

Abstract
Nonadiabatic molecular dynamics (NAMD) simulations of molecular systems require the efficient evaluation of excited-state properties, such as energies, gradients, and nonadiabatic coupling vectors. Here, we investigate the use of graphics processing units (GPUs) in addition to central processing units (CPUs) to efficiently calculate these properties at the time-dependent density functional theory (TDDFT) level of theory. Our implementation in the FermiONs++ program package uses the J-engine and a preselective screening procedure for the calculation of Coulomb and exchange kernels, respectively. We observe good speed-ups for small and large molecular systems (comparable to those observed in ground-state calculations) and reduced (down to sublinear) scaling behavior with respect to the system size (depending on the spatial locality of the investigated excitation). As a first illustrative application, we present efficient NAMD simulations of a series of newly designed light-driven rotary molecular motors and compare their S1 lifetimes. Although all four rotors show different S1 excitation energies, their ability to rotate upon excitation is conserved, making the series an interesting starting point for rotary molecular motors with tunable excitation energies.
1. Introduction
Nonadiabatic processes such as electronic excitations,1 radiationless transitions,2 and electron transfer3 are of key importance in chemistry and biology. One of the most prominent examples in these fields is the rhodopsin protein, whose chromophore undergoes a photoisomerization when exposed to light.4 This energy conversion of light to mechanical motion has inspired chemists to design synthetic light-driven rotary molecular motors,5,6 for which Bernard Feringa was awarded the Nobel Prize in chemistry in 2016 together with Jean Pierre Sauvage and Sir James Fraser Stoddart.7 The description of theses nonadiabatic processes is an ongoing challenge in modern quantum chemistry (e.g., ref (8)). This is mainly due to the fact that (1) excited states have to be taken into account and (2) the dynamics of the nuclei need to be considered.
To tackle the first challenge, several quantum-chemical methods have been developed. Examples include the complete active space self-consistent field (CASSCF) method,9 the algebraic-diagrammatic construction (ADC(2)),10 several coupled cluster methods (e.g., CC2),11 and time-dependent density functional theory (TDDFT).12,13 The latter serves (despite its well-known limitations using today’s functionals13,14) as a good compromise between effort and accuracy.15,16 As a consequence of this, not only excited-state energies but also excited-state gradients17 and nonadiabatic coupling vectors (NACVs)18−29 have been implemented to provide access to molecular properties at the TDDFT level of theory. Having excited-state energies and properties at hand, trajectory surface hopping (TSH)30,31 is a straightforward way to conduct nonadiabatic molecular dynamics (NAMD) simulations including several electronic states.
Despite many advances in the field of NAMD32−34 and their broad field of application,35−44 it remains difficult or even impossible to investigate large molecular systems. The reason for this is that TSH requires, because of its stochastic nature, a large set of independent trajectories including many time steps, which involve expensive (even at the TDDFT level of theory) excited-state energy and property calculations. One way to accelerate NAMD simulations is the use of exciton models.45 Another approach is the use of graphics processing units (GPUs) in addition to central processing units (CPUs) for the calculations. It was shown that this leads to significant speed-ups for ground-state energy and forces evaluations46−53 and was also successfully applied to excited-state energies and properties45,54−56 as well as ab initio multiple spawning57,58 and NAMD59,60 simulations.
In this work, we present efficient NAMD simulations with TDDFT energies, gradients, and NACVs calculated on hybrid CPU/GPU architectures using the FermiONs++ program package.50−52 We start with a brief summary of the theory behind TSH as well as TDDFT energies, gradients, and NACVs in Section 2 and discuss their implementation on GPUs, featuring the hybrid CPU/GPU engine52 for the two-electron integrals as well as the preselective screening procedure for the evaluation of exchange kernels.50,51 Computational details are given in Section 3. In Section 4, we discuss the accuracy and the performance of our GPU-based excited-state routines, investigating the scaling of our integral evaluations and contractions. We show timings for selected molecules containing more than 500 atoms. As a prototypical application, we investigate the photoinduced rotation of four newly designed rotary molecular motors via NAMD in Section 5, followed by a conclusion and an outlook.
2. Theory
All equations in this section use the standard notation for orbitals: i, j, k... denote occupied, a, b, c... denote virtual, and p, q, r... denote arbitrary molecular orbitals, while μ, ν, λ... denote basis functions. I, J, ... denote different electronic states. vxc and fxc are the first- and second-order exchange-correlation functional derivatives, respectively. cx is the amount of exact exchange. F and S are the Kohn–Sham and the overlap matrices, respectively. P is the ground-state density, and h is the one-electron core Hamiltonian matrix. The two-electron integrals are written using Mulliken notation (..|..). Superscript ξ denotes derivatives with respect to the nuclear coordinates, and * and † symbolize complex conjugation and adjungation, respectively. For the sake of simplicity, we use the same symbol for matrices in the atomic orbital (AO) and molecular orbital (MO) bases with different indices (e.g., Pμν vs Ppq).
2.1. Trajectory Surface Hopping
In nonadiabatic molecular dynamics (NAMD), the electronic time-dependent wave function of the system is assumed to be a linear combination of the time-independent electronic wave functions of the individual electronic states weighted by the state amplitudes (c⃗)
| 1 |
where x represents nuclear coordinates and {r} is the set of electronic coordinates. Having the state energies (ωI, see Section 2.2) and nonadiabatic coupling vectors (NACVs, τI → Jξ, see Section 2.4) at hand, it is possible to propagate the state amplitudes along with the nuclei during the molecular dynamics (MD) simulation using a unitary propagator:36
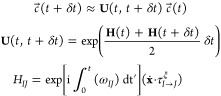 |
2 |
Here, ωJI is the energy difference between two states (ωJ – ωI). The applied δt for the propagation of c⃗ is normally 3 orders of magnitude smaller than the time step of the MD simulation (Δt).31 In the fewest-switches surface hopping algorithm,30,31c⃗ is used to calculate the probability gI → Jt → t + Δt of the system switching from its current state I to another state J at every time step:
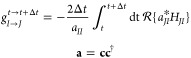 |
3 |
If a random number between zero and one exceeds g, then the MD simulation is continued on the potential energy surface of state J and the nuclear velocity is rescaled along τI → Jξ. Observables (e.g., lifetimes of states or relaxation pathways) can be drawn from ensembles of trajectories using a different set of random numbers. In the following sections, we will briefly summarize the calculation of the necessary ingredients of TSH (excited-state energies, gradients, and NACVs) at the time-dependent density functional theory (TDDFT) level of theory.
2.2. Excited-State Energies
Excitation energies can be calculated from linear response TDDFT by solving the TDDFT or the random phase approximation (RPA) equation12,61,62
| 4 |
A ± B are the orbital rotation Hessians
| 5 |
| 6 |
with ϵp being the energy of orbital p. XI and YI are the transition densities for excitation and de-excitation, respectively. Neglecting B is known as the Tamm–Dancoff approximation (TDA) of TDDFT,63 simplifying eq 4 to
| 7 |
Please note that eqs 4 and 7 are equivalent to the time-dependent Hartree–Fock (TDHF) and configuration interaction singles (CIS) equations, respectively, when cx is set equal to 1 and all exchange-correlation terms are neglected. TDA is computationally less demanding than TDDFT because fewer two-electron integrals have to be evaluated. It may also be more suitable for NAMD simulations because it is more stable (regarding its convergence) and thus typically delivers better results than TDDFT in the vicinity of conical intersections involving the ground state.39,64
TDDFT is nowadays widely used for the calculation of excitation energies, being a good compromise between accuracy and computational cost.15,16 Shortcomings are, however, the inability to calculate double excitations and the poor description of charge-transfer excitations.13,14 To tackle the latter, range-corrected functionals (e.g., ωB9765) have been introduced.
2.3. Excited-State Energy Gradients
To determine the energy gradient of an excited state I, one has to calculate the change in the excitation energy with respect to the nuclear coordinates (ωIξ). The final equation in the AO basis has been derived by Furche et al.17
| 8 |
The calculations of the relaxed difference density matrix (PI), the energy-weighted difference density matrix (WI), and the two-particle difference density matrix (ΓI) are shown in the Appendix. The first two require an iterative solution of a Z-vector equation.17RI is XI + YI or XI in the case of TDDFT or TDA, respectively.
2.4. Nonadiabatic Coupling Vectors
The NACV between two states (I and J) is defined as
| 9 |
It thus describes the change of the overlap of the wave functions of I and J with respect to the nuclear coordinates. The first formulation of the NACV involving the ground state (τ0 → Iξ) derived by Chernyak and Mukamel18 was corrected for finite basis set effects by Send et al.23 in 2010:
| 10 |
Equations for the density matrices (P0I, W0I, and Γ0I) are again given in the Appendix. They can be calculated directly from the TDDFT transition densities so that no Z-vector equation needs to be solved. Sμν[A]ξ denotes an antisymmetric overlap derivative
| 11 |
whereas γ0I is defined as
| 12 |
with LI being XI – YI or XI in the case of TDDFT or TDA, respectively.
The calculation of NACVs between two excited states has been tackled by many publications showing expressions based on linear and quadratic response at different levels of theory.25,26,29,66−71 The main conclusion is that the use of quadratic response theory leads to unphysical poles when the energy difference between excited states matches the excitation energy of another state.66−70 The use of linear response theory or the so-called pseudowave function approach25−27,29 is therefore recommended. The resulting equation in the AO basis is as follows:
| 13 |
Equations for the density matrices (PIJ, WIJ, and ΓIJ) can again be found in the Appendix. γIJ is defined as
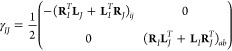 |
14 |
It was shown by Fatehi et al.25 that the antisymmetric overlap derivatives (eq 11) introduce translational variance into the NACV calculations. Neglecting these terms in eqs 10 and 13 is equivalent to adding electron-translational factors (ETFs). However, trajectory surface hopping (TSH) simulations using NACVs with and without ETFs lead to nearly identical results for larger molecular systems.72
2.5. Graphics Processing Units
Graphics processing units (GPUs) significantly accelerate quantum-chemical calculations because they allow for an efficient evaluation and contraction of two-electron integrals (eq 15) and their derivatives with respect to the nuclear coordinates (eq 16).46−52
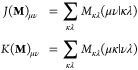 |
15 |
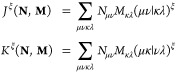 |
16 |
J and K denote the Coulomb and exchange parts, respectively, and M and N denote general density matrices. For the Coulomb part, large speed-ups can be observed when the J-engine73,74 is applied to the rearranged shell-pair data.47 To see comparable speed-ups for the exchange part, we apply an additional preselective screening (preLinK):50,51
| 17 |
| 18 |
It determines the significant shell pairs (those with an expected value above the given thresholds ϑpre and ϑpre∇) before their distribution to and calculation on the GPUs. The two-electron integral evaluation can be done even more efficiently when the workload is spread among both GPUs and CPUs.52
In ground-state calculations, M and N are solely the ground-state density (P), whereas for excited states M and N can additionally be PI, RI, or LI. However, the discussed procedure (shell pair rearrangement, J-engine, and preLinK) is still valid and can easily be adapted to excited-state routines when keeping in mind that M is not always equal to N and may be nonsymmetric.
3. Computational Details
3.1. General Remarks
The FermiONs++ program package50−52 was used for all calculations presented in this work. It was compiled using the Intel compiler (2019),75 and the Intel Math Kernel Library (MKL). Routines on AMD GPUs were compiled with the AMD APPSDK compiler. In addition, LibXC library v4.0.176,77 was used. In all calculations, we used the gm5 grid78 (with the modified Becke weighting scheme described in ref (78)) and tight thresholds for the SCF convergence (ϑSCF = 10–7 using the FP-commutator), the integrals (ϑINT = 10–10), and the Z-vector equation convergence (ϑZ = 10–5). Throughout all calculations, we neglected the symmetry of the molecules and solely calculated singlet excitations. Excited-state energies, gradients, and nonadiabatic coupling vectors (with electron transition factors) at the TDDFT level of theory were calculated using eqs 4, 8, 10, and 13, respectively.
3.2. Preparation and Calculation of Systems I, II, IIIn, IVn, V, VI, and VII
To illustrate the performance and/or accuracy of our implementation on GPUs, we use protonated formaldimine (I), the Schiff base of retinal (II), a series of linear polyethynes (IIIn) and dialkylethenes (IVn), a motorized nanocar (without “wheels”, V),79 and one (VI) and three (VII) pores of a covalent organic framework80 as example molecules. The structures of I, II, and V have been optimized at the PBE081−84/def2-SVP85,86 level of theory, while IIIn and IVn have not been optimized to maintain their linear structures. VI and VII have been prepared according to ref (80). All structures are available at https://www.cup.uni-muenchen.de/pc/ochsenfeld/download/.
TDDFT energies and properties of I, II, IIIn, IVn, V, VI, and VII were calculated at the PBE0/def2-SVP or PBE/def2-TZVP level of theory. We used tight thresholds for preLinK (ϑpre = 10–4), the preLinK gradient (ϑpre∇ = 10–10), and the TDDFT convergence (ϑTDDFT = 10–6). For I, II, IIIn, IVn, V, VI, and VII, four, six, five, three, six, seven, and seven states were taken into account, respectively.
When investigating the accuracy, we use a calculation performed on CPUs with tight thresholds (ϑTDDFT = 10–7) as a reference. To allow for a fair comparison, we employed the continuous fast multipole method (CFMM)87,88 and the LinK scheme89,90 for Coulomb and exchange kernels (and their derivatives) on CPUs when comparing CPU and GPU performance. Coulomb and exchange kernels on GPUs were calculated with the J-engine73,74 and the preLinK scheme,50,51 respectively. Timings of integral evaluations and entire routines were determined as an average over five independent calculations on two Intel Xeon CPU E5 2640 v4 @ 2.20 GHz (20 threads) CPUs and four AMD FirePro 3D W8100 GPUs. The scaling behavior is determined as the slope of the corresponding log–log plots (Supporting Information) using the timings of IIIn and IVn with n = 40, 50, 75, 100. The parallel efficiency is determined as the ratio between the measured and ideal speed-ups.
3.3. Preparation and Calculation of the Rotary Molecular Motors
The structures of the four rotary molecular machines (C, N, S, and O) have been optimized at the ωB9765/def2-SVP level of theory. Excited-state properties and timings were calculated at the TDA (ωB97/def2-SVP) level of theory using accurate thresholds (ϑpre = 10–3, ϑpre∇ = 10–10, and ϑTDDFT = 10–5). NAMD simulations were conducted at the same level of theory. The propagation of the nuclei was calculated using the Velocity Verlet algorithm.91,92 The extended Lagrangian method93 for the extrapolation of ground-state density was used to accelerate SCF convergence. Transition densities and relaxed difference densities of the previous step were used as guesses for the TDA and Z-vector equation, respectively.
Twenty initial geometries and velocities (available at https://www.cup.uni-muenchen.de/pc/ochsenfeld/download/) were drawn from a 5 ps ground-state NVT simulation (200 fs equilibration, 0.2 fs step size, velocity rescaling thermostat94) at the same level of theory. From each initial condition, five independent (different series of random numbers) NAMD simulations were conducted for 1 ps (0.2 fs step size) without equilibration, thermostat, or decoherence correction, starting at the first excited singlet state (S1). At every step of the simulation, the overall rotation and translation of the molecule was removed. Three excited states were taken into account, whereas only the coupling vector from the ground state to the first excited state was calculated and used for the propagation of the state amplitudes (eq 2) and the calculation of the hopping probability (eq 3).
4. Performance
4.1. Accuracy and Thresholds
We start with an analysis of the errors of excited-state energies and properties introduced by their calculation on GPUs and the use of the preLinK scheme51,52 for integrals and integral derivatives. Therefore, we compare calculations of the smallest Schiff base (protonated formaldimine, CH2NH2+, I) and the Schiff base of retinal (C20H30N+, II) with GPUs and preLinK to calculations on CPUs without prescreening or the continuous fast multipole method (CFMM). (For computational details, see Section 3.2.) The mean absolute errors for different thresholds are listed in Table 1.
Table 1. Mean Absolute Errors (MAE, in Atomic Units) of Excitation Energies (ωI), Gradients (ωIξ), and Nonadiabatic Coupling Vectors (τI→J) of Protonated Formaldimine (I) and the Schiff Base of Retinal (II) Calculated at the PBE0/def2-SVP Level of Theory on GPUs, Employing the preLinK Scheme and Using Different Thresholds for preLinK (ϑpre), the preLinK Gradient (ϑpre), and the TDDFT Convergence (ϑTDDFT)a.
| screening thresholds and convergence criteria | ||||||||
|---|---|---|---|---|---|---|---|---|
| ϑpre | 10–3 | 10–4 | 10–4 | 10–5 | 10–5 | 10–5 | 10–5 | 10–5 |
| ϑpre∇ | 10–11 | 10–11 | 10–11 | 10–11 | 10–11 | 10–9 | 10–10 | 10–11 |
| ϑTDDFT | 10–5 | 10–5 | 10–6 | 10–6 | 10–7 | 10–7 | 10–7 | 10–7 |
| I | ||||||||
|---|---|---|---|---|---|---|---|---|
| MAE(ωI) | 2.2 × 10–4 | 2.2 × 10–4 | 8.8 × 10–5 | 8.8 × 10–5 | 2.3 × 10–6 | 2.3 × 10–6 | 2.3 × 10–6 | 2.3 × 10–6 |
| MAE(ω2ξ) | 5.8 × 10–5 | 5.8 × 10–5 | 3.7 × 10–5 | 3.7 × 10–5 | 6.5 × 10–6 | 6.7 × 10–6 | 6.6 × 10–6 | 6.5 × 10–6 |
| MAE(τ0→1) | 5.7 × 10–4 | 5.7 × 10–4 | 1.2 × 10–4 | 1.2 × 10–4 | 9.2 × 10–5 | 8.8 × 10–5 | 9.1 × 10–5 | 9.2 × 10–5 |
| MAE(τ1→2) | 1.0 × 10–3 | 9.1 × 10–4 | 4.7 × 10–4 | 4.7 × 10–4 | 2.8 × 10–4 | 9.2 × 10–4 | 5.5 × 10–4 | 2.8 × 10–4 |
| II | ||||||||
|---|---|---|---|---|---|---|---|---|
| MAE(ωI) | 1.9 × 10–4 | 1.9 × 10–4 | 9.6 × 10–5 | 9.6 × 10–5 | 1.1 × 10–5 | 1.1 × 10–5 | 1.1 × 10–5 | 1.1 × 10–5 |
| MAE(ω2ξ) | 2.8 × 10–5 | 2.8 × 10–5 | 1.2 × 10–5 | 1.2 × 10–5 | 3.1 × 10–6 | 3.8 × 10–6 | 3.2 × 10–6 | 3.1 × 10–6 |
| MAE(τ0→1) | 1.7 × 10–3 | 1.7 × 10–3 | 7.5 × 10–4 | 7.5 × 10–4 | 2.0 × 10–4 | 2.0 × 10–4 | 2.0 × 10–4 | 2.0 × 10–4 |
| MAE(τ1→2) | 8.9 × 10–4 | 8.9 × 10–4 | 3.8 × 10–4 | 3.8 × 10–4 | 1.1 × 10–4 | 1.2 × 10–4 | 1.1 × 10–4 | 1.1 × 10–4 |
A calculation on CPUs with ϑTDDFT = 10–7 and without CFMM or preLinK is used as a reference. Throughout this work, we will use accurate (ϑpre = 10–3, ϑpre∇ = 10–10, and ϑTDDFT = 10–5) or tight thresholds (ϑpre = 10–4, ϑpre = 10–10, and ϑTDDFT = 10–6).
The choice of the preLinK threshold (ϑpre) for excited-state calculations should always depend on the applied convergence threshold for the TDDFT equation (ϑTDDFT). If the chosen threshold is too loose, then the iterative solution of the TDDFT equation does not converge (especially for large molecular systems), while a ϑpre that is too tight does not improve the result when ϑTDDFT is not adjusted accordingly (Table 1). The latter is also attributed to the small effect of preLinK on these relatively small systems.50,51 In our calculations with the FermiONs++ program package, the “ideal” ϑpre is 2 orders of magnitude larger than ϑTDDFT. A tightening of these two parameters (left side of Table 1) systematically leads to smaller errors. However, the errors in the coupling vectors of the large system do not fall below 10–4 a.u., marking the numerical limit of these second- to third-order properties. The use of the tight thresholds (ϑTDDFT = 10–6, ϑpre = 10–4) should thus, in general, be sufficient because all errors are below 10–3 a.u. A looser preLinK threshold (ϑpre = 10–3 a.u.), which is expected to give μH accuracy for ground-state properties, is still accurate for excited-state properties and could be used in extensive application calculations such as NAMD simulations, where observables are determined as ensemble averages.
The effect of the preLinK gradient threshold (ϑpre∇) on the error is not straightforward (right side of Table 1). However, we recommend it to be at least as tight as the integral threshold (ϑINT = 10–10). Because the difference between ϑpre = 10–11 and ϑpre∇ = 10–10 is negligibly small, we apply the latter throughout this work.
4.2. Scaling with the System Size
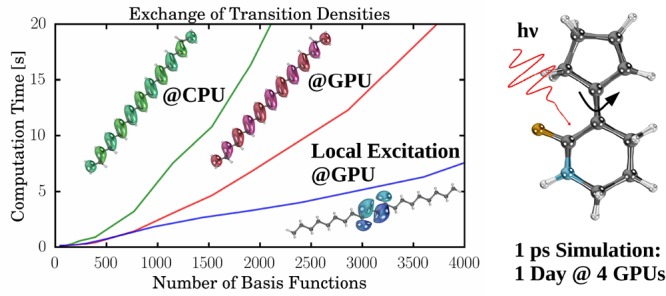
To compare timings on CPUs and GPUs and to investigate the effective scaling behavior of the excited-state integral routines on GPUs, we use integral timings of linear polyethynes (IIIn) and dialkylethenes (IVn) (for structures, see Figure 1). In Figure 2, we compare CPU and GPU timings of Coulomb (Figure 2a,c) and exchange (Figure 2b,d) calculations at the PBE0/def2-SVP level of theory. We show contractions of the ground-state density (J/K(P)), the transition density of the first excited state (J/K(X1 + Y1)) (Figure 2a,b), the integral derivatives involving the ground state and the relaxed difference density of the first excited state (Jξ/Kξ(P, P1)), and the transition density of the first excited state (Jξ/Kξ(X1 + Y1, X1 + Y1)) (Figure 2c,d). The same integrals are shown in Figure 3, where we compare the timings of IIIn and IVn on GPUs. The effective scaling behaviors of the integral evaluations are shown in the Supporting Information. Definitions of the integral contractions are given in eqs 15 and 16. Details of the calculations are given in Sections 3.1 and 3.2.
Figure 1.

Structures of the linear polyethynes (IIIn) and dialkylethenes (IVn).
Figure 2.

Timings of (a, c) Coulomb and (b, d) exchange integral evaluations (a, b; eq 15) and their derivatives with respect to the nuclear coordinates (c, d; eq 16) of polyethyne (IIIn) with n = 1–100 calculated at the PBE0/def2-SVP level of theory on CPUs (dashed lines) and GPUs (solid lines). For details on the calculations and the computational setup, see Sections 3.1 and 3.2.
Figure 3.

Timings of (a, c) Coulomb and (b, d) exchange integral evaluations (a, b; eq 15) and their derivatives with respect to the nuclear coordinates (c, d; eq 16) of polyethine (IIIn, solid line) and dialkylethene (IVn, dashed line) with n = 1–100 calculated at the PBE0/def2-SVP level of theory on GPUs. For details on the calculations and the computational setup, see Sections 3.1 and 3.2.
For the Coulomb and exchange kernels of the transition densities, we observe similar speed-ups (up to a factor of 5) as for the ground-state kernels (Figure 2a,b). The accelerations of the gradient kernels stem (nearly exclusively) from their Coulomb part, which is extremely efficient on GPUs (Figures 2c). We also see minor speed-ups for Kξ(X1 + Y1, X1 + Y1) (Figures 2d). The evaluation of Kξ(P, P1) is significantly slower because the GPU routine has to be called twice. This is due to the fact that the analytical exchange evaluation on GPUs48−51 does not exploit the full symmetry of the two-electron integral. Please note that this problem can be solved by applying a seminumerical exchange scheme, which is currently developed for GPUs in our group.95 For the largest molecule (III100), a speed-up of two for the entire determination of an excited-state gradient involving calculations of the ground-state and excited-state energy and gradient is observed. Here, it should be stressed that the linear algebra and the evaluation of the exchange-correlation kernels are performed entirely on CPUs.
When comparing the effective scaling behavior of the Coulomb integrals of IIIn (Supporting Information), one can see that the scaling of the GPU integrals is slightly larger than the scaling of the CPU integrals. This is due to the formal quadratic scaling of the J-engine employed on GPUs, in comparison to the (asymptotically linear scaling) CFMM method. This larger scaling behavior is, however, irrelevant as the prefactor of the routines is greatly reduced (Figure 2a,c). The scaling of the exchange integrals is slightly reduced by exploiting the preLinK method on GPUs. We observe ∼1.5 and ∼1.0 scalings for the integrals and the integral derivatives of these system sizes, respectively.
The excitation in IIIn is delocalized over the entire molecule, leading to similar scaling behavior for the ground-state and excited-state properties. To show the performance of a system with local excitation, we also investigate IVn, where only one double bond instead of a conjugated system is excited. This leads to massive speed-ups (Figure 3) of the excited-state exchange integrals and reduces their scaling significantly (Supporting Information). For the exchange integral derivatives, we even observe sublinear scaling behavior as a result of the preLinK screening. The effect on the Coulomb integrals is smaller, mainly because of the fact that these routines take only a few seconds even for the largest investigated molecules.
4.3. Example Calculations
To demonstrate the applicability of our excited-state properties routines in the FermiONs++ program package,50−52 we study four molecules of interest in modern excited-state research and show their timings at the PBE0/def2-SVP and PBE/def2-TZVP levels of theory on GPUs (Table 2). We investigate the Schiff base of retinal (II), a model system of the chromophore in rhodopsin,4 a nanocar79 (V) using a rotary molecular motor, and one (VI) and three (VII) pores of a covalent organic framework,80 which catalyze the formation of hydrogen from water when exposed to light. Details of the calculations and the computational setup are again listed in Sections 3.1 and 3.2.
Table 2. Molecular Structures and Computational Times (t(s)) of the Schiff Base of Retinal (II), a Motorized Nanocar (V), and One (VI) and Three (VII) Pores of a Covalent Organic Framework, Calculating Ground-State Energies (E0) and Gradients (E0ξ), Excited-State Energies (ωI) and Gradients (ωI), and Nonadiabatic Coupling Vectors (τI→J) at the PBE0/def2-SVP and PBE/def2-TZVP Levels of Theory on GPUsa.

| II | V | VI | VII | |
|---|---|---|---|---|
| formula | C20H30N+ | C51H28S2 | C144H102N30 | C312H213N69 |
| Natoms | 51 | 81 | 276 | 594 |
| PBE0/def2-SVP | ||||
| t(E0)/step | 1.3 | 4.4 | 17.8 | 67.7 |
| t(ω1)/(step and state) | 1.6 | 6.3 | 34.4 | 179.5 |
| t(E0ξ) | 12.36 | 54.5 | 140.9 | 334.3 |
| t(ω1ξ) | 65.4 | 247.9 | 668.4 | 1392.6 |
| t(τ0→1) | 24.3 | 123.4 | 375.2 | 914.8 |
| t(τ1→2) | 82.7 | 349.4 | 959.0 | 1858.0 |
| PBE/def2-TZVP | ||||
| t(E0)/step | 1.8 | 1.5 | 51.5 | 272.6 |
| t(ω1)/(step and state) | 2.1 | 1.7 | 30.2 | 95.3 |
| t(E0ξ) | 6.0 | 5.2 | 65.3 | 241.9 |
| t(ω1ξ) | 52.5 | 47.9 | 576.7 | 1942.9 |
| t(τ0→1) | 12.3 | 10.1 | 124.4 | 377.0 |
| t(τ1→2) | 56.1 | 52.7 | 523.2 | 1060.3 |
Table 2 shows that with the presented implementation on GPUs, excited-state properties and dynamics become accessible even for large systems and when applying DFT methods with exact exchange (e.g., PBE0) or triple-ζ basis sets. In our examples, the calculations of excited-state properties at the PBE/def2-TZVP level of theory are even faster than at the PBE0/def2-SVP level of theory mainly due to the fast Coulomb contractions discussed above. The only exception is t(ω1ξ) of VII, for which the Z-vector equation converges slowly at the PBE/def2-TZVP level of theory (seven instead of four iterations).
When comparing CPU and GPU timings of the entire calculations, we observe speed-ups of three and eight for VI and VII, respectively. This clearly shows that the use of GPUs becomes more attractive with increasing system size, where the integral evaluations dominate the overall computational time. However, even for relatively small molecule II we already obtain a speed-up of two. These speed-ups exclusively stem from the Coulomb and exchange calculations as the linear algebra and the evaluation of the exchange-correlation kernels are performed entirely on CPUs. The entire calculations of II and VII at the PBE0/def2-SVP level of theory take ∼5 min and ∼5 h, respectively, enabling NAMD simulations of II and the calculation of excited-state properties of VII on a reasonable time scale and yet good accuracy.
4.4. Scaling with the Computational Resources
In Figure 4, we show the parallel efficiency of J/K(XI + YI) (Figure 4a) and Jξ/Kξ(P, PI) (Figure 4b) for II, VI, and rotary molecular machine C (C10H13FN), which will be investigated in the next section using up to four GPUs. The Coulomb integrals of II and C are not analyzed because their computational time is too short.
Figure 4.

Parallel efficiency of selected GPU integral routines for the calculations of C, II, and VI. The computational time of the integral evaluation using four GPUs is given in parentheses. For details on the calculations and the computational setup, see Section 3.
Figure 4 again shows the suitability of GPUs for large molecular systems. For the time-consuming integral evaluations of VI, we observe a nearly perfect scaling of >0.9. This also indicates that adding even more GPUs will still lead to decent speed-ups of the calculations. The parallel efficiency observed in the case of the smaller molecules is lower but still remarkable considering that some computational times are <1 s. This strong scaling makes the use of GPUs also attractive for small to medium-sized molecules, as shown in the rotary molecular machine example presented in this work.
5. Illustrative Examples: Rotary Molecular Machines
As a prototypical example, we investigate the properties and the dynamics of four newly designed rotary molecular machines. For structures and definitions, see Figure 5. Similar to previous ab initio studies,41−44 our machines contain a C=N+ motif, which is also present in the chromophore of rhodopsin. Upon excitation, the molecule should rotate around the central C–C double bond. The fluoride substituent should accelerate this rotation because of the steric repulsion, whereas the puckering of the six-membered ring should influence the direction of the rotation.43,44 Here, we want to investigate the influence of the atom or group X adjacent to the central double bond on the light-driven rotation of the molecule. We use CH2, NH, S, and O as X denoted as C, N, S, and O, respectively.
Figure 5.

(a) Structures of rotary molecular machines C, N, S, and O and (b) definitions of the X–F distance (d(X–F)), the dihedral angle (γ), and the direction of rotation: clockwise (CW) and counterclockwise (CCW).
When calculating the excited-state properties and dynamics of the rotary molecular machines, we switch to the Tamm–Dancoff approximation (TDA).63 As discussed in Section 2.2, this accelerates the calculation and leads to more stable trajectories close to conical intersections.39,64 A comparison of the excited-state energies and properties at the TDA and RPA levels of theory is presented in the Supporting Information, where we show mean differences as well as plots of the difference density, the first excited-state gradient, and the NACV between the ground state and the first excited state. We observe an average difference below 10–2 a.u., with the state ordering and the shape of relaxed difference densities not being affected. The comparison of TDA and CASSCF energies (Supporting Information) also proves the suitability of TDA for the investigated problem. Trends between the systems and the energies close to the conical intersection agree remarkably well. Moreover, we apply the looser accurate thresholds (ϑpre = 10–3, ϑpre∇ = 10–10, and ϑTDDFT = 10–5) because they introduce only a maximum average error of a few 10–5 a.u. (Supporting Information). Additional details on the calculations are listed in Section 3.3.
In Figure 6, we show the excitation energies of C, N, S, and O. Changing the substituent has an influence on the bright S1 state of the rotary molecular motor. While the difference density plots look similar for all molecules, the excitation energies increase (S < C < N < O) and the directions of the gradients and NACVs change slightly. This is due to the different electronegativities of the substituents (C ≈ S < N < O) and the fact that S, N, and O atoms have free electron pairs.
Figure 6.
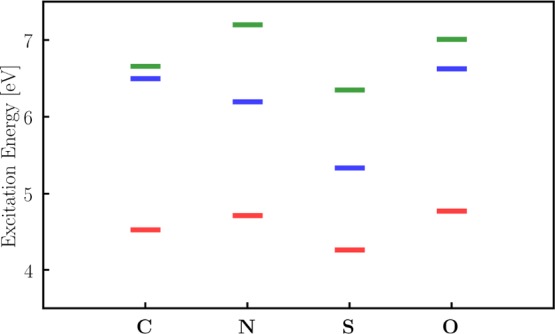
Singlet excitation energies of the four rotary molecular machines (C, N, S, and O) calculated at the TDA (wB97/def2-SVP) level of theory. For details on the calculations, see Section 3.3.
To study their effect on the light-induced rotation, we conducted NAMD simulations (105 trajectories for each rotor). In Table 3, we list the percentage of the trajectories that showed a rotation (η) as well as the ratio of clockwise and counterclockwise rotations (r). The direction of the rotation has been determined using the dihedral γ. (See the Supporting Information for plots.) The time-dependent occupation of the S1 state (calculated as an average over all trajectories) and d(X–F) for the trajectory with the fastest rotation of C, N, S, and O, respectively, are shown in Figure 7.
Table 3. Efficiency (η, Determined from the Percentage of Rotating Molecules in the NAMD Simulations) of the Four Rotary Molecular Machines (C, N, S, and O) and Ratio [r = n(CW)/n(CCW)] of CW and CCW Rotations.
| C | N | S | O | |
|---|---|---|---|---|
| η | 70% | 61% | 54% | 56% |
| r | 0.68 | 0.78 | 1.19 | 1.68 |
Figure 7.

(a) Time-dependent decay of the occupancy of the S1 state determined as a mean of all nonadiabatic molecular dynamics simulations. (b) Change in d(X–F) during one selected trajectory of C, N, S, and O, respectively, showing the rotation of the molecule around the central C–C bond. The maxima (180° rotation) are marked by the vertical dashed lines.
For all system, we observed (1) the expected rotation around the central C–C bond, which can be detected using either γ or d(X–F) and (2) the relaxation from S1 to the ground state (S0). Movies of the rotations are available at https://www.cup.uni-muenchen.de/pc/ochsenfeld/download/. The decay of the S1 population (Figure 7a) seems to correlate with the rotational speed of the corresponding rotary molecular machine (Figure 7b). C shows the fastest rotation and the fastest decay, followed by S and O, which behave nearly identically. The rotation of N is the slowest, so is the decay of the S1 occupancy. The trend in the rotational speeds (N < S < O < C) can easily be explained. O rotates slower than C because the nuclear repulsion between X and F is smaller. In the case of S, the repulsion of the S atom should be similar to that of the CH2 moiety, but the time for the rotation increases because of its larger mass. The rotation of N is even slower because of the hydrogen bond between the NH moiety and the F atom.
However, this trend cannot be observed when looking at the rotational efficiency (η, see Table 3). Although C shows the largest number of trajectories featuring a rotation (70%), the η of S and O (∼55%) is significantly smaller than the η of N (61%). The reason for this might be the slightly different coupling vectors between the ground state and the first excited state (Supporting Information). In contrast to refs (43) and (44), we do not observe an influence of the puckering of the ring on the rotation. The ratios of CW and CCW rotations (r) are close to 1 for all four rotors, and no general trend is visible in the γ plots (Supporting Information).
Changing X thus has an interesting effect on the rotary molecular machines. While the excitation energy changes significantly, the ability of the molecule to rotate around the central C–C bond is (nearly) preserved. These systems may thus be a good starting point for the design of a series of molecular rotors with tunable excitation energy. Our GPU-based routines aide these investigations by accelerating NAMD simulations even for these small systems. Because of the strong scaling (shown in Figure 4), we observe speed-ups in Coulomb and exchange integral evaluations (even when their computing time is shorter than 1 s), leading to a total speed-up of two with respect to a calculation entirely on CPUs. One trajectory took ∼1 day on two Intel Xeon CPU E5 2640 v4 @ 2.20 GHz (20 threads) CPUs and four AMD FirePro 3D W8100 GPUs.
6. Conclusions
Throughout this work, we have examined the use of graphics processing units (GPUs) for the evaluation of two-electron integrals in excited-state energies and property (gradients and nonadiabatic coupling vectors) calculations at the time-dependent density functional theory (TDDFT) level of theory. Similar to ground-state calculations, we observe that the use of GPUs along with the J-engine73,74 and the preLinK scheme50,51 leads to decent speed-ups of the integral calculations while additionally showing a reduced scaling behavior depending on the locality of the examined excitation. These speed-ups may even become larger with our currently developed seminumerical exchange scheme.95 By using GPUs, nonadiabatic molecular dynamics become more efficient for large but also small molecules (as the a result of the strong scaling) without a loss of accuracy or the introduction of further assumptions. As a first example, we investigated a series of newly designed rotary molecular machines showing that one can tune the excitation energy of these systems without losing their ability to rotate by changing the hetereocycle. For future applications, we consider extending the current implementation toward decoherence corrections96 and triplet states.97
Acknowledgments
We thank Gökçen Savaşçi for providing the optimized structures of the covalent organic framework pores (VI and VII) and Thomas Schnappinger and Prof. Regina de Vivie-Riedle (LMU Munich) for helpful discussions. Financial support was provided by the Innovative Training Network “Computational Spectroscopy in Natural Sciences and Engineering” (ITN-COSINE) and the DFG cluster of excellence (EXC 2089) “e-conversion”. C.O. acknowledges further support as a Max-Planck Fellow at the MPI-FKF Stuttgart.
Notation
All equations in this section use the same notation as in Section 2. gxc are the third order exchange correlation functional derivatives. The linear transformations H+ and H– are defined as follows:
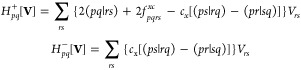 |
19 |
Excited-State Energy Gradients
The unrelaxed difference density matrix T is calculated as follows:
| 20 |
To obtain the Lagrangian multiplier ZI it is necessary to solve the following Z-vector equation:
| 21 |
| 22 |
Solving this Z-vector equation thus leads to the calculation of ZI, which is used to determine the relaxed difference density matrix (PI):
| 23 |
Using the calculated matrices, the energy-weighted difference density matrix (WI) can be formed:
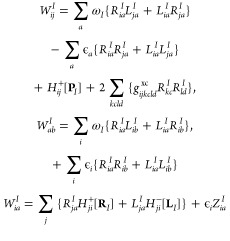 |
24 |
The calculation of the effective two-particle difference density matrix is
| 25 |
Nonadiabatic Coupling Vectors between the Ground and an Excited State
First the Lagrangian multipliers Z0I and W0I need to be determined. In order to obtain Z0I the following Z-vector equation has to be solved:
| 26 |
An iterative solution of eq 26 is not necessary. Z0I is equal to RI and the relaxed difference density matrix (P0I) is calculated as follows:
| 27 |
The calculation of W0I is presented in eq 28.
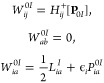 |
28 |
Γ0I is defined as
| 29 |
Nonadiabatic Coupling Vectors between Two Excited States
The unrelaxed difference density matrix (TIJ) is calculated as follows:
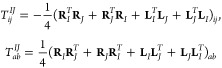 |
30 |
To obtain the Lagrangian multiplier ZIJ it is necessary to solve the following Z-vector equation:
| 31 |
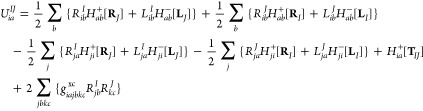 |
32 |
ZIJ is used to calculate the relaxed difference density matrix (PIJ):
| 33 |
The calculation of WIJ is
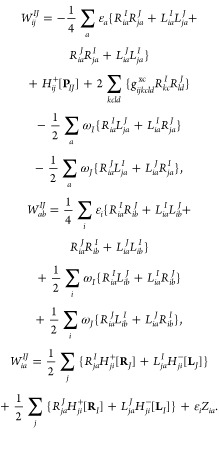 |
34 |
ΓIJ is defined as
| 35 |
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.9b00859.
log–log plots of the integral timings for the determination of the scaling behavior; validation of the use of the TDA and accurate thresholds, with excited-state energies, relaxed difference density plots, and plots of excited-state gradients and nonadiabatic coupling vectors at the TDA and RPA levels of theory; behavior of the dihedral γ during all trajectories (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Turro N. J.Modern Molecular Photochemistry; University Science Books: Sausalito, CA, 1991. [Google Scholar]
- Avouris P.; Gelbart W. M.; El-Sayed M. A. Nonradiative electronic relaxation under collision-free conditions. Chem. Rev. 1977, 77, 793–833. 10.1021/cr60310a002. [DOI] [Google Scholar]
- May V.; Kühn O.. Charge and Energy Transfer Dynamics in Molecular Science; Wiley-VCH: Weinheim, 2005. [Google Scholar]
- Lozier R. H.; Bogomolni R. A.; Stoeckenius W. Bacteriorhodopsin: a light-driven proton pump in Halobacterium Halobium. Biophys. J. 1975, 15, 955–962. 10.1016/S0006-3495(75)85875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koumura N.; Zijlstra R. W. J.; van Delden R. A.; Harada N.; Feringa B. L. Light-driven monodirectional molecular rotor. Nature 1999, 401, 152–155. 10.1038/43646. [DOI] [PubMed] [Google Scholar]
- Guentner M.; Schildhauer M.; Thumser S.; Mayer P.; Stephenson D.; Mayer P. J.; Dube H. Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor. Nat. Commun. 2015, 6, 8406. 10.1038/ncomms9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astumian R. D. How molecular motors work-insights from the molecular machinist’s toolbox: the Nobel prize in Chemistry 2016. Chem. Sci. 2017, 8, 840–845. 10.1039/C6SC04806D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreuw A. Quantum chemical methods for the investigation of photoinitiated processes in biological systems: Theory and applications. ChemPhysChem 2006, 7, 2259–2274. 10.1002/cphc.200600064. [DOI] [PubMed] [Google Scholar]
- Roos B. A new method for large-scale Cl calculations. Chem. Phys. Lett. 1972, 15, 153–159. 10.1016/0009-2614(72)80140-4. [DOI] [Google Scholar]
- Trofimov A. B.; Krivdina I. L.; Weller J.; Schirmer J. Algebraic-diagrammatic construction propagator approach to molecular response properties. Chem. Phys. 2006, 329, 1–10. 10.1016/j.chemphys.2006.07.015. [DOI] [Google Scholar]
- Christiansen O.; Koch H.; Jörgensen P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. 10.1016/0009-2614(95)00841-Q. [DOI] [Google Scholar]
- Casida M. E.Time-Dependent Density Functional Response Theory for Molecules. In Recent Advances in Density Functional Methods; Chong D. P., Ed.; Recent Advances in Computational Chemistry; World Scientific, 1995; Part 1, pp 155–192 10.1142/9789812830586_0005. [DOI] [Google Scholar]
- Casida M. E. Time-dependent density-functional theory for molecules and molecular solids. J. Mol. Struct.: THEOCHEM 2009, 914, 3–18. 10.1016/j.theochem.2009.08.018. [DOI] [Google Scholar]
- Dreuw A.; Head-Gordon M. Failure of time-dependent density functional theory for long-range charge-transfer excited states: The Zincbacteriochlorin-Bacteriochlorin and Bacteriochlorophyll-Spheroidene complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. 10.1021/ja039556n. [DOI] [PubMed] [Google Scholar]
- Dreuw A.; Head-Gordon M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 2005, 105, 4009–4037. 10.1021/cr0505627. [DOI] [PubMed] [Google Scholar]
- Laurent A. D.; Jacquemin D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. 10.1002/qua.24438. [DOI] [Google Scholar]
- Furche F.; Ahlrichs R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. 10.1063/1.1508368. [DOI] [Google Scholar]
- Chernyak V.; Mukamel S. Density-matrix representation of nonadiabatic couplings in time-dependent density functional (TDDFT) theories. J. Chem. Phys. 2000, 112, 3572. 10.1063/1.480511. [DOI] [Google Scholar]
- Baer R. Non-adiabatic couplings by time-dependent density functional theory. Chem. Phys. Lett. 2002, 364, 75–79. 10.1016/S0009-2614(02)01214-9. [DOI] [Google Scholar]
- Hu C.; Hirai H.; Sugino O. Nonadiabatic couplings from time-dependent density functional theory: Formulation in the Casida formalism and practical scheme within modified linear response. J. Chem. Phys. 2007, 127, 064103 10.1063/1.2755665. [DOI] [PubMed] [Google Scholar]
- Tavernelli I.; Tapavicza E.; Rothlisberger U. Non-adiabatic dynamics using time-dependent density functional theory: Assessing the coupling strengths. J. Mol. Struct.: THEOCHEM 2009, 914, 22–29. 10.1016/j.theochem.2009.04.020. [DOI] [Google Scholar]
- Tavernelli I.; Tapavicza E.; Rothlisberger U. Nonadiabatic coupling vectors within linear response time-dependent density functional theory. J. Chem. Phys. 2009, 130, 124107. 10.1063/1.3097192. [DOI] [PubMed] [Google Scholar]
- Send R.; Furche F. First-order nonadiabatic couplings from time-dependent hybrid density functional response theory: Consistent formalism, implementation, and performance. J. Chem. Phys. 2010, 132, 044107 10.1063/1.3292571. [DOI] [PubMed] [Google Scholar]
- Tavernelli I.; Curchod B. F. E.; Laktionov A.; Rothlisberger U. Nonadiabatic coupling vectors for excited states within time-dependent density functional theory in the Tamm-Dancoff approximation and beyond. J. Chem. Phys. 2010, 133, 194104. 10.1063/1.3503765. [DOI] [PubMed] [Google Scholar]
- Fatehi S.; Alguire E.; Shao Y.; Subotnik J. E. Analytic derivative couplings between configuration-interaction-singles states with built-in electron-translation factors for translational invariance. J. Chem. Phys. 2011, 135, 234105. 10.1063/1.3665031. [DOI] [PubMed] [Google Scholar]
- Ou Q.; Fatehi S.; Alguire E.; Shao Y.; Subotnik J. E. Derivative couplings between TDDFT excited states obtained by direct differentiation in the Tamm-Dancoff approximation. J. Chem. Phys. 2014, 141, 024114 10.1063/1.4887256. [DOI] [PubMed] [Google Scholar]
- Alguire E. C.; Ou Q.; Subotnik J. E. Calculating derivative couplings between time-dependent Hartree–Fock excited states with pseudo-wavefunctions. J. Phys. Chem. B 2015, 119, 7140–7149. 10.1021/jp505767b. [DOI] [PubMed] [Google Scholar]
- Subotnik J. E.; Alguire E. C.; Ou Q.; Landry B. R.; Fatehi S. The requisite electronic structure theory to describe photoexcited nonadiabatic dynamics: Nonadiabatic derivative couplings and diabatic electronic couplings. Acc. Chem. Res. 2015, 48, 1340–1350. 10.1021/acs.accounts.5b00026. [DOI] [PubMed] [Google Scholar]
- Ou Q.; Alguire E. C.; Subotnik J. E. Derivative couplings between time-dependent density functional theory excited states in the random-phase approximation based on pseudo-wavefunctions: Behavior around conical intersections. J. Phys. Chem. B 2015, 119, 7150–7161. 10.1021/jp5057682. [DOI] [PubMed] [Google Scholar]
- Tully J. C. Molecular dynamics with electronic transitions. J. Chem. Phys. 1990, 93, 1061–1071. 10.1063/1.459170. [DOI] [Google Scholar]
- Hammes-Schiffer S.; Tully J. C. Proton transfer in solution: Molecular dynamics with quantum transitions. J. Chem. Phys. 1994, 101, 4657–4667. 10.1063/1.467455. [DOI] [Google Scholar]
- Wang L.; Akimov A.; Prezhdo O. V. Recent Progress in Surface Hopping: 2011–2015. J. Phys. Chem. Lett. 2016, 7, 2100. 10.1021/acs.jpclett.6b00710. [DOI] [PubMed] [Google Scholar]
- Crespo-Otero R.; Barbatti M. Recent Advances and Perspectives on Nonadiabatic Mixed Quantum-Classical Dynamics. Chem. Rev. 2018, 118, 7026. 10.1021/acs.chemrev.7b00577. [DOI] [PubMed] [Google Scholar]
- Mai S.; Marquetand P.; González L. Nonadiabatic Dynamics: The SHARC Approach. WIREs Comput. Mol. Sci. 2018, 8, e1370 10.1002/wcms.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapavicza E.; Tavernelli I.; Rothlisberger U. Trajectory surface hopping within linear response time-dependent density-functional theory. Phys. Rev. Lett. 2007, 98, 023001 10.1103/PhysRevLett.98.023001. [DOI] [PubMed] [Google Scholar]
- Barbatti M.; Granucci G.; Persico M.; Ruckenbauer M.; Vazdar M.; Eckert-Maksić M.; Lischka H. The on-the-fly surface-hopping program system Newton-X: Application to ab initio simulation of the nonadiabatic photodynamics of benchmark systems. J. Photochem. Photobiol., A 2007, 190, 228–240. 10.1016/j.jphotochem.2006.12.008. [DOI] [Google Scholar]
- Barbatti M. Nonadiabatic dynamics with trajectory surface hopping method. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 620–633. 10.1002/wcms.64. [DOI] [Google Scholar]
- Tully J. C. Perspective: Nonadiabatic dynamics theory. J. Chem. Phys. 2012, 137, 22A301. 10.1063/1.4757762. [DOI] [PubMed] [Google Scholar]
- Tapavicza E.; Bellchambers G. D.; Vincent J. C.; Furche F. Ab initio non-adiabatic molecular dynamics. Phys. Chem. Chem. Phys. 2013, 15, 18336–18348. 10.1039/c3cp51514a. [DOI] [PubMed] [Google Scholar]
- Plasser F.; Crespo-Otero R.; Pederzoli M.; Pittner J.; Lischka H.; Barbatti M. Surface hopping dynamics with correlated single-reference methods: 9H-adenine as a case study. J. Chem. Theory Comput. 2014, 10, 1395–1405. 10.1021/ct4011079. [DOI] [PubMed] [Google Scholar]
- Nikiforov A.; Gamez J. A.; Thiel W.; Filatov M. Computational Design of a Family of Light-Driven Rotary Molecular Motors with Improved Quantum Efficiency. J. Phys. Chem. Lett. 2016, 7, 105–110. 10.1021/acs.jpclett.5b02575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazaryan A.; Lan Z.; Schäfer L. V.; Thiel W.; Filatov M. Surface hopping excited-state dynamics study of the photoisomerization of a light-driven fluorene molecular rotary motor. J. Chem. Theory Comput. 2011, 7, 2189–2199. 10.1021/ct200199w. [DOI] [PubMed] [Google Scholar]
- Oruganti B.; Wang J.; Durbeej B. Quantum chemical design of rotary molecular motors. Int. J. Quantum Chem. 2018, 118, e25405 10.1002/qua.25405. [DOI] [Google Scholar]
- Wang J.; Oruganti B.; Durbeej B. Light-driven rotary molecular motors without point chirality: A minimal design. Phys. Chem. Chem. Phys. 2017, 19, 6952–6956. 10.1039/C6CP08484B. [DOI] [PubMed] [Google Scholar]
- Sisto A.; Glowacki D. R.; Martinez T. J. Ab initio nonadiabatic dynamics of multichromophore complexes: A scalable graphical-processing-unit-accelerated exciton framework. Acc. Chem. Res. 2014, 47, 2857–2866. 10.1021/ar500229p. [DOI] [PubMed] [Google Scholar]
- Yasuda K. Two-electron integral evaluation on the graphics processor unit. J. Comput. Chem. 2008, 29, 334–342. 10.1002/jcc.20779. [DOI] [PubMed] [Google Scholar]
- Ufimtsev I. S.; Martínez T. J. Quantum chemistry on graphical processing units. 1. strategies for two-electron integral evaluation. J. Chem. Theory Comput. 2008, 4, 222–231. 10.1021/ct700268q. [DOI] [PubMed] [Google Scholar]
- Ufimtsev I. S.; Martínez T. J. Quantum chemistry on graphical processing units. 2. direct self-consistent-field implementation. J. Chem. Theory Comput. 2009, 5, 1004–1015. 10.1021/ct800526s. [DOI] [PubMed] [Google Scholar]
- Ufimtsev I. S.; Martínez T. J. Quantum chemistry on graphical processing units. 3. Analytical energy gradients, geometry optimization, and first principles molecular dynamics. J. Chem. Theory Comput. 2009, 5, 2619–2628. 10.1021/ct9003004. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Pre-selective screening for matrix elements in linear-scaling exact exchange calculations. J. Chem. Phys. 2013, 138, 134114. 10.1063/1.4796441. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Preselective screening for linear-scaling exact exchange-gradient calculations for graphics processing units and general strong-scaling massively parallel calculations. J. Chem. Theory Comput. 2015, 11, 918–922. 10.1021/ct501189u. [DOI] [PubMed] [Google Scholar]
- Kussmann J.; Ochsenfeld C. Hybrid CPU/GPU integral engine for strong-scaling ab initio methods. J. Chem. Theory Comput. 2017, 13, 3153–3159. 10.1021/acs.jctc.6b01166. [DOI] [PubMed] [Google Scholar]
- Peters L. D. M.; Kussmann J.; Ochsenfeld C. Efficient and accurate Born-Oppenheimer molecular dynamics for large molecular systems. J. Chem. Theory Comput. 2017, 13, 5479–5485. 10.1021/acs.jctc.7b00937. [DOI] [PubMed] [Google Scholar]
- Isborn C. M.; Luehr N.; Ufimtsev I. S.; Martínez T. J. Excited-state electronic structure with configuration interaction singles and Tamm-Dancoff time-dependent density functional theory on graphical processing units. J. Chem. Theory Comput. 2011, 7, 1814–1823. 10.1021/ct200030k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohenstein E. G.; Luehr N.; Ufimtsev I. S.; Martínez T. J. An atomic orbital-based formulation of the complete active space self-consistent field method on graphical processing units. J. Chem. Phys. 2015, 142, 224103. 10.1063/1.4921956. [DOI] [PubMed] [Google Scholar]
- Snyder J. W.; Hohenstein E. G.; Luehr N.; Martínez T. J. An atomic orbital-based formulation of analytical gradients and nonadiabatic coupling vector elements for the state-averaged complete active space self-consistent field method on graphical processing units. J. Chem. Phys. 2015, 143, 154107. 10.1063/1.4932613. [DOI] [PubMed] [Google Scholar]
- Curchod B. F. E.; Sisto A.; Martínez T. J. Ab Initio Multiple Spawning Photochemical Dynamics of DMABN Using GPUs. J. Phys. Chem. A 2017, 121, 265. 10.1021/acs.jpca.6b09962. [DOI] [PubMed] [Google Scholar]
- Snyder J. W.; Curchod B. F. E.; Martínez T. J. GPU-Accelerated State-Averaged Complete Active Space Self-Consistent Field Interfaced with Ab Initio Multiple Spawning Unravels the Photodynamics of Provitamin D3. J. Phys. Chem. Lett. 2016, 7, 2444. 10.1021/acs.jpclett.6b00970. [DOI] [PubMed] [Google Scholar]
- Hollas D.; Šištík L.; Hohenstein E. G.; Martínez T. J.; Slavíček P. Nonadiabatic Ab Initio Molecular Dynamics with the Floating Occupation Molecular Orbital-Complete Active Space Configuration Interaction Method. J. Chem. Theory Comput. 2018, 14, 339. 10.1021/acs.jctc.7b00958. [DOI] [PubMed] [Google Scholar]
- Filatov M.; Min S. K.; Kim K. W. Direct Nonadiabatic Dynamics by Mixed Quantum-Classical Formalism Connected with Ensemble Density Functional Theory Method: Application to trans-Penta-2,4-dieniminium Cation. J. Chem. Theory Comput. 2018, 14, 4499. 10.1021/acs.jctc.8b00217. [DOI] [PubMed] [Google Scholar]
- Bauernschmitt R.; Ahlrichs R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. 10.1016/0009-2614(96)00440-X. [DOI] [Google Scholar]
- Bauernschmitt R.; Häser M.; Treutler O.; Ahlrichs R. Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions. Chem. Phys. Lett. 1997, 264, 573–578. 10.1016/S0009-2614(96)01343-7. [DOI] [Google Scholar]
- Maurice D.; Head-Gordon M. Configuration interaction with single substitutions for excited states of open-shell molecules. Int. J. Quantum Chem. 1995, 56, 361–370. 10.1002/qua.560560840. [DOI] [Google Scholar]
- Cordova F.; Doriol L. J.; Casida M. E. Troubleshooting time-dependent density-functional theory for photochemical applications: Oxirane. J. Chem. Phys. 2007, 127, 164111. 10.1063/1.2786997. [DOI] [PubMed] [Google Scholar]
- Chai J. D.; Head-Gordon M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106 10.1063/1.2834918. [DOI] [PubMed] [Google Scholar]
- Li Z.; Liu W. First-order nonadiabatic coupling matrix elements between excited states: A Lagrangian formulation at the CIS, RPA, TD-HF, and TD-DFT levels. J. Chem. Phys. 2014, 141, 014110 10.1063/1.4885817. [DOI] [PubMed] [Google Scholar]
- Li Z.; Suo B.; Liu W. First order nonadiabatic coupling matrix elements between excited states: Implementation and application at the TD-DFT and pp-TDA levels. J. Chem. Phys. 2014, 141, 244105. 10.1063/1.4903986. [DOI] [PubMed] [Google Scholar]
- Ou Q.; Bellchambers G. D.; Furche F.; Subotnik J. E. First-order derivative couplings between excited states from adiabatic TDDFT response theory. J. Chem. Phys. 2015, 142, 064114 10.1063/1.4906941. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Herbert J. M. Analytic derivative couplings in time-dependent density functional theory: Quadratic response theory versus pseudo-wavefunction approach. J. Chem. Phys. 2015, 142, 064109 10.1063/1.4907376. [DOI] [PubMed] [Google Scholar]
- Parker S. M.; Roy S.; Furche F. Unphysical divergences in response theory. J. Chem. Phys. 2016, 145, 134105. 10.1063/1.4963749. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Herbert J. M. Analytic derivative couplings for spin-flip configuration interaction singles and spin-flip time-dependent density functional theory. J. Chem. Phys. 2014, 141, 064104 10.1063/1.4891984. [DOI] [PubMed] [Google Scholar]
- Liu J.; Thiel W. An efficient implementation of semiempirical quantum-chemical orthogonalization-corrected methods for excited-state dynamics. J. Chem. Phys. 2018, 148, 154103. 10.1063/1.5022466. [DOI] [PubMed] [Google Scholar]
- Reza Ahmadi G.; Almlöf J. The Coulomb operator in a Gaussian product basis. Chem. Phys. Lett. 1995, 246, 364–370. 10.1016/0009-2614(95)01127-4. [DOI] [Google Scholar]
- White C.; Head-Gordon M. A J matrix engine for density functional theory calculations. J. Chem. Phys. 1996, 104, 2620–2629. 10.1063/1.470986. [DOI] [Google Scholar]
- intel c/c++ compiler v19.0, see https://software.intel.com.
- Marques M. A. L.; Oliveira M. J. T.; Burnus T. Libxc: A library of exchange and correlation functionals for density functional theory. Comput. Phys. Commun. 2012, 183, 2272–2281. 10.1016/j.cpc.2012.05.007. [DOI] [Google Scholar]
- Lehtola S.; Steigemann C.; Oliveira M. J.; Marques M. A. Recent developments in LIBXC A comprehensive library of functionals for density functional theory. SoftwareX 2018, 7, 1–5. 10.1016/j.softx.2017.11.002. [DOI] [Google Scholar]
- Laqua H.; Kussmann J.; Ochsenfeld C. An improved molecular partitioning scheme for numerical quadratures in density functional theory. J. Chem. Phys. 2018, 149, 204111. 10.1063/1.5049435. [DOI] [PubMed] [Google Scholar]
- Morin J.-F.; Shirai Y.; Tour J. M. En Route to a motorized nanocar. Org. Lett. 2006, 8, 1713–1716. 10.1021/ol060445d. [DOI] [PubMed] [Google Scholar]
- Vyas V. S.; Haase F.; Stegbauer L.; Savasci G.; Podjaski F.; Ochsenfeld C.; Lotsch B. V. A tunable azine covalent organic framework platform for visible light-induced hydrogen generation. Nat. Commun. 2015, 6, 8508. 10.1038/ncomms9508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. 10.1103/PhysRevLett.78.1396. [DOI] [PubMed] [Google Scholar]
- Ernzerhof M.; Scuseria G. E. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029. 10.1063/1.478401. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Barone V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. 10.1063/1.478522. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs A. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Weigend F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- White C. A.; Head-Gordon M. Linear and sublinear scaling formation of Hartree–Fock-type exchange matrices. Chem. Phys. Lett. 1994, 230, 8. [Google Scholar]
- White C. A.; Johnson B. G.; Gill P. M. W.; Head-Gordon M. Linear and sublinear scaling formation of Hartree–Fock-type exchange matrices. Chem. Phys. Lett. 1996, 253, 268. 10.1016/0009-2614(96)00175-3. [DOI] [Google Scholar]
- Ochsenfeld C.; White C. A.; Head-Gordon M. Linear and sublinear scaling formation of Hartree–Fock-type exchange matrices. J. Chem. Phys. 1998, 109, 1663–1669. 10.1063/1.476741. [DOI] [Google Scholar]
- Ochsenfeld C. Linear scaling exchange gradients for Hartree–Fock and hybrid density functional theory. Chem. Phys. Lett. 2000, 327, 216–223. 10.1016/S0009-2614(00)00865-4. [DOI] [Google Scholar]
- Verlet L. Computer ″experiments″ on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. 10.1103/PhysRev.159.98. [DOI] [Google Scholar]
- Swope W. C.; Andersen H. C.; Berens P. H.; Wilson K. R. A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters. J. Chem. Phys. 1982, 76, 637–649. 10.1063/1.442716. [DOI] [Google Scholar]
- Niklasson A. M. N.; Steneteg P.; Odell A.; Bock N.; Challacombe M.; Tymczak C. J.; Holmström E.; Zheng G.; Weber V. Extended Lagrangian Born-Oppenheimer molecular dynamics with dissipation. J. Chem. Phys. 2009, 130, 214109. 10.1063/1.3148075. [DOI] [PubMed] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Laqua H.; Kussmann J.; Ochsenfeld C. Submitted for publication, 2019.
- Jaeger H. M.; Fischer S.; Prezhdo O. V. Decoherence-induced surface hopping. J. Chem. Phys. 2012, 137, 22A545. 10.1063/1.4757100. [DOI] [PubMed] [Google Scholar]
- Richter M.; Marquetand P.; González-Vázquez J.; Sola I.; González SHARC: ab Initio Molecular Dynamics with Surface Hopping in the Adiabatic Representation Including Arbitrary Couplings. J. Chem. Theory Comput. 2011, 7, 1253. 10.1021/ct1007394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.