

SUMMARY
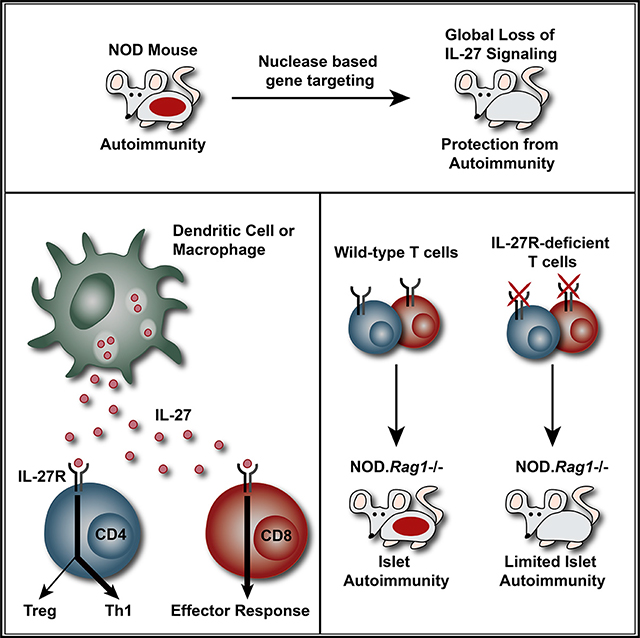
Human genetic studies implicate interleukin-27 (IL-27) in the pathogenesis of type 1 diabetes (T1D), but the underlying mechanisms remain largely unexplored. To further define the role of IL-27 in T1D, we generated non-obese diabetic (NOD) mice deficient in IL-27 or IL-27Rα. In contrast to wild-type NOD mice, both NOD.Il27−/− and NOD.Il27ra−/− strains are completely resistant to T1D. IL-27 from myeloid cells and IL-27 signaling in T cells are critical for T1D development. IL-27 directly alters the balance of regulatory T cells (Tregs) and T helper 1 (Th1) cells in pancreatic islets, which in turn modulates the diabetogenic activity of CD8 T cells. IL-27 also directly enhances the effector function of CD8 T cells within pancreatic islets. In addition to T1D, IL-27 signaling in T cells is also required for lacrimal and salivary gland inflammation in NOD mice. Our study reveals that IL-27 contributes to autoimmunity in NOD mice through multiple mechanisms and provides substantial evidence to support its pathogenic role in human T1D.
In Brief
Human genetic studies implicate IL-27 in the pathogenesis of type 1 diabetes (T1D). Ciecko et al. demonstrate that IL-27 signaling in T cells changes the balance of regulatory and effector subsets and is critical for T1D development as well as lacrimal and salivary gland inflammation in NOD mice.
Graphical Abstract
INTRODUCTION
Interleukin-27 (IL-27), a member of the IL-6/IL-12 cytokine superfamily, is a heterodimer composed of two noncovalently associated subunits: Epstein-Barr virus-induced gene 3 (EBI3) and IL-27p28 (Pflanz et al., 2002). The IL-27 receptor is also a heterodimer composed of the IL-27 receptor subunit alpha (IL-27Rα) and a signal transduction subunit, glycoprotein 130 (gp130) (Pflanz et al., 2004). IL-27 is secreted primarily by activated dendritic cells (DCs), monocytes, and macrophages, and the receptor complex is expressed on many immune cells, including T cells, B cells, DCs, macrophages, and natural killer cells (Yoshida and Hunter, 2015). IL-27 signaling activates signal transducer and activator of transcription (STAT) family proteins STAT1 and STAT3, as well as the mitogen-activated protein kinase (MAPK) pathway (Takeda et al., 2003; Lucas et al., 2003; Peters et al., 2015; Owaki et al., 2006). IL-27 has been shown to have both pro- and anti-inflammatory activities in several autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, and inflammatory bowel disease (Meka et al., 2015).
Type 1 diabetes (T1D) is an autoimmune disease characterized by the T cell-mediated destruction of insulin-producing pancreatic β cells (Serreze and Leiter, 2001). Genome-wide association studies have identified more than 50 loci significantly linked to T1D in humans (Barrett et al., 2009; Bradfield et al., 2011; Evangelou et al., 2014; Fortune et al., 2015; Todd et al., 2007; Onengut-Gumuscu et al., 2015; Cooper et al., 2008), including a region located on chromosome 16 that contains 24 protein coding genes, of which IL27 (encoding the p28 subunit) has been indicated as a strong candidate (Barrett et al., 2009; Bergholdt et al., 2012). Expression quantitative trait loci (eQTL) analysis revealed that the T1D-associated risk allele of the SNP rs4788084 located within 2 kb upstream of IL27 was linked to increased expression of GBP4 and STAT1 in human peripheral blood mononuclear cells (PBMCs) (Westra et al., 2013). Subsequently, an IL27 missense variant (rs181206) was found in strong linkage disequilibrium with rs4788084, and eQTL analysis indicated that the rs181206 variant was associated with elevated STAT1 and IRF1 transcript levels in human CD4 T cells (Kasela et al., 2017). Interestingly, this study also demonstrated that the same rs181206 variant increased IL-27 function (Kasela et al., 2017). Collectively, these genetic studies suggest the potential of IL27 allelic variants to directly affect the downstream signaling pathway, and they could have effects on T1D pathogenesis.
Previous mouse studies aimed at understanding the role of IL-27 in T1D showed a model-dependent outcome. A study in the non-obese diabetic (NOD) mouse revealed that IL-27 was expressed by activated DCs in diabetic mice, and blockade of IL-27 significantly delayed the onset of splenocyte-transferred T1D in lymphocyte-deficient NOD-scid recipients (Wang et al., 2008). In contrast, another study in which diabetes was induced by multiple injections of low-dose streptozotocin showed that IL-27 signaling conferred protection against T1D (Fujimoto et al., 2011). To better define the role of IL-27 in T1D, we generated and characterized NOD mice deficient in IL-27p28 or IL-27Rα. Our results demonstrate that IL-27 signaling in both CD4 and CD8 T cells is critical for T1D development and this cytokine directly influences differentiation and effector functions of both CD4 and CD8 T cells in pancreatic islets. In addition, we show here that IL-27 signaling in T cells is also required for lacrimal and salivary gland inflammation, indicating that its effects are not limited to β-cell autoimmunity in NOD mice.
RESULTS
IL-27 Is Required for T1D Development in NOD Mice
To study the role of IL-27 in T1D, we used zinc-finger nuclease (ZFN)-mediated mutagenesis to directly target Il27 in NOD mice (Figure S1A). Bone marrow (BM)-derived macrophages from NOD but not NOD.Il27−/− mice produced IL-27 upon stimulation with lipopolysaccharide (LPS), confirming the knockout phenotype (Figure S1B). Strikingly, both female and male NOD.Il27−/− mice were completely resistant to T1D development (Figure 1A). Compared with NOD mice, insulitis was significantly reduced in 10-week-old NOD.Il27−/− mice, and it did not further progress from 10 to 30 weeks of age (Figures 1B and 1C; Figure S1C).
Figure 1. NOD.Il27−/− Mice Are Completely Resistant to T1D.

(A) T1D incidence of NOD and NOD.Il27−/− mice. ***p < 0.005 by log rank test.
(B) Summary of insulitis in female NOD and NOD.Il27−/− mice.
(C) Summary of insulitis in male NOD and NOD.Il27−/− mice. Pancreatic islets were scored for insulitis: 0 = no infiltration, 1 = peri-insulitis, 2 = ≤25% β cell loss, 3 = between 25% and 75% β cell loss, 4 = >75% β cell loss. Each symbol represents one mouse. The horizontal bar depicts the mean. More than 30 islets were scored for each mouse. **p < 0.01 by Mann-Whitney test. NS, not significant.
(D) T1D incidence study of sublethally irradiated NOD.Il27−/− females infused with BM cells (5 × 106) from sex-matched NOD.Rag1−/− or NOD.Rag1−/−.Il27−/− mice as indicated. **p < 0.01 by log rank test.
(E and F) T1D incidence study of adoptively transferred T cell recipients.
(E) Splenic T cells (5 × 106) were isolated from 6-week-old NOD females and transferred into sex-matched NOD.Rag1−/− or NOD.Rag1−/−.Il27−/− recipients.
(F) Splenic T cells (5 × 106) were isolated from young (6- to 7-week-old) NOD, old (11- to 13-week-old) NOD, or old (14- to 15-week-old) NOD.Il27−/− mice and transferred into sex-matched NOD.Rag1−/− recipients. *p < 0.05, **p < 0.01, and ***p < 0.005 by log rank test. NS, not significant.
See also Figure S1.
Macrophages and DCs are the main sources of IL-27, but previous studies have also reported its production by activated T cells (Kimura et al., 2016; Dibra et al., 2012; Fujita et al., 2009). Therefore, we tested if introducing wild-type macrophages and DCs is sufficient for T1D development in NOD.Il27−/− mice. Sublethally irradiated NOD.Il27−/− mice were infused with NOD.Rag1−/− BM cells to allow partial reconstitution of wild-type macrophages and DCs in the presence of host Il27−/− T and B cells. Sublethally irradiated NOD.Il27−/− mice reconstituted with NOD.Rag1−/−.Il27−/− BM cells were used as the control. NOD.Il27−/− recipients of NOD.Rag1−/− but not NOD.Rag1−/−.Il27−/− BM cells progressed to diabetes (Figure 1D). These results indicate that IL-27 production by non-T and non-B cells, most likely macrophages and/or DCs, is sufficient to drive T1D progression. We then asked if IL-27 production by macrophages and/or DCs is required for T1D development by transferring NOD splenic T cells into NOD.Rag1−/− or NOD.Rag1−/−.Il27−/− recipients. NOD.Rag1−/− but not NOD.Rag1−/−.Il27−/− recipients of NOD T cells developed diabetes, indicating that IL-27 production by non-T and non-B cells is required to drive T1D (Figure 1E).
IL-27 Is Not Essential for the Development of β-Cell Autoreactive T Cells
The striking protective phenotype in NOD.Il27−/− mice prompted us to question if diabetogenic T cells are present in this strain. To test this, we transferred total splenic T cells isolated from NOD and NOD.Il27−/− mice into NOD.Rag1−/− recipients capable of producing IL-27. T cells from old (14- to 15-week-old) NOD.Il27−/− and young (6- to 7-week-old) NOD mice had similar diabetogenic activity, but both were significantly less capable of inducing T1D than those from old (11- to 13-week-old) NOD mice (Figure 1F). Thus, NOD.Il27−/− mice still harbor β-cell autoreactive T cells, but the frequency is likely lower than that in NOD mice of a similar age. These results also suggest that β-cell autoreactive T cells in NOD.Il27−/− mice are not permanently tolerized but cannot be efficiently activated and expanded. We also conclude that T cell-derived IL-27 is not required for T1D development.
IL-27 Deficiency Suppresses Islet Infiltration of DCs and T Cells
CD11b− CD103+ migratory DCs are critical for autoreactive CD8 T cell activation and expansion in pancreatic lymph nodes (PLNs) (Ferris et al., 2014). Therefore, we evaluated if IL-27 is important for the development of DC subsets in the PLN and spleen. Our analyses did not reveal a difference in the abundance of CD11b− CD103+ or other DC subsets in the PLNs and spleens of 10-week-old NOD and NOD.Il27−/− mice (Figures S2A–S2C). Furthermore, expression levels of CD40, CD80, CD86, Kd, I-Ag7, and PD-L1 on PLN DCs were comparable between NOD and NOD.Il27−/− mice (data not shown). Thus, IL-27 deficiency does not cause a general defect of DC development in NOD mice.
In NOD mice, myeloid antigen-presenting cells (APCs) are among the first immune cells to infiltrate the pancreatic islets and are required for amplification of the autoreactive T cell response (Melli et al., 2009; Ferris et al., 2016). Therefore, we analyzed whether IL-27 is important for the infiltration and accumulation of myeloid APCs and T cells in the islets. At 4 weeks of age, NOD and NOD.Il27−/− mice had a similar frequency of CD45+ cells in the islets (Figure 2A). The proportion of CD45+ cells dramatically increased in the islets of 10-week-old NOD mice, but it was only marginally increased from 4 to 10 weeks in the NOD.Il27−/− mice (Figure 2A). Likewise, NOD and NOD.Il27−/− mice had a similar low frequency of islet-infiltrating CD3+ cells at 4 weeks of age. However, by 10 weeks of age, NOD mice showed a significant accumulation of CD3+ cells in the islets, while NOD.Il27−/− mice had only a slight increase of these cells (Figure 2B). Coincidentally, at 4 weeks of age, there were no differences in the overall frequencies of F4/80+ CD11c−, F4/80+ CD11c+, or F4/80− CD11c+ APCs in the islets of NOD and NOD.Il27−/− mice (Figure 2C). However, at 10 weeks of age, the frequency of F4/80− CD11c+ DCs, which includes CD103+ migratory DCs, was significantly increased in the islets of NOD mice, whereas it was largely unchanged in the absence of IL-27 (Figure 2C). These data indicate that the overall infiltration of T cells and DCs into the pancreatic islets is significantly decreased in the absence of IL-27 signaling. Because initial T cell infiltration promotes maturation of CD11c+ APCs in islets, including upregulation of CD40 (Melli et al., 2009), we further analyzed the phenotype of islet-infiltrating myeloid APCs in NOD and NOD.Il27−/− mice. The expression of I-Ag7, Kd, and CD40 was comparable in myeloid APCs between NOD and NOD.Il27−/− mice at 4 weeks of age (data not shown). In contrast, at 10 weeks of age, myeloid APCs from NOD islets had significantly increased CD40 expression compared with NOD.Il27−/− mice despite comparable levels of I-Ag7 (Figure 2D). Reduced CD40 expression on intra-islet myeloid APCs as a result of IL-27 deficiency is consistent with the significantly decreased T cell infiltration and disease progression in NOD.Il27−/− mice.
Figure 2. IL-27 Deficiency Suppresses Islet Infiltration of DCs and T Cells.

(A) The percentages of CD45+ cells among single viable cells in the islets of 4- or 10-week-old NOD and NOD.Il27−/− females.
(B) The percentages of CD3+ T cells among CD45+ cells in the islets of 4- or 10-week-old NOD and NOD.Il27−/− females. Cells were gated on viable, single, and CD45+ cells.
(C) The percentages of F4/80+ single-positive (SP), F4/80+ CD11c+, and CD11c+ SP among myeloid APCs in the islets of 4- or 10-week-old NOD and NOD.Il27−/− females. Cells were first gated on viable, single, CD45+, and CD3− cells, followed by excluding F4/80− CD11c− cells. For (A)–(C), representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels. Summarized data are representative of two independent experiments. Each symbol represents a mouse. The horizontal line indicates the mean. *p < 0.05 by Mann-Whitney test.
(D) Representative flow cytometry plots on the left depict I-Ag7 and CD40 expression among myeloid APC subsets in the islets of 10-week-old NOD or NOD.Il27−/− females. Summarized data on the right show geometric mean fluorescence intensity of I-Ag7 or CD40 among F4/80+ SP, F4/80+ CD11c+, or CD11c+ SP APC subsets in the islet of 10-week-old NOD or NOD.Il27−/− females. Summarized data are representative of two independent experiments. Each symbol represents a mouse. The horizontal bar depicts the mean. *p < 0.05 by Mann-Whitney test.
See also Figure S2.
Activation of β-Cell Autoreactive CD8 T Cells in PLNs Is Significantly Reduced in NOD.Il27−/− Mice
Although NOD.Il27−/− mice still harbor β-cell autoreactive T cells (Figure 1F), it is possible that their activation in PLNs is reduced. To test this hypothesis, we used T cell receptor (TCR) transgenic β-cell autoreactive BDC2.5 CD4 and NY8.3 CD8 T cells (Verdaguer et al., 1997; Katz et al., 1993). We transferred eFluor670-labeled BDC2.5 CD4 or NY8.3 CD8 T cells into NOD and NOD.Il27−/− recipients and analyzed their activation in PLNs 5 days after transfer. Proliferation as well as the expression of CD44 and CD62L of adoptively transferred BDC2.5 CD4 T cells were comparable in NOD and NOD.Il27−/− mice (Figures 3A and 3B). However, both proliferation and the activation phenotype (CD44high and CD62Lneg) of transferred NY8.3 CD8 T cells were significantly reduced in the PLNs of the NOD.Il27−/− strain compared with NOD mice (Figures 3C and 3D). Thus, IL-27 deficiency significantly suppressed the activation of β-cell autoreactive CD8 T cells and had less effect on the activation of CD4 T cells in PLNs.
Figure 3. Activation of β-Cell-Specific T Cells in NOD Versus NOD.Il27−/− Recipients.

(A and B) Purified splenic BDC2.5 CD4 T cells (2 × 106) were labeled with eFluor670 proliferation dye and transferred into 8- to 9-week-old female NOD or NOD.Il27−/− recipients. PLNs were harvested 5 days post-transfer, and proliferation (A) and CD44 and CD62L expression (B) of CD4+eFluor670+ cells was analyzed using flow cytometry.
(C and D) Purified splenic NY8.3 CD8 T cells (3 × 106 to 5 × 106) were labeled with eFluor670 proliferation dye and transferred into sex-matched 7- to 9-week-old NOD or NOD.Il27−/− recipients. PLNs were harvested 5 days post-transfer, and proliferation (C) and CD44 and CD62L expression (D) of CD8+eFluor670+ cells were analyzed using flow cytometry.
(E and F) Purified splenic CD45.2+ NY8.3 CD8 T cells (5 × 106) were transferred into 7- to 9-week-old female NOD or NOD.Il27−/− recipients (expressing CD45.1). Sixteen hours after transfer, PLNs were harvested and analyzed for CD69 (E) and CD44 (F) expression on CD45.2+CD8+NRP-V7 tetramer+ cells using flow cytometry.
Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels. Summarized results are from two or three independent experiments. Each symbol represents a mouse. The horizontal bar depicts the mean. *p < 0.05 and **p < 0.01 by Mann-Whitney test. NS, not significant.
To initially test whether the decreased activation of NY8.3 CD8 T cells in NOD.Il27−/− mice is due to an impairment in the early event of antigenic stimulation, we isolated splenic CD8 T cells from NOD.Cd45.2.NY8.3 mice and transferred them into NOD and NOD.Il27−/− recipients. Sixteen hours later, the expression level of the early activation marker CD69 on transferred CD45.2+ NY8.3 CD8 T cells in the PLNs was significantly lower in NOD.Il27−/− mice than in NOD recipients (Figure 3E). Similarly, CD44 expression on transferred NY8.3 CD8 T cells was significantly reduced in the PLNs of NOD.Il27−/− recipients (Figure 3F). This result suggests that antigenic stimulation of β-cell autoreactive CD8 T cells in PLNs is reduced in the IL-27-deficient mice, likely because of decreased β-cell antigen availability as a result of limited DC infiltration in islets.
IL-27 Receptor Is Essential for T1D Development in NOD Mice
To further confirm that loss of IL-27 signaling in NOD.Il27−/− mice was responsible for diabetes protection, we used CRISPR/Cas9 to target Il27ra directly in NOD mice (Figure S1A), resulting in the absence of IL-27Rα protein (Figure S1D). NOD.Il27ra−/− mice of both sexes were also completely resistant to T1D development (Figure 4A). Compared with NOD mice, NOD.Il27ra−/− mice also had significantly decreased insulitis at 10 weeks and limited progression from 10 to 30 weeks (Figures 4B and 4C; Figure S1C). These results confirm that IL-27 signaling is essential for T1D development in NOD mice. Both IL-27 and IL-6 use the gp130 receptor subunit and induce expression of some common genes in CD4 T cells through STAT1-, STAT3-, and MAPK-dependent pathways (Pot et al., 2009; Jin et al., 2013; Hirahara et al., 2015). Therefore, we asked if IL-6 also has an important role in diabetes development in NOD mice. However, T1D development in IL-6-deficient and sufficient NOD mice was indistinguishable (Figure S3).
Figure 4. T Cell-Intrinsic IL-27 Signaling Is Essential for the Development of T1D.

(A) T1D incidence of NOD and NOD.Il27ra−/− mice. ***p < 0.005 by log rank test.
(B) Summary of insulitis in female NOD and NOD.Il27ra−/− mice.
(C) Summary of insulitis in male NOD and NOD.Il27ra−/− mice. Pancreatic islets were scored for insulitis as in Figure 1. Each symbol represents one mouse. The horizontal bar depicts the mean. **p < 0.01 by Mann-Whitney test. NS, not significant.
(D) T1D incidence study of sublethally irradiated NOD.Il27−/− or NOD.Il27ra−/− females infused with BM cells (5 × 106) from sex-matched NOD.Rag1−/− mice as indicated. **p < 0.01 by log rank test.
(E–G) Incidence of T1D in recipients of adoptively transferred T cells.
(E) Splenic T cells (5 × 106) were isolated from 6-week-old NOD females and transferred into sex-matched NOD.Rag1−/− or NOD.Rag1−/−.Il27ra−/− recipients. T1D incidence was not significantly different between recipient groups.
(F) Splenic T cells (5 × 106) were isolated from 6- to 11-week-old NOD or 11- to 15-week-old NOD.Il27ra−/− females and transferred into sex-matched NOD.Rag1−/− recipients. ***p < 0.005 by log rank test.
(G) Splenic CD4 (4 × 106) and CD8 (2 × 106) T cells were isolated from indicated 6- to 8-week-old female strains and co-transferred into sex-matched NOD.Rag1−/− recipients. **p < 0.01 and ***p < 0.005 by log rank test.
(H) In vitro suppression function of NOD and NOD.Il27ra−/− Tregs. Splenic CD4+CD25− T cells were isolated from NOD mice, labeled with CFSE, and cultured either alone or in the presence of unlabeled CD4+CD25+ Tregs at the indicated ratios. Cells were activated with soluble anti-CD3 in the presence of NOD.Rag1−/− splenocytes for 3 days, and proliferation was analyzed using flow cytometry. Representative histograms of CFSE dilution from one experiment are shown in Figure S4. Summarized data from three independent experiments are shown. The percentage suppression was calculated as [percentage of divided CD4+CD25− T cells (without Tregs) – percentage of divided CD4+CD25− (with Tregs)]/[percentage of divided CD4+CD25− T cells (without Tregs)] 3 100. Error bars represent SEM. The suppression function of NOD and NOD.Il27ra−/− Tregs was not significantly different.
(I) Incidence of T1D in recipients of adoptively transferred T cells. Splenic CD25− T cells (5 × 106) isolated from 13- to 15-week-old NOD females and splenic CD4+GITR+CD25+ Tregs (5 × 105) FACS sorted from 6- to 9-week-old NOD or NOD.Il27ra−/− females were co-transferred into sex-matched NOD.Rag1−/− recipients. ***p < 0.005 by log rank test. NS, Not significant.
See also Figures S1, S3, and S4.
T Cell-Intrinsic IL-27 Signaling Is Essential for T1D Development
Next, we used the IL-27Rα-deficient mice to determine which cell types need to respond to IL-27 for T1D development. Using the same BM chimera approach described in Figure 1D, we found that NOD.Rag1−/− BM (capable of giving rise to non-T and non-B cells that can both produce and respond to IL-27) did not induce T1D in sublethally irradiated NOD.Il27ra−/− mice (Figure 4D). In addition, NOD splenic T cells equally induced T1D in NOD.Rag1−/− and NOD.Rag1−/−.Il27ra−/− recipients (Figure 4E). Hence, the ability to respond to IL-27 by non-T and non-B cells is neither sufficient nor required for T1D development in NOD mice.
Therefore, we asked if T cells need to directly respond to IL-27 to cause T1D. Splenic T cells isolated from NOD or NOD.Il27ra−/− mice were transferred into NOD.Rag1−/− recipients to compare their diabetogenic activity. T cells isolated from NOD.Il27ra−/− mice were unable to induce T1D in the NOD.Rag1−/− recipients, indicating that T cells need to respond to IL-27 to induce T1D (Figure 4F). It is most likely that NOD.Il27ra−/− mice also harbor β-cell autoreactive T cells, as in the NOD.Il27−/− strain; however, their inability to respond to IL-27 precluded them from inducing diabetes in NOD.Rag1−/− recipients. Next, we asked if both CD4 and CD8 T cells need to respond to IL-27 for T1D development. Splenic CD4 and CD8 T cells were individually isolated from NOD or NOD.Il27ra−/− mice, mixed in a “crisscross” design, and transferred into NOD.Rag1−/− recipients. As expected, co-transfer of CD4 plus CD8 T cells from NOD mice induced a high incidence of diabetes, but co-transfer of both cell types from NOD.Il27ra−/− donors did not (Figure 4G). Co-transferred NOD CD4 and NOD.Il27ra−/− CD8 T cells or NOD.Il27ra−/− CD4 and NOD CD8 T cells were able to induce T1D but the onset was delayed and the overall incidence in both recipient groups was significantly lower than those infused with NOD CD4 and CD8 T cells (Figure 4G). Therefore, IL-27 signaling in both CD4 and CD8 T cells is important for T1D development.
Direct IL-27 Signaling Does Not Alter the Suppressive Function of Regulatory T Cells
The reduced diabetogenic activity of IL-27Rα-deficient CD4 T cells could be due to altered function of effector T cells and/or regulatory T cells (Tregs). We next determined whether the decreased diabetogenicity of IL-27Rα-deficient CD4 T cells was due to the enhanced suppressive function of IL-27Rα-deficient Tregs. First, we tested the in vitro suppressive activities of NOD and NOD.Il27ra−/− Tregs. We co-cultured NOD or NOD.Il27ra−/− Tregs (CD4+ CD25+) with NOD effector cells (CD4+ CD25−) and NOD.Rag1−/− splenocytes in the presence of anti-CD3. We found the suppressive activity of NOD and NOD.Il27ra−/− Tregs to be similar (Figure 4H; Figure S4). As the in vitro suppression assay does not completely reflect the complexity of Treg activities, we subsequently compared their in vivo functionality. Splenic Tregs (CD4+CD25+GITR+) were independently sorted from NOD and NOD.Il27ra−/− mice and co-transferred with splenic CD25-depleted NOD total T cells in a 1:10 ratio (Treg/effector) into NOD.Rag1−/− recipients. A control group received only CD25-depleted T cells and developed rapid T1D onset as expected (Figure 4I). The mice receiving CD25-depleted T cells plus either NOD or NOD.Il27ra−/− Tregs developed T1D similarly but had significantly delayed T1D onset compared with the control group (Figure 4I). Therefore, IL-27 signaling in Tregs does not directly alter their ability to suppress T1D.
IL-27 Directly Affects the Composition of CD4 T Cell Subsets in Pancreatic Islets
Although IL-27 signaling does not appear to be essential for the proliferation of β-cell autoreactive CD4 T cells in PLNs, as shown by the BDC2.5 transfer experiment (Figures 3A and 3B), it may affect their effector function in islets. To further analyze the cell-intrinsic effects of IL-27 signaling on the effector function of CD4 T cells, we directly compared wild-type and Il27ra−/− CD4 T cells in the mixed BM chimeras. We reconstituted lethally irradiated (NOD × NOD.Cd45.2)F1 recipients with an equal number of NOD.Cd45.2 and NOD.Il27ra−/− (expressing CD45.1) BM cells. Pre-diabetic chimeras were analyzed 10–12 weeks after BM reconstitution. The spleens, PLNs, and islet infiltrates of the BM chimeras were analyzed to determine if IL-27 signaling directly controls the frequency of CD4 T cells. The proportions of NOD.Cd45.2- and NOD.Il27ra−/−-derived total CD4 T cells were comparable in the spleens and PLNs, indicating that their overall reconstitution ability did not differ significantly (Figure 5A and data not shown). The frequencies of NOD.Cd45.2- and NOD.Il27ra−/−-derived total CD4 T cells were also similar in the islets (Figure 5A).
Figure 5. IL-27 Signaling Intrinsically Modulates CD4 and CD8 T Cell Subsets.

Lethally irradiated (NOD 3 NOD.Cd45.2)F1 mice were infused with equal numbers of T cell-depleted BM cells from NOD.Cd45.2 and NOD.Il27ra−/− donors. Pre-diabetic recipients were analyzed for wild-type (CD45.2+) and IL-27Rα-deficient (CD45.1+) T cell subsets 10–12 weeks after BM reconstitution.
(A) Frequencies of total CD4 and CD8 T cells in the spleens and islets of the mixed BM chimeras. Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels. Summarized results are representative of at least two independent experiments.
(B) T-bet expression in CD3+CD4+Foxp3− T cells in the spleens, PLNs, and islets of mixed BM chimeras. Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels.
(C) IFNγ production in CD3+CD4+Foxp3− T cells in the spleens, PLNs, and islets of mixed BM chimeras. Cells were stimulated with PMA and ionomycin. Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels.
(D) Frequencies of CD3+CD4+Foxp3+ Tregs in the spleens, PLNs, and islets of the mixed BM chimeras. Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the upper middle and right panels. The ratios of IL-27Rα-deficient to wild-type Treg frequencies in spleens versus islets are shown in the lower middle panel. The levels of CD25 expression are summarized in the lower right panel.
(E) T-bet expression in CD3+CD8+ T cells in the spleens, PLNs, and islets of mixed BM chimeras. Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels.
(F) IFNγ production in CD3+CD8+ T cells in the spleens, PLNs, and islets of mixed BM chimeras. Cells were stimulated with PMA and ionomycin. Representative flow cytometry profiles are shown in the left panels, and summarized results are presented in the right panels.
All summarized data in (B)–(F) are from two independent experiments. Statistical significance was determined using Wilcoxon matched-pairs signed rank test (*p < 0.05, **p < 0.01, and ***p < 0.005). NS, not significant. GMFI, geometric mean fluorescence intensity. See also Figure S5.
To further determine whether IL-27 signaling can directly control the balance of effector and regulatory CD4 T cells, we analyzed transcription factor expression and cytokine production in the mixed BM chimeras. The frequency of NOD.Il27ra−/− derived T-bet+ Foxp3−CD4 T cells was significantly reduced compared with those originating from NOD.Cd45.2 in the spleens, PLNs, and islets of the mixed BM chimeras (Figure 5B). The frequency of NOD.Il27ra−/−-derived interferon (IFN)γ-producing Foxp3− CD4 T cells was also significantly reduced in the spleens, PLNs, and islets of the mixed BM chimeras (Figure 5C). There was minimal detection of IL-17A and IL-4 production (data not shown). These results indicate that IL-27 signaling directly promotes the development of a pathogenic T helper 1 (Th1) T cell response in NOD mice. On the other hand, the frequency of NOD.Il27ra−/−-derived Tregs was significantly increased compared with those of the NOD.Cd45.2 origin in the islets and spleens but not PLNs (Figure 5D). Interestingly, the ratio of NOD.Il27ra−/− to NOD.Cd45.2-derived Tregs was significantly larger in the islets than in the spleens (Figure 5D). This observation suggests that there is a local effect of IL-27 signaling to proportionally promote Treg accumulation in the islet environment. Further supporting this possibility is the observation that the absolute number of Il27ra−/− Tregs was higher than those of the NOD.Cd45.2 origin in the pancreatic islets but not the spleens and PLNs of the mixed BM chimeras (Figure S5). There was not a difference in CD25 expression on NOD.Cd45.2- or NOD.Il27ra−/−-derived Tregs in the islets (Figure 5D), indicating that the increased frequency of NOD.Il27ra−/−-derived Tregs is not due to an enhanced ability to bind IL-2. Together these results indicate that CD4 T cell-intrinsic IL-27 signaling tips the balance between Tregs and Th1 effectors and favors the latter population in the islet environment. This is likely an important factor contributing to T1D resistance in NOD.Il27−/− and NOD.Il27ra−/− mice.
IL-27 Directly Promotes the Accumulation and Effector Function of CD8 T Cells in Pancreatic Islets
Using the same mixed BM chimera approach described above, we further defined the intrinsic effects of IL-27 signaling on CD8 T cell frequency and function. The proportions of NOD.Cd45.2- and NOD.Il27ra−/−-derived total CD8 T cells were comparable in the spleens and PLNs, indicating that their overall reconstitution ability did not differ significantly (Figure 5A and data not shown). Interestingly, the frequency of NOD.Il27ra−/−-derived CD8 T cells was significantly decreased compared with those of the NOD.Cd45.2 origin in the pancreatic islets (Figure 5A). This result indicates that CD8 T cell-intrinsic IL-27 signaling promotes their islet accumulation. To further define the intrinsic effects of IL-27 signaling on CD8 T cell function, we analyzed their T-bet expression and IFNγ production in the mixed BM chimera mice. The frequency of T-bet+ cells among total NOD.Il27ra−/−derived CD8 T cells was significantly reduced compared with those of the NOD.Cd45.2 origin in the spleens and islets but not the PLNs of the mixed BM chimeras (Figure 5E). Consistently, the frequency of NOD.Il27ra−/−-derived CD8 T cells capable of producing IFNγ was significantly reduced compared with those derived from NOD.Cd45.2 in the islets but not the spleens or PLNs of the mixed BM chimeras (Figure 5F). These results suggest that direct IL-27 signaling within pancreatic islets is important for optimal pathogenic CD8 T cell differentiation.
IL-27 Signaling in T Cells Is Required for Lacrimal and Salivary Gland Inflammation
In addition to T1D, NOD mice spontaneously develop autoimmunity of lacrimal and salivary glands and are a well-established model of Sjögren syndrome (Park et al., 2015). To determine if disruption of IL-27 signaling has a broader effect in autoimmunity, we analyzed lacrimal and salivary glands from NOD, NOD.Il27−/−, and NOD.Il27ra−/− mice. In NOD mice, Sjögren syndrome-like manifestations occur in a sex-specific manner, with males spontaneously developing lacrimal gland inflammation and females developing spontaneous salivary gland inflammation (Hunger et al., 1998, 1996; Lieberman et al., 2015; Mikulowska-Mennis et al., 2001; Takahashi et al., 1997; Toda et al., 1999). Both male lacrimal and female salivary gland inflammation were significantly reduced in NOD.Il27−/− and NOD.Il27ra−/− mice compared with age- and sex-matched wild-type NOD mice (Figures 6A and 6B). This protection was not transient as 30-week-old NOD.Il27−/− mice showed little or no lacrimal or salivary gland inflammation, with median focus scores of 0.46 foci/4 mm2 (range 0.18–2.46, n = 6) and 0 foci/4 mm2 (range 0–0.76, n = 5), respectively. Similarly, 21-weekold NOD.Il27ra−/− mice showed significantly decreased lacrimal gland inflammation compared with age- and sex-matched NOD mice (Figure 6C). Because IL-27 signaling was required for pathogenic T cells in the context of autoimmune diabetes (Figure 4F), we asked if IL-27 was similarly required for pathogenic effector T cells in the context of Sjögren syndrome-like manifestations in NOD mice. We isolated CD25-depleted splenic T cells from NOD or NOD.Il27ra−/− males and transferred them to sex-matched NOD-scid recipients. NOD T cells caused typical focal lymphocytic inflammation in NOD-scid mice, whereas recipients of NOD.Il27ra−/− T cells were protected from the development of lacrimal gland inflammation (Figure 6D). Thus, in the absence of IL-27 signaling, effector T cells failed to adequately infiltrate lacrimal glands, demonstrating that the pathogenic role of IL-27 in driving autoimmunity in NOD mice is not limited to pancreatic islets.
Figure 6. Effector T Cells Require IL-27 Signaling for Sjögren Syndrome-like Inflammation in NOD Mice.

(A and B) Quantification of inflammation of male lacrimal (A) and female salivary (B) glands from 10-week-old NOD, NOD.Il27−/−, and NOD.Il27ra−/− mice. Symbols represent individual mice, and lines are medians. p < 0.0001 (A) and p = 0.0014 (B) by Kruskal-Wallace test with Dunn’s post-test p values as indicated. *p < 0.05, **p < 0.01, and ***p < 0.005.
(C) Graph depicts quantification of inflammation of male lacrimal glands from 21-week-old NOD and NOD.Il27ra−/− mice. Symbols represent individual mice, and lines are medians. Boxed symbol represents diffuse inflammation with foci so numerous that they coalesced, preventing accurate enumeration. *p < 0.05 by Mann-Whitney test. Histology images are H&E-stained tissue sections representative of NOD (top) and NOD.Il27ra−/− (bottom) mice and demonstrate the characteristic focal inflammatory cell infiltrates that are more abundant in NOD lacrimal glands. Scale bar is 1 mm.
(D) Quantification of lacrimal gland inflammation in male NOD-scid recipients of sorted effector T cells (CD8+ and CD4+CD25− sorted together) from spleens of male NOD or NOD.Il27ra−/− mice. Symbols represent individual mice pooled from two independent transfers. Lines are medians. ***p < 0.005 by Mann-Whitney test.
DISCUSSION
In this study we aimed to better define the function of IL-27 signaling in T1D autoimmune pathogenesis. Our data show that IL-27 signaling promotes the development of insulitis and progression to T1D through multiple mechanisms. We found that IL-27 production by non-T and non-B cells was both necessary and sufficient to drive T1D progression. However, the ability of non-T and non-B cells to respond to IL-27 was neither necessary nor sufficient for T1D development. Notably, T cell-intrinsic IL-27 signaling was critical for progression to T1D. Direct IL-27 signaling in both CD4 and CD8 T cells was found to be important. CD4 T cell-intrinsic IL-27 signaling affected the balance of the Treg and Th1 effector cells and favored the latter population. Direct IL-27 signaling also promoted the accumulation of CD8 T cells in the islets and enhanced the expression of the effector molecules T-bet and IFNγ. Together, our results support a model wherein macrophage/DC-derived IL-27 promotes CD8 T cell-mediated β cell destruction directly and indirectly through modulation of CD4 T cells.
Many pro-inflammatory effects of IL-27 signaling are mediated through activation of STAT1 (Hunter and Kastelein, 2012). The completely protective phenotype of NOD.Il27−/− and NOD.Il27ra−/− mice is particularly significant considering that NOD.Stat1−/− mice are also completely protected from insulitis and T1D (Kim et al., 2007). In contrast, NOD mice deficient in other genes immediately upstream of STAT1 signaling, including Il6 (reported here), Ifng, Ifngr2, and Ifnar1, develop insulitis and T1D (Serreze et al., 2000, 2001; Quah et al., 2014). Surprisingly, NOD mice deficient in both Ifngr1 and Ifnar1 still developed T1D, although the overall incidence was reduced (Carrero et al., 2018). Both type I and type II interferon (IFN) pathways can also induce IL-27 expression (Zhang et al., 2010; Blahoianu et al., 2014; Liu et al., 2007). Together these results indicate that signaling pathways independent of type I and type II IFNs stimulate IL-27 expression leading to activation of STAT1-mediated diabetogenic activities.
Earlier studies have revealed that IL-27 signaling promotes the expression of T-bet and IFNγ production by CD4 T cells via STAT1-dependent signaling (Pflanz et al., 2002; Kamiya et al., 2004; Takeda et al., 2003). Likewise, we observed that direct IL-27 signaling promoted the accumulation of T-bet+ CD4 T cells and enhanced their production of IFNγ in the spleen, PLN, and pancreatic islets. NOD mice deficient in T-bet are protected from T1D, and T-bet-deficient BDC2.5 CD4 T cells are impaired in their ability to transfer T1D (Esensten et al., 2009). However, given the dispensable role of IFNγ signaling in T1D progression, IL-27-induced T-bet expression likely enhances the diabetogenic activity of CD4 T cells through an IFNγ-independent mechanism. Numerous studies have shown that CD4 T cells can provide help to CD8 T cells by directly activating DCs via IFNγ-dependent and IFNγ-independent mechanisms (Hivroz et al., 2012). Insulin reactive CD4 T cells and myeloid APCs are among the first cells to infiltrate the pancreatic islets (Unanue et al., 2016; Ferris et al., 2014). CD11b− CD103+ migratory DCs are critical for autoreactive CD8 T cell activation in PLNs and DC maturation in islets precedes their migration to the PLN (Melli et al., 2009; Ferris et al., 2016). Furthermore, transfer of autoreactive CD4 T cells induced maturation of migratory DCs in the pancreatic islets (Melli et al., 2009). Interestingly, we show here that in the absence of IL-27 signaling, there is limited DC and T cell infiltration in the islets and minimal expression of CD40 on myeloid APC subsets over time. Moreover, antigenic-specific activation of autoreactive CD8 T cells was suppressed in the PLN of IL-27-deficient hosts. Together, these results suggest that in the absence of IL-27 signaling, DC infiltration to the pancreatic islets and antigen trafficking to the PLN are insufficient to activate CD8 T cells and amplify the autoimmune response. Interestingly, we found that direct IL-27 signaling in myeloid APCs was not sufficient or required for T1D progression. This suggests that CD4 T cell-intrinsic IL-27 signaling promotes their ability to activate APCs in the pancreatic islets. Further experiments are required to determine whether this effect is T-bet dependent.
IL-27 can also signal through activation of STAT3 (Hunter and Kastelein, 2012). The role of STAT3 in mediating T1D is difficult to discern because of the embryonic lethality of global STAT3-deficient mice (Takeda et al., 1997). Multiple cytokines signaling through STAT3 activation have been implicated in human and NOD mouse T1D pathogenesis, including IL-2, IL-6, IL-15, and IL-21 (Tang et al., 2008; Hulme et al., 2012; Ihantola et al., 2018; Hundhausen et al., 2016; Chen et al., 2013a; Yuan et al., 2018; Spolski et al., 2008; Sutherland et al., 2009; Ferreira et al., 2015). Although IL-6 has been implicated in human T1D, we did not observe a significant role of this cytokine in diabetes development in NOD mice. It has been previously demonstrated that IL-27 signaling can upregulate IL-21 expression in both human and mouse CD4 T cells via STAT3-dependent signaling (Batten et al., 2010). Significantly, NOD.Il21−/− and NOD.Il21r−/− mice are both completely protected from T1D (Sutherland et al., 2009; Spolski et al., 2008; Chen et al., 2013b). Specifically, CD4 T cell-derived IL-21 is required for T1D progression, and IL-21R-deficient CD4 and CD8 T cells are impaired in their ability to transfer T1D (McGuire et al., 2011). Furthermore, IL-21 signaling promotes the migration and co-stimulatory functions of DCs during T1D progression (Van Belle et al., 2012). Therefore, it is conceivable that the reduced diabetogenicity of IL-27Rα-deficient CD4 T cells could be due to an inability to produce IL-21. The concept that IL-27-induced IL-21 production by CD4 T cells may drive autoimmune pathogenesis is further supported by two observations in the progression of Sjögren syndrome: (1) our observation that IL-27 signaling in T cells is required for lacrimal and salivary gland inflammation and (2) IL-21 expression in T cells correlates with the degree of lymphocytic infiltration in labial salivary glands in patients with Sjögren syndrome (Kang et al., 2011).
In addition to the indirect effect of IL-27 through CD4 T cells, IL-27 can also signal directly in CD8 T cells. Several in vitro studies have shown that IL-27 signals through STAT1 and STAT3 in CD8 T cells and promotes the expression of molecules important for an effector response, including T-bet, EOMES, IFNγ, and granzyme B (Schneider et al., 2011; Morishima et al., 2005, 2010). Yet the direct effects of IL-27 on CD8 T cells in vivo remain understudied. Pennock et al. (2014) found that direct IL-27 signaling was critical for the generation of a robust antigen-specific CD8 T cell response to subunit vaccination. However, it is unclear whether the mechanisms governing the expansion of autoreactive CD8 T cells are parallel to those observed in infectious conditions. In the present study we found CD8 T cell-intrinsic IL-27 signaling to be important for T1D progression. Although direct IL-27 signaling did not have a marked effect on the effector phenotype of CD8 T cells in the spleen or PLN, it promoted the accumulation of CD8 T cells and enhanced their expression of T-bet and IFNγ in the pancreatic islets. These results are consistent with the previous observation that CD8 T cells undergo further differentiation within the local islet environment (Graham et al., 2011, 2012). The role of IL-27 in CD8 T cells in T1D is further supported by a previous study in which islet inflammation and heightened CD8 T cell responses were observed in mice transgenically overexpressing IL-27 (Wojno et al., 2011). Additional research is warranted to determine the effect of islet-centric CD8 T cell differentiation on T1D progression and the role of intrinsic IL-27 signaling in this process.
In the context of Sjögren syndrome, the role of IL-27 has not previously been well established. In Sjögren syndrome patients, serum levels of IL-27 were elevated (Xia et al., 2012); however, whether this reflected a pathogenic process driving inflammation versus a regulatory process driven by the inflammation was not known. In another mouse model of Sjögren syndrome, overexpression of IL-27 systemically by adeno-associated viral vector-mediated gene therapy resulted in decreases in IL-17 and IL-17-producing Th17 cells (Lee et al., 2012). This was associated with improvement in some disease parameters but had less of an effect on degree of inflammation within target organs. Regardless, that study suggested a possible immunoregulatory role for IL-27 when administered later in disease. In contrast, our findings demonstrate a requirement for IL-27 early in disease development, Because the lack of IL-27 or IL-27Rα in NOD mice prevented lacrimal and salivary gland inflammation, which is the earliest recognizable hallmark of Sjögren syndrome in mice or humans. This difference in the pathogenic versus protective role for IL-27 may reflect the complex immunostimulatory and immunomodulatory effects of IL-27 along with the complex roles of different lymphocyte populations in the development and progression of Sjögren syndrome-like disease in NOD mice. It is possible that IL-17 plays a pathogenic role in progression of disease, and thus, skewing away from IL-17 production through overexpression of IL-27 may limit later disease progression.
In conclusion, we identified IL-27 as an important mediator in the pathogenesis of T1D and Sjögren syndrome, and this was attributed to the effect of IL-27 signaling on the function of CD4 and CD8 T cells. Although several mechanistic questions remain, this study highlights the potential of IL-27 to be a viable therapeutic target for the treatment of autoimmune pathologies and advances our basic understanding of the function of a human T1D candidate gene.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yi-Guang Chen (yichen@mcw.edu). All unique mouse strains generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
NOD/ShiLtJ (NOD), NOD.Cg-Tg(TcraTcrbNY8.3)1Pesa/DvsJ (NOD.NY8.3), NOD.Cg-Tg(TcraBDC2.5,TcrbBDC2.5)1Doi/DoiJ (NOD.BDC2.5), NOD.129S7(B6)-Rag1tm1Mom/J (NOD.Rag1−/−), and NOD.B6-Ptprcb/6908MrkTacJ (NOD.Cd45.2) mice were obtained from The Jackson Laboratory and subsequently maintained at the Medical College of Wisconsin (MCW) or University of Iowa (U Iowa). NOD.Il27−/− mice were generated using ZFNs as previously described (Chen et al., 2014). Constructs of the ZFN pairs specifically targeting the fourth coding exon of the mouse Il27 gene were designed, assembled, and validated by Sigma-Aldrich (target sequence CTACCACACTTCGGCCCTtccctGCCATGCTGGGAGGGCTG; ZFNs bind to each sequence shown in upper case on opposite strands). Forward (5′-AAACTTGTGAATGAAATGGAAGC-3′) and reverse (5′-TTCAACCTGATTCTGGGAG-3′) primers were used for PCR followed by Sanger sequencing. A founder heterozygous for a one bp deletion predicted to disrupt the normal reading frame and introduce a premature stop codon was identified and backcrossed to NOD. N2 heterozygous mutant mice were then intercrossed to fix the mutation to homozygosity. NOD.Il27ra−/− mice were generated using the CRISPR/Cas9 technology as previously described (Presa et al., 2018). NOD embryos were intra-cytoplasmic microinjected with 3 pl of a solution containing Cas9 mRNA and single guide RNA (sgRNA) at respective concentrations of 100 μg/ml and 50 μg/ml. The guide RNA sequence (5′-TGCTACAGCGTCGGTCCCCTGGG-3′) was designed to target the second coding exon of Il27ra. The genomic region around the targeted site was amplified by PCR using forward (5′-AACTTGACCACGGTCCTCTC-3′) and reverse (5′-AATGAGTGGGCTAGCCT GAG-3′) primers and screened by Sanger sequencing. A founder heterozygous for a 34 bp deletion predicted to disrupt the normal reading frame was identified and backcrossed to NOD. At the N2 generation, heterozygous mutant mice were intercrossed to fix the mutation to homozygosity. The generation of NOD.Rag1−/−.Il27−/− and NOD.Rag1−/−.Il27ra−/− mice was accomplished by respectively outcrossing NOD.Il27−/− and NOD.Il27ra−/− mice to the NOD.Rag1−/− strain. Similarly, the NOD.Cd45.2.NY8.3 mice were generated by outcrossing NOD.Cd45.2 to the NOD.NY8.3 strain. B6.129S2-Il6tm1Kopf/J mice (Kopf et al., 1994) obtained from The Jackson Laboratory were initially backcrossed to NOD/ShiLtJ for 10 generations and then to NOD/LtDvs for three additional generations before intercrossing to generate littermates for the diabetes incidence study. All known insulin dependent diabetes susceptibility (Idd) regions were confirmed to be of NOD origin (Driver et al., 2011). All mice were used in accordance with Institutional Animal Care and Use Committee guidelines at the MCW and U Iowa. Mice used for experiments were housed in the same facility at either the MCW or U Iowa.
METHOD DETAILS
Assessment of T1D and insulitis
Mice were tested weekly for glycosuria (Bayer Diastix ®) and considered diabetic with two consecutive readings > 250 mg/dl. Pancreata from 10-week and 30-week-old non-diabetic mice were fixed in 10% neutral buffered formalin and 4μm sections were cut, discarding 60μm in between each section. The pancreatic sections were then stained with aldehyde fuchsin followed by a hematoxylin and eosin (H&E) counterstain. At least 30 islets were scored per mouse. Insulitis scores were determined as follows: 0-no infiltration, 1-leukocytes surrounding islet but no penetration, 2-estimated loss of up to 25% of the β cells, 3-estimated loss of up to 75% of the β cells, 4-end stage, less than 25% of the β cells remaining.
Generation of bone marrow chimeras
Bone marrow (BM) cells were harvested from the tibias and femurs of NOD.Rag1−/− or NOD.Il27−/−. Rag1−/− females (7–12 weeks old). BM cells (5 × 106) were injected intravenously into sublethally irradiated (600 rads) NOD.Il27−/− or NOD.Il27ra−/− female mice (5–7 weeks old). For the generation of mixed BM chimeras, BM cells were collected from 6–9-week-old NOD.Il27ra−/− and NOD.Cd45.2 females. T cells were depleted using anti-CD3e microbeads (Miltenyi Biotec). T cell-depleted NOD.Il27ra−/− and NOD.Cd45.2 BM cells were mixed at a 1:1 ratio (2.5×106 cells each) and infused into lethally irradiated (1100 rads) 6–8-week-old (NOD × NOD.Cd45.2)F1 females.
Adoptive T cell transfer
Splenic total T cells were isolated by negative selection (Pan T cell isolation kit II, Miltenyi Biotec) from NOD (6–13 weeks old), NOD.Il27−/− (14–16 weeks old), or NOD.Il27ra−/− females (12–15 weeks old) and intravenously injected (5×106 cells) into NOD.Rag1−/−, NOD.Rag1−/−.Il27−/− or NOD.Rag1−/−.Il27ra−/− female recipients. In some experiments, splenic CD4 and CD8 T cells were independently isolated from 6–8-week-old NOD and NOD. Il27ra−/− females, mixed at a 2:1 ratio (6×106 cells total), and transferred into NOD.Rag1−/− female recipients. The purity of the transferred T cells was analyzed by flow cytometry and was consistently > 93%. To test the function of Tregs in vivo, splenic CD25 depleted total T cells were isolated from 13–15-week old female NOD mice by negative selection (Pan T cell isolation kit II, Miltenyi Biotec) with the addition of biotin conjugated anti-CD25 (7D4), (BD Biosciences). Splenic Tregs were independently isolated by fluorescence-activated cell sorting (FACS) from 6–9-week-old NOD or NOD.Il27ra−/− mice. Flow cytometry verified that similar portions (> 92%) of CD3+CD4+CD25+GITR+ splenocytes from both NOD and NOD.Il27ra −/− mice were also FOXP3+. Therefore, Tregs were sorted by gating on single, CD4+CD25+GITR+ cells using clones RM4–5, PC61 and DTA-1, respectively, and a BD FACSAria II cytometer. Splenic CD25 depleted NOD total T cells (5×106) were co-transferred with or without splenic Tregs (5×105) into NOD.Rag1−/− recipients.
In vivo cell proliferation assay
Splenic CD8 T cells or CD4 T cells were isolated by negative selection (CD8+ T cell isolation kit or CD4+ T cell isolation kit, Miltenyi Biotec) from NOD.NY8.3 or NOD.BDC2.5 mice respectively. Isolated cells were labeled with 2.5μM eFluor670 (eBioscience) in Hanks balanced salt solution (HBSS, Sigma) at 37°C for 10 minutes and washed four times with complete RPMI. Labeled CD8 T cells were intravenously injected into sex-matched NOD and NOD.Il27−/− males (3 × 106) or females (5 × 106). Labeled CD4 T cells were intravenously injected into NOD or NOD.Il27−/− females (2 × 106). Five days post-transfer, PLNs were harvested from recipients and analyzed by flow cytometry. In some experiments, splenic CD8 T cells were isolated by negative selection from NOD.Cd45.2.NY8.3 females and intravenously injected into NOD or NOD.Il27−/− females (5×106). One day post-transfer, PLNs were harvested from recipients and analyzed by flow cytometry.
Flow cytometry
Fluorochrome-conjugated antibodies specific for CD45.1 (A20), CD45.2 (104), CD3ε (145–2C11 or 17A2) or TCRβ (H57–597), CD4 (RM4–5 or GK1.5), CD8α (53–6.7), IL-27Rα (2918), CD44 (IM7), CD62L (MEL-14), Foxp3 (FJK-16 s), CD25 (PC61), T-bet (eBIO4B10), IFNγ (XMG1.2), IL-17A (eBio17B7), IL-4 (11B11), CD11b (M1–70), CD11c (N418), I-Ag7 (AMS32.1), H-2Kd (SF1–1.1.1), CD103 (2E7), F4/80 (BM8), CD49b (DX5), CD40 (3/(23)), CD80 (16–10A1), CD86 (GL1), PD-L1 (MIH5), and PDCA-1 (eBIO129c) were purchased from BD Biosciences (San Jose, CA), Bio-Legend (San Diego, CA), or Thermo Fisher Scientific (Waltham, MA). MHC class I (Kd) tetramers loaded with a mimotope peptide NRP-V7(Trudeau et al., 2003) were obtained from the National Institutes of Health Tetramer Core Facility. Single cell suspensions were prepared from the spleen, PLN, or thymus at the indicated age by passing the tissue through an 80μm Nitex screen (Dynamic Aqua Supply Ltd). For analysis of DC subsets, spleens and PLNs were digested for 30 minutes at 37°C in collagenase D (400 units/mL, Sigma) and agitated with a disposable transfer pipette to obtain a single cell suspension. Cells were washed with modified HBSS (Sigma), and then washed with FACS buffer (sodium azide (1mg/mL) and 2% fetal bovine serum (FBS, GIBCO) in phosphate buffered saline (D-PBS, GIBCO)) before staining. For analysis of islet-infiltrating cells, pancreatic islets were harvested by perfusion of the pancreas with a collagenase P solution (0.5 units/mL collagenase, Roche Diagnostics, 10mg/mL DNase, Sigma, diluted in HBSS) via the common bile duct using a 30-gauge needle. The inflated pancreata were incubated for 16 minutes at 37°C, agitated, and washed three times with HBSS plus 2% FBS. Islets were hand-picked from the pancreas suspension using a dissecting microscope and micropipette. Hand-picked islets were dissociated in non-enzymatic cell dissociation buffer (GIBCO) to obtain a single cell suspension. Cells were washed with FACS buffer before staining. For all experiments, cells were blocked with Fc block (anti-mouse CD16/CD32 clone 2.4G2, BioXCell) at room temperature for 10 minutes and then stained with the indicated antibodies for 30 minutes at 4°C. Stained cells were washed with FACS buffer. Dead cells were discriminated using 7-aminoactinomycin (7AAD, Sigma). For intracellular cytokine staining, cells were cultured at 37°C for four hours in the presence of phorbol myristate acetate (PMA, 20ng/mL, Sigma), ionomycin (1μg/mL, Sigma), and BD GolgiPlug™ (1μL/mL). Cultured cells were washed with FACS buffer before staining. For intracellular staining of cytokines and transcription factors, cells were fixed and permeabilized using the Foxp3/Transcription factor staining buffer set (eBioscience) according to the instructions. Dead cells were discriminated by fixable viability stain 575V (BD Biosciences) in the cultured samples from mixed BM chimera mice. Samples were run on the LSRII or LSRFortessa X20 cytometer (BD Biosciences). Data was analyzed with FlowJo software (Tree Star, Ashland, OR). Gating for IFNγ, IL-17A, IL-4, and T-bet were based on samples stained with isotype controls at the same concentration as the corresponding antibody.
In vitro Treg suppression assay
CD4+CD25− T cells (effectors) were isolated by negative selection and CD4+CD25+ T cells (Tregs) were isolated by the CD4+CD25+ Regulatory T cell isolation kit (Miltenyi Biotec) from the spleens of 7–9-week-old male NOD and NOD.Il27ra−/− mice. Purity of isolated cells was checked by flow cytometry and was routinely > 87% for CD4+CD25− T cells and > 90% for CD4+CD25+ T cells. Isolated CD4+CD25− T cells were labeled with 2μM CFSE (Invitrogen) in HBSS at 37°C for 10 minutes and washed four times with complete RPMI. Labeled CD4+CD25− T cells from NOD donors (5×104) were co-cultured with decreasing numbers of CD4+CD25+ T cells from NOD, or NOD.Il27ra−/− (5×104, 2.5×104, 1.25×104, and 6.25×103) and 2×105 total splenocytes from NOD.Rag1−/− mice in the presence of 1μg/mL anti-CD3 (145–211, eBioscience). Control wells containing labeled CD4+CD25− T cells plus NOD.Rag1−/− total splenocytes with or without anti-CD3 were included. Cells were incubated at 37°C for three days. Cells were then washed with FACS buffer and stained with anti-CD4 and 7AAD. Flow cytometry was used to measure the proliferation of effector T cells by dilution of CFSE. The percent suppression was calculated by [percentage of divided CD4+CD25− T cells (without Tregs) - percentage of divided CD4+CD25− (with Tregs)] / [percentage of divided CD4+CD25− T cells (without Tregs)] × 100.
IL-27 ELISA
BM was harvested from the tibias and femurs of 8–10-week-old male NOD and NOD.Il27−/− mice and cultured in non-tissue culture treated plates in the presence of 25ng/mL rmM-CSF (R&D Systems) at 37°C for seven days. The supernatant was removed, and the adherent cells were washed with PBS and incubated with non-enzymatic cell dissociation buffer (GIBCO) at 37°C for 10 minutes. Adherent cells were then lifted from the plate using a cell scraper. Cells were then stimulated for 24 hours at 37°C with 100ng/mL lipopolysaccharide (LPS). ELISA was performed using a kit (Invitrogen) according to the manufacturer’s instruction to measure IL-27 heterodimers in the cell-free supernatant.
Adoptive transfer of Sjögren syndrome
Splenic T cells from the indicated strains were enriched by magnetic sorting using a negative selection T cell purification kit according to the manufacturer’s protocol (Miltenyi Biotech Inc, Auburn, CA). Enriched T cells were then labeled with fluorophore-conjugated anti-CD8a (clone 53–6.7), anti-CD4 (clone GK1.5 or RM4–5), and anti-CD25 (clone PC61) monoclonal antibodies and subjected to FACS using an Aria II or an Aria Fusion (BD Biosciences, San Jose, CA). CD4+CD25− and CD8a+ cells were purified and collected together as effector T cells. Sorted populations were > 94% TCRα positive based on post-sort purity analyses with anti-TCRα antibody (clone H57–597) acquired on a BD LSR II (BD Biosciences). For transfers, 4×106 effector T cells were transferred intravenously via retro-orbital injection to sex-matched NOD-scid recipient mice. Seven weeks later, lacrimal glands were fixed for H&E analyses to quantify inflammation.
Histology of lacrimal and salivary glands
Exorbital lacrimal and submandibular salivary glands were fixed in buffered formalin, processed, embedded in paraffin, and sectioned. Five μm sections of paired glands were stained with H&E and inflammation was quantified by light microscopy using standard focus scoring (Barr et al., 2017). Briefly, slides were analyzed at 10x magnification by a blinded observer to determine the number of mononuclear cell foci in tissue sections of male lacrimal or female salivary glands, with a focus defined as a cluster of at least 50 mononuclear cells. Slides were scanned using PathScan Enabler IV (Meyer Instruments, Houston, TX) to obtain digital images, and tissue areas were measured using ImageJ software (US National Institutes of Health, Bethesda, MD, USA) (Schneider et al., 2012). Focus scores were calculated as number of foci per 4 mm2 tissue area. Samples with foci that were so numerous that they coalesced were designated as diffuse and assigned focus scores greater than the highest calculable value for that set of comparisons. Representative images were captured on a Leitz DM-RB research microscope with a Leica DCF700T digital camera using the Leica Application Suite X software (Leica Microsystems, Wetzlar, Germany).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined by Mann-Whitney test or Wilcoxon matched-pairs signed rank test as appropriate. Log-rank test was used for analyzing T1D incidence. Multiple group comparisons of non-normally distributed data (focus scores) were performed by Kruskal-Wallace with Dunn’s multiple comparisons post-test to compare each knockout group to wild-type. All statistical tests were performed using GraphPad Prism 7 (La Jolla, CA). p < 0.05 was considered significant: * p < 0.05, ** p < 0.01, and *** p < 0.005. Statistical details of individual experiments can be found in the figures and legends.
DATA AND CODE AVAILABILITY
This study did not generate or analyze datasets or codes.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD16/CD32 | BioXCell | Clone 2.4G2, cat#BE0307; RRID:AB_2736987 |
| Biotin anti-mouse CD25 | BD PharMingen | Clone 7D4, cat#553070; RRID:AB_394602 |
| anti-mouse CD4 (eFluor450 or PE-Cy7) | eBioscience | Clone GK1.5, cat#48–0041–82 or 25–0041–81; RRID:AB_10718983 or RRID:AB_469575 |
| APC-Cy7 anti-mouse CD4 | BD PharMingen | Clone GK1.5, cat#552051; RRID:AB_394331 |
| AF700 anti-mouse CD4 | eBioscience | Clone RM4–5, cat#56–0042–82; RRID:AB_494000 |
| eFluor450 anti-mouse CD25 | eBioscience | Clone PC61.5, cat#48–0251–82; RRID:AB_10671550 |
| PE anti-mouse GITR | eBioscience | Clone DTA-1, cat#12–5874–80; RRID:AB_465985 |
| APC-Cy7 anti-mouse CD45.1 | BioLegend | Clone A20, cat#110716; RRID:AB_313505 |
| eFluor450 anti-mouse CD45.1 | Invitrogen | Clone A20, cat#48–0453–82; RRID:AB_1272189 |
| AF700 anti-mouse CD45.2 | BioLegend | Clone 104, cat#109822; RRID:AB_493731 |
| PE-Cy7 anti-mouse CD45.2 | BD Biociences | Slone 104, cat#560696; AB_1727494 |
| Brilliant Violent 510 anti-mouse CD3ε | BioLegend | Clone 17A2, cat#100234; RRID:AB_2562555 |
| PE-Cy5 anti-mouse CD3ε | BioLegend | Clone 145–2C11, cat#100310; RRID:AB_312675 |
| BUV395 anti-mouse CD3ε | BD Bioscience | Clone 145–2C11, cat#563565; RRID:AB_2738278 |
| FITC anti-mouse CD3ε | eBioscience | Clone 145–2C11, cat#11–0031–85; RRID:AB_464883 |
| APC anti-mouse TCRβ | BD PharMingen | Clone H57–597, cat#553174; RRID:AB_398534 |
| anti-mouse CD8α (BUV395 or PE) | BD Biosciences | Clone 53–6.7, cat#563786 or 553032; RRID:AB_2732919 or RRID:AB_394570 |
| PE-Cy7 anti-mouse CD8α | Invitrogen | Clone 53–6.7, cat#25–0081–82; RRID:AB_469584 |
| PE anti-mouse IL-27Rα | BD PharMingen | Clone 2918, cat#564337; RRID:AB_2738753 |
| FITC anti-mouse CD44 | eBioscience | Clone IM7, cat#11–0441–85; RRID:AB_465046 |
| PE anti-mouse CD62L | eBioscience | Clone MEL-14, cat#12–0621–81; RRID:AB_465720 |
| APC anti-mouse Foxp3 | Invitrogen | Clone FJK-16 s, cat#17–5773–82; RRID:AB_469457 |
| PE-Cy5 anti-mouse Foxp3 | eBioscience | Clone FJK-16 s, cat#15–5773–82; RRID:AB_468806 |
| PE anti-mouse T-bet | eBioscience | Clone eBio4B10, cat#12–5825–82; RRID:AB_925761 |
| APC anti-mouse IFN-γ | BD Biosciences | Clone XMG1.2, cat#554413; RRID:AB_398551 |
| PE-Cy7 anti-mouse IL-17 | eBioscience | Clone eBIO17B7, cat# 25–7177–80; RRID:AB_10717952 |
| PE anti-mouse IL-4 | BD Biosciences | Clone 11B11, cat#562044; RRID:AB_10896652 |
| APC anti-mouse H2-Kd | eBioscience | Clone SF1–1.1.1, cat#17–5957–80; RRID:AB_1311278 |
| PE anti-mouse I-Ad | BD Biosciences | Clone AMS32.1, cat#553548; RRID:AB_394915 |
| APC anti-mouse CD103 | eBiscience | Clone 2E7, cat#17–1031–82; RRID:AB_1106992 |
| eFluor450 anti-mouse F4/80 | Invitrogen | Clone BM8, cat#48–4801–82; RRID:AB_1548747 |
| Pacific Blue anti-mouse CD49b | Biolegend | Clone DX5, cat#108918; RRID:AB_2265144 |
| PE-Cy7 anti-mouse CD40 | Biolegend | Clone 3/23, cat#124621; RRID:AB_10933422 |
| PE anti-mouse CD40 | BD Biosciences | Clone 3/23, cat#553791; RRID:AB_395055 |
| FITC anti-mouse CD80 | Invitrogen | Clone 16–10A1, cat#11–0801–82; RRID:AB_465133 |
| Pacific Blue anti-mouse CD86 | Biolegend | Clone GL-1, cat#105021; RRID:AB_493467 |
| PE anti-mouse PD-L1 | BD Biosciences | Clone M1H5, cat#558091; RRID:AB_397018 |
| PE anti-mouse PDCA-1 | eBioscience | Clone eBIO129c, cat#12–3171–82; RRID:AB_763424 |
| anti-mouse CD11c (APC or AF700) | eBioscience | Clone N418, cat#17–0114–81 or 56–0114–80; RRID:AB_469345 or RRID:AB_493993 |
| FITC anti-mouse CD11b | BD Biosciences | Clone M1/70, cat#557396; RRID:AB_396679 |
| Purified NA/LE anti-mouse CD3e | BD PharMingen | Clone 145–2C11, cat#553057; RRID:AB_394590 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MHC Class I (Kd) tetramers loaded with NRP-V7 | NIH Tetramer Core Facility (Trudeau et al., 2003) | N/A |
| eFluor670 | eBioscience | Cat#65–0840–85 |
| 7-aminoactinomycin D | Sigma | Cat#A9400–1MG |
| Foxp3/Transcription Factor staining buffer set | Invitrogen | Cat#00–5523–00 |
| Fixable Viability Stain 575V | BD Bioscience | Cat#565694 |
| Carboxyfluorescein succinimidyl ester | Invitrogen | Cat#V12883 |
| rmM-CSF | R&D Systems | Cat#416-ML |
| Critical Commercial Assays | ||
| Anti-CD3ε microbeads | Miltenyi Biotec | Cat#130–094–973 |
| Pan T cell isolation kit II | Miltenyi Biotec | Cat#130–095–130 |
| CD8+ T cell isolation kit | Miltenyi Biotec | Cat#130–104–075 |
| CD4+ T cell isolation kit | Miltenyi Biotec | Cat#130–104–454 |
| CD4+CD25+ Regulatory T cell isolation kit | Miltenyi Biotec | Cat#130–091–041 |
| IL-27 ELISA | Invitrogen | Cat#88–7274–88 |
| Experimental Models: Organisms/Strains | ||
| NOD/ShiLtJ | The Jackson Laboratory | JAX: 001976; RRID:IMSR_JAX:001976 |
| NOD.Cg-Tg(TcraTcrbNY8.3)1Pesa/DvsJ | The Jackson Laboratory | JAX: 005868; RRID:IMSR_JAX:005868 |
| NOD.Cg-Tg(TcraBDC2.5,TcrbBDC2.5)1Doi/DoiJ | The Jackson Laboratory | JAX: 004460; RRID:IMSR_JAX:004460 |
| NOD.129S7(B6)-Rag1tm1Mom/J | The Jackson Laboratory | JAX: 003729; RRID: IMSR_JAX:003729 |
| NOD.B6-Ptprcb/6908MrkTacJ | The Jackson Laboratory | JAX: 014149; RRID:IMSR_JAX:014149 |
| NOD.Il27−/− | This Manuscript | N/A |
| NOD.Il27ra−/− | This Manuscript | N/A |
| NOD.Rag1−/−.Il27−/− | This Manuscript | N/A |
| NOD.Rag1−/−.Il27ra−/− | This Manuscript | N/A |
| NOD.Cd45.2.NY8.3 | This Manuscript | N/A |
| NOD.Il6−/− | This Manuscript | N/A |
| Oligonucleotides | ||
| Il27ra sgRNA: TGCTACAGCGTCGGTCCCCTGGG | IDT | N/A |
| Forward primer for NOD.Il27−/− genotyping: AAACTTGTGAATGAAATGGAAGC | IDT | N/A |
| Reverse primer for NOD.Il27−/− genotyping: TTCAACCTGATTCTGGGAG | IDT | N/A |
| Forward primer for NOD.Il27ra−/− genotyping: AACTTGACCACGGTCCTCTC | IDT | N/A |
| Reverse primer for NOD.Il27ra−/− genotyping: AATGAGTGGGCTAGCCTGAG | IDT | N/A |
| Software and Algorithms | ||
| FlowJo 7.6.5 | FlowJo | https://www.flowjo.com |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Application Suite X | Lecia Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-id/ |
| Prism 7 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
NOD mice deficient in IL-27 or IL-27Rα are completely resistant to type 1 diabetes
T cell-intrinsic IL-27 signaling is essential for type 1 diabetes development
IL-27 directly alters the balance of Treg and effector T cells to favor the latter
T cells require IL-27 signaling for Sjögren syndrome-like inflammation in NOD mice
ACKNOWLEDGMENTS
We thank Stephanie Harleston for mouse colony management. We thank Sara Williams and Amanda Posgai for editing the manuscript. We gratefully acknowledge Xiaofang Wang for technical assistance with experimentation, tissue processing, and staining and Dr. Stanley Perlman for assistance with digital imaging. We thank Galina Petrova at the Children’s Research Institute (CRI) flow cytometry core for excellent technical assistance. We thank the CRI histology core for technical assistance. We thank The Jackson Laboratory Genetic Engineering Technologies group for technical support on this project. We thank the NIH Tetramer Core Facility for providing major histocompatibility complex (MHC) class I tetramers. This work was supported by NIH grants to S.M.L. (EY027731), A.E.C. (DK118786), A.M.G. (DK097605), D.V.S. (DK46266, DK 95735, and OD020351), and Y.-G.C. (DK107541, DK097605, AI125879, and DK121747), as well as the Children’s Miracle Network (to S.M.L.) and the American Association of Immunologists Careers in Immunology Fellowship (to S.M.L.). Some data presented herein were obtained at the Carver College of Medicine/Holden Comprehensive Cancer Center flow cytometry core research facility at the University of Iowa, which is funded through user fees and the generous financial support of the Carver College of Medicine, the Holden Comprehensive Cancer Center, the Iowa City Veterans Administration Medical Center, and the National Center for Research Resources of the NIH (1 S10 OD016199-01A1).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.010.
REFERENCES
- Barr JY, Wang X, Meyerholz DK, and Lieberman SM (2017). CD8 T cells contribute to lacrimal gland pathology in the nonobese diabetic mouse model of Sjögren syndrome. Immunol. Cell Biol 95, 684–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. ; Type 1 Diabetes Genetics Consortium (2009). Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet 41, 703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten M, Ramamoorthi N, Kljavin NM, Ma CS, Cox JH, Dengler HS, Danilenko DM, Caplazi P, Wong M, Fulcher DA, et al. (2010). IL-27 supports germinal center function by enhancing IL-21 production and the function of T follicular helper cells. J. Exp. Med 207, 2895–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergholdt R, Brorsson C, Palleja A, Berchtold LA, Fløyel T, Bang-Berthelsen CH, Frederiksen KS, Jensen LJ, Størling J, and Pociot F (2012). Identification of novel type 1 diabetes candidate genes by integrating genome-wide association data, protein-protein interactions, and human pancreatic islet gene expression. Diabetes 61, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blahoianu MA, Rahimi AA, Kozlowski M, Angel JB, and Kumar A (2014). IFN-γ-induced IL-27 and IL-27p28 expression are differentially regulated through JNK MAPK and PI3K pathways independent of Jak/STAT in human monocytic cells. Immunobiology 219, 1–8. [DOI] [PubMed] [Google Scholar]
- Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, Mentch FD, Qiu H, Glessner JT, Thomas KA, et al. (2011). A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 7, e1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero JA, Benshoff ND, Nalley K, and Unanue ER (2018). Type I and II interferon receptors differentially regulate type 1 diabetes susceptibility in male versus female NOD mice. Diabetes 67, 1830–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Feigenbaum L, Awasthi P, Butcher DO, Anver MR, Golubeva YG, Bamford R, Zhang X, St Claire MB, Thomas CJ, et al. (2013a). Insulin-dependent diabetes induced by pancreatic beta cell expression of IL-15 and IL-15Rα. Proc. Natl. Acad. Sci. U S A 110, 13534–13539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XL, Bobbala D, Rodriguez GM, Mayhue M, Chen YG, Ilangumaran S, and Ramanathan S (2013b). Induction of autoimmune diabetes in non-obese diabetic mice requires interleukin-21-dependent activation of autoreactive CD8+ T cells. Clin. Exp. Immunol 173, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, Forsberg MH, Khaja S, Ciecko AE, Hessner MJ, and Geurts AM (2014). Gene targeting in NOD mouse embryos using zinc-finger nucleases. Diabetes 63, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JD, Smyth DJ, Smiles AM, Plagnol V, Walker NM, Allen JE, Downes K, Barrett JC, Healy BC, Mychaleckyj JC, et al. (2008). Meta-analysis of genome-wide association study data identifies additional type 1 diabetes risk loci. Nat. Genet 40, 1399–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibra D, Cutrera JJ, and Li S (2012). Coordination between TLR9 signaling in macrophages and CD3 signaling in T cells induces robust expression of IL-30. J. Immunol 188, 3709–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driver JP, Serreze DV, and Chen YG (2011). Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin. Immunopathol 33, 67–87. [DOI] [PubMed] [Google Scholar]
- Esensten JH, Lee MR, Glimcher LH, and Bluestone JA (2009). T-bet-deficient NOD mice are protected from diabetes due to defects in both T cell and innate immune system function. J. Immunol 183, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelou M, Smyth DJ, Fortune MD, Burren OS, Walker NM, Guo H, Onengut-Gumuscu S, Chen WM, Concannon P, Rich SS, et al. (2014). A method for gene-based pathway analysis using genomewide association study summary statistics reveals nine new type 1 diabetes associations. Genet. Epidemiol 38, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira RC, Simons HZ, Thompson WS, Cutler AJ, Dopico XC, Smyth DJ, Mashar M, Schuilenburg H, Walker NM, Dunger DB, et al. (2015). IL-21 production by CD4+ effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia 58, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, and Unanue ER (2014). A minor subset of Batf3-dependent antigen-presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity 41, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris ST, Carrero JA, and Unanue ER (2016). Antigen presentation events during the initiation of autoimmune diabetes in the NOD mouse. J. Autoimmun 71, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortune MD, Guo H, Burren O, Schofield E, Walker NM, Ban M, Sawcer SJ, Bowes J, Worthington J, Barton A, et al. (2015). Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat. Genet 47, 839–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto H, Hirase T, Miyazaki Y, Hara H, Ide-Iwata N, Nishimoto-Hazuku A, Saris CJ, Yoshida H, and Node K (2011). IL-27 inhibits hyperglycemia and pancreatic islet inflammation induced by streptozotocin in mice. Am. J. Pathol 179, 2327–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Teng A, Nozawa R, Takamoto-Matsui Y, Katagiri-Matsumura H, Ikezawa Z, and Ishii Y (2009). Production of both IL-27 and IFN-gamma after the treatment with a ligand for invariant NK T cells is responsible for the suppression of Th2 response and allergic inflammation in a mouse experimental asthma model. J. Immunol 183, 254–260. [DOI] [PubMed] [Google Scholar]
- Graham KL, Krishnamurthy B, Fynch S, Mollah ZU, Slattery R, Santamaria P, Kay TW, and Thomas HE (2011). Autoreactive cytotoxic T lymphocytes acquire higher expression of cytotoxic effector markers in the islets of NOD mice after priming in pancreatic lymph nodes. Am. J. Pathol 178, 2716–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham KL, Krishnamurthy B, Fynch S, Ayala-Perez R, Slattery RM, Santamaria P, Thomas HE, and Kay TW (2012). Intra-islet proliferation of cytotoxic T lymphocytes contributes to insulitis progression. Eur. J. Immunol 42, 1717–1722. [DOI] [PubMed] [Google Scholar]
- Hirahara K, Onodera A, Villarino AV, Bonelli M, Sciumè G, Laurence A, Sun HW, Brooks SR, Vahedi G, Shih HY, et al. (2015). Asymmetric action of STAT transcription factors drives transcriptional outputs and cytokine specificity. Immunity 42, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hivroz C, Chemin K, Tourret M, and Bohineust A (2012). Crosstalk between T lymphocytes and dendritic cells. Crit. Rev. Immunol 32, 139–155. [DOI] [PubMed] [Google Scholar]
- Hulme MA, Wasserfall CH, Atkinson MA, and Brusko TM (2012). Central role for interleukin-2 in type 1 diabetes. Diabetes 61, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundhausen C, Roth A, Whalen E, Chen J, Schneider A, Long SA, Wei S, Rawlings R, Kinsman M, Evanko SP, et al. (2016). Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci. Transl. Med 8, 356ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunger RE, Müller S, Laissue JA, Hess MW, Carnaud C, Garcia I, and Mueller C (1996). Inhibition of submandibular and lacrimal gland infiltration in nonobese diabetic mice by transgenic expression of soluble TNF-receptor p55. J. Clin. Invest 98, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunger RE, Carnaud C, Vogt I, and Mueller C (1998). Male gonadal environment paradoxically promotes dacryoadenitis in nonobese diabetic mice. J. Clin. Invest 101, 1300–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter CA, and Kastelein R (2012). Interleukin-27: balancing protective and pathological immunity. Immunity 37, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihantola EL, Viisanen T, Gazali AM, Näntö-Salonen K, Juutilainen A, Moilanen L, Rintamäki R, Pihlajamäki J, Veijola R, Toppari J, et al. (2018). Effector T cell resistance to suppression and STAT3 signaling during the development of human type 1 diabetes. J. Immunol 201, 1144–1153. [DOI] [PubMed] [Google Scholar]
- Jin JO, Han X, and Yu Q (2013). Interleukin-6 induces the generation of IL-10-producing Tr1 cells and suppresses autoimmune tissue inflammation. J. Autoimmun 40, 28–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya S, Owaki T, Morishima N, Fukai F, Mizuguchi J, and Yoshimoto T (2004). An indispensable role for STAT1 in IL-27-induced T-bet expression but not proliferation of naive CD4+ T cells. J. Immunol 173, 3871–3877. [DOI] [PubMed] [Google Scholar]
- Kang KY, Kim HO, Kwok SK, Ju JH, Park KS, Sun DI, Jhun JY, Oh HJ, Park SH, and Kim HY (2011). Impact of interleukin-21 in the pathogenesis of primary Sjögren’s syndrome: increased serum levels of interleukin-21 and its expression in the labial salivary glands. Arthritis Res. Ther 13, R179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasela S, Kisand K, Tserel L, Kaleviste E, Remm A, Fischer K, Esko T, Westra HJ, Fairfax BP, Makino S, et al. (2017). Pathogenic implications for autoimmune mechanisms derived by comparative eQTL analysis of CD4+ versus CD8+ T cells. PLoS Genet. 13, e1006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz JD, Wang B, Haskins K, Benoist C, and Mathis D (1993). Following a diabetogenic T cell from genesis through pathogenesis. Cell 74, 1089–1100. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim HS, Chung KW, Oh SH, Yun JW, Im SH, Lee MK, Kim KW, and Lee MS (2007). Essential role for signal transducer and activator of transcription-1 in pancreatic beta-cell death and autoimmune type 1 diabetes of nonobese diabetic mice. Diabetes 56, 2561–2568. [DOI] [PubMed] [Google Scholar]
- Kimura D, Miyakoda M, Kimura K, Honma K, Hara H, Yoshida H, and Yui K (2016). Interleukin-27-producing CD4(+) T cells regulate protective immunity during malaria parasite infection. Immunity 44, 672–682. [DOI] [PubMed] [Google Scholar]
- Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, and Köhler G (1994). Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342. [DOI] [PubMed] [Google Scholar]
- Lee BH, Carcamo WC, Chiorini JA, Peck AB, and Nguyen CQ (2012). Gene therapy using IL-27 ameliorates Sjögren’s syndrome-like autoimmune exocrinopathy. Arthritis Res. Ther 14, R172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman SM, Kreiger PA, and Koretzky GA (2015). Reversible lacrimal gland-protective regulatory T-cell dysfunction underlies male-specific autoimmune dacryoadenitis in the non-obese diabetic mouse model of Sjögren syndrome. Immunology 145, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Guan X, and Ma X (2007). Regulation of IL-27 p28 gene expression in macrophages through MyD88- and interferon-gamma-mediated pathways. J. Exp. Med 204, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas S, Ghilardi N, Li J, and de Sauvage FJ (2003). IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. U S A 100, 15047–15052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire HM, Vogelzang A, Ma CS, Hughes WE, Silveira PA, Tangye SG, Christ D, Fulcher D, Falcone M, and King C (2011). A subset of interleukin-21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 34, 602–615. [DOI] [PubMed] [Google Scholar]
- Meka RR, Venkatesha SH, Dudics S, Acharya B, and Moudgil KD (2015). IL-27-induced modulation of autoimmunity and its therapeutic potential. Autoimmun. Rev 14, 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melli K, Friedman RS, Martin AE, Finger EB, Miao G, Szot GL, Krummel MF, and Tang Q (2009). Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J. Immunol 182, 2590–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikulowska-Mennis A, Xu B, Berberian JM, and Michie SA (2001). Lymphocyte migration to inflamed lacrimal glands is mediated by vascular cell adhesion molecule-1/alpha(4)beta(1) integrin, peripheral node addressin/l-selectin, and lymphocyte function-associated antigen-1 adhesion pathways. Am. J. Pathol 159, 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima N, Owaki T, Asakawa M, Kamiya S, Mizuguchi J, and Yoshimoto T (2005). Augmentation of effector CD8+ T cell generation with enhanced granzyme B expression by IL-27. J. Immunol 175, 1686–1693. [DOI] [PubMed] [Google Scholar]
- Morishima N, Mizoguchi I, Okumura M, Chiba Y, Xu M, Shimizu M, Matsui M, Mizuguchi J, and Yoshimoto T (2010). A pivotal role for interleukin-27 in CD8+ T cell functions and generation of cytotoxic T lymphocytes. J. Biomed. Biotechnol 2010, 605483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, Farber E, Bonnie JK, Szpak M, Schofield E, et al. ; Type 1 Diabetes Genetics Consortium (2015). Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet 47, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owaki T, Asakawa M, Fukai F, Mizuguchi J, and Yoshimoto T (2006). IL-27 induces Th1 differentiation via p38 MAPK/T-bet- and intercellular adhesion molecule-1/LFA-1/ERK1/2-dependent pathways. J. Immunol 177, 7579–7587. [DOI] [PubMed] [Google Scholar]
- Park YS, Gauna AE, and Cha S (2015). Mouse models of primary Sjogren’s syndrome. Curr. Pharm. Des 21, 2350–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock ND, Gapin L, and Kedl RM (2014). IL-27 is required for shaping the magnitude, affinity distribution, and memory of T cells responding to subunit immunization. Proc. Natl. Acad. Sci. U S A 111, 16472–16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Fowler KD, Chalmin F, Merkler D, Kuchroo VK, and Pot C (2015). IL-27 induces Th17 differentiation in the absence of STAT1 signaling. J. Immunol 195, 4144–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, et al. (2002). IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity 16, 779–790. [DOI] [PubMed] [Google Scholar]
- Pflanz S, Hibbert L, Mattson J, Rosales R, Vaisberg E, Bazan JF, Phillips JH, McClanahan TK, de Waal Malefyt R, and Kastelein RA (2004). WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol 172, 2225–2231. [DOI] [PubMed] [Google Scholar]
- Pot C, Jin H, Awasthi A, Liu SM, Lai CY, Madan R, Sharpe AH, Karp CL, Miaw SC, Ho IC, and Kuchroo VK (2009). Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol 183, 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presa M, Racine JJ, Dwyer JR, Lamont DJ, Ratiu JJ, Sarsani VK, Chen YG, Geurts A, Schmitz I, Stearns T, et al. (2018). A hypermorphic Nfkbid allele contributes to impaired thymic deletion of autoreactive diabetogenic CD8+ T cells in NOD mice. J. Immunol 201, 1907–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quah HS, Miranda-Hernandez S, Khoo A, Harding A, Fynch S, Elkerbout L, Brodnicki TC, Baxter AG, Kay TW, Thomas HE, and Graham KL (2014). Deficiency in type I interferon signaling prevents the early interferon-induced gene signature in pancreatic islets but not type 1 diabetes in NOD mice. Diabetes 63, 1032–1040. [DOI] [PubMed] [Google Scholar]
- Schneider R, Yaneva T, Beauseigle D, El-Khoury L, and Arbour N (2011). IL-27 increases the proliferation and effector functions of human naïve CD8+ T lymphocytes and promotes their development into Tc1 cells. Eur. J. Immunol 41, 47–59. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serreze DV, and Leiter EH (2001). Genes and cellular requirements for autoimmune diabetes susceptibility in nonobese diabetic mice. Curr. Dir. Autoimmun 4, 31–67. [DOI] [PubMed] [Google Scholar]
- Serreze DV, Post CM, Chapman HD, Johnson EA, Lu B, and Rothman PB (2000). Interferon-gamma receptor signaling is dispensable in the development of autoimmune type 1 diabetes in NOD mice. Diabetes 49, 2007–2011. [DOI] [PubMed] [Google Scholar]
- Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez-Pinzon WL, and Rabinovitch A (2001). Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J. Immunol 166, 1352–1359. [DOI] [PubMed] [Google Scholar]
- Spolski R, Kashyap M, Robinson C, Yu Z, and Leonard WJ (2008). IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc. Natl. Acad. Sci. U S A 105, 14028–14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, Grusby MJ, and von Herrath M (2009). Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes 58, 1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Ishimaru N, Yanagi K, Haneji N, Saito I, and Hayashi Y (1997). High incidence of autoimmune dacryoadenitis in male non-obese diabetic (NOD) mice depending on sex steroid. Clin. Exp. Immunol 109, 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, Kishimoto T, and Akira S (1997). Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. U S A 94, 3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW, Yoshimura A, and Yoshida H (2003). Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J. Immunol 170, 4886–4890. [DOI] [PubMed] [Google Scholar]
- Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA (2008). Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda I, Sullivan BD, Rocha EM, Da Silveira LA, Wickham LA, and Sullivan DA (1999). Impact of gender on exocrine gland inflammation in mouse models of Sjögren’s syndrome. Exp. Eye Res 69, 355–366. [DOI] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al. ; Genetics of Type 1 Diabetes in Finland; Wellcome Trust Case Control Consortium (2007). Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet 39, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau JD, Kelly-Smith C, Verchere CB, Elliott JF, Dutz JP, Finegood DT, Santamaria P, and Tan R (2003). Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J. Clin. Invest 111, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unanue ER, Ferris ST, and Carrero JA (2016). The role of islet antigen presenting cells and the presentation of insulin in the initiation of autoimmune diabetes in the NOD mouse. Immunol. Rev 272, 183–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Belle TL, Nierkens S, Arens R, and von Herrath MG (2012). Interleukin-21 receptor-mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity 36, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdaguer J, Schmidt D, Amrani A, Anderson B, Averill N, and Santamaria P (1997). Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J. Exp. Med 186, 1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Han G, Wang J, Chen G, Xu R, Wang L, Li X, Shen B, and Li Y (2008). The pathogenic role of interleukin-27 in autoimmune diabetes. Cell. Mol. Life Sci 65, 3851–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. (2013). Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojno ED, Hosken N, Stumhofer JS, O’Hara AC, Mauldin E, Fang Q, Turka LA, Levin SD, and Hunter CA (2011). A role for IL-27 in limiting T regulatory cell populations. J. Immunol 187, 266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Shen H, Zhao L, and Lu J (2012). Elevated levels of interleukin-27 in patients with Sjögren’s syndrome. Scand. J. Rheumatol 41, 73–74. [DOI] [PubMed] [Google Scholar]
- Yoshida H, and Hunter CA (2015). The immunobiology of interleukin-27. Annu. Rev. Immunol 33, 417–443. [DOI] [PubMed] [Google Scholar]
- Yuan X, Dong Y, Tsurushita N, Tso JY, and Fu W (2018). CD122 blockade restores immunological tolerance in autoimmune type 1 diabetes via multiple mechanisms. JCI Insight 3, 96600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Qian X, Ning H, Yang J, Xiong H, and Liu J (2010). Activation of IL-27 p28 gene transcription by interferon regulatory factor 8 in cooperation with interferon regulatory factor 1. J. Biol. Chem 285, 21269–21281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or codes.