

Abstract
Extra-cranial malignant rhabdoid tumors (MRTs) and cranial atypical teratoid RTs (ATRTs) are heterogeneous pediatric cancers driven primarily by SMARCB1 loss. To understand the genome-wide molecular relationships between MRTs and ATRTs, we analyze multi-omics data from 140 MRTs and 161 ATRTs. We detect similarities between the MYC subgroup of ATRTs (ATRT-MYC) and extra-cranial MRTs, including global DNA hypomethylation and overexpression of HOX genes and genes involved in mesenchymal development, distinguishing them from other ATRT subgroups that express neural-like features. We identify five DNA methylation subgroups associated with anatomical sites and SMARCB1 mutation patterns. Groups 1, 3, and 4 exhibit cytotoxic T cell infiltration and expression of immune checkpoint regulators, consistent with a potential role for immunotherapy in rhabdoid tumor patients.
In Brief
Chun et al. report similarities between the MYC subgroup of cranial and extracranial rhabdoid tumors (RTs) at genetic, gene-expression, and epigenetic levels. They identify five DNA methylation subgroups of RTs across multiple organ sites, and some subgroups exhibit increased levels of immune cell infiltration and immune checkpoint expression.
Graphical Abstract
INTRODUCTION
Rhabdoid tumors (RTs) are aggressive pediatric cancers that primarily affect infants, accounting for approximately 15% of all infant cancer incidence in the United States and United Kingdom (Packer et al., 2002; Brennan et al., 2013). RTs can arise throughout the body and are broadly classified based on the anatomical site of occurrence, i.e., atypical teratoid RTs (ATRTs) from the central nervous system (CNS) and malignant RTs (MRTs), such as RTs of the kidney (RTKs), from non-CNS tissues. Regardless of anatomical sites, RTs share pathognomonic loss of SMARCB1 (or SMARCA4 in rare cases; Versteege et al., 1998; Hasselblatt et al., 2014), which encodes a core subunit of the SWI/SNF chromatin-remodeling complex that plays critical roles in epigenetic and transcriptional regulation. Apart from SMARCB1 mutations, RTs otherwise exhibit few mutations, and in general have diploid genomes (Lee et al., 2012; Chun et al., 2016; Johann et al., 2016).
Despite being driven by SMARCB1 loss, RTs exhibit heterogeneity, with molecular subgroups identified in each of MRTs and ATRTs (Chun et al., 2016; Johann et al., 2016; Torchia et al., 2016; Nemes and Frühwald, 2018). In ATRTs, the SHH, TYR, and MYC DNA methylation subgroups have been described (Johann et al., 2016; corresponding to Groups 1, 2A, and 2B, respectively, in Torchia et al., 2016). In MRTs, two gene expression subgroups were described (Group 1 and Group 2), which exhibited ATRT-like and RTK-like gene expression profiles, respectively (Chun et al., 2016). From these studies, some genes and pathways have emerged as commonly dysregulated across subgroups, such as the expression of HOX genes and other homeobox-containing genes in the ATRT-MYC subgroup and some MRTs and genes involved in neural or neural crest development in other MRTs. The existence of these shared features stimulated our hypothesis that MRT and ATRT subgroups might share additional similarities stemming from SMARCB1/SMARCA4 loss, the identification of which might improve our understanding of RT biology and ultimately reveal much needed insights into RT therapeutic vulnerabilities.
To explore this hypothesis, we performed integrative analyses of genome, transcriptome, and epigenome profiles of 301 RTs from multiple anatomic sites to reveal consensus molecular subgroups of RTs and identify shared molecular features.
RESULTS
To facilitate comparisons across RTs, we combined our previously published ATRT and MRT datasets from 40 MRTs and 150 ATRTs (Chun et al., 2016; Johann et al., 2016) and generated additional data from 11 ATRTs and 100 MRTs. The expanded datasets consist of whole-genome sequencing (WGS), transcriptome sequencing (RNA-seq), whole-genome bisulfite sequencing (WGBS), and DNA methylation array data as well as H3K27me3 and H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) data (Table S1). In total, we analyzed data from 301 RT cases, including 161 ATRTs and 140 MRTs, of which 92 cases were from kidneys (RTKs) and 44 were from non-kidney tissues (4 cases were from unknown tissue types; Table S1).
ATRT-MYC and MRT Share Similar DNA Methylation Profiles Distinct from ATRT-SHH and -TYR
DNA methylation profiling has been used to identify molecular subgroups in many cancer types (Sturm et al., 2012; Cancer Genome Atlas Research Network, 2014b; Capper et al., 2018; Paulus, 2018). To identify and confirm molecular subgroups in RTs, we analyzed DNA methylation array data from 301 RT cases by using unsupervised clustering and dimension reduction algorithms (STAR Methods). Results from multiple algorithms substantiated the previous observation that ATRTs formed three distinct clusters (Johann et al., 2016) and revealed a distinct cluster of ATRT-MYC and MRT cases separate from ATRT-SHH and -TYR subgroups (Figures 1A and 1B). To evaluate the robustness of this clustering solution in the context of diverse cancer types, we compared DNA methylation profiles of RTs to 33 adult and 4 pediatric cancer types and 23 normal tissue types from TCGA and TARGET (n = 10,232 cases) by using an unsupervised clustering approach. MRT and ATRT-MYC again clustered together (Figure S1). Notably, RTs clustered with cancers of neural crest origin (neuroblastomas, uveal melanomas, pheochromocytomas, and paragangliomas), brain cancers (glioblastomas and low-grade gliomas) and normal brain tissues, consistent with our previous observation based on microRNA (miRNA) profiles (Chun et al., 2016).
Figure 1. Unsupervised Clustering of DNA Methylation Profiles from 140 MRTs (92 Renal, 48 Extra-Renal) and 161 ATRTs Indicate Similarity between ATRT-MYC and MRT.

(A) t-SNE analysis was performed using the top 2,000 most variably methylated CpG sites and to reveal three clusters that consisted primarily of ATRT-MYC (n = 44 cases) and MRT (n = 140 cases), ATRT-SHH (n = 64 cases), or ATRT-TYR (n = 53 cases).
(B) Unsupervised hierarchical clustering was performed using the top 1% most variably methylated CpG sites (n = 3,958) and yielded a clustering result consistent with (A). See also Figure S1 and Table S1.
ATRT-MYC and MRT Cases Can Be Further Separated into Three DNA Methylation Subgroups That Correlate with Anatomical Sites and SMARCB1 Mutation Patterns
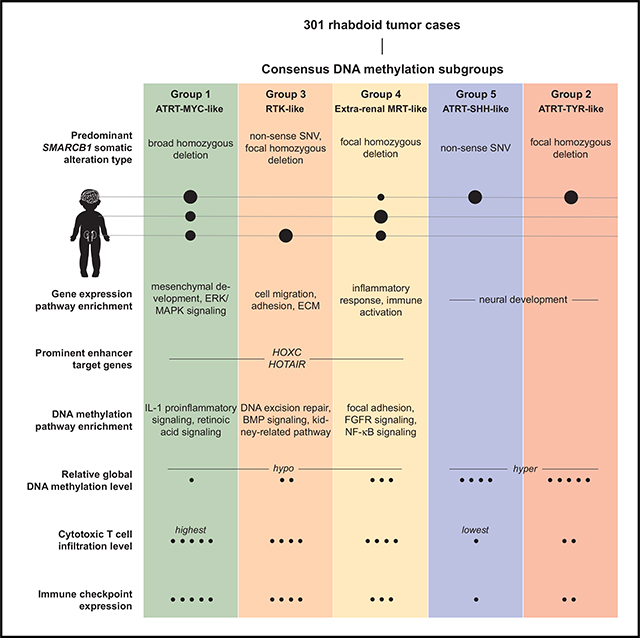
A non-negative matrix factorization (NMF) analysis (Gaujoux and Seoighe, 2010) of DNA methylation array data revealed further separation of the ATRT-MYC and MRT group into three subgroups (Groups 1, 3, and 4), which were consistently identified using hierarchical clustering and t-Distributed Stochastic Neighbor Embedding (t-SNE) methods (Figures 2A, S2A, and S2D). Although NMF results indicated that Group 1 cases could be further separated into two subgroups (Figures 2B, S2B, and S2C), we did not find molecular or clinical correlates that would support the existence of biologically relevant subgroups within Group 1. We, thus, fixed our analyses on five DNA methylation subgroups, which consisted of the previously defined ATRT-SHH and -TYR (Johann et al., 2016) and three previously undefined subgroups containing MRT and ATRT-MYC cases (Figure 2A).
Figure 2. Five DNA Methylation Subgroups of RTs from Cranial and Extra-Cranial Sites Correlate with Previously Known ATRT and MRT Subgroups, Anatomical Sites, and SMARCB1 Deletion Patterns.

(A) Unsupervised NMF analysis was performed using the top 10,000 most variably methylated CpG sites and revealed five subgroups (top). Clinical features, gene expression subgroups of MRTs, and previously characterized ATRT subgroups are shown in colored tracks (middle). Chronological age and predicted DNA methylation age (Horvath, 2013) are shown in bar plots (bottom). ATRT-SHH and Group 1 exhibited increased DNA methylation age compared to the other subgroups (Wilcoxon p values = 1.62e-05 and 6.30e-10 for ATRT-SHH and Group 1, respectively). Neither chronological age nor gender were significantly associated with the subgroups (Kruskal-Wallis p value = 0.25 and Fisher’s exact p values = 0.16 – 0.86, respectively).
(B) Cophenetic coefficients (top) and silhouette widths (bottom) for NMF cluster solutions from k = 2 to k = 15. The highest cophenetic coefficients and silhouette widths were from the NMF solutions with 5 and 6 clusters.
(C) Heatmap indicates chromosomal copy gain (indicated by red) or loss (blue), estimated using DNA methylation data, centered at the SMARCB1 locus across the five DNA methylation subgroups (n = 301 cases).
(D) Boxplot shows the mean expression levels of 74 genes (top) co-deleted with SMARCB1 across the five subgroups (n = 19 cases for Group 1, n = 41 for Group 3, n = 11 for Group 4, n = 11 for ATRT-SHH, n = 8 for ATRT-TYR) and expression levels of MIF (bottom). The asterisk indicates a significant difference (Wilcoxon p value < 0.05) between Group 1 and other RT subgroups.
See also Figures S2 and S3 and Table S2.
The “ATRT-MYC-like” Group 1 (n = 67) consisted of 32 ATRT and 35 MRT cases (19 RTKs, 12 extra-renal MRTs, and 4 cases from unknown tissue types). Nearly all (31/32) ATRTs in this group were classified as ATRT-MYC. The “RTK-like” Group 3 (n = 59) consisted of 2 ATRT and 57 MRT cases, of which 53 MRT cases were RTKs. The “extra-renal MRT-like” Group 4 (n = 59) was dominated by extra-renal MRTs, containing 11 ATRT cases (6 ATRT-MYC, 4 ATRT-SHH, and 1 ATRT-TYR) and 48 MRT cases, of which 28 cases were from extra-renal tissues. The “ATRT-TYR-like” Group 2 (n = 58) mostly consisted of ATRT-TYR cases (n = 51, the remaining cases were ATRT-MYC [n = 5] and -SHH [n = 2]). The “ATRT-SHH-like” Group 5 (n = 58) consisted of ATRT-SHH cases (n = 57; one remaining case was ATRT-TYR).
We next explored the relationship between these DNA methylation subgroups and the previously described MRT gene expression subgroups (Chun et al., 2016) and observed a significant association between “RTK-like” DNA methylation Group 3 and the RTK-like gene-expression subgroup 2 (16 out of 18 cases [89%]; Fisher’s exact p value = 0.0070; Figure S2A). In our NMF analysis that used an expanded RNA-seq dataset, including an additional 25 MRT cases (Figures S2E and S2F), we again observed a significant association between DNA methylation Group 3 and a gene expression subgroup that exclusively consisted of RTKs (24 out of 25 cases [96%]; Fisher’s exact p value = 1.07e-05; Figure S2E).
To investigate genetic alterations that might correlate with the DNA methylation subgroups, we analyzed somatic alterations using WGS data from tumor and matched normal pairs (56 MRT and 18 ATRT cases; Figure S3A). Of 26 cases with SMARCB1 deletions larger than 10 kilobases, a significant overrepresentation (14 out of 26 cases; Fisher’s exact p value = 2.12e-08; Figure S3C) was in Group 1. Group 3 and ATRT-SHH almost exclusively contained cases with somatic nonsense mutations or focal deletions of SMARCB1 (10 out of 12 cases and 11 out of 13 cases, respectively; Figures S3B and S3C). To extend our SMARCB1 copy number analyses to cases lacking WGS data, we analyzed DNA methylation data from 301 RT cases to infer copy number alterations by using the sum of methylated and unmethylated signals (Sturm et al., 2012). This analysis consistently revealed the association between larger deletions at the SMARCB1 locus and Group 1 cases (Figure 2C). As expected, genes co-deleted with SMARCB1 (74 genes) were significantly under-expressed in Group 1 compared to other subgroups that did not harbor deletions (Wilcoxon p value = 2.59e-07; Figure 2D; Table S2). Such genes included CABIN1 (a regulator of p53 and T cell receptor (TCR) signaling), SUSD2 (a tumor suppressor gene involved in G1 cell cycle arrest), SPECC1L (a regulator of craniofacial morphogenesis and cranial neural crest cell delamination; Wilson et al., 2016), and MIF (encodes a macrophage migration inhibitory factor, involved in cell-mediated immunity and inflammation; Lue et al., 2002). The association between broad SMARCB1 deletions and DNA methylation Group 1 is compatible with the notion that dysregulation of multiple genes in addition to SMARCB1 may contribute to molecular subgroups.
ATRT-MYC and MRT Exhibit Global Hypomethylation and Distinct DNA Methylation Valleys Compared to ATRT-SHH and -TYR
To compare global DNA methylation levels in MRTs and ATRTs, we analyzed WGBS data from 69 MRT and 17 ATRT cases and DNA methylation array data from 140 MRT and 161 ATRT cases. MRT and ATRT-MYC cases exhibited global DNA methylation levels that were significantly lower than ATRT-SHH and -TYR (Wilcoxon p value = 2.2e-07; Figure 3A) but comparable to normal brain tissues (from 8 adult and 2 fetal brain samples; Wilcoxon p value = 0.145; Figure S4A). However, MRTs exhibited significantly lower methylation levels in introns and non-genic regions compared to normal brain samples, indicating that these regions are abnormally hypomethylated in MRTs (Wilcoxon p values = 0.011 and 8.26e-05, respectively; Figures S4B and S4C).
Figure 3. ATRT-MYC and MRT Exhibit Similar DNA Methylation Profiles Distinct from ATRT-SHH and -TYR.

(A) Boxplot shows the distribution of mean genome-wide DNA methylation levels based on WGBS data. MRT (n = 69 cases) and ATRT-MYC (n = 3 cases) showed significant hypomethylation compared to ATRT-SHH (n = 7 cases) and -TYR (n = 7 cases; *Wilcoxon p value < 0.05).
(B) Boxplot displays the distribution of fractions of the genome covered by PMDs in MRT and ATRT-MYC, which exhibited significantly more abundant PMDs compared to ATRT-SHH and -TYR (*Wilcoxon p value < 0.05).
(C–E) Gene set enrichment of DMRs that are specific for Groups 1 (C), 3 (D), and 4 (E). The x axes indicate the statistical significance of the enrichment test. (F) Heatmap (left) shows average CpG methylation levels at the NCOR2 locus in Group-1-specific DMRs (red = 100%; blue = 0% methylation). Boxplot (right) shows significantly increased NCOR2 expression levels in Group 1 compared to other RT subgroups (*Wilcoxon p value < 0.05).
See also Figure S4 and Table S3.
Our previous study (Johann et al., 2016) showed that global hypomethylation in ATRT-MYC compared to other ATRT subgroups was linked to the prevalence of partially methylated domains (PMDs). We found that PMDs were also more abundant in MRTs compared to ATRT-SHH and -TYR, covering substantial portions of the genome (Wilcoxon p value = 0.00014; Figures 3B and S4F). In particular, MRTs in Groups 1 and 3 exhibited global hypomethylation associated with higher PMD fractions compared to ATRT-SHH and -TYR (Wilcoxon p value = 1.65-e07), whereas MRTs in Group 4 exhibited PMD fractions that were comparable to ATRT-SHH and -TYR (Figures S4D and S4E). This result indicated that although global hypomethylation is an epigenetic feature that is characteristic of most MRTs, Group 4 appears to have a distinct DNA methylation landscape.
To characterize candidate biological processes dysregulated by differential methylation across the subgroups, we identified differentially methylated regions (DMRs; average length = 1kb) and performed gene set enrichment analyses by using overexpressed genes in DMRs. Group 1 DMRs exhibited an unexpected enrichment for genes in immune-related pathways specifically related to interleukin 1-associated pro-inflammatory activities (e.g., IRAK, Toll-like receptors [TLRs], TRAF6, and JNK) that are critical for initiating innate immune responses against foreign pathogens and IRF7-associated pathways known to be activated upon viral infection (Figure 3C). We also observed a significant enrichment of upregulated genes (e.g., NCOR2, a transcriptional repressor implicated in hematological malignancies [Lin et al., 1998]) in Group-1-specific DMRs involved in retinoic acid signaling, a pathway that has not been previously associated with MRTs or ATRTs (Figure 3F; Table S3). Genes associated with Group-3-specific DMRs were enriched for DNA excision repair, BMP signaling, and pathways implicated in renal cell carcinoma development, consistent with RTK-like characteristics observed in Group 3 (Figure 3D). For Group-4-specific DMRs, the most significantly enriched pathways included focal adhesion, FGFR signaling, and nuclear factor κB (NF-κB) signaling, a key regulatory pathway for immune and inflammatory processes (Figure 3E; DiDonato et al., 2012).
ATRT-MYC and MRT Share Distinct Enhancer Landscapes Compared to Other ATRT Subgroups
We next investigated the extent of similarities between enhancer states in ATRTs and MRTs and analyzed H3K27ac ChIP-seq data from 34 MRT and 14 ATRT cases, of which 24 MRT cases were specifically profiled for this study. To robustly identify cases with similar H3K27ac profiles, we performed multiple iterations of unsupervised hierarchical clustering of enhancer elements defined by H3K27ac signal densities (STAR Methods). Across iterations, we consistently observed clustering of ATRT-MYC with MRT cases (Figure 4A), supporting the notion that ATRT-MYC and MRT share similar enhancer profiles. We also observed increased H3K27ac levels in subgroup-specific DMRs (Figure 4B), further supporting upregulation of genes in these regions (e.g., NCOR2).
Figure 4. ATRT-MYC and MRT Exhibit Distinct Enhancer Profiles.

(A) Unsupervised clustering of H3K27ac ChIP-seq read densities resulted in a cluster of ATRT-MYC cases (n = 4) and MRT cases (n = 34) indicated by green and purple bars, respectively.
(B) Line plots show the average H3K27ac signal densities of the five RT subgroups at Group-1- (including ATRT-MYC; n = 460 DMRs), Group-3- (n = 426 DMRs), and Group-4-specific DMRs (n = 280), respectively. Subgroup-specific DMRs showed the highest H3K27ac signal density levels in the respective subgroups.
(C) Mean H3K27ac density at the HOXC locus, which was specific to MRT (n = 34 cases) and ATRT-MYC (n = 4 cases) and absent in ATRT-SHH (n = 5 cases) and -TYR (n = 5 cases).
(D) Boxplots show HOXC (top) and HOTAIR (bottom) gene expression levels, which were significantly higher in MRT cases (n = 65) and ATRT-MYC (n = 6 cases) compared to ATRT-SHH (n = 11 cases) and -TYR cases (n = 8; * adjusted p values < 0.05).
(E) Unsupervised hierarchical clustering using enrichment scores of TFBS at enhancers specific to MRT (n = 312 enhancers), ATRT-MYC (n = 443 enhancers), -SHH (n = 511 enhancers), and -TYR (n = 1,385 enhancers). Heatmap colors represent the log2 enrichment scores of TFs in the enhancers. Colors next to gene names indicate known biological processes associated with TFs.
See also Table S4.
We identified 26 dense clusters of high H3K27ac signals indicative of super-enhancers that were common between ATRT-MYC and MRT. The most prominent super-enhancer was found at the HOXC locus (Figure 4C; Table S4), with genes at this locus exhibiting significant overexpression compared to ATRT-SHH and -TYR (Wilcoxon p values < 2.4e-15 for HOXC genes and DESeq adjusted p value = 3.43e-05 for HOTAIR; Figure 4D). There were 61 regular enhancer elements that were common between ATRT-MYC and MRT (Table S4) in the proximity of genes involved in epigenome modification and development, including CREBBP (encodes a histone acetyltransferase involved in embryonic development and growth control), PRDM6 (histone methyltransferase and transcriptional repressor involved in smooth muscle differentiation), and TINAGL1 (encodes an antigen associated with tubulointerstitial nephritis; also involved in proliferation and migration of cranial neural crest cells [Neiswender et al., 2017]).
We next studied enhancer-mediated transcriptional dysregulation by identifying transcription factors (TFs) that would likely bind to enhancer regions. We analyzed enrichment of TF binding sites (TFBSs) within enhancer regions that were unique to MRT, ATRT-MYC, -SHH, or -TYR, by calculating enrichment scores based on observed and expected numbers of TF motifs found in enhancer regions (STAR Methods). Unsupervised hierarchical clustering of TF motif enrichment scores showed clustering of ATRT-MYC and MRT, implying that common factors could act on such enhancers (Figure 4E). TFs known to bind to such sites included those regulating mesoderm and neural crest development, (e.g., HES7 and REST, TFs that suppress neuronal transcription programs [Bessho et al., 2001; Bruce et al., 2004]). Also enriched within ATRT-MYC and MRT was a TFBS for XBP-1, a TLR-activated TF required for production of proinflammatory cytokines (Martinon et al., 2010), corroborating our DMR analysis result (above) that indicated epigenetic dysregulation of genes involved in interleukin 1-mediated signaling. In ATRT-MYC, we observed TFBS for TFs involved in apoptosis and immune regulation, such as GMEMB1/2, RAD21, IRF5/8/9, and STAT1. IRF5/8/9 are involved in the induction of type I interferons (IFNs), inflammatory cytokines and MHC class I genes and, hence, promote immune responses involving, e.g., CD8+ cytotoxic T cells and natural killer (NK) cells. Likewise, STAT1 regulates the expression of multiple IFN target genes (Ivashkiv and Donlin, 2014). Our analyses thus indicated the unexpected possibility of immune modulation through epigenetic dysregulation in RTs.
Immune-Related Genes, HOX Genes, and Mesoderm Developmental Regulators Are Overexpressed in ATRT-MYC and MRT Compared to ATRT-SHH and -TYR
Our DNA methylation and enhancer data indicated shared epigenetic dysregulation of TFs in ATRT-MYC and MRT, potentially acting on similar gene expression programs. To identify such similarities, we performed differential gene expression analyses and identified 584 overexpressed genes and 2,500 under-expressed genes in ATRT-MYC and MRT relative to ATRT-SHH and -TYR (DESeq adjusted p values < 0.05; STAR Methods; Figure 5A). The most significantly overexpressed genes in ATRT-MYC and MRT included tissue-type-specific genes (e.g., GCG and KERA) and developmental regulators of mesoderm and mesoderm-derived tissue types (e.g., TCF21, encoding a mesoderm-specific TF, and DMP1 and MEOX2, involved in bone and vascular smooth muscle development, respectively). Notably, 26 members of all HOX gene families were likewise significantly overexpressed in ATRT-MYC and MRT. These results support the notion of dysregulated developmental programs, particularly those involved in mesodermal development, in ATRT-MYC and MRT. In contrast, ATRT-SHH and -TYR exhibited relative overexpression of genes involved in neural development (e.g., SOX1, GPR98/ADGRV1, and OTX2), suggesting more neural characteristics in these subgroups.
Figure 5. Dysregulation of Mesenchymal Development Genes Is Associated with ATRT-MYC and MRT, whereas Dysregulation of Neural Genes Is Associated with ATRT-SHH and -TYR.

(A) Volcano plot shows the statistical significance of differential expression (DE; adjusted p values < 0.05) on the y axis, and the fold change (FC) of gene expression in ATRT-MYC (n = 6 cases) and MRT (n = 65 cases) compared to ATRT-SHH (n = 11 cases) and -TYR (n = 8 cases) on the x axis. The top 20 significant DE genes, HOX genes, and genes involved in neural or mesenchymal development are labeled in colors as shown.
(B) Bar plots show the most significantly enriched pathways and adjusted enrichment p values based on analyses of 584 relatively overexpressed genes (top) and 2,500 relatively under-expressed genes (bottom) in MRTs and ATRT-MYC compared to ATRT-SHH and -TYR.
(C) Gene expression levels and H3K27ac and H3K27me3 densities (i.e., average read coverage) at the promoters of HES7 and its interactors are shown in boxplots.
(D-F) Enrichment map networks of Gene Ontology (GO) terms significantly enriched for Group-1- (D), Group-3- (E), and Group-4-specific (F) DE genes. A node size is proportional to the number of genes in the category and a node color indicates an adjusted enrichment p value. The edge thickness is proportional to a fraction of shared genes between GO terms.
Next, we used multiple pathway databases to identify functional categories enriched for differentially expressed genes (STAR Methods; Table S5). The most significantly enriched pathways, including overexpressed genes in ATRT-MYC and MRT, were developmental pathways for mesenchymal cell types and mesoderm-derived organs (Figure 5B), as well as immune-related pathways, including regulation of immune system processes and innate immune responses (adjusted p values = 1.40e-04 and 0.050, respectively; Table S5). In contrast, ATRT-SHH and -TYR exhibited significantly enriched pathways that predominantly involved neural development (Figure 5B), with ATRT-SHH further exhibiting a more neural-like gene expression program compared to ATRT-TYR (Figure S5A). Notably, we did not observe enrichment of immune-related functions in ATRT-SHH and -TYR. Increased expression of immune-related genes in ATRT-MYC and MRT was consistent with the enrichment of immune-related TFBSs (above), suggesting that ATRT-MYC and MRT might share an immune-related phenotype.
To further corroborate pathway enrichment results, we identified TF-regulatory networks consisting of TFs, putative direct target genes with corresponding TF motifs, and shared patterns of gene expression with TFs (STAR Methods; Aibar et al., 2017), integrating these by using unsupervised clustering. ATRT-MYC and MRT cases clustered together, sharing 13 common transcriptional networks distinct from ATRT-SHH and -TYR (Figure S5C). Of these, 11 involved HOX genes, of which five identified MYC as one of the putative direct HOX target genes, supporting the notion that the prominent molecular characteristics of HOX gene overexpression and dysregulation of MYC, another key characteristic of ATRT-MYC and MRT, might be linked (Figure S5C; Table S5). Another notable TF gene was HES7, a transcriptional repressor significantly overexpressed in ATRT-MYC and MRT (adjusted p value = 0.0036; Figure S5B), with binding sites that were enriched in ATRT-MYC and MRT enhancer regions (Figure 4E). Downstream target genes of HES7, such as LEF1 (implicated in co-activation of MITF and development of neural-crest-derived melanocytes; Levy et al., 2006), DUSP4 (regulator of MAPK signaling), and CTNNB1 (key component in the canonical WNT signaling pathway), exhibited significantly reduced expression in ATRT-MYC and MRT. Decreased levels of gene expression were correlated with lower H3K27ac and higher H3K27me3 levels in ATRT-MYC and MRT compared to ATRT-SHH and TYR (Figure 5C), indicating overall epigenetic dysregulation of the HES7 transcriptional network. AES, which encodes a transcriptional co-repressor of HES7 involved in neural, head mesenchyme, and ectoderm development, was, like HES7, upregulated in ATRT-MYC and MRT, further indicating distinct dysregulation of the HES7-driven transcriptional program in ATRT-MYC and MRT. TFs enriched for ATRT-SHH (e.g., NEUROD1, NHLH1, and EN2) and ATRT-TYR (OTX2 and ZIC4) included neural developmental regulators, consistent with the notion that ATRT-SHH and -TYR are more neural-like.
To determine distinct gene-expression characteristics for Groups 1, 3, and 4, we identified functional categories enriched for subgroup-specific differentially expressed genes and constructed Gene Ontology enrichment map networks. Networks of the most significantly enriched pathways for Group 1 involved early developmental processes as well as ERK/MAPK signaling (Figure 5D; Table S5). Group 3 networks also involved early developmental processes in addition to cell migration, adhesion, and extra-cellular matrix organization (Figure 5E). Group 4 networks exclusively consisted of immune-related categories (Figure 5F). To explore the association between RT subgroups and early developmental processes, we correlated transcriptome profiles of the subgroups to various progenitor cell types (Kundaje et al., 2015; Chun et al., 2016; Prescott et al., 2015). Among the subgroups, Group 1 showed the highest correlation to CD56+ mesodermal progenitor cells and Group 3 to embryonic stem cell lines (Figure S5D). ATRT-SHH showed the highest correlation to cranial neural crest cells, neuronal progenitors, and brain tissues, consistent with our observations of ATRT-SHH exhibiting the most neuronal-like characteristics among the subgroups.
Gene Expression Data Indicate Increased T Cell Presence in ATRT-MYC and Extra-Cranial MRT Subgroups
Following our analyses that indicated epigenetic modulation of genes involved in immune-related functions, we used CIBERSORT (Newman et al., 2015) to deconvolute immune cell gene expression signatures and, thus, estimate the extent of immune cell presence. To quantify overall T cell presence in each sample, we calculated a T cell score (a sum of effector T cell proportions; Figure 6A) and observed that inferred proportions of CD8+ cytotoxic T cells were among the highest in the 22 immune cell types profiled, along with tumor-associated M2 macrophages (Figures 6C and S6A; Sica et al., 2006), suggesting the involvement of both pro- and anti-tumoral immune functions in the tumor microenvironment. We observed a significant over representation of Groups 1 and 4 (Fisher’s exact p values = 0.018 and 5.13e-03, respectively), and a significant under-representation of Group 3 and ATRT-SHH (Fisher’s exact p values = 2.13e-04 and 0.031, respectively) in cases with CD8+ T cell proportions within the top 25th percentile (Figure 6B). We also noted that among such cases were two ATRT-TYR cases with abundant TBXT expression (196.4 and 35.3 Reads Per Kilobase per Million mapped reads (RPKM), median of the cohort = 0.0021 RPKM; Figure 6D), which encodes an embryonic TF (T-brachyury) that has been linked to immune responses in chordoma patients (Palena et al., 2007).
Figure 6. Gene Expression Analysis Indicates Increased T Cell Presence in RT Subgroups.

(A) Stacked bar plot shows CD8+ cytotoxic T cell proportions (yellow) and T cell scores (blue), which are based on the sum of absolute proportions of effector T cells (i.e., all T cell types except regulatory T cells [TVeg]). The samples (n = 90) are ordered based on CD8+ cytotoxic T cell proportions(and in all subsequent sub-figures in Figure 6). A subgroup of each sample is indicated in (B).
(C) Heatmap shows absolute proportions of 22 immune cell types predicted using CIBERSORT.
(D) Bar plot shows expression levels of the TBXT gene, which encodes T-brachyury.
(E-G) Heatmaps indicate expression levels of genes involved in antigen presentation and processing (E), T cell activation and homing (F), and immunosuppressive signaling (G). All genes were significantly overexpressed in cases with CD8+ T cell proportions greater than the median (adjusted p values < 0.05, except for CTLA4 [adjusted p value = 0.10]).
See also Figure S6.
To gain insight on biological processes that might contribute to increased immune activities predicted in RT subgroups, we analyzed genes involved in T cell-mediated immune responses. We found that nearly all HLA genes encoding MHC class I and II (18 out of 19 genes) were significantly overexpressed in cases with CD8+ T cell proportions greater than the median (adjusted p values < 0.05; Figure 6E). Consistent with this observation, NLRC5 and CIITA, which encode the master TFs that regulate MHC class I and II genes, were also significantly overexpressed in these cases (adjusted p values = 0.0001 and 0.0018, respectively). The increased expression of HLA genes also correlated with increased TCR diversity in these cases, represented by Shannon Wiener index scores (Welch’s t test p value = 0.012; Figure S6B; Bolotin et al., 2015; Shugay et al., 2015). The cases with increased CD8+ T cell proportions further exhibited significantly higher expression levels of key genes involved in antigen degradation, processing, and transportation (Figure 6E). Such genes included PSMB8/9/10 (which encode components of the immunoproteasome), TAP1 (encodes a component of the transporter-associated with antigen processing complex), and B2M (encodes MHC class I heavy chain). Genes involved in T cell activation, homing, and infiltration were significantly overexpressed in these cases (Figure 6F), such as TNF and IFNG (involved in T cell activation); CXCL9 and CXCL10 (encode chemokines that attract and support the influx of CD8+ T cells); and PRF1, GZMA, and GZMB (encode perforins and granzymes that are secreted by activated cytotoxic T cells). We also observed significant overexpression of CLEC9A/DNGR-1 (adjusted p value = 0.0062), which is expressed in the CD8α+ antigen-presenting dendritic cells that are associated with T cell-infiltrated tumor microenvironments (Gajewski et al., 2013). Overall, these results suggested that RTs exhibiting high CD8+ T cell proportions might have inflamed tumor microenvironments with functionally active CD8+ cytotoxic T cells. Seeking to understand how RTs might survive in such inflamed microenvironments, we analyzed genes involved in T cell inhibitory functions and observed overexpression of an important T cell inhibitory cytokine gene, IL10, and several key immune checkpoint genes (e.g., PDCD1/PD1, CD274/PD-L1, and HAVCR2/TIM3) in the cases with CD8+ T cell proportions greater than the median (adjusted p values < 0.05; Figure 6G). We also observed a significant enrichment for overexpressed genes in these cases in the Ras/ERK/MAP kinase pathway (BH adjusted p value = 1.6e-04), known to maintain clonal anergy, an immune tolerance mechanism by which lymphocytes become functionally inactivated following an antigen encounter (Schwartz, 2003). Taken together, these observations are compatible with the notion that RTs may evade the immune system by either increasing the expression of immunosuppressive programs or reducing the expression of MHC complex components.
To understand whether the level of immune cell presence is unique to RTs compared to other pediatric cancers that occur in similar anatomical sites, we compared T cell scores in RTs to those in medulloblastomas (105 cases) and Wilms tumors (130 cases; Gadd et al., 2017). We observed significantly higher proportions of T cell scores in Groups 1 and 4 and ATRT-TYR compared to medulloblastomas and Wilms tumors (Wilcoxon p values < 0.05; Figure 7A), suggesting that a subset of RTs might be more immuno-stimulated compared to other pediatric cancers of the brain and the kidney.
Figure 7. Comparison of T Cell Presence in RTs to Other Cancer Types and Validation of Increased T Cell Infiltration using IHC.

(A) Boxplot shows T cell scores across the five RT subgroups (19 cases from Group 1, 41 from Group 3, 11 from Group 4, 11 from ATRT-SHH, and 8 from ATRT-TYR), pediatric medulloblastomas (n = 105 cases), and Wilms tumors (n = 130; *Wilcoxon p values < 0.05). IHC profiling was performed on 2,979 regions selected from 185 tumor tissue slides from 35 extra-cranial MRT cases (9 from Group 1, 20 from Group 3, and 6 from Group 4) and 27 ATRT cases (10 from ATRT-MYC, 10 from ATRT-SHH, and 7 from ATRT-TYR). CD68+ myeloid cells were profiled from 915 tumor-enriched (TT), 304 peri-vascular (PV), and 297 peri-stromal (PS) regions. CD3+ lymphoid cells were profiled from 888 TT, 287 PV, and 288 PS regions.
(B and C) Scatter plots show comparisons between T cell scores and median CD3+ leukocyte densities determined for each sample using IHC (B), as well as between CD8+ T cell proportions and median CD3+CD8+ cytotoxic T cell densities determined for each sample using IHC (C; x and y axes in log10 scale). Dashed lines indicate positive linear correlations (Pearson rho = 0.540 and 0.569, linear regression p values = 0.0025 and 0.0019 for CD3+ and CD3+CD8+ cells, respectively).
(D) Boxplots show distributions of CD8+ cytotoxic T cell densities in tumor-enriched (TT), peri-stromal (PS), and peri-vascular (PV) regions (y axis, log10 scale). MRT cases in Groups 1, 3, and 4 and ATRT-MYC cases showed significantly higher CD8+ T cell densities compared to ATRT-SHH and -TYR in all regional types (Wilcoxon p values = 2.2e-16, 6.94e-15, and 3.84e-12, respectively).
(E) Examples of cases with high (top) and low (bottom) T cell infiltration revealed by multiplex IHC staining (CD3+ green; CD8+ brown). Images are at 30x magnification. Scale bars: 100 μm.
(F and G) Boxplots show distributions of overall PD-L1 + cell (F; y axis, log10 scale) and PD-L1-positive CD68+ immune cell densities (G; y axis, log10 scale). The asterisk indicates statistical significance p value < 0.05.
(H) Boxplot shows distributions of PD-L1-negative CD68+ immune cell densities, which are significantly higher in ATRT-SHH compared to other subgroups (*Dunn’s adjusted p value < 0.05).
Immunohistochemistry Confirms Increased Cytotoxic T Cell Infiltration and Immune Checkpoint Expression in MRT and ATRT-MYC
To validate our analyses and orthogonally assess the extent of immune cell infiltration in tumor tissues, we performed multiplex immunohistochemistry (IHC) profiling of 185 tumor samples from 62 patients (35 MRT cases and 27 ATRT cases) by using antibodies to identify CD8+ cytotoxic T cells (CD3+CD8+), CD4+ helper T cells (CD3+CD8−), and macrophages/microglia (CD68+). Expression of the immune checkpoint proteins, PD1 and PD-L1, was also assessed. We were able to evaluate MRT samples selected from among cases we profiled using RNA-seq or DNA methylation array data, but the ATRT samples were from a separate cohort due to a lack of availability of profiled cases. To properly assess the extent of immune cell infiltration in tumor tissues, we examined three types of regions in tumor microenvironments (total number of regions profiled = 2,979; Table S6), i.e., tumor-rich regions away from necrosis (TT; n = 1,803), peri-vascular regions surrounding vascular structures (PV; n = 591), and peri-stromal regions at the interface with benign and/or normal tissues (PS; n = 585).
Our IHC data showed higher levels of tumor-infiltrating CD3+ lymphocytes in MRT and ATRT-MYC compared to ATRT-SHH and -TYR in all regions of the tumor microenvironment (Wilcoxon p value < 2.2e-16; Figure S7A). CD3+ lymphocyte infiltration levels were consistent with our predicted effector T cell scores (Pearson rho = 0.540, linear regression p value = 0.0025; Figure 7B). CD8+ cytotoxic infiltration levels were also consistent with our predicted CD8+ proportions (Pearson rho = 0.569, linear regression p value = 0.0019; Figure 7C). Also consistent with our prediction, the majority (88.6%) of tumor-infiltrating CD3+ lymphocytes in MRT and ATRT-MYC were CD8+ cytotoxic T cells (Figures 7D and 7E; [dummy_Data S1]). In contrast, ATRT-SHH exhibited the lowest CD3+ lymphocyte and CD8+ cytotoxic T cell infiltration, whereas ATRT-TYR showed only a trend toward increased levels of CD4+ helper T cells (Figure S7C). IHC also revealed overall increased expression of PD-L1 in MRTs compared to ATRTs (Wilcoxon p value < 2.2e-16; Figure 7F). A significant increase in PD-L1-expressing CD68+ myeloid cells was also observed in MRTs compared to ATRTs (Wilcoxon p value < 2.2e-16; Figures 7G and S7B; [dummy_Data S1]). MRTs in Group 4 exhibited the highest mean density of PD1-expressing lymphocytes among RT subgroups (Wilcoxon p value = 0.0002; Figure S7D). Notably, ATRT-SHH exhibited the highest median density of PD-L1-negative CD68+ myeloid cells among the five subgroups (Kruskal-Wallis p value = 9.60e-12, Dunn’s adjusted p values against ATRT-SHH < 9.46e-03; Figure 7H).
Given the very low mutation load (and thus paucity of related neoantigens) in RTs, we sought to identify genes that may play a role in increased immunogenicity in RT subgroups. Considering other studies that linked epigenetic de-repression of endogenous retroviral elements (EREs) to anti-tumor immune responses (Chiappinelli et al., 2015; Roulois et al., 2015), we analyzed H3K27ac and DNA methylation levels of CpGs within ERE regions (LINE, SINE, LTR, and ERV from RepeatMasker; n = 3,877,818). Although we noted a significant increase in H3K27ac signals in Groups 1, 3, and 4 compared to ATRT-SHH and -TYR (Welch’s t test p value = 3.17e-05; Figure S6G), we did not observe evidence for ERE de-repression in RTs based on ERE methylation or expression levels (Welch’s t test p values > 0.05; Figures S6C–S6H). On the other hand, we identified nine known tumor antigen genes (ABCC3, CDR2, CEACAM21, CEA-CAM4, DSE, EPS8, ISG15, MUC1, and TBXT) whose expression levels correlated with T cell scores (linear regression p values < 0.05). Of these, IGS15 and TBXT were overexpressed in RTs compared to normal cell types (Figure S7E), suggesting that aberrantly expressed developmental genes such as these may be antigens in RTs.
DISCUSSION
Our integrative meta-analyses of multi-omic datasets revealed shared molecular characteristics between cranial ATRT-MYC and extra-cranial MRT at both global and local levels and enabled identification of five DNA methylation subgroups of RTs across multiple anatomical sites. Our epigenome and gene-expression analyses indicated the role of multiple early developmental states contributing to disease heterogeneity, based on mesoderm-like characteristics in subgroups consisting of MRT and ATRT-MYC, and neural-like characteristics in ATRT-SHH and -TYR. Although such characteristics may point to potential cells of origin, the observation of broad deletions of the SMARCB1 locus in Group 1 cases also presents a possibility of specific genetic alterations contributing to disease heterogeneity, although detailed functional characterizations would be required to confirm this hypothesis.
Unexpectedly, several lines of evidence described in our study supported immune modulation in RTs. ATRT-MYC and MRT showed an enrichment of TFBS in the enhancers of genes involved in type I IFN-induced responses (IRF5/8/9 and STAT1) and antigen presentation (RFX1/5 and XBP-1). Pathway enrichment analyses using subgroup-specific differentially methylated or expressed genes (e.g., UBD and AIM2) also suggested the involvement of type I IFN-mediated signaling (Thibodeau et al., 2012), NF-κB activation (Gong et al., 2010; Hornung et al., 2009), and cytosolic DNA sensing processes that mediate viral defense as well as the maturation of dendritic cells and their ability to mediate antigen presentation (Vanpouille-Box et al., 2018). Our gene expression analyses further supported the notion that a subset of RTs could exhibit increased antigen presentation contributing to creating inflammatory tumor microenvironments infiltrated with functionally active cytotoxic T cells. Although our analysis did not support the notion of epigenetically de-repressed EREs as a potential source of antigens, we did observe increased tumor antigen expression from developmentally silenced genes whose expressions are normally restricted to early embryonic stages or to specific tissue types. In addition, our data were compatible with the notion that somatic deletions affecting immune modulating genes may contribute to increased cytotoxic T cell infiltration. For example, significant under-expression of MIF due to homozygous co-deletion with SMARCB1 in Group1 cases may contribute to increased immunogenicity observed in this subgroup, as suggested by a previous study that demonstrated increased levels of CD8-induced tumor cytotoxicity in MIF double knockout mice compared to wild-type mice (Choi et al., 2012).
Increased infiltration of CD8+ cytotoxic T cells in MRT and ATRT-MYC tumors was directly validated using IHC. Such infiltration has been positively associated with survival and responses to immune checkpoint inhibition (ICI) in other cancer types (Tumeh et al., 2014; Barnes and Amir, 2017). MRTs further exhibited increased infiltration of PD-L1+CD68+ myeloid cells, which also have been associated with favorable responses to ICI (Herbst et al., 2014; Mariathasan et al., 2018). In contrast, ATRT-SHH exhibited the highest level of PD-L1-negative CD68+ myeloid cells, the presence of which has been associated with poor prognosis of ICI (Herbst et al., 2014), consistent with the observation of the lowest CD8+ T cell infiltration level observed in the ATRT-SHH subgroup.
Although ICI has emerged as a promising cancer therapy, it frequently has been described to be most effective against cancers with high mutational burdens that are thought to result in neoantigens that provide a substrate for T cell recognition (Schumacher and Schreiber, 2015; Hellmann et al., 2018). However, several recent studies indicated that mutations in the SWI/SNF complex can also increase the immunogenicity of tumors (Pan et al., 2018; Miao et al., 2018). Our observations of increased cytotoxic T cell infiltration, T cell anergy, and immunosuppressive signaling in immune-responsive MRTs and ATRT-MYC support the notion that T cells may be functionally inhibited by the effects of immune checkpoint signaling and are consistent with accumulating evidence that SWI/SNF mutations can contribute to tumor immunogenicity in ways that may enhance their vulnerability to ICI. Our analyses provoke hypotheses related to the extent of immune cell infiltration, apparent pro- and anti-tumoral immune responses in the tumor microenvironment, and the potential of immune checkpoint inhibitors applied in RT patients. Additional studies will be necessary to deduce mechanisms, but our results so far have shown epigenetic dysregulation in embryonic-development- and immune-related gene expression programs in RT subgroups, perhaps suggesting that tumors with extensive developmental gene dysregulation, which otherwise lack mutations such as RTs, may be poised for immune stimulation. These findings may thus lay the groundwork for further work to delineate whether the immune cell-inflamed phenotypes and molecular similarities between MRT and ATRT-MYC can be usefully deployed in the clinic.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marcel Kool (m.kool@kitz-heidelberg.de).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Data from 141 primary extra-cranial MRT and 161 primary cranial ATRT samples were analyzed for this paper. Data for 40 out of 141 MRT samples were generated as part of a previous report (Chun et al., 2016). In addition to these data, 29 MRT samples (26 from kidneys, 3 from soft tissues) were provided by Dr. Elizabeth Perlman (Ann and Robert H. Lurie Children’s Hospital in Chicago, USA) through the Children’s Oncology Group (COG). From COG, we received pre-therapy tumor and normal DNA from peripheral blood or kidney from rhabdoid tumors (RTs), registered on the National Wilms Tumor Study Group 5 or on COG AREN03B2 banked by the COG Biopathology Center with parental informed consent. Studies were performed with the approval of the University of British Columbia - British Columbia Cancer Agency Research Ethics Board (REB number H09–02558). 9 MRT samples (3 from the spine, 2 from kidneys, remainder from various non-renal tissues e.g., pelvis, face) were provided by Dr. Annie Huang (Hospital for Sick Children in Toronto, Canada) through the Rare Brain Tumor Consortium (RBTC). An additional 63 MRT samples (31 from kidneys, 8 from the liver, remainder from various non-renal tissues e.g., retroperitoneum, Intra-abdomen, face) were provided via the EURHAB study group, with informed consent obtained from all patients included in the study. Data for 150 out of 161 ATRT samples were generated as part of a previous report (Johann et al., 2016). 11 additional ATRT-MYC samples were provided by Dr. Martin Hasselblatt. To enable as comprehensive a study as possible for this rare tumor type, we aggregated all obtainable RT samples that passed quality criteria from COG and EURHAB studies.
For samples provided through COG, Nationwide Children’s Hospital prepared cells and nucleic acids, and shipped these materials to DKFZ for DNA methylation profiling and to Canada’s Michael Smith Genome Sciences Centre at BC Cancer (BCGSC) for whole-genome-, whole-genome bisulfite-, RNA-, and ChIP-seq. For samples provided through DKFZ, cells and nucleic acids were prepared at various sample providers’ institutions, and underwent DNA methylation profiling at DKFZ. Complete sample information, including age and sex of patient subjects, is provided in Table S1.
Tumor content was estimated for 74 cases (56 MRT and 18 ATRT cases) using whole-genome-sequencing data generated from tumor and matched normal pairs and APOLLOH software (Ha et al., 2012), as described previously (Chun et al., 2016). The median tumor purity estimated by APOLLOH was 88.31% (min = 42.78%; max = 95.04%).
METHOD DETAILS
Procedures pertaining to previously published data have been described in Chun et al. (2016) and Johann et al. (2016).
DNA Methylation Array Data Generation and Processing
DNA methylation array data from 150 primary ATRT samples were previously published (Johann et al., 2016). DNA methylation array data from 9 MRT samples from RBTC were generated using Illumina’s Infinium HumanMethylation450 BeadChip (450K) platform. Using Illumina’s Infinium MethylationEPIC (850K) platform, we generated DNA methylation data for 40 primary MRT samples that previously had been analyzed using whole-genome bisulfite sequencing (Chun et al., 2016), and also for an additional 11 ATRT and 91 MRT cases. All samples were checked for expected and unexpected genotype matches by pairwise correlation of the 65 genotyping probes on the Illumina Methylation 450K array. Raw 450K/850K data files were generated and processed as previously described (Capper et al., 2018).
Whole-Genome Library Construction and Sequencing
Whole-genome sequencing (WGS) data from pairs of 40 primary MRT and 18 primary ATRT cases, and their corresponding matched normal samples, were previously published (Chun et al., 2016; Johann et al., 2016). We generated WGS data for an additional 16 pairs of MRT and matched normal samples. WGS library construction, sequencing, and read alignment were performed as previously described in Chun et al. (2016). In brief, all primary tumor and matched normal samples underwent plate-based PCR-free WGS on the Illumina HiSeq 2500 platform to achieve the desired sequence coverage (> 30X). Sequences were aligned to the human reference genome GRCh37-lite/hg19a using the Burrows-Wheeler Aligner (BWA; version 0.5.7; Li and Durbin, 2010). Merged BAM files were marked for duplicates using Picard MarkDuplicates.jar (version 1.71).
Whole-Transcriptome Library Construction and Sequencing
Whole-transcriptome sequencing (RNA-seq) data from 40 primary MRT and 25 primary ATRT cases were previously published (Chun et al., 2016; Johann et al., 2016). We generated RNA-seq data for an additional 25 primary MRT cases. RNA-seq library construction and sequencing were performed as previously described in Chun et al. (2016). In brief, paired-end polyA+ RNA sequencing was performed preserving strand specificity on the Illumina HiSeq 2500 platform.
Whole-Genome Bisulfite-seq Library Construction and Sequencing
Whole-genome bisulfite sequencing (WGBS) data from 40 primary MRT and 17 primary ATRT cases were previously described (Chun et al., 2016; Johann et al., 2016). We generated WGBS data for an additional 29 primary MRT cases. WGBS library construction and sequencing were performed as previously described in Chun et al. (2016). In brief, fragmented bisulfite converted DNA was sequenced using paired-end 100/125 nt V3/4 sequencing chemistry on the Illumina HiSeq 2500 platform.
Chromatin Immunoprecipitation (ChIP) Library Construction and Sequencing
H3K27ac and H3K27me3 ChIP-seq data from 10 primary MRT and 14 primary ATRT cases were previously published (Chun et al., 2016; Johann et al., 2016). We generated H3K27ac and H3K27me3 ChIP-seq data for an additional 24 and 25 primary MRT cases, respectively. ChIP-seq library construction and sequencing were performed as previously described in Chun et al. (2016). In brief, samples were prepared from cross-linked tissues, from which ChIP was performed using the extracted chromatin. Fragmented chromatin DNA was sequenced using paired-end sequencing chemistry on the HiSeq 2000/2500 platforms.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mutation Analyses using Whole-Genome Sequencing Data
In addition to the previously described data, we analyzed whole-genome sequencing data from 18 pairs of ATRT and their matched normal samples, and 16 pairs of MRT and their matched normal samples to identify somatic mutations i.e., copy number alterations (CNA), single nucleotide variants (SNVs), short insertions and deletions (InDels), and structural variants such as inversions, duplications, and translocations that may lead to gene fusions. To allow data comparability, we used the same suite of software tools described in Chun et al. (2016), including Strelka (version 2.0.7; Saunders et al., 2012), SAMtools mpileup (version 0.1.17; Li et al., 2009), and MutationSeq (Ding et al., 2012) to detect somatic SNVs and InDels, APOLLOH (version 012.2014a; Ha et al., 2012) to detect regions with loss of heterozygosity (LOH), CNASeq (version 1.0.10; Jones et al., 2010) to detect CNA, and Trans-ABySS (version 1.4.10; Robertson et al., 2010) to detect structural variants such as chromosomal translocations and inversions.
Copy Number Analysis using DNA Methylation Data
We used the conumee R package (http://bioconductor.org/packages/release/bioc/html/conumee.html) to estimate the chromosomal copy number state from 450K and EPIC/850K DNA methylation array data as previously described in Johann et al. (2016). Regions with values > 0.3 were considered to have chromosome amplifications, while regions with values < −0.3 were considered to have chromosome deletions.
Analysis of RNA-Seq Data
RNA-seq read alignment and gene expression quantification for 65 MRT and 25 ATRT samples were performed using methods previously described in Chun et al. (2016). Briefly, reads were aligned to the human reference genome (version hg19/GRCh37-lite) and to exon junction sequences using BWA. JAGuaR (version 2.2.2; Butterfield et al., 2014) was used to reposition sequences mapped to exon junctions back onto the genome as gapped alignments. We calculated the sequenced base coverage across collapsed exon models to quantify gene-level expression using the gene coverage analysis software developed at Canada’s Michael Smith Genome Sciences Centre. All external RNA-seq data (i.e., neuron progenitor data from the Roadmap Epigenomics Consortium and cranial neural crest data from Prescott et al., 2015) were processed using the same software pipeline and gene annotation versions as the MRT RNA-seq data, as previously described (Chun et al., 2016).
For gene expression analyses, we selected genes that were expressed above a noise threshold of 1 RPKM in all 90 samples. 27,790 out of 58,450 genes, annotated by EnsEMBL version 69, were removed using this filter. To identify differentially expressed genes, we used the DESeq R package (version 1.14.0; R version 3.3.2; Anders and Huber, 2010) and an adjusted p value threshold of 0.05. For subsequent analyses, we further filtered low abundance transcripts that were identified to be overexpressed in one group compared to another group, but had a median expression level less than 1 RPKM. Pathway enrichment analysis was done using DAVID (version 6.8) (Huang et al., 2009), g:Profiler (Reimand et al., 2007), Metascape (Tripathi et al., 2015), and Ingenuity Pathway Analysis© tool, with an adjusted p value threshold of 0.05. We used EnrichmentMap plug-in (version 3.2.0) for Cytoscape (version 3.7.1) to visualize networks of significantly enriched Gene Ontology (GO) biological processes from g:Profiler queries. Node color represents a BH-adjusted p value of enrichment tests. The size of a node is proportional to the number of genes in a biological process. The thickness of an edge is proportional to a similarity coefficient based on the fraction of shared genes between biological processes.
To cluster RT samples based on gene expression, we first removed genes expressed below 1 RPKM in ≥ 75% of samples, and then ranked the remaining genes based on the coefficient of variation. We performed unsupervised non-negative matrix factorization (NMF) using the top 25% most variably expressed genes (n = 3,984), and considered a k value i.e., a clustering solution, at which the highest cophenetic coefficient and silhouette width were observed. We used the NMF R package (version 0.20.2; Gaujoux and Seoighe, 2010), with a default Brunet algorithm and 50 and 500 iterations for the rank survey and the clustering runs, respectively.
To deconvolute gene-expression signals originating from various immune cell types, we applied CIBERSORT analysis using the CIBERSORT R script (version 1.04; Newman et al., 2015) to gene-level RPKM data with 5,000 permutations using the absolute signature score mode. To detect T cell receptor (TCR) sequences, we used MiXCR (version 2.1.9; Bolotin et al., 2015) on FASTQ data generated from paired-end RNA-seq of 25 ATRT and 65 MRT cases, and identified reads that aligned to reference germline V, D, J, and C gene sequences from GenBank, which were then assembled for clonotype mapping i.e., construction of full-length CDR3 regions of the TCR. We then analyzed TCR β clonotypes generated from MiXCR to calculate Shannon Wiener index scores, which quantify the diversity of TCR repertoires in each sample using VDJTools (Shugay et al., 2015).
To compare gene expression levels in RTs against various normal tissue types, we used RPKM values from the Genotype-Tissue Expression dataset (GTEx version 9; number of samples = 2,500; number of normal tissue types = 52), and from normal cerebellum (n = 9) and normal kidney (n = 6) datasets.
Transcriptional network analysis consisted of three steps, as adapted from the SCENIC pipeline (Aibar et al., 2017): (1) Identify potential targets of each transcription factor (TF); (2) perform TF motif enrichment analysis to identify the putative direct targets of each TF to form a transcriptional network, and (3) score the activity of each network in each sample. Co-expression modules were constructed using the R package GENIE3 v0.99.7 (Huynh-Thu et al., 2010). The input used was a gene-expression matrix consisting of genes with >1 RPKM in all samples (n = 90; 65 MRT and 25 ATRT cases). TF-gene pairs with co-expression scores greater than 0.001 and having positive Spearman correlations were used to construct TF gene sets, each consisting of a TF and genes with co-expression patterns. TF motif enrichment analysis was then performed for each TF gene set using the R/Bioconductor package RcisTarget (version 0.99.0), which contains motif rankings for ~1800 TFs from iRegulon (Janky et al., 2014). From these, we inferred direct targets of a TF by filtering the genes that were significantly enriched for at least one of the TF’s binding motifs, and generated a TF network consisting of a TF and its putative direct targets. The activity of each TF network was quantified using the R package AUCell (version 0.99.5). An area under the recovery curve (AUC) analysis was performed for each TF network identified in a sample to quantify the proportion of genes that are present within the highest-expressing genes in each sample. To identify active TF networks in each sample, a binary score indicating network activity states (1 = on/active, 0 = off/inactive) was assigned to each sample using a threshold determined based on the distribution of AUC values across all samples (Aibar et al., 2017).
To quantify endogenous retroviral element (ERE) transcript abundance levels, we used reads that mapped to loci containing short or long interspersed nuclear elements (SINEs or LINEs, respectively) and long terminal repeat (LTR) retrotransposons including endogenous retroviruses (ERV), annotated by UCSC RepeatMasker (hg19 version). A list of 5,467,457 repeat elements and their genomic coordinates was obtained from RepeatMasker (version hg19 - Feb 2009 - RepeatMasker open-4.0.5 - Repeat Library 20140131; Smit et al., 2013). The list was filtered to remove EREs with uncertain annotations (i.e., those annotated with “?”) and EREs on the Y and non-canonical chromosomes. We further selected EREs that did not overlap with gene promoters (includes 1500 bp upstream of a TSS) and gene bodies to obtain ERE transcript abundance levels that were not likely to be confounded by “host” gene transcription (n = 3,877,818). Raw transcript abundance levels were quantified for each ERE by counting the number of reads that mapped unambiguously, with their mate reads mapping within 10kb from a read center. ERE transcript abundance levels were normalized for a library depth, and represented in reads per million reads mapped (RPM).
DNA Methylation Array Analysis
We processed the raw IDAT files using the minfi R package (version 1.20.2; Aryee et al., 2014) and applied the single-sample Noob (normal-exponential out-of-band) method to correct the background. To enable comparisons between 450K and the EPIC arrays, only the probes represented on both arrays were used for the analysis. In addition, the following filtering criteria were applied: Removal of probes targeting the X and Y chromosomes; removal of probes containing a single nucleotide polymorphism (dbSNP132Common) within five base pairs of and including the targeted CpG-site (n = 24,536), and removal of probes not mapping uniquely to the human reference genome (hg19), allowing for one mismatch (Zhou et al., 2017).
Fort-SNE analysis, the R-package tSNE (version 0.1.3) was used. Unsupervised hierarchical clustering and Consensus Clustering were carried out as described previously with varying numbers of CpG sites (Johann et al., 2016).
To assess the extent of molecular similarities and identify subgroups of RTs, we combined DNA methylation array data generated from 161 ATRT and 140 MRT samples to perform unsupervised NMF analysis. We first filtered out CpG sites that were targeted by probes annotated to be less robust (e.g., those with SNPs) according to the published annotation (Zhou et al., 2017). We also removed CpG sites with 0% methylation across all 301 samples. We considered only CpG sites on autosomes, and selected CpG sites that were positively methylated with beta-values >0.3 in at least 10% of samples, following the methods applied by TCGA (Cancer Genome Atlas Research Network, 2014a; Brat et al., 2015). The remaining CpG sites were then ranked using standard deviation, such that the most variably methylated CpG sites could be selected for downstream analyses. We performed unsupervised NMF using the top 10,000 CpG sites with a default Brunet algorithm and 50 and 500 iterations for the rank survey and the clustering runs, respectively.
To test for non-random distribution of covariates between subgroups of interest, we applied Fisher’s exact test (using the fisher.test function in R) and performed Benjamini-Hochberg multiple hypotheses testing adjustments using the p.adjust function in R to obtain adjusted p values.
We performed hierarchical clustering (Spearman correlation coefficient as the distance metric, complete linkage clustering) on 9,758 DNA methylation profiles representing 33 tumor types (n = 9,012), 23 normal tissue types (n = 746) from TCGA, and 464 DNA methylation profiles representing four pediatric tumor types (n = 452) and 12 pediatric normal brain tissues from TARGET. We also included DNA methylation profiles from 8 normal adult and 2 fetal brain samples from DKFZ. For each cancer and normal tissue type, a median beta value for CpG probes was determined (probes were previously filtered using the method described above). These median values, together with DNA methylation profiles from primary MRT and ATRT cases, were then used for the clustering analysis. The TCGA cancer types included BRCA (n = 796; Breast invasive carcinoma); LGG (n = 534; Low-grade glioma), HNSC (n = 530; Head and neck squamous cell carcinoma), THCA (n = 515; Thyroid carcinoma), PRAD (n = 503; Prostate adenocarcinoma), LUAD (n = 475; Lung adenocarcinoma), SKCM (n = 473; Skin cutaneous melanoma), UCEC (n = 439; Uterine corpus endometrial carcinoma), BLCA (n = 419; Bladder urothelial carcinoma), STAD (n = 396; Stomach adenocarcinoma), LIHC (n = 380; Liver hepatocellular carcinoma), LUSC (n = 370; Lung squamous cell carcinoma), KIRC (n = 325; Kidney renal clear cell carcinoma), COAD (n = 316; Colon adenocarcinoma), CESC (n = 309; Cervical squamous cell carcinoma and endocervical adenocarcinoma), KIRP (n = 276; Kidney renal papillary cell carcinoma), SARC (n = 265; Sarcoma), ESCA (n = 186; Esophageal carcinoma), PAAD (n = 185; Pancreatic adenocarcinoma), PCPG (n = 184; Pheochromocytoma and paraganglioma), TGCT (n = 156; Testicular germ cell tumors), GBM (n = 153; Glioblastoma multiforme), LAML (n = 140; Acute myeloid leukemia), THYM (n = 124; Thymoma), READ (n = 99; Rectum adenocarcinoma), MESO (n = 87; Mesothelioma), UVM (n = 80; Uveal melanoma), ACC (n = 80; Adrenocortical carcinoma), KICH (n = 66; Kidney chromophobe), UCS (n = 57; Uterine carcinosarcoma), DLBC (n = 48; Diffuse large B cell lymphoma), CHOL (n = 36, Cholangiocarcinoma), and OV (n = 10; Ovarian serous cystadenocarcinoma). The TARGET cancer types included CCSK (n = 11; Clear cell sarcoma of the kidneys), NBL (n = 224; Neuroblastoma), OS (n = 86; Osteosarcoma), and WT (n = 131; Wilms tumor). The level 3 TCGA and TARGET data were generated using Illumina Human Methylation 450 platform, and were obtained through the TCGA GDC Data Portal at https://portal.gdc.cancer.gov/ and the TARGET Data Portal at ftp://caftpd.nci.nih.gov/pub/OCG-DCC/TARGET/.
For hierarchical clustering, we used the hclust R package (R version 3.3.2) and clustered samples using the top 10,000 most variably methylated CpGs, using complete linkage and the Spearman correlation coefficients as the distance metrics. We also performed hierarchical clustering using the top 1% most variably methylated CpGs (n = 3,958) using complete linkage and the Pearson correlation coefficient as the distance metrics. We used the heatmap.2 function within the gplots R package (version 2.16.0) for visualization of clustering results.
Analysis of ChIP-Seq Data
Alignment of sequencing data was performed as described in Johann et al. (2016). In brief, BWA was used to align sequence reads. Duplicate reads were marked using Picard MarkTools. For enhancer and peak-centered analyses of H3K27ac and H3K27me3 data, we used MACS2 (Zhang et al., 2008) with default settings to call peaks. Peak calling was performed for each sample in the sample cohort, and peaks that were present in two or more samples were retained for analyses. Resulting peaks were merged and used for further analyses.
The signals at peaks were calculated as previously described (Hisano et al., 2013), using the “countsForRegions” function followed by scaling the counts to library size. We applied the same method to calculate H3K27ac signal at promoters, which are defined as regions ± 500 bp around the transcription start site (TSS). Peaks (enhancers) with the most variable signal across all MRT samples were chosen. For unsupervised hierarchical clustering, the top 1,500 most variable peaks over all samples were used.
For TF enrichment analyses, enhancers specific to MRTs were defined based on statistical testing of MRT enhancers versus all enhancers specific to the three ATRT subgroups characterized in our previous report (Johann et al., 2016). Briefly, we applied ANOVA with an FDR cut-off of 0.05 and required at least log2 fold change of 1.5 between MRT and ATRT enhancer signals. Nucleosome free regions (NFRs) of these specific enhancers were identified using the HOMER software (http://homer.ucsd.edu/homer/; version 4.10; Heinz et al., 2010). For TF enrichment, the ENCODE motifs were downloaded from http://compbio.mit.edu/encode-motifs/. Each motif was overlapped with the NFRs from MRT-specific enhancers. Chi-square tests were applied to identify significantly enriched TF motifs (FDR < 0.01). Enrichment values for ATRT subgroup-specific enhancers were taken from previous analyses (Johann et al., 2016).
Identification of Super-Enhancers and Target Genes of Super-Enhancers
Super-enhancers were identified using the HOMER software (http://homer.salk.edu/homer/ngs/index.html) and the findPeaks command with “-style super” option. ATRT super-enhancers had been identified previously in Johann et al. (2016). For MRT-specific super-enhancers, H3K27ac data were combined for all MRT samples and compared to ATRT subgroups. To identify super-enhancers that were common between MRT and ATRT-MYC, we compared the coordinates of super-enhancers and selected those that overlapped by at least 25% between MRT and ATRT-MYC-specific enhancers (as defined in Johann et al., 2016), but not between MRT and other ATRT subgroup-specific enhancers.
Analysis of WGBS Data
WGBS data from ATRT and MRT cases were previously published (Chun et al., 2016; Johann et al., 2016). Alignment of the data was performed as previously described (Hovestadt et al., 2014) using Bismark (Krueger and Andrews, 2011). Identification of partially methylated domains (PMDs) in MRT was performed using the same method as described in Johann et al. (2016). In brief, average methylation levels within windows of 10 kb were calculated in steps of 1 bp. Overlapping 10 kb windows with an average methylation level < 0.6 were merged, and resulting regions larger than 100 kb were termed PMDs.
To identify differentially methylated regions (DMRs), we used the bsseq R package (version 1.18.0; Hansen et al., 2012) to create the data frames for methylated reads and to calculate the whole coverage per sample based on aligned reads. A CpG site with a minimum coverage of 5 reads was selected for downstream analyses. For each sample, a bsseq object was generated, and then analyzed to identify DMRs specific for each of the five subgroups, using the callDMR function in the DSS R package (version 2.12.0; parameters used: minlen = 50, minCG = 5). To identify a gene that overlapped with DMRs, the longest transcript of a protein-coding gene was considered. For visualization of WGBS data, the GVIZR package was used (version 1.26.4; Hahne and Ivanek, 2016).
Immunohistochemistry (IHC)
Two multi-color immunohistochemical panels were stained on whole tissue slides using two staining schemes. All reagents were sourced from Biocare Medical (Pacheco, CA) unless noted otherwise. Slides were incubated overnight at 37°C, then deparaffinized manually using xylene and graded alcohols. The slides were then subjected to antigen retrieval using a Biocare decloaking chamber plus™ at 110°C for 15 minutes in Diva decloaking solution. Slides were then loaded onto a Biocare Intellipath FLX® autostainer. The first two steps of the staining schemes required blocking of endogenous peroxidase activity using peroxidased-1 dispensed using the Intellipath FLX for 5 minutes followed by blocking of non-specific binding using a blocking reagent, background sniper, for 10 minutes. All antibodies were diluted in Biocare Da Vinci Green diluent.
For multiplex IHC targeting CD3 and CD8, we used the following staining scheme: Primary antibodies of CD8 (clone C8/144b from Cell Marque) and CD3 (clone SP7 from Spring Bioscience) were combined into a cocktail diluted in Da Vinci Green diluent at 1/250 and 1/500 dilutions, respectively, which was manually added to the slides for 30 minutes. These were then followed by Mach2 Double Stain #2 polymer dispensed using the Intellipath FLX® for 30 minutes to put CD8 on IP DAB chromogen and CD3 on IP Warp Red chromogen. Following the chromogen step, slides were counterstained with CAT Hematoxylin at a 1/5 dilution and then washed and air-dried prior to coverslipping with Ecomount. The staining scheme for the multiplex IHC targeting PD1, PD-L1, and CD68 was done as follows: In the first round of staining, primary antibodies of PD-L1 (clone SP142 from Abcam) and CD68 (clone KP-1 from Biocare Medical) were combined into a cocktail diluted 1/100 in Da Vinci Green diluent, and applied to the slides for 30 minutes, followed by Mach2 Double Stain #1 polymer for 30 minutes to put CD68 on IP Ferengi Blue chromogen and PD-L1 on IP DAB chromogen. The slides then underwent a denaturation step with SDS-glycine pH2.0 at 50°C for 45 minutes (Pirici et al., 2009). In the second round of staining, we manually applied primary PD1 antibody (clone EPR4877(2) from Abcam) diluted 1/250 in Da Vinci diluent for 30 minutes, followed by Mach2 Rabbit-AP polymer for 30 minutes to put PD1 on IP Warp Red chromogen. Slides were then counterstained with CAT Hematoxylin at a 1/5 dilution and then washed and air-dried, followed by coverslipping with Ecomount.
IHC Analysis
IHC-stained slides were scanned at 10X to create whole slide scans using the Vectra 3 multispectral imaging system (Perkin Elmer, Waltham, MA). The files generated were then passed to a pathologist (B.T-C.) for selection of 15 tumor-rich (TT), 5 peri-vascular (PV), and 5 peri-stromal (PS) regions based on whole slide scans of H&E stained slides using the Pannoramic Midi system (3D Histech). Slides were then re-scanned using the Vectra 3 multispectral imaging system, generating multispectral images at 20X magnification based on the digitally annotated fields of view. Multispectral imaging enabled spectral separation between different chromogens for better visualization of images and spectral superimposition of different chromogens to identify co-expression of proteins. Multispectral images were analyzed using the inForm Image Analysis software (Perkin Elmer) to automatically identify cell phenotypes and perform cell counts. Five phenotyping algorithms were created using a training set of images (10 per algorithm) selected to recognize diverse cell phenotypes. The resulting cell counts were compared and visually validated in all cases by a researcher (H.-J.E.C). Normalized immune cell densities for each image were calculated by dividing validated cell counts by the scanned area (mm2; calculated by multiplying a number of pixels of the scanned image by 2.5 × 10−7), and were then compared across all samples. Normalized cell counts were plotted using R, and statistical significance of cell count differences was calculated using either of the Wilcoxon Mann-Whitney U or Kruskal-Wallis tests.
To enhance visibility and discrimination between IHC colors, IHC images shown in Figure 7E were adjusted to reduce the blue hematoxylin signals by 50% and were re-colored with the following pseudocolors: CD8+ signals in brown and CD3+ in green. To better visualize PD-L1+ CD68+ and PD-L1− CD68+ cells, IHC images were modified as false immunofluorescence images as shown in Figure S7B, with following pseudocolors applied: CD68+ in green, PD-L1+ in red, PD1+ in cyan, and PD-L1+CD68+ in yellow. All data analyses were performed on raw images using inForm.
Text-Mining Analysis for Identifying Putative Tumor-Associated Antigens
In order to build a list of putative tumor antigens, we used a text-mining method (Lever and Jones, 2017) to extract mentions of tumor antigens found in published abstracts and full-text papers. From PubMed abstracts and all downloadable PubMed Central articles, we extracted sentences that mention the phrase “tumor antigen” (variable spellings considered) and contain a gene name from the NCBI gene list with additional synonyms. We then used an active learning approach to annotate the sentences as to whether the gene name was a potential tumor antigen. This used the Kindred relation extraction Python package (Lever and Jones, 2017) to train a logistic regression classifier based on dependency parse-based features. Relations that the classifier found ambiguous, which were those classified differently using a bootstrapping method, were presented to an in silico annotator. This annotation provided a training set to train a final relational classifier that was applied to all relevant sentences and used to build a list of putative tumor antigens that was then reviewed manually. All code is accessible at https://github.com/jakelever/tumorantigens.
DATA AND CODE AVAILABILITY
Raw DNA methylation array data generated from ATRT samples from DKFZ have been deposited in the Gene Expression Omnibus (GEO). The accession number for the ATRT DNA methylation array data reported in this paper is GEO: GSE123601. The accession number for raw DNA methylation array data and sequence data generated from MRT samples from TARGET reported in this paper is NCBI dbGaP: phs000470, with additional data available at http://target.nci.nih.gov/dataMatrix/TARGET_DataMatrix.html. Details for other data and software availability are in the Key Resources Table. Requests for additional data and code should be directed to the Lead Contact.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD8, clone C8/144B | Esbe/Cell Marque, Rocklin, CA | Cat#108M-94; RRID: AB_1158205 |
| Rabbit anti-CD3, clone SP7 | Abcam (Supplier Spring Bioscience) | Cat#ab16669; RRID: AB_443425 |
| Mouse anti-PD-L1, clone SP142 | Abcam (Supplier Spring Bioscience) | Cat#ab228462 |
| Mouse anti-CD68, clone KP-1 | Biocare Medical (Distributed by Intermedico), Pacheco, CA | Cat#CM033 |
| Rabbit anti-PD1, clone EPR4877(2) | Abcam | Cat#ab137132 |
| Biological Samples | ||
| Primary tumor samples | Multiple tissue source sites, processed through the Biospecimen and Library Construction Core Resource | See STAR Methods and Table S1. |
| Critical Commercial Assays | ||
| Infinium MethylationEPIC BeadChip | Illumina | WG-317–1001 |
| Deposited Data | ||
| Raw DNA methylation data (ATRT cases from DKFZ) | This paper | GEO Accession: GSE123601 |
| Raw DNA methylation data (MRT cases from TARGET) | This paper | NCBI dbGaP Accession: phs000470 |
| Raw sequencing data from TARGET MRT cases | This paper | NCBI dbGaP Accession: phs000470 |
| Processed data from TARGET MRT cases | This paper | http://target.nci.nih.gov/dataMatrix/TARGETDataMatrix.html |
| Raw sequencing data previously generated from MRT cases | Chun et al., 2016 | NCBI dbGaP Accession: phs000470 |
| Processed data previously generated from MRT cases | Chun et al., 2016 | http://target.nci.nih.gov/dataMatrix/TARGETDataMatrix.html |
| Raw sequencing data previously generated from ATRT cases | Johann et al., 2016 | EGA Study Accession: EGAS00001001297 |
| Raw DNA methylation array data previously generated from ATRT cases | Johann et al., 2016 | GEO Accession: GSE70460 |
| Raw gene-expression array data previously generated from ATRT cases | Johann et al., 2016 | GEO Accession: GSE70678 |
| Software and Algorithms | ||
| BWA v0.5.7 | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/; RRID:SCR_010910 |
| Picard v1.71 | https://broadinstitute.github.io/picard/ | RRID:SCR_006525 |
| SAMtools v0.1.17 | Li et al., 2009 | http://samtools.sourceforge.net/; RRID:SCR_002105 |
| Strelka v2.0.7 | Saunders et al., 2012 | ftp://strelka@ftp.illumina.com/; RRID:SCR_005109 |
| MutationSeq | Ding et al., 2012 | http://www.shahlab.ca; RRID:SCR_006815 |
| APOLLOH v012.2014a | Ha et al., 2012 | https://shahlab.ca/projects/apolloh/; RRID:SCR_006648 |
| ABySS v1.4.10 | Robertson et al., 2010 | http://www.bcgsc.ca/platform/bioinfo/software/; RRID:SCR_010709 |
| CNASeq v1.0.10 | Jones et al., 2010 | https://www.bcgsc.ca/one/login.cgi?came_from=http%3A//www.bcgsc.ca/platform/bioinfo/software/cnaseq |
| JAGuaR v2.2.2 | Butterfield et al., 2014 | http://www.bcgsc.ca/platform/bioinfo/software/jaguar |
| DESeq R package v1.26.0 | Anders and Huber, 2010 | https://bioconductor.org/packages/release/bioc/html/DESeq.html; RRID:SCR_000154 |
| NMF R package v0.20.6 | Gaujoux and Seoighe, 2010 | https://cran.r-project.org/web/packages/NMF/index.html |
| Minfi R package v1.20.2 | Aryee et al., 2014 | https://bioconductor.org/packages/release/bioc/html/minfi.html; RRID:SCR_012830 |
| GVIZ R package v1.26.4 | Hahne and Ivanek, 2016 | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| Pheatmap R package v.1.0.10 | https://cran.r-project.org/web/packages/pheatmap/index.html | RRID:SCR_016418 |
| Bsseq R package v1.18.0 | Hansen et al., 2012 | http://www.bioconductor.org/packages/2.13/bioc/html/bsseq.html; RRID:SCR_001072 |
| HOMER v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/; RRID:SCR_010881 |
| inForm Cell Analysis | PerkinElmer | https://www.perkinelmer.com/product/inform-cell-analysis-one-seat-cls135781 |
| CIBERSORT v1.04 | Newman et al., 2015 | https://cibersort.stanford.edu/; RRID:SCR_016955 |
| MiXCR v2.1.9 | Bolotin et al., 2015 | https://mixcr.readthedocs.io/en/master/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS; RRID:SCR_013291 |
| Bismark | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/; RRID:SCR_005604 |
Supplementary Material
Highlights.
MYC subgroup of cranial RTs (ATRT-MYC) is molecularly similar to extra-cranial RTs
Five DNA methylation subgroups are identified in RTs across multiple organ sites
Groups 1, 3, and 4 exhibit cytotoxic T cell infiltration and PD1 and PD-L1 expression
ACKNOWLEDGMENTS
We thank the Library Construction, Biospecimen, Sequencing, and Bioinformatics groups at Canada’s Michael Smith Genome Sciences Centre and the German Cancer Research Center (DKFZ) for technical assistance. We thank Nationwide Children’s Hospital for providing samples through COG. We also thank the Rare Brain Tumor Consortium (RBTC; http://rarebraintumorconsortium.ca) and Dr. Annie Huang at the Hospital for Sick Children in Toronto for providing DNA methylation array data from nine MRT samples. We are grateful to Dr. Karen Novik for expert project management and Dr. Dan Jin for providing helpful suggestions on IHC analyses. We also thank Martin Krzywinski for assistance with data visualization. K.M. and B. H.N. thank the BC Cancer Foundation, the Canada Foundation for Innovation (CFI), Canada’s Networks of Centres of Excellence (BioCanRx), and Genome BC. M.A.M. gratefully acknowledges BC Cancer, the BC Cancer Foundation, the CFI, the Canada Research Chairs program, and the Canadian Institutes of Health Research (FDN-143288). H.-J.E.C. thanks the University of British Columbia for financial support through the Roman M. Babicki Fellowship in Medical Research. P.D.J. and M.K. gratefully acknowledge the Heidelberg School of Oncology and the Deutsche Forschungsgemeinschaft (DFG) (JO 1598/1–1) for funding and the EURHAB registry for providing samples. M.H. is supported by IZKF Münster (Ha3/019/15) and DFG (HA3060/5–1). M.C.F. is supported by the Deutsche Kinderkrebsstiftung. This project has been funded in whole or in part with Federal Funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. The contents of this publication do not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.013.
REFERENCES
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, and Irizarry RA (2014). Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes TA, and Amir E (2017). HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br. J. Cancer 117, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessho Y, Miyoshi G, Sakata R, and Kageyama R (2001). Hes7: a bHLH-type repressor gene regulated by Notch and expressed in the presomitic mesoderm. Genes Cells 6, 175–185. [DOI] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. ; Cancer Genome Atlas Research Network (2015). Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med 372, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan B, Stiller C, and Bourdeaut F (2013). Extracranial rhabdoid tumours: what we have learned so far and future directions. Lancet Oncol. 14, e329–e336. [DOI] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Göttgens B, and Buckley NJ (2004). Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc. Natl. Acad. Sci. USA 101, 10458–10463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield YS, Kreitzman M, Thiessen N, Corbett RD, Li Y, Pang J, Ma YP, Jones SJM, and Birol I (2014). JAGuaR: junction alignments to genome for RNA-seq reads. PLoS ONE 9, e102398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014a). Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014b). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, et al. (2018). DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. (2015). Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Kim H-R, Leng L, Kang I, Jorgensen WL, Cho C-S, Bucala R, and Kim W-U (2012). Role of macrophage migration inhibitory factor in the regulatory T cell response of tumor-bearing mice. J. Immunol 189, 3905–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HE, Lim EL, Heravi-Moussavi A, Saberi S, Mungall KL, Bilenky M, Carles A, Tse K, Shlafman I, Zhu K, et al. (2016). Genome-Wide Profiles of Extra-cranial Malignant Rhabdoid Tumors Reveal Heterogeneity and Dysregulated Developmental Pathways. Cancer Cell 29, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato JA, Mercurio F, and Karin M (2012). NF-κB and the link between inflammation and cancer. Immunol. Rev 246, 379–400. [DOI] [PubMed] [Google Scholar]
- Ding J, Bashashati A, Roth A, Oloumi A, Tse K, Zeng T, Haffari G, Hirst M, Marra MA, Condon A, et al. (2012). Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics 28, 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd S, Huff V, Walz AL, Ooms AHAG, Armstrong AE, Gerhard DS, Smith MA, Auvil JMG, Meerzaman D, Chen Q-R, et al. (2017). A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet 49, 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, and Fu YX (2013). Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, and Seoighe C (2010).A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Canaan A, Wang B, Leventhal J, Snyder A, Nair V, Cohen CD, Kretzler M, D’Agati V, Weissman S, and Ross MJ (2010). The ubiquitin-like protein FAT10 mediates NF-kappaB activation. J. Am. Soc. Nephrol 21, 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, Giuliany R, Rosner J, Oloumi A, Shumansky K, et al. (2012). Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 22, 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing genomic data using Gviz and Bioconductor. Methods Mol. Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, and Irizarry RA (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13, R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselblatt M, Nagel I, Oyen F, Bartelheim K, Russell RB, Schüller U, Junckerstorff R, Rosenblum M, Alassiri AH, Rossi S, et al. (2014). SMARCA4-mutated atypical teratoid/rhabdoid tumors are associated with inherited germline alterations and poor prognosis. Acta Neuropathol. 128, 453–456. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, Ni A, Novik JB, Mangarin LMB, Abu-Akeel M, et al. (2018). Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 33, 843–852.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano M, Erkek S, Dessus-Babus S, Ramos L, Stadler MB, and Peters AH (2013). Genome-wide chromatin analysis in mature mouse and human spermatozoa. Nat. Protoc 8, 2449–2470. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, and Fitzgerald KA (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Jones DT, Picelli S, Wang W, Kool M, Northcott PA, Sultan M, Stachurski K, Ryzhova M, Warnatz HJ, et al. (2014). Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 510, 537–541. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A,Wehenkel L, and Geurts P (2010). Inferring regulatory networks from expression data using tree-based methods. PLoS One 5, e12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janky R, Verfaillie A, Imrichová H, Van de Sande B, Standaert L, Christiaens V, Hulselmans G, Herten K, Naval Sanchez M, Potier D, et al. (2014). iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol 10, e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johann PD, Erkek S, Zapatka M, Kerl K, Buchhalter I, Hovestadt V, Jones DTW, Sturm D, Hermann C, Segura Wang M, et al. (2016). Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29, 379–393. [DOI] [PubMed] [Google Scholar]
- Jones SJM, Laskin J, Li YY, Griffith OL, An J, Bilenky M, Butterfield YS, Cezard T, Chuah E, Corbett R, et al. (2010). Evolution of an adenocarcinoma in response to selection by targeted kinase inhibitors. Genome Biol. 11, R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. ; Roadmap Epigenomics Consortium (2015). Integrative analysis of 111 reference human epigenomes. Nature 158, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, et al. (2012). A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J. Clin. Invest 122, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever J, and Jones SJM (2017). Painless Relation Extraction with Kindred. In Proceedings of the BioNLP 2017 workshop, pp. 176–183. [Google Scholar]
- Levy C, Khaled M, and Fisher DE (2006). MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med 12, 406–414. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Nagy L, Inoue S, Shao W, Miller WH Jr., and Evans RM (1998). Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature 391, 811–814. [DOI] [PubMed] [Google Scholar]
- Lue H, Kleemann R, Calandra T, Roger T, and Bernhagen J (2002). Macrophage migration inhibitory factor (MIF): mechanisms of action and role in disease. Microbes Infect. 4, 449–460. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EEIII III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, and Glimcher LH (2010). TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol 11, 411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, Norton C, Bosse D, Wankowicz SM, Cullen D, et al. (2018). Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neiswender H, Navarre S, Kozlowski DJ, and LeMosy EK (2017). Early craniofacial defects in zebrafish that have reduced function of a wnt-interacting extracellular matrix protein, Tinagl1. Cleft Palate Craniofac. J 54, 381–390. [DOI] [PubMed] [Google Scholar]
- Nemes K, and Frühwald MC (2018). Emerging therapeutic targets for the treatment of malignant rhabdoid tumors. Expert Opin. Ther. Targets 22, 365–379. [DOI] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer RJ, Biegel JA, Blaney S, Finlay J, Geyer JR, Heideman R, Hilden J, Janss AJ, Kun L, Vezina G, et al. (2002). Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J. Pediatr. Hematol. Oncol 24, 337–342. [DOI] [PubMed] [Google Scholar]
- Palena C, Polev DE, Tsang KY, Fernando RI, Litzinger M, Krukovskaya LL, Baranova AV, Kozlov AP, and Schlom J (2007). The human T-box mesodermal transcription factor Brachyury is a candidate target for T-cell-mediated cancer immunotherapy. Clin. Cancer Res 13, 2471–2478. [DOI] [PubMed] [Google Scholar]
- Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, Tsoucas D, Qiu X, Lim K, Rao P, et al. (2018). A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359, 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus W (2018). HEIRECA! The HEIdelberg REvolution of CAncer classification and what it means for neurooncology and neuropathology. Acta Neuropathol. 136, 177–179. [DOI] [PubMed] [Google Scholar]
- Pirici D, Mogoanta L, Kumar-Singh S, Pirici I, Margaritescu C, Simionescu C, and Stanescu R (2009). Antibody elution method for multiple immunohistochemistry on primary antibodies raised in the same species and of the same subtype. J. Histochem. Cytochem 57, 567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SL, Srinivasan R, Marchetto MC, Grishina I, Narvaiza I, Selleri L, Gage FH, Swigut T, and Wysocka J (2015). Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, and Vilo J (2007). g:Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, W193–W200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ, et al. (2010). De novo assembly and analysis of RNA-seq data. Nat. Methods 7, 909–912. [DOI] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. (2015). DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. [DOI] [PubMed] [Google Scholar]
- Schwartz RH (2003). T cell anergy. Annu. Rev. Immunol 21, 305–334. [DOI] [PubMed] [Google Scholar]
- Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, et al. (2015). VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLoS Comput. Biol. 11,e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sica A, Schioppa T, Mantovani A, and Allavena P (2006). Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: potential targets of anti-cancer therapy. Eur. J. Cancer 42, 717–727. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, and Green P (2013). RepeatMasker Open-4.0.2013–2015. http://www.repeatmasker.org.
- Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et al. (2012). Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22, 425–437. [DOI] [PubMed] [Google Scholar]
- Thibodeau J, Bourgeois-Daigneault M-C, and Lapointe R (2012). Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. OncoImmunology 1, 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchia J, Golbourn B, Feng S, Ho KC, Sin-Chan P, Vasiljevic A, Norman JD, Guilhamon P, Garzia L, Agamez NR, et al. (2016). Integrated (epi)-Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 30, 891–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and orthogonal integration of Influenza “OMICs” data defines a role for UBR4 in virus budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille-Box C, Demaria S, Formenti SC, and Galluzzi L (2018). Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 34, 361–378. [DOI] [PubMed] [Google Scholar]
- Versteege I, Sévenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, and Delattre O (1998). Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394, 203–206. [DOI] [PubMed] [Google Scholar]
- Wilson NR, Olm-Shipman AJ, Acevedo DS, Palaniyandi K, Hall EG, Kosa E, Stumpff KM, Smith GJ, Pitstick L, Liao EC, et al. (2016). SPECC1L deficiency results in increased adherens junction stability and reduced cranial neural crest cell delamination. Sci. Rep 6, 17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Laird PW, and Shen H (2017). Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw DNA methylation array data generated from ATRT samples from DKFZ have been deposited in the Gene Expression Omnibus (GEO). The accession number for the ATRT DNA methylation array data reported in this paper is GEO: GSE123601. The accession number for raw DNA methylation array data and sequence data generated from MRT samples from TARGET reported in this paper is NCBI dbGaP: phs000470, with additional data available at http://target.nci.nih.gov/dataMatrix/TARGET_DataMatrix.html. Details for other data and software availability are in the Key Resources Table. Requests for additional data and code should be directed to the Lead Contact.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD8, clone C8/144B | Esbe/Cell Marque, Rocklin, CA | Cat#108M-94; RRID: AB_1158205 |
| Rabbit anti-CD3, clone SP7 | Abcam (Supplier Spring Bioscience) | Cat#ab16669; RRID: AB_443425 |
| Mouse anti-PD-L1, clone SP142 | Abcam (Supplier Spring Bioscience) | Cat#ab228462 |
| Mouse anti-CD68, clone KP-1 | Biocare Medical (Distributed by Intermedico), Pacheco, CA | Cat#CM033 |
| Rabbit anti-PD1, clone EPR4877(2) | Abcam | Cat#ab137132 |
| Biological Samples | ||
| Primary tumor samples | Multiple tissue source sites, processed through the Biospecimen and Library Construction Core Resource | See STAR Methods and Table S1. |
| Critical Commercial Assays | ||
| Infinium MethylationEPIC BeadChip | Illumina | WG-317–1001 |
| Deposited Data | ||
| Raw DNA methylation data (ATRT cases from DKFZ) | This paper | GEO Accession: GSE123601 |
| Raw DNA methylation data (MRT cases from TARGET) | This paper | NCBI dbGaP Accession: phs000470 |
| Raw sequencing data from TARGET MRT cases | This paper | NCBI dbGaP Accession: phs000470 |
| Processed data from TARGET MRT cases | This paper | http://target.nci.nih.gov/dataMatrix/TARGETDataMatrix.html |
| Raw sequencing data previously generated from MRT cases | Chun et al., 2016 | NCBI dbGaP Accession: phs000470 |
| Processed data previously generated from MRT cases | Chun et al., 2016 | http://target.nci.nih.gov/dataMatrix/TARGETDataMatrix.html |
| Raw sequencing data previously generated from ATRT cases | Johann et al., 2016 | EGA Study Accession: EGAS00001001297 |
| Raw DNA methylation array data previously generated from ATRT cases | Johann et al., 2016 | GEO Accession: GSE70460 |
| Raw gene-expression array data previously generated from ATRT cases | Johann et al., 2016 | GEO Accession: GSE70678 |
| Software and Algorithms | ||
| BWA v0.5.7 | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/; RRID:SCR_010910 |
| Picard v1.71 | https://broadinstitute.github.io/picard/ | RRID:SCR_006525 |
| SAMtools v0.1.17 | Li et al., 2009 | http://samtools.sourceforge.net/; RRID:SCR_002105 |
| Strelka v2.0.7 | Saunders et al., 2012 | ftp://strelka@ftp.illumina.com/; RRID:SCR_005109 |
| MutationSeq | Ding et al., 2012 | http://www.shahlab.ca; RRID:SCR_006815 |
| APOLLOH v012.2014a | Ha et al., 2012 | https://shahlab.ca/projects/apolloh/; RRID:SCR_006648 |
| ABySS v1.4.10 | Robertson et al., 2010 | http://www.bcgsc.ca/platform/bioinfo/software/; RRID:SCR_010709 |
| CNASeq v1.0.10 | Jones et al., 2010 | https://www.bcgsc.ca/one/login.cgi?came_from=http%3A//www.bcgsc.ca/platform/bioinfo/software/cnaseq |
| JAGuaR v2.2.2 | Butterfield et al., 2014 | http://www.bcgsc.ca/platform/bioinfo/software/jaguar |
| DESeq R package v1.26.0 | Anders and Huber, 2010 | https://bioconductor.org/packages/release/bioc/html/DESeq.html; RRID:SCR_000154 |
| NMF R package v0.20.6 | Gaujoux and Seoighe, 2010 | https://cran.r-project.org/web/packages/NMF/index.html |
| Minfi R package v1.20.2 | Aryee et al., 2014 | https://bioconductor.org/packages/release/bioc/html/minfi.html; RRID:SCR_012830 |
| GVIZ R package v1.26.4 | Hahne and Ivanek, 2016 | https://bioconductor.org/packages/release/bioc/html/Gviz.html |
| Pheatmap R package v.1.0.10 | https://cran.r-project.org/web/packages/pheatmap/index.html | RRID:SCR_016418 |
| Bsseq R package v1.18.0 | Hansen et al., 2012 | http://www.bioconductor.org/packages/2.13/bioc/html/bsseq.html; RRID:SCR_001072 |
| HOMER v4.10 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/; RRID:SCR_010881 |
| inForm Cell Analysis | PerkinElmer | https://www.perkinelmer.com/product/inform-cell-analysis-one-seat-cls135781 |
| CIBERSORT v1.04 | Newman et al., 2015 | https://cibersort.stanford.edu/; RRID:SCR_016955 |
| MiXCR v2.1.9 | Bolotin et al., 2015 | https://mixcr.readthedocs.io/en/master/ |
| MACS2 | Zhang et al., 2008 | https://github.com/taoliu/MACS; RRID:SCR_013291 |
| Bismark | Krueger and Andrews, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/bismark/; RRID:SCR_005604 |