

In a morphogenic step that is exceedingly rare for nonenveloped viruses, immature rotavirus particles assemble in replication centers called viroplasms, and bud through cytoplasmic cellular membranes to acquire the outer capsid proteins for infectious particle assembly. Historically, the intracellular membranes used for particle budding were thought to be endoplasmic reticulum (ER) because the rotavirus nonstructural protein NSP4, which interacts with the immature particles to trigger budding, is synthesized as an ER transmembrane protein. This present study shows that NSP4 exits the ER in COPII vesicles and that the NSP4-containing COPII vesicles are hijacked by the cellular autophagy machinery, which mediates the trafficking of NSP4 to viroplasms. Changing the paradigm for rotavirus maturation, we propose that the cellular membranes required for immature rotavirus particle budding are not an extension of the ER but are COPII-derived autophagy isolation membranes.
KEYWORDS: ER exit sites, autophagy, rotavirus morphogenesis, viroplasms
ABSTRACT
Many viruses that replicate in the cytoplasm dramatically remodel and stimulate the accumulation of host cell membranes for efficient replication by poorly understood mechanisms. For rotavirus, a critical step in virion assembly requires the accumulation of membranes adjacent to virus replication centers called viroplasms. Early electron microscopy studies describe viroplasm-associated membranes as “swollen” endoplasmic reticulum (ER). We previously demonstrated that rotavirus infection initiates cellular autophagy and that membranes containing the autophagy marker protein LC3 and the rotavirus ER-synthesized transmembrane glycoprotein NSP4 traffic to viroplasms, suggesting that NSP4 must exit the ER. This study aimed to address the mechanism of NSP4 exit from the ER and determine whether the viroplasm-associated membranes are ER derived. We report that (i) NSP4 exits the ER in COPII vesicles, resulting in disrupted COPII vesicle transport and ER exit sites; (ii) COPII vesicles are hijacked by LC3 II, which interacts with NSP4; and (iii) NSP4/LC3 II-containing membranes accumulate adjacent to viroplasms. In addition, the ER transmembrane proteins SERCA and calnexin were not detected in viroplasm-associated membranes, providing evidence that the rotavirus maturation process of “budding” occurs through autophagy-hijacked COPII vesicle membranes. These findings reveal a new mechanism for rotavirus maturation dependent on intracellular host protein transport and autophagy for the accumulation of membranes required for virus replication.
IMPORTANCE In a morphogenic step that is exceedingly rare for nonenveloped viruses, immature rotavirus particles assemble in replication centers called viroplasms, and bud through cytoplasmic cellular membranes to acquire the outer capsid proteins for infectious particle assembly. Historically, the intracellular membranes used for particle budding were thought to be endoplasmic reticulum (ER) because the rotavirus nonstructural protein NSP4, which interacts with the immature particles to trigger budding, is synthesized as an ER transmembrane protein. This present study shows that NSP4 exits the ER in COPII vesicles and that the NSP4-containing COPII vesicles are hijacked by the cellular autophagy machinery, which mediates the trafficking of NSP4 to viroplasms. Changing the paradigm for rotavirus maturation, we propose that the cellular membranes required for immature rotavirus particle budding are not an extension of the ER but are COPII-derived autophagy isolation membranes.
INTRODUCTION
Most viruses that replicate in the cytoplasm alter the architecture of the host cell to form an intracellular environment conducive to viral replication. Viruses concentrate viral replication proteins and nucleic acid, as well as cellular proteins, to form specialized intracellular compartments known as virus factories, viral inclusions, or viroplasms. Many RNA viruses build virus factories by dramatically remodeling and accumulating host cellular membranes (1–3). The mechanisms by which these membranes accumulate and obtain a continuous supply of phospholipid remain to be fully elucidated (1, 2).
Rotavirus, the causative agent of severe gastroenteritis in young children and animals worldwide, requires host membranes for the assembly of infectious virions. Rotaviruses are nonenveloped particles that have a complex architecture consisting of three concentric capsid layers surrounding a genome of 11 double-stranded RNA (dsRNA) segments. Rotavirus dsRNA replication and immature double-layered particle (DLP) assembly occur in electron-dense viroplasms located in the cytoplasm of the infected cell. The assembly of the two outer capsid proteins, VP4 and VP7, onto immature virions to produce infectious triple-layered particles requires the rotavirus nonstructural protein NSP4 in a morphogenetic process unique for rotavirus. The C-terminal cytoplasmic domain of NSP4, amino acids 161 to 175, binds the inner coat protein (VP6) of DLPs in viroplasms (4–7). This interaction triggers the budding of the DLP through the NSP4-containing membranes where the particles become transiently enveloped. The transient lipid envelope is removed by an unknown mechanism and the outer capsid proteins, VP7 and VP4, are assembled onto the particle.
The current paradigm posits that the membranes through which immature particles bud are endoplasmic reticulum (ER) membranes because NSP4 is synthesized as an ER transmembrane glycoprotein and VP7 is a glycoprotein associated with the luminal ER membrane (8–11). In electron micrographs of rotavirus-infected cells, the ER has been described as “distended” or “dilated” or “swollen,” suggesting that viral infection alters the ER architecture (12–16). We previously demonstrated that NSP4 mediates an increase in cytoplasmic calcium that activates the cellular process of autophagy (17, 18). Autophagy is an intracellular membrane trafficking pathway and a lysosome-mediated degradation process by which cells digest their own damaged organelles and macromolecules to meet bioenergetic needs and enable protein synthesis. Rotavirus activates and exploits the autophagy process to transport the viral ER-associated proteins NSP4 and VP7 from the ER to sites of viroplasm formation in membranes that contain the autophagy marker protein LC3 II (17–19). This new evidence led us to investigate the mechanism by which NSP4 exits the ER and disrupts the ER architecture.
RESULTS
NSP4 exit from the ER is not dependent on the autophagy proteins ATG9, WIPI1, and DFCP1.
We previously reported that rotavirus infection activates the cellular process of autophagy, which is required for the temporal trafficking of NSP4 and LC3 toward viroplasms (17). This result suggested that NSP4 exits the ER to traffic to sites of viroplasm assembly. NSP4 exit from the ER may occur via the autophagy pathway or through the ER COPII vesicle transport pathway.
One possible mechanism of NSP4 exit from the ER is from phosphatidylinositol 3-phosphate [PtdIns(3)P]-enriched ER subdomains, called omegasomes, that act as cradles for autophagosome formation (20). Upon autophagy induction, the ER‑associated PtdIns(3)P-binding protein DFCP1 (double FYVE-containing protein 1, also known as ZFYVE 1 [zinc finger FYVE domain-containing protein 1]) accumulates on the ER-localized omegasome. Subsequently, the autophagy-related proteins ATG9 and WIPI1 (WD repeat domain phosphoinositide-interacting protein 1) are recruited to the omegasome at an early stage of autophagosome biogenesis (20–22).
To determine whether any of the omegasome-associated proteins—DFCP1, ATG9, or WIPI1—are involved in NSP4 exit from the ER, we examined the localization of NSP4 in rotavirus-infected cells in which each of these genes was silenced using siRNA (Fig. 1). The localization of NSP4 or yield of rotavirus (results from three experiments performed in quadruplicate) was not different in cells transfected with small interfering RNA (siRNA) targeted to DFCP1, ATG9, or WIPI1 or in cells transfected with an irrelevant siRNA.
FIG 1.

The omegasome-associated proteins WIPI1, DFCP1, and ATG9 are not required for NSP4 exit from the ER. MA104 cells were transfected with an irrelevant siRNA (A, C, and E) or siRNA to silence WIPI1 (B), DFCP1 (D), or ATG9 (E). The cells were infected with rotavirus 72 h after siRNA transfection and then fixed and permeabilized at 6 hpi. The cells were stained to detect WIPI1 (green) (A and B), DFCP1 (green) (C and D), or ATG9 (green) (E and F), NSP4 (red), viroplasms (NSP2, purple), and nuclei (blue). In panel F, an asterisk (*) denotes uninfected cells in which ATG9 was not silenced. Representative confocal images are shown. Scale bars, 5 μm. (G to I) Western blot analysis of MA104 cells transfected with an irrelevant siRNA or siRNA to silence WIPI1 (G), DFCP1 (H), or ATG9 (I) and harvested at 72 h after siRNA transfection. Immunoblots were detected with the indicated target-specific antibodies.
Lack of detecting a role for the omegasome proteins in NSP4 trafficking or rotavirus replication is not due to incomplete silencing of these proteins. Figure 1B and D show that DFCP1 and WIPI1 proteins are sufficiently silenced because they are not detected by confocal microscopy. Figure 1F shows three noninfected cells in which ATG9 is not completely silenced (indicated by asterisks), but in the rotavirus-infected cells ATG9 is completely silenced. Even when the intensity of the laser is increased on these and other infected cells, the omegasome proteins are not detected. Western blot analysis of irrelevant and omegasome-specific siRNA-transfected cells used for the infectivity assays show silencing of the target proteins indicating that the cellular proteins are sufficiently silenced in the vast majority of the cells (Fig. 1G to I). If these proteins were critical for NSP4 transport to the viroplasm and production of infectious virus, inhibition of NSP4 transport to viroplasms and a decrease in yield would have been observed; therefore, these results indicate that DFCP1, ATG9, or WIPI1 are not required for NSP4 exit from the ER and virus replication.
NSP4 exits the ER in COPII vesicles.
An alternative mechanism of NSP4 exit from the ER is through COPII vesicle transport. COPII vesicles mediate protein transport exclusively from the ER to the Golgi complex (reviewed in reference 23). The assembly of COPII vesicles on the ER membrane is an ordered process initiated by the small GTPase Sar1 (Fig. 2). Exchange of GDP for GTP on Sar1 induces the insertion of Sar1 into the ER and the subsequent recruitment of the COPII inner coat heterodimer proteins Sec23/Sec24. Sar1 interacts with Sec23, which allows Sec24 to bind the cargo protein, and concentrates these proteins into the nascent COPII vesicle. A structural cage composed of the outer coat heterotetramer proteins Sec13/Sec31 forms around the COPII vesicle, and phosphorylation of Sec31 mediated by casein kinase II (CK2) releases the vesicle into the cytoplasm. Critical for the formation of COPII vesicles is the recruitment and concentration of COPII coat proteins to discrete regions of the ER membrane called ER exit sites (ERES).
FIG 2.
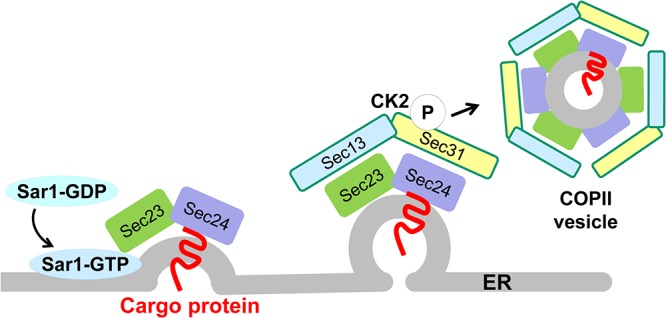
COPII-mediated vesicle formation. COPII proteins from the cytosol and cargo proteins on the membrane are recruited to discrete regions of the ER membrane, called ER exit sites. Exchange of GDP for GTP on Sar1 induces the insertion of Sar1 into the ER and the subsequent recruitment of the COPII inner coat heterodimer proteins Sec23/Sec24. Sar1 interacts with Sec23, which allows Sec24 to bind the cargo protein, and concentrates these proteins into the nascent COPII vesicle. A structural cage composed of the outer coat heterotetramer proteins Sec13/Sec31 forms around the COPII vesicle, and phosphorylation of Sec31 mediated by casein kinase II (CK2) releases the vesicle into the cytoplasm. (The figure is modified from reference 23.)
To determine whether NSP4 exits the ER in COPII vesicles, the interaction of NSP4 with Sec23, Sec24, and Sec31 was assessed by immunoprecipitation. NSP4 coimmunoprecipitated with Sec23 and Sec24 but not Sec31 (Fig. 3), demonstrating that NSP4 interacts with the inner coat, cargo-binding COPII vesicle complex Sec23/Sec24 but not the outer coat protein Sec31 and suggesting that NSP4 exit from the ER may be COPII vesicle mediated.
FIG 3.
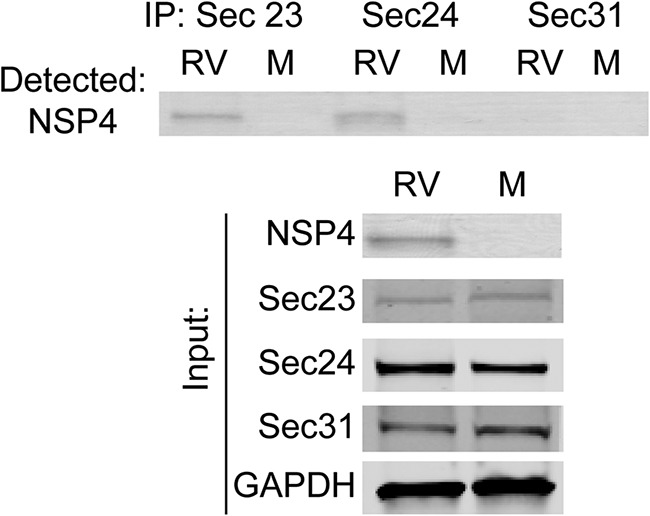
NSP4 coimmunoprecipitates with the COPII proteins Sec23 and Sec24, but not Sec31. Immunoprecipitations were performed with mock (M)- and rotavirus (RV)-infected cell lysates using antibodies against Sec23, Sec24, and Sec31 and analyzed by Western blotting. (Upper panel) A Western blot was probed for NSP4 using rabbit peptide-specific antibody αNSP4114–135. (Lower panel) NSP4, Sec23, Sec 24, Sec31, and GAPDH were detected in the input lysates using the indicated protein-specific antibodies.
The dominant-negative GDP-restricted form of Sar1 abrogates NSP4 exit from the ER.
Multiple approaches, including expression of a dominant negative protein, gene silencing, infectivity assays, and confocal microscopy, were used to confirm that NSP4 exits the ER in COPII vesicles. First, we overexpressed a dominant negative GDP-restricted form of Sar1 (Sar1T39N) that inhibits the recruitment of the Sec23/Sec24 complex to ER membranes and COPII vesicle formation (24).
To verify the effect of Sar1T39N on COPII vesicle trafficking, vesicular stomatitis virus G protein (VSV G) COPII-dependent trafficking was assessed. The ER-localized form of VSV G is glycosylated, exits the ER in COPII vesicles, and is transported to the Golgi complex for glycan modification (25). The ER form of VSV G is sensitive to the glycosidase endoglycosidase H (Endo H), while glycan modification in the Golgi complex confers resistance of the glycan on VSV G to Endo H digestion (25). VSV G, expressed in vector-transfected or Sar1T39N-expressing cells, was pulse-labeled with [35S]methionine, and then immunoprecipitated with an antibody against VSV G. The immunoprecipitates, treated or not treated with Endo H, were separated by SDS-PAGE and analyzed by autoradiography. VSV G from vector-transfected cells was resistant to Endo H digestion (Fig. 4A, left panel, filled arrow) indicating that VSV G was transported from the ER to the Golgi complex. In contrast, VSV G from Sar1T39N-expressing cells was sensitive to Endo H digestion (Fig. 4A, right panel, open arrow) indicating that VSV G was retained in the ER and confirming that Sar1T39N inhibits Sec23/24 complex recruitment and COPII vesicle transport as previously reported (26).
FIG 4.
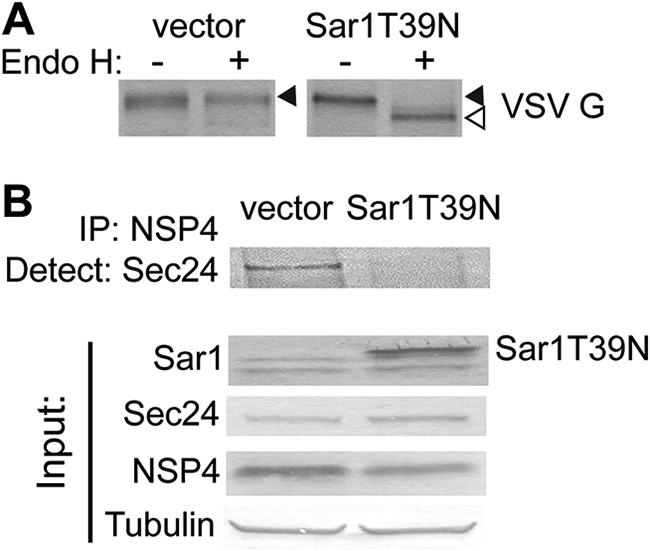
Expression of Sar1T39N abrogates NSP4 interaction with Sec24. (A) 35S-labeled VSV G was immunoprecipitated from VSV G-expressing cells transfected with vector or with the Sar1T39N-expressing plasmid. The immunoprecipitants were treated without (–) or with (+) Endo H and analyzed by autoradiography to detect VSV G. (B) NSP4 was immunoprecipitated from rotavirus-infected and vector or Sar1T39N plasmid-transfected cells using rabbit peptide-specific antibody αNSP4114–135. (Upper panel) A Western blot was probed with antibody against Sec24. (Lower panel) Sar1, Sec24, NSP4, and tubulin were detected in the input lysates using the indicated protein-specific antibodies.
We next confirmed the interaction of NSP4 with the cargo-binding protein Sec24 by immunoprecipitation from vector-transfected or Sar1T39N-expressing rotavirus-infected cell lysates. In rotavirus-infected vector-transfected cells, NSP4 coimmunoprecipitated with Sec24 (Fig. 4B, upper panel, left lane). In contrast, in rotavirus-infected cells expressing Sar1T39N, NSP4 did not coimmunoprecipitate with Sec24 (Fig. 4B, upper panel, right lane). This confirmed that NSP4 interacts with the COPII cargo-binding protein Sec24, and expression of the Sar1 dominant negative that inhibits COPII vesicle formation abrogates this interaction.
We further investigated whether Sar1T39N expression prevents NSP4 trafficking from the ER to viroplasms. ER exit sites are detected by visualizing the accumulation of COPII vesicle proteins in the ER (23). Viroplasms were identified by detecting NSP5 (Fig. 5). In uninfected cells expressing endogenous Sar1, both Sec31 and Sec23 were localized to ER exit sites indicated by strong fluorescent punctate signals (Fig. 5A, indicated by asterisks; Sec31 shown). In contrast, in infected cells where NSP4 was detected adjacent to viroplasms as expected, surprisingly few Sec31 or Sec23 puncta were visualized (Fig. 5A, Sec31 detection shown). Conversely, in rotavirus-infected Sar1T39N-expressing cells, NSP4 did not localize to viroplasms and Sec31 puncta, marking ER exit sites, are clearly detected (Fig. 5B). These results indicate that rotavirus infection disrupts the localization of Sec31 and Sec23 at ER exit sites and that NSP4 trafficking to viroplasms requires a functional Sar1 for COPII vesicle formation and NSP4 exit from the ER.
FIG 5.

Rotavirus infection disrupts COPII vesicle protein localization at ER exit sites and expression of Sar1T39N disrupts NSP4 trafficking to viroplasms. (A and B) Representative confocal images of rotavirus-infected cells transfected with empty plasmid (A) or Sar1T39N-expressing plasmid (B). Cells were probed with rabbit peptide-specific antibody αNSP4114–135, guinea pig antibody αNSP5 to detect viroplasms, and monoclonal antibody αSec31 to identify ER exit sites. Noninfected cells are indicated by an asterisk in panel A. Scale bars, 5 μm.
Silencing CK2 reduces infectious progeny production and inhibits NSP4 exit from the ER.
Casein kinase II (CK2) phosphorylation of Sec31 is required for COPII vesicle release into the cytoplasm (27). Based on our results, we hypothesized that NSP4 assembles into and exits the ER in COPII vesicles to traffic to viroplasms for infectious particle assembly. To test this hypothesis, cells were transfected with irrelevant or CK2 siRNA, and the production of infectious virus was evaluated. The yield of rotavirus progeny in CK2-silenced cells compared to irrelevant control siRNA-transfected cells was decreased by 89% (Fig. 6A), although complete silencing of CK2 was not achieved (Fig. 6B). These results indicate CK2 activity is critical for the production of infectious virus.
FIG 6.

CK2 silencing reduces the yield of rotavirus, disrupts COPII exit from the ER and NSP4 movement to viroplasms. (A) Cells transfected with irrelevant or CK2 siRNA were infected with rotavirus (3 PFU/cell). The cells and media were harvested at 24 hpi, and the progeny virus was quantified by using a fluorescent focus assay. The virus titer in CK2 siRNA-transfected (1.4 × 105 FFU/ml) cells is compared to irrelevant siRNA-transfected (1.1 × 106 FFU/ml) cells. The assay was performed twice with three replicates. Error bars represent standard deviations. (B) Confirmation of CK2 silencing by Western blotting. (C to E) Representative confocal images of rotavirus-infected cells that were transfected with an irrelevant siRNA (C) or CK2 siRNA (D and E). Cells were probed with rabbit peptide-specific antibody αNSP4114–135, guinea pig antibody αNSP5 to detect viroplasms (C to E) and monoclonal antibody αSec31 to mark ER exit sites (E). The boxed area in panel D is magnified in the right panel, as indicated. The rotavirus-infected cell is indicated by an asterisk in panel E. Scale bars: 5 μm (C and D) and 10 μm (E).
We predicted that the reduction in virus yield in CK2-silenced cells was due to the inability of NSP4 to exit the ER in COPII vesicles and traffic to viroplasms, although CK2 has many targets, and it is possible that other parts of the virus replication cycle are also influenced by silencing of CK2 activity. To determine whether silencing CK2 affected NSP4 exit from the ER and trafficking to viroplasms, the localization of Sec31, NSP4, and NSP5 in rotavirus-infected cells transfected with an irrelevant or CK2 siRNA was evaluated using confocal microscopy. In rotavirus-infected cells transfected with the irrelevant siRNA, NSP4 was detected adjacent to viroplasms (Fig. 6C). In contrast, in CK2-silenced rotavirus-infected cells, NSP4 was distributed in a reticular pattern and was not detected adjacent to viroplasms (Fig. 6D), and Sec31 was detected at ER exit sites (Fig. 6E, the infected cell is indicated by an asterisk). Altogether, these results indicate that COPII vesicles are required for NSP4 exit from the ER and that the continuous exit of NSP4 from the ER in COPII vesicles results in disruption of ER exit sites.
NSP4 exits the ER in COPII vesicles before interacting with the autophagy protein LC3.
We previously reported NSP4 mediates an elevation in cytoplasmic calcium that initiates autophagy through a calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2)-dependent mechanism. STO-609 is a specific inhibitor of CaMKK2 that abrogates NSP4-mediated autophagy and the trafficking of NSP4 to viroplasms (17). Using confocal microscopy, we previously demonstrated that NSP4 colocalizes with LC3 by 4 h postinfection (hpi), and both NSP4 and LC3 are detected surrounding viroplasms by 6 hpi (17). However, we did not assess whether NSP4 directly interacts with LC3 in our previous study.
To evaluate the interaction between NSP4 and LC3 and to determine whether NSP4 interaction with COPII vesicles is dependent on autophagy initiation, immunoprecipitation was performed with rotavirus-infected cell lysates cultured in the absence or presence of STO-609. LC3 immunoprecipitates were probed with an antibody against NSP4 and demonstrated that LC3 interacts with NSP4 (Fig. 7A, STO-609, – lane); however, this interaction was abolished in rotavirus-infected cells cultured in the presence of STO-609 (Fig. 7A, STO-609, + lane). NSP4 was detected in Sec24 immunoprecipitates as previously shown in Fig. 3, and the presence of STO-609 did not affect this interaction (Fig. 7B). These results define a sequential series of (i) NSP4 interaction with the COPII cargo-binding protein Sec24 required for NSP4 exit from the ER into the cytoplasm, followed by (ii) NSP4 interaction with LC3, which diverts the COPII vesicle from normal trafficking to the Golgi complex and toward the viroplasm.
FIG 7.
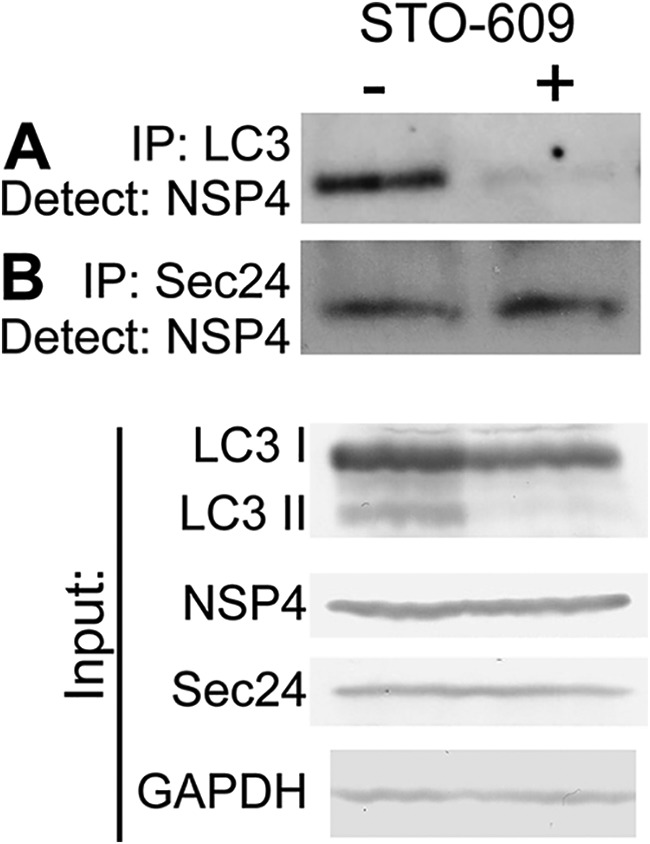
Inhibition of autophagy by STO-609 prevents NSP4 interaction with LC3 but NSP4 interaction with Sec24 is unaffected. Immunoprecipitations were performed from rotavirus-infected cells cultured in the absence (–) or presence (+) of STO-609 using antibody against LC3 (A) or Sec24 (B). The Western blots of the immunoprecipitated protein were probed with rabbit peptide-specific antibody αNSP4114–135. (Lower panel) Endogenous LC3 I and LC3 II, NSP4, Sec24, and GAPDH were detected in the input lysates using the indicated protein-specific antibodies.
Increased cytoplasmic calcium is not required for NSP4-Sec24 interaction.
We next evaluated whether the NSP4-mediated elevation in cytoplasmic calcium required to initiate autophagy is obligatory for NSP4 interaction with Sec24 by culturing cells in the absence or presence of BAPTA-AM, a cell-permeable calcium chelator. In line with our previous report (17), the yield of rotavirus progeny in BAPTA-AM compared to vehicle control-treated cells was decreased by 96% (Fig. 8A), and initiation of autophagy, determined by the detection of LC3 II by Western blotting, was abrogated by BAPTA-AM treatment (Fig. 8B, LC3 input). However, immunoprecipitation analysis showed that NSP4 interaction with Sec24 is not dependent on elevated cytoplasmic calcium as NSP4 was pulled down with Sec24 from cell lysates cultured in the absence or presence of BAPTA-AM (Fig. 8B).
FIG 8.
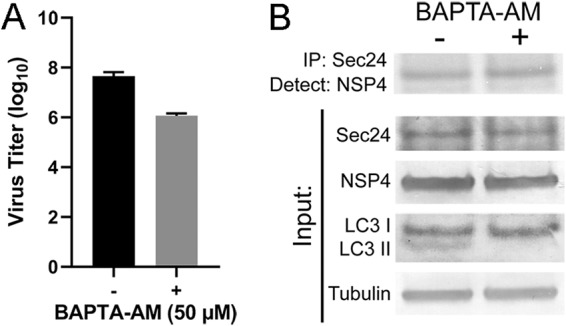
Chelating cytoplasmic calcium with BAPTA-AM reduces the yield of rotavirus but does not abrogate NSP4-Sec24 interaction. (A) MA104 cells were infected with rotavirus (MOI of 1) for 1 h. The cells were washed three times and cultured in the absence (–) or presence (+) of 50 μM BAPTA-AM. The cells and media were harvested at 24 hpi, and the progeny virus was quantified by using a fluorescent focus assay. The assay was performed twice with two or three replicates. Error bars represent standard deviations. (B) Immunoprecipitations were performed from rotavirus-infected cells cultured in the absence (–) or presence (+) of 50 μM BAPTA-AM and harvested at 8 hpi using antibody against Sec24. The Western blots of the immunoprecipitated protein were probed with rabbit peptide-specific antibody αNSP4114–135. (Lower panel) Endogenous Sec24, NSP4, LC3 I and LC3 II, and tubulin were detected in the input lysates using the indicated protein-specific antibodies.
The NSP4/LC3 membranes surrounding viroplasms no longer contain ER marker proteins SERCA or calnexin.
The current paradigm posits that the membranes through which immature particles bud are ER membranes. However, our studies demonstrate that the viroplasm-associated membranes are ER-derived COPII membranes. To determine whether the NSP4- and LC3-containing membranes adjacent to viroplasms (17) contain ER marker proteins, the localization of viroplasms, NSP4, and the ER transmembrane proteins calnexin and sarcoplasmic reticulum calcium-ATPase (SERCA) were assessed by confocal microscopy. The ER proteins were chosen based on their retention in the ER. Calnexin is an ER-transmembrane chaperone that functions to retain unfolded or unassembled N-linked glycoproteins in the ER. SERCA transfers calcium from the cytosol of the cell to the lumen of the ER. SERCA is retained in the ER by an N-terminal ER retention signal sequence (28). The localization of calnexin or SERCA, NSP4 and viroplasms was examined in rotavirus-infected cells at 6 hpi. Neither calnexin nor SERCA colocalized with NSP4 surrounding viroplasms (Fig. 9A and B) suggesting that the NSP4- and LC3-containing membranes surrounding viroplasms do not contain ER-transmembrane proteins that are markers for ER membranes. Our results confirm a previous report that calnexin localizes in the ER and not with viroplasms in rotavirus-infected cells (29).
FIG 9.

ER transmembrane marker proteins do not colocalize with NSP4 surrounding viroplasms. Rotavirus-infected cells were fixed at 6 hpi (A and B) or 4 hpi (C) and stained to detect calnexin (green) (A) or SERCA (green) (B and C), NSP4 (red), viroplasms (NSP5, purple) and nuclei (blue). In panel C, an asterisk (*) denotes infected cells in which SERCA and NSP4 colocalize. Rightmost panel C shows a magnified view of the boxed area in panel C (Merge), which shows early colocalization of SERCA with NSP4 that is subsequently lost by 6 hpi shown in panel B. Scale bars: 5 μm (A and B) and 10 μm (C).
Even though SERCA contains an N-terminal ER retention signal sequence, small NSP4/SERCA-positive puncta were detected at an early time point (4 hpi) but were no longer detected by 6 hpi (Fig. 9C). These results suggest that NSP4 is shuttled out of the ER with SERCA to form small puncta early in infection, but SERCA is subsequently lost from these puncta, possibly being recycled back to the ER.
NSP4 and VP7 traffic together in LC3 II-containing membranes to viroplasms.
Altogether, these results indicate NSP4 exits the ER in COPII vesicles. However, NSP4 and VP7 are synthesized as ER transmembrane and ER luminal-associated proteins, respectively. Because NSP4 and VP7 are required for infectious particle assembly, it is expected that these two proteins would traffic to viroplasms together. We next determined whether VP7 traffics with NSP4 to viroplasms. Analysis of rotavirus-infected cells by confocal microscopy revealed that at 4 hpi (Fig. 10A), NSP4 and VP7 form small puncta that increase in size at 5 hpi (Fig. 10B). NSP4 and VP7 are detected surrounding viroplasms at 6 hpi (Fig. 10C). These results indicate that NSP4 and VP7 exit the ER and traffic to viroplasms together.
FIG 10.

NSP4 and VP7 traffic together to viroplasms. Rotavirus-infected cells were fixed and stained at 4 hpi (A), 5 hpi (B), and 6 hpi (C) with antibody to detect VP7 (green), NSP4 (red), viroplasms (NSP5, purple) and nuclei (blue). Scale bars, 5 μm.
DISCUSSION
To gain a more comprehensive understanding of rotavirus-host interactions, it is necessary to identify and characterize the host molecules involved in the biogenesis of infectious rotavirus particles. In the current paradigm, rotavirus immature particles that assemble in viroplasms bud through ER membranes to acquire the outer capsid proteins and mature into infectious virions. We previously described that rotavirus initiates and requires the cellular process of autophagy to facilitate NSP4 transport to viroplasms (17, 18). In the present study, we present several lines of evidence that clarify the mechanism by which rotavirus NSP4 traffics to viroplasms to facilitate infectious particle assembly. First, NSP4 does not exit the ER from ER-localized omegasomes, one source for the production of autophagosomes. Silencing the autophagy- and omegasome-related proteins DFCP1, WIPI1, or ATG9 did not reduce viral yield or prevent NSP4 trafficking to viroplasms. Second, NSP4 exits the ER in COPII vesicles and COPII-mediated NSP4 egress from the ER disrupts ER exit sites. NSP4 ER exit and trafficking to viroplasms or virus production was abrogated or reduced, respectively, by inhibiting COPII vesicle formation or release. Third, LC3 interacts with NSP4 and this occurs after NSP4 exits the ER in COPII vesicles. Fourth, the ER-transmembrane marker proteins SERCA and calnexin are not detected in NSP4/LC3-containing membranes surrounding viroplasms. Altogether, we identified that NSP4-containing COPII vesicles are the source of the NSP4/LC3-containing membranes that associate with viroplasms. Furthermore, we demonstrated that NSP4 and the outer capsid protein VP7 traffic together to viroplasms.
Our findings necessitate a change in the current paradigm for the model of rotavirus morphogenesis. In the previous model, rotavirus infectious particle assembly was thought to occur in the ER due to the visualization by thin section electron microscopy of enveloped and nonenveloped particles within “swollen” ER in infected cells (12–16). In our new model of rotavirus morphogenesis (Fig. 11), (i) NSP4 and VP7 are synthesized as ER-associated proteins, (ii) NSP4 interacts with the COPII cargo-binding protein Sec24, (iii) the NSP4-containing COPII vesicles are assembled and released into the cytoplasm, where (iv) LC3 interacts with NSP4 and inserts into the NSP4-containing membranes, and (v) NSP4-LC3 and VP7 traffic to viroplasms. In this model, immature particles that are assembled in viroplasms bud through ER COPII-derived NSP4/LC3-containing membranes.
FIG 11.

Model of NSP4 COPII-mediated exit from the ER, interaction with LC3 and trafficking to viroplasms. NSP4, synthesized as an ER transmembrane protein, interacts with Sec24 and is incorporated into COPII vesicles that are released into the cytoplasm. LC3 interacts with NSP4 and inserts into the NSP4-containing, COPII-derived membranes. The LC3/NSP4-containing membranes traffic to viroplasms. Immature particles produced in the viroplasm interact with NSP4, which triggers the particles to bud through the LC3/NSP4-containing membranes, to mediate infectious particle assembly. The LC3/NSP4-containing membranes fill with virus particles and thus do not develop into autophagosomes. (The figure was modified from reference 23.)
The proposed model supports previous findings. Electron micrographs of rotavirus-infected cells in which NSP4 had been silenced with siRNA revealed viroplasms without the associated membranes, which resulted in a reduction in infectious progeny virus (30). These data support our results that NSP4 is required for the generation of the membranes that traffic to viroplasms (17). In addition, the critical role of autophagy in rotavirus infection is highlighted by the different mechanisms by which autophagy is induced: NSP4-calcium mediated activation of autophagy through CaMKK2 (17, 18) and miRNA-regulated autophagy (31, 32). Although we and others previously demonstrated that rotavirus-induced autophagy requires PI3K activity and the autophagy-initiation proteins ATG3, ATG5, and ATG7 (17, 18, 33), autophagy flux appears to be halted at an early stage of autophagy. We propose that LC3 II inserts into the ER-derived COPII vesicles as an innate immune mechanism to engulf viroplasms for degradation in a process known as xenophagy in which autophagy membranes engulf microbes or microbial components to destroy them (34–36). However, in the case of rotavirus, interaction of immature particles in the viroplasm with NSP4 triggers the particles to bud into the NSP4/LC3 II-containing membranes for infectious particle assembly. Thus, the NSP4/LC3 II-containing membranes do not form double membranes or autophagosomes but contain enveloped particles that have budded through the NSP4/LC3 II-containing membrane, as well as particles that have lost the membrane envelop and have acquired the outer capsid proteins. Therefore, we propose that the COPII-derived, NSP4/LC3 II-containing membranes are autophagy isolation membranes. Our proposed model explains why an increase in autophagosome accumulation is not observed in electron micrographs of infected cells although autophagy is initiated and required for the production of infectious virus (33).
Whereas ER exit sites and COPII vesicles have been described as a source for autophagosome biogenesis, our report identifies a specific cargo, NSP4, which is transported in COPII vesicles into the cytoplasm and is then targeted by LC3. In the budding yeast Saccharomyces cerevisiae, the transmembrane cargo Axl2 is loaded into COPII vesicles in the ER, is transferred to autophagosome intermediates, and ultimately becomes part of autophagosomal membranes (37). Thus, both of these studies provide a definitive answer to a long-standing, fundamental question regarding the mechanisms of autophagosome formation by implicating COPII vesicles as a membrane source for autophagosomes.
The source of the LC3-containing membranes in rotavirus-infected cells are from COPII vesicles, not the omegasome, since the autophagy-related proteins DFCP1 and WIPI1 are not required for NSP4 trafficking to viroplasms. Furthermore, ATG9 is not required for NSP4 ER exit, interaction with LC3 or trafficking to viroplasms. Our current understanding of ATG9 function is primarily derived from studies of starvation-induced autophagy in yeast in which the secretory pathway is largely shut down and COPII vesicles are diverted to the macroautophagy pathway, where they fuse with Golgi complex-derived vesicles containing the integral membrane protein ATG9 to form a phagophore (38, 39). However, the role of ATG9 in mammalian cells is less clear. In starved mammalian cells, ATG9 has been reported to accumulate on DFCP1-positive omegasomes, ERGIC-derived vesicles, or endosomes, but ATG9 is not a component of the autophagosome (40). Rather, it is thought that ATG9 delivers lipids required for the initiation and expansion of autophagosomes (40). Our results reveal that the source of the NSP4/LC3 II-containing membranes consist of COPII vesicles and that ATG9 is not required for the initiation or expansion of these membranes.
Our observation that rotavirus infection and NSP4 hijack COPII vesicle trafficking, resulting in disruption of ER exit sites, may have implications for viral pathogenesis. Trafficking of VSV G from the ER to the Golgi complex is disrupted in rotavirus-infected cells and this is mediated by NSP4 expression (41). Disruption of ER exit sites may mislocalize or inhibit the trafficking of other ER-synthesized proteins to other organelles or the plasma membrane, including immune factors such as cytokines. Furthermore, the perturbation of surface delivery of secreted and membrane-bound cargo molecules may contribute to the onset or persistence of rotavirus-induced diarrhea.
MATERIALS AND METHODS
Cells and virus.
Rotavirus SA114F, G3P6[1], was cultivated in MA104 cells as previously described (42).
Antibodies and chemicals.
The antibodies used in this study against NSP4 were mouse monoclonal antibody B4-2/55/17(1)/13, rabbit peptide-specific antibody αNSP4114–135 (43), and guinea pig antibody αNSP5 (44). The VP7 monoclonal antibody 60/F2 was kindly provided by Harry Greenberg (Stanford) (45). The LC3 antibody was obtained from Novus Biologicals, sarcoendoplasmic reticulum calcium transport ATPase (SERCA) and Sec24 antibodies were purchased from Abcam, calnexin antibody was obtained from Affinity Bioreagents, Sar1 antibody was purchased from Upstate, Sec23 and CK2 antibodies were purchased from Santa Cruz, Sec31 antibody was purchased from BD Transduction, and VSV G antibody was purchased from GenScript. Secondary Alexa Fluor 488-, 568-, and 633-conjugated antibodies were obtained from Invitrogen or Rockland. STO-609 was purchased from Tocris Bioscience. BAPTA-AM was purchased from EMD Millipore Corporation.
siRNA and transfection.
Small interfering RNA (siRNA) against human CK2, DFCP1/ZFYVE, WIPI, and negative-control siRNA were obtained from Santa Cruz Biotechnology, and siRNAs against human ATG9 was obtained from Cell Signaling Technology. Lipofectamine RNAiMAX transfection reagent (Invitrogen) was used for siRNA transfection. siRNA was transfected 72 h prior to rotavirus infection.
Confocal microscopy.
MA104 cells were grown on glass coverslips. After infection, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min, permeabilized with 0.5% Triton X-100 for 10 min, and blocked with 5% bovine serum albumin (BSA) in PBS for 30 min at room temperature. The indicated primary antibodies were diluted in 2.5% BSA, added to the cells on the coverslips, and incubated for 24 h at 4°C. The cells were then washed three times with PBS, and secondary Alexa Fluor-conjugated antibodies were incubated for 2 h at room temperature. Cells were washed three times with PBS, and TO-PRO-3 (Invitrogen) was added for nuclear staining. All immunofluorescence images were acquired using a Nikon A1Rs confocal laser scanning microscope. Images were obtained using sequential image acquisition (to eliminate potential channel bleed of the fluorescent signals), the laser power was set from 5 to 10%, the pinhole was set at 1.0 for the 488-nm laser, and the gain for any given image ranged from 85 to 130 (maximum possible gain, 255). Mock-infected cells were imaged using the same settings as their rotavirus-infected counterparts. All observations were carried out in the Integrated Microscopy Core, Baylor College of Medicine, Houston, TX.
Transfection, immunoprecipitation, and Western blotting.
Plasmids encoding HA-Sar1T39N and VSV glycoprotein (VSV G) were obtained from Nihal Altan-Bonnet at the National Institutes of Health (NIH). Plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen) as indicated by the manufacturer. To label VSV G, 30 μCi/ml of EasyTag [35S]methionine (Perkin-Elmer) was added to cells 24 h posttransfection for 30 min, and then the cells were harvested in radioimmunoprecipitation assay (RIPA) buffer as described previously (46). Immunoprecipitations were performed with RIPA-solubilized cell lysates using the indicated primary antibody and Pierce protein A/G-magnetic beads (Thermo Scientific) as indicated by the manufacturer. Half of the 35S-labeled VSV G immunoprecipitate was incubated with Endo H (Sigma) according to the manufacturer’s instructions. The samples were resolved on denaturing SDS-PAGE gels and imaged by autoradiography. Western blots were performed as previously described using chemiluminescent, alkaline phosphatase (NBT/BCIP) (46) or infrared LiCor methods as described by the manufacturer.
ACKNOWLEDGMENTS
This study was supported by NIH grant R01 AI080656 (to M.K.E.). This project was also supported by Advanced Technology Core Laboratories (Baylor College of Medicine), specifically, the Integrated Microscopy Core, the Mass Spectrometry Proteomics Core, and the Protein and Monoclonal Antibody Production Core with funding from P30 Cancer Center Support Grant (NCI-CA125123), P30 Digestive Disease Center (NIDDK-56338-13/15), CPRIT (RP150578), and the John S. Dunn Gulf Coast Consortium for Chemical Genomics.
REFERENCES
- 1.de Armas-Rillo L, Valera M-S, Marrero-Hernández S, Valenzuela-Fernández A. 2016. Membrane dynamics associated with viral infection. Rev Med Virol 26:146–160. doi: 10.1002/rmv.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller S, Krijnse-Locker J. 2008. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 6:363–374. doi: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Netherton C, Moffat K, Brooks E, Wileman T. 2007. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Adv Virus Res 70:101–182. doi: 10.1016/S0065-3527(07)70004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Au KS, Chan WK, Burns JW, Estes MK. 1989. Receptor activity of rotavirus nonstructural glycoprotein NS28. J Virol 63:4553–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer JC, Bergmann CC, Bellamy AR. 1989. Interaction of rotavirus cores with the nonstructural glycoprotein NS28. Virology 171:98–107. doi: 10.1016/0042-6822(89)90515-1. [DOI] [PubMed] [Google Scholar]
- 6.Taylor JA, O’Brien JA, Yeager M. 1996. The cytoplasmic tail of NSP4, the endoplasmic reticulum-localized nonstructural glycoprotein of rotavirus, contains distinct virus binding and coiled coil domains. EMBO J 15:4469–4476. doi: 10.1002/j.1460-2075.1996.tb00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Brien JA, Taylor JA, Bellamy AR. 2000. Probing the structure of rotavirus NSP4: a short sequence at the extreme C terminus mediates binding to the inner capsid particle. J Virol 74:5388–5394. doi: 10.1128/JVI.74.11.5388-5394.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stirzaker SC, Whitfeld PL, Christie DL, Bellamy AR, Both GW. 1987. Processing of rotavirus glycoprotein VP7: implications for the retention of the protein in the endoplasmic reticulum. J Cell Biol 105:2897–2903. doi: 10.1083/jcb.105.6.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arias CF, Lopez S, Espejo RT. 1982. Gene protein products of SA11 simian rotavirus genome. J Virol 41:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Both GW, Siegman LJ, Bellamy AR, Atkinson PH. 1983. Coding assignment and nucleotide sequence of simian rotavirus SA11 gene segment 10: location of glycosylation sites suggests that the signal peptide is not cleaved. J Virol 48:335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ericson BL, Graham DY, Mason BB, Hanssen HH, Estes MK. 1983. Two types of glycoprotein precursors are produced by the simian rotavirus SA11. Virology 127:320–332. doi: 10.1016/0042-6822(83)90147-2. [DOI] [PubMed] [Google Scholar]
- 12.Bishop RF, Davidson GP, Holmes IH, Ruck BJ. 1973. Virus particles in epithelial cells of duodenal mucosa from children with acute non-bacterial gastroenteritis. Lancet 2:1281–1283. doi: 10.1016/S0140-6736(73)92867-5. [DOI] [PubMed] [Google Scholar]
- 13.Petrie BL, Greenberg HB, Graham DY, Estes MK. 1984. Ultrastructural localization of rotavirus antigens using colloidal gold. Virus Res 1:133–152. doi: 10.1016/0168-1702(84)90069-8. [DOI] [PubMed] [Google Scholar]
- 14.Saif LJ, Theil KW, Bohl EH. 1978. Morphogenesis of porcine rotavirus in porcine kidney cell cultures and intestinal epithelial cells. J Gen Virol 39:205–217. doi: 10.1099/0022-1317-39-2-205. [DOI] [PubMed] [Google Scholar]
- 15.Petrie BL, Graham DY, Hanssen H, Estes MK. 1982. Localization of rotavirus antigens in infected cells by ultrastructural immunocytochemistry. J Gen Virol 63:457–467. doi: 10.1099/0022-1317-63-2-457. [DOI] [PubMed] [Google Scholar]
- 16.Esparza J, Gorziglia M, Gil F, Romer H. 1980. Multiplication of human rotavirus in cultured cells: an electron microscopic study. J Gen Virol 47:461–472. doi: 10.1099/0022-1317-47-2-461. [DOI] [PubMed] [Google Scholar]
- 17.Crawford SE, Hyser JM, Utama B, Estes MK. 2012. Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc Natl Acad Sci U S A 109:E3405–E3413. doi: 10.1073/pnas.1216539109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford SE, Estes MK. 2013. Viroporin-mediated calcium-activated autophagy. Autophagy 9:797–798. doi: 10.4161/auto.23959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrison C, Gilson T, Nuovo GJ. 2001. Histologic distribution of fatal rotaviral infection: an immunohistochemical and reverse transcriptase in situ polymerase chain reaction analysis. Hum Pathol 32:216–221. doi: 10.1053/hupa.2001.21565. [DOI] [PubMed] [Google Scholar]
- 20.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. 2008. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol 182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itakura E, Mizushima N. 2010. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N. 2013. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy 9:1491–1499. doi: 10.4161/auto.25529. [DOI] [PubMed] [Google Scholar]
- 23.Jensen D, Schekman R. 2011. COPII-mediated vesicle formation at a glance. J Cell Sci 124:1–4. doi: 10.1242/jcs.069773. [DOI] [PubMed] [Google Scholar]
- 24.Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, Cameron CE, Ehrenfeld E, van Kuppeveld FJ, Altan-Bonnet N. 2010. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davidson HW, Balch WE. 1993. Differential inhibition of multiple vesicular transport steps between the endoplasmic reticulum and trans Golgi network. J Biol Chem 268:4216–4226. [PubMed] [Google Scholar]
- 26.Kuge O, Dascher C, Orci L, Rowe T, Amherdt M, Plutner H, Ravazzola M, Tanigawa G, Rothman JE, Balch WE. 1994. Sar1 promotes vesicle budding from the endoplasmic reticulum but not Golgi compartments. J Cell Biol 125:51–65. doi: 10.1083/jcb.125.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koreishi M, Yu S, Oda M, Honjo Y, Satoh A. 2013. CK2 phosphorylates Sec31 and regulates ER-To-Golgi trafficking. PLoS One 8:e54382. doi: 10.1371/journal.pone.0054382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foletti D, Guerini D, Carafoli E. 1995. Subcellular targeting of the endoplasmic reticulum and plasma membrane Ca2+ pumps: a study using recombinant chimeras. FASEB J 9:670–680. doi: 10.1096/fasebj.9.8.7768360. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez RA, Espinosa R, Romero P, Lopez S, Arias CF. 2000. Relative localization of viroplasmic and endoplasmic reticulum-resident rotavirus proteins in infected cells. Arch Virol 145:1963–1973. doi: 10.1007/s007050070069. [DOI] [PubMed] [Google Scholar]
- 30.Lopez T, Camacho M, Zayas M, Najera R, Sanchez R, Arias CF, Lopez S. 2005. Silencing the morphogenesis of rotavirus. J Virol 79:184–192. doi: 10.1128/JVI.79.1.184-192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhyay U, Chanda S, Patra U, Mukherjee A, Rana S, Mukherjee A, Chawla-Sarkar M. 2019. Synchronized orchestration of miR-99b and let-7g positively regulates rotavirus infection by modulating autophagy. Sci Rep 9:1318. doi: 10.1038/s41598-018-38473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou Y, Geng P, Liu Y, Wu J, Qiao H, Xie Y, Yin N, Chen L, Lin X, Liu Y, Yi S, Zhang G, Li H, Sun M. 2018. Rotavirus-encoded virus-like small RNA triggers autophagy by targeting IGF1R via the PI3K/Akt/mTOR pathway. Biochim Biophys Acta Mol Basis Dis 1864:60–68. doi: 10.1016/j.bbadis.2017.09.028. [DOI] [PubMed] [Google Scholar]
- 33.Arnoldi F, De LG, Mano M, Schraner EM, Wild P, Eichwald C, Burrone OR. 2014. Rotavirus increases levels of lipidated LC3 supporting accumulation of infectious progeny virus without inducing autophagosome formation. PLoS One 9:e95197. doi: 10.1371/journal.pone.0095197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee HK, Iwasaki A. 2008. Autophagy and antiviral immunity. Curr Opin Immunol 20:23–29. doi: 10.1016/j.coi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine B. 2005. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120:159–162. doi: 10.1016/S0092-8674(05)00043-7. [DOI] [PubMed] [Google Scholar]
- 37.Shima T, Kirisako H, Nakatogawa H. 2019. COPII vesicles contribute to autophagosomal membranes. J Cell Biol 218:1503–1510. doi: 10.1083/jcb.201809032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang J, Tan D, Cai Y, Reinisch KM, Walz T, Ferro-Novick S. 2014. A requirement for ER-derived COPII vesicles in phagophore initiation. Autophagy 10:708–709. doi: 10.4161/auto.28103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graef M, Friedman JR, Graham C, Babu M, Nunnari J. 2013. ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell 24:2918–2931. doi: 10.1091/mbc.e13-07-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. 2012. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23:1860–1873. doi: 10.1091/mbc.e11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu A, Bellamy AR, Taylor JA. 2000. Immobilization of the early secretory pathway by a virus glycoprotein that binds to microtubules. EMBO J 19:6465–6474. doi: 10.1093/emboj/19.23.6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Estes MK, Graham DY, Gerba CP, Smith EM. 1979. Simian rotavirus SA11 replication in cell cultures. J Virol 31:810–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyser JM, Zeng CQ, Beharry Z, Palzkill T, Estes MK. 2008. Epitope mapping and use of epitope-specific antisera to characterize the VP5* binding site in rotavirus SA11 NSP4. Virology 373:211–228. doi: 10.1016/j.virol.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Criglar JM, Anish R, Hu L, Crawford SE, Sankaran B, Prasad BVV, Estes MK. 2018. Phosphorylation cascade regulates the formation and maturation of rotaviral replication factories. Proc Natl Acad Sci U S A 115:E12015–E12023. doi: 10.1073/pnas.1717944115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Offit PA, Blavat G. 1986. Identification of the two rotavirus genes determining neutralization specificities. J Virol 57:376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Criglar JM, Hu L, Crawford SE, Hyser JM, Broughman JR, Prasad BV, Estes MK. 2014. A novel form of rotavirus NSP2 and phosphorylation-dependent NSP2-NSP5 interactions are associated with viroplasm assembly. J Virol 88:786–798. doi: 10.1128/JVI.03022-13. [DOI] [PMC free article] [PubMed] [Google Scholar]