

Abstract
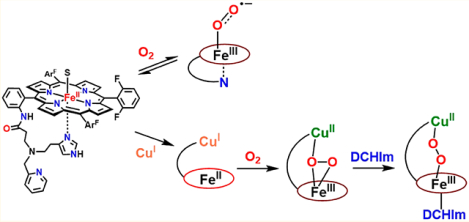
The focus of this study is in the description of synthetic heme/copper/O2 chemistry employing a hemecontaining binucleating ligand which provides a tridentate chelate for copper ion binding. The addition of O2 (−80 °C, tetrahydrofuran (THF) solvent) to the reduced heme compound (PImH)FeII (1), gives the oxy-heme adduct, formally a heme–superoxide complex FeIII−(O2•−) (2) (resonance Raman spectroscopy (rR): νO−O, 1171 cm−1 (Δ18O2, −61 cm−1); νFe−O, 575 cm−1 (Δ18O2, −24 cm−1)). Simple warming of 2 to room temperature regenerates reduced complex 1; this reaction is reversible, as followed by UV–vis spectroscopy. Complex 2 is electron paramagnetic resonance (EPR)-silent and exhibits upfield-shifted pyrrole resonances (δ 9.12 ppm) in 2H NMR spectroscopy, indicative of a six-coordinate low-spin heme. The coordination of the tethered imidazolyl arm to the heme–superoxide complex as an axial base ligand is suggested. We also report the new fully reduced heme–copper complex [(PImH)FeIICuI]+ (3), where the copper ion is bound to the tethered tridentate portion of PImH. This reacts with O2 to give a distinctive low-temperature-stable, high-spin (S = 2, overall) peroxo-bridged complex [(PImH)FeIII−(O22−)−CuII]+ (3a): λmax, 420 (Soret), 545, 565 nm; δpyrr, 93 ppm; νO−O, 799 cm−1 (Δ18O2, −48 cm−1); νFe−O, 524 cm−1 (Δ18O2, −23 cm−1). To 3a, the addition of dicyclohexylimidazole (DCHIm), which serves as a heme axial base, leads to low-spin (S = 0 overall) species complex [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b): λmax, 425 (Soret), 538 nm; δpyrr, 10.2 ppm; νO−O, 817 cm−1 (Δ18O2, −55 cm−1); νFe−O, 610 cm−1 (Δ18O2, −26 cm−1). These investigations into the characterization of the O2-adducts from (PImH)FeII (1) with/without additional copper chelation advance our understanding of the dioxygen reactivity of heme-only and heme/Cu-ligand heterobinuclear system, thus potentially relevant to O2 reduction in heme–copper oxidases or fuel-cell chemistry.
Graphical Abstract
INTRODUCTION
Cytochrome c oxidase (CcO), the terminal enzyme of the respiratory chain in the mitochondrial inner membrane from mammalian and bacterial sources, reduces dioxygen to water, coupling the redox energy to membrane proton translocation utilized for ATP synthesis.1–10 The enzyme possesses five key redox-active metal ion centers, dicopper complex CuA, which is a binuclear electron-transfer center, a low-spin heme a (cytochrome a), and the heterobinuclear (Fea3/CuB) active site, where dioxygen binding and reduction occurs.1–10 The latter (Scheme 1) consists of a high-spin heme and a neighboring (~4.5–5.0 Å) copper ion possessing three histidine imidazole nitrogen ligands, one of which is cross-linked to a tyrosine (Tyr) residue biosynthesized via a posttranslational modification.11–16 With a focus on the heme/Cu active site in varying redox or ligation states, structural aspects and the O2 reduction mechanism have been studied extensively, utilizing many spectroscopic methods,1–10 X-ray crystallography,17,18 and computational approaches.19
Scheme 1.

Proposed O−O Reductive Cleavage Mechanism by Heme–Copper Oxidases for Cytochrome c Oxidase
The current state of our understanding of early stages in the CcO O2 binding and reduction are given in Scheme 1. Molecular oxygen reacts with a fully reduced FeII⋯CuI binuclear active site, initially forming a detectable, transient, Fe−O2 (“oxy-heme”) adduct, formally a FeIII−(O2•−) (A) species possessing end-on superoxide binding. This assignment is consistent with resonance Raman (rR) spectroscopic properties, as being similar to those found in oxy-hemoglobin (νFe−O = 567 cm−1) and oxy-myoglobin (νFe−O = 570 cm−1).20,21
Intermediate A rapidly undergoes O−O cleavage to yield PM.22 A total of four electrons are required to fully reductively cleave the O−O bond of dioxygen. Three electrons are provided by the binuclear site (two from iron (FeII → FeIV) and one from CuB (CuI → CuII)). It is now accepted that the fourth electron derives from the phenol residue of the nearby His–Tyr cross-link. This phenol residue serves as an overall hydrogen atom donor (H•), leaving PM formulated as a FeIV=O/CuII−OH/Tyr• moiety, i.e., thus with a ferryl-oxo (Fea3IV=O; ν(Fe−O) = 804 cm−1), cupric-hydroxide (CuBII−OH) and a tyrosyl radical (Scheme 1).6,8,10 Also, evidence suggests that the His–Tyr cross-link enables enzyme CuB incorporation and stabilizes the precise geometry of the binuclear active site.11,16
On the basis of biochemical experiments,23 computational studies,24–27 and synthetic modeling,5,10 a peroxo-bridged heterobinuclear FeIII−(O22−)−CuII (Ip) has been predicted as an intermediate species occurring on the pathway from A to PM. This peroxo moiety critically precedes O−O bond cleavage via initial H-bonding or protonation,19 possibly through a water molecule bridge, as shown in Scheme 1.
Design and construction of heme/copper synthetic models provide critical information about structure, spectroscopy and reactivity relevant to CcO enzyme mechanism, in the usual manner that models contribute to scientific advances, allowing a breakdown and attention to particular aspects of the protein structure/function. With respect to dioxygen reactivity, reduced heme–copper assemblies (or components) have been shown to generate heme−FeIII−superoxo entities (like intermediate A, Scheme 1; see complexes from Collman28 and Naruta29 and their co-workers, Figure 1). Other studies from Naruta30 and our laboratories31,32 have revealed that peroxo-bridged heme−FeIII−(O22−)−CuII(ligand) complexes can be generated possessing three structural types10 (high-spin (HS) μ-η2:η1-peroxo (C, D),30,31,33 HS μ-η2:η2-peroxo (E),31,32 and low-spin (LS) μ−1,2-peroxo (F))10,34 (Figure 1). Collman’s investigations involving elegant binucleating (for Fe and Cu) ligands demonstrated efficient electrocatalytic four-electron four-proton O2 reduction to water (ORR)35 and stoichiometric studies where a phenol H atom donor could reductively cleave the O−O bond of a heme−O2−Cu construct.36 We have developed similar model systems with simpler compounds, i.e., complex D (Figure 1), which effect efficient electrocatalytic ORR chemistry.37,38
Figure 1.

Various complexes of heme/copper synthetic models: superoxide heme−FeIII−(O2•−)⋯CuI(ligand) complexes, (A, Collman’s group; B, Naruta’s group). Heme−FeIII−(O22−)−CuII(ligand) complexes: X-ray crystal structure of Naruta’s HS μ-η2:η1-peroxobridged complex (C: adapted from ref 30); Karlin group analog (D); μ-η2:η2-peroxo compound (E); LS heme peroxo adducts with a μ-1,2-peroxo structure type, LS-3DCHIm (F); DFT-calculated structure for [LS-4DCHIm (ArOH)] (G: adapted from ref 34), a phenol adduct.
In stoichiometric reactivity studies, detailed synthetic and theoretical/computation insights have been obtained concerning elements critical for reductive cleavage of heme–Cu bound O2 (as peroxide) intermediate. However, many details and fundamental aspects have not been elucidated or fully understood. These include the exact structural requirements for the Fe−O−O−Cu moiety, tridentate versus tetradentate Cu chelation, factors such as H-bonding to a peroxide O atoms (which one?) versus actual protonation, the origin and pKa of the proton donor, the timing of electron versus proton injection, and so on. Thus far, for heme−Cu−O2 constructs, very small changes in the local (ligand) environment may lead to absolutely varying chemistry, and there is a need to understand these effects in obtaining fundamental insights. For example, LS-3DCHIm (F) and LS-4DCHIm (G) (Figure 1), which differ by only one DCHIm Cu-ligand, behave completely differently toward proton–electron sources. The former is completely unreactive, while the latter, in the presence of an H-bonding phenol, undergoes clean peroxide O−O reductive cleavage to give water.10,19,34,39
From these perspectives, considerable further studies and advancements are needed. These include a continued effort to define the dioxygen reactivity of reduced heme–copper assemblies, while also if possible, elucidating the heme-only and (ligand)–Cu O2-chemistry. For further elaborated/new heme−O2−Cu species, subsequent defining of O−O reductive cleavage chemistry is a longer-term goal, in the contexts mentioned above.
In the present study, we report on an advanced CcO active site model system with binucleating ligand PImH; this employs a new tridentate ligand which includes a histamine moiety appended to the periphery of a fluorinated tetraphenylporphyrin. The FeII−porphyrinate complex (PImH)FeII (1), is shown in Chart 1.40 Herein, we report the oxygenation chemistry of 1, whose chemistry mimics the initial O2 binding in CcO, as well as formation of oxy-hemoglobin and -myoglobin. The generation of a heme–copper derivative of 1 has been accomplished, and oxygenation of the reduced FeII−CuI form gives a high-spin heme–peroxo–copper product. Lastly, addition of DCHIm as a heme axial ligand to the high-spin solution leads to low-spin complex. All complexes have been spectroscopically characterized by UV–vis, 2H NMR, and electron paramagnetic resonance (EPR) spectroscopies; for the O2-derived complexes, vibrational data has been obtained using rR.
Chart 1.

RESULTS AND DISCUSSION
Dioxygen Reactivity of (PImH)FeII (1).
Complex 1 reacted with dioxygen to form a low-temperature stable superoxide level intermediate, which was characterized by UV–vis, 2H NMR, rR, and EPR spectroscopies. Bubbling dry O2 through a solution of (PImH)FeII (1) {λmax = 419, 526 and 552(sh) nm} in THF at −80 °C led to new features at 424 and 541 nm which were ascribed to a (PImH)FeIII−(O2•−) (2) (Figure 2). Further characterization of the 1/O2 reaction product comes from low-temperature 2H NMR spectroscopic investigation, employing a deuterated (β-pyrrolic hydrogens) analog of 2 (2-d8) in THF solvent (Figure 3). It confirms the proposed formation of a myoglobin-like FeIII−superoxide complex. Complex 2 gives rise to a diamagnetic spectrum (δ = 9.12 ppm), consistent with the formation of (PImH)FeIII−(O2•−) (2-d8) and suggesting a low-spin (S = 0) six-coordinate iron.41 As expected for a diamagnetic species, complex 2 is EPR-silent. The literature on hemoglobin and model compounds has generally proposed that the unpaired electron of the low-spin FeIII ion is antiferromagnetically coupled with the unpaired electron of the superoxide radical anion, making the complex diamagnetic.41 Recent resonant inelastic X-ray scattering (RIXS) analyses have shown that the electronic structure more closely fits to an FeII—(O2) description for a model system (the “oxy picket-fence porphyrin”), while oxyhemoglobin has more FeIII—(O2•−) character (although not fully to the low-spin ferric level).42 Nevertheless, the latter notation is used here for clarity.
Figure 2.

(A) Reaction scheme showing reversible ferric heme superoxide (2) formation by bubbling O2(g) into a solution of fully reduced complex 1. (B) UV–vis spectra (THF at −80 °C) of the deoxygenation reaction of 1. Shown are spectra of complex 1 (black) and 2 (red).
Figure 3.

2H NMR spectra of (A) d8-(PImH)FeII (1-d8) and (B) d8-(THF)(PImH)FeIII−(O2•−) (2-d8) in THF at −80 °C. The sharp peaks at δ 3.58 and 1.73 ppm correspond to solvent THF.
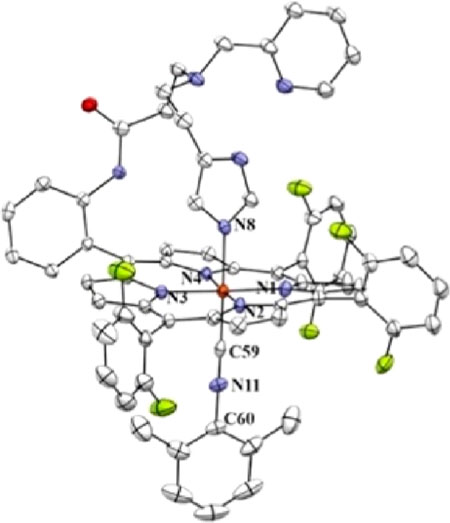
We postulate that the sixth axial ligand present in 2 is the imidazolyl group of the tridentate tether in PImH (see Chart 1). We base this conclusion on our previous X-ray crystallographic finding that in a six-coordinate iron(II) complex with PImH, also containing a strong isocyanide donor ligand DIMPI (= 2,6-dimethylphenyl isocyanide), [(PImH)FeII−DIMPI], the free imidazole group binds strongly (Fe−NIm = 2.024 Å) as the axial ligand trans to DIMPI (see diagram below).40
The O2 binding to 1 was reversible, as we have found for some other heme–Fe(II) O2-adducts with porphyrinates possessing electron-withdrawing fluorine substituents,43 where warming a solution of 2 to room temperature in a closed system allows for release of dioxygen and regeneration of (PImH)FeII (1), to the extent of ~80%. Cooling of this solution (without further addition of O2) to −80 °C leads to close to full regeneration of (PImH)FeIII−(O2•−) (2). Dioxygen binding cycling in this manner illustrated in the Figure S1. Several cycles of the reversible O2-binding process could be monitored by UV–vis spectroscopy.
Complex (PImH)FeIII−(O2•−) (2) was further characterized by rR spectroscopy (Figure 4). In the lower-frequency region, a νFe−O stretch is detected at 575 cm−1 which is similar to that of other known “oxy-heme” FeIII−(O2•−) synthetic models with a nitrogen axial ligand.10,41,44 This Fe−O stretch shifts down by 24 cm−1 upon 18O2 substitution. An oxygen isotopesensitive feature corresponding to the O−O stretch was detected at νO−O = 1171 cm−1, which shifted to 1110 cm−1 with 18O2. With the hemes that our research group has utilized, having 3 or 4 of the meso-aryl groups possessing 2,6-difluoro substituents, we have observed O−O stretches at similar frequencies (Table 1). Also consistent with the assignments is our finding that the νFe−O and νO−O of 16O/18O isotope shifts (vide supra) closely match those calculated using the harmonic oscillator model: ΔFe−O,calc (16O2/18O2) = −26 cm−1, ΔO−O,calc (16O2/18O2) = −67 cm−1. However, it is quite common that the O−O vibrational mode of dioxygen-bound heme complexes is not observable.10,28,45 Table 1 lists examples from our own investigations, along with several protein and synthetic oxy–heme (FeIII−superoxide) complexes, where νO−O is observed. A more complete listing can be found elsewhere.10
Figure 4.

Resonance Raman spectra of ferric superoxide complex (PImH)FeIII−(O2•−) (2) in frozen THF obtained at 77 K with 413 nm excitation: Fe−O and O−O stretching frequencies for the complex generated with 16O2 (blue) or 18O2 (orange). The 16O2−18O2 difference spectrum is shown in green. Other peaks which seem to be visible are thought to arise from heme core vibrations or weak coupling of intraheme bands with O2 motion. The seemingly less well-defined (based on the green difference spectrum shown here) 16O2 and 18O2 O−O stretches are readily assignable, based on analysis of the original data, in the context of extensive experience and literature.
Table 1.
rR Stretching Frequencies (cm−1) of Heme−FeIII−Superoxide Complexes
| complexa | vO-O (Δ18O2) | vFe-O (Δ18O2) | ref |
|---|---|---|---|
| Mb WT | 1103 | 578 (−29) | 46 |
| Cyt P450 WT | 1139 (−66) | 546 (−31) | 47 |
| Cyt P450 D251N | 1136 (−66) | 537 (−30) | 48 |
| (THF)(F8)FeIII−(O2•−) | 1178 (−64) | 568 (−24) | 49 |
| (6L)FeIII−(O2•−) | 1176 (−64) | 572 (−24) | 33 |
| (PIm)FeIII−(O2•−) | 1180 (−56) | 575 (−23) | 50 |
| (PImH)FeIII−(O2•−) (2) | 1171 (−61) | 575 (−24) | this work |
| [(α4Fe(CO2Me)4)O2•−] | 1004(−53) | 581 (−22) | 51 |
| 967 (−56) | |||
| [(FeIII(OPhP))−O2•−] | 1147 (−59) | 570 (−22) | 44 |
Mb WT, wild-type myoglobin; Cyt P450 WT, wild-type cytochrome P-450 monooxygenase (P450); P450 Asp-251 mutated to Asn. See Figure S2 for diagrams of the other synthetic FeIII−(O2•−) complexes.
Generation of a Reduced Heme–Cu Complex Using PImH.
Addition of 1 equiv of [CuI(CH3CN)4](B(C6F5)4) to the reduced complex (PImH)FeII (1) led to the desired reduced heterobinuclear FeII/CuI compound [(PImH)FeIICuI]+ (3), which had prominent UV–vis absorptions at 423 and 542 nm at low temperature (Figure 5). An asymmetry in pyrrole resonances of reduced complex 1 is observed in 1H NMR spectroscopy, where different chemical shifts for inequivalent porphyrin phenyl substituents leads to observation of two peaks (at δ 50 and 59 ppm; Figure 6). Addition of the copper(I) ion leads to disappearance of upfield-shifted resonances (<0 ppm) which we assign as being derived from methylene hydrogen atoms of the tethered histamine group. Since the copper ion takes up, i.e., binds to, the tridentate chelate from this complex, the imidazolyl group of the histamine arm is no longer bound to the iron center. The pyrrolic protons in 2H NMR spectroscopy at −80 °C resonate at δ 89.4 and 102.8 ppm, typical of a five-coordinate high-spin ferrous heme (Figure 7A). Complex 3 is EPR-silent.
Figure 5.

(A) Overall scheme for reactivity of [(PImH)FeIICuI]+ (3) toward O2(g) at −80 °C in THF to yield a HS peroxo species [(PImH)FeIII−(O22−)−CuII]+ (3a) and LS peroxo complex [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b) following addition of DCHIm. (B) UV–vis spectra (−80 °C) for in situ generated HS 3a (red) and LS 3b (blue) starting following oxygenation of 3 (black).
Figure 6.

1H NMR spectrum of (top) complex (PImH)FeII (1) and (bottom) (PImH)FeIICuI (3) at room temperature.
Figure 7.

2H NMR spectra of (A) in situ generation of d8-[(PImH)FeIICuI]+ (3-d8); (B) d8-[(PImH)FeIII−(O22−)−CuII]+ (3a-d8) generated by bubbling O2; (C) d8-[(DCHIm)(PImH)FeIII−(O22−)−CuII]+(3b-d8) generated by the reaction of 3a-d8 with DCHIm. The strong sharp peaks at δ 1.73 and 3.58 ppm correspond to solvent THF.
Generation of the HS Heme–Peroxo–Cu Complex.
Addition of dry O2(g) to the THF solution of fully reduced heterobinuclear heme–copper complex [(PImH)FeIICuI]+ (3) at low-temperature resulted in the formation of new species [(PImH)FeIII−(O22−)−CuII]+(3a) with UV–vis spectral features at 420, 545, and 565 nm (Figure 5B), notably with two Q-band features (500–600 nm spectral region). These characteristics are the same as those known for other previously well-characterized high-spin heme–peroxo–Cu complexes also with tridentate ligands on the copper ion. (Figure 5A).10,31,32,49,52,53
To provide more detail concerning the structure of [(PImH)FeIII−(O22−)−CuII]+(3a), we carried out rR spectroscopy (Figure 8) and found that the peroxo ligand νO−O vibration is observed at 799 cm−1; this shifts to 751 cm−1 upon 18O2 substitution. In addition, a band at 524 cm−1 (Δ18O2 = −23 cm−1) is assigned to νFe−O for this peroxide complex. Again, as discussed above for (PImH)FeIII−(O2•−) (2), observed isotope frequency shifts when using 18O2 match well with values calculated using the harmonic oscillator model (ΔO−O,calc (16O2/18O2) = −46 cm−1, ΔFe−O,calc (16O2/18O2) = −23 cm−1), corroborating our assignments. While the UV–vis features for 3a are most like high-spin heme–peroxo complexes with tridentate chelates bound to copper(II) (vide supra), we note that the νO−O value observed for 3a (799 cm−1) is much higher in energy than that found for the three other examples we have previously studied, which have tridentate ligands on the Cu(II) ion and where side-on μ-η2:η2−O22− binding to both Fe(III) and Cu(II) occurs, νO−O = 747–767 cm−1.31,32,49,54 In copper–dioxygen coordination chemistry, ligand denticity induces different O2 (as peroxide, O22−) binding modes;55–57 CuI chelates with tridentate ligands prefer a side-on binding mode with a resulting peroxide ligand, giving [(L)CuII−(μ-η2:η2-O22−)−CuII(L)]2+ binuclear complexes whereas with tetradentate ligands (L)CuI complexes react with O2 to give end-on binding, i.e., [(L)CuII−(μ−1,2-O22−)−CuII(L)]2+ species.
Figure 8.

rRaman data (16O2−18O2 difference) collected at 413 nm excitation and 77 K for HS 3a and LS 3b. Complex 2 is present as an impurity, observed as a set of 16O2/18O2 peaks at 575/550 cm−1 (marked with an asterisk).
However, these 3a vibrations (νFe−O = 524 cm−1; νO−O = 799 cm−1; Figure 8) are very close to those previously reported for the HS-TMPA heme–peroxo–copper complex (Chart 2). For this complex with tetradentate chelate bound to copper, νO−O = 804 cm−1, and the peroxo unit is bound side-on to the high-spin FeIII, but end-on to the CuII, for an overall μ-η2:η1 coordination mode that has been supported by EXAFS spectroscopy and DFT calculations.39,58 We posit that the PImH tridentate chelate induces considerable geometric distortion in the Cu(II) coordination sphere, which results in the peroxo binding to Cu in an essentially η1 fashion. Thus, we postulate that 3a possesses a μ-η2:η1-peroxo coordination, formulated as [(PImH)FeIII−(μ-η2:η1-O22−)−CuII]+, as shown in Figure 5A.
Chart 2.

On the basis of past extensive NMR spectroscopic studies on [(F8)FeIII−X−CuII(TMPA)]+ (X = O2−, OH−; TMPA = tris(2-pyridyl)methylamine),59 the peroxo analog (X = O22−),58,60 and [(2L)FeIII−(μ-η2:η2-O22−)−CuII]+32,61 (shown as E, Figure 1), the further analog with tridentate chelates for Cu ion,49,59,61,62 [(PImH)FeIII−(O22−)−CuII]+ (3a), has an overall electronic structure where S = 2, derived from antiferromagnetic coupling of high-spin FeIII (S = 5/2) and CuII (S = 1/2) centers. This assignment is further supported by its EPR-silent behavior (12 K, in THF frozen solution). Further supporting the S = 2 electronic structure assignment, 3a displays a pyrrole signal in the paramagnetic region at 93.0 ppm (−80 °C, THF; see Figure 7B), very similar to that observed for other HS complexes which are listed in Table 2. The 93.0 ppm pyrrole resonance is in the range expected for HS ferric iron.
Table 2.
2H NMR Data and Pyrrole Chemical Shifts of Heme/Copper Dioxygen Adducts or Heme–Peroxo–Copper Complexesa
Generation of a LS Heme–Peroxo–Cu Complex with PImH.
A low-spin analog to [(PImH)FeIII−(O22−)−CuII]+ (3a) was generated by the addition of axial base DCHIm in THF at −80 °C (Figure 5A). This resulted in a drastic change in the UV–vis features (Figure 5B). Relatively intense low-energy features observed at ~800–900 nm (assigned as peroxo-to-Fe LMCT bands) are characteristic of LS μ−1,2-peroxo species, as previously demonstrated for F and G (Figure 1) and in a number of other cases (see Figure S4 for 4-DCHIm and LS-AN and Chart 2 for LS-TMPA).34,39,66 The addition of DCHIm resulted in clear Soret and Q-band shifts (from those observed in 3a) to 425 and 538 nm, respectively (Figure 5B). 2H NMR spectroscopy confirms the spin state change from high-spin 3a (pyrrole signal at 93.0 ppm) to low-spin 3b (pyrrole signal at 10.2 ppm, i.e., in the diamagnetic region; see Figure 7). This behavior suggests that in 3b there exists antiferromagnetic coupling between the low-spin (S = 1/2) six-coordinate FeIII and the d9 CuII (also S = 1/2) centers, through the peroxo bridge. Complex 3b is EPR-silent (12K, THF), also consistent with antiferromagnetic coupling and a resulting S = 0 electronic structure formulation.
Resonance Raman spectroscopic interrogation of LS compound [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b) reveals the expected differences in νFe−O and νO−O stretching vibrations in comparison to HS complex [(PImH)FeIII−(O22−)−CuII]+ (3a) (Figure 8). Coordination of DCHIm to generate 3b produced two isotope-sensitive peaks: an O−O vibration at 817 cm−1 (Δ18O2 = −55 cm−1) and an Fe−O stretch at 610 cm−1 (Δ18O2 = −26 cm−1). This observed νFe−O (16O2/18O2) shift is close to expected value (ΔFe−O,calc (16O2/18O2) = −27 cm−1) in the harmonic oscillator approximation. The magnitude of the isotope shift for νO−O (= −55 cm−1) is a bit larger than the calculated value (ΔO−O,calc (16O2/18O2) = −47 cm−1); however, it is still consistent with the assignment of this stretch to that of a peroxide ligand. Changing from HS (3a) to LS (3b) dramatically increases νFe−O by 86 cm−1 (Figure 5A), while a smaller increase (18 cm−1) is observed for νO−O. Similar effects have been observed recently39,67 for DCHIm binding and indicate a change in Fe spin state and Fe–peroxo coordination geometry. The relatively high νO−O stretch for the peroxo group in 3b (compared to that for 3a) indicates a LS μ−1,2-peroxo coordination mode in the former. Thus, 3b is formulated as [(DCHIm)(PImH)FeIII−(μ−1,2-peroxo−O22−)−CuII]+ (shown in Figure 5A), based on our previous investigation and resulting established structure of LS-TMPA (Chart 2), that has been supported by DFT calculations.39
CONCLUSION
In this paper, we investigated the dioxygen reactivity of reduced heme (PImH)FeII (1) in the absence and the presence of copper ion. Reaction of 1 and O2 results in O2-bound ferrous heme compound, (PImH)FeIII−(O2•−) (2), such as that found in oxygenated myoglobin and hemoglobin. 2H NMR suggests that 2 is low-spin six-coordinate iron. The evidence points to the imidazolyl group of the appended tridentate chelate as serving as an axial ligand base. Addition of Cu(I) ion leads to a new reduced heme–copper complex (PImH)FeIICuI (3), an advanced CcO active site model system due to (i) its tridentate ligand CuI-binding and (ii) the imidazolyl ligand has a free N−H group (see below). At low temperature, dioxygen reaction with 3 gives a high-spin species [(PImH)FeIII−(μ-η2:η1O22−)−CuII]+ (3a), and addition of DCHIm as an axial ligand changes the peroxo coordination to end-on, resulting in formation of LS species with end-on peroxo coordination, [(DCHIm)(PImH)FeIII−(μ−1,2-O22−)−CuII]+(3b).
This oxygenation chemistry with (PImH)FeII (1) lays the groundwork for a better understanding of FeII/CuI/O2 reactions chemistry. Our ongoing studies are focused on investigating the reductive O−O cleavage relevant to CcO enzyme active site chemistry of heme−(O22−)−Cu complexes by employing systematically varied H+/e− sources. Also, since PImH has a built-in imidazole and tridentate system in copper chelation as occurs in CcO, another goal of our research is to mimic the post-translational modification which occurs in CcO, that being formation of a histidine-tyrosine cross-link which is functionally critical.16
EXPERIMENTAL SECTION
Materials and Methods.
All reagents and solvents purchased and used were of commercially available quality except as noted. Inhibitor-free tetrahydrofuran (THF) was distilled over Na/benzophenone under argon and deoxygenated with argon before use. The preparation and handling of air-sensitive compounds were performed under a MBraun Labmaster 130 inert atmosphere (<1 ppm of O2 and <1 ppm of H2O) glovebox filled with nitrogen. Dioxygen gas purchased from Airgas and dried by passing it through Drierite. 18O2 gas was purchased from ICON (Summit, NJ), and 16O2 gas was purchased from BOC gases (Murray Hill, NJ).
All UV–vis measurements were carried out using a Hewlett-Packard 8453 diode array spectrophotometer with HP Chemstation software and a Unisoku thermostated cell holder for low-temperature experiments. A 10 mm path length quartz cell cuvette modified with an extended glass neck with a female 14/19 joint and stopcock was used to perform all UV–vis experiments, as previously described.33,50,68 1H and 2H NMR spectra were measured on a Bruker 300-MHz NMR spectrometer at ambient or low temperatures. Chemical shifts were reported as δ (ppm) values relative to an internal standard (tetramethylsilane) and the residual solvent proton peaks. EPR spectra were recorded with a Bruker EMX spectrometer equipped with a Bruker ER 041 × G microwave bridge and a continuous flow liquid helium cryostat (ESR900) coupled to an Oxford Instruments TC503 temperature controller. Spectra were obtained at 8 K under nonsaturating microwave power conditions (ν = 9.428 GHz, microwave power = 0.201 mW, modulation amplitude = 10 G, microwave frequency = 100 kHz, and receiver gain = 5.02 × 103).
The compounds (PImH)FeII,30 the pyrrole deuterated derivative d8(PImH)FeII,30 and CuI(CH3CN)4(B(C6F5)4)35 were synthesized as previously described.
UV−Vis Spectroscopy.
[(PImH)FeIII−(O2•−)] (2). Complex [(PImH)FeIII−(O2•−)] (2) was generated in THF solution by preparing 0.01 mM (for Soret band monitoring) or 0.1 mM solutions of (PImH)FeII (1) in a 10 mm path length quartz Schlenk cuvette, which was sealed with a rubber septum in the glovebox. The cuvette was then cooled to −80 °C, and the solution was bubbled with O2 to generate the superoxide compound 2. UV−vis: λmax = 424 and 541 nm.
[(PImH)FeIICuI]+ (3), [(PImH)FeIII−(O22−)−CuII]+ (3a), and [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b). In a similar way, in the glovebox, 1 equiv of [CuI(CH3CN)4](B(C6F5)4) (from a 2 mM stock solution (THF)) was added to the 10 mm path length quartz Schlenk cuvette containing the reduced complex (PImH)FeII (1) (125 μL of a 2 mM solution) to generate complex [(PImH)FeIICuI]+ (3); the cuvette was then filled with THF up to a total volume of 2.5 mL. This cuvette was then cooled to −80 °C, and the solution was bubbled with O2 to generate the complex [(PImH)FeIII−(O22−)−CuII]+ (3a). Subsequently, addition of 1.5 equiv of DCHIm to the same cuvette results in LS complex [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b). UV–vis: [(PImH)FeIICuI]+ (3) λmax = 423, 542 nm; [(PImH)FeIII−(O22−)−CuII]+ (3a) λmax = 420, 545, 565 nm; [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b) λmax = 425, 538 nm.
EPR Spectroscopy.
(PImH)FeII (1) and [(PImH)FeIII−(O2•−)] (2). In a glovebox, 1.0 mg of (PImH)FeII (1) was dissolved in 0.5 mL of deoxygenated THF in an EPR tube. Outside the glovebox, the solution was cooled to −80 °C (acetone–dry-ice bath) and bubbled with O2 to generate the superoxide compound 2. Then, the EPR spectrum was recorded at 12 K. Both 1 and 2 were found to be EPR-silent.
[(PImH)FeIICuI]+ (3), [(PImH)FeIII−(O22−)−CuII]+ (3a), and [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b). In a similar way, in a glovebox, 1.0 mg of (PImH)FeII and 0.9 mg of CuI(CH3CN)4(B(C6F5)4) were dissolved in 0.5 mL of deoxygenated THF in an EPR tube and sealed properly. Outside the glovebox, the solution was cooled to −80 °C (acetone–dry-ice bath) and bubbled with O2 using a syringe to generate the heme–peroxo–Cu complex [(PImH)FeIII−(O22−)−CuII]+ (3a). A total of 1.5 equiv of DCHIm (3.5 mg of DCHIm in 0.5 mL of THF) was added to the reaction mixture. Then, the EPR spectrum was recorded at 12 K. Both 3a and 3b were found to be EPR-silent.
2H NMR Spectroscopy.
[d8-(PImH)FeIII−(O2•−)] (2-d8). In the glovebox, 2.6 mg of d8-(PImH)FeII (1-d8) was dissolved in 0.5 mL of deoxygenated THF in a NMR tube. The cold THF solution (at −80 °C, acetone–dry-ice bath) of 1-d8 was bubbled with O2. 2H NMR (300 MHz, THF) 2-d8: δpyrr 9.12 ppm.
[d8-(PImH)FeIICuI]+ (3-d8), [d8-(PImH)FeIII−(O22−)−CuII]+ (3a-d8), and [d8-(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b-d8). In the glovebox, a 1:1 mixture of 2.6 mg of d8-(PImH)FeII (1-d8) and 2.3 mg of [CuI(CH3CN)4](B(C6F5)4) was dissolved in 0.5 mL of deoxygenated THF in a NMR tube to generate complex [d8-(PImH)FeIICuI]+ (3-d8). The cold THF solution (at −80 °C, acetone–dry-ice bath) of 3-d8 was bubbled with O2. In the same NMR tube, 1.5 equiv of DCHIm was added to generate LS peroxo [d8-(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b-d8). 2H NMR (300 MHz, THF): Complex 3-d8, δpyrr 89.4, 102.8 ppm; 3a-d8, 93.0 ppm; 3b-d8, 10.2 ppm.
Resonance Raman Spectroscopy.
[(PImH)FeIII−(O2•−)] (2). In the glovebox, 2 mM solutions of an (PImH)FeII in THF were prepared and transferred to rR tubes and capped with tight-fitting septa. The sample tubes were placed in a cold bath (dry ice/acetone) and oxygenated using 16O2 or 18O2 gases. The oxygenated samples were set in a cold bath for 10 min, after which the sample tubes were frozen in liquid N2 and sealed by flame. rR samples were excited at 413 nm, using either a Coherent I90C–K Kr+ ion laser as the sample was immersed in a liquid-nitrogen-cooled (77 K) EPR finger Dewar (Wilmad). Power was ~2 mW at the sample, which was continuously rotated to minimize photodecomposition. The spectra were recorded using a Spex 1877 CP triple monochromator, and detected by an Andor Newton CCD cooled to −80 °C. rR [FeIII−(O2•−)] (2): νO−O, 1171 cm−1 (Δ18O2, −61 cm−1); νFe−O, 575 cm−1 (Δ18O2, −24 cm−1).
[(PImH)FeIICuI]+ (3), [(PImH)FeIII−(O22−)−CuII]+ (3a), and [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b). In the glovebox, 5 mM solutions of an equimolar mixture of (PImH)FeII and [CuI(CH3CN)4](B(C6F5)4) in THF were prepared and transferred to rR tubes and capped with tight-fitting septa. The sample tubes were placed in a cold bath (dry ice/acetone) and oxygenated using 16O2 and 18O2. The labeled gases were cooled in dry ice for 5 min and injected through the solution by using a Hamilton gastight syringe. The oxygenated samples were set in a cold bath for 10 min, after which the sample tubes were frozen in liquid N2 and sealed by flame. To the cold THF solution (at −80 °C, acetone–dry-ice bath) of 3a was added 1.5 equiv of DCHIm (3.5 mg of DCHIm in 0.5 mL of THF) for complex 3b. rR [(PImH)FeIII−(O22−)−CuII]+ (3a): νO−O, 799 cm−1 (Δ18O2, −48 cm−1); νFe−O, 524 cm−1 (Δ18O2, −23 cm−1). [(DCHIm)(PImH)FeIII−(O22−)−CuII]+ (3b): νO−O, 817 cm−1 (Δ18O2, −55 cm−1); νFe−O, 610 cm−1 (Δ18O2, −26 cm−1).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the USA National Institutes of Health (GM60353 to K.D.K. and DK031450 to E.I.S.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.9b02521.
(PImH)FeII (1) and (PImH)FeIII−(O2•−) (2) reversible dioxygen binding figure, diagrams of heme−FeIII−superoxide, high-spin heme–peroxo–copper and complexes with a low-spin μ−1,2-peroxo configuration (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Wikström MKF Proton Pump Coupled to Cytochrome c Oxidase in Mitochondria. Nature 1977, 266, 271–273. [DOI] [PubMed] [Google Scholar]
- (2).Ferguson-Miller S; Babcock GT Heme/Copper Terminal Oxidases. Chem. Rev 1996, 96, 2889–2907. [DOI] [PubMed] [Google Scholar]
- (3).Babcock GT How Oxygen Is Activated and Reduced in Respiration. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 12971–12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Peterson RL; Kim S; Karlin KD Comprehensive Inorganic Chemistry II, 2nd ed; Elsevier: Amsterdam, 2013; pp 149–177. [Google Scholar]
- (5).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yoshikawa S; Shimada A Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev 2015, 115, 1936–1989. [DOI] [PubMed] [Google Scholar]
- (7).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of Dioxygen by Copper Metalloproteins and Insights from Model Complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wikström M; Krab K; Sharma V Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev 2018, 118, 2469–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Huang X; Groves JT Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev 2018, 118, 2491–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Adam SM; Wijeratne GB; Rogler PJ; Diaz DE; Quist DA; Liu JJ; Karlin KD Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function. Chem. Rev 2018, 118, 10840–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Das TK; Pecoraro C; Tomson FL; Gennis RB; Rousseau DL The Post-Translational Modification in Cytochrome c Oxidase Is Required to Establish a Functional Environment of the Catalytic Site. Biochemistry 1998, 37, 14471–14476. [DOI] [PubMed] [Google Scholar]
- (12).Liu X; Yu Y; Hu C; Zhang W; Lu Y; Wang J Significant Increase of Oxidase Activity through the Genetic Incorporation of a Tyrosine-Histidine Cross-Link in a Myoglobin Model of Heme-Copper Oxidase. Angew. Chem., Int. Ed 2012, 51, 4312–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ravikiran B; Mahalakshmi R Unusual Post-Translational Protein Modifications: The Benefits of Sophistication. RSC Adv 2014, 4, 33958–33974. [Google Scholar]
- (14).Davidson VL Protein-Derived Cofactors Revisited: Empowering Amino Acid Residues with New Functions. Biochemistry 2018, 57, 3115–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lin YW Structure and Function of Heme Proteins Regulated by Diverse Post-Translational Modifications. Arch. Biochem. Biophys 2018, 641, 1–30. [DOI] [PubMed] [Google Scholar]
- (16).Ehudin MA; Senft L; Franke A; Ivanović- Burmazović I; Karlin KD Formation and Reactivity of New Isoporphyrins: Implications for Understanding the Tyr-His Cross-Link Cofactor Biogenesis in Cytochrome c Oxidase. J. Am. Chem. Soc 2019, 141, 10632–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Iwata S; Ostermeier C; Ludwig B; Michel H Structure at 2.8 Å Resolution of Cytochrome c Oxidase from Paracoccus Denitrificans. Nature 1995, 376, 660–669. [DOI] [PubMed] [Google Scholar]
- (18).Yoshikawa S Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science 1998, 280, 1723–1729. [DOI] [PubMed] [Google Scholar]
- (19).Schaefer AW; Kieber-Emmons MT; Adam SM; Karlin KD; Solomon EI Phenol-Induced O-O Bond Cleavage in a LowSpin Heme-Peroxo-Copper Complex: Implications for O2 Reduction in Heme-Copper Oxidases. J. Am. Chem. Soc 2017, 139, 7958–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brunner H Identification of the Iron-Ligand Vibration of Oxyhemoglobin. Naturwissenschaften 1974, 61, 129. [Google Scholar]
- (21).Van Wart HE; Zimmer J Resonance Raman Evidence for the Activation of Dioxygen in Horseradish Oxyperoxidase. J. Biol. Chem 1985, 260, 8372–8377. [PubMed] [Google Scholar]
- (22).Intermediate PM derives from an enzyme form where only the binuclear heme–Cu center is reduced, i.e., (Fea3II/CuBI). Many CcO biochemical/biophysical studies employ also PR, where four electrons are available for reactivity, i.e., where additional electrons are available also from CuA or cytochrome a3. [Google Scholar]
- (23).Poiana F; von Ballmoos C; Gonska N; Blomberg MRAA; Ädelroth P; Brzezinski P. Splitting of the O-O Bond at the Heme-Copper Catalytic Site of Respiratory Oxidases. Sci. Adv 2017, 3, e1700279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Han Du W-G; Noodleman L Density Functional Study for the Bridged Dinuclear Center Based on a High-Resolution X-Ray Crystal Structure of ba3 Cytochrome c Oxidase from Thermus Thermophilus. Inorg. Chem 2013, 52, 14072–14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Noodleman L; Han Du W-G; Fee JA; Götz AW; Walker RC Linking Chemical Electron-Proton Transfer to Proton Pumping in Cytochrome c Oxidase: Broken-Symmetry DFT Exploration of Intermediates along the Catalytic Reaction Pathway of the Iron-Copper Dinuclear Complex. Inorg. Chem 2014, 53, 6458–6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Han Du W-G; Götz AW; Yang L; Walker RC; Noodleman L A Broken-Symmetry Density Functional Study of Structures, Energies, and Protonation States along the Catalytic O-O Bond Cleavage Pathway in ba3 Cytochrome c Oxidase from Thermus Thermophilus. Phys. Chem. Chem. Phys 2016, 18, 21162–21171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schaefer AW; Roveda AC; Jose A; Solomon EI Geometric and Electronic Structure Contributions to O-O Cleavage and the Resultant Intermediate Generated in Heme-Copper Oxidases. J. Am. Chem. Soc 2019, 141, 10068–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Collman JP; Sunderland CJ; Berg KE; Vance MA; Solomon EI Spectroscopic Evidence for a Heme-Superoxide/Cu(I) Intermediate in a Functional Model of Cytochrome c Oxidase. J. Am. Chem. Soc 2003, 125, 6648–6649. [DOI] [PubMed] [Google Scholar]
- (29).Liu JG; Naruta Y; Tani F A Functional Model of the Cytochrome c Oxidase Active Site: Unique Conversion of a Heme-μ-Peroxo-CuII Intermediate into Heme-Superoxo/CuI. Angew. Chem., Int. Ed 2005, 44, 1836–1840. [DOI] [PubMed] [Google Scholar]
- (30).Chishiro T; Shimazaki Y; Tani F; Tachi Y; Naruta Y; Karasawa S; Hayami S; Maeda Y Isolation and Crystal Structure of a Peroxo-Bridged Heme-Copper Complex. Angew. Chem 2003, 115, 2894–2897. [DOI] [PubMed] [Google Scholar]
- (31).Chufán EE.; Puiu SC.; Karlin KD. Heme-Copper/Dioxygen Adduct Formation, Properties, and Reactivity. Acc. Chem. Res 2007, 40, 563–572. [DOI] [PubMed] [Google Scholar]
- (32).Kim E; Shearer J; Lu S; Moënne-Loccoz P; Helton ME;Kaderli S; Zuberbuehler AD; Karlin KD Heme/Cu/O2 Reactivity: Change in FeIII-(O22−)-CuII Unit Peroxo Binding Geometry Effected by Tridentate Copper Chelation. J. Am. Chem. Soc 2004, 126, 12716–12717. [DOI] [PubMed] [Google Scholar]
- (33).Wasser IM; Huang HW; Moënne-Loccoz P; Karlin KD Heme/Non-Heme Diiron(II) Complexes and O2, CO and NO Adducts as Reduced and Substrate-Bound Models for the Active Site of Bacterial Nitric Oxide Reductase. J. Am. Chem. Soc 2005, 127, 3310–3320. [DOI] [PubMed] [Google Scholar]
- (34).Adam SM; Garcia-Bosch I; Schaefer AW; Sharma SK; Siegler MA; Solomon EI; Karlin KD Critical Aspects of HemePeroxo-Cu Complex Structure and Nature of Proton Source Dictate Metal-O Peroxo Breakage versus Reductive O-O Cleavage Chemistry. J. Am. Chem. Soc 2017, 139, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Collman JP; Devaraj NK; Decréau RA; Yang Y; Yan Y; Ebina W; Eberspacher TA; Chidsey CED A Cytochrome c Oxidase Model Under Rate-Limiting Electron Flux. Science 2007, 315, 1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Collman JP; Decréau RA Functional Biomimetic Models for the Active Site in the Respiratory Enzyme Cytochrome c Oxidase. Chem. Commun 2008, 356, 5065–5076. [DOI] [PubMed] [Google Scholar]
- (37).Shin H; Lee DH; Kang C; Karlin KD Electrocatalytic Four-Electron Reductions of O2 to H2O with Cytochrome c Oxidase Model Compounds. Electrochim. Acta 2003, 48, 4077–4082. [Google Scholar]
- (38).Chatterjee S; Sengupta K; Hematian S; Karlin KD; Dey A Electrocatalytic O2-Reduction by Synthetic Cytochrome c Oxidase Mimics: Identification of a “Bridging Peroxo” Intermediate Involved in Facile 4e−/4H+ O2-Reduction. J. Am. Chem. Soc 2015, 137, 12897–12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Garcia-Bosch I; Adam SM; Schaefer AW; Sharma SK; Peterson RL; Solomon EI; Karlin KDA “Naked” FeIII-(O22‑)CuII Species Allows for Structural and Spectroscopic Tuning of LowSpin Heme-Peroxo-Cu Complexes. J. Am. Chem. Soc 2015, 137, 1032–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sharma SK; Kim H; Rogler PJ; Siegler MA; Karlin KD Isocyanide or Nitrosyl Complexation to Hemes with Varying Tethered Axial Base Ligand Donors: Synthesis and Characterization. JBIC, J. Biol. Inorg. Chem 2016, 21, 729–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Momenteau M; Reed CA Synthetic Heme Dioxygen Complexes. Chem. Rev 1994, 94, 659–698. [Google Scholar]
- (42).Yan JJ; Kroll T; Baker ML; Wilson SA; Decréau R; Lundberg M; Sokaras D; Glatzel P; Hedman B; Hodgson KO; Solomon EI Resonant Inelastic X-Ray Scattering Determination of the Electronic Structure of Oxyhemoglobin and Its Model Complex. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 2854–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kopf M-A; Karlin KD Dioxygen Reactivity of Reduced Heme and Heme-Copper Complexes Utilizing Tetraarylporphyrinates Tethered with Both a Pyridyl Axial Ligand and N,N-Bis[2-(2-Pyridyl)Ethyl]Amine Chelate. Inorg. Chem 1999, 38, 4922–4923. [DOI] [PubMed] [Google Scholar]
- (44).Das PK; Mittra K; Dey A Spectroscopic Characterization of a Phenolate Bound FeII-O2 Adduct: Gauging the Relative “Push” Effect of a Phenolate Axial Ligand. Chem. Commun 2014, 50, 5218–5220. [DOI] [PubMed] [Google Scholar]
- (45).Singha A; Dey A Hydrogen Atom Abstraction by Synthetic Heme Ferric Superoxide and Hydroperoxide Species. Chem. Commun 2019, 55, 5591–5594. [DOI] [PubMed] [Google Scholar]
- (46).Tsubaki M; Nagai K; Kitagawa T Resonance Raman Spectra of Myoglobins Reconstituted with Spirographis and Isospirographis Hemes and Iron 2,4-Diformylprotoporphyrin IX. Effect of Formyl Substitution at the Heme Periphery. Biochemistry 1980, 19, 379–385. [DOI] [PubMed] [Google Scholar]
- (47).Mak PJ; Denisov IG; Victoria D; Makris TM; Deng T; Sligar SG; Kincaid JR Resonance Raman Detection of the Hydroperoxo Intermediate in the Cytochrome P450 Enzymatic Cycle. J. Am. Chem. Soc 2007, 129, 6382–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Denisov IG; Mak PJ; Makris TM; Sligar SG; Kincaid JR Resonance Raman Characterization of the Peroxo and Hydroperoxo Intermediates in Cytochrome P450. J. Phys. Chem. A 2008, 112, 13172–13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kim E; Helton ME; Wasser IM; Karlin KD; Lu S; Huang H; Moënne-Loccoz P; Incarvito CD; Rheingold AL; Honecker M; Kaderli S; Zuberbuhler AD Superoxo, μ-Peroxo, and μ-Oxo Complexes from Heme/O2 and Heme-Cu/O2 Reactivity: Copper Ligand Influences in Cytochrome c Oxidase Models. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 3623–3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sharma SK; Schaefer AW; Lim H; Matsumura H; Moënne-Loccoz P; Hedman B; Hodgson KO; Solomon EI; Karlin KD A Six-Coordinate Peroxynitrite Low-Spin Iron(III) Porphyrinate Complex - The Product of the Reaction of Nitrogen Monoxide (·NO(g)) with a Ferric-Superoxide Species. J. Am. Chem. Soc 2017, 139, 17421–17430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mittra K; Chatterjee S; Samanta S; Sengupta K; Bhattacharjee H; Dey A A Hydrogen Bond Scaffold Supported Synthetic Heme FeIII-O2− Adduct. Chem. Commun 2012, 48, 10535–10537. [DOI] [PubMed] [Google Scholar]
- (52).Kim E; Chufan EE; Kamaraj K; Karlin KD Synthetic Models for Heme-Copper Oxidases. Chem. Rev 2004, 104, 1077–1133. [DOI] [PubMed] [Google Scholar]
- (53).Halime Z; Kieber-Emmons MT; Qayyum MF; Mondal B; Gandhi T; Puiu SC; Chufán EE; Sarjeant AAN; Hodgson KO; Hedman B; Solomon EI; Karlin KD HemeCopper-Dioxygen Complexes: Toward Understanding Ligand-Environmental Effects on the Coordination Geometry, Electronic Structure, and Reactivity. Inorg. Chem 2010, 49, 3629–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chufán EE; Mondal B; Gandhi T; Kim E; Rubie ND; Moënne-Loccoz P; Karlin KD Reactivity Studies on FeIII-(O22−)CuII Compounds: Influence of the Ligand Architecture and Copper Ligand Denticity. Inorg. Chem 2007, 46, 6382–6394. [DOI] [PubMed] [Google Scholar]
- (55).Que L Jr.; Tolman WB Bis(μ-Oxo)Dimetal “Diamond” Cores in Copper and Iron Complexes Relevant to Biocatalysis. Angew. Chem., Int. Ed 2002, 41, 1114–1137. [DOI] [PubMed] [Google Scholar]
- (56).Mirica LM; Ottenwaelder X; Stack TDP Structure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev 2004, 104, 1013–1046. [DOI] [PubMed] [Google Scholar]
- (57).Hatcher QL; Karlin KD Ligand Influences in Copper-Dioxygen Complex-Formation and Substrate Oxidations. Adv. Inorg. Chem 2006, 58, 131–184. [Google Scholar]
- (58).del Río D; Sarangi R; Chufan EE; Karlin KD; Hedman B; Hodgson KO; Solomon EI Geometric and Electronic Structure of the Heme-Peroxo-Copper Complex [(F8TPP)FeIII-(O22−)-CuII(TMPA)](ClO4). J. Am. Chem. Soc 2005, 127, 11969–11978. [DOI] [PubMed] [Google Scholar]
- (59).Nanthakumar A; Fox S; Murthy NN; Karlin KD Inferences from the 1H-NMR Spectroscopic Study of an Antiferromagnetically Coupled Heterobinuclear Fe(III)-(X)-Cu(II) S = 2 Spin System (X = O2−, OH−). J. Am. Chem. Soc 1997, 119, 3898–3906. [Google Scholar]
- (60).Ghiladi RA; Kretzer RM; Guzei I; Rheingold AL; Neuhold YM; Hatwell KR; Zuberbühler AD; Karlin KD (F8TPP)FeII/O2 Reactivity Studies {F8TPP = Tetrakis(2,6-Difluorophenyl)Porphyrinate(2-)}: Spectroscopic (UV-Visible and NMR) and Kinetic Study of Solvent-Dependent (Fe/O2 = 1:1 or 2:1) Reversible O2-Reduction and Ferryl Formation. Inorg. Chem 2001, 40, 5754–5767. [DOI] [PubMed] [Google Scholar]
- (61).Kim E; Helton ME; Lu S; Moënne-Loccoz P; Incarvito CD; Rheingold AL; Kaderli S; Zuberbühler AD; Karlin KD Tridentate Copper Ligand Influences on Heme-Peroxo-Copper Formation and Properties: Reduced, Superoxo, and μ-Peroxo Iron/Copper Complexes. Inorg. Chem 2005, 44, 7014–7029. [DOI] [PubMed] [Google Scholar]
- (62).Kopf M-A; Neuhold Y-M; Zuberbühler AD; Karlin KD Oxo- and Hydroxo-Bridged Heme-Copper Assemblies Formed from Acid-Base or Metal-Dioxygen Chemistry. Inorg. Chem 1999, 38, 3093–3102. [Google Scholar]
- (63).Ghiladi RA; Ju TD; Lee DH; Moënne-Loccoz P; Kaderli S; Neuhold YM; Zuberbühler AD; Woods AS; Cotter RJ; Karlin KD. Formation and Characterization of a High-Spin Heme-Copper Dioxygen (Peroxo) Complex. J. Am. Chem. Soc 1999, 121, 9885–9886. [Google Scholar]
- (64).Kim E; Kamaraj K; Galliker B; Rubie ND; Moënne-Loccoz P.; Kaderli S; Zuberbühler AD; Karlin KD Dioxygen Reactivity of Copper and Heme-Copper Complexes Possessing an Imidazole-Phenol Cross-Link. Inorg. Chem 2005, 44, 1238–1247. [DOI] [PubMed] [Google Scholar]
- (65).Ghiladi RA; Chufan EE; del Río D; Solomon EI; Krebs C; Huynh BH; Huang H.-w.; Moënne-Loccoz P; Kaderli S; Honecker M; Zuberbühler AD; Marzilli L; Cotter RJ; Karlin KD. Further Insights into the Spectroscopic Properties, Electronic Structure, and Kinetics of Formation of the Heme-Peroxo-Copper Complex [(F8TPP)FeIII-(O22−)-CuII(TMPA)]+. Inorg. Chem 2007, 46, 3889–3902. [DOI] [PubMed] [Google Scholar]
- (66).Kieber-Emmons MT; Qayyum MF; Li Y; Halime Z; Hodgson KO; Hedman B; Karlin KD; Solomon EI Spectroscopic Elucidation of a New Heme/Copper Dioxygen Structure Type: Implications for O-O Bond Rupture in Cytochrome c Oxidase. Angew. Chem., Int. Ed 2012, 51, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Ehudin MA; Schaefer AW; Adam SM; Quist DA; Diaz DE; Tang JA; Solomon EI; Karlin KD Influence of Intramolecular Secondary Sphere Hydrogen-Bonding Interactions on Cytochrome c Oxidase Inspired Low-Spin Heme-Peroxo-Copper Complexes. Chem. Sci 2019, 10, 2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ghiladi RA; Huang H; Moënne-Loccoz P; Stasser J; Blackburn NJ; Woods AS; Cotter RJ; Incarvito CD; Rheingold AL; Karlin KD Heme-Copper/Dioxygen Adduct Formation Relevant to Cytochrome c Oxidase: Spectroscopic Characterization of [(6L)FeIII-(O22−)-CuII]+. JBIC, J. Biol. Inorg. Chem 2005, 10, 63–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.