

Abstract
It is known that smooth, hydrophobic solid surfaces exhibit low ice adhesion values, which have been shown to approach a lower ice adhesion strength limit (~150 kPa) defined by the water receding contact angle. To overcome this limit, we have designed self-lubricating icephobic coatings by blending polydimethylsiloxane (PDMS)-poly(ethylene glycol) (PEG) amphiphilic copolymers into a polymer coating matrix. Such coatings provide low ice adhesion strength values (~50 kPa) that can substantially reduce the lower bound of the ice adhesion strength achieved previously on smooth, hydrophobic solid surfaces. Different molecular mechanisms are responsible for the low ice adhesion strength attained by these two approaches. For the smooth hydrophobic surfaces, an increased water depletion layer thickness at the interface weakens the van der Waals’ interactions between the ice and the polymeric substrate. For the self-lubricating icephobic coatings, the PEG component of the amphiphilic copolymer is capable of strongly hydrogen bonding with water molecules. The surface hydrogen-bonded water molecules do not freeze, even at substantial levels of subcooling, and therefore serve as a self-lubricating interfacial liquid-like layer that helps to reduce the adhesion strength of ice to the surface. The existence of non-frozen water molecules at the ice-solid interface is confirmed by solid-state nuclear magnetic resonance (NMR) spectroscopy.
Keywords: icephobic, ice adhesion, hydrophobic, water depletion layer, nonfrozen quasi-liquid layer, self-lubricating, solid-state NMR, delayed freezing
Graphical Abstract
INTRODUCTION
The formation and accumulation of ice on a substrate can affect the operational performance, and/or cause significant damage to many different types of structures, such as airplane wings, ships, wind turbine blades, power lines, vehicles, buildings and solar panels1–3. Ice accretion changes the shape and roughness of the wind turbine blade, leading to power loss and even to mechanical failures. Many wind turbines are designed to reduce power or shut off completely if too much ice accumulation occurs4. A common practice for deicing is to spray iced structures, such as airplanes, with deicing fluids that contain an ethylene glycol component. Such deicing fluids basically lower the freezing point of water and thereby cause the ice formed on the structures to melt. However, this approach requires frequent applications, can be expensive, and can be detrimental to the environment1, 3. Some recently developed systems that employ porous coatings impregnated with freezing point depressants have led to moderate success in reducing the ice adhesion strength on a substrate5–6. This approach can reduce the frequency of reapplication of the impregnated deicing liquids. Researchers have also designed slippery, liquid-infused porous surfaces (SLIPS) with a low surface tension liquid layer present as a lubricating layer, providing anti-icing and anti-frost properties to the surfaces7. Recently, researchers have prepared organogels, e.g., silicone elastomers imbibed with organic solvents or silicone oils, for icephobic coatings, yielding remarkable low ice adhesion values8–11. However, for both SLIPS and organogels, the mechanical robustness of these coatings and liquid retention within these coatings remain to be improved9, 12.
Another appealing approach is to design solid surface coatings that have robust anti-icing properties without the need for periodic reapplication of deicing or lubrication liquids. Ideally, ice would have minimal adhesion strength on these coatings such that any frozen water that accretes on the interface could be removed by its own weight or other additional natural forces such as wind. Exploring this approach, Meuler et al. have studied the relationship between water wettability and ice adhesion3. The practical work of adhesion for water is characterized by the water receding contact angle: , where is the practical work of adhesion per unit area for water on a surface, is the surface tension of water, and is the receding water contact angle3, 13. Meuler et al. found that measurements of the average strength of ice adhesion varied nearly linearly with the practical work of adhesion for the liquid water. This suggests that maximizing the receding contact angle of a water drop on a surface coating will minimize the ice adhesion. The lowest ice adhesion (~150 kPa) achieved by Meuler et al. was a polymeric nanocomposite coating that contains fluorodecyl polyhedral oligomeric silsesquioxane (Fluorodecyl POSS) and poly(ethyl methacrylate) (20:80 percent by weight), which has the largest receding contact angle () measured on smooth hydrophobic surfaces. Further reduction in ice adhesion strength requires the introduction of surface texture to a hydrophobic surface, leading to a superhydrophobic surface. However, a superhydrophobic surface tends to lose its surface texture during repeating deicing events14–16. Superhydrophobic surfaces can also sometimes significantly increase ice adhesion if ice is able to grow into the surface texture15, 17–19.
It is known that water can exist in a non-frozen state below the normal freezing point of 0 °C20. Polar fishes and insects produce antifreeze-proteins (AFPs) that bind to the surface of nascent ice crystals, preventing further growth at temperatures within a characteristic thermal hysteresis range21. Therefore, those fishes and insects can survive in icy cold climates. Researchers have attached AFPs to different substrates to achieve an anti-icing effect22–23. However, the surface grafted AFPs seem to only delay the temporal formation of ice on these surfaces22–23. The ice adhesion strength on such surfaces has not been tested. Moreover, AFPs are expensive, and tend to denature and lose their function under acidic pH or high temperatures24. These attributes limit the use of AFPs for practical anti-icing surface coatings. Therefore, it is very attractive to explore the use of inexpensive synthetic polymers that have the capability to bind significant amounts of water which do not freeze at or below 0 °C. The design goal of surface coatings containing these synthetic polymers would be to produce a thin layer of non-frozen water at the interface between ice and the underlying substrate. The layer of non-frozen water can then serve as a lubrication layer to reduce the ice adhesion strength. This approach may be advantageous compared to SLIPS or organogels coatings, in which lubricating liquids need be externally reapplied periodically. Researchers have synthesized polymers, such as cross-linked poly(acrylic acid), that can bind strongly with water molecules. These polymers have shown initial success in reducing the ice adhesion strength24–27. However, the existence of non-frozen water at the interface has only been speculated. These polymers usually also involve multistep synthesis processes and can only be applied to certain substrates to restrict swelling. There is thus an unmet demand for anti-icing polymer coatings that can be applied readily to a wide variety of substrates and that can be readily scaled up to cover large surface areas. Such polymer coatings should significantly reduce ice adhesion on the substrate and also must be affordable.
In this work, we design scalable, self-lubricating icephobic coatings by utilizing commercially-available copolymers embedded in a polymer coating matrix. By comparing ice adhesion measurements on this new low ice adhesion coating system with the previously established hydrophobic low ice adhesion coating system, we illustrate the different molecular mechanisms that are responsible for low ice adhesion on these two different classes of surfaces.
EXPERIMENTAL SECTION
Materials and Sample Preparation
Polydimethylsiloxane (PDMS) elastomer precursor (Sylgard 184) was obtained from Dow Corning, Auburn MI. Polydimethylsiloxane (PDMS)-poly(ethylene glycol) (PEG) copolymers (CMS-226, DBP-732, CMS-221 and DBE-224) were obtained from Gelest Inc. For the PDMS elastomer coating, Sylgard 184 base and cross-linker were mixed thoroughly in a 10:1 ratio by weight, followed by degassing via vacuum to remove air bubbles, and spin-coating at 2000 rpm for 60 s to reach a thickness of about 20 microns. The spin-coated samples were then baked at 80 °C for 24 h to achieve complete curing. For the PDMS-PEG film, all the processing conditions were the same except that 1 or 5 wt% PDMS-PEG was blended in the PDMS elastomer precursor during the mixing step. The final thickness of the PDMS+PDMS-PEG films was also about 20 micron. For CMS-626 and DBP-732, blending of 1 or 5 wt% PDMS-PEG barely changed the viscosity of the PDMS elastomer precursor so no dilution was necessary for achieving the same film thickness by applying the same spin-coating parameters. For CMS-221 and DBE-224, a significant increase in the viscosity of the PDMS elastomer precursor was observed. Therefore, the mixture was diluted with chloroform (Sigma-Aldrich) to a concentration of 40–60% by weight so the same film thickness of about 20 microns was achieved with the same spin-coating parameters.
Poly(ethyl methacrylate) (PEMA, Mw=515 kg/mol, Sigma-Aldrich), poly(methyl methacrylate) (PMMA, Mw=540 kg/mol, Scientific Polymer Products), poly(n-butyl methacrylate) (PBMA, Mw=337 kg/mol, Sigma-Aldrich), Fluorodecyl polyhedral oligomeric silsesquioxane (POSS, a gift from NBD Nanotechnologies Inc.) and Asahiklin (AK225, Asahi Glass Company, Tokyo Japan) were used as received. Polymer solutions (with a solid concentration of 20 mg/mL) were prepared by dissolving PEMA, PMMA, PBMA, and the PEMA/fluorodecyl POSS mixture (80:20 by weight) in Asahiklin, respectively. Thin (∼200–300 nm) coatings were deposited at room temperature on silicon substrates via a spin coating process. About 0.2 mL of a polymer solution was placed on top of each substrate and the sample was spun at 1,000 rpm for 30 s. Then the coated samples were thermally annealed at 60 °C for at least 2 h. Polystyrene (PS, Mw=35,000, Sigma-Aldrich), poly(vinyl phenol) (PVPh, Mw=25,000, Sigma-Aldrich), and poly(styrene-b-ethylene oxide) (PS-PEO, Mn=51,000-b-11,500, Polymer Source) were used as received. PS and PS-PEO were dissolved in toluene (Sigma-Aldrich) to prepare 3 wt% PS and PS-PEO solutions, respectively. PVPh was dissolved in 1,4-dioxane (Sigma-Aldrich) to prepare a PVPh solution with a solid concentration of 3 wt%. About 0.2 mL of one solution was placed on top of each clean silicon substrate and the sample was spun at 1,000 rpm for 30 s. The resulting film thickness was about 100 nm. These samples were dried in a vacuum oven to completely remove the solvent. For guar gum coated samples, the silicon substrates were first treated with oxygen plasma (PDC-32G, Harrick Scientific Products, Inc.) for 10 min at 150 mTorr. After this step, these substrates were immersed in a methyl ethyl ketone (MEK, 99+% A.C.S. reagent, Sigma-Aldrich) solution containing 0.1% (w/v) poly(glycidyl methacrylate) (PGMA, Mw = 25 kDa, Polysciences) for 20 s. These samples were then placed in a 110 °C oven for 30 min to covalently bond PGMA to the substrate. After cooling to ambient temperature, the PGMA-coated substrates were immersed in a 20 mg/mL aqueous solution of gaur gum (G4129, obtained from Sigma-Aldrich,) for 20 min. The guar gum coated substrates were dried at ambient temperature and again placed in the oven at 110 °C for 30 min to induce a chemical reaction between the residual epoxide groups present in PGMA and the hydroxyl groups on guar.
Epoxy resin (PB140653, Epoxy Technology Inc. Billerica, MA) was mixed thoroughly with 1 wt% DBE-224 before spin coating onto a clean silicon substrate (1×1 inch) at 2000 rpm for 60 s. The as-spun samples were then cured by UV exposure for 90 s with a Dymax ECE 5000 flood UV lamp. Pure epoxy resin was processed in the same conditions to prepare the control samples. Polyurethane precursor (Clear flex 95, Smooth-on Inc. Macungie PA) was mixed in a A:B=1:1.5 weight ratio, together with 1 wt% DBE-224. The mixture was degassed using a vacuum pump to remove the bubbles introduced during the mixing step. Next, the mixture was spin coated onto clean silicon substrates at 2000 rpm for 60 s. These samples were then cured for 24 h at room temperature followed by another 6 h at 60 °C. Pure polyurethane was processed in the same conditions to prepare the control samples. Fluorinated polyurethane coatings were prepared in a similar manner as for polyurethane. Fluorinated polyurethane precursor (Luxecolor 4FVBA-800, obtained from Helicity Technologies, Inc.) was mixed in a ratio 10:0.8 by weight of fluorinated polyol solution to polyisocyanate crosslinker (Desmodur N3400), together with 1 wt% DBE-224 in the final solid film. The mixture was spin coated on clean silicon substrates at 2000 rpm for 60 s. Then these samples were cured for 24 h at room temperature followed by another 24 h at 60 °C. Pure fluorinated polyurethane was processed in the same conditions to prepare the control samples.
Wetting Properties Characterization
Contact angles of deionized water (18 MΩ/cm, Millipore) on polymer coatings were measured using a ramé-hart Model 590 goniometer.
Advancing angles () were measured as water was supplied via a syringe while receding angles () were measured as water was removed via a syringe. The total drop volume is 5 μL and the pump dispense speed is 0.2 μL/s. Measurements were taken over three or more different locations on each surface, and the reported values are listed in Table 1 and Table 3 in the format of average ± standard deviation.
Table 1.
Measured Water Contact Angles and Average Shear Strengths of Ice Adhesion
| Surfaces | Advancing angle (θadv)(°) |
Receding angle (θrec)(°) |
Ice adhesion strength (kPa) |
|---|---|---|---|
| Clean silicon wafer | 35±1 | 10±2 | 869±43 |
| PEMA | 79±1 | 67±1 | 672±47 |
| PEMA-POSS(80/20) | 122±1 | 117±1 | 151±5 |
| PMMA | 105±1 | 64±1 | 737±154 |
| PBMA | 89±1 | 73±1 | 519±4 |
| PDMS 10:1 | 118±1 | 82±2 | 317±16 |
| PDMS+PDMS-PEG* | 112±2 | 70±1 | 117±7 |
| PS | 92±2 | 82±2 | 683±44 |
| PVP | 74±1 | 9±2 | 902±62 |
| PS-PEG | 83±1 | 60±1 | 558±18 |
| Guar gum | 51±1 | 0° | 1321±11 |
PDMS-PEG copolymer is CMS-626 (Gelest Inc.), blended into a PDMS elastomer film at 1wt% ratio.
Table 3.
Measured Water Contact Angles and Average Shear Strengths of Ice Adhesion for PDMS elastomer Coatings Containing PDMS-PEG Copolymers
| Surfaces | Advancing Angle (θadv)(°) |
Receding Angle (θrec)(°) |
Ice adhesion Strength (kPa) |
|---|---|---|---|
| PDMS + 1 wt% CMS-626 | 112±2 | 70±1 | 117±7 |
| PDMS + 5 wt% CMS-626 | 126±2 | 59±1 | 94±8 |
| PDMS + 1 wt% DBP-732 | 112±1 | 76±1 | 181±7 |
| PDMS + 5 wt% DBP-732 | 126±2 | 59±2 | 180±16 |
| PDMS + 1 wt% CMS-221 | 122±1 | 86±1 | 99±3 |
| PDMS + 5 wt% CMS-221 | 126±2 | 68±1 | 46±9 |
| PDMS + 1 wt% DBE-224 | 120±2 | 80±2 | 57±9 |
| PDMS + 5 wt% DBE-224 | 126±1 | 71±1 | 51±14 |
Ice Adhesion Measurements
Deionized water was poured into plastic cuvettes (10 mm × 10 mm × 45 mm). Coated substrates were clamped onto a base plate and then mounted against the tops of the cuvettes. The assembly was inverted and bolted to a Peltier cooling plate whose surface was thermostatted at a target temperature (−15 °C). The top sample holder was then removed. The water was frozen for at least 3 h before starting the ice adhesion test. The ice adhesion test apparatus is illustrated in Figure 1. The probe of the force transducer was propelled at 0.1 mm/s into the side of each cuvette until the ice column detached from the test surface. In the current tests, we maintained a constant separation height , which is the distance between the applied force location and the ice-substrate interface (shown in Figure 1). The maximum force is recorded and converted into the ice adhesion strength using the known cross-sectional area of the ice-substrate interface in each cuvette. To minimize water condensation from ambient air and the formation of frost on the cooled surfaces, the ice adhesion setup as shown in Figure 1 was placed in a glove box filled with dry nitrogen.
Figure 1.

A schematic illustration showing the ice adhesion test apparatus. Coated substrates were mounted to a Peltier cooling plate whose surface was thermostatted at a target temperature (−15 °C). Plastic cuvettes (10 mm × 10 mm × 45 mm) filled with deionized water were inverted on the substrate surface and the water was frozen for at least 3 h before starting the ice adhesion test. The probe of the force transducer was propelled at 0.1 mm/s into the side of each cuvette until the ice column detached from the test surface, and the maximum fracture force is recorded.
Three to six samples for each type of polymer coating were prepared and the ice adhesion was tested on these samples under the same condition (−15 °C) throughout the experiment. The measured ice adhesion strength is reported in the format of an average value ± standard deviation, as listed in Table 1.
Nuclear Magnetic Resonance (NMR) Spectroscopy
NMR experiments were conducted on a Bruker Avance III HD 400 MHz spectrometer using a 4 mm MAS probe. 1H radiofrequency field strengths of 62.5 kHz were used. 1H chemical shifts were referenced to the Hγ peak of phosphocholine in a DOPC: DOPG: cholesterol membrane at 3.26 ppm on the TMS scale.
Spectra and relaxation data were collected using a Hahn echo experiment28. Echo times ranging from 0 to 1 s were used for T2 decay curves. Samples were spun at a MAS frequency of 4 kHz. A recycle delay of 5 s was used. Due to the freezing hysteresis observed with water in contact with the polymers being studied, variable temperature experiments were only conducted by decreasing the sample temperature. The first temperature point was taken at 276 K. The temperature was then lowered to 268 K for three hours before obtaining more spectra. Spectra were then obtained every three hours, with a 5 K drop for each successive temperature point, until there was no longer any NMR detectable liquid water.
Freezing Delay and Differential Scanning Calorimetry (DSC) Measurement
To observe the influence of the PDMS-PEG copolymer on heterogeneous surface ice nucleation, a water drop of 300 μL was placed on a silicon substrate that was coated with a 20 μm thick PDMS elastomer film containing 1 wt% DBE-224 PDMS-PEG copolymer. The silicon substrate was placed on a Peltier cooling plate at −15 °C. A water drop of the same volume on a silicon substrate coated with a 20 μm thick PDMS elastomer film placed on the same Peltier cooling plate was used as the control sample. The freezing process was recorded with a digital camera.
DSC was performed using a DSC Q200 (TA Instruments). An accurately weighed (10 mg) water microdrop was placed into an aluminum cup coated with a thin PDMS elastomer film or a thin PDMS elastomer film containing 1 wt% DBE-224 PDMS-PEG copolymer and subsequently sealed. An empty cup was used as reference. The experiment consisted of two runs: the first cooling step from 30 to −60 °C to freeze the water and the second step from −60 to 30 °C to melt the ice. The experiments were run at a scanning rate of 3 K/min.
RESULTS AND DISCUSSION
Water contact angles and ice adhesion measurements (–15 °C, 0.1 mm/s probe velocity) for 11 tested surfaces are summarized in Table 1. We note that we prepared our samples by spin coating method, so our samples have rather smooth surfaces with their root-mean-square roughness in the range of a few nanometers except for the PDMS+PDMS-PEG surfaces which have slight larger root-mean-square surface roughness of ~40 nm. The small variance in surface roughness has negligible effect on the water contact angles (The Wenzel roughness is smaller than 1.1 for all surfaces). How the roughness influences the ice adhesion strength will be discussed later. Other than thermal melting of ice from the surfaces, removing ice from a surface is essentially a fracture problem. Using the same ice adhesion test apparatus as in Meuler et al.3 as shown in Figure 1, we push the frozen sample of area off a surface by a linear stage and record the maximum force for fracturing the ice. The ice adhesion strength as measured by the maximum force depends on how the crack propagates–the modes of cracking (Three modes of fracture are depicted in Figure S1). In our experimental setup, the fracture occurs in a combination of Mode I crack (a tensile stress normal to the plane of the crack) and Mode II crack (a shear stress acting parallel to the plane of the crack and perpendicular to the crack front). The ratio depends on the distance “H“ between the applied force and the ice-substrate interface, as illustrated in Figure 1. In the current tests, we maintain a small and constant In this small gap limit, the fracture is primarily Mode II crack. The average strength of ice adhesion is defined as the maximum fracture force divided by the ice-substrate contact area. Notably, a PDMS elastomer film containing 1 wt% PDMS-PEG copolymer (CMS-626, Gelest Inc.) shows a remarkably low value of the ice adhesion strength, which is even lower than that on the PEMA/POSS surface, i.e., the lowest ice adhesion value previously achieved on smooth, hydrophobic solid surfaces, although the PDMS+PDMS-PEG surface has a much lower water receding angle. We note that we allowed our samples to freeze for at least 3 h before performing the ice adhesion measurements. In experiments we tested ice adhesion strength for the same surfaces after freezing time in the range of 3–8 h and no significant difference in the value of ice adhesion strength was observed. However, whether the ice adhesion strength will change over even longer freezing time (e. g. a day or longer) remains to be tested.
We also note that incorporating 1 wt% PDMS-PEG copolymer (CMS-626, Gelest Inc.) in the PDMS elastomer film can significantly reduce the ice adhesion strength value from 317±16 kPa on a PDMS elastomer film to 117±7 kPa. The active component that leads to low ice adhesion strength is the PDMS-PEG copolymer. The structure of this copolymer is shown in Scheme 1. To examine the influence of the molecular weight and composition, as well as the loading ratio of the active component on the ice adhesion strength, we have also obtained three other PDMS-PEG copolymers from Gelest Inc. They all have the same general molecular structure as depicted in Scheme 1. Their molecular weight and composition, as well as their physical properties, are listed in Table 2 together with the CMS-626 PDMS-PEG copolymer. We blended each of these PDMS-PEG copolymers into PDMS elastomer films separately at a weight ratio of 1 % or 5 %. The measured water contact angles and the ice adhesion strengths on these surfaces are listed in Table 3. All of these surfaces show comparable or even lower ice adhesion strength values compared to the PEMA/POSS surface. Increasing the PDMS-PEG copolymer loading ratio in PDMS elastomer films from 1 wt% to 5 wt% does not result in a significant change in the ice adhesion strength except for the CMS-221 PDMS-PEG copolymer that has a relatively low molecular weight and a low non-siloxane composition. The two PDMS-PEG molecules containing lower non-siloxane (or PEG) composition are water insoluble, and show lower ice adhesion strength regardless of their molecular weight. The PDMS elastomer film containing 1 wt% DBE-224 PDMS-PEG copolymer shows an ice adhesion strength value among the lowest values we have achieved for all the systems tested. PDMS elastomer films containing 5% CMS-221 or DBE-224 PDMS-PEG copolymer showed even lower ice adhesion strength values. However, these PDMS films are less mechanically robust. Without apparently compromising the mechanical properties of the PDMS elastomer, the DBE-224 PDMS-PEG copolymer is selected as the most effective polymer to use in the subsequent characterizations.
Scheme 1.

The molecular structure of PDMS-PEG copolymers
Table 2.
Molecular Weight and Composition of PDMS-PEG Copolymers in Use
| Product code |
Wt% Non-Siloxane |
Glycol capping (R) |
Viscosity cSt. |
Molecular Weight |
Water Solubility |
|---|---|---|---|---|---|
| CMS-626* | 65 | OH | 550–650 | 4,500–5,500 | Yes |
| DBP-732** | 65–70 | OMe | 1800 | 20,000 | Yes |
| CMS-221* | 20–25 | OH | 125–150 | 4,000 | No |
| DBE-224*** | 25 | OMe | 400 | 10,000 | No |
100% EG,
EG/PG (40/60),
~10 mole% EG substituted
Compared to the common practice of using deicing fluids to remove ice from surfaces, one unique advantage of the passive coating approach is the reduction/elimination of reapplications during multiple icing events. Therefore, it is also critical for such passive coatings to have robust anti-icing performance during subsequent deicing tests. Ice adhesion strengths for different surfaces in 3 repeated icing tests are shown in Figure 2. For all the surfaces tested, there were no significant changes in the ice adhesion strength after three separate trials. To test the long-term durability of our icephobic surfaces, many more icing/deicing cycles are still called for14–16, 29. Lower ice adhesion (~50–110 kPa) was achieved on PDMS+PDMS-PEG coatings (PDMS+CMS-626 and PDMS+DBE-224) than on the PEMA-POSS coating (~150 kPa), on which the lowest ice adhesion for smooth, hydrophobic solid surfaces has been reported previously. Icing, snowing, and raining conditions often occur simultaneously, and are known collectively as a “wintry mix”. Rainwater can extract and remove water-soluble PDMS-PEG copolymers from the coating, leading to deterioration in anti-icing properties. Indeed, the water washed PDMS+CMS-626 coating had a slightly higher ice adhesion strength than the non-washed counterparts, indicating some removal of the active component (CMS-626) due to its water solubility. No apparent change in ice adhesion is observed on the PDMS+DBE-224 coating after washing thanks to the water insolubility of DBE-224, which may help eliminate the necessity of reapplications.
Figure 2.

The ice adhesion strength for different surfaces in 3 repeated icing tests. For all the surfaces tested, there are no significant changes in the ice adhesion strength during the 3 tests. Lower ice adhesion was achieved on PDMS+PDMS-PEG coatings (PDMS+626 and PDMS+224) than on PEMA-POSS coating. The washed PDMS+626 coating has slightly higher ice adhesion strength than the non-washed counterparts. No apparent change in ice adhesion is observed on the PDMS+224 coating after washing.
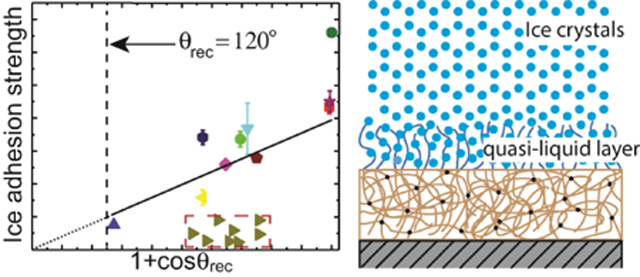
Next, following Meuler et al.3, we plot the average strength of ice adhesion as a function of the relevant water contact angle parameter that scales with the practical work of adhesion for the liquid water through . All the data points listed in Table 1 and Table 3 are shown in Figure 3. Noticeably, except for the guar gum coated surface, the rest of the data points fall into two groups. One group follows a linear trend line through the origin as predicted by the empirical relationship proposed by Meuler et al. Fitting these points gives rise to a linear relationship . This value of the prefactor is close (within 15% difference) to the value Meuler et al. obtained. Another group of data points show that the ice adhesion strengths on PDMS+PDMS-PEG coated substrates are significantly lower than what is predicted by the linear trend line, as highlighted by the red, dashed rectangle. This dramatic reduction indicates that different mechanisms of ice adhesion are coming into play and suggests that there is a previously unexplored region of parameter design space for achieving low ice adhesion surfaces. The ice adhesion strength on the guar gum coated substrate is much higher than predicted by the empirical linear relationship. This deviation can be explained by the different failure mechanism that we observed. The frozen cuvettes tend to fail cohesively on the guar gum coated substrate and leave small shards of ice remaining on the substrate, instead of being detached completely at the ice-substrate interface.
Figure 3.

The ice adhesion strength is plotted as a function of the water contact angle parameter that scales with the practical work of adhesion for water for 17 different polymer coatings and a bare clean silicon wafer. The empirical relationship between the ice adhesion strength and the practical work of adhesion for water proposed by Meuler et al. can accurately capture the test results, except for two sets of data points that are apparently off the trend line: the guar gum coated substrate and the PDMS+PDMS-PEG coated substrates (8 different substrates represented by right triangular symbols.).
Because of the wide range of possible test conditions and different definitions of icephobicity, surface characteristics can have contradictory effects on anti-icing performance18. The ice adhesion strength can be influenced by surface elasticity (soft vs. hard), surface topography (smooth vs. rough), and liquid extent (dry vs. wet). All the surface coatings we have investigated are hard coatings (in a glassy state with a Young’s modulus on the order of 1 GPa) except for the PDMS or PDMS+PDMS-PEG coatings, which are soft elastomers with a Young’s modulus on the order of 1 MPa in freezing conditions thanks to the low glass transition temperature of PDMS (~−125 °C). If we use low ice adhesion strength as the criterion to define surface icephobicity, we can consider surface characteristics that may affect the adhesion strength. According to the Griffith criterion for fracture, the fracture stress () is proportional to the square root of the composite modulus at the interface (): 30. Therefore, lower modulus substrates give rise to lower ice adhesion strengths. As shown in Figure 3, soft substrates including the PDMS elastomer and PDMS+PDMS-PEG elastomer coatings have lower ice adhesion strengths than predicted by the linear trend described by Meuler et al. for smooth hydrophobic surfaces. Low-modulus PDMS elastomer coatings for low ice adhesion surfaces have been reported previously10, 31_ENREF_10. Here we show that adding a small amount of PDMS-PEG copolymers into the PDMS matrix can further significantly reduce the ice adhesion strength to values even lower than that of the hydrophobic PEMA/POSS surface. Another factor that influences the ice adhesion strength is the surface roughness. AFM measurements show that the Sylgard 184 PDMS (10:1 mixing ratio) film, prepared by spin-coating at 2000 rpm and subsequent fully-curing at 80 °C, has a root-mean-square surface roughness of ~4 nm. Blending 1 wt% PDMS-PEG copolymer (DBE-224) in the Sylgard 184 PDMS and preparing the film with the same procedure, increases the root-mean-square surface roughness to ~40 nm (Figure S2a) which is due to the phase separation between the PDMS matrix and the PDMS-PEG copolymers. Depending on which of the two effects described below is dominant, the increased surface roughness can either increase or decrease ice adhesion strength. It may contribute to the mechanical interlocking between the ice and the substrate and thereby increase the ice adhesion strength32. On the other hand, it can seed interfacial adhesion defects and therefore help to reduce the fracture stress33. The third surface attribute that influences the ice adhesion strength is the extent of the coating liquid character (often referred to as “dry” vs. “wet” coating). We measure low ice adhesion strength on two very different types of surfaces: the PEMA/POSS surface and the PDMS+PDMS-PEG surfaces. The hydrophobic PEMA/POSS surface stays dry during the icing event, whereas the PDMS+PDMS-PEG surfaces may become hydrated in the icing/deicing experiment due to the strong interaction between PEG and water molecules, implying markedly different icephobic mechanisms.
To determine the mechanisms of low ice adhesion strength on the two different types of surfaces, it is important to interrogate how water molecules interact with these two types of surfaces. For the hydrophobic surfaces, both experiments and simulations have identified the existence of a density-depleted region between the water and the hydrophobic surfaces34–36. The thickness of the depletion layer (, as indicated in Figure 4a) grows as the hydrophobicity of the surface increases, represented by the increasing water contact angle. On a hydrophobic surface, the interaction between water/ice and the substrate will be dominated by van der Waal’s forces32. The adhesion strength is given by
| (1) |
where A~10−19 J is the Hamaker constant and is the thickness (typically in the range of 0.1–1 nm) of the depletion layer37. As surface hydrophobicity increases, increases and decreases. This molecular picture agrees qualitatively with the empirical relationship () proposed by Meuler et al based on direct measurements of ice adhesion strength on different surfaces. The Fluoro-POSS/PEMA surface is the surface known to have the highest intrinsic hydropbobicity as measured by a value of the receding contact angle of water . Therefore, the depletion layer thickness between water and such a surface will be the largest (on the order of 1 nm), which gives rise to the lowest ice adhesion values on smooth, hydrophobic solid surfaces. The estimated adhesion strength from equation (1) gives , which is much higher than the measured ice adhesion strength. This is because equation (1) does not take into account the defect size at the interface in the experiments, which can significantly lower the interfacial fracture strength.
Figure 4.

Proposed mechanisms responsible for low ice adhesion on two distinct types of surfaces: (a) the increased thickness (D) of the water depletion layer leading to reduced van der Waal’s interaction between the ice and the hydrophobic surface; (b) non-frozen quasi-liquid-layer (QLL) on a PDMS+PDMS-PEG surface lubricating the contact and reducing the adhesion strength between ice and the solid surface.
For the PDMS+PDMS-PEG surfaces, 1 wt% PDMS-PEG copolymer is blended into silicone elastomer precursor (Sylgard 184 PDMS 10: 1 by weight mixing ratio) prior to spin coating on a silicon wafer substrate and subsequent curing to form an elastomer film. In air, PDMS components saturate at the surface of the elastomer film to lower the surface energy. However, when in contact with water, the extremely low glass transition temperature of PDMS () allows for the rearrangement of the surface molecules38–39. PEG chains will preferentially segregate to the interface to interact with water molecules and lower the interfacial energy of the total system38. PEG molecules are known to bind strongly with water molecules through hydrogen bonding40–41. As shown in Figure 4b, the hydrogen bonded water molecules can form a thin hydration layer (or “quasi-liquid like” (QLL) layer) at the interface, which serves as a self-lubricating layer to reduce the ice adhesion on the coated substrates.
Strong hydrogen-bonding interactions between water molecules and PEG chains suppress the formation and growth of ice crystals within the hydration layer. The free energy of water freezing point depression is given by the Gibbs–Helmholtz equation
| (2) |
where is the Gibbs free energy, is the molar fusion energy of ice melting, and is the suppressed freezing point42. The freezing of water in the hydration layer is penalized by the favorable energy of mixing between PEG chains and water molecules. PEG chains strongly hydrogen bond with water molecules. The energy of mixing for this strongly associating system cannot be estimated by the classical Flory-Huggins theory. One of the limitations of the classic Flory-Huggins theory is that it assumes there are no energetically preferred arrangements of polymer segments and solvent molecules in the solution. For the PEG-water system, strong hydrogen bonding interaction significantly reduces the configurational entropy of bonded water molecules. Therefore extended Flory-Huggins models have been formulated43–44. One simple approach is to consider an effective Flory-Huggins parameter that depends on the temperature and the polymer volume fraction43, so the free energy of mixing can still be calculated following the classical Flory-Huggins model
| (3) |
where is the free energy of mixing, is the ideal gas constant, is the temperature, is the polymer volume fraction in the hydration layer, is the degree of polymerization, and are the molar volume of water and the polymer repeating units, respectively. Balancing the freezing point depression energy with the energy of demixing
| (4) |
and assuming that 45*, , and 44, 46(according to the experimental conditions and literature values for water-PEG system), we can estimate a water freezing point depression of . We note that this estimate of a 23 K freezing point depression is based on the assumption of a constant polymer volume fraction in the hydration layer. In actuality, the real hydration layer comprises of multiple layers40, 44. Water molecules in the loosely bounded outer layer freeze first. So the polymer volume fraction in the hydration layer increases as the temperature is lowered during the freezing process. As we show below, this gradual freezing process of the hydration layer can be observed from the relative 1H peak intensity measured in our nuclear magnetic resonance (NMR) experiments.
To confirm the existence of the non-frozen quasi-liquid-layer (QLL) at the interface between the bulk water and the PDMS+PDMS-PEG coating, we turned to the use of solid-state NMR spectroscopy. NMR is an ideal candidate for identifying trace amounts of non-frozen water because solid ice is invisible to conventional 1H NMR experiments due to its extremely long T1 and short T2 relaxation times, while liquid water produces an easily detectable 1H signal47–48. 1H spectra were first measured for the PDMS+water and PDMS+PDMS-PEG+water samples at 276 K to characterize bulk water properties before the start of the freezing process. Both samples show water 1H chemical shifts of 5.03 ppm, with a linewidth of 0.4 ppm and with similar lineshapes. After decreasing the temperature to 268 K and stabilizing for three hours, the water 1H signal intensity of both samples decreased to only ~1% of the intensities at 276 K (Figure 5).
Figure 5.

1H spectra of PDMS+water (a) and PDMS+PDMS-PEG+water (b) samples. The water and PDMS peaks are observed at 5 and 0 ppm, respectively. Approximately 99% of the liquid water was frozen into ice after holding the sample at 268 K for three hours.
Beginning at 268 K in the temperature decremented experiments, sharp features with linewidths of 5 Hz are observed in the water peak that were not present in the 276 K spectra (Figure 6). We attribute these features to water trapped in microenvironments at the polymer-water interface with different hydrogen-bonding networks that are unable to exchange with other microenvironments. In contrast, the 276 K spectra show a broad peak composed of overlapping peaks with linewidths of 10–30 Hz, indicating fast exchange of all water above the bulk water freezing point. Solid-state NMR studies of the interaction of water with proteins, phospholipid membranes, and plant cell wall polysaccharides have been previously reported 49–52. Most of these experiments relied on magnetization transfer from water protons to protons in the biomolecules and then to 13C enriched nuclei. However, these techniques are not applicable to the unlabeled polymers studied here due to the low natural abundance of 13C. In these previous studies of proteins and polysaccharides, when the sample temperature was lowered below the bulk water freezing point, no qualitative differences in the water lineshapes was observed, in contrast to the water observed in the current polymeric samples. In our experience, linewidths narrower than 10 Hz were not observed in any of the previous studies and usually only up to two water peaks were observed, indicating that there are no distinct microenvironments free of exchange on the relevant NMR time scales.
Figure 6.

1H spectra of PDMS+water (a) and PDMS+PDMS-PEG+water (b) collected between 276 K and 253 K. The dominant water peaks used for analysis of relaxation times are labeled A (a) and B (b). The PEG hydroxyl peak can be seen in (b) after magnifying the spectra greater than 500 fold. The peak labelled C in (b) is due to exchange of protons between water that is hydrogen bonded to PEG and the PEG hydroxyl on a time scale that is faster than the NMR experiments conducted.
The water 1H chemical shifts of the PDMS polymers increase with decreasing temperature. This trend is consistent with previous studies of supercooled water 53, and can be attributed to stronger hydrogen bonding at lower temperatures, deshielding the water protons. Angell et al. observed an average 1H chemical shift change of −0.01 ppm/K in the range from 273 to 263 K, which is identical to our observed chemical shift changes for the dominant water peaks in the two samples between 268 and 253 K. Below 263 K, the 1H chemical shift change with temperature became more nonlinear, with a negative slope increasing in magnitude. We used the most intense water peaks for the PDMS+water and the PDMS+PDMS-PEG+water samples, labelled A and B (Figure 6), for comparing the T2 relaxation data. Assignment of these two peaks throughout the temperature range can be slightly ambiguous due to the clustering of multiple sharp peaks. However, neighboring peaks have similar 1H T2 relaxation times within experimental uncertainty. Thus, assignment ambiguity does not affect the conclusions drawn from the relaxation data.
There are two sets of peaks in the PDMS+PDMS-PEG+water spectra (Figure 6b) that are distinctly absent from the PDMS+water spectra (Figure 6a). The first is the PEG hydroxyl peak at 4.5 ppm54. This peak does not become visible until the spectrum is magnified 500 fold because PEG only makes up 0.25% of the total mass of the polymer in this sample. The second set of peaks that are present in the PDMS+PDMS-PEG+water spectra but not the PDMS+water spectra are the peaks ~0.2 ppm upfield of the main water peaks. We label the dominant peak in this set peak C (Figure 6b), and measured its T2 relaxation time. We attribute these upfield peaks to water protons that exchange with the PEG hydroxyl protons on timescales faster than the 1H chemical shift differences between water and PEG, which is approximately 120 s-1. The lower temperature sensitivity of the peak C 1H chemical shift compared to peaks A and B is consistent with previous observations that strong hydrogen bonds have a smaller temperature sensitivity to chemical shift 55. This observations confirms the existence of non-frozen water even at −20 °C, which serves as a self-lubricating interfacial layer, and is responsible for the low ice adhesion strength on the PDMS-PEG surfaces. We note that as the temperature is lowered below 263 K, the signal intensity of the non-frozen water (indicating the amount of non-frozen water) quickly decreases. At a critical degree of subcooling, the non-frozen water will significantly decrease and may completely freeze eventually, which is responsible for the sudden steep increase in the ice adhesion strength observed on such types of self-lubricating surfaces26.
The freezing point depression arises primarily from the colligative behavior of PEG chains in water. The unique hydrogen-bonding associations between PEG and water molecules lead to highly ordered water molecules in the hydration layer40. The structured water molecules in the quasi-liquid layer have a much lower configurational entropy compared to bulk water56, which leads to a tremendous increase in the water viscosity. Experimentally, it has been observed that the local viscosity of water hydrogen bonded with PEG is six orders of magnitude larger than that of bulk water57. In our current system, we estimate the viscosity of non-frozen water by performing 1H T2 relaxation measurements. The T2 relaxation decays, shown in Figure 7, cannot be fit to a single-exponential or a biexponential function for all the peaks across the temperature range of 268 K to 253 K. We attribute this multiexponential character to complex interactions of water trapped in microenvironments during the freezing process as well as chemical exchange of water with PEG. To allow uniform fitting of all the peaks at all measured temperatures, we thus used a stretched exponential function,
| (5) |
which allows us to use a single time constant to characterize each decay curve. The exponential β denotes the extent of T2 distribution: β = 1 indicates a single-exponential fit, while smaller β values reflect broader distributions of time constants (See Table S1 for tabulated values of T2 and β).
Figure 7.

T2 relaxation decay curves corresponding to the peaks labelled A (a), B (b), and C (c) from Figure 6. Stretched exponential fits are plotted for the three temperatures shown: 268 K, 263 K, and 258 K. Peak C (c) has the strongest temperature dependence on temperature, as can be seen by the much faster signal decay at 258 K compared to 263 K or 268 K.
1H T2 relaxation times can be related to molecular motions and viscosity using theories developed by Bloembergen et al.58 and Debye59. In this Bloembergen-Purcell-Pound (BPP) theory, T2 relaxation times are related to rotational correlation times according to:
| (6) |
where γ is the 1H gyromagnetic ratio, ħ is the reduced Planck constant, r is the internuclear distance between interacting dipoles, τc is the rotational correlation time, and is the 1H Larmor frequency at the given magnetic field strength. At a 400 MHz Larmor frequency, due to the correlation times of 10–100 ps for water below its freezing point, the second and third terms in the above equation can be neglected, leaving the following proportionality:
| (7) |
Using a modified version of the correlation time in Debye’s theory of dielectric dispersion in polar liquids58–59, the rotational correlation time can be related to viscosity by the expression:
| (8) |
where η is the liquid viscosity, kB is the Boltzmann constant, and T is absolute temperature.
Equations (7) and (8) indicate that 1H T2 relaxation times are proportional to temperature and inversely proportional to viscosity. As expected, the measured water 1H T2 times decreased monotonically for all three peaks with decreasing temperature, indicating a slowing down of molecular motions and increased rotational correlation times. Water populations existing at the same temperature but with different T2 relaxation times have different viscosities. We calculated τc and η using Eq. 6 and Eq. 8, assuming that the 1H-1H internuclear distance in liquid water is 1.5 Å (Table S2). At 268 K, the calculated viscosity associated with Peak C, 0.034 Pa·s, is twice that of Peak A, 0.013 Pa·s, and about 1.5 times that of Peak B, 0.024 Pa·s. Moreover, the water viscosity associated with Peak C is more sensitive to temperature changes than the peaks associated with other local water microenvironments: at 258 K, the water viscosity associated with Peak C is 0.64 Pa·s, which is more than an order of magnitude larger than that of Peaks A or B, 0.022 and 0.077 Pa·s, respectively, at the same temperature.
Supercooled water has previously been observed to be a fragile liquid that exhibits non-Arrhenius behavior. A thermodynamic analysis of this behavior is provided by Ito et al.60. In our PDMS + PDMS-PEG system, plotting ln(T2−1) versus 1000/T (Figure 8) shows non-Arrhenius behavior and does not fit the simple model:
| (9) |
where τc,0 is the rotational correlation time at a reference temperature T0, Ea is the activation energy of molecular rotational motion, and R is the ideal gas constant. However, useful information can still be extracted from this plot, even if an explicit expression for the activation energy cannot. Peaks A and B have a similar temperature dependence of ln(T2−1), suggesting that the energy barrier for motion in these water populations is similar. The most striking feature of this plot is the larger slope observed for peak C compared to peaks A and B. This strong temperature dependence for ln(T2−1) indicates a higher barrier to motion for water molecules described by peak C. We attribute this large motional barrier to a highly viscous quasi-liquid layer of water that is strongly hydrogen-bonded to PEG at the interface between ice and polymer. Although it is difficult to comment on the nonlinearity with only three data points, Peak C appears to have a much more significant non-Arrhenius behavior than peaks A or B, further suggesting that this population of water is in a viscous environment with a stronger hydrogen bonding network than we observe for peaks A or B. It has previously been demonstrated that the ice nucleation rate is inversely proportional to the viscosity of water ()61. The much higher local viscosity associated with Peak C will significantly lower the freezing rate at the water/PEG-functionalized interface.
Figure 8.

Natural log of T2−1 versus inverse temperature plot for Peaks A, B, and C. The steeper slope for Peak C indicates a higher activation energy for molecular motion compared to peaks A or B.
The water interaction with the PDMS-PEG polymer is distinct from the water interaction with antifreeze proteins. Siemer et al.62 used relaxation and 2D correlation solid-state NMR experiments to investigate the water structuring mechanism of antifreeze proteins. Utilizing 13C indirect detection of 1H T1 relaxation times, 1H cross saturation experiments, and 2D 13C-1H correlation experiments, they showed that the antifreeze protein (AFP III) directly interacts with both ice and water, while the control protein, ubiquitin, is surrounded by a liquid hydration shell and is thus shielded from ice. Thus, AFP III binds to small ice crystals to retard nucleation and hence inhibit bulk ice formation and deposition. This mechanistic interaction differs from the mechanism of the icephobic PDMS-PEG polymers found here. Instead of retarding bulk-ice formation, the PDMS-PEG coating maintains a quasi-liquid water layer between the ice and polymer so that any bulk ice that forms does not strongly adhere to the polymer surface and can be easily removed by shear.
In addition to low ice adhesion strength, delayed ice nucleation can be another criterion for determining the icephobicity of a surface18. Ice nucleation is delayed on the PDMS-PEG functionalized surface in comparison to that on the PDMS functionalized surface. As shown in supplementary Movie 1 (A side view image is shown in Figure 9a), when a water drop in contact with a coated silicon substrate is cooled down to , heterogeneous ice nucleation starts at the liquid-solid interface and the crystallization front propagates into the water drop as heat is transferred from the water droplet to the substrate. The unfrozen part of the drop remains a spherical cap as dictated by the liquid-vapor surface tension. The drop expands in the vertical direction as the ice forms due to volumetric expansion, which, in combination with the influence of surface tension, leads to a freezing singularity at the tip63. There have been debates in experiments about ice nucleation at the solid-liquid-vapor three-phase contact line or at the solid-liquid interface56, 64–65. In our experiment, change in turbidity is first observed at the three-phase contact line during freezing (Movie 1), which may indicate ice nucleation at the contact line. Incorporation of 1 wt% PDMS-PEG copolymer in the PDMS coating doesn’t seem to change this behavior. To examine the exact location of ice nucleation, more careful experiment using properly positioned high-speed camera is required. Ice nucleation from a bulk water drop (with volume of 300 μL) is delayed for 2–3 min on the PDMS surface that contains 1 wt% PDMS-PEG copolymer in comparison to that on the PDMS functionalized surface. Such delayed ice nucleation in water microdrops can also be observed and quantified using differential scanning calorimetry (DSC) measurements and the results are shown in Figure 9b. The numbered arrows indicate the cooling and subsequent heating runs at a scanning rate of 3 K/min. On the PDMS surface, as the sample is cooled below 0 °C, the water is supercooled until ice formation at −19.9 °C which releases a large amount of latent heat of freezing and causes rising of the sample temperature. Then the sample is cooled again by the DSC instrument to −60 °C. In the subsequent heating run, the formed ice melts at 0 °C. On the PDMS surface that contains 1 wt% PDMS-PEG copolymer, the ice formation is delayed to −23.0 °C, whereas the ice melts at almost the same temperature (0 °C). Delayed ice nucleation provides an additional benefit in developing icephobic coatings if supercooled water can be shed from the surface prior to freezing.
Figure 9.

Delayed ice nucleation on the PDMS+PDMS-PEG functionalized surface (a) in large water droplets in contact with a cold substrate () and (b) in micro-water droplets by differential scanning calorimetry (DSC).
Considering that only 1 wt% of PDMS-PEG (with the PEG component being 25 wt% within this copolymer) is embedded in the PDMS elastomer film, it is quite remarkable that such a small amount of PEG (0.25 wt% of the total sample mass) provides such a significant delay in the ice nucleation on the surface. Several factors can contribute to this delay in the nucleation kinetics. First, the increased surface roughness can play a role66. Compared to the PDMS elastomer that has a root-mean-square roughness of nm, AFM measurements show that blending 1 wt% PDMS-PEG copolymer into the PDMS elastomer increases the dry surface roughness to nm (Figure S2a). When in contact with water, the mobile chains at the PDMS+PDMS-PEG surface can also rearrange their molecular configuration. However, a tapping mode AFM in water experiment found that the surface roughness of the wet sample remains the same as the dry surface roughness ( nm in Figure S2b). Classic nucleation theory shows that at a given temperature, an ice nucleus must reach a critical size , for freezing to start, where and represent respectively the ice-water interfacial energy and the free energy associated with the water freezing point depression per unit volume. At the experimental temperature , and , where is the volumetric fusion energy of ice melting67, and the ice melting point is . Using these values, the size of a critical ice nucleus is expected to be . As shown in Figure S2b, the feature size on our PDMS-PEG surface is on the micron scale and the mean square roughness is about 40 nm. Therefore we expect the local surface mean radius of curvature to be much larger than . For such types of surface, Poulikakos et al. have shown that the nucleation temperature and nucleation delay are relatively insensitive to the surface roughness66, 68. Therefore, changes in surface roughness due to the incorporation of PDMS-PEG are unlikely to result in the observed delayed onset of icing. Second, the quasi-liquid layer also has reduced heat conductivity so the rate of heat transfer from the bulk water to the substrate is also lowered69. However, considering the nanometer scale thickness of the quasi-liquid layer, the freezing-delay effect due to the lowered heat conductivity is presumably small. Third, as discussed in the NMR T2 analysis, the increased water viscosity in the quasi-liquid layer can significantly reduce the ice nucleation rate56–57, 61. When viewed holistically, the major factor for freezing-delay is expected to be the increased viscosity of the water microenvironments that we have measured in the quasi-liquid layer at the interface.
While PDMS elastomer coatings incorporating PDMS-PEG copolymers show low ice adhesion, PDMS or silicone elastomers have not typically been used as coating materials applied to airplanes, wind turbines, power lines, vehicles, or construction structures due to their poor mechanical properties and abrasion resistance. To explore the broad utility and effectiveness of our current strategy, we have also blended 1 wt% DBE-224 PDMS-PEG copolymer into a range of other matrix materials including epoxy resin, polyurethane, or fluoro-polyurethane, which are typically used as hard coatings, top coatings, or anti-reflection coatings in the aforementioned applications. Water contact angles and ice adhesion strengths are measured using the same experimental procedures as described previously, and the results are reported in Table 4. Blending only 1wt% DBE-224 into each continuous matrix reduces the ice adhesion strength to about 1/3 of the ice adhesion strength observed on each type of coating. It is expected that the incorporation of 1 wt% DBE-224 won’t appreciably change the mechanical properties of these coatings. Therefore, the interfacial mechanism for reducing ice adhesion that we have elucidated in this work can have broad applications in coating airplane wings, wind turbine blades, power lines, and vehicles.
Table 4.
Measured Water Contact Angles and Average Shear Strengths of Ice Adhesion for Commercial Coatings and Commercial Coatings Containing 1 wt% PDMS-PEG Copolymers
| Surfaces | Advancing Angle (θadv) |
Receding Angle (θrec) |
Ice adhesion Strength (kPa) |
|---|---|---|---|
| Epoxy | 75±1° | 44±1° | 404±48 |
| Epoxy + 1 wt% DBE-224 |
101±1° | 73±2° | 153±9 |
| Polyurethane | 83±1° | 46±1° | 582±93 |
| Polyurethane + 1 wt% DBE-224 |
83±1° | 34±1° | 185±4 |
| Fluoro-Polyurethane | 96±1° | 69±1° | 686±203 |
| Fluoro-Polyurethane + 1 wt% DBE-224 |
99±1° | 52±2° | 148±48 |
Conclusions
We have designed scalable, self-lubricating icephobic coatings by blending commercially available amphiphilic copolymers into a polymer coating matrix. Such coatings provide low ice adhesion strength values that are comparable to, or in some cases, much lower than what has previously been achieved using smooth hydrophobic solid surfaces. We have compared and contrasted the molecular mechanisms that are responsible for the low ice adhesion values observed on these two types of surfaces. For the hydrophobic surfaces, the increased thickness of the water depletion layer at the interface weakens the van der Waal’s interaction between the ice and the underlying substrate. The existence of such a water depletion layer at the interface has been confirmed previously both experimentally and by molecular dynamics simulations. By contrast, in the amphiphilic coatings, the addition of a small mass fraction of PDMS-PEG copolymer helps promote the retention of a viscous lubricating liquid-like layer at the interface. The surface-segregated PEG component can strongly hydrogen bond with water molecules. The resulting hydrogen-bonded water does not freeze even at substantial levels of subcooling, and therefore serves as a self-lubricating interfacial layer that helps to reduce the adhesion strength of ice to the surface. The existence of non-frozen water at the ice-solid interface is confirmed by solid-state nuclear magnetic resonance (NMR) spectroscopy. NMR T2 relaxation analysis allows us to quantify the high viscosity of the non-frozen water molecules hydrogen bonded to PEG, which is also expected to contribute to the delayed heterogeneous ice nucleation on our coatings–another attractive property for producing icephobic surfaces. Our method thus provides a passive anti-icing mechanism without the need for impregnating a porous matrix with liquid phases that can leach into the environment, causing water or land pollution. The low mass fraction of copolymer required (1 wt%) means that such coatings can be easily and inexpensively retrofitted to existing structures such as airplane wings and wind turbine blades by simply blending the active amphiphilic copolymers into the current protective coatings, conferring up to three fold reduction in ice adhesion for a 1 wt% addition.
Supplementary Material
Acknowledgement
Dayong Chen, Robert E. Cohen, and Gareth H. McKinley are financially supported by Army Research Office, Contract W911NF-13-D-0001. We thank Professor Michael F. Rubner and Dr. Srinivas Prasad Bengaluru Subramanyam for their helpful suggestions in the ice adhesion test. Martin D. Gelenter and Mei Hong are partially supported by NIH Grant GM066976.
Footnotes
Considering the similarity between surface grafted polymer chains in a good solvent and rearranged PEG component in the hydration layer of our sample, we assume the averaged polymer volume fraction in the hydration layer is similar to that () of surface grafted polymer chains in a good solvent.
References
- 1.Kreder MJ; Alvarenga J; Kim P; Aizenberg J, Design of anti-icing surfaces: smooth, textured or slippery? Nat. Rev. Mater 2016, 1, 15003. [Google Scholar]
- 2.Lv J; Song Y; Jiang L; Wang J, Bio-inspired strategies for anti-icing. ACS Nano 2014, 8, 3152–3169. [DOI] [PubMed] [Google Scholar]
- 3.Meuler AJ; Smith JD; Varanasi KK; Mabry JM; McKinley GH; Cohen RE, Relationships between water wettability and ice adhesion. ACS Appl. Mater. Interfaces 2010, 2, 3100–3110. [DOI] [PubMed] [Google Scholar]
- 4.Dalili N; Edrisy A; Carriveau R, A review of surface engineering issues critical to wind turbine performance. Renew. Sustainable Energy Rev 2009, 13, 428–438. [Google Scholar]
- 5.Sun X; Damle VG; Liu S; Rykaczewski K, Bioinspired Stimuli-Responsive and Antifreeze-Secreting Anti-Icing Coatings. Adv. Mater. Interfaces 2015, 2 1400479. [Google Scholar]
- 6.Ozbay S; Yuceel C; Erbil HY, Improved Icephobic Properties on Surfaces with a Hydrophilic Lubricating Liquid. ACS Appl. Mater. Interfaces 2015, 7, 22067–22077. [DOI] [PubMed] [Google Scholar]
- 7.Wilson PW; Lu W; Xu H; Kim P; Kreder MJ; Alvarenga J; Aizenberg J, Inhibition of ice nucleation by slippery liquid-infused porous surfaces (SLIPS). Phys. Chem. Chem. Phys 2013, 15, 581–585. [DOI] [PubMed] [Google Scholar]
- 8.Zhu L; Xue J; Wang Y; Chen Q; Ding J; Wang Q, Ice-phobic coatings based on silicon-oil-infused polydimethylsiloxane. ACS Appl. Mater. Interfaces 2013, 5, 4053–4062. [DOI] [PubMed] [Google Scholar]
- 9.Urata C; Dunderdale GJ; England MW; Hozumi A, Self-lubricating organogels (SLUGs) with exceptional syneresis-induced anti-sticking properties against viscous emulsions and ices. J. Mater. Chem. A 2015, 3, 12626–12630. [Google Scholar]
- 10.Golovin K; Kobaku SP; Lee DH; DiLoreto ET; Mabry JM; Tuteja A, Designing durable icephobic surfaces. Sci. Adv 2016, 2, e1501496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim P; Wong T-S; Alvarenga J; Kreder MJ; Adorno-Martinez WE; Aizenberg J, Liquid-infused nanostructured surfaces with extreme anti-ice and anti-frost performance. ACS Nano 2012, 6, 6569–6577. [DOI] [PubMed] [Google Scholar]
- 12.Subramanyam SB; Rykaczewski K; Varanasi KK, Ice adhesion on lubricant-impregnated textured surfaces. Langmuir 2013, 29, 13414–13418. [DOI] [PubMed] [Google Scholar]
- 13.Gao L; McCarthy TJ, Teflon is hydrophilic. Comments on definitions of hydrophobic, shear versus tensile hydrophobicity, and wettability characterization. Langmuir 2008, 24, 9183–9188. [DOI] [PubMed] [Google Scholar]
- 14.Lazauskas A; Guobienė A; Prosyčevas I; Baltrušaitis V; Grigaliūnas V; Narmontas P; Baltrusaitis J, Water droplet behavior on superhydrophobic SiO 2 nanocomposite films during icing/deicing cycles. Mater. Charact 2013, 82, 9–16. [Google Scholar]
- 15.Kulinich S; Farhadi S; Nose K; Du X, Superhydrophobic surfaces: are they really ice-repellent? Langmuir 2010, 27, 25–29. [DOI] [PubMed] [Google Scholar]
- 16.Boinovich LB; Emelyanenko AM; Ivanov VK; Pashinin AS, Durable icephobic coating for stainless steel. ACS Appl. Mater. Interfaces 2013, 5, 2549–2554. [DOI] [PubMed] [Google Scholar]
- 17.Varanasi KK; Deng T; Smith JD; Hsu M; Bhate N, Frost formation and ice adhesion on superhydrophobic surfaces. Appl. Phys. Lett 2010, 97, 234102. [Google Scholar]
- 18.Sojoudi H; Wang M; Boscher N; McKinley G; Gleason K, Durable and scalable icephobic surfaces: similarities and distinctions from superhydrophobic surfaces. Soft Matter 2016, 12, 1938–1963. [DOI] [PubMed] [Google Scholar]
- 19.Maitra T; Jung S; Giger ME; Kandrical V; Ruesch T; Poulikakos D, Superhydrophobicity vs. Ice Adhesion: The Quandary of Robust Icephobic Surface Design. Adv. Mater. Interfaces 2015, 21500330. [Google Scholar]
- 20.Rosenberg R, Why is ice slippery? Physics Today 2005, 58, 50–55. [Google Scholar]
- 21.Garnham CP; Campbell RL; Davies PL, Anchored clathrate waters bind antifreeze proteins to ice. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 7363–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esser-Kahn AP; Trang V; Francis MB, Incorporation of antifreeze proteins into polymer coatings using site-selective bioconjugation. J. Am. Chem. Soc 2010, 132, 13264–13269. [DOI] [PubMed] [Google Scholar]
- 23.Gwak Y; Park J.-i.; Kim M; Kim HS; Kwon MJ; Oh SJ; Kim Y-P; Jin E, Creating Anti-icing Surfaces via the Direct Immobilization of Antifreeze Proteins on Aluminum. Sci. Rep 2015, 5, 12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hederos M; Konradsson P; Borgh A; Liedberg B, Mimicking the properties of antifreeze glycoproteins: Synthesis and characterization of a model system for ice nucleation and antifreeze studies. J. Phys. Chem. B 2005, 109, 15849–15859. [DOI] [PubMed] [Google Scholar]
- 25.Dou R; Chen J; Zhang Y; Wang X; Cui D; Song Y; Jiang L; Wang J, Anti-icing coating with an aqueous lubricating layer. ACS Appl. Mater. Interfaces 2014, 6, 6998–7003. [DOI] [PubMed] [Google Scholar]
- 26.Chen J; Dou R; Cui D; Zhang Q; Zhang Y; Xu F; Zhou X; Wang J; Song Y; Jiang L, Robust prototypical anti-icing coatings with a self-lubricating liquid water layer between ice and substrate. ACS Appl. Mater. Interfaces 2013, 5, 4026–4030. [DOI] [PubMed] [Google Scholar]
- 27.Chernyy S; Järn M; Shimizu K; Swerin A; Pedersen SU; Daasbjerg K; Makkonen L; Claesson P; Iruthayaraj J, Superhydrophilic Polyelectrolyte Brush Layers with Imparted Anti-Icing Properties: Effect of Counter ions. ACS Appl. Mater. Interfaces 2014, 6, 6487–6496. [DOI] [PubMed] [Google Scholar]
- 28.Hahn EL, Spin Echoes. Phys. Rev 1950, 80, 580–594. [Google Scholar]
- 29.Kulinich S; Honda M; Zhu A; Rozhin A; Du X, The icephobic performance of alkyl-grafted aluminum surfaces. Soft Matter 2015, 11, 856–861. [DOI] [PubMed] [Google Scholar]
- 30.Griffith AA, The phenomena of rupture and flow in solids. Phil. Trans. R. Soc. A 1921, 221, 163–198. [Google Scholar]
- 31.Hoover K; Watson C; Putnam J; Dolan R; Bonarrigo B; Kurz P; Weisse M, Erosion resistant anti-icing coatings. Patent US20070254170 A1: 2007.
- 32.Menini R; Farzaneh M, Advanced icephobic coatings. J. Adhes. Sci. Technol 2011, 25, 971–992. [Google Scholar]
- 33.Nosonovsky M; Hejazi V, Why superhydrophobic surfaces are not always icephobic. ACS Nano 2012, 6, 8488–8491. [DOI] [PubMed] [Google Scholar]
- 34.Poynor A; Hong L; Robinson IK; Granick S; Zhang Z; Fenter PA, How water meets a hydrophobic surface. Phys. Rev. Lett 2006, 97, 266101. [DOI] [PubMed] [Google Scholar]
- 35.Chattopadhyay S; Uysal A; Stripe B; Ha Y.-g.; Marks TJ; Karapetrova EA; Dutta P, How water meets a very hydrophobic surface. Phys. Rev. Lett 2010, 105, 037803. [DOI] [PubMed] [Google Scholar]
- 36.Janecek J; Netz RR, Interfacial water at hydrophobic and hydrophilic surfaces: Depletion versus adsorption. Langmuir 2007, 23, 8417–8429. [DOI] [PubMed] [Google Scholar]
- 37.Israelachvili JN, Intermolecular and Surface Forces: Third Edition Academic press: 2011. [Google Scholar]
- 38.Synytska A; Biehlig E; Ionov L, Adaptive PEG–PDMS Brushes: Effect of Architecture on Adhesiveness in Air and under Water. Macromolecules 2014, 47, 8377–8385. [Google Scholar]
- 39.Zhang L; Zhang Z; Wang P, Smart surfaces with switchable superoleophilicity and superoleophobicity in aqueous media: toward controllable oil/water separation. NPG Asia Mater. 2012, 4 (2), e8. [Google Scholar]
- 40.Israelachvili J, The different faces of poly (ethylene glycol). Proc. Natl. Acad. Sci. U.S.A 1997, 94, 8378–8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H; Alcaraz ML; Rubner MF; Cohen RE, Zwitter-wettability and antifogging coatings with frost-resisting capabilities. ACS Nano 2013, 7, 2172–2185. [DOI] [PubMed] [Google Scholar]
- 42.Atkins P, Physical Chemistry. 6th. Oxford University Press: 1998. [Google Scholar]
- 43.Huang L; Nishinari K, Interaction between poly (ethylene glycol) and water as studied by differential scanning calorimetry. J. Polym. Sci. Part B Polym. Phys 2001, 39, 496–506. [Google Scholar]
- 44.Dormidontova EE, Role of competitive PEO-water and water-water hydrogen bonding in aqueous solution PEO behavior. Macromolecules 2002, 35, 987–1001. [Google Scholar]
- 45.Feng W; Nieh M-P; Zhu S; Harroun TA; Katsaras J; Brash JL, Characterization of protein resistant, grafted methacrylate polymer layers bearing oligo (ethylene glycol) and phosphorylcholine side chains by neutron reflectometry. Biointerphases 2007, 2, 34–43. [DOI] [PubMed] [Google Scholar]
- 46.Bae Y; Shim J; Soane D; Prausnitz J, Representation of vapor–liquid and liquid–liquid equilibria for binary systems containing polymers: applicability of an extended Flory–Huggins equation. J. Appl. Polym. Sci 1993, 47, 1193–1206. [Google Scholar]
- 47.Kuntz ID; Brassfield TS; Law GD; Purcell GV, Hydration of Macromolecules. Science 1969, 163, 1329–1331. [DOI] [PubMed] [Google Scholar]
- 48.Kvlividze V; Kiselev V; Kurzaev A; Ushakova L, The mobile water phase on ice surfaces. Surf. Sci 1974, 44, 60–68. [Google Scholar]
- 49.Li S; Su Y; Luo W; Hong M, Water−Protein Interactions of an Arginine-Rich Membrane Peptide in Lipid Bilayers Investigated by Solid-State Nuclear Magnetic Resonance Spectroscopy. J. Phys. Chem. B 2010, 114, 4063–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo W; Hong M, Conformational Changes of an Ion Channel Detected Through Water−Protein Interactions Using Solid-State NMR Spectroscopy. J. Am. Chem. Soc 2010, 132, 2378–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.White PB; Wang T; Park YB; Cosgrove DJ; Hong M, Water–Polysaccharide Interactions in the Primary Cell Wall of Arabidopsis thaliana from Polarization Transfer Solid-State NMR. J. Am. Chem. Soc 2014, 136, 10399–10409. [DOI] [PubMed] [Google Scholar]
- 52.Williams JK; Hong M, Probing membrane protein structure using water polarization transfer solid-state NMR. J. Magn. Reson 2014, 247, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Angell CA; Shuppert J; Tucker JC, Anomalous properties of supercooled water. Heat capacity, expansivity, and proton magnetic resonance chemical shift from 0 to −38%. J. Phys. Chem 1973, 77, 3092–3099. [Google Scholar]
- 54.Dust JM; Fang ZH; Harris JM, Proton NMR characterization of poly(ethylene glycols) and derivatives. Macromolecules 1990, 23, 3742–3746. [Google Scholar]
- 55.Baxter NJ; Williamson MP, Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 1997, 9, 359–369. [DOI] [PubMed] [Google Scholar]
- 56.Jung S; Dorrestijn M; Raps D; Das A; Megaridis CM; Poulikakos D, Are superhydrophobic surfaces best for icephobicity? Langmuir 2011, 27, 3059–3066. [DOI] [PubMed] [Google Scholar]
- 57.Kim HI; Kushmerick JG; Houston JE; Bunker BC, Viscous “interphase” water adjacent to oligo (ethylene glycol)-terminated monolayers. Langmuir 2003, 19, 9271–9275.. [Google Scholar]
- 58.Bloembergen N; Purcell EM; Pound RV, Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phy. Rev 1948, 73, 679–712. [Google Scholar]
- 59.Debye PJW, Polar Molecules. New York: The Chemical Catalog Company, Inc, 1929. [Google Scholar]
- 60.Ito K; Moynihan CT; Angell CA, Thermodynamic determination of fragility in liquids and a fragile-to-strong liquid transition in water. Nature 1999, 398, 492–495. [Google Scholar]
- 61.Li K; Xu S; Chen J; Zhang Q; Zhang Y; Cui D; Zhou X; Wang J; Song Y, Viscosity of interfacial water regulates ice nucleation. Appl. Phys. Lett 2014, 104, 101605. [Google Scholar]
- 62.Siemer AB; Huang K-Y; McDermott AE, Protein–ice interaction of an antifreeze protein observed with solid-state NMR. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 17580–17585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marin AG; Enriquez OR; Brunet P; Colinet P; Snoeijer JH, Universality of tip singularity formation in freezing water drops. Phys. Rev. Lett 2014, 113, 054301. [DOI] [PubMed] [Google Scholar]
- 64.Gurganus C; Kostinski AB; Shaw RA, High-speed imaging of freezing drops: still no preference for the contact line. J. Phys. Chem. C 2013, 117, 6195–6200. [Google Scholar]
- 65.Fu QT; Liu EJ; Wilson P; Chen Z, Ice nucleation behaviour on sol–gel coatings with different surface energy and roughness. Phys. Chem. Chem. Phys 2015, 17, 21492–21500. [DOI] [PubMed] [Google Scholar]
- 66.Eberle P; Tiwari MK; Maitra T; Poulikakos D, Rational nanostructuring of surfaces for extraordinary icephobicity. Nanoscale 2014, 6, 4874–4881. [DOI] [PubMed] [Google Scholar]
- 67.Lide DR, CRC Handbook of Chemistry and Physics, Internet Version 2007, (87th Edition), Taylor and Francis, Boca Raton, FL, 2007. [Google Scholar]
- 68.Schutzius TM; Jung S; Maitra T; Eberle P; Antonini C; Stamatopoulos C; Poulikakos D, Physics of icing and rational design of surfaces with extraordinary icephobicity. Langmuir 2014, 31, 4807–4821. [DOI] [PubMed] [Google Scholar]
- 69.Foster KR; Cheever E; Leonard JB; Blum FD, Transport properties of polymer solutions. A comparative approach. Biophys. J 1984, 45, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.