

SUMMARY
In the eukaryotic cell, spliceosomes assemble onto pre-mRNA cotranscriptionally. Spliceosome assembly takes place in the context of the chromatin environment, suggesting that the state of the chromatin may affect splicing. The molecular details and mechanisms through which chromatin affects splicing, however, are still unclear. Here, we show a role for the histone methyltransferase Set2 and its histone modification, H3K36 methylation, in pre-mRNA splicing through high-throughput sequencing. Moreover, the effect of H3K36 methylation on pre-mRNA splicing is mediated through the chromodomain protein Eaf3. We find that Eaf3 is recruited to intron-containing genes and that Eaf3 interacts with the splicing factor Prp45. Eaf3 acts with Prp45 and Prp19 after formation of the precatalytic B complex around the time of splicing activation, thus revealing the step in splicing that is regulated by H3K36 methylation. These studies support a model whereby H3K36 facilitates recruitment of an “adapter protein” to support efficient, constitutive splicing.
Graphical Abstract
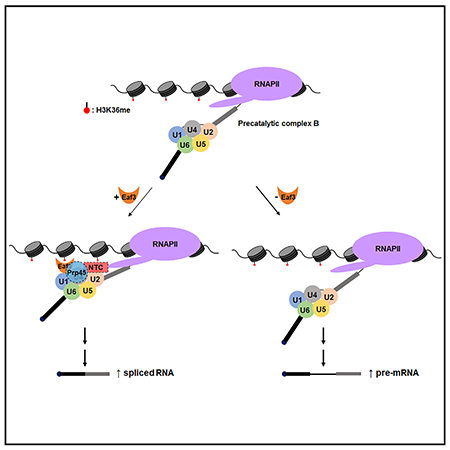
In Brief
Leung et al. demonstrate that H3K36 trimethylation facilitates efficient pre-mRNA splicing through the association of chromodomain protein Eaf3. Eaf3 binds to methylated H3K36 at intron-containing genes to stabilize association of the splicing factor Prp45 and regulate proper cotranscriptional spliceosome assembly.
INTRODUCTION
RNA splicing is a critical and highly regulated process in eukaryotic gene expression. RNA polymerase II (Pol II) catalyzes the synthesis of protein-coding genes to produce unspliced precursor mRNA (pre-mRNA). These genes contain intervening sequences that are removed during pre-mRNA splicing by the spliceosome, a dynamic macromolecular machine composed of five ribonucleoprotein subunits (U1, U2, U4, U5, and U6 small nuclear ribonucleoproteins [snRNPs]) and many associated protein cofactors (Will and Lührmann, 2011). Components of the spliceosome assemble around the splice site consensus sequences located at both ends of the intron. Accurate pre-mRNA splicing by the spliceosome is crucial, and many human diseases are associated with splicing defects or misregulation (Singh and Cooper, 2012).
Alternative splicing allows a single gene to have two or more mature mRNA variants, thus expanding protein diversity in eukaryotes. Alternative splicing occurs in ~95% of human genes, while splicing occurs less frequently in the budding yeast Saccharomyces cerevisiae, whose genome has few annotated introns (Ares et al., 1999; Pan et al., 2008). Nonetheless, the spliceosome is highly conserved; over 30% of the messages in yeast are derived from intron-containing genes (ICGs) (Ares et al., 1999), and splicing is regulated in response to changes in environmental conditions (Johnson and Vilardell, 2012), making this biochemically and genetically tractable organism a good model system for studying the basic mechanisms of splicing.
Recent studies have shown that pre-mRNA splicing and transcription are closely linked. Components of the spliceosome have been shown to associate with pre-mRNA as it is being actively transcribed by Pol II, and splicing of many introns is completed before transcription termination (Lacadie et al., 2006; Oesterreich et al., 2016; Pandya-Jones and Black, 2009). A variety of molecular approaches have been used to show that U1, U2, and the U4/U6*U5 tri-snRNP are recruited to the nascent RNA in a stepwise manner (Görnemann et al., 2005; Gunderson and Johnson, 2009; Kotovic et al., 2003). Pol II elongation rates can also affect splicing. Pausing of Pol II at the terminal exon usually promotes splicing, and slowing of the polymerase can allow inclusion of exons that contain weak splice sites by decreasing the synthesis of competing, downstream splice sites (de la Mata et al., 2003; Howe et al., 2003). In some cases, slow elongation rates can also allow for the recruitment of splicing repressors, which leads to exon skipping (Jonkers and Lis, 2015). There is also evidence in yeast and mammals that there may be an optimal elongation rate for splicing of individual introns (Fong et al., 2014; Neves et al., 2017).
Recently, a new layer of complexity has been added to our understanding of cotranscriptional splicing. Specifically, changes in chromatin affect splicing (Hnilicová and Staněk, 2011; Luco and Misteli, 2011). The N-terminal “tails” of histones extend out from the face of the nucleosome and undergo posttranslational modification. It has been well established that, in mammals, histone modification and positioning of nucleosomes play significant roles in regulating gene activity (Kouzarides, 2007). Nucleosomes are preferentially found on exons, and different sets of histone modifications have been identified at exons and at introns (Andersson et al., 2009; Tilgner et al., 2009). In yeast, deletion of the histone acetyltransferase Gcn5 affects cotranscriptional recruitment of U2 snRNP components (Gunderson and Johnson, 2009; Gunderson et al., 2011); H2B ubiquitylation stimulates recruitment of the early splicing factors (Hérissant et al., 2014); and histone variant H2A.Z is required for efficient splicing (Neves et al., 2017; Nissen et al., 2017).
Studies have previously implicated H3K36 methylation (H3K36me) in regulating alternative pre-mRNA splicing in human cells (Luco et al., 2010; Simon et al., 2014). In yeast and metazoans, H3K36me3 occupies the gene body of actively transcribed genes (Bannister et al., 2005; Krogan et al., 2003). In mammals, worms, and flies, H3K36me3 has been shown to be enriched on exons relative to introns, suggesting a link between this histone modification and alternative splicing (Andersson et al., 2009; Huff et al., 2010; Kolasinska-Zwierz et al., 2009; Schwartz et al., 2009; Spies et al., 2009). It has also been reported that H3K36me is important for proper splicing of a reporter construct in budding yeast (Sorenson et al., 2016). Nonetheless, despite its conservation and likely conserved role in pre-mRNA splicing, the mechanism by which this histone mark regulates pre-mRNA splicing has remained elusive.
Here, we investigate the role of the histone methyltransferase Set2 and its associated chromatin mark, H3K36me, in determining splicing outcomes. Through high-throughput RNA sequencing (RNA-seq), we identify transcripts whose efficient splicing is dependent on the presence of H3K36 methylation. To understand the mechanism by which H3K36me affects splicing, we have analyzed molecular events downstream of H3K36me and have identified the chromodomain-containing protein Eaf3 as a mediator of the interaction between the splicing machinery and H3K36 methylation. Loss of Eaf3 leads to defective splicing of the same genes whose splicing is sensitive to the presence of H3K36me. Moreover, Eaf3 physically interacts with the splicing factor Prp45, the yeast ortholog of human SKIP. Deletion of EAF3 leads to inefficient recruitment of Prp45 to ICGs, suggesting that Eaf3 is important for cotranscriptional spliceosome assembly. These studies provide a mechanistic basis for a highly conserved histone modification in RNA splicing.
RESULTS
Defective Splicing Is Observed in set2Δ and H3K36A Mutants
To determine how H3K36me regulates pre-mRNA splicing, we assayed yeast cells in which SET2, the gene that encodes the histone methyltransferase for H3K36, is deleted. We also use the histone point mutant strain, H3K36A, to verify that the splicing changes we observe are due to loss of histone methylation as opposed to a non-histone target. Western blot analysis confirmed complete loss of H3K36me3 in the set2Δ and H3K36A mutants (Figure 1A). To observe genome-wide changes in splicing upon loss of H3K36me, total RNA was isolated from wild-type, set2Δ, and H3K36A cells, and stranded RNA-seq was performed.
Figure 1. H3K36 Methylation Is Required for Efficient Pre-mRNA Splicing.
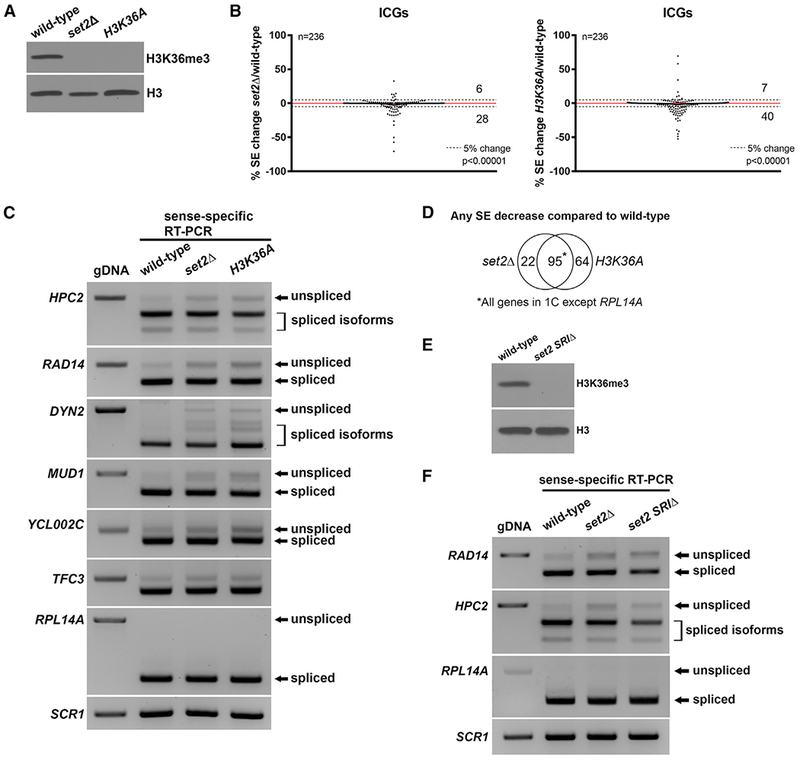
(A) H3K36me3 is absent in the set2Δ and H3K36A mutants. Whole cell extracts from wild-type, set2Δ, and H3K36A cells were subjected to western blotting.
(B) Changes in splicing efficiencies (SEs) of ICGs upon loss of H3K36me represented in a scatterplot. Dashed lines represent a 5% change in SE in the mutant compared to wild-type. Overall splicing decreases in both set2Δ and H3K36A compared with wild-type (chi-square test, p value indicated). Numbers indicate number of ICGs above and below the dashed lines.
(C) RT-PCR validation of splicing changes observed in RNA-seq analysis. ICGs shown display a ≥ 5% decrease in SE in both set2Δ and H3K36A compared with wild-type. RPL14A is a gene that does not show a change in SE. Products were analyzed on a 1.8% agarose gel.
(D) Venn diagram displaying significant overlap of ICGs that display any SE decrease in set2Δ and H3K36A compared with wild-type cells (p < 0.0001, chi-square test).
(E) H3K36me3 is absent in a set2 SRIΔ mutant. Whole cell extracts from wild-type and set2 SRIΔ were subjected to western blotting.
(F) RT-PCR analysis of splicing changes in set2 SRIΔ cells. Products were analyzed on a 1.8% agarose gel. gDNA, genomic DNA. SCR1 is a loading control.
We observe a decrease in the splicing efficiency (SE) of a group of ICGs in both the set2Δ and H3K36A mutants (p < 0.0001) compared with wild-type cells (Figure 1B) suggesting that loss of H3K36me leads to decreased SE. Intron accumulation was verified by RT-PCR analysis; representative genes are shown (Figure 1C). To rule out the possibility that the SE changes are due to changes in splicing of MUD1, a gene that encodes a splicing factor and shows intron accumulation, protein levels of endogenously HA-tagged Mud1 were examined. Mud1 levels are similar in set2Δ compared with wild-type cells (Figure S1A). Furthermore, SEs of all other splicing factor ICGs do not change in either the set2Δ or H3K36A mutants compared with wild-type cells in our RNA-seq data. RT-PCR analysis of splicing factor encoding ICGs show neither expression nor splicing changes in the H3K36A mutant; a representative example is shown in Figure S1B. Because of strict filtering, we knew it was possible that lowly expressed genes that might still display a change in splicing were filtered out. Therefore, we relaxed the read count requirement, and upon reanalysis of the RNA-seq data, we observed a larger set of ICGs that displayed splicing defects in both set2Δ and H3K36A; a representative RT-PCR analysis is shown (Figure S1C). There is significant overlap of the ICGs that show a decrease in SE in set2Δ and H3K36A compared with wild-type cells (p < 0.0001), indicating that the splicing defects observed are due to loss of H3K36me and not loss of the Set2 protein itself (Figure 1D).
Set2 associates with the phosphorylated carboxyl-terminal domain (CTD) of Pol II via its SRI domain and methylates histones cotranscriptionally (Li et al., 2002; Xiao et al., 2003). To determine whether loss of Set2 binding to the CTD affects splicing, we generated a set2 SRIΔ mutant. We confirmed loss of H3K36me3 in the set2 SRIΔ mutant (Figure 1E). RT-PCR of the genes RAD14 and HPC2 revealed splicing defects similar to those observed in the set2Δ mutant (Figure 1F). This further suggests that the splicing defects we observed are not due to non-histone targets of Set2 and that cotranscriptional methylation of H3K36 is important for splicing. It has been previously reported that deletion of the SRI domain of Set2 selectively abolishes H3K36me3 but not H3K36me1 or H3K36me2 (Jha and Strahl, 2014). Therefore, it is likely that the splicing defects we observed in our set2Δ and H3K36A mutants are due to loss of H3K36me3.
We also analyzed two other H3K36 point mutants, H3K36R and H3K36Q, and both confer splicing defects consistent with that of H3K36A (Figure S1D), further indicating that this residue is important for proper splicing. Moreover, since H3K36Q mimics constitutive acetylation, these data suggest that the lack of methylation underlies the splicing defect.
H3K36 Methylation Effects on Splicing Are Unlikely to Be Due to Transcription Defects
Because spliceosome assembly and subsequent splicing catalysis are cotranscriptional, the elongation properties of Pol II can affect splicing (Alexander et al., 2010; de la Mata et al., 2003; Fong et al., 2014). To address whether the observed splicing changes in set2Δ and H3K36A cells are due to changes in transcription, we calculated the reads per kilobase of transcript per million mapped reads (RPKM) of all sense transcripts from the stranded RNA-seq data. The RNA abundance of all genes, including splicing factor genes and snRNAs, is strongly correlated between the set2Δ and H3K36A mutants and wild-type (rs = 0.9556 and rs = 0.9051, respectively) (Figure S2A). Expression of ICGs is also strongly correlated between the mutants and wild-type (Figure S2B). In addition, RNA abundance of all genes in set2Δ and H3K36A cells are also very strongly correlated (rs = 0.9744), suggesting that the expression profiles of the two mutants are nearly identical (Figure S2C). Recent studies indicate that changes in expression of the highly expressed ribosomal protein intron-containing genes (RP ICGs) leads to changes in splicing because of competition between pre-mRNAs for the spliceosome in yeast (Munding et al., 2013; Awad et al., 2017; Venkataramanan et al., 2017). However, expression of RP ICGs is not significantly altered in the mutants (Figure S2B). When we compare the SE change of ICGs that show a greater than two-fold decrease in expression to those that show a less than two-fold decrease in expression in the H3K36A mutant compared with wild-type, we do not observe a significant difference in SE between the two groups (Figure S2D). Finally, we performed chromatin immunoprecipitation sequencing (ChIP-seq) on the elongating form of Pol II (phospho-S2), in wild-type and H3K36A cells. We observed a weak correlation (rs = −0.2371) between SE change and Pol II occupancy change on all ICGs in H3K36A compared with wild-type cells, suggesting that the splicing defects observed cannot be easily explained by transcription defects (Figure S2E).
Loss of Chromodomain Protein Eaf3 Leads to Defective Pre-mRNA Splicing
We performed ChIP-seq on H3K36me3 in wild-type cells and observed that H3K36me3 is enriched on ICGs compared with intronless genes (Figure 2A). For example, when we compared the H3K36me3 profile of intron-containing HPC2 to that of an intronless gene with similar expression and gene length (PUT4), we observed more H3K36me3 throughout the HPC2 gene (Figure 2B). We considered the possibility that a protein binds methyl marks on histones to facilitate recruitment of the splicing machinery. In mammals, MRG15, the yeast homolog of Eaf3, has been suggested to regulate alternative splicing via binding to H3K36me3 (Iwamori et al., 2016; Luco et al., 2010). Eaf3 contains a chromodomain that allows it to bind to methylated H3K36 and is a subunit of the Rpd3S histone deacetylase complex and the NuA4 histone acetyltransferase complex in yeast (Reid et al., 2004). We hypothesized that Eaf3 may recruit the splicing machinery to the chromatin and that the splicing changes in the set2Δ and H3K36A cells are due to loss of binding of Eaf3 to H3K36. To test this hypothesis, EAF3 was deleted and stranded RNA-seq was performed to assess splicing. We observe a widespread decrease in SE (p = 0.0077) (Figure 2C). RT-PCR confirmed the splicing changes observed from the RNA-seq analysis for the same subset of genes (Figure 2D). There is a significant (p < 0.0001) overlap between ICGs that show any decrease in SE in eaf3Δ and H3K36A compared with wild-type (Figure 2E). ChIP analysis of endogenously HA-tagged Eaf3 (Eaf3-HA) reveals significant decrease in Eaf3 occupancy on ICGs in the set2Δ mutant; HPC2 and RAD14 are shown here (Figure 2F). Levels of Eaf3-HA protein are identical between wild-type and set2Δ cells (Figure 2G). When we performed ChIP of Eaf3-HA on ECM33, a gene that does not show a splicing defect in the absence of H3K36me, we did not observe a significant decrease in Eaf3-HA occupancy, suggesting that loss of Eaf3 on an ICG may be correlated with a decrease in splicing of that ICG (Figure 2H, left). We also analyzed Eaf3-HA occupancy ± SET2 on an intronless gene, ADH1, and observe only a modest and not significant decrease of Eaf3 on the gene (Figure 2H, right). These results suggest that Eaf3 requires other proteins in addition to H3K36me to stabilize its interaction with chromatin, but that its loss, nonetheless, has a deleterious effect on splicing.
Figure 2. Eaf3 Is Required for Efficient Pre-mRNA Splicing.
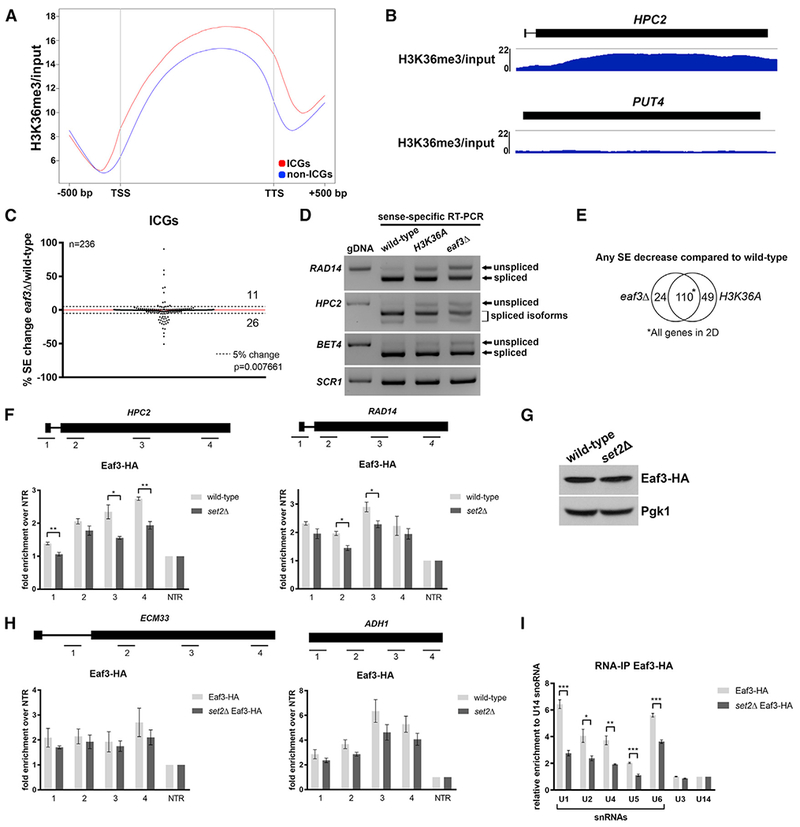
(A) Metagene plot of H3K36me3 ChIP-seq enrichment across the transcribed region and 500 nucleotides (nt) upstream of the transcription start site (TSS) and 500 nt downstream of the transcription termination site (TTS) of ICGs and non-ICGs.
(B) Genome-browser tracks of H3K36me3 ChIP-seq enrichment on ICG HPC2 (RPKM: 7.355; length: 1962 bp) and intronless gene PUT4 (RPKM: 7.658; length: 1884 bp).
(C) Scatterplot of percentage change in splicing efficiency (SE) of all ICGs in eaf3Δ over wild-type. Dashed lines represent a 5% change in SE. Numbers indicate number of ICGs above and below the dashed lines.
(D) RT-PCR validation of splicing changes observed in the eaf3Δ mutant. Products were run on a 1.8% agarose gel. SCR1 is a loading control.
(E) Venn diagram displaying significant overlap of ICGs that display any SE decrease in eaf3Δ and H3K36A compared to wild-type cells (p < 0.0001, chi-square test).
(F) Occupancy of Eaf3-HA on ICGs HPC2 (left) and RAD14 (right) relative to a non-transcribed region (NTR) on chromosome V (Chr. V) in wild-type (light gray bars) and set2Δ cells (dark gray bars).
(G) Western blot analysis of Eaf3-HA protein levels in wild-type and set2Δ cells. Pgk1 is a loading control.
(H) Occupancy of Eaf3-HA on ICG ECM33 relative to a NTR on Chr. V in wild-type and set2Δ cells (left). Occupancy of Eaf3-HA on intronless gene ADH1 relative to a NTR on Chr. V in wild-type and set2Δ cells (right).
(I) Spliceosomal snRNAs co-immunoprecipitated with Eaf3. Bar graph depicting the five spliceosomal snRNAs that are immunoprecipitated with Eaf3 in wild-type and set2Δ cells under non-crosslinking conditions. U3 and U14 are snoRNAs. Bars represent the average of 3 biological replicates. Error bars represent the SEM. p values were determined by 2-tailed unpaired t test. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
To determine whether Eaf3 interacts with the spliceosome, we performed RNA immunoprecipitation (RIP) without crosslinking on HA-tagged Eaf3. Eaf3-HA was immunoprecipitated and qPCR was performed to determine whether any of the spliceosomal snRNAs co-precipitate. Interestingly, we observed enrichment of the snRNAs relative to other small RNA controls (Figure 2I). Loss of Set2 results in decreased binding between Eaf3-HA and the five snRNAs, indicating that this interaction is mediated through interaction between Eaf3 and chromatin, perhaps before release of U1 snRNA before spliceosome activation (Figure 2I). To determine whether the binding of snRNAs to Eaf3 was specific, we also performed RIP with HA-tagged Chd1, another yeast chromodomain protein. In contrast to Eaf3, we did not observe a strong interaction between Chd1-HA and snRNAs (Figure S3A).
Previous studies have implicated Eaf3 in recruitment of the Rpd3S complex, which includes Rpd3, to deacetylate the histone H3 tail (Keogh et al., 2005). Moreover, Eaf3 also recruits the NuA4 histone acetyltransferase, which acetylates the four conserved lysines on the histone H4 tail, to chromatin (Sathianathan et al., 2016). We considered the possibility that the inability to acetylate the histone H4 tail or deacetylate the histone H3 tail might underlie the splicing defect observed in the eaf3Δ mutant. To test this, we performed RT-PCR to analyze splicing of selected ICGs in eaf3Δ, rpd3Δ, and H4K5,8,12,16R cells. Neither deletion of RPD3 nor mutation of H4 phenocopy the splicing defects observed in the eaf3Δ mutant (data not shown).
RNA abundance in the eaf3Δ mutant is strongly correlated with RNA abundance in the H3K36A mutant, which suggests that their overall expression profiles are nearly identical (rs = 0.9754) (Figure S3B). Lastly, we performed ChIP-seq on Pol II (phospho-S2) and, similar to the H3K36A mutant, we did not observe a correlation (rs = −0.09173) between change in SE and change in the occupancy of the elongating Pol II on ICGs in the eaf3Δ mutant (Figure S3C).
Eaf3 Is Required for the Efficient Cotranscriptional Recruitment of Splicing Factor Prp45
Eaf3 has been previously reported to physically interact with the splicing factor Prp45 using a high-throughput yeast two-hybrid screen (Albers et al., 2003). We confirmed this physical interaction by performing a co-immunoprecipitation (coIP) between Eaf3-HA and endogenously Myc-tagged Prp45 (Prp45-Myc) (Figure 3A). This result is consistent with physical interaction between the two proteins, although we suspected that the interaction was weak and/or transient. To address this and confirm that the interaction occurred in vivo, we performed crosslinking of whole cells followed by coIP (Figure 3B). We next analyzed the cotranscriptional recruitment of Prp45 in the absence of Eaf3. Spliceosome assembly has been shown to occur in a stepwise manner cotranscriptionally by ChIP-qPCR, and ChIP signals are routinely used as a proxy for the timing of association of splicing factors with the pre-mRNA (Kotovic et al., 2003; Görnemann et al., 2005; Gunderson and Johnson, 2009; Gunderson et al., 2011). The recruitment profile of Prp45 has recently been reported using this approach (Hálová et al., 2017). We performed ChIP on endogenously tagged Prp45 (Prp45-HA) in wild-type, set2Δ, and eaf3Δ cells on HPC2 and RAD14, two genes that displayed strong splicing defects in the mutants. In wild-type cells, we observed a Prp45 ChIP pattern similar to what has been previously described (Figure 3C). Deletion of Eaf3 leads to decreased Prp45-HA occupancy compared to wild-type, indicating less stable association of Prp45-HA on both HPC2 and RAD14 (Figure 3C). In the set2Δ mutant, we observed an almost identical reduction in Prp45-HA binding to the eaf3Δ mutant (Figure 3C); Prp45-HA protein levels are identical in wild-type and the two mutants (Figure 3D). This suggests that the inefficient recruitment of Prp45 to HPC2 and RAD14 in set2Δ is due to loss of Eaf3 binding to chromatin. ChIP of Prp45 on a gene that does not show a splicing defect in the absence of eaf3Δ or set2Δ, shows no change in Prp45 occupancy (Figure 3E, left), indicating that, for this gene, Prp45 occupancy is not sensitive to Set2 or Eaf3. The intronless gene ADH1 shows negligible Prp45 occupancy which does not appear to be affected by Set2 or Eaf3 (Figure 3E, right).
Figure 3. Eaf3 Is Required for the Proper Cotranscriptional Recruitment of Prp45.
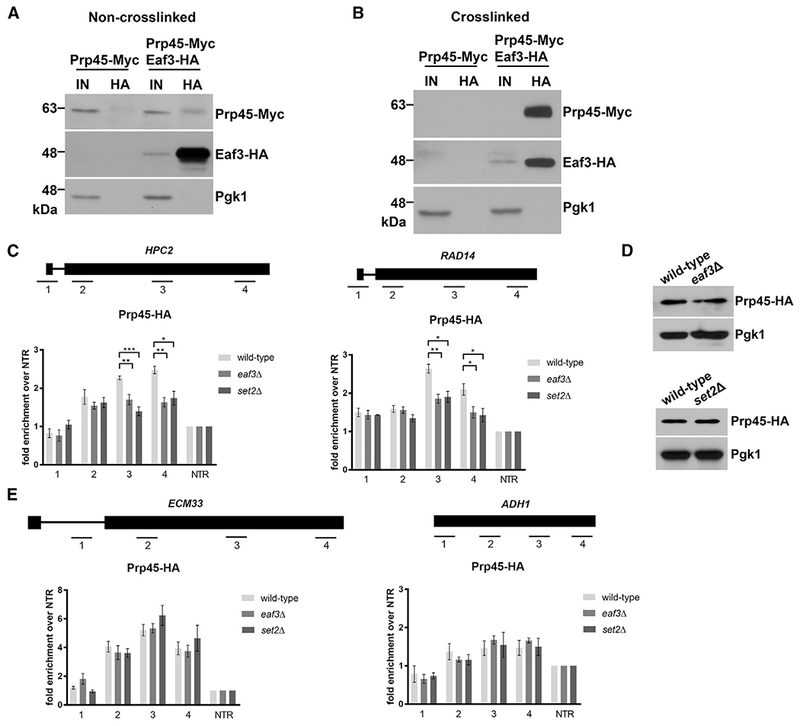
(A) Non-crosslinked co-immunoprecipitation (coIP) of Eaf3-HA and Prp45-Myc. Eaf3-HA was immunoprecipitated with an anti-HA antibody. Input is 2% of total lysate used for pull-down. Products were analyzed by western blotting with an anti-HA antibody and anti-Myc antibody.
(B) Crosslinked coIP of Eaf3-HA and Prp45-Myc. Whole cell extracts were crosslinked with 1% for maldehyde. Eaf3-HA was immunoprecipitated with an anti-HA antibody. Input is 2% of total lysate used for pull-down. Products were analyzed by western blotting with an anti-HA antibody and anti-Myc antibody.
(C) Occupancy of Prp45-HA on ICGs HPC2 (left) and RAD14 (right) relative to a non-transcribed region (NTR) on Chr. V in wild-type (light gray bars), eaf3Δ (medium gray bars), and set2Δ (dark gray bars).
(D) Western blot analysis of Prp45-HA protein levels in wild-type and eaf3Δ (top), and set2Δ (bottom) cells. Pgk1 is a loading control.
(E) Occupancy of Prp45-HA on ICG ECM33 and intronless gene ADH1 relative to a NTR on Chr. V. in wild-type, eaf3Δ, and set2Δ. Bars represent the average of at least 3 biological replicates. Error bars represent the SEM. p values were determined by 2-tailed unpaired t test. *, p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Efficient Cotranscriptional Recruitment of Prp19 Depends on the Presence of Eaf3
Prp45 has been shown to interact with components of the nineteen complex (NTC) (Fabrizio et al., 2009), a complex important for spliceosome activation. Moreover, Prp45 has been shown to be necessary for NTC recruitment early in cotranscriptional spliceosome assembly (Hálová et al., 2017). Because efficient recruitment of Prp45 depends on Eaf3, we hypothesized that recruitment of the NTC would also be negatively affected by the absence of Eaf3. We endogenously HA-tagged Prp19 and determined that protein levels of Prp19 do not change in an eaf3Δ mutant (Figure 4A). We then performed ChIP of Prp19-HA in wild-type and eaf3Δ cells and observed a significant decrease in recruitment of Prp19-HA to ICGs HPC2 and RAD14 (Figure 4B) in the absence of Eaf3. Similar to what was observed when we ChIP Prp45 (Figure 3E), there was no change of occupancy of Prp19-HA on ECM33 (Figure 4C).
Figure 4. Prp19 Is Inefficiently Recruited to ICGs in the Absence of Eaf3.
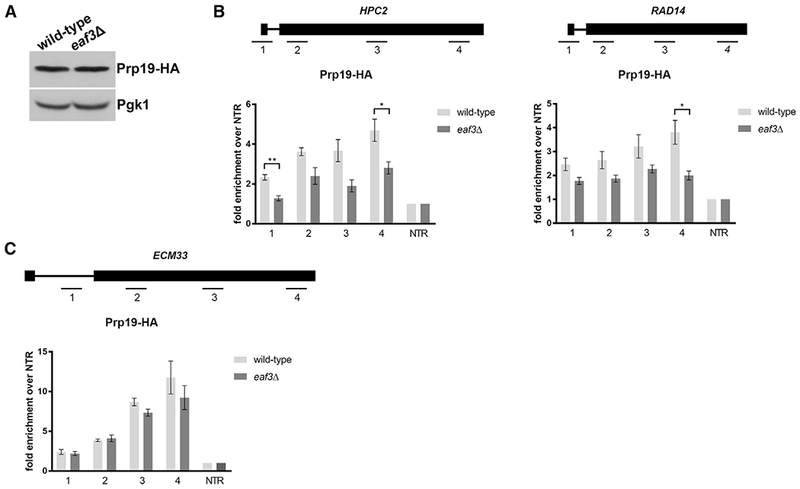
(A) Western blot analysis of Prp19-HA protein levels in wild-type and eaf3Δ cells. Pgk1 is a loading control.
(B) Occupancy of Prp19-HA on ICGs HPC2 (left) and RAD14 (right) relative to a non-transcribed region (NTR) on Chr. V in wild-type (light gray bars) and eaf3Δ (medium gray bars).
(C) Occupancy of Prp19-HA on ICG ECM33 relative to a NTR on Chr. V. in wild-type and eaf3Δ cells. Bars represent the average of at least 3 biological replicates. Error bars represent the SEM. p values were determined by 2-tailed unpaired t test. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
The NTC plays a critical role in catalytic activation of the spliceosome and associates with the spliceosome along with the U4/U6*U5 tri-snRNP around the time of U4/U6 unwinding and concomitant release of U1 and U4 (Fabrizio et al., 2009). Prp45 physically interacts with the NTC and co-purifies with the precatalytic complex B (Albers et al., 2003; Fabrizio et al., 2009), suggesting that the NTC, as well as NTC-associated protein Prp45, facilitates the transition from the precatalytic complex B to the activated complex BACT. Eaf3 also appears to play an important role in these steps (Figure 5).
Figure 5. Eaf3 Recognizes H3K36 Methylation to Regulate Pre-mRNA Splicing.
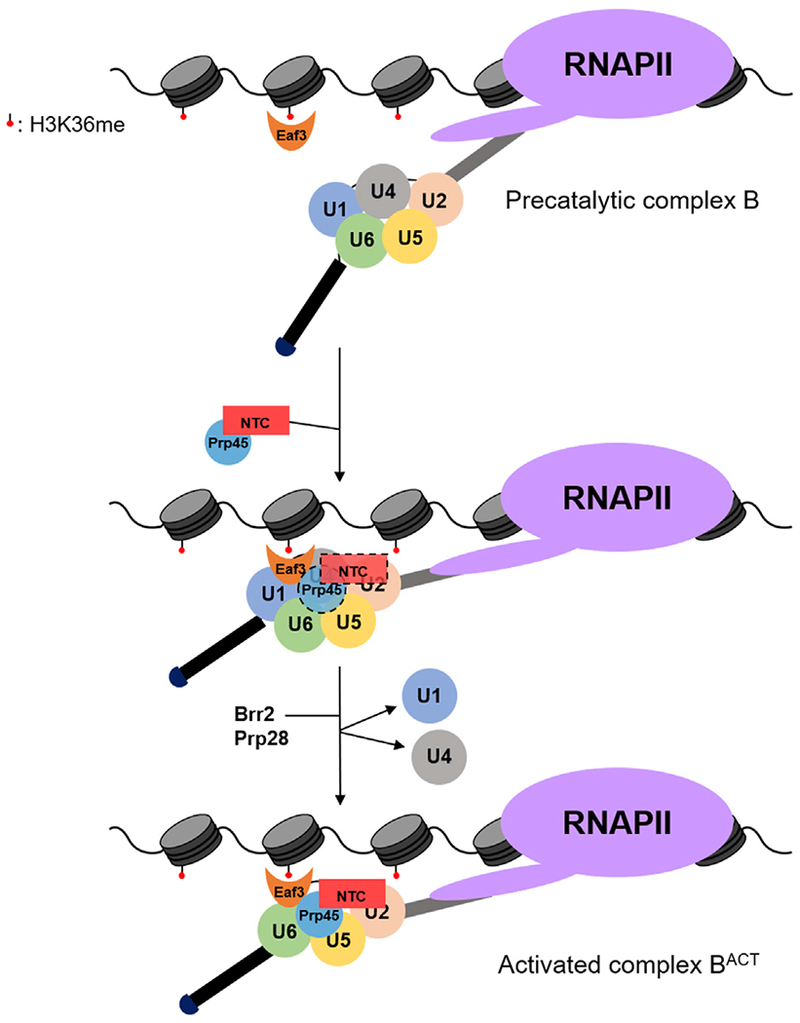
Eaf3 recruits Prp45 and the nineteen complex (NTC) to the precatalytic complex B to promote spliceosome activation. The putative NTC and Prp45 interaction with the spliceosome prior to U1 and U4 snRNP release may be transient (indicated by dashed lines).
DISCUSSION
Posttranslational modifications on histones have been implicated in the regulation of RNA splicing (Andersson et al., 2009; Hérissant et al., 2014; Hnilicová and Staněk, 2011). H3K36me3 was first reported to regulate exon selection in the FGFR2 gene in human cells via recruitment of chromatin-associated protein MRG15 and polypyrimidine tract-binding protein (PTB) (Luco et al., 2010). Nonetheless, the sometimes subtle, gene-specific splicing effects and incomplete overlap between MRG15- and PTB-splicing dependent events left open the possibility that there were other effects of H3K36me3 on splicing that remained to be elucidated. Another report suggested that PWWP domain-containing protein Psip1/p52 binds to H3K36me3 and recruits SR-containing proteins and splicing factors to regulate alternative splicing of a specific set of genes (Pradeepa et al., 2012). Studies have also linked H3K36 methylation to genome-wide splicing effects, although the underlying mechanisms have been less clear (Guo et al., 2014; Simon et al., 2014). H3K36me has also been implicated in regulating splicing in yeast via a gene expression reporter (Sorenson et al., 2016). This reporter screen made use of the intron in RPL28, an efficiently spliced ICG (Sorenson and Stevens, 2014), which does not uncover the effects of H3K36me loss on splicing genome-wide or its role in expression of genes that are less efficiently spliced.
H3K36 Methylation Is Required for Constitutive Splicing in Yeast
Here we show that H3K36me3 is required for efficient constitutive pre-mRNA splicing genome-wide (Figure 1B). We observed that H3K36 methylation is central to core splicing events and not exclusively to exon inclusion events. Our data are consistent with a recruitment model by which H3K36me recruits the chromodomain protein Eaf3 to mediate the interaction between chromatin and the spliceosome. Indeed, deletion of EAF3 phenocopies the splicing of the same genes that are sensitive to loss of H3K36me (Figure 2E).
One of the additional reported functions of H3K36 methylation is to suppress cryptic and antisense transcription within gene bodies through recruitment of the Rpd3S complex (Keogh et al., 2005). Since Eaf3 is also a component of the Rpd3S complex, it is tempting to speculate that antisense transcription might affect splicing of the sense transcript. However, from our stranded RNA-seq analysis, we did not observe a significant correlation between antisense transcription and changes in SE (data not shown). Although antisense transcription does not seem to affect RNA splicing, we observe several antisense transcripts that are spliced, albeit rather inefficiently (Douglass et al., 2019).
Eaf3 Interaction with Prp45 Affects Its Role in Splicing
We reveal a mechanism by which the chromodomain-containing protein Eaf3 binds methylated H3K36 and interacts physically with Prp45 (Figures 3A and 3B) to facilitate stable Prp45 association during cotranscriptional spliceosome assembly (Figure 3C). Recent cryo-EM structures of S. cerevisiae and S. pombe spliceosomes have revealed that Prp45 is an intrinsically disordered protein that spans over 150 Å and stabilizes the catalytic center through extensive contacts with several proteins and snRNAs (Wan et al., 2016; Yan et al., 2015; Yan et al., 2016). We propose that because Prp45 is found throughout the splicing process and after spliceosome assembly (Albers et al., 2003), Eaf3 is well positioned to link the spliceosome to chromatin until the splicing reaction is completed (Figure 5).
Interestingly, the human ortholog of Prp45 is SKIP, a protein involved in both transcription and splicing (Brès et al., 2005; Makarov et al., 2002; Nagai et al., 2004; Neubauer et al., 1998). In yeast, Prp45 fused with a DNA-binding domain has been shown to activate transcription of a reporter gene (Martínková et al., 2002). Like SKIP, it is possible that Prp45 bridges transcription initiation and pre-mRNA processing but may depend on Eaf3 or other factors for its binding to chromatin since Prp45 lacks many of the binding motifs found in SKIP.
Eaf3 Is Required for the Proper Cotranscriptional Recruitment of Prp19
All five spliceosomal snRNAs immunoprecipitate with Eaf3, suggesting that, perhaps, its role in splicing occurs when all five snRNPs are present. Since we show Eaf3 interacts with Prp45, which is associated with the NTC, we hypothesize that Eaf3 acts between the precatalytic complex B and the activated complex BACT. Indeed, in the absence of Eaf3, cotranscriptional recruitment of Prp19 to ICGs HPC2 and RAD14 is diminished (Figure 4B). We propose a model whereby Eaf3 stabilizes Prp45 and NTC interactions with the precatalytic complex B spliceosome to facilitate major structural rearrangements within the spliceosome, including the irreversible loss of U4 that occurs immediately after NTC association (Hoskins et al., 2016).
Prp19 has been implicated in a variety of processes, in addition to splicing, in both yeast and higher eukaryotes. These include genome maintenance, recruitment of ubiquitylated proteins to the proteasome, liquid droplet formation, and importantly, transcription elongation (Chanarat and Sträßer, 2013). This raises the possibility that Eaf3′s role in directing NTC activity may extend beyond effects on splicing, a question we are currently addressing.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tracy L. Johnson (tljohnson@ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The S. cerevisiae strains used in this study are listed in Table S1. All strains were grown in YPD (1% yeast extract, 2% peptone, and 2% dextrose) at 30°C. All strains are derived from BY4741. Individual deletion strains were obtained from GE Dharmacon. 3HA- and 13Myc-endogenous C-terminal tagged strains were generated by homologous recombination following PCR amplification from the pFA6a-3HA-kanMX6, pFA6a-3HA-His3MX6, and pFA6A-13Myc-kanMX6 plasmids as described previously (Longtine et al., 1998).
METHOD DETAILS
RNA Isolation
RNA was isolated from yeast during log-phase (OD600 = 0.5–0.6) by hot phenol:chloroform:isoamyl alcohol (PCA) extraction with SDS as described previously (Ares, 2012). Ethanol precipitation was performed to concentrate the RNA.
RNA-seq Library Preparation and Analysis
Stranded RNA-seq libraries were prepared using the TruSeq Stranded Total RNA Kit (Illumina). Prior to library preparation, isolated total RNA from 2 biological replicates was treated with DNase I (Roche) and was subsequently depleted of ribosomal RNA using the Ribo-Zero Gold rRNA Removal Kit (Illumina). 100 bp single-end reads (Illumina HiSeq 4000) were aligned to the sacCer3 assembly (UCSC 2011) using TopHat 2.0.14 (Trapnell et al., 2009) with a maximum of 2 read mismatches, 0 splice mismatches, and a minimum anchor of 8 nucleotides. An average of 38,326,464 reads were obtained per sample, of which an average of 32,533,221 reads were mapped (84.9% of input). RPKMs were calculated by dividing read counts that aligned within a gene by the length of the gene in kilobases per million mapped reads. For intron-containing genes, exonic reads were quantitated by aligned reads that mapped entirely in an exon as defined by the Ares Lab Yeast Intron Database. Unspliced reads were defined as reads mapped within an annotated intron and reads mapped partially in an intron. Spliced reads were defined as splice junction reads. Introns that contained annotated snoRNAs were omitted from the splicing analysis. Spliced and unspliced read counts were normalized by the total number of unique alignments that can contribute to its count value. For spliced reads, this is simply read length minus one (99). For unspliced reads, we normalized by dividing by the intron length plus the read length minus one. Splicing efficiency (SE) was calculated using the formula: . SEs for all ICGs are listed in Table S2. For splicing analysis, ICGs with less than 3 total spliced reads between biological replicates were filtered out. For the relaxed read count splicing reanalysis, we eliminated the requirement for 3 total spliced reads and added a 0.01 normalized spliced pseudocount for ICGs that had a 0 spliced read count. Genes with an RPKM of less than 1 were filtered out prior to all data analysis.
RT-PCR Analysis
Following total RNA isolation, 20 μg of RNA was treated with DNase I (Roche). 500 ng-1 μg of DNase-treated RNA was used for cDNA synthesis. Sense-specific cDNA was generated using Maxima Reverse Transcriptase (ThermoFisher Scientific) and gene-specific primers. Gene-specific primers and RT primers are listed in Table S3. 1–2 μL of cDNA was used as the template in a 50 μL PCR reaction. Primers that flank the intron were used to observe splicing changes. The PCR products were run on a 1.8% agarose gel. Agarose gels were stained with ethidium bromide (ThermoFisher Scientific). Gels were then imaged on Image Lab (Bio-Rad).
Western Blot Analysis
50 mL of log-phase yeast were pelleted and resuspended in 500 μL of lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate) with protease inhibitors. An equal volume of 0.5 mm acid-washed glass beads (BioSpec) was added to the cell resuspension and the samples were lysed in a cell disruptor for 5 minutes at 4°C. Proteins were separated in a 10% SDS-PAGE gel for Eaf3-HA, Prp45-HA, and Prp45-Myc and a 15% SDS-PAGE gel for histone H3 and histone H3K36me3. HA blots were probed with anti-HA antibody (clone 12CA5, Roche), Myc blots were probed with anti-Myc antibody (clone 9E10, Roche), histone H3 blots were probed with anti-histone H3 (clone ab1791, Abcam), and histone H3K36me3 blots were probed with anti-histone H3K36me3 (clone ab9050, Abcam).
ChIP-seq Library Preparation and ChIP-seq Analysis
ChIP-seq has been described previously (Morselli et al., 2015) with the following modifications. 1% formaldehyde was added to 50 OD of yeast cells and gently rotated for 15 minutes at room temperature. 125mM glycine was then added to the cells and rotated for 5 minutes at room temperature to quench the crosslinking reaction. Cells were pelleted at 4°C at 4000 rpm and washed twice with 25 mL of ice-cold PBS. The pellet was resuspended in 400 μL of ice cold lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate). An equal volume of 0.5 mm acid-washed glass beads was added, and the resuspension was placed in a cell disruptor at 4°C for 5 minutes. The lysates were then sonicated using a Covaris S2 programmed to run 16 cycles (total sonication time: 8 minutes, duty cycle: 5%, intensity: 5%). DNA was sonicated to an average size of 300 bp. Following sonication, the samples were centrifuged for 10 minutes at 4°C. The supernatant was collected, and 10 μL from each sample of the was collected for input and stored at −20°C. 50 μL of each sample was incubated overnight at 4°C with 4 μg of anti-H3K36me3 (clone ab9050, Abcam) or anti-RNAPII S2P (clone ab5095, Abcam). The samples were incubated for 2 hours at 4°C with Protein A Dynabeads (ThermoFisher Scientific). The beads were washed with low salt buffer (140 mM NaCl), high salt buffer (500 mM NaCl), LiCl buffer, and TE (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) buffer. Beads were resuspended with elution buffer and incubated for 10 min at 65°C. Following elution, all tubes were incubated at 65°C overnight to reverse crosslinks. An RNase cocktail mix was added to all samples and incubated for 1 hour at 37°C. Proteinase K was added and the samples were incubated for 1 hour at 60°C. Sample were purified using 1.8 volumes of AMPure XP beads (Beckman Coulter). The real-time reaction was performed using iTaq Universal SYBR (Bio-Rad) on a CFX96 Real-Time PCR system (Bio-Rad). For ChIP-seq analysis, an Ovation Ultralow DR kit (NuGEN) was used to prepare the libraries using 1 ng of purified DNA. An Illumina HiSeq 4000 system was used for sequencing using 50 bp single-end reads. Reads were aligned to the sacCer3 (UCSC 2011) reference genome using Bowtie 0.12.8 (Langmead et al., 2009) with up to 2 mismatches. Peak calling was performed using Model-Based Analysis of ChIP-Seq (MACS2) (Zhang et al., 2008). A q-value cutoff of 0.05 was used when identifying significantly enriched regions of the genome using MACS2. Metagene profiles and heatmaps were generated using SeqPlots 3.7 (Stempor and Ahringer, 2016).
ChIP-qPCR Analysis
50 mL of log-phase yeast cells were crosslinked with 1% formaldehyde for 15 minutes at room temperature. The reaction was then quenched with 125mM glycine for 5 minutes at room temperature. Cells were pelleted and washed twice with 25 mL of ice-cold PBS and resuspended in 400 μL of lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate). The cells were then lysed by bead beating at 4°C for 5 minutes. The lysates were then sonicated using a Branson Digital Sonifier to run 15 cycles (total sonication time: 7.5 minutes, amplification: 15%) to an average size of 300 bp. The samples were then clarified for 10 minutes at 4°C. 10 μL from each sample of the was collected for input and 700 μL was incubated overnight at 4°C with 4 mg of anti-HA (clone 12CA5, Roche). 50 μL of GammaBind G Sepharose beads (GE Healthcare) were added to each sample and incubated for 2 hours at 4°C. The beads were washed four times in lysis buffer and twice in TE. The washed beads were then resuspended with elution buffer and incubated for 10 min at 65°C. All samples were incubated at 65°C overnight to reverse crosslinks. RNase A (ThermoFisher Scientific) was added to all samples and incubated for 1 hour at 37°C. Proteinase K (Roche) was added and the samples were incubated for 1 hour at 60°C. Following proteinase K treatment, DNA was extracted by column purification. Purified DNA was used for qPCR analysis using gene-specific primers (Table S3). The real-time reaction was performed using iTaq Universal SYBR (Bio-Rad) on a CFX96 Real-Time PCR system (Bio-Rad).
RNA Immunoprecipitation
150 mL of log-phase yeast were pelleted and resuspended in 1 mL of lysis buffer with RiboLock RNase inhibitor (ThermoFisher Scientific) and protease inhibitors. An equal volume of acid-washed beads was added to the resuspension. The cells were then lysed in a cell disruptor for 5 minutes at 4°C. The lysate was then centrifugated at 13,000 rpm at 4°C for 20 minutes. 10 μL of clarified lysate was removed as input. 40 μL of Pierce anti-HA magnetic beads (ThermoFisher Scientific) was added to the 1 mL of clarified lysate and rotated at 4°C overnight. Beads were washed once with lysis buffer and three times with TBS 0.05% tween. Acidic elution was performed, and samples were neutralized with Tris-HCl pH 8.5. The samples were treated with DNase I (Roche) and acid phenol: chloroform extraction was performed to isolate immunoprecipitated RNA. cDNA was synthesized from the extracted RNA using the Maxima First Strand cDNA Synthesis Kit (ThermoFisher Scientific). qPCR analysis for pull-down of snRNAs and control RNAs was performed using iTaq Universal SYBR (Bio-Rad) on a CFX96 Real-Time PCR system (Bio-Rad). Primers for qPCR analysis are listed in Table S3.
Co-immunoprecipitation and crosslinked co-immunoprecipitation
150 mL of log-phase yeast were crosslinked in 1% formaldehyde for 15 minutes at room temperature and quenched in 125 mM glycine for 5 minutes at room temperature for the crosslinked co-immunoprecipitation. Non-crosslinked and crosslinked cultures were then pelleted and resuspended in 1 mL of lysis buffer with protease inhibitors. Acid-washed beads were added to the resuspension and samples were lysed in a cell disruptor for 5 minutes at 4°C. The lysates were centrifuged at 13,000 rpm at 4°C for 20 minutes to remove the insoluble material. 1 mL of lysate was precleared with 50 μL of GammaBind G Sepharose beads (GE Healthcare). 10 μL of precleared lysate was removed as input. 4 μg of anti-HA antibody (clone: 12CA5, Roche) was added to the 700 μL of clarified lysate and rotated at 4°C overnight. Samples were incubated with 25 μL of GammaBind G Sepharose beads (GE Healthcare) for two hours at 4°C. Beads were then washed five times with lysis buffer and eluted by boiling with 2X SDS-PAGE loading buffer. Crosslinked samples were incubated at 95°C for 30 minutes to reverse crosslinks.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data in bar graphs represent the average of at least 3 biological replicates. Error bars represent the standard error of the mean (SEM). P values were determined by 2-tailed unpaired Student’s t test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Chi-square test was used in RNA-seq splicing analysis to determine the statistical significance of changes in SE in the mutants compared to wild-type. Spearman’s rank correlation was used to measure the association of gene expression between wild-type and mutants. Statistical analyses were performed in Microsoft Excel (Version 1812) or Prism 7 (GraphPad).
DATA AND CODE AVAILABILITY
The accession number for the RNA-seq and ChIP-seq data reported in this paper is GEO: GSE120051.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K36me3 | Abcam | Cat# ab9050; RRID:AB_306966 |
| Anti-Histone H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Anti-HA (12CA5) | Roche | Cat# 11583816001; RRID:AB_514505 |
| Anti-Myc (9E10) | Roche | Cat# 11667149001; RRID:AB_390912 |
| Anti-RNAPII S2P | Abcam | Cat# ab5095; RRID:AB_304749 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bovine Serum Albumin | Rockland | Cat# BSA-50 |
| Non-fat Dry Milk | Genesee Scientific | Cat# 20-241 |
| Agencourt AMPure XP | Beckman Coulter | Cat# A63880 |
| cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat# 4693159001 |
| Formaldehyde | Fisher Scientific | Cat# BP531-500 |
| iTaq Universal SYBR® Green Supermix | Bio-Rad | Cat# 172-5121 |
| GammaBind G Sepharose | GE Healthcare | Cat# 17-0885-01 |
| Pierce Anti-HA Magnetic Beads | ThermoFisher Scientific | Cat# 88836 |
| Maxima Reverse Transcriptase | ThermoFisher Scientific | Cat# EP0742 |
| RiboLock RNase Inhibitor | ThermoFisher Scientific | Cat# EO0382 |
| RNase A | ThermoFisher Scientific | Cat# AM2270 |
| Proteinase K | Roche | Cat# 03115828001 |
| DNase I recombinant | Roche | Cat# 4716728001 |
| Glass Beads | BioSpec | Cat# 11079105 |
| Protein A Dynabeads | ThermoFisher Scientific | Cat# 10001D |
| HiBind DNA columns | VWR | Cat# 95043-212 |
| UltraPure Ethidium Bromide | ThermoFisher Scientific | Cat# 15585011 |
| Critical Commercial Assays | ||
| TruSeq Stranded mRNA Library Preparation Kit | Illumina | Cat# RS-122-2101 |
| Ribo-Zero Gold rRNA Removal Kit (Yeast) | Illumina | Cat# MRZY1324 |
| Ovation Ultralow DR Kit | NuGEN | Cat# 0344 |
| Deposited Data | ||
| RNA-seq and ChiP-seq data | This study | GEO: GSE120051 |
| Experimental Models: Organisms/Strains | ||
| Yeast strains, see Table S1 | This study | N/A |
| Oligonucleotides | ||
| Primers for PCR, RT-PCR, and qPCR, see Table S3 | This study | N/A |
| Recombinant DNA | ||
| pFA6a-3HA-kanMX6 | Longtine et al., 1998 | N/A |
| pFA6a-3HA-HIS3MX6 | Longtine et al., 1998 | N/A |
| pFA6A-13Myc-kanMX6 | Longtine et al., 1998 | N/A |
| Software and Algorithms | ||
| Prism 7 | GraphPad | N/A |
| TopHat 2.0.14 | Trapnell et al., 2009 | https://ccb.jhu.edu/software/tophat/index.shtml |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS |
| SeqPlots 3.7 | Stempor and Ahringer, 2016 | https://bioconductor.org/packages/release/bioc/html/seqplots.html |
| Bowtie 0.12.8 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Microsoft Excel (Version 1812) | Microsoft | N/A |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
Highlights.
H3K36 methylation is necessary for efficient constitutive pre-mRNA splicing
The chromodomain protein Eaf3 links the spliceosome and H3K36 methylation
Eaf3 physically interacts with the splicing factor Prp45
Eaf3 is required for proper cotranscriptional spliceosome assembly
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences (GM-085474 to T.L.J.), as well as the Cellular and Molecular Biology Training Program Ruth L. Kirschstein National Research Service Award (GM007185 to C.S.L.), the Whitcome Predoctoral Fellowship (to C.S.L.), and the UCLA Dissertation Year Fellowship (to C.S.L.). M.S.P. was supported by the Russian Foundation for Basic Research grants 17-29-06056 and 18-29-01027 and Russian Science Foundation grant 19-44-02027.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interest.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.100.
REFERENCES
- Albers M, Diment A, Muraru M, Russell CS, and Beggs JD (2003). Identification and characterization of Prp45p and Prp46p, essential pre-mRNA splicing factors. RNA 9, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander RD, Innocente SA, Barrass JD, and Beggs JD (2010). Splicing-dependent RNA polymerase pausing in yeast. Mol. Cell 40, 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Enroth S, Rada-Iglesias A, Wadelius C, and Komorowski J (2009). Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome Res. 19, 1732–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ares M (2012). Isolation of total RNA from yeast cell cultures. Cold Spring Harb. Protoc 2012, 1082–1086. [DOI] [PubMed] [Google Scholar]
- Ares M Jr., Grate L, and Pauling MH (1999). A handful of intron-containing genes produces the lion’s share of yeast mRNA. RNA 5, 1138–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad AM, Venkataramanan S, Nag A, Galivanche AR, Bradley MC, Neves LT, Douglass S, Clarke CF, and Johnson TL (2017). Chromatin-remodeling SWI/SNF complex regulates coenzyme Q6 synthesis and a metabolic shift to respiration in yeast. J. Biol. Chem 292, 14851–14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, and Kouzarides T (2005). Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem 280, 17732–17736. [DOI] [PubMed] [Google Scholar]
- Brès V, Gomes N, Pickle L, and Jones KA (2005). A human splicing factor, SKIP, associates with P-TEFb and enhances transcription elongation by HIV-1 Tat. Genes Dev 19, 1211–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanarat S, and Sträßer K (2013). Splicing and beyond: the many faces of the Prp19 complex. Biochim. Biophys. Acta 1833, 2126–2134. [DOI] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, and Kornblihtt AR (2003). A slow RNA polymerase II affects alternative splicing in vivo. Mol. Cell 12, 525–532. [DOI] [PubMed] [Google Scholar]
- Douglass S, Leung CS, and Johnson TL (2019). Extensive splicing across the Saccharomyces cerevisiae genome. bioRxiv. 10.1101/515163. [DOI] [Google Scholar]
- Fabrizio P, Dannenberg J, Dube P, Kastner B, Stark H, Uriaub H, and Lührmann R (2009). The evolutionarily conserved core design of the catalytic activation step of the yeast spliceosome. Mol. Cell 36, 593–608. [DOI] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, and Bentiey DL (2014). Pre-mRNA splicing is facilitated by an optimal RNA poiymerase II elongation rate. Genes Dev. 28, 2663–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Görnemann J, Kotovic KM, Hujer K, and Neugebauer KM (2005). Cotranscriptional spliceosome assembly occurs in a stepwise fashion and requires the cap binding complex. Mol. Cell 19, 53–63. [DOI] [PubMed] [Google Scholar]
- Gunderson FQ, and Johnson TL (2009). Acetylation by the transcriptional coactivator Gcn5 plays a novel role in co-transcriptional spliceosome assembly. PLoS Genet. 5, e1000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson FQ, Merkhofer EC, and Johnson TL (2011). Dynamic histone acetylation is critical for cotranscriptional spliceosome assembly and spliceosomal rearrangements. Proc. Natl. Acad. Sci. USA 108, 2004–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, et al. (2014). BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol. Cell 56, 298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hálová M, Gahura O, Převorovský M, Cit Z, Novotný M, Vaientová A, Abrhámová K, Půta F, and Folk P. (2017). Nineteen complex-related factor Prp45 is required for the early stages of cotranscriptional spliceosome assembly. RNA 23, 1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hérissant L, Moehle EA, Bertaccini D, Van Dorsseiaer A, Schaeffer-Reiss C, Guthrie C, and Dargemont C (2014). H2B ubiquitylation moduiates spliceosome assembly and function in budding yeast. Biol. Cell 106, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnilicová J, and Staněk D (2011). Where splicing joins chromatin. Nucleus 2, 182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins AA, Rodgers ML, Friedman LJ, Gelles J, and Moore MJ (2016). Single molecule analysis reveals reversible and irreversible steps during spliceosome activation. eLife 5, e14166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe KJ, Kane CM, and Ares M Jr. (2003). Perturbation of transcription elongation influences the fidelity of internal exon inclusion in Saccharomyces cerevisiae. RNA 9, 993–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JT, Plocik AM, Guthrie C, and Yamamoto KR (2010). Reciprocal intronic and exonic histone modification regions in humans. Nat. Struct. Mol. Biol 17, 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamori N, Tominaga K, Sato T, Riehle K, Iwamori T, Ohkawa Y, Coarfa C, Ono E, and Matzuk MM (2016). MRG15 is required for pre mRNA splicing and spermatogenesis. Proc. Natl. Acad. Sci. USA 113, E5408–E5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha DK, and Strahl BD (2014). An RNA poiymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun 5, 3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TL, and Vilardell J (2012). Regulated pre-mRNA splicing: the ghost writer of the eukaryotic genome. Biochim. Biophys. Acta 1819, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, and Lis JT (2015). Getting up to speed with transcription elongation by RNA poiymerase II. Nat. Rev. Mol. Cell Biol 16, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podoiny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. (2005). Cotranscriptional set2 methylation of histone H3 iysine 36 recruits a repressive Rpd3 complex. Cell 123, 593–605. [DOI] [PubMed] [Google Scholar]
- Kolasinska-Zwierz P, Down T, Latorre I, Liu T, Liu XS, and Ahringer J (2009). Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet 41, 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotovic KM, Lockshon D, Boric L, and Neugebauer KM (2003). Cotranscriptional recruitment of the U1 snRNP to intron-containing genes in yeast. Mol. Cell. Biol 23, 5768–5779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, et al. (2003). Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol 23, 4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacadie SA, Tardiff DF, Kadener S, and Rosbash M (2006). In vivo commitment to yeast cotranscriptional splicing is sensitive to transcription elongation mutants. Genes Dev. 20, 2055–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Moazed D, and Gygi SP (2002). Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J. Biol. Chem 277, 49383–9388. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Luco RF, and Misteli T (2011). More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr. Opin. Genet. Dev 21, 366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, and Misteli T (2010). Regulation of alternative splicing by histone modifications. Science 327, 996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov EM, Makarova OV, Urlaub H, Gentzel M, Will CL, Wilm M, and Lührmann R (2002). Small nuclear ribonucleoprotein remodeling during catalytic activation of the spliceosome. Science 298, 2205–2208. [DOI] [PubMed] [Google Scholar]
- Martínková K, Lebduska P, Skružný M, Folk P, and Půta F (2002). Functional mapping of Saccharomyces cerevisiae Prp45 identifies the SNW domain as essential for viability. J. Biochem 132, 557–563. [DOI] [PubMed] [Google Scholar]
- Morselli M, Pastor WA, Montanini B, Nee K, Ferrari R, Fu K, Bonora G, Rubbi L, Clark AT, Ottonello S, et al. (2015). In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. eLife 4, e06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munding EM, Shiue L, Katzman S, Donohue JP, and Ares M Jr. (2013). Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol. Cell 51, 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai K, Yamaguchi T, Takami T, Kawasumi A, Aizawa M, Masuda N, Shimizu M, Tominaga S, Ito T, Tsukamoto T, and Osumi T (2004). SKIP modifies gene expression by affecting both transcription and splicing. Biochem. Biophys. Res. Commun 316, 512–517. [DOI] [PubMed] [Google Scholar]
- Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, and Mann M (1998). Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet 20, 46–50. [DOI] [PubMed] [Google Scholar]
- Neves LT, Douglass S, Spreafico R, Venkataramanan S, Kress TL, and Johnson TL (2017). The histone variant H2A.Z promotes efficient cotranscriptional splicing in S. cerevisiae. Genes Dev. 31, 702–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissen KE, Homer CM, Ryan CJ, Shales M, Krogan NJ, Patrick KL, and Guthrie C (2017). The histone variant H2A.Z promotes splicing of weak introns. Genes Dev. 31, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oesterreich FC, Herzel L, Straube K, Hujer K, Howard J, and Neugebauer KM (2016). Splicing of Nascent RNA Coincides with Intron Exit from RNA Polymerase II. Cell 165, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, and Blencowe BJ (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Pandya-Jones A, and Black DL (2009). Co-transcriptional splicing of constitutive and alternative exons. RNA 15, 1896–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa MM, Sutherland HG, Ule J, Grimes GR, and Bickmore WA (2012). Psip1/Ledgfp52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 8, e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JL, Moqtaderi Z, and Struhl K (2004). Eaf3 regulates the global pattern of histone acetylation in Saccharomyces cerevisiae. Mol. Cell. Biol 24, 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathianathan A, Ravichandran P, Lippi JM, Cohen L, Messina A, Shaju S, Swede MJ, and Ginsburg DS (2016). The Eaf3/5/7 Subcomplex Stimulates NuA4 Interaction with Methylated Histone H3 Lys-36 and RNA Polymerase II. J. Biol. Chem 291, 21195–21207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Meshorer E, and Ast G (2009). Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol 16, 990–995. [DOI] [PubMed] [Google Scholar]
- Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, Ho TH, Kuan PF, Jonasch E, Furey TS, et al. (2014). Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 24, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RK, and Cooper TA (2012). Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med 18, 472–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson MR, and Stevens SW (2014). Rapid identification of mRNA processing defects with a novel single-cell yeast reporter. RNA 20, 732–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorenson MR, Jha DK, Ucles SA, Flood DM, Strahl BD, Stevens SW, and Kress TL (2016). Histone H3K36 methylation regulates pre-mRNA splicing in Saccharomyces cerevisiae. RNA Biol. 13, 412–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, and Burge CB (2009). Biased chromatin signatures around polyadenylation sites and exons. Mol. Cell 36, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stempor P, and Ahringer J (2016). SeqPlots-Interactive software for exploratory data analyses, pattern discovery and visualization in genomics. Wellcome Open Res. 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilgner H, Nikolaou C, Althammer S, Sammeth M, Beato M, Valcárcel J, and Guigó R (2009). Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol 16, 996–1001. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramanan S, Douglass S, Galivanche AR, and Johnson TL (2017). The chromatin remodeling complex Swi/Snf regulates splicing of meiotic transcripts in Saccharomyces cerevisiae. Nucleic Acids Res. 45, 7708–7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Yan C, Bai R, Huang G, and Shi Y (2016). Structure of a yeast catalytic step I spliceosome at 3.4 Å resolution. Science 353, 895–904. [DOI] [PubMed] [Google Scholar]
- Will CL, and Lührmann R (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol 3, a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, and Strahl BD (2003). Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 17, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Hang J, Wan R, Huang M, Wong CC, and Shi Y (2015). Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 349, 1182–1191. [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, and Shi Y (2016). Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 353, 904–911. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq and ChIP-seq data reported in this paper is GEO: GSE120051.