

Poor sleep in older adults is associated with aging of the brain’s resident innate immune cells and impaired cognition.
Abstract
Sleep disruption is associated with cognitive decline and dementia in older adults; however, the underlying mechanisms are unclear. In rodents, sleep disruption causes microglial activation, inhibition of which improves cognition. However, data from humans are lacking. We studied participants in two cohort studies of older persons—the Rush Memory and Aging Project and the Religious Orders Study. We assessed sleep fragmentation by actigraphy and related this to cognitive function, to neocortical microglial marker gene expression measured by RNA sequencing, and to the neocortical density of microglia assessed by immunohistochemistry. Greater sleep fragmentation was associated with higher neocortical expression of genes characteristic of aged microglia, and a higher proportion of morphologically activated microglia, independent of chronological age- and dementia-related neuropathologies. Furthermore, these were, in turn, associated with worse cognition. This suggests that sleep fragmentation is accompanied by accelerated microglial aging and activation, which may partially underlie its association with cognitive impairment.
INTRODUCTION
Cognitive impairment and dementia constitute a growing public health concern. The global prevalence of dementia is estimated at 35.6 million individuals and is predicted to nearly double in 20 years (1). Measures to prevent or delay cognitive impairment and dementia are urgently needed.
Sleep disruption may contribute to cognitive impairment and dementia and their biological substrates in older adults (2). In experimental studies, sleep deprivation is associated with altered expression of genes associated with inflammation and altered immune function (3). Immune dysregulation may, in turn, contribute to cognition-related disease processes, including Alzheimer’s disease (AD) (4–6). These findings raise the possibility that sleep disruption may contribute to cognitive impairment through an immune mechanism. Microglia, the resident innate immune cells of the central nervous system, may play a key role in this. A role for microglia in AD has been implicated by genetic studies (5). Moreover, in rodents, chronic sleep restriction or deprivation can alter the immune signaling milieu in a way known to trigger changes in microglial function and can lead to morphologic microglial activation (7, 8), and inhibiting this can improve cognition (8). However, data relating sleep, microglial biology, and cognition in humans are lacking—an important gap given differences in microglial biology between mice and humans (9). The few studies that have examined human immune gene expression in relation to sleep disruption have focused on peripheral blood and have shown increases in inflammatory, immune, and stress response gene expression (3). However, data from the human brain are lacking, impeding the development of therapies to mitigate the deleterious impact of sleep disruption on cognition and dementia.
To address this gap, we studied participants in the Rush Memory and Aging Project (MAP) and Religious Orders Study (ROS). We quantified sleep fragmentation by actigraphy, the continuous measurement of movement using a wristwatch-like accelerometer, and related this to cognition, neocortical microglial gene expression quantified by RNA sequencing, and neocortical microglial density and morphologic activation assessed by immunohistochemistry. We tested the hypotheses that greater sleep fragmentation is associated with higher expression of microglial marker genes and a higher density of morphologically activated microglia and that these are, in turn, associated with worse cognition.
RESULTS
Study participants
We studied 685 adults (>65 years old; 265 with Alzheimer’s dementia at the last available assessment and 420 without) participating in two cohort studies of older persons—the MAP and the ROS. Clinical and postmortem characteristics of the participants are summarized in Table 1, and the overlap between subsets of these participants is shown in fig. S1A.
Table 1. Characteristics of the study population.
NA, not applicable.
| Characteristic |
Participants with RNA sequencing and actigraphy |
Participants with RNA sequencing and cognitive assessment |
Participants with immunohistochemistry for microglia and actigraphy |
| Number | 152 | 621 | 156 |
| Age at last visit (years) | 88.7 [85.9, 92.6] | 87.8 [83.6, 92.1] | 88.5 [85.5, 92.1] |
| Age at death (years) | 89.6 [86.6, 93.3] | 89.0 [84.8, 93.1] | 89.4 [86.3, 92.9] |
| Female sex | 102 (67.1%) | 396 (63.8%) | 105 (67.3%) |
| European descent | 150 (98.7%) | 611 (98.4%) | 153 (98.1%) |
| Years of education | 14.0 [12.0, 16.0] | 16.0 [14.0, 18.0] | 14.0 [12.0, 16.0] |
| Mini Mental State Examination Score | 25.5 [19.1, 28.0] | 25.0 [18.0, 28.0] | 27.3 [24.8, 28.5] |
| Composite global cognition | −0.62 [−1.41, −0.01] | −0.67 [−1.55, −0.06] | −0.48 [−1.24, 0.05] |
| Clinical diagnosis AD | 51 (33.6%) | 246 (39.6%) | 45 (28.8%) |
| Time from last cognitive assessment to death (years) |
0.7 [0.4, 1.0] | 0.7 [0.4, 1.0] | 0.7 [0.4, 1.1] |
| Average sleep fragmentation (kRA) | 0.027 [0.024, 0.032] | NA | 0.027 [0.025, 0.032] |
| Number of actigraphy recordings | 2.0 [1.0, 3.0] | NA | 2.0 [1.0, 39.0] |
| Time from last actigraphy to death (years) |
1.3 [0.6, 2.3] | NA | 1.4 [0.6, 2.5] |
| Postmortem interval (years) | 6.3 [5.1, 7.8] | 5.9 [4.4, 8.4] | 6.4 [5.2,8.3] |
| AD pathology summary score | 0.58 [0.18, 1.00] | 0.56 [0.16, 1.03] | 0.56 [0.17,0.97] |
| NIA-Reagan pathological AD (intermediate/high) |
96 (63.2%) | 375 (60.4%) | 93 (59.6%) |
| Presence of Lewy body pathology | 33 (21.7%) n = 147 | 125 (20.1%) n = 599 | 34 (21.8%) n = 149 |
| Macroscopic infarcts present | 21 (13.8%) | 76 (12.2%) | 23 (14.7%) |
| Microscopic infarcts present | 30 (19.7%) | 96 (15.5%) | 22 (14.1%) |
| Extralimbic TDP-43 pathology | 83 (54.6%) n = 150 | 269 (43.3%) n = 533 | 86 (55.1%) n = 155 |
| Presence of hippocampal sclerosis | 12 (7.9%) | 45 (7.2%) n = 616 | 13 (8.3%) |
| Density of microglia | 190.8 [150.4, 225.6] | 182.4 [141.4, 217.0] | 191.1 [144.0, 237.9] |
| % Stage I microglia | 95.9 [94.0, 98.3] | 95.9 [94.0, 98.4] | 95.8 [93.9, 98.4] |
| % Stage II microglia | 3.8 [2.0, 5.0] | 3.8 [2.3, 5.1] | 3.9 [2.3, 5.2] |
| % Stage III microglia | 1.3 [0.9, 1.9] | 1.5 [1.0, 2.3] | 1.4 [1.0, 2.1] |
Sleep fragmentation is associated with aging and activation of microglia
We quantified antemortem sleep fragmentation by actigraphy and postmortem dorsolateral prefrontal cortex gene expression by RNA sequencing in 152 MAP participants. We considered the association between antemortem sleep fragmentation and the expression of sets of microglial marker genes from three published sources: the HuMi_Aged gene set (10) and the Galatro gene set (9) derived from postmortem human brain tissue, and the NeuroExpresso cortical microglial gene set derived from rodents (11). We considered gene expression at the individual gene level. We also computed a summary z score by taking the average normalized gene expression across all genes in that set. Despite the incomplete overlap between gene sets (fig. S1B), their composite expression levels were highly correlated (Pearson R = 0.95 to 0.98).
We first examined the HuMi_Aged gene set. In linear regression models adjusted for age, sex, education, time between last actigraphy and death, postmortem interval, RNA quality (RIN) score, and proportion of ribosomal bases, the expression levels of 352 HuMi_Aged genes were associated with sleep fragmentation at an unadjusted threshold of P = 0.05 (Fig. 1A and table S1). Of these, 279 showed positive associations such that greater sleep fragmentation was associated with higher expression, while 73 showed negative associations such that greater sleep fragmentation was associated with lower expression. Eight were associated with sleep fragmentation at a Bonferroni adjusted threshold of P = 0.00005 (ARID5A, CISH, TNFRSF18, SELL, IRF7, PLAUR, SLC11A1, and RPS19); all were expressed at higher levels with greater sleep fragmentation. Next, to capture the collective change in expression of the HuMi_Aged gene set at the bulk tissue level in each participant, we generated a composite measure of microglial marker gene expression by taking the mean normalized expression of all genes in the HuMi_Aged set as described previously (10). In a linear regression model adjusted for age, sex, education, and the same technical covariates, greater antemortem sleep fragmentation was associated with higher composite expression of HuMi_Aged microglial genes (estimate = +0.066 standard units of expression per 0.01 unit difference in kRA; SE = 0.027; P = 0.014; Fig. 1B). Each 0.01 unit increase in kRA, representing roughly 1 SD, was associated with an effect on the composite expression of microglial marker genes (HuMi_Aged gene set) equivalent to 4.5 years of aging [95% confidence interval (CI), 0.9 to 10.5),
Fig. 1. Antemortem sleep fragmentation and expression of microglia marker genes.
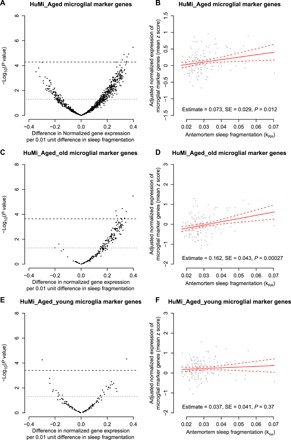
(A and B) HuMi_Aged gene set. (C and D) HuMi_Aged genes enriched in aged microglia (HuMi_Aged_Old gene set). (E and F) HuMi_Aged genes enriched in young microglia (HuMi_Aged_Young gene set). (A, C, and E) Volcano plots of −log10(P value) versus effect size for normalized gene expression as a function of antemortem sleep fragmentation, controlling for age at death, sex, education, and methodological covariates. Each dot represents a single gene. Dotted line indicates unadjusted P < 0.05. Dashed line indicates Bonferroni corrected P < 0.05. (B, D, and F) Partial residual plot of microglial gene expression summary score as a function of antemortem sleep fragmentation adjusted for age, sex, education, and methodological covariates. Y axis is the composite expression for the gene set calculated as described in the text. X axis is average antemortem sleep fragmentation. Each dot represents a single sample. Solid line indicates the predicted composite gene expression for an average participant. Dotted lines indicate 95% CIs on the prediction.
To ensure that these results were not specific to the HuMi_Aged gene set, we repeated these analyses using the Galatro and NeuroExpresso gene sets. Despite incomplete overlap between these gene sets (fig. S1B), results were similar (fig. S2). Similar results were seen when we repeated these analyses considering only those genes that were shared between all three gene sets (fig. S3, A and B) and those genes that were unique to each gene set (fig. S3, C to H), supporting the robustness of these findings. Sleep-wake fragmentation can accompany circadian rhythm dysfunction. Therefore, we repeated the above analyses using an actigraphically derived nonparametric metric of circadian regularity, interdaily stability, that has previously been shown to be abnormal in AD (12). Unlike parametric cosinor-based methods, this does not assume that the activity rhythm conforms to any particular shape. We found no association between interdaily stability and the expression of microglial marker genes (fig. S4), suggesting that the association between sleep fragmentation and microglial marker gene expression is not primarily driven by differences in circadian rhythmicity. To examine the specificity of these findings, we repeated these analyses using astrocyte marker genes from the NeuroExpresso gene set (fig. S5A and table S1). No astrocyte marker genes were associated with sleep fragmentation after Bonferroni correction, and there was no statistically significant association between sleep fragmentation and composite expression of astrocytic marker genes (estimate = +0.021 standard units of gene expression per 0.01 unit difference in kRA; SE = 0.040; P = 0.61; fig. S5B).
The transcriptional phenotype of human microglia changes with age (10). To examine for a differential effect of sleep on age-related microglial transcriptional programs, we repeated the above analyses considering separately those HuMi_Aged microglial marker genes previously identified as being enriched in aged or in young microglia (10, 13). The composite expression of genes characteristic of aged microglia (HuMi_Aged_Old gene set) was positively correlated with age (estimate = +0.009 standard units of gene expression per 1 year of age; SE = 0.003; P = 0.002). Of the 117 genes identified as being enriched in aged microglia, 115 were expressed at higher levels with greater sleep fragmentation at an uncorrected P < 0.05, and 7 (ARID5A, IRF7, SLC11A1, RPS19, CYTH4, HAMP, and RHBDF2) were associated with sleep fragmentation after Bonferroni correction (Fig. 1C; table S1), independent of chronological age at death. Of these, three (CYTH4, HAMP, and RHBDF2) were not significant after Bonferroni correction in our primary analyses of the HuMi_Aged gene set. The reason for this is that there are fewer genes in the HuMi_Aged_Old set, resulting in a less stringent Bonferroni cutoff. In addition to these gene-level findings, composite expression of HuMi_Aged_Old genes was strongly associated with sleep fragmentation (estimate = +0.161 standard units of gene expression per 0.01 unit difference in kRA; SE = 0.041; P = 0.00014; Fig. 1D). To contextualize this, each 0.01 unit greater kRA, corresponding to roughly 1 SD, had an effect on the expression of aged microglial marker genes (HuMi_Aged_Old gene set) equivalent to 7.7 additional years (95% CI, 4.0 to 16.1) of chronological age, suggesting that sleep fragmentation is a better predictor of microglial transcriptional aging than chronological age. Conversely, of the 26 genes previously identified as being associated with young microglia (HuMi_Aged_Young gene set), and meeting quality control criteria in our study, more were expressed at lower levels with greater sleep fragmentation (16 of 26) than were expressed at higher levels with greater sleep fragmentation (10 of 26) (Fig. 2E; table S1), and there was no significant relationship between the composite expression of genes characteristic of young microglia and sleep fragmentation (estimate = +0.015 standard units of expression per 0.01 unit difference in kRA; SE = 0.038; P = 0.69; Fig. 2F). Health behaviors such as smoking and alcohol consumption may plausibly influence sleep and microglial biology. However, the association between sleep fragmentation and the composite expression of genes characteristic of aged microglia remained significant in models controlling for history of smoking and consumption of alcohol.
Fig. 2. Antemortem sleep fragmentation and expression of aging-associated microglial genes—Participants with and without AD pathology.
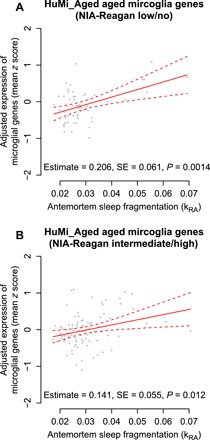
Samples with (A) low or no and (B) intermediate or high NIA-Reagan AD classification. Partial residual plot of microglial gene expression summary score as a function of antemortem sleep fragmentation adjusted for age, sex, education, and methodological covariates. Y axis is the composite expression for the gene set calculated as described in the text. X axis is average antemortem sleep fragmentation. Each dot represents a single sample. Solid line indicates the predicted composite gene expression for an average participant. Dotted lines indicate 95% CIs on the prediction.
A number of brain pathologies, including AD pathology, Lewy body pathology (characteristics of Parkinson’s disease), and infarcts, are associated with sleep fragmentation (14–16). However, the association between greater sleep fragmentation and higher composite expression of genes characteristic of aged microglia remained significant in models controlling for the burden of AD pathology, Lewy body pathology, macroinfarcts, microinfarcts, TDP-43 pathology (characteristics of frontotemporal dementia), or hippocampal sclerosis, alone or in combination (estimate = +0.17 standard units of expression per 0.01 unit difference in kRA; SE = 0.04; P = 0.0001; table S2, model H). Moreover, the association between sleep fragmentation and the expression of genes characteristic of aged microglia (HuMi_Aged_Old gene set) was not significantly different between individuals with and without AD pathology as defined by an National Institute on Aging (NIA)–Reagan classification of intermediate or high (interaction P = 0.82; Fig. 2). Thus, greater sleep fragmentation is associated with higher expression of genes characteristic of aged microglia independent of the presence of AD and other dementia-associated brain pathologies.
Sleep disruption is associated with microglial activation in model organisms (7, 8). We related antemortem sleep fragmentation to neocortical microglial density assessed by immunohistochemistry in 156 MAP participants with quantification of both. In linear regression models adjusted for age at death, sex, education, time between last actigraphy and death, and postmortem interval, we found no significant association between greater sleep fragmentation and the total density of microglia (P = 0.66; Fig. 3A). Next, we examined separately microglia in different stages of morphologic activation: stage I (resting), stage II (activated), and stage III (phagocytic) (fig. S6). We observed a greater proportion of morphologically activated (stages II and III) microglia in association with greater sleep fragmentation (estimate = +0.055, SE = 0.026, P = 0.034; Fig. 3B). This suggests that sleep fragmentation is associated with morphological microglial activation.
Fig. 3. Antemortem sleep fragmentation and density of cortical microglia identified by immunohistochemistry.
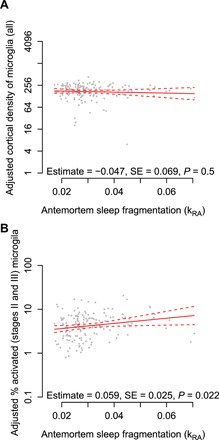
Average of counts in the mid-frontal and inferior temporal cortices. Partial residual plot of total microglial density (A) or proportion of stages II and II microglia (B) as a function of antemortem sleep fragmentation adjusted for age, sex, education, and postmortem interval. Each dot represents a single participant. Solid line indicates the predicted microglial count for an average participant. Dotted lines indicate 95% CIs on the prediction.
We next explored the relationship between composite expression of genes characteristic of aged microglia and morphologic microglial activation quantified by immunohistochemistry. To do so, we analyzed data from the 96 MAP participants who had antemortem quantification of sleep fragmentation and assessment of microglia by both immunohistochemistry and RNA sequencing. In a linear regression model adjusted for age, sex, education, time from last actigraphy and death, postmortem interval of autopsy, RNA RIN score, and proportion of ribosomal bases, the association between sleep fragmentation and the composite expression of aging-associated microglial genes (HuMi_Aged_Old gene set) remained significant (estimate = +0.15 standard units of expression per 1 SD difference in kRA; SE = 0.05; P = 0.0023; table S3). The overall density of microglia was not associated with composite expression of genes characteristic of aged microglia. In contrast, the proportion of activated microglia was associated with the expression of genes characteristic of aged microglia (Spearman R = 0.27, P = 0.009), although this association was somewhat attenuated when controlling for age, sex, and other covariates as above (estimate = +0.08 standard units of expression per 1 SD difference in the proportion of morphologically activated microglia; SE = 0.05, P = 0.12; table S3). The association between sleep fragmentation and the composite expression of genes characteristic of aged microglia was minimally attenuated when controlling for the overall density of microglia or for the proportion of morphologically activated microglia (estimate = +0.14, SE = 0.05, P = 0.004; table S3, model D), suggesting that the association between sleep fragmentation and an aged microglial transcriptional phenotype is not solely due to morphologically defined microglial activation. By contrast, in a model with the proportion of morphologically activated microglia as the outcome, when we controlled for the composite expression of genes characteristic of aged microglia, the effect of sleep fragmentation was attenuated (table S3, model G), which is consistent with a scenario where the association between sleep fragmentation and microglial morphologic activation is mediated in part by a shift toward an aged microglial transcriptional phenotype (fig. S7).
Microglial transcriptional aging and activation are associated with cognitive impairment
We next examined whether higher expression of genes characteristic of aged microglia was associated with cognition proximate to death. To do so, we considered data from the 621 ROS and MAP participants with available dorsolateral prefrontal cortex (DLPFC) RNA sequencing and at least one cognitive assessment. We found that higher composite expression of genes characteristics of aged microglia from the HuMi_Aged_Old gene set was associated with poorer composite global cognition proximate to death, adjusted for age, sex, education, time from last clinical assessment to death, postmortem interval, RNA RIN score, and proportion of ribosomal bases (estimate = −0.254 standard units of composite global cognition per 1 standard unit of gene expression; SE = 0.097; P = 0.0095; Fig. 4A). To contextualize this, each SD greater expression of genes characteristic of aged microglia was equivalent to an additional 3.4 years of chronological age (95% CI, 0.8 to 7.3) in its association with cognition. This association remained significant when controlling for dementia-associated brain pathologies, including the burden of AD pathology, Lewy body pathology, macroinfarcts, microinfarcts, TDP-43 pathology, or hippocampal sclerosis, alone or in combination (P < 0.05; table S4), indicating that the association between the expression of genes characteristic of aged microglia and cognition is not solely accounted for by the presence of these brain pathologies. Moreover, this association remained significant when controlling for source cohort (MAP versus ROS) and did not differ between source cohorts (interaction, P = 0.65).
Fig. 4. Relation of cognition to microglial gene expression and sleep fragmentation.
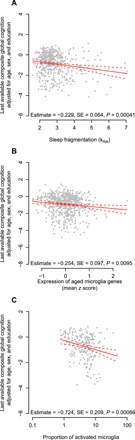
Partial residual plot of composite global cognitive summary score as a function of average antemortem sleep fragmentation (A), composite expression of genes enriched in aged microglia (HuMi_Aged_Old) (B), or proportion of stages II and III microglia (C) adjusted for age, sex, education, and methodological covariates. Each dot represents a single participant. Solid line represents the predicted cognition for an otherwise average participant. Dotted lines indicate 95% CIs on the prediction.
We then examined whether a greater proportion of morphologically defined activated microglia was associated with differences in cognition. To do so, we considered data from the 224 ROS and MAP participants with quantification of microglial activation by immunohistochemistry. In a linear model adjusted for age, sex, education, and technical covariates, a greater proportion of activated microglia was associated with poorer composite global cognition proximate to death (estimate = −0.72, SE = 0.21, P = 0.00066; Fig. 4B).
Next, we examined the relationship between sleep fragmentation, cognitive performance, dementia-related brain pathologies, and microglial aging and activation. In concordance with previous results from our laboratory and others (14, 17), in 480 deceased participants with actigraphy, greater sleep fragmentation was associated with poorer composite global cognition proximate to death (estimate = −0.23 standard units of cognition per 0.01 unit difference in kRA; SE = 0.06; P = 0.0004; Fig. 4C).
Similar results were seen when the analysis was limited to those participants with RNA sequencing data (n = 152; table S4, model I) or with immunohistochemical quantification of microglial density (n = 156; table S4, model M). These associations were partially attenuated in models controlling for the composite expression of genes characteristic of aged microglia (table S4, model J) or for the proportion of morphologically activated microglia (table S4, model N). They were also partially attenuated in models controlling for dementia-associated brain pathologies (AD pathology, Lewy body disease, macroinfarcts, microinfarcts, TDP-43 pathology, and hippocampal sclerosis; table S4, models K and O). In models controlling for both the dementia-associated brain pathologies and for the microglial measures (table S4, models L and P), the association between sleep fragmentation and composite global cognition proximate to death was no longer significant. This is compatible with a scenario in which microglial transcriptional aging or morphological activation partially mediates the association between sleep fragmentation and poor cognition, independent of the presence of dementia-related brain pathologies.
Last, we examined to what extent these relationships differed between those with and without AD pathology. Greater sleep fragmentation was associated with worse cognitive performance in individuals with or without Alzheimer’ disease pathology (interaction P = 0.90; Fig. 5, A and B). However, a pathological diagnosis of AD was a significant modifier of the association between the expression of genes characteristic of aged microglia and cognition (interaction, P = 0.037). Whereas greater microglial gene expression was strongly associated with cognition in those with a pathological diagnosis of AD, this effect was attenuated in the absence of AD pathology (Fig. 5, C and D). Considered another way, high levels of expression of genes characteristic of aged microglia amplified the effect of AD pathology on cognition, with a 1.5-fold greater effect of AD pathology in those in the highest versus lowest quartile of microglial gene expression (Fig. 5, E and F).
Fig. 5. Relation of cognition to microglial gene expression and sleep fragmentation in participants with and without AD pathology.
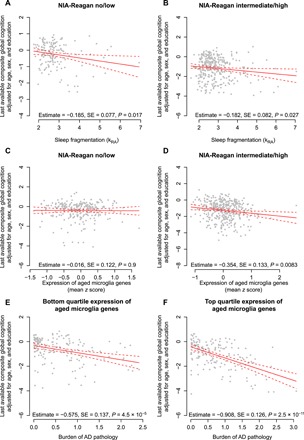
Partial residual plots of composite global cognitive summary score as a function of average antemortem sleep fragmentation (A and B) or the composite expression of genes enriched in aged microglia (HuMi_Aged_Old) (C and D) adjusted for age, sex, education, and methodological covariates. Each dot represents a single participant. Solid line represents the predicted cognition for an otherwise average participant. Dotted lines indicate 95% CIs on the prediction. Subjects with (A and C) low or no or (B and D) intermediate or high NIA-Reagan AD pathological classification. (E and F) Partial residual plot of composite global cognitive summary score as a function of the burden of AD pathology (composite of amyloid plaque and neurofibrillary tangle pathology) adjusted for age, sex, education, and methodological covariates. Subjects with (E) bottom quartile or (F) top quartile expression of genes characteristic of aged microglia. Each dot represents a single participant. Solid line represents the predicted cognition as a function of microglial gene expression for an otherwise average participant. Dotted lines indicate 95% CIs on the prediction.
DISCUSSION
In this cross-sectional study of older community-dwelling adults, greater sleep fragmentation was associated with higher expression of genes characteristic of aged microglia and a greater density of morphologically activated microglia. The transcriptional changes were independent of chronological age, density of microglia, and dementia-related brain pathologies and were not completely accounted for by the increased density of morphologically activated microglia. Moreover, greater expression of genes characteristic of aged microglia was associated with worse cognition and partially accounted for the association between sleep fragmentation and worse cognition. These findings raise the possibility that microglial aging and activation may be a consequence of sleep fragmentation and may link sleep fragmentation to poor cognition in older adults.
Methodological considerations
In this study, we used actigraphy to assess sleep fragmentation. This has several advantages over other approaches to quantifying sleep fragmentation, such as self-report and polysomnography. Self-reported sleep measures can be confounded by misperception and poor recall, particularly in individuals with dementia or cognitive impairment, and do not always correlate well with objective sleep measures (18). In contrast, the metric of sleep fragmentation used in this study correlates well with polysomnographic measures of sleep fragmentation (14). Meanwhile, unlike in-laboratory polysomnography, actigraphy minimally perturbs natural sleep behavior and can be performed for multiple days in the home environment, enabling collection of data from populations of individuals, such as those with dementia and other medical conditions, who may not tolerate sleeping in the laboratory environment.
In this study, we examined microglial biology using two complementary approaches—RNA sequencing to assess differences in the expression of microglial marker genes and immunohistochemistry with stereological counts to assess differences in microglial density and morphology. RNA sequencing allowed us to simultaneously examine hundreds of microglial marker genes rather than being limited to a handful of markers. Moreover, it allowed us to quantify changes in microglial transcriptional phenotype in the absence of changes in microglial density or morphology, as may occur when there are microglial transcriptional changes that may not manifest via morphological changes in activation but nonetheless affect microglial function. To ensure that our results were not sensitive to the choice of microglial marker genes and to enhance cross-species generalizability, we used three different gene sets from two different species, human (9, 10) and mouse (11), all of which yielded concordant results. One limitation of bulk tissue RNA sequencing is that it cannot distinguish between higher microglial gene expression due to greater density of microglia or transcriptional changes at the cellular level in the absence of differences in cell density. To distinguish these possibilities, we used immunohistochemistry to quantify microglial density in a subset of participants and adjusted for this in our analyses. Another limitation of bulk tissue RNA sequencing is that it cannot distinguish between microglial expression of microglial marker genes and ectopic expression of these genes in other cell types. To guard against this, we examined only marker genes that were previously shown to be highly enriched in microglia. Moreover, our summary metric of microglial gene expression incorporated hundreds of marker genes and would have been relatively insensitive to ectopic expression of a handful of microglial marker genes in other cell types. Notwithstanding this, our results will need to be confirmed in future studies using cell-specific sequencing approaches, such as single-cell RNA sequencing.
Sleep fragmentation is associated with greater aged microglial gene expression and density of activated microglia
We have shown that greater sleep fragmentation in older adults is associated with higher expression of microglial marker genes. Similar results were not seen when we used a panel of astrocytic marker genes or when we used an actigraphic marker of circadian rhythmicity, supporting the specificity of these associations. These transcriptional changes were also independent of total microglial density, suggesting that the gene expression results were driven by transcriptional activation rather than an increase in microglial number. The microglial marker gene that showed the strongest association with sleep fragmentation was adenine-thymine (AT)–rich interactive domain 5a (ARID5A), which plays a role in mRNA stabilization and transcriptional regulation of interleukin-6 (IL-6) (19). Ectopic ARID5A overexpression results in increased sensitivity and production of significantly higher levels of IL-6, while ARID5A deficiency inhibits IL-6 elevation in lipopolysaccharide-treated mice (19). Together, these results indicate that ARID5A plays an important role in the promotion of inflammatory processes and increased ARID5A expression results in enhanced inflammatory responses to insults. In concordance with this role, previous studies have shown that low-level sustained microglial activation can lead to abnormal responses to a secondary insult (7). Thus, chronic sleep loss, through microglial priming and possibly the function of ARID5A, may predispose the brain to exaggerated immune responses. Other microglial marker genes that were associated with sleep fragmentation at a Bonferroni significant level have functions in cytokine signaling and secretion [cytokine inducible SH2 containing protein (CISH), rhomboid 5 homolog 2 (RHBDF2), and interferon regulatory factor 7 (IRF7)], lymphocyte interaction and activation [tumor necrosis factor receptor superfamily member 18 (TNFRSF18) and selectin L (SELL)], iron transport [hepcidin antimicrobial peptide (HAMP) and solute carrier family 11 member 1 (SLC11A1)], protein encoding, sorting and membrane trafficking [cytohesin 4 (CYTH4) and ribosomal protein S19 (RPS19)], and plasmin formation [plasminogen activator, urokinase receptor (PLAUR)]. However, further studies are needed to clarify their functions in general and in relation to sleep fragmentation and microglial function.
We saw a particularly strong association between sleep fragmentation and genes characteristic of aged microglia, independent of chronological age, an effect that was independent of measured dementia-associated neuropathologies and present in those with and without AD pathology. Moreover, while we saw no association between sleep fragmentation and overall microglial density, greater sleep fragmentation was associated with a greater density of morphologically activated (stages II and III) microglia. Findings from model organisms support a relationship between microglial aging and activation (20, 21). Our data are compatible with a model whereby sleep fragmentation–related microglial aging predisposes to morphological activation (fig. S7). We were unable to determine the extent to which the population of morphologically activated microglia overlapped with the population of microglia exhibiting greater expression genes characteristic of aged microglia because we performed RNA sequencing on bulk tissue. Experiments using single-cell RNA sequencing are needed to explore this further and definitively link morphologically defined activation to a transcriptional profile.
Our findings are compatible with three scenarios: microglial aging and activation may lead to sleep fragmentation, sleep fragmentation may lead to microglial aging and activation, or both may be caused by other brain changes, such as dementia-related brain pathologies. Microglial aging and/or activation is associated with a number of dementia-related brain pathologies, including AD pathology (22, 23), Lewy body pathology (24), TDP-43 pathology (25), and hippocampal sclerosis (26), several of which are also associated with sleep fragmentation (14–16). Our data indicate that the associations between sleep fragmentation and microglial aging and activation are independent of the burden of these pathologies and do not differ between individuals with and without AD pathology. However, it is possible that other unmeasured brain pathologies may be causing both sleep fragmentation and microglial changes.
Since we analyzed microglial marker gene expression and morphological activation only at a single time point (postmortem), we are unable to definitively determine the causal direction of the association between sleep fragmentation and differences in microglial biology. Studies using positron emission tomography (PET) imaging to longitudinally assess microglial activation, for instance, with translocator protein ligands (27), may clarify the temporal sequence of sleep fragmentation and microglial activation. Notwithstanding this temporal uncertainty, a body of experimental data from model organisms supports a causal link between sleep disruption and microglial activation as noted above. Several mechanisms may be involved. Sleep fragmentation and resultant prolonged wake may cause increases in levels of synaptic debris resulting in the activation of microglia, which participate in the removal of this debris (7). At least some of the individuals in our study with high levels of sleep fragmentation may have had sleep apnea. It has been previously reported that chronic intermittent hypoxia may induce NADPH (reduced form of nicotinamide adenine dinucleotide phosphate) oxidase activation in the brain and accumulation of reactive oxygen species leading to enhanced cellular inflammatory responses and reduced expression of genes required to maintain synaptic structure and function, resulting in pathological neurons that synthesize proinflammatory cytokines, activate microglia, and promote a cycle of oxidative injury, microglial activation, and proinflammatory gene expression (4). Future studies simultaneously measuring sleep apnea and sleep fragmentation are needed to determine whether the association between sleep fragmentation and microglial aging and activation is driven by the subset of participants with sleep apnea or whether this applies to sleep fragmentation more broadly. More generally, studies simultaneously measuring sleep fragmentation and other potential contributors to sleep fragmentation, such as environmental factors (noise and light), sleep disorders, and physical symptoms, are needed to identify the specific causes of sleep fragmentation most strongly related to microglial biology. Moreover, studies are also needed to identify triggers for microglial transcriptional aging and activation in the absence of sleep fragmentation. While genetic and neuropathological factors have been identified as predictors of microglial activation (28), there may be many other as-of-yet unidentified factors of importance.
An aged microglial transcriptional phenotype is associated with cognitive function
We found that higher expression of genes characteristics of aged microglia is associated with cognitive impairment, independent of chronological age. In rats, aging is associated with an increase in microglial activation and associated impairment of hippocampal long-term potentiation (6). Aging primes or sensitizes microglia to have an amplified and exaggerated neuroinflammatory response when activated (29). Microglial activation, in turn, may play an important role in the development or spreading of tau pathology and associated memory deficits (30).
One mechanism through which microglia may affect cognition is by direct modulation of synaptic function. Development of an aged microglial transcriptional phenotype may predispose to formation of activated microglia that can modulate synaptic formation, function, and plasticity by elevating and prolonging production of a range of soluble factors such as inflammatory mediators (such as IL-1β), trophic factors, neurotransmitters, and neuromodulators (31), and by reducing key downstream mediators (such as Arc and brain-derived neurotrophic factor). Reactive oxygen species are part of the neuroinflammatory cascade and can play a prominent role in the development of age-related declines in long-term potentiation (32). In some models, microglial activation can induce a rapid increase of spontaneous excitatory postsynaptic current (ePSC) frequency, through a metabotropic glutamate receptor 5–dependent mechanism, by binding of adenosine triphosphate to purinergic receptors located on astrocytes (33). This increase in ePSC frequency may lead to synaptic dysfunction and cognitive impairment.
In our study, the association between microglial aging and cognitive impairment was particularly significant in individuals with AD pathology, in whom greater expression of genes characteristic of aged microglia amplified the association between the burden of AD pathology and cognitive impairment. This suggests a complex two-hit model where greater sleep fragmentation is associated with higher expression of genes characteristic of aged microglia, irrespective of the presence or absence of AD pathology, but the subsequent impact of microglial transcriptional aging on cognition is greatest in those who also have AD pathology, in whom microglial transcriptional aging amplifies the cognitive impact of AD pathology.
Future directions and clinical implications
Considered as a whole, our data are compatible with a model in which sleep fragmentation may contribute to microglial aging and activation, and this may be an important mechanism underlying the link between sleep fragmentation, cognitive decline, and dementia in older adults. If true, this suggests that targeting microglial aging and/or associated activation may improve cognition in the context of sleep fragmentation. However, additional experiments are first needed. First, experiments are needed to confirm that modifying sleep fragmentation (for instance, by treating sleep disorders) can affect in vivo markers of microglial state assessed by PET or in the cerebrospinal fluid (CSF) [e.g., CSF TREM2 (34)]. Second, experiments are needed to examine the impact of targeting microglial biology on cognition in older adults. In rats following sleep deprivation, inhibiting microglial activity with minocycline improves spatial memory and hippocampal neurogenesis (8). Minocycline has also been shown to prevent the age-related attenuation of long-term potentiation in the rat hippocampus (6). Meanwhile, the tumor necrosis factor–α (TNF-α) synthesis inhibitor 3,6′-dithiothalidomide has been shown to significantly reverse hippocampus-dependent cognitive deficits induced by chronic neuroinflammation (35). However, there are limited data concerning the impact of similar agents on the cognitive impairment associated with sleep fragmentation in humans. Experiments evaluating these effects are needed before proceeding to clinical trials. Last, we studied exclusively older adults, and further studies of younger cohorts are needed to determine whether our findings are generalizable to younger populations.
MATERIALS AND METHODS
Study participants
We analyzed data from participants in two prospective observational community-based cohort studies of older persons (the MAP and the ROS). Characteristics of the study participants are in Table 1. Both studies follow similar clinical and postmortem data collection facilitating their joint analyses. The ROS is a longitudinal study of aging in Catholic brothers, nuns, and priests from across the United States (36). The MAP is a community-based study of aging in the greater Chicago area that enrolls participants with diverse backgrounds and socioeconomic status from continuous care retirement communities throughout northwestern Illinois, as well as from individual homes across the Chicago metropolitan area (36). Participants in both studies are free of known dementia at study enrollment and agree to brain donation upon death. For our primary analyses relating sleep fragmentation and microglial gene expression, we considered all MAP participants with available DLPFC RNA sequencing data meeting quality control criteria (see below) and at least one actigraphic recording. Of 952 deceased MAP participants as of June 2018, 301 had available DLPFC RNA sequencing data meeting quality control criteria, of which 152 also had actigraphy before death. For our analyses relating sleep fragmentation to cortical microglial density, we considered all deceased MAP participants with at least one actigraphic recording and with immunohistochemical assessment of microglial density. Of 952 deceased MAP participants as of June 2018, 224 had assessment of microglial density, of which 156 had actigraphy before death. For our analyses relating expression of microglial genes to cognition, we considered all ROS and MAP participants with available DLPFC RNA sequencing data and with at least one cognitive assessment. All 1765 deceased MAP and ROS participants had had at least one cognitive assessment. Of these, 621 had RNA sequencing data meeting quality control criteria. The overlap of these sets is depicted in fig. S1A.
Statement of ethics approval
The ROS and MAP studies were approved by the Institutional Review Board of Rush University Medical Center. Both studies were conducted in accordance with the latest version of the Declaration of Helsinki, and all participants provided written informed consent and signed an anatomic gift act for brain donation and a repository consent for data sharing.
Assessment of sleep fragmentation and circadian disruption
As described more fully in Supplementary Materials and Methods, sleep fragmentation was assessed biennially in the MAP cohort only by up to 10 days of ambulatory actigraphy using the metric kRA, which as previously published correlated well with polysomnographic measures of sleep fragmentation (14). We also quantified circadian regularity using the interdaily stability metric (12).
In keeping with prior work with other risk factors (37), where more than one measurement was available for an actigraphic measure in a given individual, we took the average of all available measurements. This best reflected the cumulative impact of sleep fragmentation and circadian disruption across the duration of the study while minimizing the effects of individual measurements, which could be reflective of transient acute illness. Because many dementia-related pathologies accumulate over years, we believe that the cumulative burden of sleep fragmentation over the course of the study is likely to have a greater influence on dementia-related pathologies than a single measurement proximate to death.
Assessment of cognition
As described in the Supplementary Materials and Methods, ROS and MAP participants underwent annual structured cognitive assessments consisting of 19 tests from which a composite measure of global cognitive function was computed. We used cognition proximate to death as our primary cognitive endpoint, as it is the cognitive measurement closest in time to the measurement of pathology.
Assessment of other clinical covariates
Age, sex, educational level, smoking history, and alcohol consumption were assessed as described in the Supplementary Materials and Methods.
Assessment of DLPFC microglial and astrocytic gene expression
As described in the Supplementary Materials and Methods, RNA sequencing was performed on blocks of DLPFC from a subset of ROS and MAP participants. These data are available through the synapse.org AMP-AD Knowledge Portal (www.synapse.org; SynapseID: syn3388564). Data from 621 ROS and MAP samples met all quality control criteria and were included in these analyses. For each gene, we computed a normalized expression level by subtracting the mean expression for that gene across all samples and dividing by the SD.
We identified marker genes using data from three sources: the HuMi_Aged gene set (10) and the Galatro gene set (9) derived from postmortem human brain tissue, and the NeuroExpresso cortical microglial and astrocytic gene set derived from rodents (11). For the HuMi_Aged gene set, which was derived from participants in the ROS and MAP cohorts, we also considered separately the subset of microglial genes identified as enriched in aged (HuMi_Aged_Old) versus young (HuMi_Aged_Young) microglia, as derived from a published comparison of the HuMi_Aged gene set (10) with the gene set determined by Zhang et al. (13). For the HuMi_Aged_Old and HuMi_Aged_Young gene sets, we included only those genes that were also part of the primary HuMi_Aged gene set. For the NeuroExpresso gene set, we considered only those genes with human homologs as identified by Homologene (www.ncbi.nlm.nih.gov/homologene). For all gene sets, we considered only those genes that were detectable at nonzero levels in all our samples. HuMi_Aged genes (964 of 1175), HuMi_Aged_Old genes (217 of 238), HuMi_Aged_Young genes (128 of 162), Galatro genes (728 of 1297), and NeuroExpresso microglial genes (172 of 289) met these criteria and were included in these analyses, resulting in a total of 1225 unique genes. In addition, 199 of 302 NeuroExpresso astrocytic genes met these criteria and were included in a separate analysis. These genes and their set membership are depicted in table S1 and fig. S1B. For each of the five microglial gene sets (HuMi_Aged, HuMi_Aged_Old, HuMi_Aged_Young, Galatro, and NeuroExpresso) and the astrocytic gene set (NeuroExpresso_Astro), we computed a summary z score by taking the average normalized gene expression across all genes in that set, as previously described (10).
Quantification of microglial density
As described previously (28, 38), an automated Leica Bond immunostainer (Leica Microsystems Inc., Bannockborn, IL) with anti-human human leukocyte antigen-DP, DQ, and DR antibodies (1:100; clone CR3/43, DakoCytomation, Carpinteria, CA) was used to perform immunohistochemistry for microglia. Individual cells were classified into three groups by an investigator blinded to the other clinical, pathological, and transcriptomic data: stage I (thin ramified processes), stage II (plump cytoplasm and thicker processes), and stage III (appearance of macrophages) (fig. S6). We have shown that the classification approach used in this study correlates well with human genetic variants relevant to microglial biology and/or AD (28, 38), as well as with human cognition and AD pathology (28). The Stereo Investigator 8.0 software program was used to place a 1000 μm by 750 μm sampling grid over the region, and the program was engaged to sample 4.0% of the region with a 200 μm by 150 μm counting frame at ×400 magnification at interval grid intersection points. Using separate tags for stages I, II, and III microglia, the operator marked each individual microglia in each counting frame. These counts were then upweighted by the stereology software to estimate total number of microglia (stages I, II, and III) in the mid-frontal and inferior temporal cortices. Areas occupied by discrete cavitated infarcts, which were relatively rare, were excluded from analysis. Composite average densities of microglia in each region were obtained by averaging data from the two adjacent blocks of tissue (0.5 to 1.0 cm apart). Our data suggest that microglial counts are highly correlated between the mid-frontal and inferior temporal cortices (R = 0.87 for log-transformed total microglial density; R = 0.70 for log-transformed density of activated microglia). Moreover, studies in humans have shown similar levels of microglial activation (39, 40) and morphologic change (41) in different cortical regions in AD and Parkinson’s disease. Therefore, the mean densities in the mid-frontal and inferior temporal cortices were averaged to obtain summary measures for the cortical densities of stages I, II, and III microglia. These were log2 transformed before analysis. In addition to this, we computed the percentage of cells classified as stages I, II, and III microglia. These were log10 transformed before analysis.
Other neuropathological evaluation
As described in the Supplementary Materials and Methods, decedents underwent a structured autopsy. We generated a continuous summary measure of the burden of AD pathology based on counts of neuritic and diffuse plaques and neurofibrillary tangles on Bielschowsky silver staining (42). For a categorical pathological diagnosis of AD, cases were classified as no AD, low likelihood AD, intermediate likelihood AD, or high likelihood AD based on the NIA-Reagan criteria (43); a participant was considered to have a pathological diagnosis of AD if his or her NIA-Reagan classification was “intermediate likelihood” or “high likelihood.” As described in the Supplementary Materials and Methods, we also identified macroscopic infarcts, microscopic infarcts, Lewy body pathology, TDP-43 pathology, and hippocampal sclerosis.
Statistical analyses
For each analysis, only observations with complete data for that analysis were used. First, we used multivariable linear regression models to relate the expression of each marker gene in the HuMi_Aged gene set (10) to average sleep fragmentation before death, adjusted for age at death, sex, years of education, time between last actigraphic recording and death, postmortem interval, RNA integrity number, and proportion of ribosomal bases. Then, instead of considering each gene individually, we computed a composite measure for the expression of all genes in the set by taking the mean of the normalized expression across all genes, and we related this measure to antemortem sleep fragmentation as above. Next, we repeated these analyses using the Galatro and NeuroExpresso microglial gene sets (9, 11). In the sensitivity analyses, we then repeated these analyses considering only those genes that were shared between all three microglial gene sets, and then those genes that were not shared between the three microglial gene sets. We also repeated our primary analyses considering a nonparametric marker of circadian regularity, interdaily stability, in lieu of sleep fragmentation. Next, we repeated our primary analyses using the NeuroExpresso astrocyte gene set in lieu of the microglial gene sets.
Of the genes in the HuMi_Aged gene set (10), a subset was previously identified as being enriched in aged versus young microglia, and another as being enriched in young versus aged microglia by comparison to the Zhang dataset (13). We repeated the above analyses separately considering these two sets of genes (HuMi_Aged_Old and HuMi_Aged_Young). Finding that aging-enriched microglial genes were particularly strongly associated with antemortem sleep fragmentation, we focused on these genes in subsequent analyses.
To assess for confounding by AD and other brain pathologies, we augmented the above models with terms for the burden of AD pathology, presence of Lewy body pathology, presence of macroscopic and microscopic infarcts, presence of TDP-43 pathology in nonlimbic areas, and presence of hippocampal sclerosis. We also examined for effect modification by a pathological diagnosis of AD and analyzed separately those with and without a pathological diagnosis of AD.
We next examined the association between sleep fragmentation and both the overall density of microglia and the proportion of morphologically activated microglia quantified by immunohistochemistry and stereology using linear regression equations adjusted for age at death, time between last clinical assessment and death, sex, years of education, and postmortem interval.
Then, we examined the relationship between microglial gene expression and the density of microglia and proportion of morphologically activated microglia assessed by immunohistochemistry. We first considered linear regression models relating the outcome of composite expression of microglial genes characteristic of aged microglia (HuMi_Aged_Old gene set) to sleep fragmentation, overall microglial density, the proportion of activated microglia, or all of these in combination. Then, we considered linear regression models relating the proportion of morphologically activated microglia to sleep fragmentation, microglial gene expression, or both. These models were all adjusted for age at death, time between last actigraphy and death, sex, years of education, postmortem interval, proportion of ribosomal bases, and RNA RIN score.
We next used multivariable linear regression to relate the last available composite global cognitive score before death to average antemortem sleep fragmentation, controlling for age, sex, and education. Then, in all ROS and MAP participants with available cognitive and RNA sequencing data, we related the last available composite global cognitive score to the composite expression of HuMi_Aged aged microglial genes at death, controlling for age, sex, education, postmortem interval, time between last assessment and death, RNA RIN score, and proportion of ribosomal bases. Next, in all ROS and MAP participants with immunohistochemical quantification of morphologically activated microglia, we related the last available composite global cognitive score to the proportion of activated microglia at death, controlling for age, sex, education, and postmortem interval. Then, we considered multivariable linear regression models relating last available composite global cognitive score before death to average antemortem sleep fragmentation, first controlling for the burden of AD pathology, presence of Lewy body pathology, presence of macroscopic and microscopic infarcts, presence of TDP-43 pathology in nonlimbic areas, and presence of hippocampal sclerosis, and then additionally controlling for the composite expression of HuMi_Aged genes characteristic of aged microglia or for the density of activated microglia. Last, we considered the presence of a pathological diagnosis of AD as an effect modifier, considering the associations between sleep and cognitive impairment and between microglial gene expression and cognitive impairment, separately in those with and without AD pathology.
Supplementary Material
Acknowledgments
We acknowledge the participants in the ROS and MAP cohorts and their families. Funding: This study was funded by the NIH (www.nih.gov) (grants R01AG052488, RF1AG036042, U01AG46152, R01AG048015, U01AG061356, P30AG010161, RF1AG15819, R01AG017917, R01AG047976, R01AG056352, R01AG024480, R01NS78009, R01AG042210, UH2NS100599, R01NS089674, and R01AG043617), the Canadian Institutes of Health Research (www.cihr-irsc.gc.ca/) (MOP125934 and MSH136642), the Robert C. Borwell Endowment Fund, and a CREMS studentship from the University of Toronto. Author contributions: Conceptualization: A.S.P.L. Data curation: J.A.S., A.S.P.L., P.L.D.J., D.A.B., and A.S.B. Formal analysis: A.S.L., S.T., L.Y., and K.K. Funding acquisition: A.S.P.L., E.M.B., J.A.S., D.A.B., A.S.B., and P.L.D.J. Investigation: P.L.D.J., J.A.S., A.S.B., and D.A.B. Methodology: A.S.P.L., J.A.S., P.L.D.J., D.A.B., and A.S.B. Project administration: A.S.P.L., J.A.S., P.L.D.J., D.A.B., and A.S.B. Resources: D.A.B., P.L.D.J., A.S.B., and A.S.P.L. Software: A.S.P.L., L.Y., and S.T. Supervision: A.S.P.L., J.A.S., P.L.D.J., D.A.B., and A.S.B. Validation: A.S.P.L., J.A.S., D.A.B., A.S.B., and P.L.D.J. Visualization: A.S.P.L. and K.K. Writing (original draft): K.K. Writing (review and editing): M.O., S.T., L.Y., E.M.B., J.A.S., A.S.B., D.A.B., P.L.D.J., and A.S.P.L. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The RNA sequencing data are available through the synapse.org AMP-AD Knowledge Portal (www.synapse.org; SynapseID: syn3388564) pending scientific review and a completed data transfer agreement. Requests for the RNA sequencing data should be submitted to www.synapse.org. The remainder of the data are available through the Rush Alzheimer's Disease Center Resource Sharing Hub pending scientific review and a completed data transfer agreement. Requests for these data should be submitted to www.radc.rush.edu/.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax7331/DC1
Supplementary Materials and Methods
Fig. S1. Analytic subsets and microglial gene sets.
Fig. S2. Antemortem sleep fragmentation and expression of microglia marker genes in Galatro and NeuroExpresso microglial gene sets.
Fig. S3. Antemortem sleep fragmentation and expression of shared and unshared microglial marker genes from the HuMi_Aged, Galatro, and NeuroExpresso microglial gene sets.
Fig. S4. Antemortem circadian regularity and expression of microglial marker genes.
Fig. S5. Antemortem sleep fragmentation and expression of astrocytic marker genes.
Fig. S6. Representative photomicrographs of microglial morphological stages.
Fig. S7. Summary diagram.
Table S1. Antemortem sleep fragmentation and expression of microglia and astrocyte marker genes: Gene level results.
Table S2. Antemortem sleep fragmentation and composite expression of aging-enriched microglial genes: Adjustment for neuropathology.
Table S3. Antemortem sleep fragmentation, composite expression of genes characteristic of aged microglia, and proportion of activated microglia.
Table S4. Sleep fragmentation, expression of microglial genes, microglial activation, and composite global cognition proximate to death.
REFERENCES AND NOTES
- 1.Reitz C., Brayne C., Mayeux R., Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 7, 137–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bubu O. M., Brannick M., Mortimer J., Umasabor-Bubu O., Sebastião Y. V., Wen Y., Schwartz S., Borenstein A. R., Wu Y., Morgan D., Anderson W. M., Sleep, cognitive impairment, and Alzheimer’s disease: A systematic review and meta-analysis. Sleep 40, 10.1093/sleep.zsw032, (2017). [DOI] [PubMed] [Google Scholar]
- 3.Möller-Levet C. S., Archer S. N., Bucca G., Laing E. E., Slak A., Kabiljo R., Lo J. C. Y., Santhi N., von Schantz M., Smith C. P., Dijk D.-J., Effects of insufficient sleep on circadian rhythmicity and expression amplitude of the human blood transcriptome. Proc. Natl. Acad. Sci. U.S.A. 110, E1132–E1141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sochocka M., Diniz B. S., Leszek J., Inflammatory response in the CNS: Friend or foe? Mol. Neurobiol. 54, 8071–8089 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tansey K. E., Cameron D., Hill M. J., Genetic risk for Alzheimer’s disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 10, 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffin W. S., Inflammation and neurodegenerative diseases. Am. J. Clin. Nutr. 83, 470S–474S (2006). [DOI] [PubMed] [Google Scholar]
- 7.Bellesi M., de Vivo L., Chini M., Gilli F., Tononi G., Cirelli C., Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J. Neurosci. 37, 5263–5273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wadhwa M., Prabhakar A., Ray K., Roy K., Kumari P., Jha P. K., Kishore K., Kumar S., Panjwani U., Inhibiting the microglia activation improves the spatial memory and adult neurogenesis in rat hippocampus during 48 h of sleep deprivation. J. Neuroinflammation 14, 222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Galatro T. F., Holtman I. R., Lerario A. M., Vainchtein I. D., Brouwer N., Sola P. R., Veras M. M., Pereira T. F., Leite R. E. P., Möller T., Wes P. D., Sogayar M. C., Laman J. D., den Dunnen W., Pasqualucci C. A., Oba-Shinjo S. M., Boddeke E. W. G. M., Marie S. K. N., Eggen B. J. L., Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 20, 1162–1171 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Olah M., Patrick E., Villani A.-C., Xu J., White C. C., Ryan K. J., Piehowski P., Kapasi A., Nejad P., Cimpean M., Connor S., Yung C. J., Frangieh M., McHenry A., Elyaman W., Petyuk V., Schneider J. A., Bennett D. A., De Jager P. L., Bradshaw E. M., A transcriptomic atlas of aged human microglia. Nat. Commun. 9, 539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancarci B. O., Toker L., Tripathy S. J., Li B., Rocco B., Sibille E., Pavlidis P., Cross-laboratory analysis of brain cell type transcriptomes with applications to interpretation of bulk tissue data. eNeuro 4, ENEURO.0212-17.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witting W., Kwa I. H., Eikelenboom P., Mirmiran M., Swaab D. F., Alterations in the circadian rest-activity rhythm in aging and Alzheimer’s disease. Biol. Psychiatry 27, 563–572 (1990). [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Sloan S. A., Clarke L. E., Caneda C., Plaza C. A., Blumenthal P. D., Vogel H., Steinberg G. K., Edwards M. S. B., Li G., Duncan J. A. III, Cheshier S. H., Shuer L. M., Chang E. F., Grant G. A., Gephart M. G. H., Barres B. A., Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim A. S., Kowgier M., Yu L., Buchman A. S., Bennett D. A., Sleep fragmentation and the risk of incident alzheimer’s disease and cognitive decline in older persons. Sleep 36, 1027–1032 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sohail S., Yu L., Schneider J. A., Bennett D. A., Buchman A. S., Lim A. S. P., Sleep fragmentation and Parkinson’s disease pathology in older adults without Parkinson’s disease. Mov. Disord. 32, 1729–1737 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim A. S., Yu L., Schneider J. A., Bennett D. A., Buchman A. S., Sleep fragmentation, cerebral arteriolosclerosis, and brain infarct pathology in community-dwelling older people. Stroke 47, 516–518 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonnet M. H., Cognitive effects of sleep and sleep fragmentation. Sleep 16, S65–S67 (1993). [DOI] [PubMed] [Google Scholar]
- 18.Lauderdale D. S., Knutson K. L., Yan L. L., Liu K., Rathouz P. J., Self-reported and measured sleep duration: How similar are they? Epidemiology 19, 838–845 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higa M., Oka M., Fujihara Y., Masuda K., Yoneda Y., Kishimoto T., Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a. Proc. Natl. Acad. Sci. U.S.A. 115, E1214–E1220 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norden D. M., Godbout J. P., Review: Microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Streit W. J., Sammons N. W., Kuhns A. J., Sparks D. L., Dystrophic microglia in the aging human brain. Glia 45, 208–212 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Friedman B. A., Srinivasan K., Ayalon G., Meilandt W. J., Lin H., Huntley M. A., Cao Y., Lee S. H., Haddick P. C. G., Ngu H., Modrusan Z., Larson J. L., Kaminker J. S., van der Brug M. P., Hansen D. V., Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 22, 832–847 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Mathys H., Adaikkan C., Gao F., Young J. Z., Manet E., Hemberg M., De Jager P. L., Ransohoff R. M., Regev A., Tsai L.-H., Temporal tracking of microglia activation in neurodegeneration at single-cell resolution. Cell Rep. 21, 366–380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mackenzie I. R., Activated microglia in dementia with Lewy bodies. Neurology 55, 132–134 (2000). [DOI] [PubMed] [Google Scholar]
- 25.Sloan S. A., Barres B. A., Glia as primary drivers of neuropathology in TDP-43 proteinopathies. Proc. Natl. Acad. Sci. U.S.A. 110, 4439–4440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beach T. G., Woodhurst W. B., MacDonald D. B., Jones M. W., Reactive microglia in hippocampal sclerosis associated with human temporal lobe epilepsy. Neurosci. Lett. 191, 27–30 (1995). [DOI] [PubMed] [Google Scholar]
- 27.Perrone M., Moon B. S., Park H. S., Laquintana V., Jung J. H., Cutrignelli A., Lopedota A., Franco M., Kim S. E., Lee B. C., Denora N., A novel PET imaging probe for the detection and monitoring of translocator Protein 18 kDa expression in pathological disorders. Sci. Rep. 6, 20422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsky D., Roostaei T., Nho K., Risacher S. L., Bradshaw E. M., Petyuk V., Schneider J. A., Saykin A., Bennett D. A., De Jager P. L., Neuropathological correlates and genetic architecture of microglial activation in elderly human brain. Nat. Commun. 10, 409 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rawji K. S., Mishra M. K., Michaels N. J., Rivest S., Stys P. K., Yong V. W., Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139, 653–661 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asai H., Ikezu S., Tsunoda S., Medalla M., Luebke J., Haydar T., Wolozin B., Butovsky O., Kügler S., Ikezu T., Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 18, 1584–1593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patterson S. L., Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1β, BDNF and synaptic plasticity. Neuropharmacology 96, 11–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGahon B. M., Martin D. S., Horrobin D. F., Lynch M. A., Age-related changes in LTP and antioxidant defenses are reversed by an α-lipoic acid-enriched diet. Neurobiol. Aging 20, 655–664 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Pascual O., Ben Achour S., Rostaing P., Triller A., Bessis A., Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc. Natl. Acad. Sci. U.S.A. 109, E197–E205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gisslén M., Heslegrave A., Veleva E., Yilmaz A., Andersson L.-M., Hagberg L., Spudich S., Fuchs D., Price R. W., Zetterberg H., CSF concentrations of soluble TREM2 as a marker of microglial activation in HIV-1 infection. Neurol. Neuroimmunol. Neuroinflamm. 6, e512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belarbi K., Jopson T., Tweedie D., Arellano C., Luo W., Greig N. H., Rosi S., TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflammation 9, 23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett D. A., Buchman A. S., Boyle P. A., Barnes L. L., Wilson R. S., Schneider J. A., Religious orders study and rush memory and aging project. J. Alzheimers Dis. 64, S161–S189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buchman A. S., Schneider J. A., Wilson R. S., Bienias J. L., Bennett D. A., Body mass index in older persons is associated with Alzheimer disease pathology. Neurology 67, 1949–1954 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Bradshaw E. M., Chibnik L. B., Keenan B. T., Ottoboni L., Raj T., Tang A., Rosenkrantz L. L., Imboywa S., Lee M., Korff A. V., Initiative T. A. D. N., Morris M. C., Evans D. A., Johnson K., Sperling R. A., Schneider J. A., Bennett D. A., De Jager P. L., CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egensperger R., Kösel S., von Eitzen U., Graeber M. B., Microglial activation in Alzheimer disease: Association with APOE genotype. Brain Pathol. 8, 439–447 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edison P., Ahmed I., Fan Z., Hinz R., Gelosa G., Ray Chaudhuri K., Walker Z., Turkheimer F. E., Brooks D. J., Microglia, amyloid, and glucose metabolism in Parkinson’s disease with and without dementia. Neuropsychopharmacology 38, 938–949 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paasila P. J., Davies D. S., Kril J. J., Goldsbury C., Sutherland G. T., The relationship between the morphological subtypes of microglia and Alzheimer’s disease neuropathology. Brain Pathol. 29, 726–740 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bennett D. A., Wilson R. S., Schneider J. A., Evans D. A., Aggarwal N. T., Arnold S. E., Cochran E. J., Berry-Kravis E., Bienias J. L., Apolipoprotein E ε4 allele, AD pathology, and the clinical expression of Alzheimer’s disease. Neurology 60, 246–252 (2003). [DOI] [PubMed] [Google Scholar]
- 43.National Institute on Aging and Reagan Institute Working Group of Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease , Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol. Aging 18, S1–S2 (1997). [PubMed] [Google Scholar]
- 44.Lim A. S., Yu L., Costa M. D., Buchman A. S., Bennett D. A., Leurgans S. E., Saper C. B., Quantification of the fragmentation of rest-activity patterns in elderly individuals using a state transition analysis. Sleep 34, 1569–1581 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson R. S., Schneider J. A., Arnold S. E., Bienias J. L., Bennett D. A., Conscientiousness and the incidence of Alzheimer disease and mild cognitive impairment. Arch. Gen. Psychiatry 64, 1204–1212 (2007). [DOI] [PubMed] [Google Scholar]
- 46.McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E. M., Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS—ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology 34, 939–944 (1984). [DOI] [PubMed] [Google Scholar]
- 47.Schneider J. A., Wilson R. S., Bienias J. L., Evans D. A., Bennett D. A., Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology 62, 1148–1155 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Arvanitakis Z., Leurgans S. E., Barnes L. L., Bennett D. A., Schneider J. A., Microinfarct pathology, dementia, and cognitive systems. Stroke 42, 722–727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nag S., Yu L., Capuano A. W., Wilson R. S., Leurgans S. E., Bennett D. A., Schneider J. A., Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann. Neurol. 77, 942–952 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lim A. S., Klein H. U., Yu L., Chibnik L. B., Ali S., Xu J., Bennett D. A., De Jager P. L., Diurnal and seasonal molecular rhythms in human neocortex and their relation to Alzheimer’s disease. Nat. Commun. 8, 14931 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mostafavi S., Gaiteri C., Sullivan S. E., White C. C., Tasaki S., Xu J., Taga M., Klein H.-U., Patrick E., Komashko V., McCabe C., Smith R., Bradshaw E. M., Root D. E., Regev A., Yu L., Chibnik L. B., Schneider J. A., Young-Pearse T. L., Bennett D. A., de Jager P. L., A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat. Neurosci. 21, 811–819 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Levin J. Z., Yassour M., Adiconis X., Nusbaum C., Thompson D. A., Friedman N., Gnirke A., Regev A., Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat. Methods 7, 709–715 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adiconis X., Borges-Rivera D., Satija R., DeLuca D. S., Busby M. A., Berlin A. M., Sivachenko A., Thompson D. A., Wysoker A., Fennell T., Gnirke A., Pochet N., Regev A., Levin J. Z., Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johnson W. E., Li C., Rabinovic A., Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaax7331/DC1
Supplementary Materials and Methods
Fig. S1. Analytic subsets and microglial gene sets.
Fig. S2. Antemortem sleep fragmentation and expression of microglia marker genes in Galatro and NeuroExpresso microglial gene sets.
Fig. S3. Antemortem sleep fragmentation and expression of shared and unshared microglial marker genes from the HuMi_Aged, Galatro, and NeuroExpresso microglial gene sets.
Fig. S4. Antemortem circadian regularity and expression of microglial marker genes.
Fig. S5. Antemortem sleep fragmentation and expression of astrocytic marker genes.
Fig. S6. Representative photomicrographs of microglial morphological stages.
Fig. S7. Summary diagram.
Table S1. Antemortem sleep fragmentation and expression of microglia and astrocyte marker genes: Gene level results.
Table S2. Antemortem sleep fragmentation and composite expression of aging-enriched microglial genes: Adjustment for neuropathology.
Table S3. Antemortem sleep fragmentation, composite expression of genes characteristic of aged microglia, and proportion of activated microglia.
Table S4. Sleep fragmentation, expression of microglial genes, microglial activation, and composite global cognition proximate to death.