

SUMMARY
Plasma cells (PC) are found in the CNS of multiple sclerosis (MS) patients, yet their source and role in MS remains unclear. We find that some PC in the CNS of mice with experimental autoimmune encephalomyelitis (EAE) originate in the gut and produce immunoglobulin A (IgA). Moreover, we show that IgA+ PC are dramatically reduced in the gut during EAE, and likewise, a reduction in IgA-bound fecal bacteria is seen in MS patients during disease relapse. Removal of plasmablast (PB) and/or PC resulted in exacerbated EAE that was normalized by the introduction of gut-derived IgA+ PC. Furthermore, mice with an over-abundance of IgA+ PB and/or PC were specifically resistant to the effector stage of EAE, and expression of interleukin (IL)-10 by PB and/or PC was necessary and sufficient to confer resistance. Our data show that IgA+ PB and/ or PC mobilized from the gut play an unexpected role in suppressing neuroinflammation.
INTRODUCTION
After initial encounter with antigen, B cells can differentiate into plasmablasts (PB) and plasma cells (PC). PB are short-lived effector cells whereas PC are long-lived mediators of lasting humoral immunity (Nutt et al., 2015). Studies in mice indicate that PC reside in niches that are rich in survival factors such as interleukin (IL)-6, BAFF, and APRIL (Chu and Berek, 2013).
Treatment of relapsing-remitting multiple sclerosis (RRMS) with antibodies that deplete CD20+ B cells prevents the formation of new inflammatory lesions in the CNS (Hauser et al., 2008). However, this therapy does not target CD20neg PC, nor does it impact oligoclonal immunoglobulin bands found in the cerebral spinal fluid of MS patients (Piccio et al., 2010). In contrast, treatment with atacicept (TACI-Ig), an agent that neutralizes both APRIL and BAFF, not only reduces circulating B cells but also decreases serum antibody titers (Tak et al., 2008). Surprisingly, however, treatment of RRMS patients with atacicept resulted in dose-dependent disease exacerbations (Kappos et al., 2014) and promoted the development of MS in optic neuritis patients (Sergott et al., 2015).
Although IgG is the main isotype in serum, a significant number of antibody secreting cells circulating in the peripheral blood secrete IgA (Mei et al., 2009). There is evidence that IgA-producing cells specific for gut-encountered antigens can be found outside the gut. For example, Rotavirus and cholera toxin-specific IgA+ B cells (Jaimes et al., 2004; Lemke et al., 2016) and commensal-specific IgA+ PC (Wilmore et al., 2018) have been detected in the blood and the bone marrow. Curiously, IgA-producing cells can be found in unexpected places such as in prostate and liver tumors (Shalapour et al., 2015, 2017), atherosclerotic plaques (Iverson et al., 2006), and in generally immune-privileged locations like the MS brain (Stern et al., 2014). Collectively, these results demonstrate that IgA-producing cells can access the circulation as well as inflamed, damaged, or cancerous tissues. Definitive experiments are required to understand the role and source of IgA-producing cells in disease pathology
In this study, we sought to understand the source and function of PB and/or PC found in the CNS during neuroinflammation. We discovered that gut-derived IgA+ B cells are mobilized from the gut and subsequently attenuate inflammation in the CNS. Thus, while IgA-producing PC produce large quantities of anti-commensal Ab in the gut during homeostasis, our results provoke a re-consideration of the role of these cells during autoimmune disease.
RESULTS
IgA-Producing Cells Are Detected in the CNS during EAE
The divergent results of clinical trials testing anti-CD20 and TACI-Ig as MS treatments prompted us to re-assess the role of PB and/or PC during neuroinflammation. To this end, we used the MOG35–55 experimental autoimmune encephalomyelitis (EAE) animal model for kinetic, phenotypic, and functional studies. Because B cell differentiation into PB and/or PC is driven by the Prdm1 gene and the subsequent upregulation of Blimp1 protein (Minnich et al., 2016), we induced EAE in Prdm1yfp mice to unambiguously monitor the presence of PB and/or PC in the brain (Br) and spinal cord (Sc) at different stages of disease (Figure S1A). To corroborate our findings, we also induced EAE in a fate-map mouse that enables tracking of previously antigen-activated B cells and thus have expressed activation-induced cytidine deaminase (Aicdyfp mice) (Crouch et al., 2007) (Figure S1B). We measured expression of CD138 (a PC marker), B220 (relatively downregulated on PC), and Ki67 (PC are typically non-proliferative and Ki67–). A representative gating strategy for PB and/or PC is shown in Figure S1C. Of note, although CD138 is expressed to varying degrees on PB and PC, we found that CD138 was dynamically modulated on BM-resident PB and/or PC during the course of disease (Figure S1D). Due to the lack of distinguishing markers, we used the PB and/or PC designator to describe Prdm1-YFP+ or Aicd-YFP+B cells throughout this study.
Focusing first on the chronic stage of disease, we observed that Prdm1-YFP+ cells in the Brand Sc exhibited low expression of B220, were Ki67dim, and were CD138low. In comparison, Prdm1-YFP+ cells in the BM expressed variable levels of B220, were also Ki67dim and CD138+. Prdm1-YFP+ cells from the draining axillary lymph nodes expressed the highest levels of B220, were Ki67+ and CD138high (Figure 1A), similar to what has been previously described in the spleen (Pracht et al., 2017).
Figure 1. IgA+ PB and/or PC Are Detected in the Brain and Spinal Cord during EAE.

(A) Expression of indicated markers on Prdm1-YFP+ cells in the Br, Sc, BM, and LN during the chronic phase of EAE. Grey filled histograms represent the FMO for each stain and empty histograms represent specific Ab staining as determined by flow cytometry.
(B) Absolute numbers of Prdm1−YFP+B220−/dim cells in the Br and Sc at different time points of EAE determined by flow cytometry.
(C) Representative contour plots of intracellular IgA and IgG expression by Prdm1−YFP+B220−/dim cells determined by flow cytometry.
(D) Summary of frequency data obtained by flow cytometry.
(E) Number of IgA or IgG ASC in the Sc and Br determined by two-color ELISPOT of unimmunized (UI) or EAE WT mice (chronic phase).
(F) Number of IgA or IgG ASC in the Br determined by two-color ELISPOT of UI mice or IgA−/− chimeric EAE mice.
(A–E) Experiments were repeated 3 times with at least 5 mice per group. (F) Two experiments were performed with at least 6 mice per group. *p < 0.05, **p < 0.01, NS (not significant) by Mann-Whitney test with mean and SD depicted. Corresponding EAE data for IgA−/− chimeric EAE mice are found in Table S1 and Figure S6A.
See also Figure S1.
We observed a significant increase in absolute numbers of Prdm1-YFP+B220int/− cells in both the Br and Sc at the peak of EAE and an even greater increase during the chronic phase of the disease compared with unimmunized mice, where PB and/or PC were undetectable (Figure 1B). To characterize the Ig isotype of CNS-resident Prdm1-YFP+B220int/− cells, we performed intracellular dual flow cytometry staining for IgG and IgA at the chronic stage of EAE. While the majority of Prdm1-YFP+B220int/− cells were not class-switched (IgG−IgA− double negative cells), IgG class-switched cells could be detected in the CNS. Surprisingly, a significant portion of Prdm1-YFP+− B220lnt/− cells stained positive for intracellular IgA (Figures 1C and 1D) and confirmatory results were obtained using Aicd-creYFPfl/fl mice (Figures S1E and S1F).
To validate the presence of class-switched PB and/or PC in the CNS during EAE, we measured the frequency of IgG and IgA antibody-secreting cells (ASC) in the CNS by ELISPOT. Unlike flow cytometry, ELISPOT assays incorporate a 24-hr in vitro culture period that may skew results toward PB and/or PC that have the best survival capacity in vitro. Nevertheless, consistent with our flow cytometric findings, we observed significant increases in frequencies of both IgG and IgA ASC in the CNS during the chronic phase of EAE compared with unimmunized mice (Figure 1E).
Because IgA is predominantly produced at mucosal surfaces, the presence of IgA+ PB and/or PC in the CNS was somewhat surprising. To confirm this result, we ruled out potential artifactual detection of IgA+ ASC in the ELISPOT assay by examining IgA ASC in the CNS of Jh−/− chimeric mice that received a transplant of IgA−/− BM. Accordingly, while IgG+ ASC were detected in the brain of chimeric IgA−/− EAE mice during EAE, IgA+ ASC were undetectable (Figure 1F), thus confirming the specificity of our assay.
Given that PB and/or PC are detected in the CNS during EAE, we next determined whether these cells impact the clinical course of the disease. To do this, we generated PB and/or PC-deficient Cd19crePrdm1fl/fl and AicdcrePrdm1fl/fl mice and confirmed PB and/or PC-deficient status by immunofluorescence (Figure S1G) and by measuring serum Ig levels by ELISA (Figure S1H). Interestingly, we found that PB and/or PC-deficient mice exhibited exacerbated EAE (Figures S1I and S1J) and higher mortality (Table S1) compared with littermate controls.
In summary, while CD138lowKi67lowYFP+B220int/− PB and/or PC are not observed in the CNS during the steady state, PB and/or PC that produce IgG and IgA accumulate in the Br and Sc during EAE, and the absence of PB and/or PC results in increased EAE severity.
PB and/or PC Are Reduced in the Gut during EAE, and Add-Back of Gut-Derived PB and/or PC Attenuates Disease
Recently it has been shown that IgA+ PC exhibit immunosuppressive functions in both prostate and liver tumor microenvironments (Shalapouretal., 2015, 2017). Because we observed IgA+ PB and/or PC in the inflamed CNS and PB and/or PC-deficient mice exhibit exacerbated EAE, we hypothesized that IgA+ PB and/or PC may be specifically responsible for suppressing neuroinflammation. We focused on the gut as a possible source of CNS-infiltrating IgA+ PB and/or PC, because the gut is the largest reservoir of IgA-producing cells in the body. A significant decrease in the number of CD138+Aicd-YFP+ PB and/or PC in the small intestinal lamina propria (SILP) was observed during the chronic phase of EAE, suggesting that the homeostasis or localization of gut-resident PB and/or PC is somehow altered during EAE (Figures 2A–2C).
Figure 2. PB and/or PC Are Diminished in the Gut during EAE, and Transfer of Gut-Derived Blimp+ Cells to PB and/or PC-Deficient Mice Reduces Disease Severity.

(A) Representative immunofluorescence images of Aicd-YFP small intestines stained with DAPI, anti-CD8α, and anti-CD138 and visualized for YFP at the pre-onset or chronic phase of EAE.
(B) Quantification of CD138+ cells using the frequency of CD8+ cells as an unchanged denominator.
(C) Quantification of YFP+ cells using the frequency of CD8+ cells as an unchanged denominator.
(D and E) EAE clinical scores of PB and/or PC adoptive transfer in (D) CD19crePrdm1fl/fl recipients or (E) Jht−/− recipients, with adoptive transfer time-points indicated by arrows.
(F) Detection of adoptively transferred Prdm1-YFP+ cells in the Br, BM, and LN of PB and/or PC-deficient recipient mice.
Experiments in (A) were repeated 3 times with 3–8 mice per group. Experiments in (D) were repeated 5 times with 3–4 mice per group. Experiments in (E) were repeated 3 times with 3–5 mice per group. *p < 0.05, **p < 0.01 using two-way ANOVA test or Mann-Whitney test with mean and SD depicted. Scale bar depicted in (A) is 200 μm.
To test our hypothesis that gut IgA+ PB and/or PC may suppress neuroinflammation, we performed adoptive transfers of SILP-derived B220+CD5− B cells versus IgA+Ki67B220− PB and/or PC (Figure S2). Comparing with PBS treatment, a delay in disease onset and reduced EAE severity were observed when SILP-derived Prdm1-YFP+B220− cells were introduced into PB and/or PC-deficient mice. However SILP-derived Prdm1-YFP−B220+ cells had no effect on EAE (Figure 2D; Table S1). Similar results were observed upon transfer of Prdm1-YFP+B220− cells into B cell-deficient (Jht−/−) EAE mice (Figure 2E). At the chronic phase of the disease, we detected Prdm1-YFP+ cells in the Br, BM, and the LN of recipient mice (Figure 2F), confirming that the transferred cells could reach these tissues. In summary, the SILP contains PB and/or PC that, when adoptively transferred, can reach the CNS and are sufficient to reduce the clinical symptoms of EAE.
IgA+ B Cells from the SILP Recirculate to Distal Tissues
Because we detected a decrease in IgA+ B cells in the gut concomitantly with an increase in IgA-producing cells in the CNS during EAE, we asked in a non-transfer scenario whether gut-resident IgA+ B cells have the capacity to recirculate to other tissues. To test this, we devised an ELISPOT assay that enabled us to detect Rotavirus (RV)-specific B cells, because RV specifically infects mature enterocytes in the small intestine and provokes a robust local IgA response (Franco and Greenberg, 1999).
We first tested whether RV-specific IgA ASC could be recovered in other mucosal tissues such as the lung. To do this, we used a dual infection approach whereby mice were first orally infected with RV (primary infection) followed 30 days later by intranasal infection with Influenza (secondary infection). RV+Flu experimental mice were compared with 3 different control groups: RV and Flu naive (uninfected “UI”), primary infection alone (“RV-only”), and secondary infection alone (“Flu-only”) (see schematic in Figure S3A). Consistent with previous findings (Youngman et al., 2002), we found that RV was cleared from the feces at 7 days post-infection (dpi) and the RV-specific IgA response peaked at day 12 (Figure S3B), at which point RV-specific IgA ASC were found in both the SILP and the BM (Figure S3C). Of note, no differences in weight loss were observed in “Flu-only” versus “RV+Flu” mice in response to Flu infection (Figure S3D).
As expected, RV-specific IgA ASC (but not RV-specific IgG ASC) were detected at high frequencies in the gut post-Flu infection in both “RV-only” and “RV+Flu” mice (Figure 3A). We also observed a high frequency of RV-specific IgA ASC in the BM in “RV-only” and “RV+Flu” mice. In line with previously published work (Youngman et al., 2002), we also detected RV-specific IgG ASC in the BM, but these were present at low levels compared to RV-specific IgA ASC (Figure 3B). Lastly, IgA ASC were also detected in the lungs of “RV+Flu” mice as early as 1.5 dpi (Figure 3C). This suggests that inflammation in the lungs may rapidly attract RV-specific IgA ASC to migrate from the gut to the lungs. Interestingly, a low frequency of RV-specific IgA ASC was also observed in the lungs of “RV-only” mice (Figure 3C). This suggests that there may be a low level of homeostatic trafficking of RV-specific IgA ASC in the absence of lung inflammation.
Figure 3. IgA+ B Cells from the SILP Recirculate to Distal Tissues.

(A) RV-specific IgA and IgG ASC in the SILP of uninfected (UI) mice or mice infected with RV and/or Flu.
(B) As in (A) but assessing RV-specific IgA and IgG ASC in the BM.
(C) As in (A) but assessing RV-specific IgA and IgG ASC in the lungs. These experiments were repeated 5 times, and the graphs depicted contain all pooled data (n = 7–15 mice per group). *p < 0.05, **p < 0.01, ***p < 0.001 (Mann-Whitney test with mean and SD depicted.
(D) Schematic of parabiosis experimental design. Parabiosis experiments were conducted over 60 days with RV-only (“A”), and UI (“B”) mice. A and B mice were subsequently paired together.
(E) Fecal samples from each mouse were collected and tested for RV antigen via ELISA. Mice infected with RV cleared the virus by D12 and were subsequently paired with their parabiont partner on D15 post-infection (p.i.).
(F) Sample ELISPOTs from each collected tissue. Except for the first vertical panel, Parabiont 1 was RV-infected and Parabiont 2 was UI. Both pairs in the first vertical panel were uninfected.
(G) Total RV-specific IgA ASC/1×106 cells in the SILP of parabionts.
(H) Total RV IgA ASC/1×106 cells in BM of parabionts. Note that due to the highly damaged state of the gut at the peak of influenza infection at D6 post-flu infection, only limited numbers of cells could be isolated from the gut thus D6 was not included in our analysis. These experiments were repeated 2 times, and the graphs depicted contain all pooled data (n = 4 pairs of mice per group). *p < 0.05 using Mann-Whitney test with mean and SD depicted.
See also Figures S3 and S4.
In summary, these results demonstrate that IgA ASC previously generated in response to an intestinal pathogen 30 days prior can be rapidly recruited to the lungs upon Flu infection.
RV-Specific IgA ASC Can Access the Circulatory System and Populate the CNS
Because a low frequency of RV-specific IgA+ ASC was noted in the lungs at steady state in “RV-only” mice, we next determined if IgA+ B cells could access the circulation in resting mice. To test this, we employed a parabiosis technique whereby we surgically attached mice previously infected with RV (group A) to uninfected mice (group B, Figure 3D). Because group A was allowed to clear the infection prior to surgery (Figure 3E), this effectively separated RV priming events in one mouse from migration events in a second mouse. Analysis of paired animals revealed that RV-specific IgA ASC were present in the gut and BM of both the RV infected and RV-naive parabionts (Figures 3F–3H). Although there were fewer RV-specific IgA ASC in the gut and BM of group B mice compared to their group A partners, these data nevertheless demonstrate that RV-specific IgA ASC can access the circulation in the steady state.
To confirm our findings, we used an alternative experimental approach of tissue-directed photo-conversion. Specifically, upon exposure to violet light, cells expressing a Kaede transgene are irreversibly converted from green to red (Tomura et al., 2008). We therefore photoconverted the length of the small intestine of Kaede-Tg mice and 3 days later used flow cytometry to measure the number of Kaede-Red+ cells at the site of photoconversion (SILP) as well as in the BM (see representative gating strategy in Figure S4A). While significant photoconversion of SILP-resident cells was observed in Kaede-Tg mice, Kaede-Red+ cells were undetectable in non-transgenic or sham surgery controls. Moreover, photoconversion of mesenteric LN cells was minimal (Figure S4B) demonstrating that photoconversion was specific to the small intestine. Of the Kaede-Red+ cells in the SILP, we observed IgA+B220+, IgA+B220−, and IgA−B220+ cells with the majority being IgA+B220− (Figure S4C). Importantly, we also observed IgA+B220+ Kaede-Red+ B cells in the BM (Figure S4D), demonstrating that IgA+ B cells photoconverted in the gut can be recovered from the BM in the steady state.
To relate our findings back to EAE, we examined the CNS for presence of RV-specific IgA ASC by inducing EAE in RV-infected mice (see schematic in Figure 4A). We validated the clearance of RV prior to EAE (Figure S4E) and confirmed that a priori infection with RV does not impact the severity of EAE (Figure S4F). Using this approach, we found that RV-specific IgA ASC were absent from the CNS in the steady state (Figure 4B). However, during peak and chronic EAE we observed RV-specific IgA ASC in the CNS (Figures 4B, 4C, and 4F). In the SILP of “RV+EAE” mice we observed a significant reduction in RV-specific IgA ASC at the peak of disease compared to control mice (Figure 4D), and a similar finding was observed in the BM at the chronic phase (but not the peak) of EAE (Figure 4E).
Figure 4. RV-Specific IgA+ ASC Migrate to the CNS during MOG35-55-Induced EAE.

(A) Schematic representation of RV-EAE experiments.
(B) RV-specific IgA ASC in the Br of unimmunized (UI) mice or mice infected with RV and/or immunized with MOG35–35.
(C) As in (B) but assessing RV-specific IgA ASC in the Sc.
(D) As in (B) but assessing RV-specific IgA ASC in the SILP.
(E) As in (B) but assessing RV-specific IgA ASC in the BM.
(F) Representative RV-specific IgA ASC ELISPOT from Br, Sc, SILP, and BM of RV-EAE mice at the chronic stage of the disease.
(G) Representative images of commensal-reactive IgA ASC derived from the SILP of naive WT, Germ Free, and Cd19crePrdm1fl/fl mice. PBS-coated wells were used as a negative control for the assay.
(H) Quantitative summary of the results shown in (G).
(I) Quantification of commensal-reactive IgA ASC derived from sorted Prdm1-YFP+ B220− or Prdm1-YFP− B220+ cells from the SILP of Prdm1YFP mice.
(J) Representative images of commensal-reactive IgA ASC from results depicted in (I).
(K) Quantification of commensal-reactive IgA ASC in the BM (top) and brain (bottom) from UI and EAE (chronic) WT mice.
(L) Representative images of commensal-reactive IgA ASC from results depicted in (K). These experiments were repeated 3 times.
The number of mice depicted in (B)–(E) are as follows: UI (n = 7), RV (n = 6), EAE (n = 6), RV EAE peak (n = 4), and RV EAE chronic (n = 5). The number of mice depicted in (H), (I), and (K) are n = 4–7 for each group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 using Mann-Whitney test with mean and SD depicted.
See also Figure S4.
Taken together, gut-derived, RV-specific IgA+ B cells can access the circulatory system and home to the CNS during EAE.
Commensal-Reactive IgA ASC Populate the CNS during EAE
To ascertain if our findings are generalizable to gut-intrinsic commensal microbes, we developed a commensal ELISPOT assay. Application of single-cell suspensions from the SILP of wild-type (WT) mice to plates coated with heat-killed autologous fecal matter resulted in the detection of commensal-reactive IgA ASC in WT, but not germ-free mice or Cd19crePrdm1fl/fl mice (Figures 4G and 4H). Moreover, pre-sorting Prdm1-YFP+B220− cells from the SILP of Prdm1yfp reporter mice resulted in a significant enrichment of commensal-reactive IgA ASC in the ELISPOT assay, whereas minimal IgA ASC were observed in ELISPOT wells containing Prdm1-YFP−B220+ cells, indicating that the majority of commensal-reactive IgA ASC detected in this assay are PB and/or PC (Figures 4I and 4J).
Next, we determined if we could detect commensal-reactive IgA ASC in the BM and in the CNS. Consistent with a recent report (Wilmore et al., 2018), we were able to detect commensal-reactive IgA ASC in the BM. We also found commensal-specific IgA ASC in the Br during the chronic phase of EAE, but not at steady state (Figures 4K and 4L). In summary, commensal-reactive IgA PB and/or PC can be found in extra-intestinal sites such as the BM and the inflamed Br.
IgA-Binding of Fecal Bacteria Is Reduced in Patients with MS during an Acute Relapse
To assess the generalizability of our findings to human MS, we examined IgA content in the gut lumen of MS patients. Recently, bacterial flow cytometry (BUGFlow) has been used to assess IgA binding of bacteria (Palm et al., 2014). We therefore measured IgA-binding of autologous gut bacteria as an indirect measure of gut-resident IgA+ PB and/or PC in a cohort of healthy control versus MS patients. The MS cohort included patients in remission versus those experiencing an acute relapse (Figure 5A; Table S2). 16S rRNA gene sequencing of IgA-bound bacteria (IgA-SEQ) from human control samples confirmed that this method captured a broad range of both aerobic and anaerobic bacteria including anaerobes of the phyla Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria, among others (data not shown). Quantification of IgA-bound bacteria revealed a non-statistically significant trend toward higher IgA-binding in MS patients during remission compared to healthy controls by BUGFlow (Figures 5B and 5C) and ELISA (Figure 5D). Interestingly, actively relapsing patients exhibited significantly lower percentages of IgA-bound gut bacteria compared to patients in remission (Figures 5B–5D) indicating potential egress of IgA-producing cells from the gut during an inflammatory relapse.
Figure 5. IgA-Binding of Bacteria in the Gut Is Reduced in MS Patients during an Acute Relapse.

(A) Study overview: fresh-frozen fecal samples of 33 patients with clinically Isolated syndrome (CIS) or relapslng-remlttlng multiple sclerosis (RRMS) (n = 22 during disease remission, n = 11 during an acute relapse) and 32 healthy controls were collected. Fecal bacteria were isolated and IgA-binding of autologous gut bacteria was quantified by bacterial flow cytometry (BUGFlow) and ELISA.
(B) BUGFlow: bacteria were identified based on forward and side scatter and the IgA-positive population was defined based on an isotype control. Shown is an example of staining of fecal bacteria from an RRMS patient with an isotype control (left) as well as with anti-IgA during remission (middle) and during relapse (right).
(C) Percentage of fecal-bound IgA by BUG Flow: 1 × 106 fecal bacteria were stained with anti-IgA and % of IgA binding was assessed by flow cytometry. Values shown are replicates from 2 experiments.
(D) Quantification of fecal-bound IgA by ELISA: 1 × 106 fecal bacteria were assessed for IgA binding using a commercially available quantitative IgA ELISA assay. Values shown are mean values of duplicate measurements. *p < 0.05, t test with Welch correction with mean and SEM depicted.
See also Table S2.
A Commensal Microbe that Elevates Systemic IgA Attenuates EAE
Given that EAE is associated with the presence of gut-derived IgA+ PB and/or PC in the CNS, and that transfer of gut-derived IgA+ PB and/or PC attenuates EAE, commensal microbes that increase systemic levels of IgA+ PB and/or PC should reduce the severity of EAE. To test this, we supplemented an already established microbiota with a singular IgA-promoting commensal microbe, Trichomonas musculis (T.mu) (Chudnovskiy et al., 2016). Compared to T.mu− mice, T.mu+ colonized mice exhibited less severe EAE with lower incidence of disease (Figures S5A and S5B), and reduced inflammation and demyelination in the spinal cord (Figures S5C–S5E). T.mu+ colonized mice also exhibited with serum and fecal IgA (Figure S5G) and an increase in the frequency of IgA ASC in the gut, bone marrow, and brain (Figure S5G). Although the frequency of CNS-resident Th17 and Th1 cells was unaltered by colonization of T.mu, the frequency of GM-CSF producing CD4+ T cells was significantly reduced (Figure S5H). Thus, although T.mu has been shown to elevate Th1 and Th17 responses in the gut (Chudnovskiy et al., 2016), colonization with T.mu protects against severe EAE concomitant with an elevation of IgA ASC in the brain.
Assessment of PC-Derived Factors Involved in EAE Suppression
The accumulation of RV-specific and commensal-reactive IgA ASC in the brain during EAE prompted us to determine if IgA antibodies may play a role in ameliorating disease. However, we found that irradiated B cell deficient (Jht−/−) mice reconstituted with IgA−/− BM exhibited roughly similar EAE severity as cohoused Jht−/− mice that received WT BM, although a slight acceleration in onset in the IgA−/− recipient group resulted in a modest increase in cumulative score (Figure S5I). These experiments suggest that IgA itself does not play a primary role in dictating the severity of EAE.
Recent work has shown that PB and/or PC can express immunoregulatory proteins such as IL-10 and IL-35 that impact the immune response during disease (Shen et al., 2014). Thus, we hypothesized that PB and/or PC may suppress ongoing inflammation in the CNS independent of IgA production. We focused mainly on IL-10 due to its critical role in attenuating EAE (Matsumoto et al., 2014; Shen et al., 2014) and accordingly tested if gut-resident IgA+ cells were a significant source of IL-10. Interestingly, we found that IgA+ B cells in the SILP showed evidence of IL-10 immunofluorescence staining that was not present in IL-10−/− mice. Moreover, IL-10 staining was greatly reduced in the SILP of PB and/or PC-deficient mice (Figure 6A).
Figure 6. PC-Derived Factors that Are Involved in EAE Suppression.
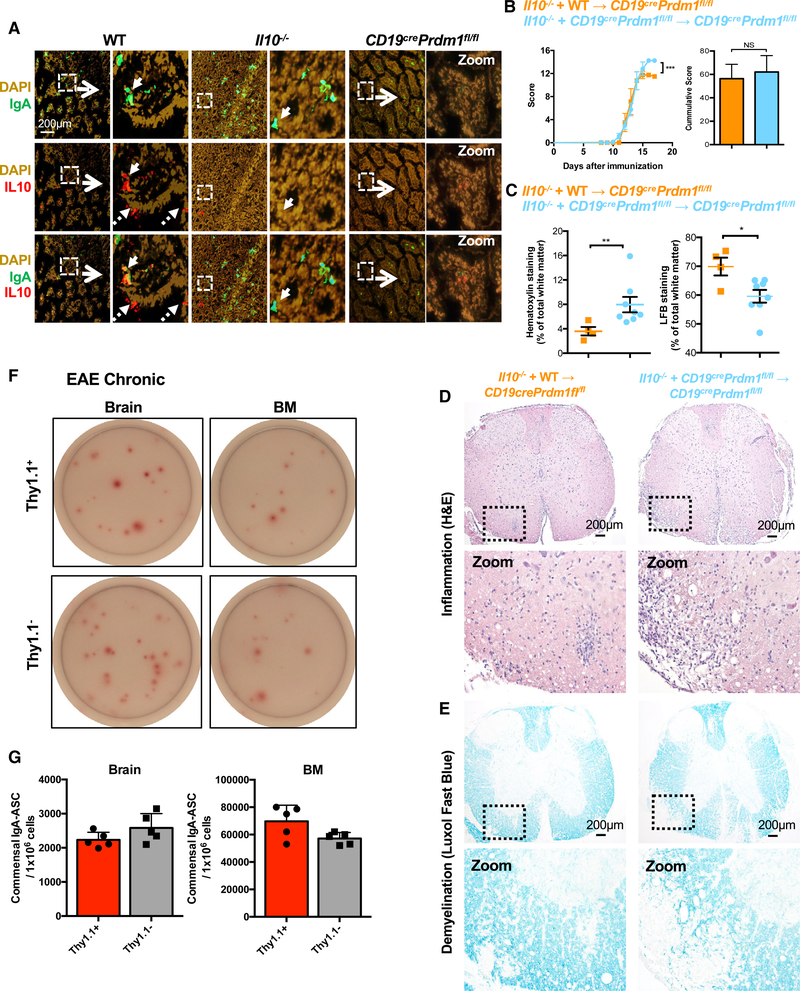
(A) Representative immunofluorescence images of WT and IL-10−/− small intestines stained with DAPI, anti-IgA, and anti-IL-10.
(B) Mixed BM chimeras whereby PB and/or PC-deficient mice were reconstituted with and 80/20 mixture of PB and/or PC-deficient + IL-10−/− BM and control chimeras reconstituted with an 80/20 mixture of WT + IL-10−/− BM were subjected to EAE and clinical score was measured overtime. Cumulative score is shown on the right.
(C) Quantitative analysis of H&E (left) and LFB (right) staining of thoracic spinal cord sections derived from mixed BM chimeras.
(D and E) Representative images of H&E (D) and LFB (E) stains from thoracic spinal cord sections derived from mixed BM chimeras acquired at4× magnification using a light microscope to visualize inflammation and demyelination.
(F) Representative images of commensal-reactive IgA ASC pre-sorted as Dump− (CD4, CD8, F4/80 negative) and subsequently sorted as Thy1.1+ or Thy1.1- cells from the brain (7,500 cells/well) or BM (150 cells/well) during the chronic phase of EAE. Note that the pre-sort step results in an enrichment in these number of spots detected.
(G) Quantification of commensal-reactive IgA ASC from (F).
Experiments in (A)were repeated two times with at least 5 mice per group, with IL10−/− staining control being performed once. Experiments in (B) were performed three times. Experiments in (C)–(E) were performed three times with 4–8 mice per group. Experiments in (F) and (G) were performed three times with 5 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 using two-way ANOVA test for(B) and Mann-Whitney test for(C) and (G) with mean and SD depicted, (C) mean and SEM depicted. Scale bar depicted in (A) and (D) is 200 μm.
See also Figures S5 and S6 and Table S1.
Next, we assessed whether PB and/or PC-intrinsic IL-10 was required for suppression of EAE by generating mixed BM chimeric mice whereby PB and/or PC cannot produce IL-10 but other hematopoietic cells retain this capacity. Compared with control chimeras, we found that mixed chimeric mice that harbor IL-10−/− PB and/or PC exhibited exacerbated EAE (Figure 6B) compared to control mixed BM chimeric mice (Table S1). Due to the severity of disease, we were unable to carry out the experiment to the chronic stage of EAE. We thus confirmed our clinical findings by immunohistochemistry and noted that BM chimeric mice with PB and/or PC unable to produce IL-10 exhibited significantly increased immune infiltration (Figures 6C and 6D) and demyelination (Figures 6C and 6E) of the spinal cord.
In order to analyze cells that are actively transcribing Il10, we next used 10BiT mice that report the expression of Il10 via cell surface Thy1.1 expression. 10BiT mice have been used to query IL-10 expression in a number of cell types, with IL-10 protein production only associated with Thy1.1+ cells and not Thy1.1− cells (Cunningham et al., 2016). Flow cytometric analysis revealed surface Thy1.1 protein on IgA+B220− cells in the SILP of 10BiT mice, confirming our immunofluorescence findings (Figure S5J). Next, we isolated all cells expressing Il10 mRNA by sorting Thy1.1+ versus Thy1.1 cells from the Br and the BM during the chronic phase of EAE. Sorted cells were then plated in commensal ELISPOT plates and developed with anti-IgA. We found that ~50% of commensal-reactive IgA ASC in the BM and the Br were derived from the Thy1.1+ fraction (Figures 6F and 6G).
We (Fritz et al., 2011) and others (Matsumoto et al., 2014; Shen et al., 2014) have found that PB and/or PC express other immunomodulatory molecules beyond IL-10, including IL-35 and iNOS. This suggests that PB and/or PC may employ a variety of mechanisms to attenuate EAE. Because Nos2−/− mice exhibit exacerbated EAE (Fenyk-Melody et al., 1998), we assessed a possible PB and/or PC-intrinsic role for iNOS in suppressing EAE using BM chimeras. Compared to control chimeric mice, deletion of Nos2 in B cells (Figure S6A) or specifically in PB and/or PC (Figure S6B) resulted in modestly exacerbated EAE and higher mortality (phenocopying Nos2−/− → Nos2−/− BM chimeras, see Table S1). This phenotype was accompanied by enhanced spinal cord inflammation and demyelination (Figures S6C–S6E).
In summary, while IgA itself is not sufficient to suppress EAE, commensal-reactive IgA ASC express IL-10, and the expression of IL-10 by PB and/or PC (and to a lesser extent iNOS) is required to reduce the symptoms of EAE.
BAFF-Transgenic Mice Are Highly Resistant to the Effector Phase of EAE
We have previously shown that mice overexpressing BAFF (BAFF-Tg mice) have an expanded pool of IgA+ PC in the SILP accompanied by commensal-reactive IgA in the serum (McCarthy et al., 2011). Because we found that SILP-derived Prdm1-YFP+B220− cells could ameliorate EAE, we hypothesized that BAFF-Tg mice would also exhibit an altered response to EAE.
Consistent with our observations in WT mice, we observed a dramatic decrease in IgA+CD138+ cells during the chronic stage of EAE (Figures 7A–7C). Interestingly, unlike WT mice where IgA and IgG ASC were virtually undetectable in the CNS of unimmunized mice, BAFF-Tg mice exhibited a baseline level of IgA ASC in the Br and Sc at steady state that was not further elevated during EAE (Figures 7D and 7E). In contrast, like WT mice, IgG ASC were only detected in the CNS of BAFF-Tg mice during EAE, but not in the steady state.
Figure 7. BAFF-Tg Mice Are Resistant to MOG35–55-Induced EAE via IL-10.
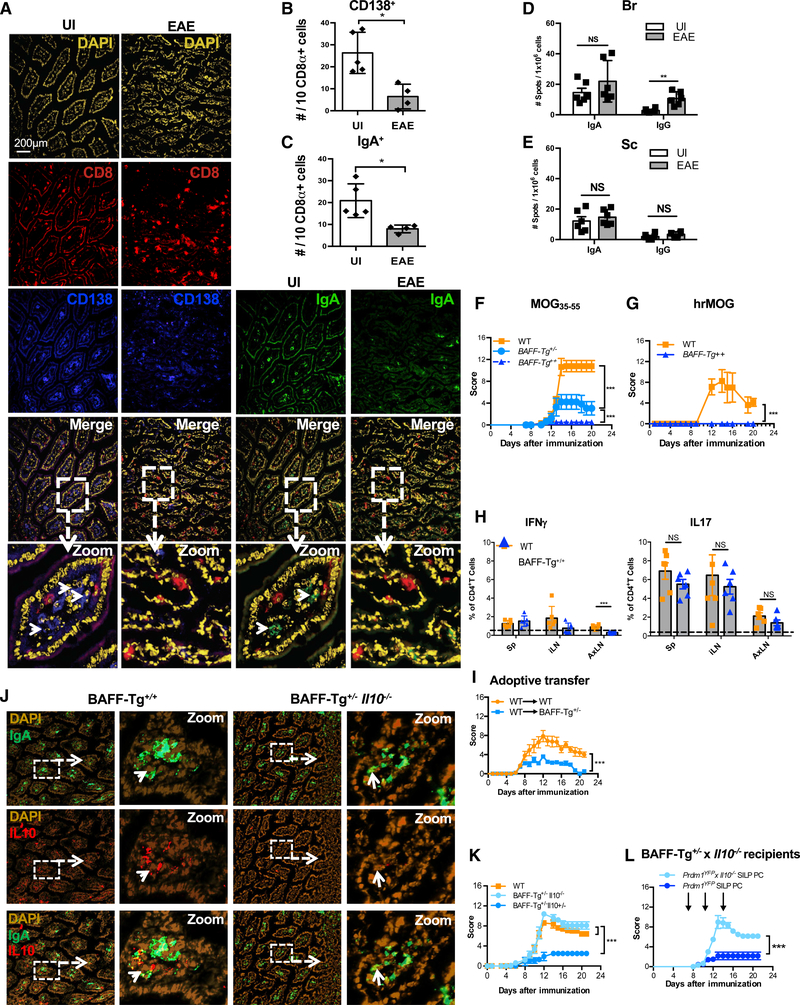
(A) Representative immunofluorescence images of the SILP of BAFF-Tg+/+ mice before (UI) and during the chronic phase of MOG35–55-induced EAE stained with DAPI, anti-CD138, and anti-IgA.
(B) Quantification of CD138+ cells in the SILP using CD8+ cells as an unchanged denominator.
(C) Quantification of IgA+ cells in the SILP using CD8+ cells as an unchanged denominator.
(D and E) Number of IgA or IgG ASC enumerated by ELISPOT analysis in the (D) Br and (E) Sc of BAFF-Tg+/+ mice during MOG35–55-induced EAE (chronic phase) compared with UI BAFF+/+-Tg mice.
(F) Clinical scores of BAFF+/+-Tg, BAFF+/−-Tg, and WT littermates after immunization with MOG35–55.
(G) Clinical scores of BAFF+/+ and WT mice after immunization with hrMOG.
(H) Frequency of MOG35–55 IFNγ+ CD4+ T cells (left) or IL-17 CD4+ T cells (right) from WT or BAFF+/−-Tg littermates with chronic EAE.
(I) EAE Clinical scores of adoptive transfer EAE whereby pre-primed T cells were transferred into WT or BAFF+/−-Tg recipient mice.
(J) Representative immunofluorescence images of BAFF+/+-Tg and BAFF+/−-Tg x IL-10−/− small intestines stained with DAPI, anti-IgA, and anti-IL-10.
(K) Clinical scores of BAFF+/−-Tg, BAFF+/−-Tg x IL-10−/−, and WT littermates after immunization with MOG35–55.
(L) EAE Clinical scores of Baff+/−-Tg IL-10−/− recipients, after transfer of Prdm1YFP-IL-10−/− gut PC or Prdm1YFP gut PC.
Experiments in (A)–(F) and (K)–(L) were repeated 3 times with at least 4 mice per group. The experiment in (G) was repeated two times with at least 4–5 mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 using two-way ANOVA test for (F), (G), (I), (K), and (L) and Mann-Whitney test for (B)–(E) and (H) with mean and SD depicted. Scale bar depicted in (A) is 200 μm.
Strikingly, we found that BAFF-Tg+/+ mice immunized with MOG35–55 do not develop clinical symptoms of disease compared to co-caged WT littermates, and only mild disease was observed in mice with only one copy of the BAFF transgene (BAFF-Tg+/−) (Figure 7F). These results were also confirmed in a separate vivarium (Table S1). To assess whether BAFF-mediated resistance to MOG35–55 EAE was generalizable to other EAE models, we immunized BAFF-Tg+/+ mice with full-length re-combinant human MOG (rhMOG), a protein that induces pathogenic anti-MOG auto-antibodies that promote CNS injury. BAFF-Tg+/+ mice were likewise resistant to rhMOG-induced disease (Figure 7G).
To determine whether overexpression of BAFF and a surplus of IgA+ PB and/or PC in these mice affects the priming versus the effector stage of EAE, we examined the production of cytokines by CD4+ T cells in the draining lymph nodes and spleen of MOG35–55 immunized BAFF-Tg+/+ mice during the pre-onset period of the disease. Similar levels of CD4+IFNγ+, and CD4+IL-17+ T cells were detected in WT and BAFF-Tg+/+ mice, with a reduction in CD4+IFNγ+ T cells only being observed in the axillary lymph nodes of BAFF-Tg+/+ mice (Figure 7H). Similar results were found upon immunization with rhMOG (data not shown). In contrast, defects were observed in the effector phase of the disease in BAFF-Tg mice. Specifically, while adoptive transfer of pre-primed T cells from WT donor mice immunized with MOG35–55 peptide resulted in EAE when T cells were transferred into WT recipients, transfer of the same T cells into BAFF-Tg+/− recipients resulted in significantly attenuated EAE (Figure 7I). This was accompanied by a non-significant trend of reduced frequency and numbers of IFNγ+CD4+ and IL-17+CD4+ T cells, and a significant reduction in the frequency of GM-CSF+ CD4+ T cells, in the CNS (Figure S7A). A trend toward increased frequency of commensal-reactive IgA ASC (measured by ELISPOT) in the CNS was also noted (Figure S7B), as well as a significant reduction in inflammation and demyelination in the spinal cord (Figures S7C–S7E).
Last, we determined whether TACI (TNFRSF13b), a receptor for BAFF and APRIL that is highly expressed by PB and/or PC (Pracht et al., 2017), provides protective signals during EAE. To test this, Tnfrsf13−/− mice were immunized with MOG35–55 peptide and assessed for clinical parameters of EAE. Similar to PB and/or PC-deficient mice, Tnfrsf13b−/− mice also show evidence of exacerbated EAE compared to WT controls (Figure S7D).
In summary, BAFF-Tg mice, which harbor IgA-producing cells in the CNS during the steady state, are highly resistant to both MOG35–55 and rhMOG-induced EAE, possibly due to TACI-derived signals. Moreover, whereas the priming response induced by MOG immunization is relatively normal, the effector phase of EAE is attenuated in BAFF-Tg mice compared to WT mice.
Gut-Derived IgA PB and/or PC Promote Resistance to EAE in BAFF-Tg Mice via IL-10
We hypothesized that IL-10 would be involved in EAE resistance in BAFF-Tg mice. Indeed, BAFF-Tg+/+ mice showed evidence of IL-10 expression by SILP-resident IgA+ cells (Figures 7J and S7G), and compared to WT mice, IgA+IL-10+ cells were even more abundant in the BAFF-Tg+/+ gut (Figure S7H). Also, like WT mice, we found evidence of IL-10 expression by a portion of IgA+B220+ PB and/or PC in the brain of BAFF-Tg+/+ mice during EAE (Figure S7I).
To test whether IL-10 is required for EAE resistance in BAFF-Tg mice, we generated BAFF-Tg+/−IL-10−/−, BAFF-Tg+/−IL- 10+/−, and WT littermates. Interestingly, BAFF-Tg+/−IL-10−/− mice developed EAE disease similar to what was observed in WT littermate controls (Figure 7K; Table S1), demonstrating that IL-10 is necessary for BAFF-driven resistance to EAE. To ascertain if IgA-producing SILP-derived PB and/or PC were a relevant source of IL-10 in BAFF-Tg mice, we adoptively transferred Prdm1-YFP+ PC from the SILP of Prdm-1yfp mice versus Prdm1-YFP+ × Il10−/− mice into BAFF-Tg+/−Il10−/− recipient mice. We found that only IL-10-sufficient gut-derived Prdm1-YFP+ cells could confer disease relief in BAFF-Tg+/−//10−/− recipient mice (Figure 7L), demonstrating that intestinal PB and/or PC provide an essential source of IL-10 that is sufficient to suppress EAE.
In summary, BAFF-Tg, but not BAFF-Tg+/−//10−/− mice, are highly resistant to EAE, and BAFF-Tg mice harbor a significant number of IgA+ cells in the SILP that express IL-10. Moreover, SILP-derived PB and/or PC are a sufficient source of IL-10 for the attenuation of EAE clinical symptoms in BAFF-Tg+/−.//10−/− mice.
DISCUSSION
In this study, we characterized the kinetics, source, and function of IgA+ B cells entering the CNS during EAE. Using multiple approaches, we found that gut-derived IgA+ B cells are capable of accessing the periphery and the inflamed CNS, and MS patients exhibit a reduction in fecal bacteria-bound IgA during MS relapses, perhaps due to egress of IgA-producing cells out of the gut. Consistent with a previous report (Matsumoto et al., 2014), we found that IL-10 expression by PB and/or PC was both necessary and sufficient for the suppression of EAE. Here, we found that commensal-reactive IgA+ PB and/or PC are a relevant source of IL-10 in the CNS during EAE. We propose that intestinal IgA+ B cells, one of the biggest reservoirs of lymphocytes in the body, represent a population of regulatory cells that can be recruited to inflamed tissues independent of their B cell receptor specificity. Mono-colonization with specific microbes has been shown to promote T-regulatory function during EAE (Cekanaviciute et al., 2017; Ochoa-Repáraz et al., 2010). The finding that colonization with T.mu in the context of a pre-existing microbiota is associated with EAE protection provides hope for developing treatments for MS that mobilize immunosuppressive IgA+ PB and/or PC.
It is important to note that our ELISPOT experiments that detect RV-specific or commensal-reactive IgA ASC cannot distinguish between IgA+ PC versus memory IgA+ B cells that have homed to extra-intestinal tissues. However, the bulk of our data demonstrate that the immunosuppressive B cells in the context of EAE are PB and/or PC. In support of this, we find that gut-derived B220+ cells fail to attenuate EAE, that PB and/or PC are the main source of IL-10 in the gut at steady state, and deletion of Il10 or Nos2 specifically in PB and/or PC results in exacerbated EAE. These data, in conjunction with recent findings in the spleen (Lino et al., 2018), support the notion that immunosuppressive PB and/or PC are a key source of IL-10.
Interestingly, although sorted as B220− and confirmed as intracellular IgA+Ki67−, adoptively transferred gut derived PB and/or PC exhibited altered B220/Ki67 status when transferred to PB and/or PC-deficient recipient mice. Current dogma posits that cell-cycle exit is irreversible for PC (Amanna and Slifka, 2010). However, gene expression profiling studies have shown that a variety of cell-cycle regulators are expressed during the transition of human PB to long-lived PC, suggesting the potential for cell-cycle re-entry (Slocombe et al., 2013), and perhaps this is particularly evident in the context of strong inflammation, as what would be observed during EAE.
Emerging studies hint at the existence of dynamic migration of lymphocytes between the gut and the periphery (Wilmore et al., 2018). Our work now adds to this accumulating evidence by demonstrating that gut-derived IgA ASC can localize to the inflamed mouse CNS. We hypothesize that inflammation in the CNS “changes the rules” allowing IgA+ PC to migrate out of their niches. In support of this concept, gut-derived intra-epithelial lymphocytes can be detected in the EAE CNS (Kadowaki et al., 2016). This may be because the gut environment itself is altered during inflammation in the periphery. Indeed, intestinal barrier function is compromised during EAE and MS (Buscarinu et al., 2017; Nouri et al., 2014), and in our EAE mice we noted visible gaps in the DAPI+ epithelial cells of the villi (Figures 2 and 7), suggesting that the gut may be damaged during EAE.
In general, a “firewall” constrains IgA responses to commensal bacteria to the mucosal immune system, and we previously reported that this firewall is compromised in BAFF-Tg mice (McCarthy et al., 2011). In the current study, we show that in WT mice, RV-specific IgA ASC can access the circulation even in the steady state, suggesting a firewall breach even without BAFF overexpression. It is possible that there is a low level of permeability of the firewall during steady state that we failed to detect using the western blot techniques in our previous study comparing WT and BAFF-Tg mice. Indeed, in humans, anti-commensal IgA titers are observed at low levels in the serum of healthy individuals (Haas et al., 2011). Nevertheless, the egress of IgA+ ASC out of the gut during steady state is likely not a frequent event because, although RV-specific IgA ASC are observed in uninfected parabionts, they are present at greatly reduced numbers compared to the RV-infected parabiont. However, it is important to note that the number of IgA ASC we detect in extra-intestinal tissues is likely an under-estimate given the reported problems of cell recovery in many tissues (Steinert et al., 2015).
The mechanism of IgA+ PC migration out of the gut during EAE is not known but could conceivably involve the upregulation of PC-intrinsic chemokine receptors. For example, it has been recently shown that IgA+ PC are localized in areas of the EAE spinal cord that express BAFF, APRIL, and CXCL12, suggesting that CXCR4 expression on gut-derived PB and/or PC may drive migration to regions of the CNS that are enriched for PC survival factors (Pollok et al., 2017). Alternatively, the loss of molecules that anchor PC to their supportive niche environments could also provoke PC egress from the gut. Indeed, we found that CD138, which mediates tethering of PC to collagen I, was down-regulated on PB and/or PC in the BM and in the CNS during EAE. Further analysis is required to assess the mechanism(s) that facilitate IgA+ B cell recruitment to the CNS.
We found that highly potent, encephalitogenic T cells generated in WT donor mice have limited capacity to elicit EAE disease in BAFF-Tg recipients. This suggests that BAFF acts at the effector phase of EAE to limit inflammation. Indeed, CD4+ T cells producing GM-CSF, a cytokine that is both necessary and sufficient to induce and maintain the effector stage of EAE (Spath et al., 2017), are significantly reduced in the CNS of BAFF-Tg recipient mice as well as in mice colonized with T.mu. One potential caveat to our findings is that a separate study showed that BAFF-Tg mice exhibited exacerbated EAE correlating with an increase in Th17 cells in the draining LN (Zhou et al., 2011). It is possible that differences in vivarium-associated microbiota, which we found can impact the frequency of IgA+ ASC in the CNS, may have resulted in divergent results that would be worthy of further study.
In summary, our findings demonstrate that gut-derived IgA+ PB and/or PC play an important role in dampening neuroinflammation. Although pathogenic memory B cells are targeted by anti-CD20 (Jelcic et al., 2018), IgA+ PB and/or PC will be spared, potentially explaining the robust and rapid efficacy of anti-CD20 treatment in MS. Moreover, our results provide a rational explanation for the MS disease exacerbations provoked by atacicept (TACI-Ig) (Kappos et al., 2014), and the exacerbated EAE we observed in TACl−/− mice mirrors this finding.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jennifer Gommerman (jen.gommerman@utoronto.ca). Certain materials are shared with academic and non-profit research organizations for research and education purposes only under an MTA to be discussed in good faith with the recipient.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
Prdm1fl/fl mice (Jackson Labs) were backcrossed with C57BL/6 mice (Charles River), AicdCrexYFPfl/fl mice (courtesy of Rafael Casellas) (Crouch et al., 2007) or Cd19Cre+/− mice (Jackson Labs) to generate PC conditional knockout mice. Prdm1yfp mice were purchased from Jackson labs and subsequently interbred to maintain the line. BAFF-Transgenic (BAFF-Tg) mice (obtained from Drs. Ann Ranger and Jeff Browning, Biogen Inc) (McCarthy et al., 2011) were backcrossed with WT mice (Charles River) and subsequently interbred as BAFF-Tg+/+ or BAFF-Tg+/− mice. BAFF-Tg+/+ were crossed with Il10−/− to generate littermates for experiments. Igh-Jtm1Cgn (Jht−/−), IgA−/− and Nos2−/− mice were used in some cases to generate BM chimeras. Tnfsf13b−/− and corresponding littermate controls were housed at the University of Melbourne. Il10-Thy1.1 transcriptional reporter (10BiT) mice were provided by Dr. David Brooks at the University Health Network with permission from Casey Weaver. Kaede mice were generated as described (Tomura et al., 2008) and provided by Dr. Andrew Luster’s lab at Harvard University and subsequently interbred to maintain the line. The genotypes of the above models were confirmed by PCR or qPCR. All animals were housed in a specific pathogen free, closed caging system and provided with a standard irradiated chow diet (Envigo Teklad (2918)), acidified water (reverse-osmosis and UV-sterilized) and housed under a 12-hr light cycle. All animal experiments were conducted with ethical approval from the University of Toronto, Faculty of Medicine animal care committee and the University of Melbourne animal care committee. The majority of the experiments were performed first in separately caged mice and then were repeated with co-caged and/or littermate mice to confirm our results.
Bone Marrow Chimeras
In order to develop single or mixed-BM chimeras, BM cells from Il10−/−, Nos2−/− or IgA−/− mice were used as donors alone or in combination with B cell deficient Jht−/−, WT, or Cd19cre Prdm1fl/fl BM cells, to reconstitute B cell deficient Jht−/− irradiated recipient mice thus creating BM chimeras in which B cells or PC are unable to produce IL10, iNOS or IgA. All the experiments in Bone Marrow Chimeras were performed in females, 6–8 weeks old.
Induction of EAE
To induce EAE, female mice, 6–8 weeks of age, were immunized subcutaneously on day 0 with 100 μg of MOG35–55 peptide or 100 μg of recombinant human MOG1–120 (rhMOG) and 500 μg of H37Ra (DIFCO Laboratories) emulsified in incomplete Freund’s adjuvant (BD, Biosciences). Mice were subcutaneously injected with 100μl of emulsion in 3 previously disinfected locations on the back for a total of 300μl of injected material per mouse. Pertussis toxin (PTX - List Biological Laboratories) was subsequently injected intraperitonally twice in 500μL PBS, on days 0 and 2, 500ng per injection. Due to high mortality in some strains, we increased daily nursing to improve survival by placing cages on heat pads with Pure-o’Cel paperless bedding, diet was supplemented with breeder mash (Envigo Teklad (2919)) to help mice gain or maintain body weight, and subcutaneous injections of 1ml-1.5ml of lactated Ringer’s solution, as well as 5% dextrose for caloric supplementation were provided 1–3 times daily depending on symptoms and signs of dehydration. Disease incidence was typically 100%. This mouse model does not relapse, but rather displays a chronic form of EAE. Clinical signs of EAE were assessed daily (blinded) with a modified 0–16 clinical scoring system to better evaluate paralysis on each limb individually. This composite scale provides superior sensitivity compared to the typical 5-point scale system, taking into account paralysis of each of the 4 limbs separately (Pikor et al., 2015). In some experiments, the traditional 5-point scale was compared with the 16-point scale, and we found no differences in terms of EAE disease trajectory or in comparing different groups of mice (Table S1).
Onset of disease was defined as a score ≥ 1, for at least two consecutive days. Peak phase was defined as the time point with the maximum score during disease. Chronic phase was defined as a consistent decrease in the peak score, persisting for at least two consecutive days. A cumulative clinical score was calculated for each mouse by adding the daily scores from the day of onset until the end of the experiment.
For some experiments we also performed the conventional 6-score scale since the experiments were performed in other laboratories or in order to compare results by using both scales. In those cases, scores were assigned as follows: 0 = asymptomatic, = loss of tail tone, 2 = hind limb weakness, 3 = partial hind limb paralysis, 4 = complete hind limb paralysis, and 6 = moribund.
Influenza Infection
Influenza strain A/Puerto Rico/8/1934(H1N1), also known as “PR8,” was kindly provided by Dr. Tania Watts, University of Toronto. Stock vials were diluted (sterile) between 5×103 to 106 TCID50/mouse depending on the experiment in no more than 30 μl/vial. Diluted stocks were kept sterile and on ice for the duration of the infection procedure. Female and male 6–8 weeks old mice were anesthetized with 5% isofluorane at an induction volume of 3L/min. Mice were removed from the isofluorane mask and using a P20 pipette and sterile tips, mice were given 15 μL of diluted virus per nostril. Mice were administered more isofluorane between each nostril dose to ensure mice did not wake up mid-infection. Mice were placed back into their cage to revive. Weight was monitored daily. Mash food was given when mice lost approximately 10% of their body weight, and fluids were given on approximately day 4.
Rotavirus Infection
Female mice, 6–8 weeks old received 100 μL of 1.33% of Sodium Bicarbonate (Na2CO3) prior to infection with Rotavirus (RV) to neutralize stomach acid. Mice were then inoculated by oral gavage with 100 μL 104 ID50 of EC virus (1:100 of the virus stock 107 ID50/ml in Medium199 (Sigma)), and then monitored for RV clearance by measuring RV antigen in stool. To detect RV antigen with an enzyme linked immunosorbent assay (ELISA), NUNC maxi-sorp 96-well plates were coated with rabbit-α-RV (ABD Serotech, CAT #AHP1360) diluted 1:2000 in TNC (10mM Tris, 100mM NaCl, 1mM CaCl2, Ph 7.4) overnight at 4°C. Plates were then blocked with 5% BLOTTO (5% skim milk powder in TNC) for 2 hr at 37°C. Plates were dumped and patted dry. Fecal samples were added in duplicate at a dilution of 1/2 in 1% BLOTTO, and incubated for 2 hr at 37°C. Plates were washed 5 times with ~200 μL 0.1% TWEEN20/TNC. 50 μl/well of mouse-α-RV monoclonal antibody was added at 1:200 and incubated for 1 hr at 37°C. Plates were washed 3 times as before. 50 μl/well of goat-α-mouse-IgG2b-HRP (Southern Biotech, CAT #1090–05) was added at 1:1000 and incubated for 30 min at 37°C. Plates were washed 3 times as before and developed with 50 μL of 3,3’,5,5’-Tetramethylbenzidine (Bioshop) and stopped with 50 μL H2SO4. Plates were read on a photo-spectrometer at 450nm. An internal control for the RV ELISA was generated using fecal samples combined from several mice and tested for abundance of the RV Ag.
Tritrichomonas musculis Colonization
Purification of T.mu was performed as described (Chudnovskiy et al., 2016). Briefly, the cecal content of T.mu containing mice was harvested into 20ml of sterile PBS at 4°C and filtered through a 70μm cell strainer. Filtered cecal content was then spun at 1400rpm for 7min at 4°C. The supernatant was discarded and the pellet was washed twice with 40ml of sterile PBS. The pellet was then re-suspended in 5ml of 40% percoll and overlaid on 5ml of 80% percoll. The 40/80% percoll (1X sterile PBS) was made from a 90% percoll solution diluted with 10X sterile PBS. The 40/80% percoll gradient centrifugation step was performed at 2500rpm for 20min at 20°C. The percoll interface containing T.mu was collected and spun down at 1400rpm for 7min at 4°C. The pellet was then resuspended in 5ml of sterile PBS and filtered again through a 70μm cell strainer. T.mu were then sorted into sterile PBS on a BD Influx using the 100μm nozzle at 27psi at 4°C. Purity was > 99%. Sorted T.mu were then spun down at 1400rpm for 7min at 4°C and the pellet was resuspended in sterile PBS. Two million sorted T.mu were orally gavaged into 6–8 week old C57BL/6, female mice (Charles River) immediately after the sort. Mice were subjected to EAE three weeks post-infection. To quantify efficiency of infection, cecums were harvested and cut longitudinally. Cecal content was resuspended in 10ml of sterile PBS. Trophozoites were counted using a hemocytometer.
Human study participants and fecal sample collection
A total of 34 female (Average age: 40 years old) and 31 male (Average age: 44 years old) patients with clinically isolated syndrome (CIS) or relapsing-remitting multiple sclerosis (RRMS) (n = 33) (during remission n = 22, during relapse n = 11) and healthy controls (n = 32) were recruited at the University of California, San Francisco (UCSF) MS Center. Fecal samples were collected, shipped within 24 hr at 4°C and stored at −80°C prior to bacterial isolation. All individuals signed a written informed consent in accordance with the sampling procedure approved by the UCSF School of Medicine Institutional Review Board.
Diagnosis of RRMS was made based on the 2010 McDonald criteria (Polman et al., 2011). All patients were treatment naive for disease-modifying therapies (n = 32) or off immunomodulatory treatment for more than 6 years (n = 1) prior to fecal sampling and did not receive steroids within at least 30 days prior to sampling, and fecal samples were collected before administration of steroids or immunomodulatory treatment (Table S2). The diagnosis of relapse was made based on clinical assessment of new/exacerbated symptoms and gadolinium enhancement on magnetic resonance imaging. Clinical disease severity was assessed using the Expanded Disability Status Scale (EDSS) (Neurostatus) (Kappos et al., 2015; Kurtzke, 1983). Age-matched controls sharing the same household were used where available. The inclusion criteria specified no use of antibiotics or cancer therapeutics 3 months prior to the study and excluded a history of autoimmune disorders in healthy controls. Detailed clinical and epidemiological information on patients and controls is available in Table S2.
METHOD DETAILS
Tissue Harvesting
In all cases, mice were euthanized using a CO2 tank attached to an animal cage with a flowmeter to regulate CO2 pressure to 2–31 per minute as per our animal facility’s standard operating procedures. Subsequently, mice were perfused with 30ml of cold PBS (Sigma) via the left ventricle while cutting the right atrium to allow fluid escape. For lung, because we ultimately wished to measure RV-specific IgA ASC in the lungs, we adapted our perfusion protocol to achieve better perfusion of the lungs by pushing the needle up through the left ventricle to the left atrium to preferentially target the pulmonary circulation. Following perfusion the lungs were removed and placed in cold PBS in a 6-well plate. Using a 70μm cell strainer and a 5ml syringe insert, lungs were mashed through the cell strainer mesh to make a single cell suspension. Cells were spun down at 1200rpm for 5 min and resuspended in 2ml of 80% Percoll (GE Healthcare) and 2ml of 40% Percoll was laid gently on top. Tubes were centrifuged at 1600 rpm (585 RCFxg) for 25 min to separate the fat on the top layer, lymphocytes at the midpoint between the 80% and 40% solutions, and debris in the bottom pellet. Approximately 1ml was collected from the midline to collect the lymphocytes and placed in a new tube with PBS and centrifuged at 1200rpm for 5 min. Cells were washed once more and then used for downstream applications.
For BM isolation, following euthanasia and perfusion, the skin of the mouse’s leg was removed and the leg was detached at the hip joint. Using scissors and gauze, the muscle tissue was cleaned off the femur and tibia. The ends of each bone that forms the joint were removed and both the femur and tibia were placed in a punctured 0.6ml PCR tube placed at the opening of a 1.5ml microtube with 200 μL of PBS (Sigma). Samples were pulse spun such that the marrow from each bone was collected at the bottom of the microtube. The bones were discarded and the BM pellets were resuspended in RBC lysis buffer (155mM NH4Cl, 12mM NaHCO3, 0.1mM EDTA) for 5 min on ice. Lysis buffer containing the cells was transferred to a 50mLtube containing 10ml of PBS to stop the lysis reaction and the cells were spun down at 1200rpm for 5 min. Samples were washed once more using the same method. Cells were kept on ice until ready for further use.
Blood was collected from the saphenous leg vein into capillary tubes. Samples were spun down at 10,000 rpm (9,000 RCFxg) for 5–7 min and serum was collected and transferred into autoclaved 0.6ml microtubes. All samples were frozen at −20°C until further use. For fecal matter preparation, 2–3 pellets of fecal matter were collected in 1.5ml microtubes. All fecal matter was frozen at −20°C until further use then subsequently thawed and weighed prior to downstream applications.
Axillary and inguinal Lymph Nodes (AxLN or iLN) as well as spleen (Sp) were collected and placed into cold PBS in a 6-well plate. Using a 70 μm cell strainer and the back of an insert of a 5ml syringe, LNs were mashed through the mesh of the cell strainer to make a single cell suspension. Red blood cell lysis was performed at this point for spleen only. Cells were washed once in cold PBS and centrifuged at 1200rpm (329 RCFxg) for 5 min. Cells were kept on ice until ready for further use.
Brain (Br) and spinal cord (Sc) were homogenized first using a 70 μm strainer and a 10ml syringe, then cell suspensions were incubated with the addition of 60 μg/ml DNaseI for 45 min at 37°C. We avoided using Collagenase D for Br digestion because it resulted in decreased PC recovery/viability in our hands. Lymphocytes were purified using a 30% Percoll solution and resuspended in PBS for counting and flow cytometry staining or in complete RPMI media (10% FBS, L-glutamine, sodium pyruvate, penicillin G, streptomycin sulfate, and β-mercaptoethanol) for ELISPOT assay.
SILP preparations were performed as previously described (Fritz et al., 2011). Briefly, small intestines were dissected and cleaned in situ of mesenteric fat and Peyer’s patches were removed. Small pieces of the intestine were then thoroughly washed and EDTA solution was used to remove IELs. The remaining LP fraction was then digested with collagenase IV (Sigma-Aldrich, USA) and lymphocytes were enriched by Percoll gradient (GE Healthcare, Sweden). Given the inherent variability in gut preparations, we enumerated cellular compartments based on frequency rather than absolute numbers and complemented these findings with immunofluorescence.
Flow Cytometry
Cells were washed with ice-cold PBS containing 2% FBS (Wisent Inc, Canada) and prior to antibody staining a live/dead stain was applied using a fixable aqua dead cell stain kit (Molecular Probes). Subsequently, cells were incubated with 1 mg/ml of a rat anti-mouse CD16/CD32 antibody (“Fc-block,” clone: 2.4G2 made in house) to block non-specific staining for 15 min at 4°C. Predetermined concentrations of fluorochrome labeled antibodies were then added in a total volume of 100μl, thoroughly mixed with the cells and incubated for 15 min at 4°C. The following antibodies were used in different combinations among 3 panels, all of which were purchased from Ebiosciences unless otherwise noted: rat anti-mouse CD19-APC (1D3), rat anti-mouse CD4-PECy7 (GK1.5), CD8-PECy7, F4/80-PECy7 (collectively used as a “dump” gate), rat anti-mouse CD45R (B220)-eFluor450 (RA3–6B2), and rat anti-mouse CD138-PercpCy5.5 (281–2, BD Biosciences). After washing with FACS buffer, cells were fixed and permeabilized using a cytofix/cytoperm kit from BD Biosciences according to the manufacturer’s protocol. Intracellular staining was then performed for 30 min at 4°C using the following antibodies: rat anti-mouse IgA-biotin (11–44-2) followed by Streptavidin-APC-eF780, Ki67-eFluor450, IgG-BV450 or IgA-FITC. In the case of T cell cytokine detection, the following antibodies were used: anti-mouse IFNγ PE, anti-mouse IL17 PerCPCy5.5 and anti-mouse GM-CSF FITC. Cells were then washed twice with Perm/Wash buffer and resuspended in FACS buffer prior to flow cytometric acquisition using either a Fortessa or an LSR-II instrument (BD Biosciences). Acquired data was analyzed and processed using FlowJo (Tree Star Inc.).
Immunofluorescence microscopy
Small intestinal tissues were obtained from mouse dissection before EAE induction (D0), during the peak of the disease (D15), and during the chronic phase (D21 or D23). Briefly, after euthanasia and perfusion, small intestines were removed and placed on plastic wrap to clean out the fecal content. At the midpoint, curved forceps were used to clear the feces from the intestine and a 1cm piece was cut out, avoiding the Peyer’s patches. The piece was placed in a histology tray and covered in OCT (Sakura Finetek) and was frozen in 2-methyl butane cooled on dry ice. Trays were wrapped in aluminum foil and stored at −80°C until further use. 5 μm frozen sections were subsequently cut with a Leica CM3050 cryostat in preparation for acetone fixation and staining. For a more comprehensive examination of the tissues, five slides were prepared from each sample with two sections on each slide. Defrosted and PBS-re-hydrated tissues were subjected to Fc receptor blocking with Superblock solution in TBS (Thermo Scientific, USA) followed by primary antibody staining with rat-anti-mouse antibodies. Data depicted were obtained by staining with IgA-FITC, CD138-PE, and CD8α-APC, although some experiments used CD138-APC with CD8α-PE, or IL10 PE. After nuclear staining with DAPI, the mounted slides were visualized by microscopy using a LeicaDMRA2 microscope with OpenLab software where each fluorochrome was assigned a color (DAPI = yellow, YFP/FITC = green, APC = blue, PE = red). Single RGB images were saved as TIFF files. In Photoshop (version CC), we cropped the original images using a 1000px x 1000px marquis and zoom images were derived from these cropped images using a 250px x 250px marquis. Subsequently, brightness was adjusted for each fluorochrome uniformly across samples. TIFFS were then merged as overlays on ImageJ 1.15 s (National Institute of Health, USA) and using the cell counter function, the number of CD138+ or IgA+ cells per every 10 CD8α+ cells was quantified. We confirmed first, that numbers of CD8α+ cells in the SILP did not change during EAE by Immunofluorescence (not shown). In case of IL10/IgA counts, number of cell counts were normalized according to the area. In order to account for the variability among sections within a tissue, a minimum of two to six images (technical replicates) on each biological replicate were acquired from each sample and an average cell count was taken for the final analysis.
Hematoxylin & Eosin and Luxol Fast Blue staining
Mice whose spinal columns were harvested for histology were euthanized with CO2 and intracardially perfused with PBS. The spinal columns were excised and post-fixed in 10% buffered formalin for 1 week prior to processed into paraffin. Seven micron paraffin coronal sections of mouse spinal cord were mounted on Superfrost Plus glass slides (Knittel Glass) and dried overnight at 37°C. Paraffin sections were deparaffinated in xylene and rehydrated through a series of ethanol. Histology was performed using standard Hematoxylin & Eosin (H&E) to visualize immune cell infiltration, and Luxol Fast Blue (LFB), which stains myelin and allow us to visualize areas of demyelination. RGB images of H&E and LFB stains from thoracic spinal cord sections were acquired at 4X magnification using a light microscope (Zeiss Axioscope). Quantitative analysis of stains was performed using ImageJ 1.15 s in a blinded manner. The RGB images were separated into single color channels using the color deconvolution plugin. Single color channel for hematoxylin and LFB were subjected to thresholding followed by the Area Fraction measurement on the region of interest (total white matter area). Staining is expressed as percentage stained area of total white matter area.
ELISPOT analysis
Membrane plates (Milipore HA clear plates, sterile 0.45um surfactant-free mixed cellulose ester membrane MSHAS4510) were coated (sterile) with goat-α-mouse -Ig diluted 1:1000 for the total Ig ELISPOT (Southern Biotechnologies #1010–01, 1mg/ml) diluted 1:1000, or coated with rabbit-anti-RV polyclonal antibody diluted 1:1000 for the RV-specific Ig ELISPOT (ABD Serotech, CAT #AHP1360) diluted 1:1000, and placed at 4° C overnight. For the commensal-reactive ELISPOT assay, high binding ELISPOT plates (Multiscreen HTS) were coated with autologous heat killed inactivated Fecal matter (1mg/ml). Plates were blocked the next morning with 10% FBS/RPMI (Sigma) for at least 2 hr while tissues were being prepared. Starting with 1 million cells, single cell suspensions were loaded onto the plate at serial 2-fold dilutions in FBS/RPMI, and left overnight. Cells were removed the next morning and plates were washed with 0.1% TWEEN-20/PBS 3x, leaving the third wash in the plate while rotating on an orbital shaker for 15 min and washed twice more. HRP conjugated IgA or IgG detection antibodies were subsequently added. In some cases, two-color ELISPOTs were done with the simultaneous addition of IgA-HRP and IgG-AP. Plates were washed again as before and developed while covered with aluminum foil for at least 9 min or until spots were visible using ImmPACT AEC Peroxidase (for HRP-conjugated Ab) (Vector Laboratories, CAT #SK-4205) or Vector Blue (for AP-conjugated Ab) (Vector Lavoratories, CAT #SK-4300) substrates. For two-color ELISPOTS, plates were first developed with AEC, washed with distilled water, rinse once with Tris-HCl pH8.2–8.5 buffer, and developed again with Vector Blue. Plates were dried overnight and spots were counted using a light microscope taking into account the original cell dilution.
Parabiosis: Surgical Attachment of Two Female Mice
Sterile technique was used and animals were kept warm with a heating pad throughout the surgery. The entire experiment was conducted according to a published methodology (Rauch et al., 2012). Mice were weight-matched and caged for 1 week together prior to surgery to acclimate. On surgery day, analgesia was administered (meloxicam, 1mg/kg). Mice were anesthesized with isofluorane (2%−3% mixed with oxygen). After shaving the corresponding lateral aspects of each mouse, matching skin incisions were made from the olecranon to the knee joint of each mouse, and the subcutaneous fascia was bluntly dissected to create approximately 1/2 cm of free skin. The latissimus dorsi and abdominal external oblique muscles on each mouse were split. The peritoneal cavities were not penetrated. The olecranon was attached by a single monofilament suture and tie, and the dorsal and ventral skins were approximated by continuous suture. Because the union joins the bones, there was no need to use a flexible cohesive bandage post-surgery. This method gave firm support to both animals, which prevented the strain on the sutures of the skin and abdominal walls. To alleviate pain, buprenorphine (0.1mg/kg, s.c.) was administered every 12 hr for the first 72 hr post procedure. Wounds generally healed within a few days. The parabionts were placed in a cage (1 parabiont pair/cage) for the remainder of the experiment (1 month).
Abdominal Surgery for Intestinal Photoconversion in Kaede mice
In order to induce photoconversion of cells in the small intestine in Kaede mice, abdominal surgery to expose the intestines was necessary. At 6–8 weeks of age, survival surgery was performed where an incision was made on the midline of the abdomen to carefully expose the small intestines of Kaede or control mice (non-Kaede C57BL/6 mice). Hair was removed from the abdomen with an electric shaver one day prior to surgery. Mice were anesthetized with 2%−3% isofluorane and their skin was subsequently sterilized with iodine/alcohol twice each and alternating using sterile gauze. Sterile technique was used throughout the surgery and mice were kept on a heat pad throughout. A drape with a hole in the middle was placed over the mouse and an incision was made into the abdomen, through the hole. A piece of sterile aluminum foil was placed on top of the sterile drape and used to cover the abdomen, in order to protect the surrounding tissue from exposure to the violet light. The small intestine was carefully pulled through the incision on top of the foil and using a handheld LED light, the intestines were exposed to violet light for 2× 90 s for a total of 3 min, with the light source ~15cm away. These conditions were selected based on pilot experiments (data not shown). We noted that if the light source was too close or maintained for too long next to tissue, significant tissue necrosis occurred. Conversely, having the light source too far away did not allow for efficient photoconversion. During photoconversion, warmed, sterile PBS was continuously applied to the intestine with a syringe to keep the tissue hydrated until it was replaced back into the abdomen. The muscle layer and skin layer was closed with 4–0 absorbable interrupted sutures. The mouse was given 5 mg/kg Ketoprofen subcutaneously and 0.1mg/kg Buprenorphine subcutaneously before surgery and a second dose of Buprenorphine at the end of the day. The mice received subsequent equivalent doses of Ketoprohen once per day and Buprenorpine twice per day for one additional day after surgery. An LSR Fortessa flow cytometry machine from BD Bioscience at the University of Toronto Immunology Flow Cytometry Facility was used to analyze tissues from Kaede mice. The machine contained a laser and filter that was able to visualize photoconverted Kaede-Red, which has a similar wavelength to mCherry at 561nm. Unconverted Kaede-Green was visualized using the FITC channel with a wavelength of 488nm. Since the Kaede fluorescent protein is under transcriptional control of actin, Kaede-Red+ populations were Kaede-Red+ and Kaede-Greenint.
PB and/or PC Transfer
Prdm1-YFP SILP cells were sorted based on the expression of YFP separating two populations based on singlets (FSC-W versus FSC-H) followed by a generous lymphocyte gate (FSC-A versus SSC-A), followed by live cell gating (Aqua-) followed by elimination of irrelevant cells (Dump-: The “dump” gate are cells that stained positive for CD19-APC (1D3), rat anti-mouse CD4-PECy7 (GK1.5), CD8-PECy7, F4/80-PECy7) then sorted as YFP+B220- (PB and/or PC) using a FACS ARIA Sorter machine. Purity of samples was confirmed by post-sort analysis as well as the fact that most of the sorted cells were IgA+, Ki67- (Figure S2). Prdm1fl/fl x Cd19cre or Jht−/− mice received IV tail injections of B cells or PB and/or PC at the onset of EAE and 2 more injections spaced 3–4 days apart thereafter and compared with PBS only controls. Depending on the experiment, mice received intravenous injections of 4000–10000 YFP+B220- PB and/or PC at the onset of the disease and 2 more injections every 3–4 days thereafter, until they reached disease peak. We injected more B cells than PB and/or PC in order to prove the stringency of PB and/or PC efficacy compared to a separate cell subset from the same anatomical compartment. Mice were scored daily for evidence of clinical disease until the chronic phase of the disease, and then tissues were harvested to confirm presence of YFP+ cells by FACS.
Adoptive transfer EAE in C57BL6 mice
Donor C57BL/6 mice were actively immunized with 100 μg MOG35–55 peptide emulsified in CFA (Difco). At day 9 after immunization, lymphocytes from the spleen, axillary, inguinal, and cervical lymph nodes were collected and cultured in vitro for 72–84 hr in complete T cell media (RPMI1640, 10% fetal bovine serum (GIBCO), 100 U/ml penicillin, 100 μg/ml streptomycin, 1X Glutamax (GIBCO), 1X MEM non-essential amino acid (GIBCO); 1mM sodium pyruvate (Sigma), 10mM HEPES (Lonza), and 50 μM β-mercaptoethanol). For T cell skewing, MOG35–55 (20 μg/ml), rmIL-6 (20 ng/ml, Peprotech), rmIL-12p70 (3 ng/ml, Peprotech), rmIL-23 (20 ng/ml, R&D), and human TGFb (4 ng/ml, Peprotech) were added to the media. 40 million cells from cultured lymphocytes were transferred intraperitoneally to C57BL/6 or BAFF-Tg recipient mice. At day 2 post-transfer, recipients also received 200ng of pertussis toxin.
Isolation of Fecal Bacteria from human samples
Total bacteria were isolated from patient or control stool samples by suspending ~100 mg stool sample in 1.5 mL Phosphate Buffered Saline (PBS), passing it three times through a 40 μm cell strainer and washing twice with 1.5 mL PBS by spinning at 8000x g. Bacteria were quantified photometrically at an optical density of 600 nm (OD600) and stored at a concentration of 109 CFU/ml in a solution of 30% glycerol in Luria Bertani (LB) medium.
Bacterial Flow Cytometry
Fecal-bound IgA was assessed using bacterial flow cytometry for IgA-bound bacteria (BUGFlow). BUGFlow was performed as previously described (Palm et al., 2014). In brief, one million isolated fecal bacteria per individual were washed in 1 mL staining buffer (PBS containing 1% (w/v) Bovine Serum Albumin (BSA, Sigma) pelleted (5 min, 8,000x g, 4°C), and blocked for 20 min in 300 μL blocking buffer (20% Normal Mouse Serum (Jackson ImmunoResearch) in staining buffer) on ice. Next, samples were stained with 100ul staining buffer containing PE-conjugated anti-human IgA (1:10, Miltenyi Biotec clone IS11–8E10) or isotype control (mouse IgG1-PE, 1:10, Miltenyi Biotec) for 30 min on ice. Samples were then washed 3 times with staining buffer and fixed in 2% paraformaldehyde (PFA) in PBS at 4°C before flow cytometric analysis on a Beckton Dickinson (Viladomiu et al., 2017) LSR Fortessa. Bacteria were gated based on forward and side scatter and the gating strategy was verified by SYTOBC (Invitrogen). An isotype control was used to identify the stained population. Analysis of IgA binding was done using FlowJo (v10.1).
Quantitative IgA ELISAs (human and mouse)
For human IgA, total IgA-bound bacteria (low- and high-affinity IgA) were quantified by ELISA. One million isolated fecal bacteria per individual were coated in duplicates and probed for IgA using a commercially available quantitative ELISA Assay (Human IgA ELISA Kit, Abcam). For mouse IgA, 100–200μl of mouse blood was collected and serum was isolated using Microvette CV 300 Z serum separation tubes (SARSTEDT Germany) according to the manufacturer’s protocol. Serum was stored at −80°C. Fecal pellets were collected, weighed and 500μl of sterile PBS was added. Pellets were vortexed on max speed at 4°C for 10min. The dissociated pellet was spun at 12000rpm for 10min and the fecal water supernatant was collected. Fecal water was stored at −80°C. Immunoglobulins were absorbed on high-bound Nunc MaxiSorp ELISA plates (BioLegend) and anti-IgA ELISA was performed using anti-mouse IgA-HRP antibody (Southern Biotech).
Statistical Analysis
Statistical analyses were performed using Graphpad Prism software 7 (GraphPad Software Inc.). The variability of distribution was assessed by Shapiro-Wilk normality test. Student’s t test was used for data with a normal distribution. The Mann-Whitney U test was used for non-Gaussian distributed data and 2-way ANOVA test was used to evaluate differences in clinical disease over time between groups. All tests were performed using 2-tailed analysis. Log-rank (Mantel-Cox test) was performed for evaluating differences in survival over time. Significance cutoff was set at p < 0.05 at a 95% confidence level. Most graphs provide mean values with SD unless stated differently.
DATA AND SOFTWARE AVAILABILITY
A detailed list of the experiments and the clinical data from patients to support our results are listed in Tables S1 and S2. Original unprocessed data can be accessed via Mendeley Data: https://doi.org/10.17632/zks84bv2wj.1
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Anti-Mouse IgA-HRP | Southern Biotech | RRID:AB_2714213 |
| Goat Anti-Mouse Ig, Human ads-UNLB | Southern Biotech | RRID:AB_2687542 |
| Goat Anti-Mouse IgG-AP | Southern Biotech | RRID:AB_2692322 |
| Rat Anti-Mouse CD19 APC | ThermoFisher | RRID:AB_1659676 |
| Rat Anti-Mouse CD4 PECy7 | ThermoFisher | RRID:AB_469578 |
| Rat Anti-Mouse CD8 PeCy7 | ThermoFisher | RRID:AB_469584 |
| Rat Anti-Mouse F4/80 PeCy7 | ThermoFisher | RRID:AB_469653 |
| Rat Anti-Mouse B220 ef450 | ThermoFisher | RRID:AB_154876 |
| Rat Anti-Mouse CD138 PerCp Cy5.5 | Biolegend | RRID:AB_2561600 |
| Rat Anti-Mouse IgA Biotin | ThermoFisher | RRID:AB_466863 |
| Streptavidin APCef780 | ThermoFisher | RRID:AB_10366688 |
| Rat Anti-Mouse Ki67 ef450 | ThermoFisher | RRID:AB_11149124 |
| Rat Anti-Mouse IgG BV421 | Biolegend | RRID:AB_10900419 |
| Rat Anti-Mouse IgA FITC | Southern Biotechnology | Cat. #1165–02 |
| Goat Anti-Mouse IgA PE | Southern Biotechnology | Cat, #1106–09 |
| Rat Anti-Mouse CD19 BV605 | Biolegend | RRID:AB_11204087 |
| Rat Anti-Mouse CD138 APC | BD | RRID:AB_1645216 |
| Rat Anti-Mouse CD8 PE | eBioscience | RRID:AB_465531 |
| Mouse anti-human IgA PE | Miltenyi | RRID:AB_1036158 |
| Mouse IgG1 PE Isotype Control | Miltenyi | RRID:AB_871707 |
| Chemical Peptides and recombinant proteins | ||
| hrMOG | Produced in house | N/A |
| MOG35–55 | Canpeptide | AA Sequence: MEVGWYRSPFSRVVHLYRNGK |
| PMA | Sigma Aldrich | Cat. #8139–5mg |
| Ionomycim | Sigma Aldrich | Cat. #I9657–1mg |
| Brefeldin A | eBiosciences | Cat. #00–4506-51 |
| Pertussin Toxin | List Biological Laboratory | Cat. #181 |
| M. Tuberculosis H37 Ra | BD | Cat. #231141 |
| Commercial Assays | ||
| Mouse IFNg Elisa kit | Invitrogen | Cat. #88–7314-88 |
| Mouse IL17 Elisa Kit | Invitrogen | Cat. #88–7371-88 |
| Human IgA Elisa | Abcam | Cat. #Ab137980 |
| Deposited Data | ||
| Raw Data File for EAE Clinical Scores | Mendeley Data Set | https://doi.org/10.17632/zks84bv2wj.1 |
| Other | ||
| AEC Peroxidase Substrate Kit | Vector Laboratories | Cat. #SK-4200 |
| Vector Blue Substrate Kit | Vector Laboratories | Cat. #SK-5300 |
| Collagenase IV | Sigma | Cat. #C5138 |
| Collagenase D | Roche | Cat. #11088882001 |
| Live/Dead Aqua Dye | Invitrogen | Cat. #L34957 |
| SYTO BC | Invitrogen | Cat.#S34855 |
| Fix/Perm Buffer Kit | BD | Cat. #554714 |
| DNase I | Roche | Cat. #10104154001 |
| OCT | Thermo Fisher Scientific | Cat. #6769006 |
| Percoll | Thermo Fisher Scientific | Cat. #17–0891-01 |
| TMB | Bioshop | Cat. #333.100 |
| Luxol Fast Blue | Sigma | Cat. #S3382 |
| Lithium Carbonate | Sigma | Cat. #L4283 |
| Harris Haematoxylin | Sigma | Cat. #HHS16 |
| Eosin Y | Sigma | Cat. #E4382 |
| Entellan | Merck Millipore | Cat. #107961 |
| High Binding Elispot Plates | Millipore | Cat. #M5IPS4W10 |
| Regular Elispot Plates | Millipore | Cat. #M5HA545 |
| Adjuvant Incomplete Freund | BD | Cat. #263910 |
ACKNOWLEDGMENTS
We would like to acknowledge Dr. Andy Luster for sending us the Kaede mice, with permission from Drs. Yoshihiro Miwa and Tomura Kanagawa and permission from Dr. Casey Weaver to use 10BiT mice. We also thank Dr. Rafael Case-llas for AID-YFP mice and Dr. Harry Greenberg for kindly providing the EC Rotavirus. We are grateful for the expertise of Dionne White in the Faculty of Medicine Flow Cytometry facility and for our colleagues in the Faculty of Medicine Division of Comparative Medicine for assistance with EAE experiments. We would like to acknowledge Dr. Bruce Cree, Dr. Jennifer Graves and Sneha Singh for patient recruitment and Drs. Xiaoyuan Zhou and Chris Graham Fenton for computational expertise. A.M. is supported by an Operating Grant from the Canadian Institutes of Health Research (CIHR 388337) and is the Tier 2 Canadian Research Chair in Mucosal Immunology. J.L.G., A.B.-O., and A.P. are supported by a collaborative team grant (“B cells in MS”) from the MS Society of Canada Research Foundation, and A.P. is also supported by a Tier 1 Canada Research Chair in Multiple Sclerosis. A.-K.P. is supported by a Swiss National Science Foundation fellowship (P2SKP3_164938/1 and P300PB_177927/1) and a National Multiple Sclerosis Society fellowship (NMSS Kathleen C. Moore Fellowship: FG-1708–28871). D.A. is supported by NIH(R01-AI113543). D.G.B. receives funding from the Canadian Institutes of Health Research (CIHR) Foundation Grant (FDN148386) and from NIH (AI085043SSZ). S.S.Z. receives research receives funding from the NIH (RO1 NS092835 and R21 NS108159–01), the NMSS (RG1701–26628), The Maisin Foundation, Biogen, and Celgene. S.E.B. holds the Heidrich Family and Friends endowed Chair in Neurology at UCSF and is supported by the US National MS Society (CA1072-A-7), the US Department of Defense (W81XWH-15–1-0652), and the Valhalla Charitable Foundation. J.L.G.’s lab is supported by a CIHR Foundation grant (FDN1552) and an MS Society of Canada grant (EGID3194).
J.L.G. is a consultant for Roche (Canada) and currently holds grants with Novartis, EMD Serono, and Roche. S.S.Z. is Deputy Editor of Neurology, Neuroimmunology and Neuroinflammation. He has served as a consultant and received honoraria from Biogen-Idec, EMD-Serono, Genzyme, Novartis, Roche/Genen-tech, and Teva Pharmaceuticals, Inc. He has served on Data Safety Monitoring Boards for Lilly, BioMS, Teva, and Opexa Therapeutics. A.B.O. has received consulting fees and/or grant support from Actelion/Jansen, Atara Biotherapeutics, Biogen Idec, Celgene/Receptos, Genentech/Roche, MAPI, Med- immune, Merck/EMD Serono, Novartis, and Sanofi-Genzyme.
Footnotes
DECLARATION OF INTERESTS
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.11.035.
REFERENCES
- Amanna IJ, and Slifka MK (2010). Mechanisms that determine plasma cell lifespan and the duration of humoral immunity. Immunol. Rev 236, 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscarinu MC, Cerasoli B, Annibali V, Policano C, Lionetto L, Capi M, Mechelli R, Romano S, Fornasiero A, Mattei G, et al. (2017). Altered intestinal permeability in patients with relapsing-remitting multiple sclerosis: A pilot study. Mult. Scler 23, 442–46. [DOI] [PubMed] [Google Scholar]
- Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, Kanner R, Bencosme Y, Lee YK, Hauser SL, et al. (2017). Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 114, 10713–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, and Berek C. (2013). The establishment of the plasma cell survival niche in the bone marrow. Immunol. Rev 251, 177–188. [DOI] [PubMed] [Google Scholar]
- Chudnovskiy A, Mortha A, Kana V, Kennard A, Ramirez JD, Rahman A, Remark R, Mogno I, Ng R, Gnjatic S, et al. (2016). Host-protozoan interactions protect from mucosal infections through activation of the inflammasome. Cell 167, 444–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montaño C, Feigenbaum L, Wilson P, Janz S, Papavasiliou FN, and Casellas R. (2007). Regulation of AID expression in the immune response. J. Exp. Med 204, 1145–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CR, Champhekar A, Tullius MV, Dillon BJ, Zhen A, de la Fuente JR, Herskovitz J, Elsaesser H, Snell LM, Wilson EB, et al. (2016). Type I and type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 12, e1005356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyk-Melody JE, Garrison AE, Brunnert SR, Weidner JR, Shen F, Shelton BA, and Mudgett JS (1998). Experimental autoimmune encephalomyelitis is exacerbated in mice lacking the NOS2 gene. J. Immunol 160, 2940–2946. [PubMed] [Google Scholar]
- Franco MA, and Greenberg HB (1999). Immunity to rotavirus infection in mice. J. Infect. Dis 179 (Suppl 3), S466–S469. [DOI] [PubMed] [Google Scholar]
- Fritz JH, Rojas OL, Simard N, McCarthy DD, Hapfelmeier S, Rubino S, Robertson SJ, Larijani M, Gosselin J, Ivanov II, et al. (2011). Acquisition of a multifunctional IgA+ plasma cell phenotype in the gut. Nature 481, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Zimmermann K, Graw F, Slack E, Rusert P, Ledergerber B, Bossart W, Weber R, Thurnheer MC, Battegay M, et al. ; Swiss HIV Cohort Study (2011). Systemic antibody responses to gut commensal bacteria during chronic HIV-1 infection. Gut 60, 1506–1519. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, et al. ; HERMES Trial Group (2008). B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med 358, 676–688. [DOI] [PubMed] [Google Scholar]
- Iverson GM, von Mühlen CA, Staub HL, Lassen AJ, Binder W, and Norman GL (2006). Patients with atherosclerotic syndrome, negative in anti-cardiolipin assays, make IgA autoantibodies that preferentially target domain 4 of beta2-GPI. J. Autoimmun 27, 266–271. [DOI] [PubMed] [Google Scholar]
- Jaimes MC, Rojas OL, Kunkel EJ, Lazarus NH, Soler D, Butcher EC, Bass D, Angel J, Franco MA, and Greenberg HB (2004). Maturation and trafficking markers on rotavirus-specific B cells during acute infection and convalescence in children. J. Virol 78, 10967–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, Madjovski A, Ruhrmann S, Faigle W, Frauenknecht K, et al. (2018). Memory B cells activate brain-homing, autoreactive CD4(+) T cells in multiple sclerosis. Cell 175, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki A, Miyake S, Saga R, Chiba A, Mochizuki H, and Yamamura T. (2016). Gut environment-induced intraepithelial autoreactive CD4(+) T cells suppress central nervous system autoimmunity via LAG-3. Nat. Commun 7, 11639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Hartung H-P, Freedman MS, Boyko A, Radü EW, Mikol DD, Lamarine M, Hyvert Y, Freudensprung U, Plitz T, and van Beek J; ATAMS Study Group (2014). Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 13, 353–363. [DOI] [PubMed] [Google Scholar]
- Kappos L, D’Souza M, Lechner-Scott J, and Lienert C. (2015). On the origin of neurostatus. Mult. Scler. Relat. Disord 4, 182–185. [DOI] [PubMed] [Google Scholar]
- Kurtzke JF (1983). Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology 33, 1444–1452. [DOI] [PubMed] [Google Scholar]
- Lemke A, Kraft M, Roth K, Riedel R, Lammerding D, and Hauser AE (2016). Long-lived plasma cells are generated in mucosal immune responses and contribute to the bone marrow plasma cell pool in mice. Mucosal Immunol. 9, 83–97. [DOI] [PubMed] [Google Scholar]
- Lino AC, Dang VD, Lampropoulou V, Welle A, Joedicke J, Pohar J, Simon Q, Thalmensi J, Baures A, Fluhler V, et al. (2018). LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity 49, 120–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, Kallies A, Nutt SL, Sakaguchi S, Takeda K, et al. (2014). Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity 41, 1040–1051. [DOI] [PubMed] [Google Scholar]
- McCarthy DD, Kujawa J, Wilson C, Papandile A, Poreci U, Porfilio EA, Ward L, Lawson MA, Macpherson AJ, McCoy KD, et al. (2011). Mice overexpressing BAFF develop a commensal flora-dependent, IgA-associated nephropathy. J. Clin. Invest 121, 3991–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei HE, Yoshida T, Sime W, Hiepe F, Thiele K, Manz RA, Radbruch A, and Dörner T. (2009). Blood-borne human plasma cells in steady state are derived from mucosal immune responses. Blood 113, 2461–2469. [DOI] [PubMed] [Google Scholar]
- Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, Tarakhovsky A, Nutt SL, Jaritz M, and Busslinger M. (2016). Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat. Immunol 17, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouri M, Bredberg A, Weström B, and Lavasani S. (2014). Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS ONE 9, e106335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Hodgkin PD, Tarlinton DM, and Corcoran LM (2015). The generation of antibody-secreting plasma cells. Nat. Rev. Immunol 15, 160–171. [DOI] [PubMed] [Google Scholar]
- Ochoa-Repáraz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, and Kasper LH (2010). A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 3, 487–495. [DOI] [PubMed] [Google Scholar]
- Palm NW, deZoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158,1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, and Cross AH (2010). Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch. Neurol 67, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikor NB, Astarita JL, Summers-Deluca L, Galicia G, Qu J, Ward LA, Armstrong S, Dominguez CX, Malhotra D, Heiden B, et al. (2015). Integration of Th17- and lymphotoxin-derived signals initiates meningeal-resident stromal cell remodeling to propagate neuroinflammation. Immunity 43, 1160–1173. [DOI] [PubMed] [Google Scholar]
- Pollok K, Mothes R, Ulbricht C, Liebheit A, Gerken JD, Uhlmann S, Paul F, Niesner R, Radbruch H, and Hauser AE (2017). The chronically inflamed central nervous system provides niches for long-lived plasma cells. Acta Neuropathol. Commun 5, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, et al. (2011). Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol 69, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracht K, Meinzinger J, Daum P, Schulz SR, Reimer D, Hauke M, Roth E, Mielenz D, Berek C, Côrte-Real J, et al. (2017). A new staining protocol for detection of murine antibody-secreting plasma cell subsets by flow cytometry. Eur. J. Immunol 47, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, Tiglao E, Figueiredo JL, Iwamoto Y, Theurl I, et al. (2012). Innate response activator B cells protect against microbial sepsis. Science 335, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergott RC, Bennett JL, Rieckmann P, Montalban X, Mikol D, Freuden-sprung U, Plitz T, and van Beek J; ATON Trial Group (2015). ATON: results from a Phase II randomized trial of the B-cell-targeting agent atacicept in patients with optic neuritis. J. Neurol. Sci 351, 174–178. [DOI] [PubMed] [Google Scholar]
- Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, Willimsky G, Ammirante M, Strasner A, Hansel DE, et al. (2015). Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 521, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, Vrbanac AF, Li W, Perkins A, Matsutani T, et al. (2017). Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 551, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, et al. (2014). IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 507, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slocombe T, Brown S, Miles K, Gray M, Barr TA, and Gray D. (2013). Plasma cell homeostasis: the effects of chronic antigen stimulation and inflammation. J. Immunol 191, 3128–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, and Becher B. (2017). Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity 46, 245–260. [DOI] [PubMed] [Google Scholar]
- Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, and Masopust D. (2015). Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern JN, Yaari G, Vander Heiden JA, Church G, Donahue WF, Hint-zen RQ, Huttner AJ, Laman JD, Nagra RM, Nylander A, et al. (2014). B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med 6, 248ra107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tak PP, Thurlings RM, Rossier C, Nestorov I, Dimic A, Mircetic V, Rischmueller M, Nasonov E, Shmidt E, Emery P, and Munafo A. (2008). Atacicept in patients with rheumatoid arthritis: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating, single- and repeated-dose study. Arthritis Rheum. 58, 61–72. [DOI] [PubMed] [Google Scholar]
- Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, and Kanagawa O. (2008). Monitoring cellular movement in vivo with photo-convertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. USA 105, 10871–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, et al. (2017). IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17- dependent inflammation. Sci. Transl. Med 9, eaaf9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, Cole SD, Misic AM, Beiting DP, and Allman D. (2018). Commensal microbes induce serum IgA responses that protect against polymicrobial sepsis. Cell Host Microbe 23, 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman KR, Franco MA, Kuklin NA, Rott LS, Butcher EC, and Greenberg HB (2002). Correlation of tissue distribution, developmental phenotype, and intestinal homing receptor expression of antigen-specific B cells during the murine anti-rotavirus immune response. J. Immunol 168, 2173–2181. [DOI] [PubMed] [Google Scholar]
- Zhou X, Xia Z, Lan Q, Wang J, Su W, Han YP, Fan H, Liu Z, Stohl W, and Zheng SG (2011). BAFF promotes Th17 cells and aggravates experimental autoimmune encephalomyelitis. PLoS ONE 6, e23629. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed list of the experiments and the clinical data from patients to support our results are listed in Tables S1 and S2. Original unprocessed data can be accessed via Mendeley Data: https://doi.org/10.17632/zks84bv2wj.1