

Abstract
Fluorescence in situ hybridization (FISH) is currently the gold-standard assay to detect recurrent genomic abnormalities of prognostic significance in multiple myeloma (MM). Since most translocations in MM involve a position effect with heterogeneous breakpoints, we hypothesize that FISH has the potential to miss translocations involving these regions. We evaluated 70 bone marrow samples from patients with plasma cell dyscrasia by FISH and whole-genome mate-pair sequencing (MPseq). Thirty cases (42.9%) displayed at least one instance of discordance between FISH and MPseq for each primary and secondary abnormality evaluated. Nine cases had abnormalities detected by FISH that went undetected by MPseq including 6 tetraploid clones and three cases with missed copy number abnormalities. In contrast, 19 cases had abnormalities detected by MPseq that went undetected by FISH. Seventeen were MYC rearrangements and two were 17p deletions. MPseq identified 36 MYC abnormalities and 17 (50.0% of MYC abnormal group with FISH results) displayed a false negative FISH result. MPseq identified 10 cases (14.3%) with IgL rearrangements, a recent marker of poor outcome, and 10% with abnormalities in genes associated with lenalidomide response or resistance. In summary, MPseq was superior in the characterization of rearrangement complexity and identification of secondary abnormalities demonstrating increased clinical value compared to FISH.
Subject terms: Cancer genomics, Cancer genetics
Introduction
Multiple myeloma (MM) is a plasma cell neoplasm (PCN) representing the second most common hematopoietic malignancy and accounts for ~20% of all hematologic cancer related deaths in the United States1. During the last decade there have been remarkable improvements in the treatment of patients with MM that have resulted in increased survival, including immunomodulatory compounds, proteasome inhibitors, and immunotherapeutic approaches such as monoclonal antibodies2. Paralleling the advances in novel therapeutic strategies, characterization of the genomic complexities of MM have significantly improved with the implementation of next-generation sequencing (NGS), thus enabling the identification of novel single nucleotide variants (SNV), structural rearrangements and copy number abnormalities (CNA)3–11. Comprehensive genomic characterization studies such as the Multiple Myeloma Research Foundation (MMRF) CoMMpass Trial and other research studies are necessary for the discovery of novel variants of clinical significance that may lead to improved treatment approaches and prognostication strategies12,13.
In contrast to the use of genome-wide NGS strategies employed in the research/investigational trial setting, most clinical genomics laboratories rely upon traditional cytogenetic methodologies such as conventional chromosome studies and fluorescence in situ hybridization (FISH) to characterize recurrent cytogenetic abnormalities of prognostic significance. High-risk cytogenetic abnormalities as defined by the Mayo Clinic mSMART 3.0 algorithm14 include t(4;14), t(14;16), t(14;20) translocations, 17p deletions and 1q gains, while standard-risk cytogenetic abnormalities include hyperdiploidy (gains of odd-numbered chromosomes), t(11;14) and t(6;14) translocations15,16. A limited number of laboratories evaluate for MYC and t(6;14) rearrangements, and detection of IGK and IgL rearrangements is not routinely performed in the clinical setting15. Although FISH assays have high sensitivity, are relatively inexpensive compared to NGS techniques and provide input for risk stratification17, several limitations exist. They allow for the interrogation of only the regions for which FISH probes are available and multiple FISH probes are needed in order to be comprehensive, with each probe requiring a resource-consuming validation. More importantly, FISH has the potential to miss cryptic abnormalities, including rearrangements that result in a position effect due to juxtaposition of enhancers near oncogenes18–22. Since many translocations identified in MM involve a position effect (i.e., IGH and MYC) with heterogeneous breakpoints10,23,24 and some CNAs may be cryptic, we hypothesize that some clinical FISH probes used in the characterization of PCNs have a high rate of false negative FISH results.
To test this hypothesis, we evaluated the performance of a genome wide mate-pair sequencing (MPseq) assay in comparison to FISH panel testing for MM. Since MPseq utilizes long input DNA (2–5 Kb) followed by circularization and fragmentation to the size of paired-end fragments (200–500 bp) that are sequenced at reduced depth, this assay is designed to detect structural rearrangements and CNAs throughout the genome resulting in a cost-effective strategy amenable to a clinical genomics laboratory. Furthermore, as MPseq has higher resolution than FISH and is not limited to specific genomic footprints for interrogation, this assay could provide an alternative technique to comprehensively detect structural rearrangements and CNAs in a single assay. Herein we describe the performance, along with the added clinical utility, of MPseq in 70 samples previously characterized by FISH to detect chromosome rearrangements and CNAs in patients with a PCN.
Methods
Patient samples
All samples were referred to the Mayo Clinic Genomics Laboratory as part of routine clinical testing and further evaluated by MPseq as part of a Mayo Clinic Institutional Review Board approved study. There were multiple sources of samples obtained either from fresh or frozen whole bone marrow (BM), or from fixed cell pellets (FCP) from an abnormal BM chromosome study. Some specimens had undergone plasma cell enrichment from fresh whole BM that was either flow sorted or subjected to CD138+ magnetic-enrichment from patients that had an abnormal plasma cell FISH result. Additional methodology, including conventional chromosome analysis and flow cytometry are included in supplemental data.
Fluorescence in situ hybridization
Plasma cell proliferative disorder FISH (PCPDF) of immunoglobulin (cIg)-stained positive PCs studies were performed as previously described25 using the following probes to detect primary and secondary MM abnormalities: monosomy 13 or 13q deletion (Abbott Molecular, Abbott Park, IL), monosomy 17 or TP53 deletion (Abbott Molecular), trisomy 3, 7, 9 or 15 (Abbott Molecular), 1q gain (in house, custom developed), MYC rearrangement (Abbott Molecular), IGH rearrangement (in house, custom developed), t(11;14) CCND1/IGH (Abbott Molecular), t(4;14)(p16.3;q32) FGFR3/IGH (Abbott Molecular), t(6;14)(p21;q32) CCND3/IGH (Abbott Molecular), t(14;16)(q32;q23) IGH/MAF (Abbott Molecular), and t(14;20)(q32;q12) IGH/MAFB (Abbott Molecular). The PCN FISH panel is indicated in supplemental Table 1 with footprints and probe source shown in supplemental Table 2.
Plasma cell enrichment
BM cells (20 × 106) were lysed in ACK lysis buffer for 5 min. This was followed by 2 wash steps in PBS (lyse-wash procedure) and the cell pellet was re-suspended in 3% BSA/PBS. 10 × 106 cells were then incubated for 15 min with the following antibodies: CD19-PerCP 5.5 (clone SJ25C1, BD Biosciences), CD38-APC (clone REA671, Miltenyi Biotec), CD45-BB515 (clone HI30, BD Biosciences), CD56-PE-Cy7 (clone NCAM16.2, BD Biosciences), CD138-BV421 (clone MI15, BD Biosciences), and CD319-PE (clone REA150, Miltenyi Biotec). The specimen was centrifuged and re-suspended in 1.5 mL of PBS. Sorting was performed on BD FACSMelody cell sorter (BD Biosciences, San Jose, CA). Sorting streams were defined for each case separately, using gates to include CD138-positive, CD319-positive, CD38-bright, CD56-positive and/or CD45-negative plasma cells, and separate them from normal plasma cells. A minimum of 2 × 105 cells were collected, with the purity of at least 95%, verified by Kaluza software (Beckman Coulter Life Sciences, Indianapolis, IN). In some cases, plasma cells were separated by positive selection using CD138-coated magnetic beads (MACS; Miltenyi Biotec, CA) in a RoboSep system (STEMCELL Technology, Canada) as described in Jang et al.26.
DNA extraction and library preparation
DNA extraction and mate pair library preparation methods have been previously described18,27,28. Briefly, DNA was isolated using either the Qiagen Puregene extraction kit (for samples < 2 mL), Autopure LS Automated high quality DNA extraction (for samples > 2 mL) or the QIAmp Tissue kit for fixed cell pellet samples. DNA was processed using the Illumina Nextera Mate Pair library preparation kit and sequenced on the Illumina HiSeq 2500 in rapid run mode as described in Aypar, et al.18. Pooled libraries were hybridized onto a flow cell (2 samples per lane) and sequenced using 101-basepair reads and paired end sequencing.
Structural variant bioinformatics pipeline and visualization
The sequencing data was analyzed for the detection of structural variants (SVs), which are large genomic changes (>30Kb) that involve breakpoint junctions and/or CNAs. The sequencing data was mapped to the reference genome (GRCh38) using BIMA29 and the output was analyzed using SVAtools. This set of algorithms can detect and report the breakpoint locations of both junctions and CNAs at high resolution and accuracy (Schematic in supplemental Fig. 1)18,27,28. Junctions and CNVs were graphically illustrated using genome, junction and region plots as previously described18,27,30.
Results
Patient characteristics
A total of 70 cases referred to the Mayo Clinic Genomics Laboratory for routine clinical PCN FISH testing were selected for further evaluation by MPseq (Tables 1, 2 and supplemental Table 3). Criteria for inclusion included the type of primary cytogenetic abnormality to ensure representation of each recurring rearrangement and sample source to evaluate various methods of sample attainment, including PC enrichment (Tables 1, 2). The median age was 66 years (range 42–88) demonstrating male predominance with a 1.3:1 (M:F) ratio. Fifty-seven cases (81.4%) had either a diagnosis of MM (N = 35) or a reason for referral (RFR) of MM or PCN indicated at the time of clinical testing (N = 22) (Tables 1, 2). Of thirty-five cases with complete clinical data, 13 (37.1%) were newly diagnosed (ND) and 22 (62.9%) had relapsed and/or refractory disease (RR).
Table 1.
Patient cohort.
| Site | Sex | Age (years) | Dx or RFRa | ND RR | % PC | Light chain | Sample type | Primary abnormality (FISH) | FISH | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | MCL | M | 77 | MMa | U | 85 | Kappa | FCP | 11;14 | nuc ish(MYC,RB1,LAMP1)x1,(CCND1-XT,IGH-XT)x3(CCND1-XT con IGH-XTx2) |
| 2 | MAYO | F | 65 | MM | ND | 23 | Lambda | Sort | 11;14 | nuc ish(TP73x2,1q22x3),(5′MYCx2,3’MYCx1)(5′MYC con 3′MYCx1),(CCND1-XT,IGH-XT)x3(CCND1-XT con IGH-XTx2) |
| 3 | MCL | F | 70 | PCNa | U | 19 | Kappa | Sort | 11;14 | nuc ish(MYCx2)(5′MYC sep 3′MYCx1),(CCND1-XT,IGH-XT)x3(CCND1-XT con IGH-XTx2)/(CCND1-XT,IGH-XT)x4(CCND1-XT con IGH-XTx3),(RB1,LAMP1)x1,(TP53x1,D17Z1x2) |
| 4 | MAYO | M | 71 | MM | ND | 37 | Lambda | Sort | 11;14 | nuc ish(TP73x2,1q22x3),(CCND1-XT,IGH-XT)x4–5(CCND1-XT con IGH-XTx3–4)/(CCND1-XTx1,CCND1-XT amp,IGH-XTx1,IGH-XT amp)(CCND1-XT amp con IGH-XT amp),(RB1,LAMP1)x1 |
| 5 | MCL | M | 69 | MMa | U | 50 | Kappa | Sort | 11;14 | nuc ish(TP73x2,1q22x5),(D3Z1,D9Z1,D15Z4)x3,(CCND1-XT,IGH-XT)x3(CCND1-XT con IGH-XTx2),(RB1,LAMP1)x1 |
| 6 | MAYO | M | 63 | MM | RR | 28 | Kappa | Sort | 11;14 | nuc ish(TP73x1,1q22x3–4)/(TP73x2,1q22x6),(D3Z1,D7Z1,D9Z1,D15Z4)x4,(5′MYCx3,3′MYCx2)(5′MYC con 3′MYCx2)/(5′MYCx6,3′MYCx4)(5′MYC con 3′MYCx4),(CCND1-XT,IGH-XT)x3(CCND1-XT con IGHx2)/(CCND1-XT,IGH-XT)x4(CCND1-XT con IGHx3)/(CCND1-XT,IGH-XT)x5(CCND1-XT con IGHx4),(TP53x1,D17Z1x2) |
| 7 | MAYO | F | 83 | MM | ND | 37 | Kappa | Sort | 11;14 | nuc ish(TP73x2,1q22x3),(CCND1-XT,IGH-XT)x3(CCND1-XT con IGH-XTx2) |
| 8 | MCL | M | 63 | MMa | U | 73 | Kappa | Fresh | 11;14 | nuc ish(CCND1-XTx3,IGH-XTx2)(CCND1-XT con IGHx2)/(CCND1-XTx5,IGH-XTx4)(CCND1-XT con IGHx4),(TP53x1,D17Z1x2) |
| 9 | MCL | M | 69 | MM | U | 65 | Kappa | Fresh | 11;14 | nuc ish(TP73,1q22,MYC,RB1,LAMP1,TP53,D17Z1)x4,(D3Z1,D7Z1,D9Z1,D15Z4)x3–4, (CCND1-XTx6,IGH-XTx7)(CCND1-XT con IGH-XTx4) |
| 10 | MCL | M | 77 | MMa | U | 68 | Kappa | Fresh | 11;14 | nuc ish(CCND1-XT,IGH-XT)x4(CCND1-XT con IGH-XTx3),(TP53x1,D17Z1x2) |
| 11 | MAYO | M | 58 | PCL | RR | 58 | Lambda | Frozen | 11;14 | nuc ish(CCND1-XT,IGH-XT)x4(CCND1-XT con IGH-XTx3),(RB1x1,LAMP1x2) |
| 12 | MAYO | F | 77 | MM | ND | 50 | Kappa | Frozen | 11;14 | nuc ish(CCND1-XTx3,IGH-XTx2)(CCND1-XT con IGH-XTx1) |
| 13 | MAYO | F | 59 | AL | ND | 5 | Lambda | CD138+ | 11;14 | nuc ish(CCND1-XTx3),(IGH-XTx3),(CCND1-XT con IGH-XTx2) |
| 14 | MAYO | F | 63 | PCPD | U | 5 | Lambda | CD138+ | 11;14 | nuc ish(CCND1-XTx3,IGH-XTx2)(CCND1-XT con IGH-XTx1),(RB1,LAMP1)x1 |
| 15 | MAYO | F | 54 | MM | ND | 45 | Kappa | CD138+ | 11;14 | nuc ish(CCND1-XTx2),(IGH-XTx2),(CCND1-XT con IGH-XTx1)/(CCND1-XTx2),(IGH-XTx3),(CCND1-XT con IGH-XTx1) |
| 16 | MCL | F | 67 | IgA gammopathya | U | 40 | Lambda | FCP | 4;14 | nuc ish(TP73x2,1q22x3–4),(FGFR3,IGH)x3(FGFR3 con IGHx2),(5′MYCx3,3′MYCx2)(5′MYC con 3′MYCx2),(RB1,LAMP1)x1 |
| 17 | MAYO | M | 75 | MM | RR | 13 | Kappa | Sort | 4;14 | nuc ish(TP73x2,1q22x3),(FGFR3,IGH)x3(FGFR3 con IGHx2),(MYCx2)(5′MYC con 3′MYCx1),(RB1,LAMP1)x1 |
| 18 | MCL | M | 73 | MMa | U | 34 | Kappa | Sort | 4;14 | nuc ish(TP73x2,1q22x3),(FGFR3,IGH)x3(FGFR3 con IGHx2),(RB1,LAMP1)x1 |
| 19 | MCL | M | 68 | Monoclonal gammopathya | U | 20 | Lambda | Sort | 4;14 | nuc ish(TP73x2,1q22x3),(D3Z1,D9Z1,D15Z4)x3,(FGFR3,IGH)x3(FGFR3 con IGHx2),(RB1,LAMP1)x1 |
| 20 | MCL | M | 72 | PCL | U | 70 | Kappa | Fresh | 4;14 | nuc ish(TP73x2,1q22x4),(FGFR3,IGH)x3(FGFR3 con IGHx2),(MYCx2)(5′MYC sep 3′MYCx1),(RB1,LAMP1)x1,(TP53x1,D17Z1x2) |
| 21 | MCL | M | 42 | MMa | U | 89 | Kappa | Fresh | 4;14 | nuc ish(TP73x2,1q22x3),(FGFR3,IGH)x3(FGFR3 con IGHx2),(MYCx4)(5′MYC sep 3′MYCx1),(RB1,LAMP1)x1,(TP53x2,D17Z1x1) |
| 22 | MAYO | M | 57 | MM | RR | 41 | Kappa | Fresh | 4;14 | nuc ish(TP73x2,1q22x3),(FGFR3,IGH)x3(FGFR3 con IGHx2),(5’MYCx2,3′MYCx1)(5′MYC con 3′MYCx1),(RB1,LAMP1)x1,(TP53x1,D17Z1x2)/(TP73x4,1q22x6),(D3Z1x3),(FGFR3,IGH)x4(FGFR3 con IGHx3)/(FGFR3,IGH)x5(FGFR3 con IGHx4),(D7Z1,D9Z1,D15Z4)x3–4,(5′MYCx4,3′MYCx2)(5’MYC con 3’MYCx2),(CCND1-XTx4),(RB1,LAMP1)x2,(TP53x2,D17Z1x4) |
| 23 | MAYO | M | 78 | MM | RR | 77 | Lambda | Frozen | 4;14 | nuc ish(TP73x2,1q22x3),(FGFR3,IGH)x2(FGFR3 con IGHx1),(RB1,LAMP1)x1,(TP53x1,D17Z1x2) |
| 24 | MCL | M | 63 | MMa | U | 13 | Kappa | Sort | 14;16 | nuc ish(TP73x2,1q22x3),(D9Z1,RB1,LAMP1)x1,(IGHx4,MAFx3)(IGH con MAFx2) |
| 25 | MAYO | F | 60 | MM | ND | 31 | Kappa | Sort | 14;16 | nuc ish(TP73x2,1q22x3),(RB1,LAMP1)x1,(IGHx3,MAFx2)(IGH con MAFx2) |
| 26 | MAYO | F | 80 | MM | RR | 25 | Kappa | CD138+ | 14;16 | nuc ish(D3Z1,D9Z1,p53,D17Z1)x3/(Rb1,LAMP1,D15Z4)x1/(IGHx2),(c-MAFx3),(IGH con c-MAFx1) |
| 27 | MAYO | F | 67 | MM | ND | 60 | Kappa | CD138+ | 14;16 | nuc ish(IGHx3),(MAFx3),(IGH con MAFx2) |
| 28 | MCL | M | 75 | MMa | U | 27 | Kappa | FCP | 14;20 | nuc ish(TP73x4,1q22x6–8),(5′MYC,3′MYC)x4(5′MYC con 3′MYCx2),(RB1,LAMP1)x2,(IGHx6,MAFBx4)(IGH con MAFBx2),(TP53,D17Z1)x4 |
| 29 | MCL | M | 88 | MMa | U | 56 | Kappa | FCP | 14;20 | nuc ish(MAFBx2,IGHx3)(MAFB con IGHx2),(RB1,LAMP1)x1 |
| 30 | MCL | F | 65 | R/O MMa | U | 63 | Kappa | Fresh | 14;20 | nuc ish(MYCx2)(5′MYC sep 3′MYCx1),(RB1,LAMP1)x1,(IGH,MAFB)x3(IGH con MAFBx2)/(IGHx4,MAFBx3)(IGH con MAFBx2) |
| 31 | MAYO | F | 74 | MM | ND | 5 | Kappa | Fresh | 14;20 | ish(TP73x3,1q22x6),(D3Z1,D9Z1,D15Z4,TP53,D17Z1)x4,(D7Z1x5),(MYCx3–4),(CCND1-XTx4–5),(RB1,LAMP1)x2,(IGHx3,MAFBx4)(IGH con MAFBx1) |
| 32 | MCL | M | 74 | MMa | U | N/A | N/A | FCP | 6;14 | nuc ish(TP73,1q22)x3,(CCND3x2,IGHx1)(CCND3 con IGHx1),(RB1,LAMP1)x1,(TP53x1,D17Z1x2) |
| 33 | MCL | M | 51 | R/O MM, hypercalcemia, renal failurea | U | N/A | N/A | FCP | 6;14 | 84,XX,der(Y;16)(q10;p10)x2,i(1)(q10)x2,–2,–4,–4,t(6;14)(p21.1;q32)x2,−8,t(8;14) (q24.1;q32),−10,−11,der(14)t(8;14)6/46,XY14 nuc ish,(CCND3x2,IGHx1)(CCND3 con IGHx1) only CCND3/IGH FISH performed on FCP sample |
| 34 | MCL | F | 58 | MMa | U | 21 | Kappa | FCP | Hyper + IGH sep | nuc ish(TP73x2,1q22x3),(D7Z1,MYC)x3,(RB1,LAMP1,D15Z4)x1,(3′IGHx2,5’IGHx1)(3′IGH con 5′IGHx1)/(IGHx2)(3’IGH sep 5′IGHx1),(TP53x1,D17Z1x2) |
| 35 | MCL | F | 54 | MMa | U | 31 | Kappa | FCP | Hyper + IGH sep | nuc ish(D3Z1,D9Z1,D15Z4)x3,(FGFR3,CCND3,MAF,MAFB)x1,(MYCx2)(5′MYC sep 3′MYCx1),(CCND1-XTx4–5),(3′IGHx3,5′IGHx2)(3′IGH con 5′IGHx2)/(IGHx3)(3′IGH sep 5′IGHx2) |
| 36 | MAYO | M | 82 | MM | RR | 7 | Lambda | Sort | Hyper + IGH sep | nuc ish(TP73x2,1q22x3),(CCND3,D7Z1,D9Z1)x3,(3′IGHx2,5′IGHx1)(3′IGH con 5′IGHx1) |
| 37 | MAYO | M | 61 | MM | RR | 27 | Kappa | Sort | Hyper + IGH sep | nuc ish(5′MYCx2,3′MYCx1)(5′MYC con3′MYCx1)/(D9Z1x3–4)/(CCND1-XTx3)/(3′IGHx2,5′IGHx1)(3′IGH con 5′IGHx1)/(3′IGHx3,5′IGHx2)(3′IGH con 5′IGHx2) |
| 38 | MAYO | F | 58 | MM | RR | 11 | Lambda | Sort | Hyper + IGH sep | nuc ish(TP73x2,1q22x4–5),(D3Z1,D7Z1,D9Z1,CCND1-XT,TP53,D17Z1)x3,(RB1x1,LAMP1x2),(3′IGHx2,5’IGHx1)(3′IGH con 5’IGHx1) |
| 39 | MCL | M | 64 | MM | U | 20 | Kappa | Fresh | Hyper + IGH sep | nuc ish(TP73x2,1q22x3),(MYC,D9Z1,CCND1-XT,D15Z4)x3,(3′IGHx3,5′IGHx2)(3′IGH con 5′IGHx2) |
| 40 | MCL | M | 80 | MM | U | 81 | Lambda | Fresh | Hyper + IGH sep | nuc ish(TP73x2,1q22x3),(FGFR3,MAF,MAFB)x1,(CCND3,D9Z1,D15Z4)x3,(3′IGHx3,5′IGHx2)(3′IGH con 5′IGHx2),(TP53x1,D17Z1x2) |
| 41 | MCL | M | 60 | MMa | U | 98 | Kappa | Fresh | Hyper + IGH sep | nuc ish(TP73x3,1q22x2),(D3Z1,CCND3,CCND1-XT,D15Z4)x3,(MYCx2)(5′MYC sep 3′MYCx1),(RB1,LAMP1,MAF)x1,(3′IGHx4–5,5′IGHx4)(3’IGH con 5′IGHx3),(TP53x1,D17Z1x2) |
| 42 | MAYO | M | 70 | MM | RR | 19 | Kappa | Sort | Hyper + IGH sep | nuc ish(D3Z1,D7Z1,D9Z1,D15Z4)x3,(5′MYCx3,3′MYCx2)(5′MYC con 3′MYCx2),(CCND1-XT amp),(3′IGHx3,5′IGHx2)(3′IGH con 5′IGHx2) |
| 43 | MAYO | F | 56 | PCPD | U | 4 | Indeterminate | Sort | Hyper | nuc ish(CCND1-XTx3),(RB1,LAMP1)x1 |
| 44 | MAYO | M | 67 | MM | RR | 12 | Kappa | Sort | Hyper | nuc ish(CCND1-XTx3),(RB1,LAMP1,IGH,TP53,D17Z1)x1 |
| 45 | MCL | M | 76 | MMa | U | 14 | Kappa | Sort | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,D9Z1,CCND1-XT)x3,(MYCx2)(5′MYC sep 3′MYCx1),(D15Z4x3–4) |
| 46 | MAYO | M | 66 | MM | ND | 19 | Kappa | Sort | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,MYC,CCND1-XT,D15Z4)x3,(D9Z1x3–4) |
| 47 | MAYO | M | 72 | MM | ND | 10 | Kappa | Sort | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,MYC)x3,(D7Z1,D9Z1)x3–4,(RB1,LAMP1,IGH,D15Z4)x1 |
| 48 | MCL | F | 77 | Anemia, coagulation defecta | U | 10 | Kappa | Sort | Hyper | nuc ish(TP73x2,1q22x3),(D7Z1,D9Z1,D15Z4)x3,(RB1x1,LAMP1x2) |
| 49 | MCL | M | 73 | R/O MMa | U | 35 | Kappa | Sort | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,D7Z1,D9Z1,CCND1-XT)x3,(D15Z4x3–4) |
| 50 | MCL | F | 64 | MMa | U | 48 | Kappa | Fresh | Hyper | nuc ish(CCND1-XTx4),(IGHx3),(MAFx1) |
| 51 | MAYO | F | 49 | MM | RR | 84 | Lambda | Fresh | Hyper | nuc ish(TP73x2,1q22x3–4),(D3Z1,D9Z1)x3,(5′MYCx4–5,3′MYCx1)(5’MYC con 3′MYCx1),(CCND1-XT,D15Z4)x3–4,(TP53x1,D17Z1x2) |
| 52 | MCL | F | 75 | PCPDa | U | 73 | Lambda | Fresh | Hyper | nuc ish(D3Z1,D7Z1,D9Z1)x3,(RB1,LAMP1,IGH)x1,(TP53x1,D17Z1x2) |
| 53 | MCL | F | 66 | MMa | U | 52 | Kappa | Fresh | Hyper | nuc ish(D3Z1,D9Z1,CCND1-XT,D15Z4)x3 |
| 54 | MCL | M | 65 | MMa | U | 52 | Lambda | Fresh | Hyper | nuc ish(TP73x2,1q22x3–4),(D3Z1,D7Z1,D9Z1,IGH,D15Z4,TP53,D17Z1)x3,(CCND1-XTx3–4),(RB1,LAMP1)x1/(RB1x2,LAMP1x1) |
| 55 | MCL | M | 57 | Solitary plasmacytoma | U | 99 | Kappa | Fresh | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,D9Z1)x3,(CCND1-XT,D15Z4)x3–4,(RB1x1,LAMP1x2) |
| 56 | MCL | F | 56 | MMa | U | 34 | Kappa | Fresh | Hyper | nuc ish(D3Z1,D7Z1,CCND1-XT)x3,(D9Z1,D15Z4)x3–4 |
| 57 | MAYO | M | 61 | MM | RR | 55 | Kappa | Frozen | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,D7Z1,D15Z4)x3,(D9Z1)x3–4,(RB1x1,LAMP1x2) |
| 58 | MAYO | M | 59 | MM | RR | 79 | Indeterminate | Frozen | Hyper | nuc ish(TP73x2,1q22x3),(D3Z1,CCND1-XT)x3 |
| 59 | MAYO | M | 74 | MM | RR | 60 | Kappa | CD138+ | Hyper | nuc ish(D3Z1,D7Z1,D9Z1,D15Z4)x3 |
| 60 | MAYO | M | 53 | MM | ND | 90 | Kappa | CD138+ | Hyper | nuc ish(D3Z1,D9Z1,CCND1-XT,D15Z4),(IGH)x1 |
| 61 | MAYO | F | 61 | MM | RR | 25 | Kappa | CD138+ | Hyper | nuc ish(D3Z1,D7Z1,D9Z1,CCND1)x3/(Rb1,LAMP1)x1/(D15Z4x3–4) nuc ish(TP73x2,CKS1Bx3),(MYCx3) |
| 62 | MAYO | M | 48 | MM | RR | 75 | Lambda | CD138+ | Hyper | nuc ish(D3Z1,D9Z1,CCND1-XT,D15Z4)x3/(IGHx1) |
| 63 | MAYO | F | 61 | MM | RR | 5 | Kappa | CD138+ | Hyper | nuc ish(D9Z1,CCND1-XT,D15Z4)x3 |
| 64 | MAYO | F | 48 | MM | RR | 70 | Kappa | CD138+ | Hyper | nuc ish(D3Z1,D7Z1,CCND1-XT)x3/(D9Z1,D15Z4)x3–4 |
| 65 | MAYO | F | 66 | MM | RR | 25 | Lambda | CD138+ | Hyper | nuc ish(CCND1-XT)x3 |
| 66 | MCL | F | 70 | MM | U | 39 | Lambda | Sort | Tetraploid | nuc ish(TP73x2,1q22x4–6),(D3Z1,TP53,D17Z1)x4,(D7Z1,MYC,D9Z1,CCND1-XT,RB1,LAMP1,IGH,D15Z4)x3–4 |
| 67 | MCL | M | 74 | Gammopathya | U | 36 | Lambda | Sort | Monosomy 13/14 | nuc ish(TP73x1,1q22x2),(RB1,LAMP1,IGH)x1 |
| 68 | MAYO | M | 58 | MM | ND | 15 | Lambda | CD138+ | Monosomy 15 | nuc ish(D15Z4x1) |
| 69 | MAYO | F | 62 | AL | RR | 5 | Lambda | CD138+ | Monosomy 15 | nuc ish(D15Z4x1) |
| 70 | MAYO | F | 68 | AL | RR | 9 | Kappa | CD138+ | Normal | Normal |
Cohort of 70 patients evaluated. MCL Mayo Clinic Laboratories, F female, M male, Dx diagnosis, RFR reason for referral, AL amyloidosis, ND newly diagnosis, RR relapsed/refractory, U unknown status. aIndicates only RFR is available. % PC from flow cytometry
Only abnormal FISH results are indicated in the ISCN
Table 2.
Patient characteristics
| Total (N = 70) | |
|---|---|
| Characteristic | N (%) |
| Sex | |
| Male | 40 (57.1) |
| Female | 30 (42.9) |
| Age | |
| Median | 66 years |
| Range | 42–88 years |
| 40–49 | 4 (5.7) |
| 50–59 | 14 (20.0) |
| 60–69 | 26 (37.1) |
| 70–79 | 21 (30.0) |
| 80–89 | 5 (7.1) |
| Diagnosis or RFR | |
| MM, PCN diagnosis or RFR | 57 (81.4) |
| Amyloidosis | 3 (4.3) |
| Plasma cell leukemia | 2 (2.9) |
| Plasma cell proliferative disorder | 3 (4.3) |
| Other | 5 (7.1) |
| Site | |
| Mayo Clinic-local | 37 (52.9) |
| Mayo Clinic Laboratories-outside | 33 (47.1) |
| PC percentage (N = 68) | |
| Median | 35.5% |
| Range | 4–99% |
| 4–19 | 19 (27.9) |
| 20–39 | 19 (27.9) |
| 40–59 | 11 (16.2) |
| 60–79 | 12 (17.6) |
| 80–99 | 7 (10.3) |
| Sample type | |
| No enrichment | 31 (44.3) |
| Fixed cell pellet (FCP) | 8 (11.4) median PCs (35.5) |
| Fresh sample | 18 (25.7) median PCs (66.5) |
| Frozen sample | 5 (7.1) median PCs (58.0) |
| Enrichment | 39 (55.7) |
| Flow sorting | 24 (34.3) median PCs (19.5) |
| CD138+ magnetic | 15 (21.4) median PCs (25.0) |
| Light chain | |
| Kappa | 46 (65.7) |
| Lambda | 20 (28.6) |
| Indeterminate or unknown | 4 (5.7) |
| Primary cytogenetic abnormality | |
| t(11;14) | 15 (21.4) |
| t(4;14) | 8 (11.4) |
| t(14;16) | 4 (5.7) |
| t(14;20) | 4 (5.7) |
| t(6;14) | 2 (2.9) |
| Hyperdiploid only | 23 (32.9) |
| Hyperdiploid with an unknown IGH rearrangement | 9 (12.9) |
| Tetraploid without primary abnormality | 1 (1.4) |
| Monosomy 13/14 alone | 1 (1.4) |
| Monosomy 15 alone | 2 (2.9) |
| Normal | 1 (1.4) |
| Conventional chromosome study | |
| Not performed | 28 (40.0) |
| Performed | 42 (60.0) |
| Normal or loss of Y | 27 (38.6 of total, 64.3 of performed) |
| Abnormal with PC abnormalities | 14 (20.0 of total, 33.3 of performed) |
| Abnormal with non-PC abnormalities | 1 (1.4 of total, 2.4 of performed) |
Patient characteristics of the 70 patients within the cohort evaluated
MM abnormalities identified by FISH
Recurrent primary MM cytogenetic abnormalities identified by FISH in samples 1–65 were t(11;14) (21.4%), t(4;14) (11.4%), t(14;16) (5.7%), t(14;20) (5.7%), t(6;14) (2.9%), and hyperdiploidy (45.7%) either without an IGH rearrangement (32.9%) or with an IGH rearrangement that did not involve CCND1, FGFR3, MAF, MAFB or CCND3 (12.9%) (Tables 1, 2). Five samples (cases 66–70) had undefined primary abnormalities including one case of tetrapoloidy with a relative 1q gain, one case with monosomies 13 and 14, two cases with monosomy 15 by FISH and a single case with normal FISH results in a patient with a diagnosis of amyloidosis (Tables 1, 2). Conventional chromosome studies were performed on 42 (60.0%) cases and an abnormal PC clone was identified in 33.3% of the cases with chromosome studies performed (Supplemental Table 3).
We have previously determined tumor content requirements for MPseq requiring 10% tumor for the detection of structural rearrangements and 25% tumor for the detection of CNAs18. Since variable and sometimes low clonal PC percentages can be identified in the BM aspirates of patients with NDMM31, we performed two enrichment strategies for samples with low PCs including magnetic enrichment or flow sorting. For some samples, no enrichment was performed. Thirty-nine (55.7%) samples with a median 23.0% PCs were subjected to either flow sorting (N = 24) or CD138 + magnetic bead for PC enrichment (N = 15). For the remaining 31 samples (44.3%) with a median 58% PC, no PC enrichment was performed.
Identification of recurrent, primary cytogenetic abnormalities using MPseq
To determine the accuracy of MPseq in comparison to our PCN FISH panel (Supplemental Table 1) in the detection of recurrent, primary MM abnormalities (IGH rearrangement and/or hyperdiploidy), we analyzed DNA extracted from either a fixed cell pellet (FCP) from a chromosome study (n = 8), from fresh (n = 18) or frozen (n = 5) BM aspirates or from fresh BM specimens that had been flow sorted (n = 24) or subjected to CD138 + magnetic enrichment (n = 15) (Supplemental Fig. 1, Tables 1, 2). For samples 1–65, MPseq confirmed the primary abnormality identified by FISH in each case demonstrating 100% concordance between both assays for the classification of recurrent, primary cytogenetic abnormalities (Fig. 1). For those cases without evidence of a recurrent, primary abnormality (samples 66–69), MPseq did not identify tetraploidy in case 66 and monosomy 15 in cases 68–69, but identified monosomies 13 and 14 in case 67 and confirmed no recurrent abnormality in case 70 with normal FISH results. As a negative control, no recurrent primary MM abnormalities (MM specific IGH rearrangements and/or hyperdiploidy with gains of odd numbered chromosomes) were identified by MPseq in a previously described cohort of 88 patients with a reason for referral of acute myeloid leukemia (data not shown)18.
Fig. 1. Concordance between MPseq and FISH.

In bold indicates primary cytogenetic abnormalities. Cytogenetic risk applied to all cases: H: high and S:standard. ND: Newly diagnosed, RR: relapsed/refractory. For case 33, there was no FISH for MYC BAP, but detection of t(8;14) and t(6;14) was achieved using chromosome studies and CCND3/IGH rearrangement confirmed by FISH. For case 37, there was a history of trisomies 9,11,15, and IGH separation in an older sample. For cases 61 and 65 there was evidence of hyperdiploidy by FISH in older samples. Highlighted in yellow is a single case with a difference in cytogenetic risk between MPseq and FISH.
Comparison of MPseq to FISH for detection of recurrent, secondary abnormalities
For each primary and secondary abnormality that was identified by either MPseq or FISH, 40 cases (57.1%) displayed concordance between FISH and MPseq. Thirty cases (42.9%) displayed at least one instance of discordance between FISH and MPseq (Figs. 1, 2a). Nine of these 30 discordant cases had abnormalities detected by FISH that went undetected by MPseq (Figs. 1, 2a, “FISH advantage”). Of these nine cases, six had a tetraploid clone that was not detectable by MPseq and in three cases MPseq failed to detect CNAs that were identified by FISH (trisomy 3, trisomy 9 and 1q gain). In six cases, FISH identified a CNA involving chromosome 15 that was not confirmed by MPseq. These abnormalities included monosomy 15 identified by FISH without evidence of monosomy 15 by MPseq (cases 26, 47, 68, 69), or one (case 34) or two (case 52) copies of chromosomes 15 identified by FISH in cases with trisomy 15 identified by MPseq (Figs. 1, 2a). In contrast, 19 of the 30 discordant cases had abnormalities detected by MPseq that went undetected by FISH (Figs. 1, 2a, “MPseq advantage”). Of these 19 cases, 17 were MYC rearrangements and two were 17p deletions (cases 4 and 21), including a 17p translocation involving the TP53 gene in one case (Figs. 1, 2a).
Fig. 2. Discordance summary and MYC breakpoint locations.

a Total number of cases with evidence of MPseq advantage, FISH advantage and polymorphism of chromosome 15. b The location of breakpoints in the MYC locus across all cases are depicted as vertical lines (black if the MYC alteration was detected by FISH, light gray if it was not tested by FISH, and red if it was undetected by FISH). In cases where multiple breakpoints were found in the MYC locus, the lines are connected by an arc. The locations of the MYC BAP probes used for FISH detection are shown at the top (5′ in red, 3′ in green) and gene locations are shown in the middle (forward strand in light blue, reverse strand in pink).
Increased detection rate of MYC rearrangements by MPseq
From 70 total cases, we identified 36 cases (51.4%) that displayed a MYC rearrangement by MPseq (Fig. 1). Of these 36 cases, 34 had FISH data evaluating the MYC locus. Seventeen cases (50.0% of MYC abnormal group with FISH results) displayed a false negative MYC FISH result where a MYC rearrangement was identified by MPseq, but was negative by FISH (Fig. 1). The most common partner gene/enhancer segment identified were IGH (n = 7), FAM46C (n = 5), IGK (n = 4), NSMCE2 (n = 4), TXNDC5 (n = 4) and IGL (n = 4) (Table 3). Of the 36 MYC rearrangements, multiple mechanisms resulting in positioning of MYC near enhancer sequences including small insertions, inversions, simple, balanced or complex translocations were identified (Table 3). The most common method of rearrangement identified in 15 cases included a small insertion of enhancer sequences near the MYC gene or, alternately, the insertion of MYC near enhancer sequences. These insertions typically involve the duplication of genetic material of similar size at both the source location and the insertion location, whereby the source DNA is inserted between flanking duplications at the insertion location32 (Fig. 2b). Thirteen of these 15 insertion cases co-occurred with hyperdiploidy (hyperdiploidy only or hyperdiploidy with IGH separation) and two of these cases were identified by FISH studies. Of the 17 MYC cases that were missed by FISH, 11 represented these small insertions (Table 3).
Table 3.
Genetic information of secondary alterations involving MYC.
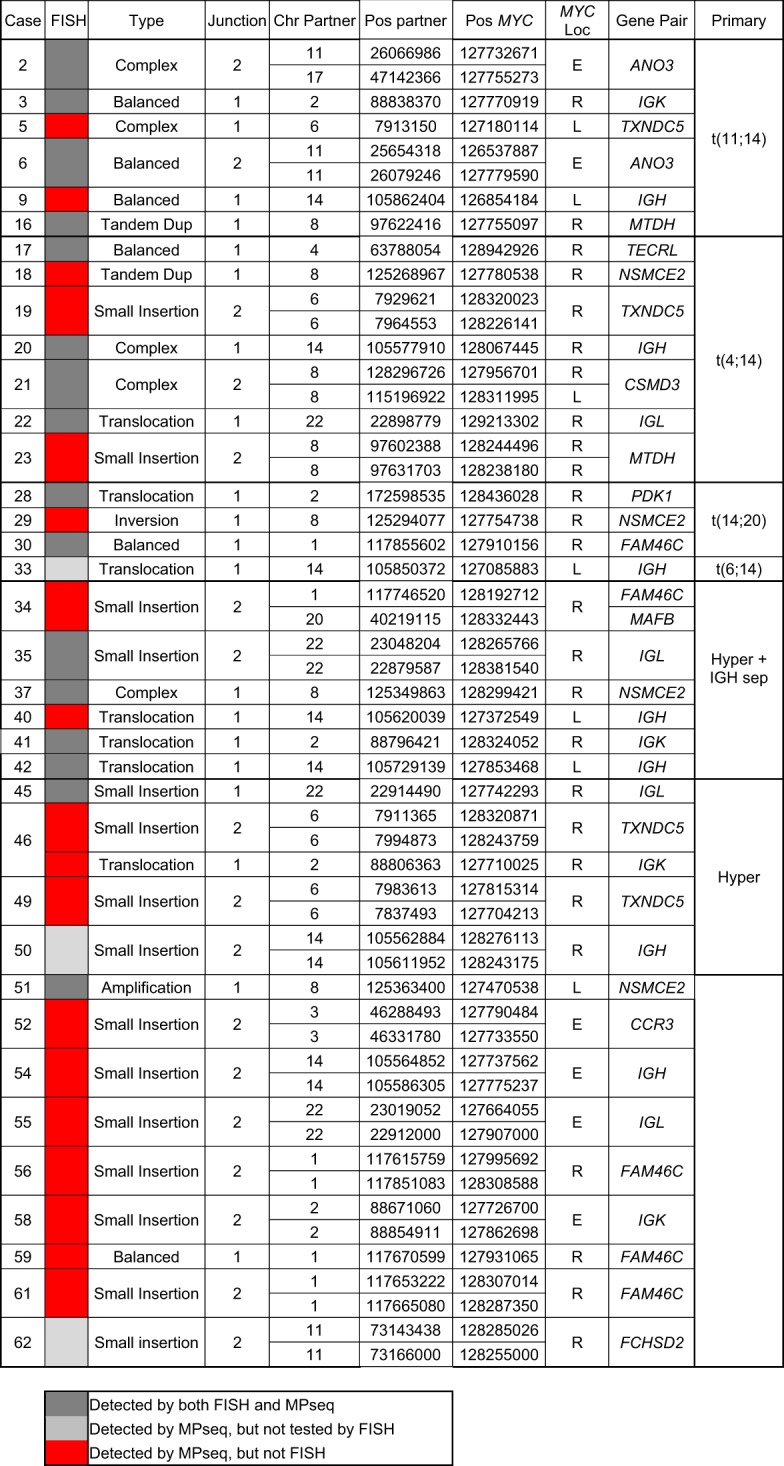
For each case where a secondary alteration involving MYC was found, the relevant genomic information is provided for the junction(s). The case column is the case number. The FISH column indicates whether or not the MYC FISH test detected the secondary alteration (dark gray–detected by both FISH, light gray–detected by MPseq but not tested by FISH and red–detected by MPseq only). The type column is the type of alteration involved with MYC classified as either a balanced event, a tandem duplication, a translocation, an inversion, part of an amplification, part of a small insertion motif, a complex event, or ND where it was not possible to definitively classify the alteration. The Junction column is the number of junctions involved directly in the alteration, either 1 or 2. The Chr Partner and Pos Partner columns are the chromosome and position location (GRCh38) of the partner breakpoints that are part of alteration. The Pos MYC and MYC Loc columns give the position of the breakpoint in the MYC locus and whether the alteration is to the left, right, or encompassing (L, R, or E) the MYC gene, respectively. The Gene Pair column is the gene that is found at or near the partner breakpoint location. The Primary column is the primary alteration for the case
Detection of additional genomic alterations by MPseq that are not evaluated by FISH
We next evaluated for the presence of rearrangements involving non-recurrent IGH MM partners (excluding CCND1, FGFR3, MAF, MAFB or CCND3) and the IGK and IGL loci by MPSeq. There were 19 additional IGH rearrangements identified in 18 cases (25.7% of cohort) with partner chromosomes at 8q24.21 (MYC) (n = 7) as the only recurrent rearrangement (Table 4, Fig. 3). Of the nine cases classified as “hyperdiploidy with IGH separation”, an IGH partner was identified in six cases, while the other three cases had a loss within the IGH locus. Two cases (cases 50 and 54) classified as hyperdiploidy without an IGH rearrangement to one of the common partner chromosomes had the “small insertion” type of MYC/IGH rearrangement (Tables 3, 4, Fig. 3). There were three cases with a CCND1 rearrangement to a locus other than IGH (IGK/CCND1 in case 43, IGL/CCND1 in case 4 and BRINP3/CCND1 in case 57) that had additional copies of CCND1 observed by FISH in case 4 and 43. FISH for CCND1 was not performed in case 57 and in case 4, the signal pattern for CCND1 was scored as amplification (Table 4, Figs. 1, 3).
Table 4.
IGH, IGK, and IGL partner genes.
| Case | IGH partner chromosome | Putative gene target | Primary abnormality |
|---|---|---|---|
| 5 | 14q24.3 | BATF | 11;14 |
| 6 | 19p13.2 | TYK2 | 11;14 |
| 8 | 11q14.1 | RAB39 | 11;14 |
| 9 | 1p35.3 | PTPRU | 11;14 |
| 9 | 8q24.21 | MYC | 11;14 |
| 10 | 20q11.21 | COMMD7 | 11;14 |
| 11 | 22q13.1 | POLR2F | 11;14 |
| 15 | 2p24.3 | MYCN | 11;14 |
| 20 | 8q24.21 | MYC | 4;14 |
| 30 | 5p15.33 | TERT | 14;20 |
| 33 | 8q24.21 | MYC | 6;14 |
| 34 | 7q32.1 | Unknown | Hyper + IGH sep |
| 36 | Xq32.33 | MTMR1 | Hyper + IGH sep |
| 37 | 14q24.3 | DPF3 | Hyper + IGH sep |
| 40 | 8q24.21 | MYC | Hyper + IGH sep |
| 41 | 9p13.2 | PAX5 | Hyper + IGH sep |
| 42 | 8q24.21 | MYC | Hyper + IGH sep |
| 50 | 8q24.21 | MYC | Hyper |
| 54 | 8q24.21 | MYC | Hyper |
| Case | IGK partner chromosome | Putative gene target | Primary abnormality |
|---|---|---|---|
| 3 | 8q24.21 | MYC | 11;14 |
| 41 | 8q24.21 | MYC | Hyper + IGH sep |
| 43 | 11q13.3 | CCND1 | Hyper |
| 46 | 8q24.21 | MYC | Hyper |
| 58 | 8q24.21 | MYC | Hyper |
| Case | IGL partner chromosome | Putative gene target | Primary abnormality |
|---|---|---|---|
| 4 | 11q13.3 | CCND1 | 11;14 |
| 22 | 8q24.21 | MYC | 4;14 |
| 35 | 8q24.21 | MYC | Hyper + IGH sep |
| 39 | 3q26.2 | MECOM | Hyper + IGH sep |
| 45 | 8q24.21 | MYC | Hyper |
| 50 | 17q25.1 | GRB2 | Hyper |
| 55 | 8q24.21 | MYC | Hyper |
| 56 | 8q24.22 | ST3GAL1/NDRG1 | Hyper |
| 61 | 8q24.22 | ST3GAL1/NDRG1 | Hyper |
| 63 | 8q24.22 | ST3GAL1/NDRG1 | Hyper |
Partner genes associated with IGH, IGK, and IGL showing cytogenetic location and putative target genes. Hyper: Hyperdiploidy only. Hyper+IGH sep: Hyperdiploidy with IGH separation
Fig. 3. Detection of additional abnormalities by MPseq in relation to each primary and secondary abnormality.

In bold indicates primary abnormalities. Cytogenetic risk applied to all cases: H: high and S:standard. ND: Newly diagnosed, RR: relapsed/refractory.
There were five cases with IGK rearrangements (7.1% of cohort) mainly with partner chromosome 8q24.21 (MYC) (n = 4) and a single case with partner chromosome at 11q13.3 (CCND1) (Table 4, Fig. 3). In addition, 10 cases (14.3% of cohort) had IGL rearrangements with partner chromosomes at 8q24.21 (MYC) (n = 4), 11q13.3 (CCND1) (n = 1), 8q24.22 (n = 3) (putative target ST3GAL1/NDRG1), 3q26.2 (MECOM) (n = 1) and 17q25.1 (GRB2) (n = 1) (Table 4, Fig. 3). Of these 15 cases with either an IGK or IGL rearrangement, 12 (80.0%) co-occurred with hyperdiploidy (hyperdiploidy only or hyperdiploidy with IGH separation).
We explored alterations in additional genes contributing to dysregulation of multiple pathways such as WNT or NF-kB signaling including genes CYLD at 16q12.1, BIRC2 and BIRC3 at 11q22,2, NFKB1 at 4q24, NFKB2 at 10q24.32, TRAF2 at 9q34.3, TRAF3 at 14q32.32 and MAP3K14/NIK at 17q21.31 or other tumor suppressor genes such as CDKN2C (p18) at 1p32.3 or TENT5C/FAM46C at 1p1233,34 (Table 5, Fig. 3). Twenty-five cases (35.7%) had an alteration in TENT5C/FAM46C with 6 cases with translocations (five of these to MYC) and 19 cases had a heterozygous deletion of TENT5C/FAM46C ranging in size from 3.4 Mb to 120 Mb. Nineteen cases (27.1%) had alterations in CDKN2C and/or FAF1. Fourteen were heterozygous deletions involving CDKN2C ranging in size from 587 Kb to 120 Mb, four cases had focal biallelic CDKN2C and FAF1 deletions (Supplemental Fig. 2A, case #5) and one case had a heterozygous 655 Kb FAF1 deletion without a CDKN2C deletion (Table 5, Fig. 3). Ten cases had deletions of TRAF3 with 5 as heterozygous deletions and five as biallelic deletions (Supplemental Fig. 2B, case #62) and a single case had a 92.9 Kb heterozygous deletion of TRAF2 (Table 5, Fig. 3). Twenty-eight cases had deletions of CYLD (40% of cohort) with 24 cases having heterozygous deletions ranging in size from 634 Kb to 90.3 Mb with the majority representing large 16q deletions and four cases will smaller biallelic deletions (Table 5, Fig. 3, Supplemental Fig. 2C, case #40). Additional alterations in MAP3K14 were observed in four cases (three as heterozygous deletions and one as a 735 Kb gain), heterozygous deletion of NFKB1 in seven cases, heterozygous deletion of NFKB2 in 6 cases and a heterozygous and homozygous BIRC2 and BIRC3 deletions in two separate cases (Table 5, Fig. 3).
Table 5.
Abnormalities of additional genes of clinical significance
| Case | CYLD | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 1 | HD | 16p13.3–16q24.3 | 0–90338345 | 90338345 | 11;14 |
| 2 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 11;14 |
| 4 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 11;14 |
| 6 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 11;14 |
| 8 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 11;14 |
| 9 | HD | 16q21.1–16q24.3 | 50093000–89129000 | 39036000 | 11;14 |
| 15 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 11;14 |
| 24 | BD | 16q12.1–16q12.1 | 50232040–50913020 | 680980 | 14;16 |
| 25 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 14;16 |
| 28 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 14;20 |
| 29 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 14;20 |
| 30 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | 14;20 |
| 33 | BD | 16q12.1–16q12.1 | 50777028–50812200 | 35172 | 6;14 |
| 35 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper + IGH sep |
| 37 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper + IGH sep |
| 40 | BD | 16q12.1–16q12.2 | 50376741–52630833 | 2254092 | Hyper + IGH sep |
| 41 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper + IGH sep |
| 42 | HD | 16q12.1–16q12.1 | 50193000–50827000 | 634000 | Hyper + IGH sep |
| 44 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper |
| 48 | HD | 16q12.1–16q12.2 | 50123000–55838000 | 5715000 | Hyper |
| 49 | BD | 16q12.1–1612.1 | 50290162–51082053 | 791891 | Hyper |
| 50 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper |
| 52 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper |
| 54 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper |
| 61 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper |
| 63 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Hyper |
| 66 | HD | 16q11.2–16q24.3 | 46454000–90338000 | 43884000 | Tetraploid |
| 67 | HD | 16p13.3–16q24.3 | 0–90338345 | 90338345 | Monosomy 13/14 |
| Case | BIRC2 and BIRC3 | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 6 | HD | 11q14.1–11q22.3 | 79621665–108999346 | 29377681 | 11;14 |
| 21 | BD | 11q22.1–11q22.2 | 101044665–102389301 | 1344636 | 4;14 |
| Case | TENT5C/FAM46C | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 5 | HD | 1p22.3–1p12 | 87833886–119707445 | 31873559 | 11;14 |
| 6 | HD | 1p36.33–1p12 | 1–119990000 | 119989999 | 11;14 |
| 9 | HD | 1p35.3–1p12 | 29234000–119991000 | 90757000 | 11;14 |
| 15 | HD | 1p32.3–1p12 | 51107000–119761000 | 68654000 | 11;14 |
| 21 | HD | 1p31.1–1p12 | 77992000–119733000 | 41741000 | 4;14 |
| 23 | HD | 1p34.2–1p12 | 42342000–121700000 | 79358000 | 4;14 |
| 24 | HD | 1p13.3–1p12 | 110882000–120028000 | 9146000 | 14;16 |
| 28 | HD | 1p32.1–1p12 | 58536000–119985000 | 61449000 | 14;20 |
| 29 | HD | 1p31.1–1p12 | 75850000–118934000 | 43084000 | 14;20 |
| 30 | Translocation to MYC | 1p12 | 117855602 | N/A | 14;20 |
| 32 | HD | 1p31.1–1p12 | 70504000–119991000 | 49487000 | 6;14 |
| 33 | HD | 1p36.33–1p12 | 1–119982000 | 119981999 | 6;14 |
| 34 | Translocation to MYC and MAFB | 1p12 | 117611301;117746520 | N/A | Hyper + IGH sep |
| 35 | HD | 1p33–1p12 | 49064000–119989000 | 70925000 | Hyper + IGH sep |
| 37 | HD | 1p22.2–1p12 | 89819000–118483000 | 28664000 | Hyper + IGH sep |
| 40 | HD | 1p13.1–1p12 | 116616000–119990000 | 3374000 | Hyper + IGH sep |
| 53 | HD | 1p31.1–1p12 | 77694018–119983000 | 42288982 | Hyper |
| 56 | Translocation to MYC | 1p12 | 117615759;117851083 | N/A | Hyper |
| 58 | Translocation to IL16 | 1p12 | 117592488;117745524 | N/A | Hyper |
| 59 | Translocation to MYC | 1p12 | 117670599 | N/A | Hyper |
| 60 | HD | 1p22.1–1p12 | 92935000–119981000 | 27046000 | Hyper |
| 61 | Translocation to MYC | 1p12 | 117653222;117665080 | N/A | Hyper |
| 64 | HD | 1p31.3–1p12 | 67860000–118597000 | 50737000 | Hyper |
| 66 | HD | 1p36.33–1p12 | 1–119990000 | 119989999 | Tetraploid |
| 67 | HD | 1p36.33–1p12 | 1–119991000 | 119990999 | Monosomy 13/14 |
| Case | CDKN2C and FAF1 | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 5 | BD | 1p32.3–1p32.3 | 50884258–51012825 | 128567 | 11;14 |
| 6 | HD | 1p36.33–1p12 | 1–119990000 | 119989999 | 11;14 |
| 9 | HD | 1p35.3–1p12 | 29234000–119991000 | 90757000 | 11;14 |
| 10 | HD | 1p32.3–1p32.3 | 50402893–50989867 | 586974 | 11;14 |
| 15 | BD | 1p32.3–1p32.3 | 50599579–51106763 | 507184 | 11;14 |
| 21 | HD | 1p32.2–1p31.1 | 50276000–73879000 | 23603000 | 4;14 |
| 22 | BD | 1p32.3–1p32.3 | 50924750–50971658 | 46908 | 4;14 |
| 23 | HD | 1p34.2–1p12 | 42342000–121700000 | 79358000 | 4;14 |
| 24 | HD FAF1 only | 1p33–1p32.3 | 49951000–50606000 | 655000 | 14;16 |
| 33 | HD | 1p36.33–1p12 | 1–119982000 | 119981999 | 6;14 |
| 34 | HD | 1p32.3–1p12 | 50750000–117611000 | 66861000 | Hyper + IGH sep |
| 35 | HD | 1p33–1p12 | 49064000–119989000 | 70925000 | Hyper + IGH sep |
| 40 | HD | 1p33–1p13.3 | 49723000–109237000 | 59514000 | Hyper + IGH sep |
| 51 | HD | 1p33–1p31.3 | 50018770–65125485 | 15106715 | Hyper |
| 52 | BD | 1p32.3–1p32.3 | 50925212–51007221 | 82009 | Hyper |
| 58 | HD | 1p32.3–1p13.3 | 50467681–107513627 | 57045946 | Hyper |
| 60 | HD | 1p34.1–1p32.2 | 45515071–55049538 | 9534467 | Hyper |
| 66 | HD | 1p36.33–1p12 | 1–119990000 | 119989999 | Tetraploid |
| 67 | HD | 1p36.33–1p12 | 1–119991000 | 119990999 | Monosomy 13/14 |
| Case | MAP3K14 | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 2 | HD | 17q21.31–17q21.32 | 44943000–47142000 | 2199000 | 11;14 |
| 36 | HD | 17q21.31–17q21.31 | 44583000–45982000 | 1399000 | Hyper + IGH sep |
| 44 | HD | 17p13.3–17q21.31 | 1–45734287 | 45734286 | Hyper |
| 45 | Gain | 17q21.31–17q21.31 | 45191000–45926000 | 735000 | Hyper |
| Case | NFKB1 or NFKB2 | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 6 | HD NFKB1 | 4q13.2–4q26 | 65930271–115942883 | 50012612 | 11;14 |
| 23 | HD NFKB1 | 4p14–4q35.2 | 36402000–189875000 | 153473000 | 4;14 |
| 33 | HD NFKB1 | 4p16.3–4q35.2 | 1–190214555 | 190214554 | 6;14 |
| 34 | HD NFKB2 | 10q24.1–10q26.3 | 97005000–133797422 | 36792422 | Hyper + IGH sep |
| 40 | HD NFKB1 | 4p16.3–4q35.2 | 1–190214555 | 190214554 | Hyper + IGH sep |
| 44 | HD NFKB1 | 4q13.3–4q31.3 | 73221000–150520000 | 77299000 | Hyper |
| 46 | HD NFKB2 | 10q24.32–10q24.33 | 101899000–103362000 | 1463000 | Hyper |
| 47 | HD NFKB2 | 10q24.32–10q24.32 | 102148932–102721927 | 572995 | Hyper |
| 51 | HD NFKB1 | 4p16.3–4q35.2 | 1–190214555 | 190214554 | Hyper |
| 65 | HD NFKB2 | 10q24.32–10q25.1 | 102399071–104266176 | 1867105 | Hyper |
| 66 | HD NFKB1 | 4p16.3–4q35.2 | 1–190214555 | 190214554 | Tetraploid |
| 66 | HD NFKB2 | 10p15.3–10q26.3 | 1–133797422 | 133797421 | Tetraploid |
| 67 | HD NFKB2 | 10q11.21–10q26.3 | 42354000–133797422 | 91443422 | Monosomy 13/14 |
| Case | TRAF2 or TRAF3 | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 8 | HD TRAF3 | 14q22.3–14q32.33 | 56254000–104990000 | 48736000 | 11;14 |
| 23 | HD TRAF3 | 14q11.2–14q32.33 | 19958000–105864169 | 85906169 | 4;14 |
| 32 | BD TRAF3 | 14q32.32–14q32.32 | 102754161–102809688 | 55527 | 6;14 |
| 37 | HD TRAF3 | 14q24.3–14q32.33 | 77344000–105590563 | 28246563 | Hyper + IGH sep |
| 44 | HD TRAF3 | 14q21.1–14q32.33 | 39707000–107043718 | 67336718 | Hyper |
| 47 | HD TRAF3 | 14q32.32–14q32.32 | 102845410–102902550 | 57140 | Hyper |
| 52 | BD TRAF3 | 14q32.32–14q32.32 | 102722216–102790013 | 67797 | Hyper |
| 60 | BD TRAF3 | 14q32.32–14q32.32 | 102741220–102888391 | 147171 | Hyper |
| 62 | BD TRAF3 | 14q32.31–14q32.32 | 102680377–102913558 | 233181 | Hyper |
| 67 | BD TRAF3 | 14q32.32–14q32.32 | 102841855–102878463 | 36608 | Monosomy 13/14 |
| 67 | HD TRAF2 | 9q34.3–9q34.3 | 136828276–136921241 | 92965 | Monosomy 13/14 |
| Case # | CRBN or IKZF1 or IKZF3 | Location | Breakpoints | Size (bp) | Primary |
|---|---|---|---|---|---|
| 8 | HD IKZF3 | 17q12–17q21.31 | 35371000–44480000 | 9109000 | 11;14 |
| 17 | HD CRBN | 3p26.3–3p26.2 | 2738159–3194829 | 456670 | 4;14 |
| 22 | HD CRBN | 3p26.3–3q22.1 | 1–130531000 | 130530999 | 4;14 |
| 28 | HD CRBN | 3p26.3–3p25.2 | 1–12659000 | 12658999 | 14;20 |
| 41 | HD CRBN | 3p26.3–3p24.1 | 1–28213000 | 28212999 | Hyper + IGH sep |
| 44 | HD IKZF3 | 17p13.3–17q21.31 | 1–45734287 | 45734286 | Hyper |
| 58 | HD CRBN | 3p26.3–3p26.1 | 1–7110000 | 7109999 | Hyper |
| 58 | IKZF1 Gain + insertion to 10q25.2 | 17p12.2 | 50207542–50430511 | 222969 | Hyper |
Abnormalities of genes of known clinical significance in MM
Large gains of chromosome material are not indicated
HD heterozygous deletion, BD biallelic deletion indicated in bold, cytogenetic band and location in GRCh38
Evaluation for loss of function alterations of genes that have been associated with lenalidomide response or resistance (CRBN, IKZF1 and IKZF3) identified 10.0% of the cohort had either a CRBN, IKZF1 and IKZF3 gene alteration (Table 5, Fig. 3, Supplemental Fig. 2D, case #58). Specifically, five cases had a heterozygous deletion of CRBN ranging in size from 457 Kb to 130.5 Mb including case #58 that had both a 7.1 Mb deletion encompassing CRBN and a 223 Kb IKZF1 duplication with insertion of IKZF1 into 10q25.2. Two cases had a heterozygous deletion that included IKZF3 (9.1 Mb and 45.7 Mb).
Discussion
Most clinical laboratories employ FISH analysis of CD138 enriched plasma cells as the preferred methodology in order to identify recurrent primary and secondary genomic abnormalities of prognostic and therapeutic significance in patients with PCNs15. The majority of these laboratories utilize only a limited FISH panel with many focusing on high risk abnormalities defined by the revised International Staging System (R-ISS) including 1q gain, t(4;14), t(14;16) or 17p deletion15. Some laboratories have incorporated the use of chromosomal microarray analysis in the detection of CNAs such as hyperdiploidy, 17p deletions and 1q gains, however microarray studies are unable to identify balanced structural rearrangements necessitating the use of other methodologies in the detection of IGH rearrangements15. It has also become increasingly apparent that some FISH probes, such as those targeting MYC rearrangements, display evidence of false negative results18–22. In addition, FISH panels for PCNs are variable between individual laboratories, provide a limited view of the whole genome and may not always reflect genomic complexity. Given that multiple research studies and investigational trials have used NGS based techniques to identify CNAs, SNVs along with structural rearrangements, we sought to explore the feasibility of employing an NGS technique in the detection of CNAs and structural rearrangements as a FISH replacement assay within a clinical genomics laboratory.
We describe the performance and added utility of a whole genome NGS based strategy, MPseq, in comparison to the current gold standard FISH approach in the evaluation of patients with PCNs. While MPseq and FISH displayed equal performance in the ability to classify the presence or absence of a recurrent, primary cytogenetic subtype (i.e. hyperdiploidy or specific IGH rearrangement), MPseq was superior compared to FISH in the characterization of rearrangement complexity, identification of secondary abnormalities, resolution of atypical FISH results and identification of novel abnormalities of prognostic significance not targeted by traditional FISH panels. Many samples chosen for this study had a high plasma cell burden (median 36% PCs) and ~33% of cases were obtained from fresh or frozen samples that did not require enrichment.
An advantage to using a whole genome NGS technique like MPseq is the ability to identify rearrangements using an unbiased approach. Other laboratories have developed and validated NGS methodologies utilizing target-enrichment approaches for PCNs allowing a custom target pull down of limited genomic regions13,35–37. While these targeted approaches have reduced cost and simplified analysis workflows, a genome wide approach utilizing long-insert whole genome sequencing employed by the MMRF CoMMpass Study in their Seq-FISH analysis has demonstrated improved sensitivity with similar specificity in relation to clinical FISH testing38. Although MPseq is similar to Seq-FISH with regard to a whole genome sequencing approach, a significant limitation to the current MPseq strategy is the inability to identify SNVs. This limitation can be resolved with deeper and faster sequencing, coupled with reduced sequencing costs. An integrated genomic analysis incorporating structural variation, CNAs, and SNVs together may lead to enhanced prognostication13. Of practical consideration is the ~two-fold increased cost and “turn-around-time” of reporting of clinical grade testing for MPseq compared to a comprehensive FISH panel; although we anticipate over time the cost and time of reporting for NGS approaches will continue to be reduced.
Another limitation to the use of MPseq is the inability to identify rearrangements in highly repetitive regions of the genome containing constitutive heterochromatin such as those involving telomeres, centromeres, and in regions near the centromeres of chromosomes 1, 9, and 16 and in the Y chromosome27. This limitation may be reflected by the inability of MPseq to identify apparent trisomies in 2 cases (cases 5 and 42) with evidence of hyperdiploidy. Case 5 displayed a gain of a structurally abnormal chromosome 3 by conventional chromosome studies. Since the centromere regions that are targeted by the FISH probes are not covered by MPseq, it is unclear whether a small gain or presence of a polymorphism of these regions are present without evidence of a bona fide trisomy or whether the trisomy was present at a subclonal level below the limit of detection by MPseq (<25% for CNAs)18. Polymorphisms of the acrocentric chromosome 15 have also been reported39 and are observed in FISH analysis of PCNs in our laboratory (data not shown). Discrepancies involving chromosome 15 are present in 6 of 70 cases in this study demonstrated by either a monosomy 15 FISH result with normal chromosome 15 s by MPseq or either a normal or monosomy 15 FISH result with trisomy 15 by MPseq. Since MPseq does not rely on detection of only the centromere region like FISH, analysis of copy number changes throughout the whole chromosome can be useful to interpret the presence or absence of a trisomy. On the other hand, any missed hyperdiploid cases may be of less relevance as hyperdiploidy can be detected by flow cytometric methods40.
MPseq is not currently used in the detection of copy-neutral LOH or ploidy. As a result, MPseq did not identify any cases as tetraploid, an abnormality found in approximately 6% of patients with MM that has been associated with high-risk genomic abnormalities and with poor prognosis in NDMM41. Six patients in our cohort had evidence of tetraploidy and 5 of these cases had tetraploidy in combination with high risk cytogenetics (Fig. 1), which were identified by MPseq. For case 39, it is unclear why MPseq failed to identify a 1q gain. No evidence of a duplication involving chromosome 1q (chr. 1:155122503–155571708) was identified by MPseq and based on FISH data, the 1q gain did not appear to be subclonal, however this sample was extracted from unsorted bone marrow with 20% clonal plasma cells which may have contributed to this missed abnormality. Subclonal CNAs and cases with low tumor load have a risk of being missed by MPseq, a risk that also exists when performing FISH.
Of the 30 discordant cases, 19 cases had abnormalities identified by MPseq that were missed by FISH, the majority involving insertional events near the MYC gene region. In total, MYC rearrangements were found in 51.4% of the cases in our cohort which includes NDMM and RRMM. This is consistent with previous reports identifying MYC rearrangements in 35% of NDMM with increased frequency in RRMM10,42. MYC rearrangements have been reported as subclonal events and are associated with disease progression3,11,43. Approximately 66% of MYC rearrangements have been found in association with non-Ig partners resulting in juxtaposition to enhancer sequences promoting aberrant MYC gene expression, which may be targeted by BRD4 inhibitors in MM10. Identification of MYC rearrangements using a break-apart probe strategy resulted in a 50.0% false negative rate in our patient cohort. Whether these false negative insertion cases have the same prognostic implication as other MYC rearrangements remains unknown.
Two cases had deletions of the TP53 gene region that were not identified by FISH. For cases 4 and 21, MPseq identified a deletion of TP53 (5.6 Mb in case 4, 2.7 Mb in case 21). Interestingly, for case 21, MPseq also identified a translocation involving TP53 (to 4q32.1). Both cases were scored as having two copies of TP53 by FISH and represent false negative results due the location of the deletion in relation to the FISH footprint in case 4 and the TP53 translocation in combination with the deletion in case 21. Although these cases had missed high risk abnormalities, the mSMART risk did not change since those cases also had additional high risk abnormalities [1q gain for cases 4 and t(4;14) for case 21]. For case 4, a separate NGS assay analyzing SNVs identified a pathogenic TP53 mutation [Chr17(GRCh37):g.7577111 G > T; NM_001126113.2(TP53):c.827 C > A; p.Ala276Asp] located in the DNA-binding domain and in vitro functional data predicts that this variant results in non-functional p53 protein44. TP53 has been found to be mutated in 3–16% of NDMM6,45–47 with a higher frequency in RRMM48–50. TP53 mutations in combination with 17p deletions are associated with double hit MM with reduced overall, progression-free and relapse-free survival51. Therefore, this missed TP53 deletion fails to identify the presence of a likely double hit MM in patient 4. The combination of a rearrangement and deletion also likely represents a double hit MM abnormality in case 21.
The CCND1/IGH dual color, dual fusion probe set is used to identify CCND1/IGH rearrangements. However, three copies of CCND1 in the absence of IGH fusion can indicate trisomy 11 or non-IGH CCND1 rearrangements. MPseq identified three CCND1 rearrangements including case 4 (IGL/CCND1), case 43 (IGK/CCND1) described more fully in Peterson, et al.52 and case 57 (BRINP3/CCND1). FISH also identified amplification of CCND1 in case 4, three copies of CCND1 in case 43 and a normal signal pattern for CCND1 in case 57. The CCND1 rearrangement identified in case 57 was a complex translocation between 1q31.1 and 11q13.3 consisting of four junctions and deletions of ~100 kb at both ends. Through this complex event, CCND1 is brought into close proximity to the 3′ end of BRINP3, while the balancing set of junctions brings the 5′ end of BRINP3 near 11q24.3. Additionally, the derivative chromosome containing CCND1 has been copied. Overall this would result in three copies of CCND1, two of which have been translocated near the 3′ end of BRINP3. This case demonstrates how MPseq is able to determine complex rearrangements involving important genes without prior knowledge of the junction partner location.
Although immunoglobulin lambda rearrangements have been recently reported in association with poor prognosis3, light chain rearrangements are typically not evaluated in the diagnostic work up of MM in most clinical genomics laboratories. Using MPseq data, we identify 10 cases (14.3% of entire cohort) with IGL rearrangements with five of these cases with standard risk cytogenetic results. IGL rearrangements and other focal deletions of clinical significance are typically not evaluated by FISH. Given the high rate of false-negative MYC rearrangements and inability to appreciate all abnormalities of clinical significance, we demonstrate that MPseq has increased clinical value compared to FISH in characterizing genomic abnormalities in PCNs.
Supplementary information
Acknowledgements
Research reported in this publication was supported with contributions from the Mayo Clinic Department of Laboratory Medicine and Pathology with support from the Marion Schwartz Career Development Award in Multiple Myeloma, Center for Individualized Medicine and the National Cancer Institute of the National Institutes of Health under Award Number P50CA186781 from the Mayo Clinic Multiple Myeloma Specialized Program of Research Excellence. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest
S.K.: AbbVie, Celgene, Janssen, Merck, Novartis, Roche, Amgen, Sanofi, and Takeda (research funding, consulting) and Adaptive (honoraria). K.S.: consultant for Bristol-Myers Squibb, Celgene, Amgen, Janssen, Takeda, and Roche. Algorithms described in this paper are licensed to WholeGenome LLC, owned by G.V.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: James Smadbeck, Jess F. Peterson
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41408-019-0255-z).
References
- 1.Howlader N, et al. Improved estimates of cancer-specific survival rates from population-based data. J. Natl Cancer Inst. 2010;102:1584–1598. doi: 10.1093/jnci/djq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rajkumar SV. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018;93:981–1114. doi: 10.1002/ajh.25117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barwick BG, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat. Commun. 2019;10:1911. doi: 10.1038/s41467-019-09555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman MA, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egan JB, et al. Whole-genome sequencing of multiple myeloma from diagnosis to plasma cell leukemia reveals genomic initiating events, evolution, and clonal tides. Blood. 2012;120:1060–1066. doi: 10.1182/blood-2012-01-405977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohr JG, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller A, et al. High somatic mutation and neoantigen burden are correlated with decreased progression-free survival in multiple myeloma. Blood Cancer J. 2017;7:e612. doi: 10.1038/bcj.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker BA, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132:587–97.. doi: 10.1182/blood-2018-03-840132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walker BA, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat. Commun. 2015;6:6997. doi: 10.1038/ncomms7997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Affer M, et al. Promiscuous MYC locus rearrangements hijack enhancers but mostly super-enhancers to dysregulate MYC expression in multiple myeloma. Leukemia. 2014;28:1725–35.. doi: 10.1038/leu.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misund K. K., et al. MYC dysregulation in the progression of multiple myeloma. Leukemia (2019). https://www.ncbi.nlm.nih.gov/pubmed/31439946. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 12.Manier S, et al. Genomic complexity of multiple myeloma and its clinical implications. Nat. Rev. Clin. Oncol. 2017;14:100–13.. doi: 10.1038/nrclinonc.2016.122. [DOI] [PubMed] [Google Scholar]
- 13.Bolli N, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32:2604–16.. doi: 10.1038/s41375-018-0037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajkumar S. V. mSMART stratification for myeloma and risk-adapted therapy. www.msmart.org
- 15.Pugh TJ, et al. Assessing genome-wide copy number aberrations and copy-neutral loss-of-heterozygosity as best practice: an evidence-based review from the Cancer Genomics Consortium working group for plasma cell disorders. Cancer Genet-Ny. 2018;228:184–96.. doi: 10.1016/j.cancergen.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Kumar SK, Rajkumar SV. The multiple myelomas - current concepts in cytogenetic classification and therapy. Nat. Rev. Clin. Oncol. 2018;15:409–21.. doi: 10.1038/s41571-018-0018-y. [DOI] [PubMed] [Google Scholar]
- 17.Sonneveld P, et al. Treatment of multiple myeloma with high-risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127:2955–2962. doi: 10.1182/blood-2016-01-631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aypar U, et al. Mate pair sequencing improves detection of genomic abnormalities in acute myeloid leukemia. Eur. J. Haematol. 2019;102:87–96. doi: 10.1111/ejh.13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.King Rebecca L., McPhail Ellen D., Meyer Reid G., Vasmatzis George, Pearce Kathryn, Smadbeck James B., Ketterling Rhett P., Smoley Stephanie A., Greipp Patricia T., Hoppman Nicole L., Peterson Jess F., Baughn Linda B. False-negative rates for MYC fluorescence in situ hybridization probes in B-cell neoplasms. Haematologica. 2018;104(6):e248–e251. doi: 10.3324/haematol.2018.207290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson JF, et al. Characterization of a cryptic IGH/CCND1 rearrangement in a case of mantle cell lymphoma with negative CCND1 FISH studies. Blood Adv. 2019;3:1298–302.. doi: 10.1182/bloodadvances.2019031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson Jess F., Pitel Beth A., Smoley Stephanie A., Smadbeck James B., Johnson Sarah H., Vasmatzis George, Kearney Hutton M., Greipp Patricia T., Hoppman Nicole L., Baughn Linda B., Ketterling Rhett P. Use of mate-pair sequencing to characterize a complex cryptic BCR/ABL1 rearrangement observed in a newly diagnosed case of chronic myeloid leukemia. Human Pathology. 2019;89:109–114. doi: 10.1016/j.humpath.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Peterson Jess F., Pitel Beth A., Smoley Stephanie A., Smadbeck James B., Johnson Sarah H., Vasmatzis George, Koon Sarah J., Webley Matthew R., McGrath Mary, Bayerl Michael G., Baughn Linda B., Rowsey Ross A., Ketterling Rhett P., Greipp Patricia T., Hoppman Nicole L. Detection of a cryptic NUP214/ABL1 gene fusion by mate-pair sequencing (MPseq) in a newly diagnosed case of pediatric T-lymphoblastic leukemia. Molecular Case Studies. 2019;5(2):a003533. doi: 10.1101/mcs.a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker BA, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood. Cancer J. 2014;4:e191. doi: 10.1038/bcj.2014.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker BA, et al. Characterization of IGH locus breakpoints in multiple myeloma indicates a subset of translocations appear to occur in pregerminal center B cells. Blood. 2013;121:3413–3419. doi: 10.1182/blood-2012-12-471888. [DOI] [PubMed] [Google Scholar]
- 25.Baughn LB, et al. Differences in genomic abnormalities among African individuals with monoclonal gammopathies using calculated ancestry. Blood Cancer J. 2018;8:96. doi: 10.1038/s41408-018-0132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang JS, et al. Molecular signatures of multiple myeloma progression through single cell RNA-Seq. Blood Cancer J. 2019;9:2. doi: 10.1038/s41408-018-0160-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson SH, et al. SVAtools for junction detection of genome-wide chromosomal rearrangements by mate-pair sequencing (MPseq) Cancer Genet. 2018;221:1–18. doi: 10.1016/j.cancergen.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Smadbeck James B., Johnson Sarah H., Smoley Stephanie A., Gaitatzes Athanasios, Drucker Travis M., Zenka Roman M., Kosari Farhad, Murphy Stephen J., Hoppman Nicole, Aypar Umut, Sukov William R., Jenkins Robert B., Kearney Hutton M., Feldman Andrew L., Vasmatzis George. Copy number variant analysis using genome-wide mate-pair sequencing. Genes, Chromosomes and Cancer. 2018;57(9):459–470. doi: 10.1002/gcc.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drucker TM, et al. BIMA V3: an aligner customized for mate pair library sequencing. Bioinformatics. 2014;30:1627–1629. doi: 10.1093/bioinformatics/btu078. [DOI] [PubMed] [Google Scholar]
- 30.Gaitatzes A, Johnson SH, Smadbeck JB, Vasmatzis G. Genome U-Plot: a whole genome visualization. Bioinformatics. 2018;34:1629–34.. doi: 10.1093/bioinformatics/btx829. [DOI] [PubMed] [Google Scholar]
- 31.Lee N, et al. Discrepancies between the percentage of plasma cells in bone marrow aspiration and BM biopsy: Impact on the revised IMWG diagnostic criteria of multiple myeloma. Blood Cancer J. 2017;7:e530. doi: 10.1038/bcj.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Demchenko Y, et al. Frequent occurrence of large duplications at reciprocal genomic rearrangement breakpoints in multiple myeloma and other tumors. Nucleic Acids Res. 2016;44:8189–8198. doi: 10.1093/nar/gkw527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu YX, et al. Loss of FAM46C promotes cell survival in myeloma. Cancer Res. 2017;77:4317–27.. doi: 10.1158/0008-5472.CAN-16-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jimenez C, et al. A next-generation sequencing strategy for evaluating the most common genetic abnormalities in multiple myeloma. J. Mol. Diagn. 2017;19:99–106. doi: 10.1016/j.jmoldx.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Bolli N, et al. A DNA target-enrichment approach to detect mutations, copy number changes and immunoglobulin translocations in multiple myeloma. Blood Cancer J. 2016;6:e467. doi: 10.1038/bcj.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White BS, et al. A multiple myeloma-specific capture sequencing platform discovers novel translocations and frequent, risk-associated point mutations in IGLL5. Blood Cancer J. 2018;8:35. doi: 10.1038/s41408-018-0062-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldsmith Scott R., Fiala Mark A., Dukeman James, Ghobadi Armin, Stockerl-Goldstein Keith, Schroeder Mark A., Tomasson Michael, Wildes Tanya M., Vij Ravi. Next Generation Sequencing-based Validation of the Revised International Staging System for Multiple Myeloma: An Analysis of the MMRF CoMMpass Study. Clinical Lymphoma Myeloma and Leukemia. 2019;19(5):285–289. doi: 10.1016/j.clml.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Davila-Rodriguez MI, et al. Constitutive heterochromatin polymorphisms in human chromosomes identified by whole comparative genomic hybridization. Eur. J. Histochem. 2011;55:e28. doi: 10.4081/ejh.2011.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sidana S, et al. Rapid assessment of hyperdiploidy in plasma cell disorders using a novel multi-parametric flow cytometry method. Am. J. Hematol. 2019;94:424–30.. doi: 10.1002/ajh.25391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sidana S, et al. Tetraploidy is associated with poor prognosis at diagnosis in multiple myeloma. Am. J. Hematol. 2019;94:E117–E20.. doi: 10.1002/ajh.25420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shou Y, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc. Natl Acad. Sci. USA. 2000;97:228–233. doi: 10.1073/pnas.97.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glitza IC, et al. Chromosome 8q24.1/c-MYC abnormality: a marker for high-risk myeloma. Leuk. Lymphoma. 2015;56:602–607. doi: 10.3109/10428194.2014.924116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kato S, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl Acad. Sci USA. 2003;100:8424–8429. doi: 10.1073/pnas.1431692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolli N, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014;5:2997. doi: 10.1038/ncomms3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kortum KM, et al. Longitudinal analysis of 25 sequential sample-pairs using a custom multiple myeloma mutation sequencing panel (M(3)P) Ann. Hematol. 2015;94:1205–1211. doi: 10.1007/s00277-015-2344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker BA, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. J. Clin. Oncol. 2015;33:3911–3920. doi: 10.1200/JCO.2014.59.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weinhold N, et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood. 2016;128:1735–1744. doi: 10.1182/blood-2016-06-723007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong W, et al. An analysis of the clinical and biologic significance of TP53 loss and the identification of potential novel transcriptional targets of TP53 in multiple myeloma. Blood. 2008;112:4235–4246. doi: 10.1182/blood-2007-10-119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chng WJ, et al. Clinical significance of TP53 mutation in myeloma. Leukemia. 2007;21:582–584. doi: 10.1038/sj.leu.2404524. [DOI] [PubMed] [Google Scholar]
- 51.Walker BA, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2019;33:159–70.. doi: 10.1038/s41375-018-0196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peterson JF, et al. Whole genome mate-pair sequencing of plasma cell neoplasm as a novel diagnostic strategy: a case of unrecognized t (2; 11) structural variation. Clin. Lymphoma, Myeloma Leuk. 2019;19:598–602. doi: 10.1016/j.clml.2019.06.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.