

Abstract
Histone deacetylase 6 (HDAC6) is emerging as a target for inhibition in therapeutic strategies aimed at treating cancer, neurodegenerative disease, and other disorders. Among the metal-dependent HDAC isozymes, HDAC6 is unique in that it contains two catalytic domains, CD1 and CD2. CD2 is a tubulin deacetylase and a tau deacetylase, and the development of HDAC6-selective inhibitors has focused exclusively on this domain. In contrast, there is a dearth of structural and functional information regarding CD1, which exhibits much narrower substrate specificity in comparison with CD2. As the first step in addressing the CD1 information gap, we now present X-ray crystal structures of seven inhibitor complexes with wild-type, Y363F, and K330L HDAC6 CD1. These structures broaden our understanding of molecular features important for catalysis and inhibitor binding. The active site of HDAC6 CD1 is wider than that of CD2, which is unexpected in view of the narrow substrate specificity of CD1. Amino acid substitutions between HDAC6 CD1 and CD2, as well as conformational differences in conserved residues, define striking differences in active site contours. Catalytic activity measurements with HDAC6 CD1 confirm the preference for peptide substrates containing C-terminal acetyllysine residues. However, these measurements also show that CD1 exhibits weak activity for peptide substrates bearing certain small amino acids on the carboxyl side of the scissile acetyllysine residue. Taken together, these results establish a foundation for understanding the structural basis of HDAC6 CD1 catalysis and inhibition, pointing to possible avenues for the development of HDAC6 CD1-selective inhibitors.
Graphical abstract
INTRODUCTION
The metal-dependent histone deacetylases (HDACs) serve critical roles in the regulation of protein function by catalyzing the hydrolysis of acetyllysine side chains to generate lysine and acetate.1–4 Lysine acetylation-deacetylation cycles are implicated in a wide variety of biological processes such as metabolism,5,6 cell signaling,7 and epigenetics,8,9 so the facile deacetylation of acetyllysine is critically important in the chemistry of the cell. Notably, the disruption of balanced acetylation-deacetylation cycles is associated with certain pathologies, such as cancer and neurodegenerative disease.10,11 The development and use of HDAC inhibitors to modulate acetylation-deacetylation cycles has been a validated therapeutic strategy10–14 ever since the approval of suberoylanilide hydroxamic acid (SAHA, formulated as Vorinostat) for the treatment of cutaneous T-cell lymphoma in 2006.15–17
Consisting of a single polypeptide chain of 1215 amino acids, human histone deacetylase 6 (HDAC6; Uniprot Q9UBN7) is the largest member of the metal-dependent HDAC family.18,19 HDAC6 is unique among the greater HDAC family in that it contains two catalytic domains,19–21 designated CD1 and CD2, in addition to a ubiquitin binding domain.22 Residues important for the chemistry of catalysis are conserved in both catalytic domains, including Zn2+ ligands, tandem histidine residues serving general base-general acid functions, and a tyrosine residue that assists the Zn2+ ion in polarizing the scissile carbonyl of acetyllysine for nucleophilic attack by a Zn2+-bound solvent molecule.4,23–25
While CD2 functions to catalyze the deacetylation of cytosolic protein substrates such as α-tubulin26,27 and Tau,28–30 the catalytic function of CD1 is more enigmatic. Initial studies demonstrated that both CD1 and CD2 exhibited activity in the deacetylation of histone substrates, but only CD2 exhibited tubulin deacetylase activity, suggesting alternate substrate specificities for each catalytic domain.19,31 Additional studies suggested that both domains were required for deacetylase activity,32 but only CD2 was catalytically active.33 Curiously, a more recent report suggested that CD1 is an E3 ubiquitin ligase,34 and an even more recent report suggested that CD1 deacetylates the RNA helicase DDX3X.35 However, these measurements were made in cell extracts and not purified enzymes, so the structural mechanistic bases of these activities are not established.
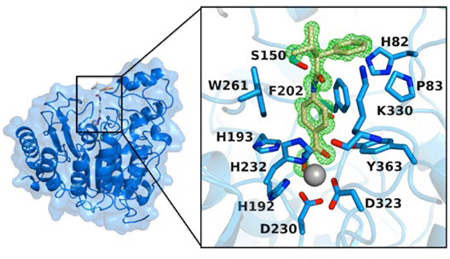
Recombinant expression of protein constructs corresponding to individual CD1 and CD2 domains of human and zebrafish HDAC6 conclusively demonstrated that both catalytic domains exhibit lysine deacetylase activity.36 However, the CD1 domain exhibits very narrow specificity for peptide substrates containing a C-terminal acetyllysine residue. A free α-carboxylate group appears to enable substrate binding in the CD1 active site due to the presence of a “gatekeeper” residue, K330 (zebrafish HDAC6 CD1 numbering; Uniprot F8W4B7), that is unique to HDAC6 CD1 orthologs. This residue appears as a leucine in other HDAC isozymes except for HDAC8, where it is a methionine, and HDAC10, where it is a glutamate.
The X-ray crystal structures of HDAC6 CD2 from Homo sapiens (human),36 and HDAC6 CD1 and CD2 from Danio rerio (zebrafish)36,37 were recently reported. Since then, numerous crystal structures of HDAC6 CD2-inhibitor complexes have illuminated active site features contributing to isozyme-selective inhibition.38–43 However, although more than 50 crystal structures of HDAC6 CD2-inhibitor complexes have been reported to date, the structures of only 2 different HDAC6 CD1-inhibitor complexes are currently available.36,37 Given the observed structural differences between the active site clefts of HDAC6 CD1 and CD2,36,37 as well as the conclusive determination of catalytic activity and narrow substrate specificity for HDAC6 CD1,36 an understanding of inhibitor binding determinants in the active site of this unusual domain is needed to inform the design and evaluation of selective high-affinity inhibitors. In turn, such inhibitors might be useful for probing the biological function of HDAC6 CD1.
To address this dearth of structural information, we now report seven new crystal structures of HDAC6 CD1-inhibitor complexes. Inhibitor structures are illustrated in Figure 1. These structures include complexes formed with wild-type, K330L, or Y363F HDAC6 CD1 (Y363 is the catalytic tyrosine). Additionally, we report catalytic activity measurements of HDAC6 CD1 with a series of acetyllysine-containing peptide substrates. While we previously demonstrated that HDAC6 CD1 cannot readily tolerate alanine or proline residues following the scissile acetyllysine residue,36 we show here that HDAC6 CD1 exhibits low catalytic activity with substrates containing other small residues following the scissile acetyllysine. Even so, maximal catalytic activity is observed only for substrates bearing a free α-carboxylate group.
Figure 1.

Inhibitors studied in complexes with HDAC6 CD1.
MATERIALS AND METHODS
Reagents.
In general, all chemicals were purchased from Fisher Scientific, Millipore Sigma, or Hampton Research and used without further purification. Trichostatin A was purchased from Millipore Sigma. Resminostat and (S)-N-hydroxy-4-(3-methyl-2-phenylbutanamido)-benzamide (AR-42) were purchased from Selleck Chemicals. Givinostat was purchased from Tocris Bioscience.
Protein purification.
HDAC6 CD1 from D. rerio (henceforth simply HDAC6 CD1) was recombinantly expressed using a pET-28a(+) vector by Genscript. This construct utilizes a NdeI/BAMHI cloning site and contains kanamycin bacterial resistance. By nature of the pET-28a(+) vector, a His tag is located at both the N-terminus and C-terminus with an N-terminal thrombin cleavage site. This procedure differs from that initially reported by Hai and Christianson.36
Protein was expressed using Escherichia coli One Shot BL21(DE3) cells (Invitrogen) and grown in 2xYT medium in the presence of 50 μg/mL ampicillin. Cells were grown at 37° C and 250 RPM in an Innova 40 incubator shaker until OD600 reached approximately 0.80. The temperature was then reduced to 18° C until OD600 reached 1.0. At this point, cells were supplemented with 400 μM isopropyl β-L-1-thiogalactopyranoside (IPTG; Gold Biotechnology) and were grown for an additional 18 h at 250 RPM. Finally, cells were centrifuged for 20 min at 5,000 RPM using a Sorvall LYNX 6000 centrifuge and cell pellets were stored at −80° C until further use.
The cell pellet was thawed and re-suspended in 100 mL of Buffer A [50 mM K2HPO4 (pH 8.0), 1 mM tris(2-carboxyethyl)phosphine (TCEP), 300 mM NaCl, 30 mM imidazole, 5% glycerol]. Additionally, 2 protease inhibitor tablets, 0.1 mg/mL lysozyme, and 50 μg/mL DNAse were added to the solution. The cells were then lysed by sonication. Cell lysate was centrifuged for 1 h at 15,000 RPM using a Sorval LYNX 6000 centrifuge and then applied to a 5-mL pre-equilibrated HisTrap HP column.
The His-tagged HDAC6 CD1 protein bound to the HisTrap HP column and was eluted using Buffer B [50 mM K2HPO4 (pH 8.0), 1 mM TCEP, 300 mM NaCl, 300 mM imidazole, 5% glycerol]. The CD1-containing fractions were concentrated to 5 mL using a 15-mL centrifugal filter unit with a molecular weight cut-off of 10 kDa. The protein was then filtered using a 0.22-μM Millex-GV filter unit prior to being loaded onto a HiLoad Superdex 26/600 200 pg column. The column was pre-equilibrated with 360 mL of Buffer C [50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (pH 7.5), 100 mM KCl, 1 mM TCEP, 5% glycerol]. The 5-mL sample of CD1 was injected at a rate of 1 mL/min, and 5-mL fractions were collected. Pure CD1 protein was confirmed by SDS-PAGE, concentrated to approximately 10 mg/mL, and stored at −80° C.
Catalytic activity measurements.
To measure the rate of lysine deacetylation catalyzed by CD1, a discontinuous liquid chromatography-mass spectrometry (LC-MS) assay was implemented as previously established for HDAC6.36 Upon deacetylation of the acetyllysine-containing peptide substrate by HDAC6 CD1 and incubation with dansyl chloride, the liberated primary amino group of product lysine is dansylated and thereby enables detection and quantification using LC-MS. No mass shift indicates no activity.
To briefly summarize the procedure, 5 μL of HDAC6 CD1 (final enzyme concentration = 1 μM) in assay buffer [20 mM HEPES (pH 7.5), 100 mM NaCl, 5 mM KCl, 1 mM MgCl2] was combined with 35 μL of substrate solution (final substrate concentration = 1 mM). After 60 min at 25°C, each reaction was quenched with 10 μL of NaHCO3 (1.6 M, pH 10.0) and 40 μL dansyl chloride (10 mM in acetonitrile). Each reaction mixture was allowed to equilibrate for 60 min at 50 °C, after which it was analyzed by LC-MS using a Waters SQD equipped with an Acquity UPLC (Waters, Milford, MA, USA). Specific activities were calculated using a standard curve. Assays were run in triplicate.
Crystallization.
All HDAC6 CD1-inhibitor complexes were crystallized by the sitting-drop vapor diffusion method using a Mosquito crystallization robot (TTP Labtech). Typically, a 100-nL drop of protein solution [10 mg/mL HDAC6 CD1, 50 mM HEPES (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM TCEP, and 2 mM inhibitor] was added to a 100-nL drop of precipitant solution and equilibrated against 80 μL of precipitant solution in the well reservoir. Prior to flash-cooling, all crystals were soaked with 20% ethylene glycol for cryoprotection. All complexes were crystallized at 4 °C. X-ray diffraction data were collected on Northeastern Collaborative Access Team (NE-CAT) beamlines 24-ID-C and 24-ID-E at the Advanced Photon Source (APS).
For cocrystallization of the HDAC6 CD1–Trichostatin A complex, the precipitant solution was 0.2 M ammonium tartrate dibasic (pH 7.0) and 20% PEG 3350. For cocrystallization of the HDAC6 CD1–AR-42 complex, the precipitant solution was 0.2 M ammonium tartrate dibasic (pH 7.0) and 20% PEG 3350. For cocrystallization of the Y363F HDAC6 CD1–Trichostatin A complex, the precipitant solution was 0.2 M sodium malonate (pH 7.0) and 20% PEG 3350. For cocrystallization of the Y363F HDAC6 CD1–AR-42 complex, the precipitant solution was 0.2 M sodium phosphate dibasic dihydride and 20% PEG 3350. For cocrystallization of the K330L HDAC6 CD1–AR-42 complex, the precipitant solution was 0.2 M magnesium formate dihydrate and 20% w/v PEG 3350. For cocrystallization of the K330L HDAC6 CD1–Resminostat complex, the precipitant solution was 0.1 M ammonium citrate tribasic (pH 7.0) and 12% w/v PEG 3350. For cocrystallization of the K330L HDAC6 CD1–Givinostat complex, the precipitant solution was 0.2 M magnesium formate dihydrate and 20% w/v PEG 3350.
Data reduction, phasing, and structure refinement.
The CCP4 program suite44 was used for data reduction. Data were indexed using the CCP4 program iMosflm45 while Aimless46 was utilized for data scaling. The initial electron density map of each enzyme-inhibitor complex was phased by molecular replacement using the atomic coordinates of HDAC6 CD1 (PDB 5EEF)36 as a search probe with Phaser.47 The interactive graphics program Coot48 was used to build and manipulate atomic models of each enzyme-inhibitor complex. Crystallographic refinement was performed using Phenix.49 Final refined structures were validated using MolProbity50 prior to deposition in the Protein Data Bank (www.rcsb.org). All data collection, reduction, and refinement statistics are recorded in Table 1.
Table 1.
Data Collection and Refinement Statistics for HDAC6 CD1–Inhibitor Complexesa
| Data collection | HDAC6 CD1– Trichostatin A |
HDAC6 CD1– AR–42 |
Y363F HDAC6 CD1– Trichostatin A |
Y363F HDAC6 CD1–AR-42 |
K330L HDAC6 CD1–AR-42 |
K330L HDAC6 CD1– Resminostat |
K330L HDAC6 CD1–Givinostat |
|---|---|---|---|---|---|---|---|
| Space group | P 21 | P 21 21 21 | P 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 | P 21 21 21 |
| a,b,c (Å) | 53.0 124.2 55.5 | 54.7 61.0 121.6 | 52.8 124.4 55.6 | 54.5 60.5 121.9 | 55.0 60.6 121.8 | 67.2 102.6 112.9 | 55.1 60.4 122.1 |
| α,β,γ (°) | 90.0 114.0 90.0 | 90.0 90.0 90.0 | 90.0 114.4 90.0 | 90.0 90.0 90.0 | 90.0 90.0 90.0 | 90.0 90.0 90.0 | 90.0 90.0 90.0 |
| R mergeb | 0.064 (0.546) | 0.140 (0.995) | 0.076 (0.578) | 0.052 (0.747) | 0.075 (0.222) | 0.145 (2.032) | 0.094 (0.359) |
| Rpimc | 0.041 (0.350) | 0.062 (0.445) | 0.048 (0.378) | 0.022 (0.316) | 0.032 (0.094) | 0.062 (0.880) | 0.040 (0.150) |
| CC 1/2d | 0.997 (0.753) | 0.986 (0.776) | 0.995 (0.731) | 1.000 (0.872) | 0.996 (0.977) | 0.997 (0.502) | 0.995 (0.952) |
| Redundancy | 3.4 (3.4) | 6.0 (5.8) | 3.5 (3.1) | 6.6 (6.5) | 6.5 (6.6) | 6.5 (6.2) | 6.5 (6.7) |
| Completeness (%) | 99.1 (98.6) | 99.7 (99.8) | 94.5 (87.9) | 99.9 (99.3) | 100.0 (99.9) | 99.8 (99.6) | 99.7 (99.7) |
| I/σ | 9.9 (2.0) | 7.2 (2.0) | 10.1 (1.8) | 19.2 (2.2) | 15.2 (6.8) | 9.8 (1.9) | 12.4 (5.6) |
| Refinement | |||||||
| Resolution (Å) | 50.6–1.65 (1.70–1.65) | 43.0–1.09 (1.12–1.09) | 62.2–1.27 (1.31–1.27) | 54.2–1.44 (1.49–1.44) | 42.9–1.40 (1.44–1.40) | 50.3–1.58 (1.63–1.58) | 42.9–1.40 (1.45–1.40) |
| No. reflections | 77846 (7694) | 169488 (16796) | 162601 (15378) | 73857 (7242) | 81863 (8131) | 106860 (10560) | 80865 (7985) |
| R work/Rfreee | 0.187/0.222 (0.267/0.301) | 0.183/0.190 (0.275/0.282) | 0.182/0.196 (0.264/0.269) | 0.174/0.196 (0.258/0.271) | 0.166/0.184 (0.195/0.234) | 0.184/0.208 (0.252/0.286) | 0.152/0.169 (0.166/0.198) |
| No. atomsf | |||||||
| Protein | 5466 | 2856 | 5536 | 2816 | 2843 | 5603 | 2880 |
| Ligand | 50 | 38 | 50 | 30 | 26 | 90 | 46 |
| Solvent | 193 | 415 | 563 | 266 | 384 | 497 | 432 |
| Average B factors (Å2) | |||||||
| Protein | 25 | 13 | 17 | 20 | 15 | 19 | 13 |
| Ligand | 29 | 15 | 17 | 22 | 13 | 27 | 22 |
| Solvent | 28 | 25 | 27 | 30 | 27 | 29 | 27 |
| R.m.s. deviations | |||||||
| Bond lengths (Å) | 0.006 | 0.004 | 0.005 | 0.005 | 0.004 | 0.006 | 0.004 |
| Bond angles (°) | 0.81 | 0.83 | 0.77 | 0.80 | 0.81 | 0.85 | 0.82 |
| Ramachandran plotg | |||||||
| Favored | 97.04 | 97.20 | 96.90 | 96.35 | 97.19 | 96.06 | 97.19 |
| Allowed | 2.96 | 2.80 | 3.10 | 3.65 | 2.81 | 3.80 | 2.81 |
| Outliers | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.10 | 0.00 |
Values in parentheses refer to the highest-resolution shell indicated.
Rmerge = ∑hkl∑i|Ii,hkl − ⟨I⟩hkl|/∑hkl∑iIi,hkl, where ⟨I⟩hkl is the average intensity calculated for reflection hkl from replicate measurements.
Rp.i.m.= (∑hkl(1/(N-1))1/2∑i|Ii,hkl − ⟨I⟩hkl|)/∑hkl∑i Ii,hkl, where ⟨I⟩hkl is the average intensity calculated for reflection hkl from replicate measurements and N is the number of reflections.
Pearson correlation coefficient between random half-datasets.
Rwork = ∑||Fo| − |Fc||/∑|Fo| for reflections contained in the working set. |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree is calculated using the same expression for reflections contained in the test set held aside during refinement.
Per asymmetric unit.
Calculated with MolProbity.
RESULTS
Crystal structure determinations.
The 1.65 Å-resolution crystal structure of the HDAC6 CD1–Trichostatin A complex reported herein crystallizes in monoclinic space group P21 and is isomorphous with the previously reported crystal structure of this complex determined at lower (2.15 Å) resolution. There are no major conformation differences between these two structures, and the root-mean-square deviation (rmsd) is 0.20 Å for 306 Cα atoms in monomer A. The simplified HDAC6 CD1 construct used in the current study represents a significant improvement over previously reported constructs in terms of the 0.5 Å-resolution improvement in the diffraction quality of crystals compared with HDAC6 CD1 derived from the fusion prepared with maltose binding protein.36 Additionally, the simplified construct does not undergo slow proteolysis as observed for the CD1-CD2 didomain construct expressed in baculovirus-infected Sf9 cells.37 The improved expression and purification scheme for HDAC6 CD1 consistently yields large quantities of pure protein that is more amenable to crystallization experiments.
Inhibitor binding interactions are identical in the 1.65 Å resolution structure and the 2.15 Å resolution structure. Briefly, the catalytic Zn2+ ion is chelated by the inhibitor hydroxamate group with bidentate coordination geometry (Figure 2a). The Zn2+-bound hydroxamate C=O group also accepts a hydrogen bond from Y363, while the Zn2+-bound hydroxamate N–O– group accepts a hydrogen bond from H192. The hydroxamate NH group also donates a hydrogen bond to H193. Interestingly, electron density corresponding to the side chain of gatekeeper residue K330 is poor in the 1.65 Å resolution structure, so the side chain of this residue is not modeled beyond the Cβ atom. This contrasts with the 2.15 Å resolution structure, in which the amino group of the K330 side chain donates a hydrogen bond to the ketone carbonyl of the inhibitor.
Figure 2.

(a) Polder omit maps of the HDAC6 CD1–Trichostatin A complex (PDB 6UO2; monomer B, inhibitor contoured at 5.5 σ; K330 is contoured at 3.5 σ). Atoms are color-coded as follows: C = light blue (monomer B), light gray (monomer A), or wheat (inhibitor), N = blue, O = red, and Zn2+ = gray sphere. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Superposition of the HDAC6 CD1–Trichostatin A complex (monomer B, color-coded as in (a)) and the HDAC6 CD2–Trichostatin A complex (PDB 5WGI; monomer A, C = light gray (protein) or dark gray (inhibitor). W78 of CD1 is conserved as W459 in CD2, and this residue adopts alternate conformations in CD1 (“in”) and CD2 (“out”). The “out” conformation of W459 at the mouth of the HDAC6 CD2 active site enables the binding of polyethylene glycol fragment PEG-821.
Differences in the binding conformation of Trichostatin A to HDAC6 CD1 and CD2 have been noted previously in the first structure determinations of HDAC6,36,37 and these differences are maintained in view of the 1.65 Å-resolution structure of the HDAC6 CD1–Trichostatin A complex reported herein and the 1.05 Å-resolution structure of the HDAC6 CD2-Trichostatin A complex recently reported by Porter and colleagues (PDB 5WGI) (Figure 2b).38 Differences in the protein structure are largely confined to W78, the side chain of which moves toward the bound inhibitor in the HDAC6 CD1 complex. Differences in inhibitor binding are manifest in a ~4 Å-shift of the dimethyaminophenyl capping group toward W78.
The 1.09 Å-resolution crystal structure of the HDAC6 CD1–AR-42 complex is the highest resolution structure of HDAC6 CD1 determined to date and exemplifies the high quality crystals that can be prepared with the simplified protein construct developed for the current study. This complex crystallizes in previously unobserved orthorhombic space group P212121 with one monomer in the asymmetric unit. The hydroxamate moiety of the inhibitor chelates the catalytic Zn2+ ion nearly symmetrically (Zn2+---O1 distance = 2.1 Å and Zn2+---O2 distance = 2.2 Å) (Figure 3). The Zn2+-bound hydroxamate C=O group additionally accepts a hydrogen bond from Y363, and the Zn2+-bound hydroxamate N–O– group accepts a hydrogen bond from H192; the hydroxamate NH group donates a hydrogen bond to H193.
Figure 3.

Polder omit map of the HDAC6 CD1–AR-42 complex (PDB 6UO3; inhibitor contoured at 5.0 σ; K330 contoured at 2.5 σ). Atoms are color-coded as follows: C = light blue (monomer A), light gray (symmetry mate), or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
The amide carbonyl oxygen in the capping group of AR-42 accepts a hydrogen bond from the side chain of S150. Interestingly, the corresponding residue also appears in HDAC6 CD2 as S531, where it accepts a hydrogen bond from the backbone NH group of the scissile acetyllysine residue of the substrate.36 These serine residues are unique to the active sites of HDAC6 CD1 and CD2 in comparison with other HDAC isozymes. Notably, HDAC6 CD2-selective inhibitors form hydrogen bonds with S531,36,38,40,42,43 so S150 may similarly serve as a target in the design of HDAC6 CD1-selective inhibitors.
The amide carbonyl group of AR-42 forms two water-mediated hydrogen bonds in the active site of HDAC6 CD1: one with the side chain amino group of K330 and another to the imidazole Nε-H group of Zn2+ ligand H232. Water-mediated hydrogen bonds with corresponding Zn2+ ligand H614 of HDAC6 CD2 are similarly observed in the binding of certain inhibitors to this catalytic domain.36,38,40 The phenyl capping group of the inhibitor is oriented toward H82 and P83, while the isopropyl group is oriented towards solvent. Notably, the benzyl group of AR-42 is nestled in the aromatic crevice formed by W261 and F202 where it makes favorable offset π-π interactions. This aromatic crevice contrasts with that of the CD2 domain, where it is defined by two phenylalanine residues (F583 and F643).39,40
The 1.27 Å-resolution crystal structure of the Y363F HDAC6 CD1–Trichostatin A complex contains two monomers in the asymmetric unit (Figure 4a). Crystals form in monoclinic space group P21 and are isomorphous with crystals of the wild-type enzyme-inhibitor complex. The amino acid substitution does not cause any significant structural rearrangements, and the rmsd of 300 Cα atoms between the wild-type and Y363F HDAC6 CD1 structures is 0.16 Å. The phenolic hydroxyl group of Y363 is important for catalysis and inhibitor binding in metal-dependent deacetylases. However, absent a hydrogen bond interaction with Y363 in the Y363F variant, it is notable that inhibitor binding is otherwise generally identical to that observed in the active site of the wild-type enzyme (Figure 4b).
Figure 4.

(a) Polder omit map of the Y363F HDAC6 CD1–Trichostatin A complex (PDB 6UO4; monomer A, contoured at 4.5 σ; F363 contoured at 4.5 σ). Atoms are color-coded as follows: C = light blue (monomer A), light gray (monomer B), or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Superposition of the Y363F HDAC6 CD1–Trichostatin A complex (color-coded as in (a)) with the wild-type HDAC6 CD1–Trichostatin A complex (C = light gray (protein) or dark gray (inhibitor); PDB 6UO2). The inhibitor binding conformation is essentially identical in both structures and is not affected by the amino acid substitution.
Even so, some structural differences are observed for certain active site residues. Most prominent is the 66° rotation of F363 compared with Y363 in the wild-type enzyme-inhibitor complex, suggesting flexibility for this residue (Figure 4b). A water molecule fills the void created by the conformational change of F363. This water molecule accepts a hydrogen bond from the backbone NH group of G362 and donates a hydrogen bond to the hydroxamate C=O group of Trichostatin A. Additionally, K330 is characterized by well-defined electron density, but it does not form a hydrogen bond with the inhibitor. The dimethylamino moiety of the capping group also forms a water-mediated hydrogen bond with H82.
The 1.44 Å-resolution crystal structure of the Y363F HDAC6 CD1–AR-42 complex contains one monomer in the asymmetric unit. Crystals form in orthorhombic space group P212121 and are isomorphous with crystals of the wild-type enzyme-inhibitor complex. The amino acid substitution does not cause any significant structural rearrangements, and the rmsd of 313 Cα atoms between the wild-type and Y363F HDAC6 CD1 structures is 0.15 Å. Intriguingly, however, hydroxamate-Zn2+ coordination geometry is monodentate, with metal coordination achieved only by the hydroxamate N–O– group (Figure 5). A Zn2+-bound water molecule additionally forms a hydrogen bond with the carbonyl group of the hydroxamate moiety. This contrasts with bidentate hydroxamate-Zn2+ coordination geometry in the wild-type enzyme-inhibitor complex (Figure 3). Possibly, the loss of the hydrogen bond between Y363 and the hydroxamate carbonyl group in the Y363F mutant influences the denticity of hydroxamate-Zn2+ coordination. A hydrogen bond with the phenolic hydroxyl group of Y363 orients and activates the scissile carbonyl of acetyllysine for catalysis,23–25 so it is conceivable that a hydrogen bond with Y363 also helps to orient the hydroxamate carbonyl of certain inhibitors.
Figure 5.

Polder omit map of the Y363F HDAC6 CD1–AR-42 complex (PDB 6UO5; contoured at 6.0 σ; F363 is contoured at 3.0 σ). Atoms are color-coded as follows: C = light blue (monomer A), light gray (symmetry mate), or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
The side chain of F363 is rotated by 70° compared with Y363 in the wild-type enzyme-inhibitor complex (Figure 5). A water molecule occupies the resulting void, and this water molecule accepts a hydrogen bond from the backbone NH group of G362 and donates a hydrogen bond to the Zn2+-bound hydroxamate N–O– group. Additionally, the side chain of S150 adopts two alternative conformations, each of which is within hydrogen bonding distance to the amide NH group of AR-42. Since K330 is unique to HDAC6 CD1 among all metal-dependent HDAC isozymes, hydrogen bond interactions with this residue – either direct or water-mediated – could be targeted in the development of HDAC6 CD1-specific inhibitors. As observed in the enzyme-inhibitor complex with the wild-type enzyme, the benzyl group of AR-42 is nestled in the aromatic crevice formed by W261 and F202 where it makes favorable offset π-π interactions.
The 1.40 Å-resolution crystal structure of the K330L HDAC6 CD1–AR-42 complex contains one monomer in the asymmetric unit. Crystals form in orthorhombic space group P212121 and are isomorphous with crystals of the wild-type enzyme-inhibitor complex. The amino acid substitution does not cause any significant structural rearrangements, and the rmsd of 334 Cα atoms between the wild-type and K330L HDAC6 CD1 structures is 0.08 Å. The K330L substitution makes the active site more like that of HDAC6 CD2, and this substitution confers more relaxed substrate specificity.36 Notably, K330L is located in the vicinity of the L1 pocket, where in HDAC6 CD2 the majority of inhibitor capping groups bind.36,38–43
The K330L substitution does not perturb the inhibitor binding conformation (Figure 6). Bidentate hydroxamate-Zn2+ coordination geometry is retained, and the inhibitor capping group moiety binds in the same pocket as observed in complexes with wild-type and Y363F HDAC6 CD1 complexes. Therefore, although L330 is not the naturally occurring residue in the active site of HDAC6 CD1, its substitution does not perturb inhibitor binding conformations. However, its substitution enables cocrystallization of HDAC6 CD1 with additional inhibitors that do not behave similarly well in cocrystallization experiments with wild-type HDAC6 CD1.
Figure 6.

(a) Polder omit map of the K330L HDAC6 CD1–AR-42 complex (PDB 6UO7; contoured at 6.0 σ; L330 is contoured at 3.0 σ). Atoms are color-coded as follows: C = light blue (monomer A), light gray (symmetry mate), or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Superposition of the K330L HDAC6 CD1–AR-42 complex (color-coded as in (a)) with the wild-type HDAC6 CD1–AR-42 complex (C = light gray (protein) or dark gray (inhibitor); PDB 6UO3). The inhibitor binding conformation is essentially identical in both structures and is not affected by the amino acid substitution.
The 1.58 Å-resolution crystal structure of the K330L HDAC6 CD1–Resminostat complex contains two monomers in the asymmetric unit of a second, new orthorhombic space group. Electron density for a single conformation L330 is well defined in both monomers (Figure 7). As observed in most of the structures of enzyme-inhibitor complexes described herein, the ionized hydroxamate moiety coordinates to Zn2+ in bidentate fashion.
Figure 7.
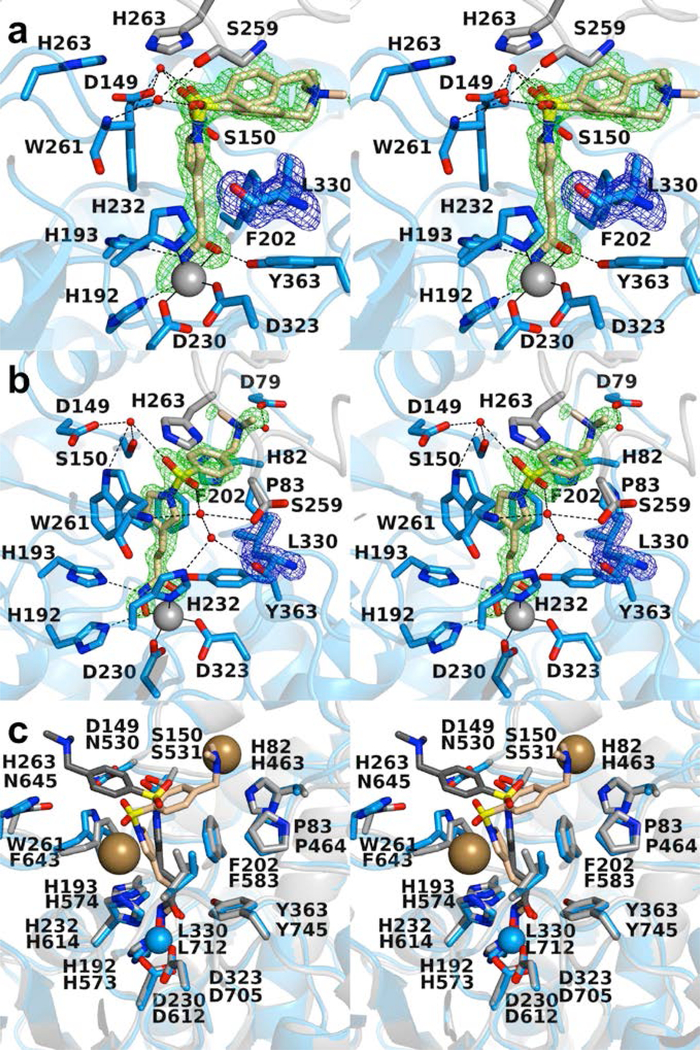
(a) Polder omit map of the K330L HDAC6 CD1–Resminostat complex (PDB 6UOB; monomer A, contoured at 3.5 σ). L330 is contoured at 3.5 σ. Atoms are color-coded as follows: C = light blue (monomer A), light gray (monomer B), or wheat (inhibitor), N = blue, O = red, S = yellow, Zn2+ =gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Polder omit map of the K330L HDAC6 CD1–Resminostat complex (PDB 6UOB; monomer B, contoured at 5.0 σ). L330 is contoured at 3.5 σ. Atoms are color-coded as follows: C = light blue (monomer B), light gray (symmetry mate), or wheat (inhibitor), N = blue, O = red, S = yellow, Zn2+ =gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (c) Superposition of the K330L HDAC6 CD1–Resminostat complex (monomer B, color-coded as in (a)) and the wild-type HDAC6 CD2–Resminostat complex (monomer B, C = light gray (protein) or dark gray (inhibitor), I– = large sand-colored spheres; PDB 6PZR). The inhibitor capping group binds in alternate locations in each active site. The inhibitor binding conformation in the HDAC6 CD2 active site appears to be influenced by the binding of two iodide ions.
There is slight variation of enzyme-inhibitor interactions between monomers. The dimethylamino capping moiety is oriented toward F202 in both monomers. In monomer A, the dimethylamino group exhibits two alternative conformations separated by a 53° rotation. The sulfonyl group forms water-mediated hydrogen bonds with the backbone carbonyl of symmetry mate residue S259 in addition to forming water-mediated hydrogen bonds with D149, S150, and W261. In monomer B, the dimethylamino group exhibits only one conformation with a water-mediated hydrogen bond to D79. The sulfonyl group forms water-mediated hydrogen bonds to symmetry mate residue S259 in addition to forming water-mediated hydrogen bonds to D149, S150, H232, W261, and the backbone carbonyl of L330.
It is interesting to compare the binding conformation of Resminostat to HDAC6 CD2 (PDB 6PZR) and K330L HDAC6 CD1. Superposition of the two structures shows that the inhibitor capping group occupies alternate locations in each structure (Figure 7c). Two iodide ions accompany inhibitor binding to HDAC6 CD2 and appear to influence the binding conformation of the capping group.
The 1.40 Å-resolution crystal structure of the K330L HDAC6 CD1–Givinostat complex contains one monomer in the asymmetric unit. Givinostat is the bulkiest inhibitor cocrystallized with HDAC6 CD1 to date and makes extensive interactions in the active site. The hydroxamate moiety coordinates to the catalytic Zn2+ ion in characteristic bidentate fashion, and the aromatic ring of the phenylhydroxamate moiety nestles in the aromatic cleft defined by W261 and F202 (Figure 8). While the diethylamino capping moiety is slightly disordered based on weak or broken electron density, the diethylamino group clearly donates a hydrogen bond with symmetry mate residue E97. Additional hydrogen bond interactions are observed between the inhibitor amide and S150 as well as a water-mediated hydrogen bond between the inhibitor amide and Zn2+ ligand H232.
Figure 8.

Polder omit map of the K330L HDAC6 CD1–Givinostat complex (PDB 6UOC; monomer A, contoured at 3.0 σ; L330 contoured at 3.0 σ). Atoms are color-coded as follows: C = light blue (monomer A), light gray (symmetry mate), or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and water molecules = smaller red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
Catalytic activity measurements.
It has previously been demonstrated36 that zebrafish HDAC6 CD1 and human HDAC6 CD1 exhibit optimal catalytic activity with substrates containing C-terminal acetyllysine residues such as Ac-GAKAc-CO2–. The measurement of steady-state kinetics with this substrate yields catalytic efficiencies (kcat/KM) of 8600 M−1s−1 and 1500 M−1s−1 with zebrafish HDAC6 CD1 and human HDAC6 CD1, respectively. In comparison, using the corresponding amidated substrate Ac-GAKAc-CONH2, catalytic efficiency with zebrafish HDAC6 CD1 is reduced to 600 M−1s−1, and activity with human HDAC6 CD1 is reduced below measurable levels. Of note, the zebrafish enzyme exhibits much weaker catalytic activity with chromophoric substrates containing a C-terminal aminomethylcoumarin (AMC) moiety, such as Ac-RGKAc-AMC; no catalytic activity is observed by the human enzyme with these substrates. This narrow substrate specificity is attributed to steric effects resulting from K330 in the HDAC6 CD1 active site.36
Here, based on specific activity measurements using the C-terminal acetyllysine substrates Ac-GAKAc-CO2– and Ac-AKAc-CO2–, we confirm as a positive control the narrow substrate specificity for maximal zebrafish HDAC6 CD1 activity. However, we also report weak catalytic activity against substrates containing amino acid residues on the carboxyl side of the scissile acetyllysine residue (Table 2; due to weak specific activity measured for most substrates, we did not perform full steady-state kinetic analyses). Smaller residues, such as glycine or cysteine, but also including isoleucine, can be accommodated at this position in HDAC6 CD1 substrates. Even so, the observed catalytic activity against the best of these alternate substrates is at least 2–5-fold lower than that observed with Ac-AKAc and Ac-GAKAc.
Table 2.
Catalytic activity of HDAC6 CD1 and CD2 with selected peptide substrates
| Catalytic Domain | Substrate | Specific Activity (nmol product nmol−1 enzyme min−1) |
|---|---|---|
| CD1 | Ac-GAKAc | 62 ± 5 |
| CD2 | Ac-GAKAc | 11.0 ± 0.5 |
| CD1 | Ac-AKAc | 55 ± 4 |
| CD2 | Ac-AKAc | 20.0 ± 0.2 |
| CD1 | Ac-KAcCGS-NH2 | 25 ± 2 |
| CD2 | Ac-KAcCGS-NH2 | 8.0 ± 0.1 |
| CD1 | Ac-KAcIGS-NH2 | 17 ± 1 |
| CD2 | Ac-KAcIGS-NH2 | 16.0 ± 0.8 |
| CD1 | Ac-RGKAcG-NH2 | 12 ± 1 |
| CD1 | Ac-GKGKAcGS-NH2 | 12 ± 5 |
| CD1 | Ac-GKAcGKGS-NH2 | 8 ± 1 |
| CD1 | Ac-GKGKAcGS | 5 ± 0.4 |
| CD1 | Ac-GKAcGKGS | 2 ± 0.2 |
| CD1 | Ac-LVKAcG | 7 ± 0.4 |
| CD1 | Ac-LVKAcGI-NH2 | 1.00 ± 0.02 |
| CD1 | Ac-ELVKAcGIL-NH2 | 1.00 ± 0.07 |
| CD1 | Ac-QPKKAc | 6 ± 1 |
| CD1 | Ac-TKAcP | 1.00 ± 0.07 |
| CD1 | Ac-KAcKIET-NH2 | 1.0 ± 0.1 |
| CD1 | Ac-KAcPVD-NH2 | 1.00 ± 0.01 |
All measurements represent mean ± average of absolute deviations from the mean in a given set of data. Ac-AKAc and Ac-GAKAc served as positive controls (Kac = acetyllysine). All measurements were run in triplicate at 25° C.
Peptides Ac-KAcCGS-NH2 and Ac-KAcIGS-NH2 derive from the HDAC6 substrate tau.28–30 The KXGS motifs are located within the microtubule-binding domain of tau and are important for microtubule stabilization.51 While HDAC6 CD2 exhibits activity against these tau-derived substrates, it is interesting to note that HDAC6 CD1 exhibits comparable or better activity (Table 2). While speculative, perhaps HDAC6 has two functional catalytic domains to enable simultaneous hydrolysis reactions with two different acetyllysine containing substrates, such as tau and the α-domain of tubulin in the microtubule. It is unknown whether substrate binding and catalysis in one catalytic domain might influence substrate binding and catalysis in the other domain through some sort of allosteric effect. Although tau is thought to bind on the exterior of the microtubule,52 some evidence suggests that it can also bind in the lumen of the microtubule,53 where HDAC6 CD2 functions to deacetylate K40 of α-tubulin.37,54
Peptide Ac-RGKAcG-NH2 derives from cytoplasmic dynein 1 heavy chain 1.55,56 Given that the linker between CD1 and CD2 in full-length human HDAC6 contains a dynein motor protein binding segment,18,19 we were curious to see if HDAC6 CD1 might exhibit catalytic activity against the dynein-based peptide or other peptides containing a –KAcG– segment. However, HDAC6 CD1 exhibits lower activity against these substrates.
DISCUSSION
HDAC6 is unique among all metal-dependent HDAC isozymes in that it contains two fully functional catalytic domains CD1 and CD2, and these catalytic domains exhibit striking differences in substrate specificity and inhibitor binding. While HDAC6 CD2 is the most-studied of the two, given its identification as a tubulin deacetylase26,27 and as a tau deacetylase,28–30 the biological function of HDAC6 CD1 has remained enigmatic. We expect that in-depth structural characterization of HDAC6 CD1 will further illuminate structure-function relationships that, in turn, will inform the design and development of selective inhibitors. As the first step toward this goal, we have presented an array of seven HDAC6 CD1–inhibitor complexes to facilitate the study of affinity interactions in the active site of HDAC6 CD1. While these inhibitors are “pan-HDAC” inhibitors, i.e., they are not particularly selective for the inhibition of HDAC6 over other HDAC isozymes, structural analyses of their complexes with HDAC6 nonetheless yield valuable information regarding potential interactions that could be targeted in the design of second-generation inhibitors with improved properties of affinity and selectivity.
The Zn2+ binding site and catalytic residues are identical in HDAC6 CD1 and CD2, so the chemical mechanism of amide bond hydrolysis must be identical in both catalytic domains: the Zn2+ ion and the catalytic tyrosine residue polarize the scissile carbonyl of acetyllysine for nucleophilic attack by a Zn2+-bound water molecule, assisted by tandem histidine residues that serve general base-general acid functions.23–25 However, there are nearby structural differences, including the aromatic crevice that accommodates the linker moiety of most inhibitors. In HDAC6 CD2, this crevice is formed by F583 and F643, whereas in HDAC6 CD1 this crevice is formed by F202 and W261. Since aromatic linkers of inhibitors bind in this crevice and make favorable offset π-π interactions, the slightly larger aromatic crevice of HDAC6 CD1 may be capable of accommodating modified aromatic substituents.
On the opposite side of the aromatic crevice in the active site of HDAC6 CD1 is K330, which confers specificity for acetyllysine substrates bearing a free α-carboxylate group.36 It is thought that the additional steric bulk of this residue, in comparison with the leucine residue at this position in HDAC6 CD2, accounts for differences in the binding conformations of Trichostatin A to each catalytic domain (Figure 2b).36,37 While the side chain of K330 is mainly disordered in this complex (Figure 2a), its steric influence must nevertheless contribute to the shift in the capping group of Trichostatin A. This shift is further accommodated by the conformational difference observed for W78 in HDAC6 CD1 compared to HDAC6 CD2.
Direct comparison of the active site contours of HDAC6 CD1 and CD2 reveals notable differences that could impact inhibitor binding. For example, D149, H263, and W261 in HDAC6 CD1 appear as N530, N645, and F643 in HDAC6 CD2 (Figure 7c). An additional amino acid substitution includes K330 in HDAC6 CD1, which appears as L712 in HDAC6 CD2 (Figure 9). It is striking, too, that conformational differences in conserved residue W78/W459 also contribute to differences in the active site contours. Overall, the active site cleft of HDAC6 CD1 is wider than that of HDAC6 CD2, which contrasts with the narrow substrate specificity exhibited by HDAC6 CD1. Since the K330L substitution relaxes this narrow substrate specificity,36 K330 must play an important role in substrate binding; similarly, it could play an important role in inhibitor binding and selectivity.
Figure 9.

(a) Molecular surface of HDAC6 CD1 (light blue). (b) Molecular surface of HDAC6 CD2 (light gray). Residues K330 and L712 differ between the catalytic domains, giving rise to different contours at the mouth of the active site.
With regard to inhibitor binding conformations in the active sites of wild-type and mutant HDAC6 CD1 enzymes, it is important to note similar binding conformations regardless of whether K330 is substituted with leucine (Figure 6b) or whether catalytic tyrosine Y363 is substituted with phenylalanine (Figure 4b). Since we were unable to prepare crystalline complexes of wild-type HDAC6 CD1 with all inhibitors described in this work, it is reassuring that inhibitor cocrystallization studies with Y363F and K330L HDAC6 CD1 are likely to yield structures of complexes that are quite similar to those involving the wild-type enzyme.
The specific activity measurements recorded in Table 2 confirm the narrow substrate specificity of zebrafish HDAC6 CD1, which exhibits maximal catalytic activity with acetyllysine substrates bearing a free α-carboxylate group.36 However, weaker catalytic activity is also observed for HDAC6 CD1 with substrates containing internal acetyllysine residues, such as Ac-KAcCGS-NH2, which derives from the tau protein. The specific activity of HDAC6 CD1 with this tau-based substrate is only three-fold weaker than that measured with the best substrate, Ac-GAKAc. Therefore, it is clear that zebrafish HDAC6 CD1 can accommodate substrates in which the scissile acetyllysine residue is flanked by smaller amino acids. This is consistent with the identification of human HDAC6 CD1 substrates containing internal acetyllysine residues using acetylome peptide microarrays in an elegant study reported by Barinka and colleagues.57 Even so, our previously reported studies with a human HDAC6 CD1-CD2 di-domain construct (with the CD2 domain inactivated by mutagenesis) indicate stricter substrate specificity for C-terminal acetyllysine residues compared with zebrafish HDAC6 CD1.36 Since Barinka and colleagues used full-length human HDAC6 in their studies,57 activity differences may result from the use of full-length human HDAC6 versus the truncated CD1-CD2 di-domain construct in enzyme assays.
CONCLUDING REMARKS
While the precise biological function of HDAC6 CD1 remains unknown, the current study confirms our previous work36 showing that catalytic activity exhibits very narrow substrate specificity, with maximal catalytic activity observed for acetyllysine substrates bearing a free α-carboxylate group. Modification of the α-carboxylate group by amidation, or peptide bond formation with an additional amino acid, compromises catalytic activity. Weak catalytic activity is measured with certain substrates bearing a smaller residue on the carboxyl side of acetyllysine, so the requirement for a free α-carboxylate group on acetyllysine is not absolute for weak activity. The identification of glycine residues preceding the scissile acetyllysine residue in HDAC6 CD1 substrates identified in microarray analyses57 is consistent with a general observation that smaller and flexible residues flanking acetyllysine appear to facilitate HDAC6 CD1 activity.
While the overall active site contour of HDAC6 CD1 is wider than that of HDAC6 CD2 (Figure 9), one side of the HDAC6 CD1 active site is constricted by K330. This residue contributes to the catalytic specificity for C-terminal acetyllysine substrates.36 Our crystal structures reported herein show that K330 can interact with bound inhibitors, but it can also be disordered and thereby serve as a simple steric block. We cannot rule out the possibility that HDAC6 CD1 may utilize an as-yet undiscovered non-peptide substrate in vivo.
Structural differences between the active sites of HDAC6 CD1 and CD2, such as the aromatic crevice and the mouth of the active site in the vicinity of W78, lead to different binding orientations for Trichostatin A. While different binding conformations are also observed for Resminostat, binding to HDAC6 CD2 may also be influenced by two iodide ions that also bind in the active site of this complex. Nevertheless, sufficient structural differences are found in the active site of HDAC6 CD1 that could serve as the basis for the design and development of isozyme-selective inhibitors. Such inhibitors could prove useful in elucidating the biological function of HDAC6 CD1. We will report our results to this end in due course.
Acknowledgments
We thank the Northeastern Collaborative Access Team (NE-CAT) beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on beamline 24-ID-C is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding
We thank the National Institutes of Health for grants GM49758 (D.W.C.) in support of this research.
Footnotes
ASSOCIATED CONTENT
Accession Codes
The atomic coordinates and crystallographic structure factors of CD1 complexes with the inhibitors shown in Figure 1 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes as follows: HDAC6 CD1–Trichostatin A complex, 6UO2; HDAC6 CD1–AR-42 complex, 6UO3; Y363F HDAC6 CD1–Trichostatin A complex, 6UO4; Y363F HDAC6 CD1–AR-42 complex, 6UO5; K330L HDAC6 CD1–AR-42 complex, 6UO7; K330L HDAC6 CD1–Resminostat complex, 6UOB; K330L HDAC6 CD1–Givinostat complex, 6UOC.
The authors declare no competing financial interests.
References
- 1.Kouzarides T (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Norvell MA (2000) Rise of the rival. Science 327, 964–965. [DOI] [PubMed] [Google Scholar]
- 3.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, and Mann M (2009) Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325, 834–840. [DOI] [PubMed] [Google Scholar]
- 4.López JE, Sullivan ED, and Fierke CA (2016) Metal-dependent deacetylases: cancer and epigenetic regulators. ACS Chem. Biol. 11, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, and Guan KL (2010) regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP (2010) Acetylation of metabolic enzymes coordinated carbon source utilization and metabolic flux. Science 327, 1004–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhary C, Weinert BT, Nishida Y, Verdin E, and Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550. [DOI] [PubMed] [Google Scholar]
- 8.Berger SL, Kouzarides T, Shiekhattar R, and Shilatifard A (2009) An operational definition of epigenetics. Genes Dev. 23, 781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delcuve GP, Khan DH, and Davie JR (2012) Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin. Epigenet. 12, 4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arrowsmith CH, Bountra C, Fish PV, Lee K, and Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 11, 384–400. [DOI] [PubMed] [Google Scholar]
- 11.Falkenberg KJ, and Johnstone RW (2014) Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 13, 673–691. [DOI] [PubMed] [Google Scholar]
- 12.West AC, and Johnstone RW (2014) New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 124, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma N, Luo Y, Wang Y, Liao C, Ye WC, and Jiang S (2015) Selective histone deacetylase inhibitors with anticancer activity. Curr. Top. Med. Chem. 16, 415–426. [DOI] [PubMed] [Google Scholar]
- 14.Dokmanovic M, Clarke C, and Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 5, 981–989. [DOI] [PubMed] [Google Scholar]
- 15.Richon VM, Webb Y, Merger R, Sheppard T, Jursic B, Ngo L, Civoli F, Breslow R, Rifkind RA, and Marks PA (1996) Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. U. S. A. 93, 5705–5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks PA (2007) Discovery and development of SAHA as an anticancer agent. Oncogene 26, 1351–1356. [DOI] [PubMed] [Google Scholar]
- 17.Mann BS, Johnson JR, Cohen MH, Justice R, and Pazdur R (2007) FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12, 1247–1252. [DOI] [PubMed] [Google Scholar]
- 18.Verdel A, and Khochbin S (1999) Identification of a new family of higher eukaryotic histone deacetylases: coordinate expression of differentiation-dependent chromatin modifiers. J. Biol. Chem. 274, 2440–2445. [DOI] [PubMed] [Google Scholar]
- 19.Grozinger CM, Hassig CA, and Schreiber SL (1999) Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. U. S. A. 96, 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Gilquin B, Khochbin S, and Matthias P (2006) Two catalytic domains are required for protein deacetylation. J. Biochem. 281, 201–204. [DOI] [PubMed] [Google Scholar]
- 21.Zou H, Wu Y, Navre M, and Sang BC (2006) Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun. 341, 45–50. [DOI] [PubMed] [Google Scholar]
- 22.Hook SS, Orian A, Cowley SM, and Eisenman RN (2002) Histone deacetylase 6 binds polyubiquitin through its zinc finger (PAZ domain) and copurifies with deubiquitinating enzymes. Proc. Natl. Acad. Sci. U. S. A. 99, 13425–13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lombardi PM, Cole KE, Dowling DP, and Christianson DW (2011) Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 21, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolfson NA, Pitcairn CA, and Fierke CA (2013) HDAC8 substrates: histones and beyond. Biopolymers 99, 112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter NJ, Christianson DW (2019) Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Op. Struct. Biol. 59, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, and Yao TP (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, and Matthias P (2003) HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 22, 1168–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, and Gan L (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 23, 953–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noack M, Leyk J, and Richter-Landsberg C (2014) HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia 62, 535–547. [DOI] [PubMed] [Google Scholar]
- 31.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, and Schreiber SL (2003) Domain-selective small molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U. S. A. 100, 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Gilquin B, Khochbin S, and Matthias P (2006) Two catalytic domains are required for protein deacetylation. J. Biol. Chem. 281, 2401–2404. [DOI] [PubMed] [Google Scholar]
- 33.Zou H, Wu Y, Navre M, and Sang B-C (2006) Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun. 341, 45–50. [DOI] [PubMed] [Google Scholar]
- 34.Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, Hu C, Tong D, Haakenson J, Wang C, Zhang S, Pavlovicz RE, Jones A, Schmidt K,H, Tang J, Dong H, Shan B, Fang B, Radhakrishnan R, Glazer PM, Matthias P, Koomen J, Seto E, Bepler G, Nicosia SV, Chen J, Li C, Gu L, Li GM, Bai W, Wang H, and Zhang X (2014) HDAC6 deacetylases and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol Cell 55, 31–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito M, Hess D, Eglinger J, Fritsch AW, Kreysing M, Weinert BT, Choudhary C, and Matthias P (2019) Acetylation of intrinsically disordered regions regulates phase separation. Nat. Chem. Biol. 15, 51–61. [DOI] [PubMed] [Google Scholar]
- 36.Hai Y, and Christianson DW (2016) Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, and Matthias P (2016) Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 12, 748–754. [DOI] [PubMed] [Google Scholar]
- 38.Porter NJ, Mahendran A, Breslow R, and Christianson DW (2017) Unusual zinc binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U. S. A. 114, 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porter NJ, Wagner FF, and Christianson DW (2018) Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6. Biochemistry 57, 3916–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porter NJ, Osko JD, Diedrich D, Kurz T, Hooker JM, Hansen FK, and Christianson DW (2018) Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-zinc denticity. J. Med. Chem. 61, 8054–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porter NJ, Shen S, Barinka C, Kozikowski AP, and Christianson DW (2018) Molecular basis for the selective inhibition of histone deacetylase 6 by a mercaptoacetamide inhibitor. ACS Med. Chem. Lett. 9, 1301–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhatia S, Krieger V, Groll M, Osko JD, Reβing N, Ahlert H, Borkhardt A, Kurz T, Christianson DW, Hauer J, and Hansen FK (2018) Discovery of the first-in-class dual histone deacetylase-proteasome inhibitor. J. Med. Chem. 61, 10299–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackwitz MKW, Hamacher A, Osko JD, Held J, Schöler A, Christianson DW, Kassack MU, and Hansen FK (2018) Multicomponent synthesis and binding mode of imidazo[1,2- α]pyridine-capped selective HDAC6 inhibitors. Org. Lett. 20, 3255–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Cryst. D 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011) iMOSFLM: a new graphical interface for diffraction image processing with MOSFLM. Acta Cryst. D 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution? Acta Cryst. D 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Cryst. D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. D 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cook C, Carlomagno Y, Gendron TF, Dunmore J, Scheffel K, Stetler C, Davis M, Dickson D, Jarpe M, DeTure M, and Petrucelli L (2014) Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 23, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, and Zweckstetter M (2015) Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. U. S. A. 112, 7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kar S, Fan J, Smith MJ, Goedert M, and Amos LA (2003) Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J. 22, 70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soppina V, Herbstman JF, Skiniotis G, and Verhey KJ (2012) Luminal localization of α-tubulin K40 acetylation by cryo-EM analysis of Fab-labeled microtubules. PLOS One 7, e48204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roberts AJ, Kon T, Knight PJ, Sutoh K, and Burgess SA (2013) Functions and mechanics of dynein motor proteins. Nat. Rev. Mol. Cell Biol. 14, 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grotjahn DA, and Lander GC (2019) Setting the dynein motor in motion: new insights from electron tomography. J. Biol. Chem. 294, 13202–13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kutil Z, Skultetyova L, Rauh D, Meleshin M, Snajdr I, Novakova Z, Mikesova J, Pavlicek J, Hadzima M, Baranova P, Havlinova B, Majer P, Schutkowski M, Barinka C (2019). The unraveling of substrate specificity of histone deacetylase 6 domains using acetylome peptide microarrays and peptide libraries. FASEB J. 33, 4035–4045. [DOI] [PubMed] [Google Scholar]