

Abstract
BP180, also termed collagen XVII, is a hemidesmosomal transmembrane glycoprotein expressed in basal keratinocytes, and functions as a cell-matrix adhesion molecule in the dermal-epidermal junction of the skin. Its function other than cell-matrix adhesion remains unclear. We generated a mouse strain with BP180 dysfunction (termed ∆NC16A), which develops spontaneous skin inflammation accompanied by an influx of myeloid derived suppressor cells (MDSCs). We utilized the B16 mouse melanoma model to demonstrate that BP180 dysfunction in either skin or basal keratinocytes promotes MDSC influx into skin and tumor progression. MDSC depletion reduced tumor progression in ∆NC16A mice, demonstrating a critical role for BP180 dysfunction-driven MDSCs in melanoma progression. This study provides the first direct evidence that BP180, a cell-cell matrix adhesion molecule, possesses anti-tumor function through modulating infiltration of MDSCs. Basal keratinocytes actively participate in skin microenvironment changes caused by BP180 dysfunction. ∆NC16A mice could be a new animal model to study the melanoma microenvironment.
Keywords: BP180/collagen XVII, keratinocytes, melanoma, microenvironment, MDSC
INTRODUCTION
BP180, also known as collagen XVII, is a transmembrane glycoprotein of the hemidesmosome 1. The intracellular region of BP180 is linked to the intermediate filament network, and its extracellular portion is anchored into the basement membrane zone (BMZ) through interacting with extracellular matrix proteins 2,3,4. In the skin, BP180 serves as a critical cell-cell matrix adhesion molecule to maintain the integrity in dermal-epidermal junctions. Loss of BP180 function either by mutations in the BP180 gene in the genetic disease termed junctional epidermolysis bullosa (JEB) or by autoantibodies against BP180 in the autoimmune disease bullous pemphigoid (BP) leads to subepidermal blistering in humans and mice 5,6. BP autoantibodies mainly target the extracellular non-collagenous 16A (NC16A) domain of BP180 7,8. Antibodies against the human NC16A and the corresponding mouse NC14A of BP180 are pathogenic in antibody passive transfer models of BP 9,10.
Currently, the only established function of BP180 is as a cell/ECM adhesion molecule, which is supported by the disease JEB, caused by mutations in the BP180 gene COL17A1 11. Initially, BP180 null (Col17a1−/−) mice were developed as a tool by two groups to investigate the function of BP180 in vivo 9,12. One line, termed Col17m− were developed by Dr. Emi Nishimura et al in Japan 9. With this mouse model, Dr. Nishimura’s group found BP180 plays a crucial role in the aging of hair follicle stem cells (HFSC), and maintains a functional niche for melanocyte stem cells through the aging process of HFSC 13,14. However, considering that more than 80% of all Col17m− mice died before two weeks after birth, this imposed a significant limitation on using BP180 null mice to investigate the function of BP180 in the context of disease progression. Dr. Kaisa Tasanen’s group in Finland also developed a BP180 null mouse called Col17a1−/− 12. The Col17a1−/− was developed with a similar strategy compared to Dr. Nishimura’s group, and also showed a similar mortality rate; 90% of them died within two weeks of birth. Therefore, the high mortality rate of BP180 null mice severely restricts its application in investigating BP180 function in vivo.
Since the 1980s, multiple reports have shown that altered BP180 expression is associated with various types of skin cancers, including squamous cell carcinoma (SCC) 15,16,17, basal cell carcinoma (BCC) 18, and melanoma 19. Untransformed melanocytes do not express BP180; when transformed, melanoma cells acquire the expression of BP180 19. Epidermal keratinocytes can modulate the metastasis of nearby melanoma cells 20; however, whether BP180 in basal keratinocytes is involved in melanoma and/or other skin cancer progression remains to be determined.
To develop the BP passive transfer mouse model, our lab generated a humanized mouse with the mouse NC14A domain being replaced with the human NC16A domain (termed hNC16A or WT mice). Our lab purposely flanked the NC16A-encoding sequence with loxP sites 10. As we crossed the humanized NC16A mice with mice carrying different promoter-driven Cre genes, we generated systemic or conditional NC16A deficient mice (termed ∆NC16A) 21. The ∆NC16A mice can live up to one year after birth, which provided a valuable tool to investigate a role of BP180 other than as a cell/ECM adhesion molecule 21. We previously reported that ∆NC16A mice develop spontaneous skin inflammation 21 and inflammation had been consider as one of the hallmark of cancer22,23. Therefore, in this study, we test the hypothesis that BP180 in basal keratinocyte may affect melanoma progression. When injected with B16 melanoma cells, ∆NC16A mice showed accelerated tumor progression. Our findings provide the first direct evidence that BP180 plays a role in modulating the tumor microenvironment and melanoma progression.
RESULTS
The generation of the BP180 dysfunctional ΔNC16A Mice.
We generated humanized NC16A mice (termed WT mice) by replacing the mouse NC14A domain with the human NC16A counterpart to study the disease mechanisms of BP10. The NC16A domain is encoded by exons 18 and 19 of the BP180 gene, which were flanked by lox-P sites (Figure 1A). When crossed with germline Cre mice, Cre recombination removes the loxP-flanked exons 18 and 19 and maintains the remaining reading frame, resulting in mice expressing NC16A domain truncated BP180 (termed ΔNC16A). Lack of the NC16A domain in ΔNC16A mice was confirmed by genotyping (Figure 1B), immunoblotting (Figure 1C) and immunofluorescence staining (Figure 1D). Similar to previously described mice lacking the NC14A domain 24, ΔNC16A mice showed no clinical phenotypes after birth but began to gradually develop various clinical phenotypes starting eight weeks after birth, which includes: reduced body size/weight, hair loss, depigmentation and skin inflammation (Figure 1E and Figure 1F, panel b). We will only focus on the aspect of skin inflammatory microenvironment and melanoma progression in this report.
Figure 1. Generation of BP180 dysfunctional ΔNC16A mice.
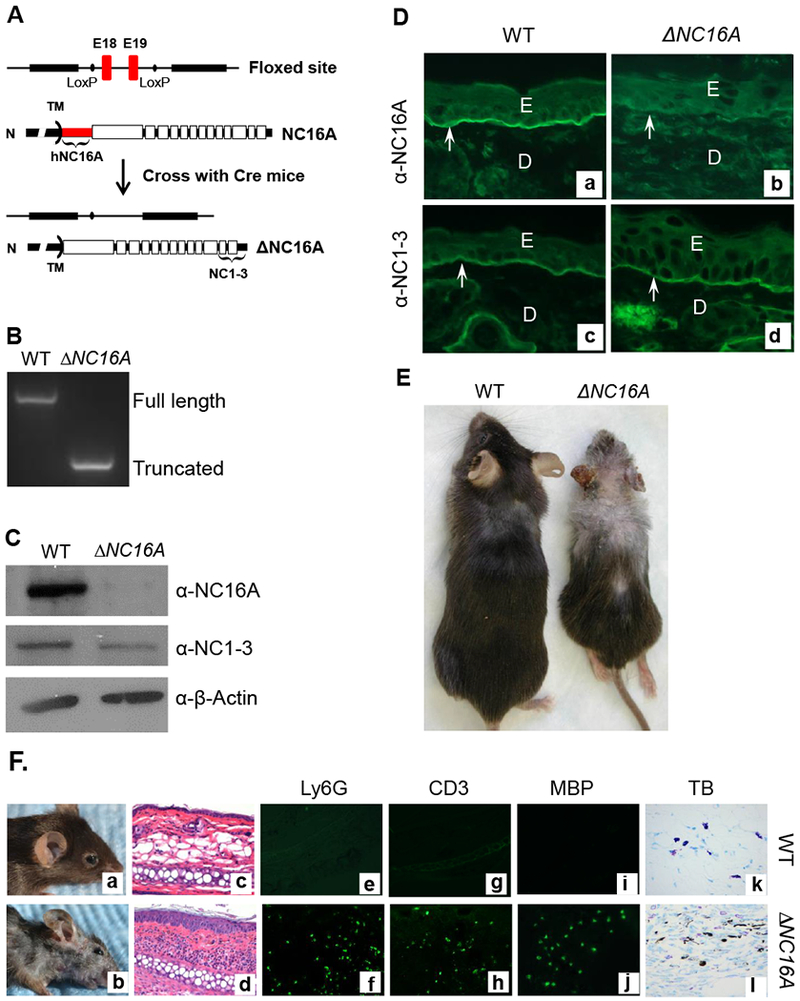
A, Humanized NC16 mice (WT) with NC16A-encoding exons 18 and 19 (red) flanked by loxP sites (9) were crossed with germline Cre mice, resulting in mice expressing NC16A truncated BP180 (termed ΔNC16A). Antibodies against NC16A or NC1-3 domain were used to distinguish the full-length or NC16A truncated BP180. B, Genomic DNA PCR analysis confirmed the absence of the NC16A-encoding exons in ΔNC16A mice. C, Immunoblotting of skin protein extracts detected a full-length BP180 in WT and a NC16A-truncated BP180 in ΔNC16A mice. D, Immunofluorescence (IF) of skin revealed that anti-NC16A antibody stained the BMZ of only WT skin, while anti-NC1-3 antibody stained the BMZ of both WT and ΔNC16A skin, confirming the NC16A-truncated BP180 were also localized in the BMZ of ΔNC16A mouse skin (panel d). E, epidermis; D, dermis; arrows, BMZ. E. ΔNC16A mice develop several physiological phenotypes, which include reduced size, depigmentation and hair loss starting at 8 weeks after birth. F. Clinical examination found that ΔNC16A mice started to develop spontaneous skin inflammation at around 8 weeks after birth (panel b). Histological examination revealed an inflammatory infiltrate and epidermal hyperplasia in ΔNC16A mice (panel d). Lesional skin of ΔNC16A mice showed significantly increased infiltrating neutrophils (Ly6G positive, panel e and f), T cells (CD3 positive, panel g and h) and eosinophils (MBP positive, panel i and j) by indirect IF and mast cells by toluidine blue (TB) staining (panel k and l).
ΔNC16A Mice Exhibit an Influx of Immune Cells into skin
Histological examination revealed minor skin inflammation starting at the age of 8 weeks after birth, accompanied with increased epidermal thickness and a low degree of dermal-epidermal separation in the skin (Figure 1F, panel d); infiltration of immune cells (Figure 1F, panel d), including neutrophils (Figure 1E, panel f), T cells (Figure 1E, panel h), eosinophils (Figure 1E, panel j) and mast cells (Figure 1F, panel l).
BP180 is also expressed in many tissues/organs other than skin keratinocytes 25. To determine whether the influx of immune cells seen in ∆NC16A was caused by deletion of NC16A in the skin, we generated tamoxifen inducible Cre-NC16A mice (termed TamCre-NC16A mice) by crossing NC16A mice with tamoxifen inducible Cre mice. When treated with tamoxifen topically, TamCre-NC16A mice became skin-specific ΔNC16A (termed skinΔNC16A) mice and was confirmed by immunoblotting and immunofluorescence (Supplemental Figure 1). Skin-specific ΔNC16A mice also exhibit the increased skin infiltration of neutrophils, MCs, T cells and eosinophils, starting day 14 post tamoxifen treatment (Supplemental Figures 2). WT mice treated with the same dose of tamoxifen did not develop immune cell influx, which rules out that the influx of immune cells is caused by tamoxifen (Supplemental Figure 2A). Myeloperoxidase (MPO) assay results further confirmed that skin-specific NC16A deletion is sufficient to promote the influx of neutrophils (Supplemental Figure 2B and 2C)
To determine whether ΔNC16A directly promotes immune cells influx into skin through basal keratinocytes, we crossed NC16A mice with K14Cre mice to generate basal keratinocyte-specific ΔNC16A (termed K14Cre/ΔNC16A) mice (Supplemental Figure 3). Like ΔNC16A and skinΔNC16A mice, K14Cre/ΔNC16A also showed a significantly increased influx of MCs, neutrophils, T cells and eosinophils (Supplemental Figure 3).
Mice Lacking NC16A Exhibit Accelerated B16 Melanoma Progression
Inflammatory tumor microenvironment is one of the hallmarks of cancer 22. It is still unclear whether untransformed non-immune cells may play a role in tumor progression by modulating the tumor microenvironment. Skin is known to be the largest immune-protective organ in our body, and keratinocytes play a crucial role in modulating inflammation and defense against host infection in the skin 26,27. To our knowledge, there are very limited reports showing that keratinocytes may play a role in the tumor microenvironment 20,28. It had been reported that chronic skin inflammation is a risk factor for skin cancer initiation, and also promotes a microenvironment which favors tumor progression in skin 29,30,31. Because ΔNC16A triggers immune cell influx through basal keratinocytes, we hypothesized that BP180 in basal keratinocytes plays a role in melanoma progression through modulating immune cells in skin. To test this hypothesis, C57BL/6J and ΔNC16A mice were injected with B16 melanoma cells (1×105 cells, s.c) into the flank and monitored for tumor progression for 3 weeks. Melanoma grew significantly faster in ΔNC16A mice compared to C57BL/6J mice (Figure 2A and 2B), and ΔNC16A mice showed a reduced survival (Figure 2C).
Figure 2. ΔNC16A mice show significantly increased melanoma progression in both flank and ear injection models.
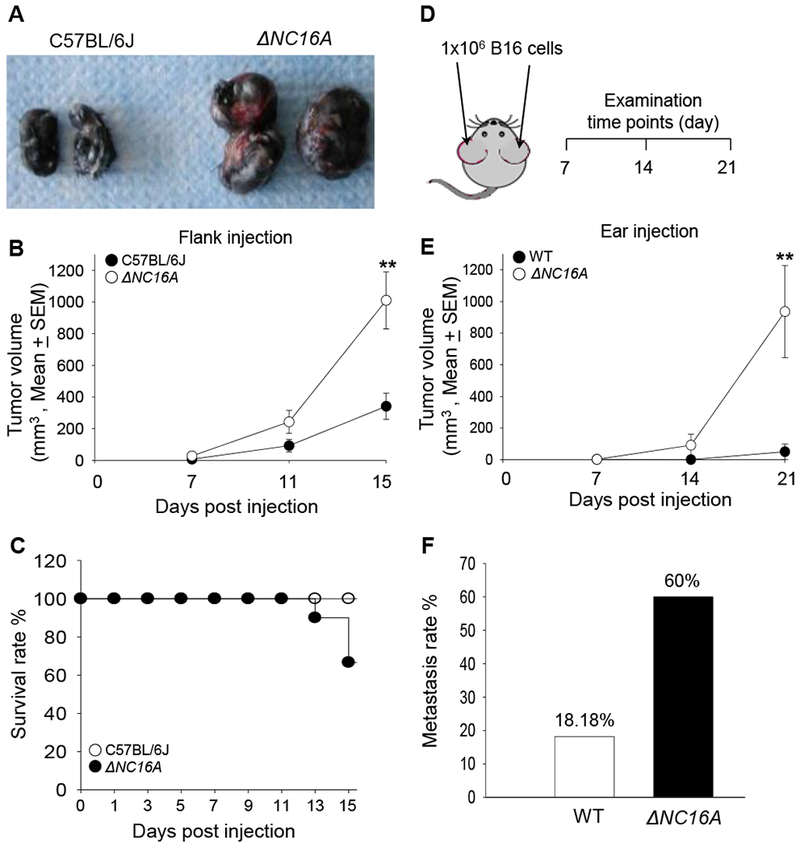
Eight weeks old of C57BL/6J and ΔNC16A mice were injected at the flank (A-C) or ear (D-F) with 1×105 or 1×106 B16 melanoma cells, respectively, and examined at different time points post injection. In the flank model, ΔNC16A mice developed significantly larger size of tumors (A), significantly increased tumor growth rate starting from day 11 (B) (** p<0.05, Scheirer-Ray-Hare test; n=9, for both WT and ΔNC16A mice.) and reduced survival rate at day 15 post injection (C).Using the cox regression model, we obtain an expected 2.55 fold (95% CI 0.66-9.89) the hazard ratio of NC16A comparing to the wild-type group, with p-value of 0.156 in the likelihood-ratio test. In the ear model (D), ΔNC16A mice showed significantly faster tumor growth starting day 14 (E) (** p<0.01, Scheirer-Ray-Hare test) and significantly increased rate of developing neck lymphatic metastasis at day 21 post melanoma cell injection (p<0.05, Fisher’s Exact Test) (F) For ear injection mode, n=10 for ΔNC16A, n=22 for WT).
Mouse melanocytes are mainly distributed in hair follicles, while human melanocytes are located at the basal keratinocyte layer 32. The only mouse sites that are compatible to human skin in terms of melanocyte distribution are on the ears and tail 33,32. Injection of melanoma cells into the ear has been considered a more clinically relevant model for human melanoma progression and metastasis 34,35. In addition, we switch to hNC16A (WT) mice instead of C57BL/6J mice due to they are more genetically relevant to ΔNC16A mice 21. We injected B16 melanoma cells (1 × 106 cells) into the ears of hNC16A (WT) and ΔNC16A mice and monitored the tumor growth locally on the ears and metastasis on the neck for 3 weeks (Figure 2D). Tumor volumes were significantly increased in ΔNC16A mice compared to WT mice starting at day 14 and day 21 after injection (Figure 2E). 60% of ΔNC16A mice developed lymphatic metastasis compared to none of the WT at 21 days post injection (Figure 2F). Taken together, these results suggest that BP180 plays a role in melanoma progression.
Skin local and basal keratinocyte conditional ΔNC16A promotes melanoma progression.
Increased melanoma progression in ΔNC16A mice could be caused by local BP180 dysfunction and/or global deletion of NC16A leading to reduced systemic tumor suppression. To distinguish these, the same TamCre-NC16A mice were topically treated at the left ear with tamoxifen and right ear with vehicle. Ten days later, both ears were injected with the same number of B16 melanoma cells (1×106 cells). Tamoxifen-treated left ears showed significantly increased tumor growth and metastasis, whereas the control right ear showed no visible tumor growth 21 days after melanoma injection (Figure 3A). Thus, local BP180 dysfunction affects B16 melanoma progression. To elucidate whether local skin BP180 dysfunction is sufficient enough to promote melanoma progression, we expanded our experiments to compare the tumor progression between WT mice treated with Tamoxifen (Control) and Tam-CreNC16A (SkinΔNC16A) mice. SkinΔNC16A developed a significantly increased tumor volume (Figure 3B) and more lymphatic metastasis compared to the control mouse ears at 21 days post melanoma cell injection (77% of SkinΔNC16A vs. 20% of Control ears, Figure 3C). Melanoma progression in skin-specific ΔNC16A mice is similar to whole body ΔNC16A mice (Figure 3B and 3C). These results suggest that BP180 local dysfunction is sufficient to promote B16 melanoma progression in vivo.
Figure 3. Skin-and basal keratinocyte-specific BP180 dysfunction are sufficient to promote B16 melanoma progression.
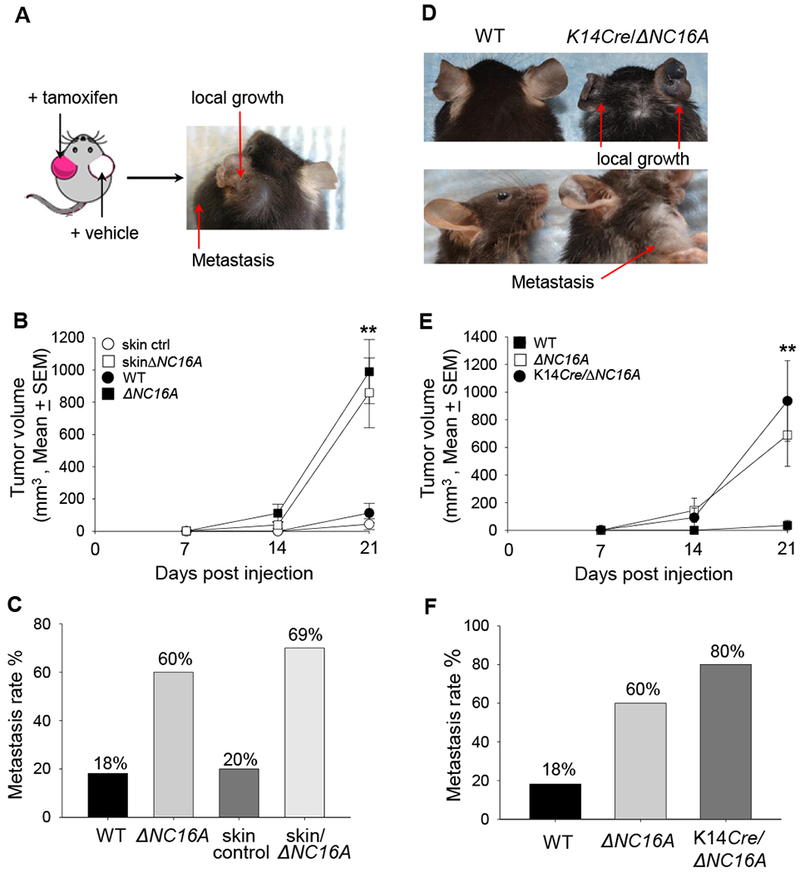
A, TamCre-NC16A mice were treated at the left ear with tamoxifen (red) and right ear with vehicle control (white). B16 melanoma cells (1× 106cells) were injected into both tamoxifen-and vehicle-treated ears 10 days post tamoxifen treatment. At 21 days post melanoma injection, only the tamoxifen-treated ear developed local tumor and lymphatic metastasis. B, Tumors volumes in tamoxifen-treated ears were significantly increased starting from day 14 post melanoma cell injection compared to tamoxifen-treated WT ears (skin control) (** p<0.01, Scheirer-Ray-Hare test; n=13 for skinΔNC16A, n=15 for skin control). C, A significantly larger proportion (69%) of tamoxifen-treated TamCre-NC16A mouse (skinΔNC16A) ears also developed lymphatic metastasis as compared to 20% of skin control at 21 days post injection. (p<0.05, Fisher’s Exact Test; n=13 for skinΔNC16A, n=15 for skin control). D-F, Ears of 8 weeks old basal keratinocyte-specific ΔNC16A (K14Cre/∆NC16A) mice were injected with B16 melanoma cells (1×106 cells) and monitored for 21 days. K14Cre/∆NC16A mice developed significantly larger tumors at day 21 (D), and the significantly increased tumor growth started at day 14 (** p<0.01, Scheirer-Ray-Hare test; n=8 for K14Cre/∆NC16A mice, n=22 for WT) (E). In addition, 80% of K14Cre/∆NC16A mice developed lymphatic metastasis as compared to 18% of WT mice at day 21 post melanoma injection (Fisher’s Exact Test, p<0.01 n=8 for K14Cre/∆NC16A mice, n=22 for WT) (F).
To test whether BP180 dysfunction in basal keratinocytes is sufficient to promote melanoma progression, K14Cre/ΔNC16A mice were injected at the ear with B16 melanoma cells. Both tumor volumes (Figure 3D and 3E) and metastasis (Figure 3F) in K14Cre/ΔNC16A mice were comparable to whole body ΔNC16A mice, but significantly increased compared to WT mice starting day 14 after injection. Therefore, we concluded that BP180 dysfunction promotes tumor progression through the modulating of the skin tumor microenvironment by basal keratinocytes.
ΔNC16A mice exhibit increased MDSC infiltration in the skin.
To investigate how BP180 dysfunction in skin can leads to increased melanoma progression, we deployed cytokine array to explore the microenvironment of the ∆NC16A mice skin. Skin cytokine profiles showed that there is a significant increased level of various cytokines and chemokines in the skin∆NC16A mice compared to control mice (Figure 4A), which includes CXCL1 (KC), CXCL2 and CCL2 (monocyte chemoattractant protein 1(MCP1)). CXCL1, CXCL2 and CCL2 are chemokines known to attract myeloid derived suppressor cells (MDSCs) 36,37. ELISA assay of skin lysates confirmed significantly increased CXCL1, CXCL2 and CCL2 in the skin∆NC16A mice compared to control mice (Figure 4B–D). K14Cre/∆NC16A and ∆NC16A skin also exhibited a significant increase in these three chemokines (Supplemental Figure 4). Therefore, NC16A deletion in basal keratinocytes are sufficient to cause increased levels of CXCL1, CXCL2 and CCL2 in vivo. To investigate whether keratinocytes are the source of the increased chemokines, we isolated and cultured keratinocytes from ΔNC16A and NC16A sufficient WT mice. We found there is a significant increase in the amount of CXCL1 in the condition media of ΔNC16A keratinocytes as compared to WT control (Figure 4E). Taken together, NC16A deletion in basal keratinocytes significantly upregulates expression of CXCL1, CXCL2 and CCL2 in skin (in vivo) and expression/release of CXCL1 in vitro.
Figure 4. Dysfunction of BP180 significantly increase the level of MDSC attracting chemokines in the skin.
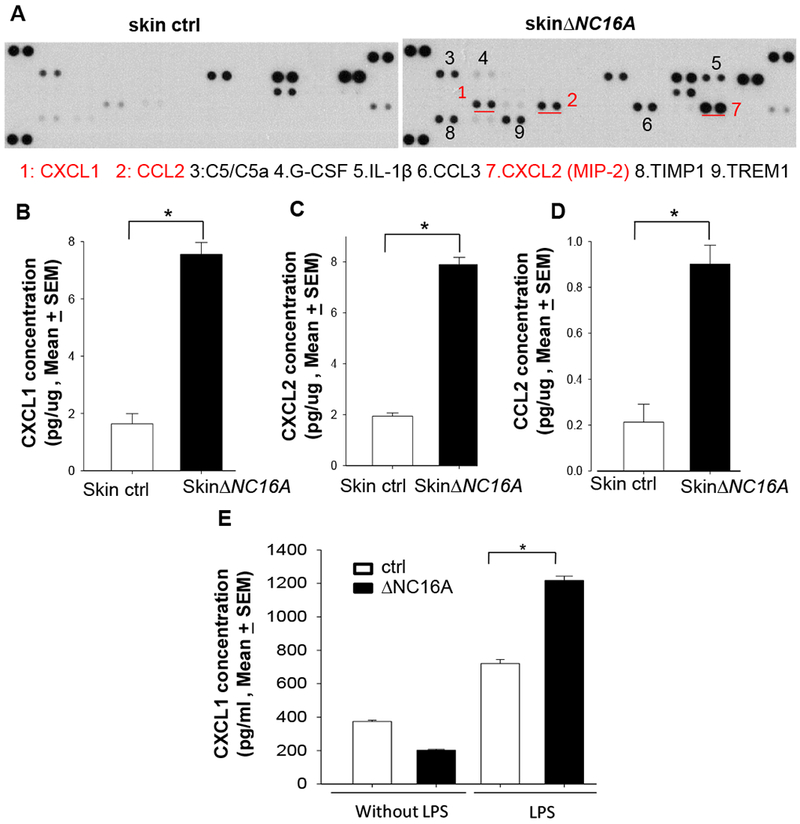
A. Inflammatory cytokine array showed that skin∆NC16A mice exhibited a significant increase in the level of various cytokines and chemokines, including MDSC attracting CXCL1 (KC), CXCL2 and CCL2 (MCP1). B-D ELISA assay confirmed increased levels of CXCL1 (B), CXCL2 (C) and CCL2 (D) in ∆NC16A skin compared to controls. E. LPS treatment (5ug/ml) for 4 hr significantly increases the level of CXCL1 in the condition media of ∆NC16A keratinocyte culture compared to the BP180 functional sufficient control keratinocytes. (*p<0.05, Mann–Whitney U test; n>=5 for each group).
MDSCs play an important role in the progression of the various solid tumors, including melanoma 38 . MDSCs are characterized as CD11b+Gr1+ myeloid cells in mice 36. They are a heterogeneous mixture of the immature myeloid cells, and are composed by two groups, Ly6G expressing polymorphonuclear like MDSC (PMN-MDSCs, previously also known as granulocytic-MDSCs) and monocytic like MDSCs (M-MDSCs) 39,40. PMN-MDSCs express MPO 36. MDSCs in the microenvironment promote tumor progression by suppressing immune surveillance, enhancing angiogenesis and promoting metastasis 41,42. Knowing increased skin infiltration of neutrophils (Supplemental Figure 2B and 2C) and increased levels of MDSC chemoattractants (Figure 4 and Supplemental Figure 4) in mice lacking NC16A, we used flow cytometry analysis to determine and quantify MDSCs in the skin of ΔNC16A, skinΔNC16A and K14Cre/ΔNC16A mice. All mice lacking NC16A showed a significantly increased influx of MDSCs (Figure 5A), with PMN-MDSCs being predominant (Figure 5B), and a smaller population of M-MDSCs (Figure 5C). Both PMN-MDSCs and M-MDSCs isolated from K14Cre/ΔNC16A mice exhibited the ability to suppress T-cell proliferation (Supplemental Figure 5). Q-RT-PCR found there is a significant increase in Arginase 1 and iNOS, the two major immunoregulatory proteins expressed by MDSCs 43, in the skin of ∆NC16A mice (Supplemental Figure 6). These results show that BP180 dysfunction leads to significantly increased PMN-MDSC infiltration.
Figure 5. Whole body-, skin- and basal keratinocyte-specific ΔNC16A mice have increased infiltration of MDSCs in the skin.
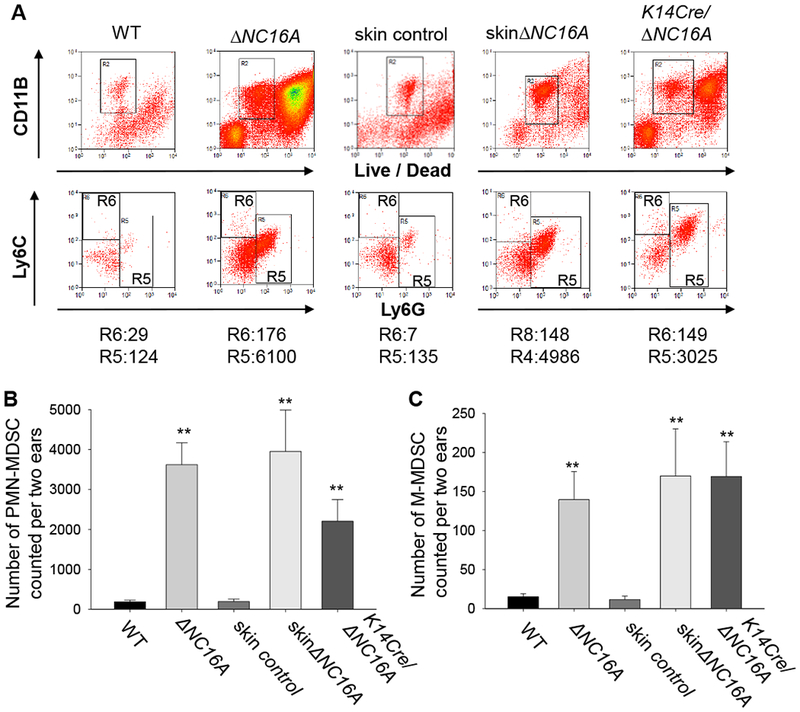
MDSCs in the skin of 8 weeks old WT, whole body ΔNC16A and basal keratinocyte-specific ΔNC16A (K14Cre/ΔNC16A) mice were identified and quantified by flow cytometry. Skin control and skinΔNC16A mouse was prepared through 14 days post tamoxifen treatment on the ear of WT and TamCre-NC16A mice. Polymorphonuclear-MDSCs (PMN-MDSCs) are CD11b+Ly6G+ Ly6Clow, while monocytic MDSCs (M-MDSCs) are CD11b+Ly6G−Ly6C+. A. A significantly increased number of PMN-MDSCs (B) and M-MDSCs (C) are present in the skin of ΔNC16A, skinΔNC16A and K14Cre/∆NC16A mice compared to WT mice. (*p<0.05 for K14Cre/ΔNC16A vs WT and **p<0.01 for the other two comparisons for both (B) and (C), Mann–Whitney U test, n=3 for K14Cre/∆NC16A, n=6 KO and skinΔNC16A groups, and n=8 for WT and skin control groups.)
MDSC influx plays a significant role in the melanoma progression in ΔNC16A mice
To determine whether infiltrating MDSCs play a key role in melanoma progression or innocent bystanders in ∆NC16A mice, skin∆NC16A mice were treated with anti-Gr1 antibody to deplete Gr1 positive MDSCs, (Gr1+ cells include both Ly6G+ and 6C+ populations). As expected, the skin∆NC16A mice treated with anti-Gr1 antibody showed significantly reduced Gr1 positive immune cells in the skin (Figure 6A). Importantly, anti-Gr1 antibody treatment significantly reduced melanoma tumor volume (Figure 6B) and the rate of developing metastasis (Figure 6C). These results demonstrate that MDSC infiltration is crucial for accelerated melanoma progression caused by BP180 dysfunction. In conclusion, BP180 dysfunction in basal keratinocytes leads to a tumor-favoring inflammatory microenvironment in local skin and MDSCs play a vital role in this microenvironment.
Figure 6. Anti-Gr1 antibody mediated myeloid cells depletion significantly reduce Gr1 positive immune cells in the skin of ΔNC16A mice.
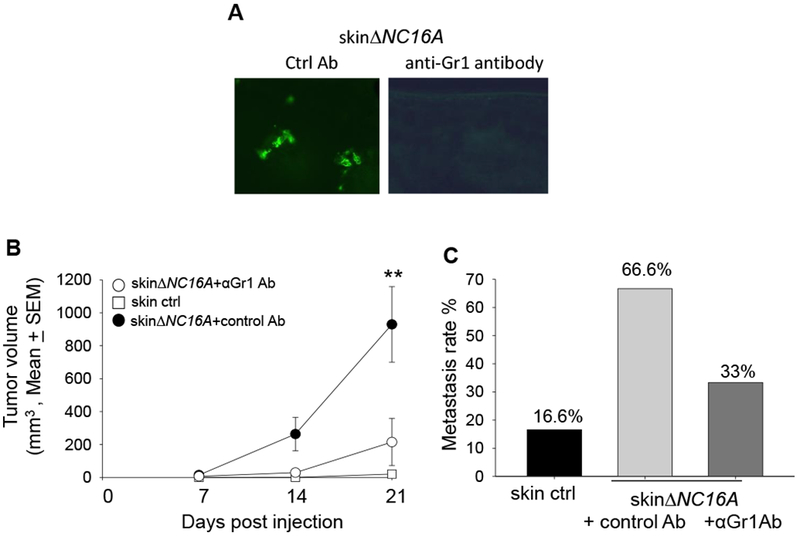
(A) Anti-Gr1 antibody or control antibody was injected to TamCre-NC16A mice two weeks after Tamoxifen treatment. IF result showed that myeloid cells are significantly reduced in the skin Gr1 antibody injected mice 24 hrs after antibody injection. MDSC depletion (i.p. injection anti-Gr1 antibody) significantly reduce the tumor volume starting from day 14 (B) (**p <0.01, Scheirer-Ray-Hare test; n=6 for skin∆NC16A+αGr1 Ab, n=9 for skin∆NC16A+control Ab). In addition, MDSC depletion reduced the rate of developing neck metastasis (C) in skinΔNC16A mice (p= 0.31, Fisher’s exact test; n=6 for skin∆NC16A+αGr1 Ab, n=9 for skin∆NC16A+control Ab).
DISCUSSION
In this study, we found that mice with dysfunctional BP180 exhibit MDSC influx in skin, and accelerated melanoma progression when challenged with B16 mouse melanoma cells. Since the 1980s, multiple reports have shown that altered BP180 expression is associated with various types of skin cancers, including squamous cell carcinoma (SCC) 15,44,17, basal cell carcinoma (BCC) 18, and melanoma 19. Unlike the previous reports that focus on BP180 expression in transformed cancer cells, we showed that BP180 in basal keratinocytes can play a significant role in melanoma progression in vivo. To our knowledge, it is the first report demonstrating the following concepts: 1) the hemidesmosomal protein BP180 plays a critical role in melanoma progression in vivo, hence JEB patients with BP180 mutations may have an increased chance to develop melanoma; and 2) skin basal keratinocytes play a role in the tumor microenvironment by modulating MDSC influx into the skin.
JEB is a rare autosomal recessive disorder caused by defects in genes encoding hemidesmosomal proteins and hemidesmosome-associated proteins including α6β4 integrin, BP230, BP180, laminin 332 and collagen VII 45,46. BP180 mutations lead to the partial or complete loss of BP180 function, causing JEB, which is commonly termed as non-Herlitz JEB 47,48. From a study based on 43 European non-Herlitz JEB patients, the patient group exhibited a wide spectrum of clinical variabilities, from mild to severe phenotypes of JEB symptoms 48. In general, the milder forms of JEB are associated with missense or splice-site mutations and the presence of truncated BP180 protein in the skin 48. The symptoms of non-Herlitz JEB caused by BP180 mutations include itch and the infiltration of proinflammatory immune cells into the skin 49, which share phenotypical similarity with our ∆NC16A mice and the reported NC14A deficient mice 24. In addition, ∆NC16A mice also exhibit minor dermal-epidermal separations starting at around eight weeks after birth, which further indicates that lacking the NC16A domain of BP180 significantly jeopardizes the normal cell/ECM adhesive function of BP180. These clinical studies support the clinical relevance of our ∆NC16A mouse model as the pathological consequence of BP180 dysfunction in JEB patients 49.
Due to the rarity of all EB diseases with a prevalence being less than eight per one million population, and melanoma only accounting for less than 5% of all skin cancers, only limited case reports showed an increased risk of melanoma associated with any types of EB diseases 50,51. But, JEB is linked to significantly increased susceptibility to squamous cell carcinoma (SCC) 52,51. A study based on clinical data collected between 1986~2002 in the US also found children with recessive dystrophic EB have increased susceptibility to melanoma 50. Nevertheless, our animal model findings do provide the reverse-genetic evidence, implicating that the melanoma in patients with BP180 deficiency, such as JEB, may progress more severely, compared to patients with functional BP180. Future clinical studies with more cases of JEB associated with melanoma may provide more definite evidence to support our conclusion.
Our study not only highlights the role of BP180, but also points out an important role for basal keratinocytes in modulating the tumor microenvironment. It had been reported that epidermal keratinocytes can modulate the metastasis of nearby melanoma cells 20; however, whether keratinocytes play a role in the tumor microenvironment remains unclear. To our knowledge, this is the first report showing that basal keratinocytes directly participate in melanoma progression through modulating MDSCs in skin. Whether other hemidesmosomal proteins in basal keratinocytes also affect the tumor microenvironment remains to be determined. It needs to be pointed out that altered expression or deficiency of BP180 may play different roles in the initiation vs. growth phase of melanoma and other skin cancers, since B16 melanoma cells injected into ∆NC16A mice express BP180. Future studies would directly address this scenario.
It is generally accepted that MDSC migration and accumulation in tumor is mediated by tumor-derived factors 38,53. It is unclear whether basal keratinocytes or hemidesmosomal proteins, such as BP180, affect MDSC migration and accumulation in the tumor microenvironment. To our knowledge, our findings are the first report showing that basal keratinocytes and BP180 play a role in tumor progression through MDSCs. The BP180 dysfunction-caused CXCL1 release from keratinocyte could be one of the mechanisms for the increased MDSC influx (Figure 4E). However, we also found at least other two MDSCs chemoattractants CXCL2 and CCL2 are also increased in the skin of ∆NC16A mice (Figure 4B_D and Supplemental Figure 4). Therefore, it is highly unlikely that BP180 dysfunction promote melanoma progression only through CXCL1 Future studies will determine the exact pathway(s) on how BP180 dysfunction in keratinocytes triggers MDSC influx into skin which promotes melanoma progression.
In summary, we uncovered that the cell-cell matrix adhesion molecule BP180 also functions as a key regulator of MDSC influx and MDSC-driven melanoma progression. We also found that BP180 dysfunction leads to a spontaneous skin inflammation, and increased influx of MDSCs is the mechanism underlying the increased melanoma progression (Figure 6). Our present findings also suggest that any skin inflammation accompanied with a MDSC influx should be able to promote tumor growth. Our present studies did not determine potential functional interplay between general inflammation and MDSC infiltration. Future studies are need to address this scenario. Basal keratinocytes, when losing the NC16A domain, are the cell source to either directly or indirectly increase the amount of MDSC attracting chemokines and MDSCs. Future studies are need to investigate whether NC16 deficient basal keratinocytes directly promote MDSC influx or indirectly, through working with other local cells, upregulate MDSC influx and MDSC-driven melanoma progression. With this new melanoma model, further elucidating the role of BP180 in MDSC influx at the molecular level will help to better understand the mechanisms of melanoma progression and to develop new therapeutic strategies for the disease.
MATERIALS AND METHODS
Generation of mice
Different strains of humanized NC16 mice and ΔNC16A mice were generated 21,10. To generate skin-specific ΔNC16A mice, NC16A mice were crossed with UBC-Cre-ERT2 mice (Jackson Lab #008085). The TamCre-NC16A mice (ERCre+NC16Afl/fl mice) when treated topically with tamoxifen (Sigma, 25 μl of 10 mg/ml in 62% EtOH/sunflower oil mixture), become skin-specific ΔNC16A (skinΔNC16A) mice. To generate basal keratinocyte-specific ΔNC16A mice (K14Cre/ΔNC16 or K14Cre+NC16Afl/fl), NC16A mice were crossed with Krt-14 promoter driven Cre mice (Jackson Lab #004782). All of the mice used in experiments are between 8~12 weeks after birth. All the mice were bred, housed and experiments done at the University of North Carolina at Chapel Hill Animal Facility in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill.
Murine melanoma models with flank and ear injection of B16 melanoma cells
The B16-F10 murine melanoma cell line was acquired from ATCC (CRL6475) and cultured in DMEM media (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. For the flank injection model, 8 week old mice were injected subcutaneously (s.c) into the side flank with 1×105 cells in 100 μl of HBSS buffer. For the ear injection model, B16 melanoma cells (1×106) were injected intradermally (i.d) into the ears of 8 week old mice in 20 μl of HBSS buffer. Tumor volume was measured with a digital caliper and calculated using the following equation: ½ x (length) x (width) (width). Mice were terminated 21 days after the initial melanoma cell injection. Lymphatic metastasis was monitored by examining tumor growth on the draining lymph nodes of the ears on the neck 35. No randomization of mice is used in this study. For skin control and ΔNC16A, we ear tagged the mice with or without cre for blind analysis and treated with the same dose of tamoxifen and the same amount of tumor cells for injection. We checked the ear tag at the end of experiments after assessing the outcome. Therefore, we were blinded to the cre background of the mice during the experiments.
Myeloid cell depletion was accomplished by the intraperitoneal injection (i.p) of 200mg of RB6–8C5 rat anti mouse Gr1 antibody (Biolegend #108402) as previously described 54. Normal rat IgG at the same dose (Biolegend #400602) was used as control. The injection schedule was day-1, day 0 and then biweekly.
Keratinocyte Culture.
Primary keratinocyte isolation and the generation of ΔNC16A keratinocyte culture were described before 55,21. Briefly, keratinocytes were isolated from the skin of TamCre-NC16A neonatal mice 48 hours after birth and maintained in E medium with 15% FBS and 50 μM CaCl2 (low Ca2+ medium). E medium is in house produced and provided by Dr. Scott E Williams from UNC-Chapel Hill. Both WT and ΔNC16A keratinocytes were stimulated with LPS (5ug/ml in media). Supernatants were collected 4hrs after LPS stimulation and used for ELISA analysis.
Cytokine array analysis and ELISA
Skin lysates from three age and sex matched skin controls, (WT mice treated with tamoxifen), and skinΔNC16A (TamCre-NC16A mice treated with tamoxifen) mice were extracted with PBS and analyzed with a Proteome Profiler Mouse Cytokine Array Kit, Panel A as recommended by the Manufacturer (R&D Systems, ARY006). In addition, skin lysate was also used for ELISA from R&D Systems with the procedure recommended by the manufacturer (R&D Systems, CXCL1 DY453, CCL2 DY479 and CXCL2 DY279).
MPO activity assay
Infiltrating granulocytic myeloid cells in the skin were quantified by measuring the enzymatic activity of the cell marker myeloperoxidase (MPO) 56. Briefly, skin samples were extracted by homogenization in an extraction buffer containing 0.1 M Tris-Cl (pH 7.6), 0.15 M NaCl, and 0.5% hexadecyl trimethylammonium bromide. MPO activity in the supernatants was measured by the change in optical density at 460 nm and was then normalized with the protein concentrations of skin extract supernatants (expressed as mU/mg protein + SE).
Analysis of the infiltrating inflammatory cells in the skin by flow cytometry and MDSC/T cell suppression assay.
Isolation, identification, and quantification of skin inflammatory cells were performed as described 57. Briefly, ear skin was obtained and rinsed in 70% ethanol, and then was cut into 1×4-mm sheets and submerged in a 6-well plate containing RPMI 1640 with 25 mM HEPES, 10% heat-inactivated FBS and penicillin/streptomycin. The plate was incubated at 37°C for 4 hours in a 5% CO2 incubator. The cells spontaneously emigrating out of the skin sections were pooled for flow cytometry analysis. Cells were identified by characteristic size (forward scatter) and granulosity (side scatter) combined with Sytox Orange Dye staining to distinguish live and dead cells (Thermo Fisher S34861). Antibody recognizing CD11b, Ly6G and Ly6C were also used for flow cytometry analysis (Biolegend 101230, 124614, 128008). MDSC isolation and suppression assays were performed as described before58. All of the flow cytometry analysis were performed using Beckman Coulter CyAn ADP in UNC-CH Flow Cytometry Core Facility. Data were analyzed with Summit 4.4 software.
Statistical analysis
The tumor volume data are expressed as mean + SEM. Fisher’s exact test is used for metastasis rate comparisons, and the Cox regression model is used for survival analysis in Figure 2C. The rest of the data analysis is performed using the Mann–Whitney U test and the Scheirer-Ray-Hare test, which are rank-based tests used in the place of the Student’s t-test and two-way ANOVA, respectively, so that the normality and homoscedasticity assumptions are not required for the tests. All the statistical analyses are performed using the R statistical software.
Details of Immunoblotting and immunofluorescence, RNA isolation and q-RT-PCR. MDSC/T cell suppression assay and Statistical analysis are described in the Supplemental Material and Method.
Supplementary Material
ACKNOWLEDGEMENT
We thank Dr. Dennis Loop for providing the Krt-14 promoter driven Cre mice. We thank Dr. Donna Culton and Susan McCray for assistance in Flow Cytometry. We thank Dr. Yisong Wan and Dr. Jenny PY Ting for their assistance. We thank the UNC Flow Cytometry Core Facility, which is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center, for their assistance in flow cytometry analyses.
Grant Support
This work was supported by the NIH (R01 AI40768 and R01 AR072694 to Z. Liu), the UNC Cancer Center Research Award (to Z. Liu) and UNC Graduate School Dissertation Completion Fellowship (to BJ. Hwang) and P01CA206980 (to N. Thomas).
Abbreviation
- BP
Bullous Pemphigoid
- JEB
junctional epidermolysis bullosa
- WT
wild type
- SCC
squamous cell carcinoma
- BCC
basal cell carcinoma
- IB
immunoblotting
- IF
immunofluorescence
- MDSC
Myeloid-derived suppressor cells.
- PMN-MDSC
Polymorphonuclear-Myeloid-derived suppressor cells.
- M-MDSC
Monocytic-Myeloid-derived suppressor cells
- MPO
Myeloperoxidase
Footnotes
The authors declare no potential conflicts of interest.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
Supplementary information is availabel at Oncogene’s website.
References
- 1.Hammers CM, Stanley JR. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu Rev Pathol Mech Dis 2016; 11: 175–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koster J, Geerts D, Favre B, Borradori L, Sonnenberg A. Analysis of the interactions between BP180, BP230, plectin and the integrin alpha6beta4 important for hemidesmosome assembly. J Cell Sci 2003; 116: 387–399. [DOI] [PubMed] [Google Scholar]
- 3.Hopkinson SB, Jones JC. The N terminus of the transmembrane protein BP180 interacts with the N-terminal domain of BP230, thereby mediating keratin cytoskeleton anchorage to the cell surface at the site of the hemidesmosome. Mol Biol Cell 2000; 11: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Margadant C, Frijns E, Wilhelmsen K, Sonnenberg A. Regulation of hemidesmosome disassembly by growth factor receptors. Curr Opin Cell Biol 2008; 20: 589–596. [DOI] [PubMed] [Google Scholar]
- 5.Leighty L, Li N, Diaz LA, Liu Z. Experimental models for the autoimmune and inflammatory blistering disease, Bullous pemphigoid. Arch Dermatol Res 2007; 299: 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGrath JA, Galalica B, Christiano AM, Li K, Owaribe K, McMillian JR et al. Mutation in the 180-kD bulleus pemphigoid antigen (BPAG2), a hemidesmosomal transmembreane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat Genet 1995; 11: 83–86. [DOI] [PubMed] [Google Scholar]
- 7.Van den Bergh F, Eliason SL, Burmeister BT, Giudice GJ. Collagen XVII (BP180) modulates keratinocyte expression of the proinflammatory chemokine, IL-8. Exp Dermatol 2012; 21: 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giudice GJ, Emery DJ, Diaz LA. Cloning and primary structural analysis of the bullous pemphigoid autoantigen BP180. J Invest Dermatol 1992; 99: 243–250. [DOI] [PubMed] [Google Scholar]
- 9.Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR et al. Humanization of autoantigen. Nat Med 2007; 13: 378–383. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Sui W, Zhao M, Li Z, Li N, Thresher R et al. Subepidermal blistering induced by human autoantibodies to BP180 requires innate immune players in a humanized bullous pemphigoid mouse model. J Autoimmun 2008; 31: 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonkman MF, Jong MCJM De, Heeres K, Pas HH, Meer JB Van Der, Owaribe K et al. 180-kD Bullous Pemphigoid Antigen ( BP180 ) Is Deficient in Generalized Atrophic Benign Epidermolysis Bullosa. 1982; : 1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurskainen T, Moilanen J, Sormunen R, Franzke C-W, Soininen R, Loeffek S et al. Transmembrane collagen XVII is a novel component of the glomerular filtration barrier. Cell Tissue Res 2012; 348: 579–88. [DOI] [PubMed] [Google Scholar]
- 13.Tanimura S, Tadokoro Y, Inomata K, Binh NT, Nishie W, Yamazaki S et al. Hair follicle stem cells provide a functional niche for melanocyte stem cells. Cell Stem Cell. 2011; 8: 177–187. [DOI] [PubMed] [Google Scholar]
- 14.Matsumura H, Mohri Y, Binh NT, Morinaga H, Fukuda M, Ito M et al. Hair follicle aging is driven by transepidermal elimination of stem cells via COL17A1 proteolysis. Science 2016; 351: 1–14. [DOI] [PubMed] [Google Scholar]
- 15.Parikka M, Kainulainen T, Tasanen K, Väänänen A, Bruckner-Tuderman L, Salo T. Alterations of collagen XVII expression during transformation of oral epithelium to dysplasia and carcinoma. J Histochem Cytochem 2003; 51: 921–9. [DOI] [PubMed] [Google Scholar]
- 16.Parikka M, Nissinen L, Kainulainen T, Bruckner-Tuderman L, Salo T, Heino J et al. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration-A novel role for αIIb integrin and tirofiban. Exp Cell Res 2006; 312: 1431–1438. [DOI] [PubMed] [Google Scholar]
- 17.Stelkovics E, Korom I, Marczinovits I, Molnar J, Rasky K, Raso E et al. Collagen XVII/BP180 protein expression in squamous cell carcinoma of the skin detected with novel monoclonal antibodies in archived tissues using tissue microarrays and digital microscopy. Appl Immunohistochem Mol Morphol 2008; 16: 433–41. [DOI] [PubMed] [Google Scholar]
- 18.Parikka M Altered expression of collagen XVII in ameloblastomas and basal cell carcinomas. J Oral Pathol 2001; : 589–595. [DOI] [PubMed] [Google Scholar]
- 19.Krenacs T, Kiszner G, Stelkovics E, Balla P, Teleki I, Nemeth I et al. Collagen XVII is expressed in malignant but not in benign melanocytic tumors and it can mediate antibody induced melanoma apoptosis. Histochem Cell Biol 2012; 138: 653–667. [DOI] [PubMed] [Google Scholar]
- 20.Golan T, Messer AR, Amitai-Lange A, Melamed Z, Ohana R, Bell RE et al. Interactions of Melanoma Cells with Distal Keratinocytes Trigger Metastasis via Notch Signaling Inhibition of MITF. Mol Cell 2015; 59: 664–676. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Hwang B-J, Liu Z, Li N, Lough K, Williams SE et al. BP180 dysfunction triggers spontaneous skin inflammation in mice. Proc Natl Acad Sci 2018; 115: 6434–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan D, Weinberg RA, Pan KH, Shay JW, Cohen SN, Taylor MB et al. Hallmarks of Cancer: The Next Generation. Cell. 2011; 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 23.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009; 30: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 24.Hurskainen T, Kokkonen N, Sormunen R, Jackow J, Löffek S, Soininen R et al. Deletion of the Major Bullous Pemphigoid Epitope Region of Collagen XVII Induces Blistering, Autoimmunization, and Itching in Mice. J Invest Dermatol 2014; : 1–8. [DOI] [PubMed] [Google Scholar]
- 25.Aho S, Uitto J. 180-kD Bullous Pemphigoid Antigen / Type XVII Collagen : Tissue-Specific Expression and Molecular Interactions With Keratin 18. 1999; 367: 356–367. [DOI] [PubMed] [Google Scholar]
- 26.Di Meglio P, Perera GK, Nestle FO. The Multitasking Organ: Recent Insights into Skin Immune Function. Immunity 2011; 35: 857–869. [DOI] [PubMed] [Google Scholar]
- 27.Bangert C, Brunner PM, Stingl G. Immune functions of the skin. Clin Dermatol 2011; 29: 360–376. [DOI] [PubMed] [Google Scholar]
- 28.Van Kilsdonk JWJ, Bergers M, Van Kempen LCLT, Schalkwijk J, Swart GWM. Keratinocytes drive melanoma invasion in a reconstructed skin model. Melanoma Res 2010; 20: 372–380. [DOI] [PubMed] [Google Scholar]
- 29.Tang L, Wang K. Chronic Inflammation in Skin Malignancies. J Mol Signal 2016; 11: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ortiz ML, Kumar V, Martner A, Mony S, Donthireddy L, Condamine T et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17–producing CD4 + T cells. J Exp Med 2015; 212: 351–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lund AW, Medler TR, Leachman SA, Coussens LM. Lymphatic vessels, inflammation, and immunity in skin cancer. Cancer Discov 2016; 6: 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bleehen S Normal pigmentation, radiation and response to sun exposure In: Champion R, Burns D, Breathnach S (eds). Textbook of Dermatology. Blackwell-Science: London, 1998, pp 1765–7. [Google Scholar]
- 33.Silvers WK. The coat colors of mice: A model for mammalian gene action and interaction In: Altman PL, K DD, editors. Inbred and Genetically Defined Strains of Laboratory Animals, Part1 Mouse and Rat. 1979; : 1979. [Google Scholar]
- 34.Rozenberg GI, Monahan KB, Torrice C, Bear JE, Sharpless NE. Metastasis in an orthotopic murine model of melanoma is independent of RAS/RAF mutation. Melanoma Res 2010; 20: 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bobek V, Kolostova K, Pinterova D, Kacprzak G, Adamiak J, Kolodziej J et al. A clinically relevant, syngeneic model of spontaneous, highly metastatic B16 mouse melanoma. Anticancer Res 2010; 30: 4799–4804. [PubMed] [Google Scholar]
- 36.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol 2016; 37: 208–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood 2008; 111: 5457–5466. [DOI] [PubMed] [Google Scholar]
- 38.Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer 2013; 13: 739–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J Immunol 2008; 181: 5791–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016; 7: 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer 2008; 8: 618–31. [DOI] [PubMed] [Google Scholar]
- 42.Condamine T, Ramachandran I, Youn J-I, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med 2015; 66: 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrand-Rosenberg S, Sinha P. MDSCs: Linking Inflammation and Cancer. J Immunol 2009; 182: 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parikka M, Nissinen L, Kainulainen T, Bruckner-Tuderman L, Salo T, Heino J et al. Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration--a novel role for alphaIIb integrin and tirofiban. Exp Cell Res 2006; 312: 1431–8. [DOI] [PubMed] [Google Scholar]
- 45.Fine J-D. Inherited Epidermolysis Bullosa. Orphanet J Rare Dis 2010; 5: 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bubier JA, Sproule TJ, Alley LM, Webb CM, Fine J-D, Roopenian DC et al. A mouse model of generalized non-Herlitz junctional epidermolysis bullosa. J Invest Dermatol 2010; 130: 1819–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fine JD. Inherited epidermolysis bullosa: Past, present, and future. Ann. N. Y. Acad. Sci. 2010; 1194: 213–222. [DOI] [PubMed] [Google Scholar]
- 48.Kiritsi D, Kern JS, Schumann H, Kohlhase J, Has C, Bruckner-Tuderman L. Molecular mechanisms of phenotypic variability in junctional epidermolysis bullosa. J Med Genet 2011; 48: 450–457. [DOI] [PubMed] [Google Scholar]
- 49.Mabuchi E, Umegaki N, Murota H, Nakamura T, Tamai K, Katayama I. Oral steroid improves bullous pemphigoid-like clinical manifestations in non-Herlitz junctional epidermolysis bullosa with COL17A1 mutation. Br J Dermatol 2007; 157: 596–598. [DOI] [PubMed] [Google Scholar]
- 50.Fine JD, Johnson LB, Weiner M, Li KP, Suchindran C. Epidermolysis bullosa and the risk of life-threatening cancers: The National EB Registry experience, 1986–2006. J Am Acad Dermatol 2009; 60: 203–211. [DOI] [PubMed] [Google Scholar]
- 51.Mallipeddi R Epidermolysis bullosa and cancer. Clin Exp Dermatol 2002; 27: 616–623. [DOI] [PubMed] [Google Scholar]
- 52.Fine J-D. Squamous cell carcinoma and junctional epidermolysis bullosa. J Am Acad Dermatol 2012; 66: 856–857. [DOI] [PubMed] [Google Scholar]
- 53.Youn J-I, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol 2010; 40: 2969–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Deventer HW, Burgents JE, Wu QP, Woodford RMT, Brickey WJ, Allen IC et al. The inflammasome component Nlrp3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res 2010; 70: 10161–10169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Williams SE, Beronja S, Pasolli HA, Fuchs E. Asymmetric cell divisions promote Notch-dependent epidermal differentiation. Nature 2011; 470: 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen R, Ning G, Zhao M, Fleming MG, Diaz L a, Werb Z et al. Mast cells play a key role in neutrophil recruitment in experimental bullous pemphigoid. J Clin Invest 2001; 108: 1151–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen R, Fairley J a, Zhao M-L, Giudice GJ, Zillikens D, Diaz L a et al. Macrophages, but not T and B lymphocytes, are critical for subepidermal blister formation in experimental bullous pemphigoid: macrophage-mediated neutrophil infiltration depends on mast cell activation. J Immunol 2002; 169: 3987–92. [DOI] [PubMed] [Google Scholar]
- 58.Bruce DW, Stefanski HE, Vincent BG, Dant TA, Reisdorf S, Bommiasamy H et al. Type 2 innate lymphoid cells treat and prevent acute gastrointestinal graft-versus-host disease. J Clin Invest 2017; 127: 1813–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.