a conformational change of the L1 loop. The pS39 HDAC8 simulation reveals that this lost interaction alters the position of the bound substrate. The substrate does not fully interact with the canonical substrate binding surface and instead is shifted into a channel between K33 and Y306. In this simulation, K36 interacts with D29 and the location of the side chain of the L1 loop residue, K33, is altered.
Substrate binding is typically oriented in part by Y306 and K33. However, in the absence of the hydrogen bond interaction between Y306 and the K33 backbone carbonyl, the substrate is shifted in the active site between these residues. This was the most significant difference observed in the simulations. The inhibitor-bound crystal structure does not show this altered Y306/K33 interaction and the inhibitor is positioned in the typical Zn(II)-bound orientation such that the carbonyl forms a hydrogen bond with Y306 (Figure 2B). The fact that the crystal structure and the simulation provide somewhat different visualizations of ligand-bound S39E HDAC8 may be due to several factors. Mainly, the inhibitor is small and interacts primarily with the active site, limiting interactions with the HDAC8 peptide binding groove outside of the active site tunnel. Additionally, some residues (e.g. K33) cannot be directly compared to the simulated structure because they were not resolved in the crystal structure determination, suggesting significant mobility. Finally, while the crystal structure of S39E HDAC8 demonstrates structural difference and mimics phosphorylation, glutamate is not identical to phosphoserine and thus some differences between E39 and pS39 are expected.
S39E HDAC8 exhibits a 9- to 230-fold decrease in catalytic efficiency toward peptide substrates compared to wild type, consistent with previous data indicating that phosphorylation decreases HDAC8 activity. However, the alteration in substrate selectivity by the phosphomimetic mutant suggests that phosphorylation also regulates the targeting of specific cellular substrates for deacetylation catalyzed by HDAC8. The simulations and crystal structure suggest that the decrease in catalytic activity and change in substrate selectivity that accompanies perturbation of S39 arise from the consequent reordering of and/or disorder in the L1 loop flanking the active site cleft and subsequent changes to the ligand binding surface. Structural differences in this loop presumably influence its affinity for substrates and inhibitors binding to the active site. Assuming that the mutation does not change the kinetic mechanism of this enzyme, KM is proposed to reflect the substrate binding affinity and kcat reflects the rate of hydrolysis of the acetyl-lysine.36 Therefore, the increase in KM observed for Fe(II)-constituted S39E HDAC8 compared to wild-type HDAC8 is consistent with the perturbation in the L1 loop affecting substrate affinity.36 Furthermore, the decrease in kcat indicates that the mutation also decreases catalysis of the hydrolytic step. This may be due to a direct effect on the reactivity of the metal-water nucleophile via structural changes propagated by the altered D29-S39 interaction, which could also explain the increase in the metal dissociation rates (koff) of Zn(II) and Fe(II) observed for S39E HDAC8. Additionally, as the substrate selectivity between the two metals was altered, the change in kcat could also be linked to the alteration in the site of the bound peptide that leads to differential positioning of the acetyl-lysine amide bond relative to the nucleophilic metal-bound water molecule. However, the kcat value was only accurately determined for the Fe(II)-constituted enzyme while the structure and simulations used Zn(II)-HDAC8, and activity data demonstrate that S39E HDAC8 and wild-type HDAC8 are activated to different extents depending on the identity of the catalytic metal ion.36, 71
Taken together, these data provide insight into the residue interactions (i.e. S39/D29) that lead to perturbation of the kinetic properties by S39 phosphorylation. Further study is needed to parse how the phosphorylation, substrate selectivity, and metal-dependence of HDAC8 are interconnected. The role and regulation of phosphorylated HDAC8 in the cell is unclear, however, the peptide selectivity data suggest that phosphorylation both decreases catalytic activity and alters the cellular protein targets. Further examination of the cellular effect of phosphorylation will provide insight into the regulation of deacetylation and inform drug discovery, as phosphorylation-dependent protein-protein interactions may present targeting approaches for small molecule therapeutics.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NSLS for access to beamline X29 for X-ray crystallographic data collection. We thank Drs. Ted Huston and Lubomír Dostál for performing ICP-MS to verify solutions were free of metals. We thank Dr. Ningkun Wang and Oleta T. Johnson for collaboration on protein purification. We thank Noah A. Wolfson who helped with the substrate specificity assay using the NADH-coupled assay.
Funding Sources
We thank the National Institutes of Health for Grants 5-R01-GM-040602 (C.A.F.) and 5-R01-GM-49758 (D.W.C.), the University of Michigan Chemistry-Biology Interface (CBI) training program NIH grant 5T32GM008597 (C.A.C.), Cellular Biotechnology training program (CBTP) NIH grant T32GM008353 (K.W.L.) and Rackham Graduate School (C.A.C. and K.W.L.) for funding.
ABBREVIATIONS
- AMBER
assisted model building with energy refinement
- AMC
7-amino-4-methylcoumarin
- CAD
carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase protein
- CoA
coenzyme A
- CSRP2BP
cysteine rich protein 2 binding protein
- EDTA
ethylenediaminetetraacetic acid
- GROMACS
Groningen machine for chemical simulations HDAC, histone deacetylase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- hEST1B
human Est1p-like protein B
- Hsp70
heat shock protein 70
- ICP-MS
inductively coupled plasma mass spectrometry
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LARP1
La-related protein 1
- MD
molecular dynamics
- NADH
nicotinamide adenine dinucleotide
- MYH1
myosin 1
- PDB
protein databank
- PEG
polyethylene glycol
- PKA
protein kinase A
- PME
particle mesh Ewald
- PTM
post-translational modification
- Q-TOF HPLC MS
quadrupole time-of-flight high performance liquid chromatography mass spectrometry
- RMS
root mean square
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SMC3
structural maintenance of chromosomes protein 3
- TCEP
tris(2-carboxyethyl)phosphine
- TLS
translational, libration, screw
- VMD
visual molecular dynamics
- WT
wild type
Footnotes
ACCESSION CODES
The atomic coordinates and the crystallographic structure factors of S39E HDAC8 in complex with Droxinostat have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 5BWZ.
| Protein | Full Name | Species | UniProt IDa |
|---|---|---|---|
| HDAC1 | histone deacetylase 1 | human | Q13547 |
| HDAC2 | histone deacetylase 2 | human | Q92769 |
| HDAC3 | histone deacetylase 3 | human | O15379 |
| HDAC4 | histone deacetylase 4 | human | P56524 |
| HDAC5 | histone deacetylase 5 | human | Q9UQL6 |
| HDAC6 | histone deacetylase 6 | human | Q9UBN7 |
| HDAC7 | histone deacetylase 7 | human | Q8WUI4 |
| HDAC9 | histone deacetylase 9 | human | Q9UKV0 |
| HDAC8 | histone deacetylase 8 | human | Q9BY41 |
| HDAC8 | histone deacetylase 8 | mouse | Q8VH37 |
| HDAC8 | histone deacetylase 8 | bovine | Q0VCB2 |
| HDAC8 | histone deacetylase 8 | African clawed frog | Q6GPA7 |
| HDAC8 | histone deacetylase 8 | zebrafish | Q7SXM0 |
| HDAC8 | histone deacetylase 8 | blood fluke | A5H660 |
| PKA | protein kinase A | human | P17612 |
| EST1B | EST1-like protein B | human | Q9UPR3 |
| Hsp70 | heat shock protein 70 | human | P0DMV8 |
| H3 | histone H3 | human | P68431 |
| SMC3 | structural maintenance of chromosomes protein 3 | human | Q7SXM0 |
| CSR2BP | cysteine-rich protein 2-binding protein | human | P68431 |
| LARP1 | La-related protein 1 carbamoyl-phosphate synthetase 2, aspartate | human | Q6PKG0 |
| CAD | transcarbamylase, and dihydroorotase fusion protein | human | Q6PKG0 |
| MYH1 | myosin-1 | human | P12882 |
| p53 | cellular tumor antigen p53 | human | P04637 |
REFERENCES
- 1.Brandl A; Heinzel T; Kramer OH (2009) Histone deacetylases: salesmen and customers in the post-translational modification market. Biol. Cell 101, 193–205. [DOI] [PubMed] [Google Scholar]
- 2.Eom GH; Kook H (2014) Posttranslational modifications of histone deacetylases: Implications for cardiovascular diseases. Pharmacol. Ther 143, 168–180. [DOI] [PubMed] [Google Scholar]
- 3.Tsai SC; Seto E (2002) Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem 277, 31826–31833. [DOI] [PubMed] [Google Scholar]
- 4.Lee H; Rezai-Zadeh N; Seto E (2004) Negative Regulation of Histone Deacetylase 8 Activity by Cyclic AMP-Dependent Protein Kinase A. Mol. Cell. Biol 24, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottomley MJ; Lo Surdo P; Di Giovine P; Cirillo A; Scarpelli R; Ferrigno F; Jones P; Neddermann P; De Francesco R; Steinkuhler C; Gallinari P; Carfi A (2008) Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J. Biol. Chem 283, 26694–26704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.UniProt Consortium, T. (2018) UniProt: the universal protein knowledgebase. Nucleic Acids Res 46, 2699–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simoben C; Robaa D; Chakrabarti A; Schmidtkunz K; Marek M; Lancelot J; Kannan S; Melesina J; Shaik T; Pierce R; Romier C; Jung M; Sippl W (2018) A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules 23, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarti A; Oehme I; Witt O; Oliveira G; Sippl W; Romier C; Pierce RJ; Jung M (2015) HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol. Sci 36, 481–492. [DOI] [PubMed] [Google Scholar]
- 9.Kannan S; Melesina J; Hauser A-T; Chakrabarti A; Heimburg T; Schmidtkunz K; Walter A; Marek M; Pierce RJ; Romier C; Jung M; Sippl W (2014) Discovery of Inhibitors of Schistosoma mansoni HDAC8 by Combining Homology Modeling, Virtual Screening, and in Vitro Validation. J. Chem. Inf. Model 54, 3005–3019. [DOI] [PubMed] [Google Scholar]
- 10.Galasinski SC; Resing KA; Goodrich JA; Ahn NG (2002) Phosphatase inhibition leads to histone deacetylases 1 and 2 phosphorylation and disruption of corepressor interactions. J. Biol. Chem 277, 19618–19626. [DOI] [PubMed] [Google Scholar]
- 11.Pflum MK; Tong JK; Lane WS; Schreiber SL (2001) Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J. Biol. Chem 276, 47733–47741. [DOI] [PubMed] [Google Scholar]
- 12.Cai R; Kwon P; Yan-Neale Y; Sambuccetti L; Fischer D; Cohen D (2001) Mammalian histone deacetylase 1 protein is posttranslationally modified by phosphorylation. Biochem. Biophys. Res. Commun 283, 445–453. [DOI] [PubMed] [Google Scholar]
- 13.Eom GH; Cho YK; Ko J-H; Shin S; Choe N; Kim Y; Joung H; Kim H-S; Nam K-I; Kee HJ; Kook H (2011) Casein Kinase-2α1 Induces Hypertrophic Response by Phosphorylation of Histone Deacetylase 2 S394 and its Activation in the Heart. Circulation 123, 2392–2403. [DOI] [PubMed] [Google Scholar]
- 14.Karolczak-Bayatti M; Sweeney M; Cheng J; Edey L; Robson SC; Ulrich SM; Treumann A; Taggart MJ; Europe-Finner GN (2011) Acetylation of Heat Shock Protein 20 (Hsp20) Regulates Human Myometrial Activity. J. Biol. Chem 286, 34346–34355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waltregny D; Glénisson W; Tran SL; North BJ; Verdin E; Colige A; Castronovo V (2005) Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J 19, 966–968. [DOI] [PubMed] [Google Scholar]
- 16.Waltregny D; de Leval L; Glénisson W; Ly Tran S; North BJ; Bellahcène A; Weidle U; Verdin E; Castronovo V (2004) Expression of Histone Deacetylase 8, a Class I Histone Deacetylase, Is Restricted to Cells Showing Smooth Muscle Differentiation in Normal Human Tissues. Am. J. Pathol 165, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang AH; Kruhlak MJ; Wu J; Bertos NR; Vezmar M; Posner BI; Bazett-Jones DP; Yang XJ (2000) Regulation of histone deacetylase 4 by binding of 14–3-3 proteins. Mol. Cell. Biol 20, 6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grozinger CM; Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14–3-3-dependent cellular localization. Proc. Natl. Acad. Sci. U. S. A 97, 7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X; Song S; Liu Y; Ko SH; Kao HY (2004) Phosphorylation of the histone deacetylase 7 modulates its stability and association with 14–3-3 proteins. J. Biol. Chem 279, 34201–34208. [DOI] [PubMed] [Google Scholar]
- 20.Kao HY; Verdel A; Tsai CC; Simon C; Juguilon H; Khochbin S (2001) Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J. Biol. Chem 276, 47496–47507. [DOI] [PubMed] [Google Scholar]
- 21.Ha CH; Kim JY; Zhao J; Wang W; Jhun BS; Wong C; Jin ZG (2010) PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U. S. A 107, 15467–15472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang C-WJ; Lee L; Yu D; Dao K; Bossuyt J; Bers DM (2013) Acute β-Adrenergic Activation Triggers Nuclear Import of Histone Deacetylase 5 and Delays Gq-induced Transcriptional Activation. J. Biol. Chem 288, 192–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Somoza JR; Skene RJ; Katz BA; Mol C; Ho JD; Jennings AJ; Luong C; Arvai A; Buggy JJ; Chi E; Tang J; Sang BC; Verner E; Wynands R; Leahy EM; Dougan DR; Snell G; Navre M; Knuth MW; Swanson RV; McRee DE; Tari LW (2004) Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334. [DOI] [PubMed] [Google Scholar]
- 24.Dowling DP; Gantt SL; Gattis SG; Fierke CA; Christianson DW (2008) Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry 47, 13554–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dowling DP; Gattis SG; Fierke CA; Christianson DW (2010) Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 49, 5048–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vannini A; Volpari C; Gallinari P; Jones P; Mattu M; Carfi A; De Francesco R; Steinkuhler C; Di Marco S (2007) Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep 8, 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitehead L; Dobler MR; Radetich B; Zhu Y; Atadja PW; Claiborne T; Grob JE; McRiner A; Pancost MR; Patnaik A; Shao W; Shultz M; Tichkule R; Tommasi RA; Vash B; Wang P; Stams T (2011) Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem 19, 4626–4634. [DOI] [PubMed] [Google Scholar]
- 28.Cole KE; Dowling DP; Boone MA; Phillips AJ; Christianson DW (2011) Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J. Am. Chem. Soc 133, 12474–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vannini A; Volpari C; Filocamo G; Casavola EC; Brunetti M; Renzoni D; Chakravarty P; Paolini C; De Francesco R; Gallinari P; Steinkuhler C; Di Marco S (2004) Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. U. S. A 101, 15064–15069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H; Sengupta N; Villagra A; Rezai-Zadeh N; Seto E (2006) Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell. Biol 26, 5259–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haider S; Joseph CG; Neidle S; Fierke CA; Fuchter MJ (2011) On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg. Med. Chem. Lett 21, 2129–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kunze MB; Wright DW; Werbeck ND; Kirkpatrick J; Coveney PV; Hansen DF (2013) Loop interactions and dynamics tune the enzymatic activity of the human histone deacetylase 8. J. Am. Chem. Soc 135, 17862–17868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Decroos C; Clausen DJ; Haines BE; Wiest O; Williams RM; Christianson DW (2015) Variable Active Site Loop Conformations Accommodate the Binding of Macrocyclic Largazole Analogues to HDAC8. Biochemistry 54, 2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weerasinghe SVW; Estiu G; Wiest O; Pflum MKH (2008) Residues in the 11 Å Channel of Histone Deacetylase 1 Promote Catalytic Activity: Implications for Designing Isoform-Selective Histone Deacetylase Inhibitors. J. Med. Chem 51, 5542–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Decroos C; Bowman CM; Moser JA; Christianson KE; Deardorff MA; Christianson DW (2014) Compromised structure and function of HDAC8 mutants identified in Cornelia de Lange Syndrome spectrum disorders. ACS Chem. Biol 9, 2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gantt SL; Gattis SG; Fierke CA (2006) Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 45, 6170–6178. [DOI] [PubMed] [Google Scholar]
- 37.Wolfson NA Determining HDAC8 Substrate Specificity. Dissertation, University of Michigan, 2014. [Google Scholar]
- 38.Wegener D; Wirsching F; Riester D; Schwienhorst A (2003) A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol 10, 61–68. [DOI] [PubMed] [Google Scholar]
- 39.Wegener D; Hildmann C; Riester D; Schwienhorst A (2003) Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal. Biochem 321, 202–208. [DOI] [PubMed] [Google Scholar]
- 40.Ku HH (1966) Notes on the use of propagation of errors formulas. J. Res. Natl. Inst. Stan 70C, 263–273. [Google Scholar]
- 41.Wolfson NA; Pitcairn CA; Sullivan ED; Joseph CG; Fierke CA (2014) An enzyme-coupled assay measuring acetate production for profiling histone deacetylase specificity. Anal. Biochem 456, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baumann M; Stürmer R; Bornscheuer UT (2001) A High-Throughput-Screening Method for the Identification of Active and Enantioselective Hydrolases. Angew. Chem. Int. Ed 40, 4201–4204. [DOI] [PubMed] [Google Scholar]
- 43.Kim B; Pithadia AS; Fierke CA (2015) Kinetics and thermodynamics of metal-binding to histone deacetylase 8. Protein Sci 24, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khoury GA; Thompson JP; Smadbeck J; Kieslich CA; Floudas CA (2013) Forcefield_PTM: Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput 9, 5653–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khoury GA; Smadbeck J; Tamamis P; Vandris AC; Kieslich CA; Floudas CA (2014) Forcefield_NCAA: ab initio charge parameters to aid in the discovery and design of therapeutic proteins and peptides with unnatural amino acids and their application to complement inhibitors of the compstatin family. ACS Synth. Biol 3, 855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J; Wang W; Kollman PA; Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model 25, 247–260. [DOI] [PubMed] [Google Scholar]
- 47.Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA (2004) Development and testing of a general amber force field. J. Comput. Chem 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- 48.Jakalian A; Jack DB; Bayly CI (2002) Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem 23, 1623–1641. [DOI] [PubMed] [Google Scholar]
- 49.Case DA; Berryman JT; Betz RM; Cerutti DS; Cheatham TEI; Goetz AW; Homeyer N; Izadi P; Janowksi P; Kaus J; Kovalenko A; Lee TS; LeGrand S; Li P; Luchko T; Luo R; Madej B; Merz KM; Monard G; Needham P; Nguyen H; Nguyen HT; Omelyan I; Onufriev A; Roe DR; Roitberg A; Salomon-Ferrer R; Simmerling CL; Swails J; Walker RC; Wang J; Wolf RM; Wu X; York; Darden TA; Cheatham TE; Simmerling CL; Wang J; Duke RE; Luo R; Walker RC; Zhang W; Merz KM; Roberts B; Hayik S; Roitberg A; Seabra G; Swails J; Walker RC; Wang J; Wolf RM; Xu X; York DM; Kollman PA, AMBER 15. University of California, San Francisco: 2015. [Google Scholar]
- 50.Joung IS; Cheatham TE 3rd (2008) Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 112, 9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Darden T; York D; Pedersen L (1993) Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys 98, 10089–10092. [Google Scholar]
- 52.Ryckaert J-P; Ciccotti G; Berendsen HJC (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 23, 327–341. [Google Scholar]
- 53.Berendsen HJC; Postma JPM; van Gunsteren WF; DiNola A; Haak JR (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys 81, 3684–3690. [Google Scholar]
- 54.Ohnmacht SA; Marchetti C; Gunaratnam M; Besser RJ; Haider SM; Di Vita G; Lowe HL; Mellinas-Gomez M; Diocou S; Robson M; Šponer J; Islam B; Barbara Pedley R; Hartley JA; Neidle S (2015) A G-quadruplex-binding compound showing anti-tumour activity in an in vivo model for pancreatic cancer. Sci. Rep 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harvey MJ; Giupponi G; Fabritiis GD (2009) ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput 5, 1632–1639. [DOI] [PubMed] [Google Scholar]
- 56.Lindahl E; Hess B; van der Spoel D (2001) GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model 7, 306–317. [Google Scholar]
- 57.Pronk S; Páll S; Schulz R; Larsson P; Bjelkmar P; Apostolov R; Shirts MR; Smith JC; Kasson PM; van der Spoel D; Hess B; Lindahl E (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Humphrey W; Dalke A; Schulten K (1996) VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 59.DeLano WL The PyMOL Molecular Graphics System, Schrodinger, LLC, 2008. [Google Scholar]
- 60.Fernandez-Recio J; Totrov M; Abagyan R (2003) ICM-DISCO docking by global energy optimization with fully flexible side-chains. Proteins 52, 113–117. [DOI] [PubMed] [Google Scholar]
- 61.Otwinowski Z; Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 62.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta. Crystallogr. D. Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P; Lohkamp B; Scott WG; Cowtan K (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wood TE; Dalili S; Simpson CD; Sukhai MA; Hurren R; Anyiwe K; Mao X; Suarez Saiz F; Gronda M; Eberhard Y; MacLean N; Ketela T; Reed JC; Moffat J; Minden MD; Batey RA; Schimmer AD (2010) Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol. Cancer Ther 9, 246–256. [DOI] [PubMed] [Google Scholar]
- 65.Gurard-Levin ZA; Kim J; Mrksich M (2009) Combining mass spectrometry and peptide arrays to profile the specificities of histone deacetylases. ChemBioChem 10, 2159–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wolfson NA; Pitcairn CA; Fierke CA (2013) HDAC8 substrates: Histones and beyond. Biopolymers 99, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deardorff MA; Bando M; Nakato R; Watrin E; Itoh T; Minamino M; Saitoh K; Komata M; Katou Y; Clark D; Cole KE; De Baere E; Decroos C; Di Donato N; Ernst S; Francey LJ; Gyftodimou Y; Hirashima K; Hullings M; Ishikawa Y; Jaulin C; Kaur M; Kiyono T; Lombardi PM; Magnaghi-Jaulin L; Mortier GR; Nozaki N; Petersen MB; Seimiya H; Siu VM; Suzuki Y; Takagaki K; Wilde JJ; Willems PJ; Prigent C; Gillessen-Kaesbach G; Christianson DW; Kaiser FJ; Jackson LG; Hirota T; Krantz ID; Shirahige K (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alam N; Zimmerman L; Wolfson NA; Joseph CG; Fierke CA; Schueler-Furman O (2016) Structure-Based Identification of HDAC8 Non-histone Substrates. Structure 24, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Olson DE; Udeshi ND; Wolfson NA; Pitcairn CA; Sullivan ED; Jaffe JD; Svinkina T; Natoli T; Lu X; Paulk J; McCarren P; Wagner FF; Barker D; Howe E; Lazzaro F; Gale JP; Zhang YL; Subramanian A; Fierke CA; Carr SA; Holson EB (2014) An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem. Biol 9, 2210–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castaneda CA; Wolfson NA; Leng KR; Kuo YM; Andrews AJ; Fierke CA (2017) HDAC8 substrate selectivity is determined by long- and short-range interactions leading to enhanced reactivity for full-length histone substrates compared with peptides. J. Biol. Chem 292, 21568–21577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Castaneda CA; Lopez JE; Joseph CG; Scholle MD; Mrksich M; Fierke CA (2017) Active Site Metal Identity Alters Histone Deacetylase 8 Substrate Selectivity: A Potential Novel Regulatory Mechanism. Biochemistry 56, 5663–5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.