

Abstract
Over the last 20 years, there has been an increased appreciation of the long-term sequelae of acute kidney injury (AKI) and the potential development of chronic kidney disease (CKD). Several pathophysiological features have been proposed to mediate the AKI to CKD progression including maladaptive alterations in tubular, interstitial, inflammatory and vascular cells. These alterations likely interact to culminate in the progression to CKD. In this article we focus primarily on evidence of vascular rarefaction secondary to AKI, and the potential mechanisms by which rarefaction occurs in relation to other alterations in tubular and interstitial compartments. We further focus on the potential that rarefaction contributes to renal hypoxia. Consideration the role of hypoxia in the AKI to CKD transition focuses on experimental evidence of persistent renal hypoxia following AKI and experimental maneuvers to evaluate the influence of hypoxia, per se, in progressive disease. Finally, consideration of methods to evaluate hypoxia in patients is provided with the suggestion that non-invasive measurement of renal hypoxia may provide insight into progression in post AKI patients.
Keywords: Fibrosis, peritubular capillaries, inflammation, renal hypoxia
Background
Acute kidney injury (AKI) is characterized by an abrupt loss of glomerular filtration rate, often associated with altered renal blood flow and increased local and systemic inflammation.1 The leading causes of AKI are ischemia reperfusion (I/R) injury,2 which may occur secondary to major surgery such as cardiac surgery (particularly when requiring cardiopulmonary bypass),3 sepsis4 and exposure to nephrotoxic agents.5 It is estimated that approximately 5% to 10% of critically ill patients suffer from AKI 6 and every year around 600,000 new cases of AKI are reported in the United States.7
While recovery of renal function is expected in the majority of surviving patients following AKI, epidemiological studies have shown that such patients are more likely to develop chronic kidney disease (CKD) compared with patients without a history of AKI.8,9 Using data from the U.S. Department of Veterans Affairs, Amdur et al. reported that patients developing AKI following myocardial infarction or pneumonia showed a decline in renal function and an increased risk of developing stage-4 CKD versus similar patients without AKI.9 In addition, Ishani et al. reported that AKI in U.S. Medicare patients was associated with increased risk of end-stage renal disease (ESRD), which was significantly enhanced in patients with a prior history of CKD.8 In a meta-analysis, Coca et al. reported that AKI was associated with an increased risk of CKD by 8.8-fold and ESRD by approximately 3.1-fold versus patients without prior AKI.10 These reports suggest that AKI predisposes the development of CKD. However, most of the information on pathophysiological mechanisms of the AKI-to-CKD transition derives from studies using animal models of renal injury.
The pathophysiology of AKI to CKD
Following AKI, CKD may progress because of incomplete or maladaptive tissue repair, setting in motion processes promoting the development of interstitial fibrosis.11 Alterations in renal structure that may promote development of CKD following acute injury include nephron loss, incomplete tubular repair, changes in interstitial cell composition, inflammation and vascular rarefaction (Figure 1). These alterations do not occur in isolation and interact to direct progressive CKD. With regard to capillary rarefaction, we have suggested that limitations in renal perfusion may enhance renal hypoxia and fuel progression of CKD.12 In this article, we summarize recent developments in our understanding of maladaptive repair responses that contribute to the AKI-CKD transition. We focus on the genesis of capillary rarefaction following both AKI and in progressive CKD. We consider the potential role of hypoxia, and consider how future efforts may be pursued to translate data from preclinical modes to the patient.
Figure 1. Hypothesized role of renal hypoxia in the progression of acute kidney injury to chronic kidney disease.
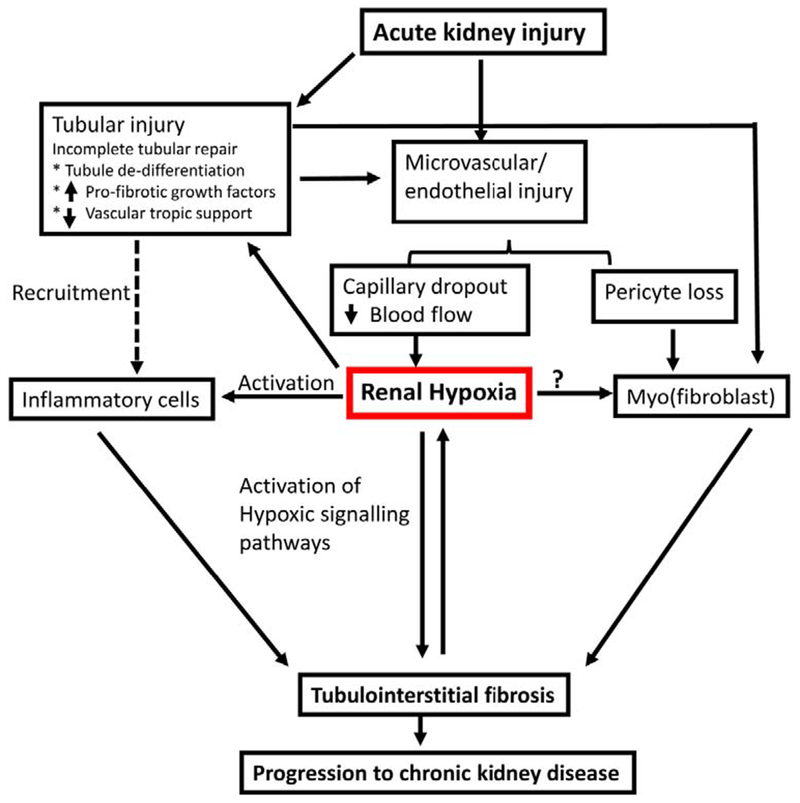
Acute kidney injury is associated with damage to both tubular and vascular compartments. Tubular injury results in increased expression of profibrotic cytokines and growth factors and decreased expression of vascular factors such as VEGF in tubular cells may lead to renal microvascular loss. The reduction of peritubular capillaries restricts downstream blood flow from glomerular capillaries and contributes to the development of renal hypoxia, while also being associated with activation of pericytes toward a fibrogenic myofibroblast phenotype. Hypoxia influences pro-fibrotic and inflammatory activity of tubular and inflammatory cells. Myofibroblast profibrotic signaling is influenced by factors produced from hypoxic tubules or inflammatory cells or may be influenced directly by hypoxia (?). Progressive interstitial fibrosis exacerbates renal hypoxia by increasing oxygen diffusion distance between peritubular capillaries and parenchyma forming a pathological cycle promoting progressive chronic kidney disease.
Adaptive and maladaptive epithelial repair following AKI
Recovery of renal function is expected following AKI in most in surviving patients, while subsets of patients may develop renal insufficiency and progress to CKD.1 Generally, recovery from AKI is associated with the resolution of renal perfusion and repair of the damaged epithelial cells, typically the most prominent morphological feature following injury.13 Increasing the severity of AKI may tilt the balance toward maladaptive and inefficient repair while adaptive repair may become limited.11 Not surprisingly, in models of ischemia reperfusion (I/R), increasing ischemic time results in both greater initial tubular damage as well as a greater degree of renal fibrosis one month following reperfusion.14
Efficient tubular repair results from recovery of sub-lethally damaged cells and/or proliferation of damaged de-differentiated cells to repopulate the tubular basement membrane.15 Re-differentiation and hypertrophy of recovered or new cells helps to re-establish normal tubular architecture. However, areas of tubular atrophy or evidence of persistent de-differentiation have been observed following I/R injury. In rats, the frequency of these structures is enhanced when injury occurs in the setting of reduced renal mass, becoming more common following I/R with a 50% or 75% reduction in renal mass.16 While complete tubular atrophy reduces total nephron number, focal lesions around atrophic tubules are sharply demarcated and fibrosis does not progress from these areas, suggesting that atrophic nephrons do not directly contribute to progressive interstitial fibrosis.16,17
In contrast, Venkatachalam and colleagues have proposed that small clusters of proximal tubular epithelial cells that fail to completely re-differentiate may promote interstitial fibrosis.18 These undifferentiated cells can be identified by expression of vimentin and are associated with increased expression of pro-fibrotic growth factors including: transforming growth factor-β1 (TGF- β1), platelet derived growth factor-B (PDGF-B) and connective tissue growth factor (CTGF), when compared with fully differentiated tubules.17,19,20 These secreted growth factors have been proposed to act in a paracrine fashion on interstitial cells i.e., pericytes or fibroblasts, found in close proximity.18
The activation of cell cycle activity is important for effective recovery of the tubule, while persistent activation of the cell cycle, identified by activity of c-Jun kinase (JNK), has been associated with development of interstitial fibrosis.19,21 A potential role for cell cycle activity in fibrosis was addressed in studies using various murine models of renal injury including moderate reversible I/R injury, severe I/R injury, aristolochic acid nephropathy and unilateral ureteral obstruction (UUO).19 Interestingly, the models associated with a greater degree of fibrosis displayed a greater number of cells in G2/M cell-cycle arrest, identified by phosphorylation with histone H3. These cells produce higher levels of pro-inflammatory and pro-fibrotic cytokines, including TGF-β and CTGF when compared with cells in G0/G1 after I/R injury.19 Interestingly, blockade of JNK bypassed G2/M arrest and reduced fibrosis following bilateral renal I/R injury.19,22
Taken together, persistent de-differentiation and/or G2/M cell cycle arrest are considered to be important drivers of renal fibrosis following injury. We suggest that physiological factors related to re-establishment of perfusion may influence the success or failure of tubular repair, and therefore the fibrotic nature of tubules following AKI. Specifically, peritubular capillaries, which provide nutrient and oxygen for tubules, are compromised following AKI and may lead to failed tubular recovery (Figure 1). Support for this idea comes from studies of rats subjected to renal micro-embolization injury following infusion of acrylic microspheres into the renal artery.23 Tubules from rats at 4 weeks following injection demonstrated heterogeneous patchy areas of undifferentiated, vimentin-positive cells, which expressed PDGF-B, localized adjacent to expanded interstitium. In contrast, vimentin-negative cells, even in the same tubule, did not express PDGF-B.23 Thus, disruption of capillary flow may impair efficient tubular recovery and promote a persistent pro-inflammatory/pro-fibrotic phenotype.
Capillary dropout after AKI
In contrast to the renal tubular system, the renal microvascular system has a poor regenerative capacity and impairments in microvascular structure have been proposed to contribute or hasten progressive CKD.12,24 Observations of Basile and colleagues demonstrated significant and essentially permanent reductions of peritubular capillary (PTC) density in rats following bilateral I/R injury (Figure 2).24 In their study, Microfil® silicon rubber was infused into rat kidneys between 4 to 40 weeks after renal I/R injury. Despite evidence of recovery of renal function and tubular structure, capillary density was reduced in both the cortex (~25% - 30%) and the inner stripe of the outer medulla (~35% - 40%).24 The finding of reduced capillary density following AKI has been confirmed by other investigators using a variety of different methods (Table 1). For example, Kramann et al. utilized fluorescence micro-angiography and reported a reduction of the total perfused area in both renal cortex and medulla after severe I/R-induced AKI in mice.25 The reduced perfusion was associated with a reduction in both the density of vessels as well as the diameter of individual surviving PTCs.25 Several others have utilized immunohistochemical staining to detect changes in vascular density. Studies using antibodies against CD31, RECA (rat endothelial cell antigen), MECA32 (mouse endothelial cell antigen) and cablin, have all shown acute and sustained reductions in PTC density following AKI (Table 1). Labeling of endothelial cells using a transgenic approach by crossing Tie2-Cre mice with a floxed reporter line, has also been used to demonstrate an overall reduction in PTC following AKI.26 Capillary rarefaction has been demonstrated in other experimental animal models of AKI including, UUO and folic acid nephropathy.27,28 and has also been demonstrated in human patients after delayed graft function.29,30
Figure 2. Micrographs of Microfil®- perfused kidney sections processed from rats kidney at 4 weeks after sham-operation (A) or post ischemia reperfusion injury (B).
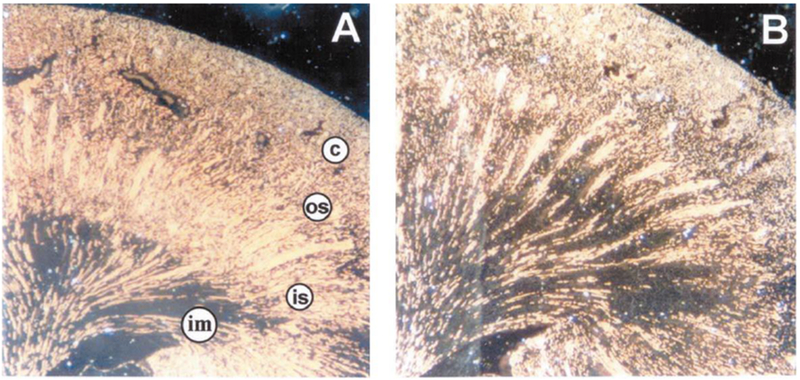
Note, renal capillaries are filled with Microfil which appears as bright yellow against a dark background. In post ischemic kidneys, capillary density is reduced in both the cortex and the medulla. Cortex (c), outer stripe of the outer medulla (os), inner stripe of the outer medulla (is), and inner medulla (im) are shown. Images were reproduced from Basile et al. with permission.24
Table 1.
Evidence of capillary drop out following AKI
| Model/Condition | Method | Observation | Citation |
|---|---|---|---|
| Rat I/R | Microfil | 30-50% reduction | (Basile et al 2001)24 |
| Rat I/R | CD31/PECAM staining Cablin staining | ~30% reduction by Cablin staining, preserved by VEGF 121 | (Basile et al 2011)26, (Leonard et al 2008)33 |
| Rat I/R | CD31 staining | 90% loss of CD31 staining SS31 | (Liu et al 2014)35 |
| Rat I/R | RECA staining | Reduced by > 50%, preserved by EPC microvesicles | (Cantaluppi et al 2012)36 |
| Rat I/R | JG12 staining | ~50% reduction; exacerbated by reduced renal mass | (Polichnowski et al 2014)16 |
| Mouse I/R | Flurescence micro-angiography | 40% reduction in capillaries at 8 weeks; reduced size of remaining vessels. ~60% decrease in total perfusion area | (Kramann et al 2014)25 |
| Mouse I/R | Meca-32 staining and Micro-CT | 20-43% reductions; tight correlation with Meca-32 and Micro-CT; inversely correlated with collagen I staining | (Ehling et al 2016)32 |
| Mouse I/R | Cablin staining | 30-40% after 4 weeks MMP9 loss prevented loss of cablin staining following IR | (Lee et al 2011)37 |
| Mouse I/R | CD31 staining | Reduction in staining by 20%, preserved by MSC | (Xing et al 2014)38 |
| Mouse I/R | CD31 staining | 15-20% rarefaction; some evidence that endothelial proliferation is enhanced in Id1 overexpressing mice | (Lee et al 2011)39 |
| DTR transgenic mouse | CD31 staining | Dramatic reduction in CD31 staining associated with fibrosis | (Grgic et al 2012)40 |
| Human DGF | CD31/CD34 co-staining | Reduced peritubular capillaries in biopsy 3 months post DGF | (Steegh et al 2011)30 |
| Human DGF | vWF staining | Evidence of peritubular capillary endothelial cells damage in sustained acute renal failure | (Kwon et al 2008)29 |
Abbreviation: I/R, ischemia reperfusion; PECAM, platelet endothelial cell adhesion molecule; VEGF, vascular endothelial growth factor; RECA, rat endothelial cell antigen; EPC, endothelial progenitor cells; JG12, endothelial aminopeptidase P in renal tissue; Meca-32, endothelial cell antigen; Micro-CT, micro computed tomography; MMP-9, matrix metalloproteinase-9; MSC, mesenchymal stem cells; Id1, inhibitor of DNA-binding/differentiation proteins; DTR, diphtheria toxin receptor; DGF, delayed graft function, Vwf, von Willebrand Factor.
It should be recognized that all of these approaches have limitations and caution should be exercised in interpreting results from these studies. Measurements that rely on staining a putative endothelial marker are subject to changes in protein expression, which could be altered in response to sub-lethal injury and not reflective of actual loss of vessels. For example, CD31/platelet endothelial cell adhesion molecule, one of the most widely used endothelial cells (EC) markers, may be transiently down-regulated in response to interferon gamma or TNF-α.31 The loss of staining may therefore overestimate PTC loss, especially in the early post-AKI period. By extension, a subsequent increase in CD31 expression may not distinguish between recovery of sub-lethally damaged cells versus the generation of new endothelial cells due to repair. In contrast, techniques based on vascular filling (Microfil, micro computed tomography (microCT), microangiography) are fraught with technical and biological considerations. In particular, the ability to fill a vessel may be difficult if it is constricted or congested. Since both of these complications are present in the early stages of AKI,1 reduced filling may not necessarily reflect rarefaction. Despite these experimental concerns, the abundance of reports using different experimental approaches strongly suggests that renal microvascular density is compromised following acute kidney injury.
While the majority of studies indicate persistent rarefaction following AKI, few studies have carefully evaluated the time course of this response. One notable exception provided by Ehling et al. used MECA32 staining to quantify renal microvessels following I/R in mice.32 Reduced vessel density was detectable as early as 1 day post I/R, but progressed steadily up to 14 days and remained stable thereafter. This timing is noteworthy as it suggests that renal capillary rarefaction develops slowly, simultaneous to the time normally associated with repair of renal tubules.32 Moreover, these data imply that a window of opportunity during the repair phase could be envisaged to attenuate vascular rarefaction. Several approaches, including treatment with vascular endothelial growth factor (VEGF)-12133 or cartilage oligomeric matrix protein/angiopoietin-134 provide protection against microvessel loss following AKI.
The mechanisms mediating PTC rarefaction are not completely understood. Surprisingly, evidence of apoptotic cell death within the endothelium is minimal. Horbelt et al., using a Tie2-GFP mouse, could not detect endothelial apoptosis by TUNEL staining following renal I/R injury, while injection of an anti-CD95 antibody abundantly labeled TUNEL+EC in kidney; thus EC apoptosis may be context dependent.41 Conversely, ECs express markers of mesenchymal cells for at least 7 days following I/R in rats, suggesting endothelial-mesenchymal transition (EndoMT).26 The findings of fate-tracing studies using Tie2-Cre mice crossed with YFP reporter mice are consistent with activation of EndoMT in the setting of AKI.26 Live multiphoton imaging studies 2 weeks following I/R demonstrated focal regions where peritubular vessels were devoid of blood flow.26 The YFP-positive cells were thickened and lacked the normal endothelial appearance observed in sham-operated controls.26 Moreover, there was little evidence of endothelial proliferation following injury suggesting that EndoMT, combined with a lack of adequate endothelial repair mechanisms, contributes to PTC loss following AKI.
Renal microvascular rarefaction may be due in part to alterations in the balance of positive and negative trophic factors in the AKI microenvironment. Transient elevations of anti-angiogenic factors have been reported following AKI.1,42 VEGF-A, an angiogenic factor, is expressed in renal tubular epithelial cells and might bind to its receptor located on endothelial cells of adjacent PTCs.43 This tubulo-vascular crosstalk is crucial for maintenance of PTC density in kidney. In mice expressing the diphtheria toxin receptor in proximal tubules, administration of the toxin induces both proximal tubule injury and a reduction in capillary density.40 Consistent with this, Dimke et al. demonstrated that genetic ablation of VEGF-A in mouse renal tubules also decreased PTC density.43
Several studies have demonstrated that renal VEGF expression is compromised early after AKI in rats 33,44 and is associated with reduction of PTC density.33 VEGF expression is reduced in other chronic injury models associated with fibrosis.45,46 The loss of VEGF expression is counter-intuitive since other HIF-related genes are prominently activated in response to I/R injury.44 However, it was suggested that reduced VEGF expression reflects the undifferentiated phenotype of injured tubules.16 Regardless, the transient reduction in VEGF may participate in vascular rarefaction since treatment of rats with VEGF-121 during the early post-ischemic period ameliorates capillary rarefaction and subsequent CKD.33
Vascular stability is also provided by the tight interplay between pericytes and endothelial cells.47 Several recent studies have generated evidence that the distance of pericytes from ECs increases within hours of renal I/R injury. This detachment or disruption of the normal pericyte-EC structural relationship results in migration of pericytes from the perivascular to the interstitial space and activation of these cells to extracellular matrix producing myofibroblasts.48-51 Evidence suggests that these activated myofibroblasts represent the primary cell type responsible for interstitial fibrosis in response to injury.48,49,52 Pericyte activation is also thought to destabilize the PTC network. When compared with pericytes from non-injured kidneys, injury-activated kidney pericytes/myofibroblasts express anti-angiogenic factors such as ADAMTS-1 and these cells disrupt endothelial 3-D networks in vitro.53 Evidence for a critical role for proper perictye-EC interaction in PTC structure was provided by a study in which ablation of Gli1+ pericytes promoted endothelial cell damage, a reduction in PTC number, focal hypoxia and subsequent evidence of damage to the adjacent tubular epithelium.54
Capillary loss, hypoxia and progressive CKD
Renal distribution and consumption of oxygen is heterogeneous in nature.55 The unique microvasculature network of the kidney creates a gradient of oxygen tension between cortex and medulla such that the partial pressure of oxygen in the cortex varies between 30-50 mmHg and approximately 10-20 mmHg in the renal medulla. Therefore, the renal medulla is generally considered to be on the brink of hypoxic injury.56 In the kidney, peritubular capillaries run parallel to renal tubules and provide nutrients and oxygen to the adjacent tubules in a manner that fulfills metabolic demand.57
Renal oxygen tension is determined by the balance between renal oxygen delivery and oxygen consumption.58 Pathological processes that decrease renal blood flow and oxygen delivery, or processes that increase renal oxygen consumption (e.g., by increasing glomerular filtration rate), could impair renal oxygenation, and promote renal damage.59 Clinical agents such as polyene antibiotics, nonsteroidal anti-inflammatory drugs, and radiographic contrast agents that reduce renal blood flow or increased tubular workload may exacerbate renal hypoxia and worsen the kidney injury.56 Amelioration of renal hypoxia by increasing medullary blood flow or reducing metabolic demand is thought to provide protection to kidneys from hypoxic damage.12,60
It has been two decades since Fine and colleagues proposed renal tissue hypoxia as a mediator of progressive kidney disease.61-63 Reductions in PTC density could impair post-glomerular perfusion and contribute to renal hypoxia.64 Reductions in PTC density have been reported in virtually every model of renal disease associated with interstitial fibrosis, including, but not limited to, hypertension, aging, diabetes and reduced renal mass.12 Capillary rarefaction was identified in biopsies from human patients with CKD of different etiologies and the degree of rarefaction strongly correlated with interstitial fibrosis and was predictive of organ failure.65 Decreased renal oxygen tension has been proposed as a factor predisposing the development of tubulointerstitial fibrosis by activating various profibrotic signaling pathways (described further below). A vicious cycle of progressive tubulointerstitial fibrosis would be expected as a decrease in oxygen tension is exacerbated by developing fibrosis as the oxygen diffusion distance from capillaries to parenchyma expands.61-63
Effect of hypoxia on fibrotic pathways
An essential feature of the chronic hypoxia hypothesis is the putative role that low oxygen levels play in activating pro-fibrogenic pathways. The activation of the hypoxia inducible factor (HIF-1α and HIF-2α) pathways appears central to the activities of tubular, interstitial and inflammatory cells. HIFs are oxygen sensitive transcription factors, which represent key mediators of the cellular adaptive response to hypoxia, and much has been written on the effects of HIF on resistance and recovery from renal injury.66–68 In this pathway, molecular oxygen levels are sensed by prolyl-hydroxylase-2 (PHD-2), which facilitates modification of transcription factors HIF-1α and HIF-2α by von Hippel-Lindau tumor suppressor protein.68 With adequate oxygen levels, PHD-2 targets the α-subunits of HIF toward ubiquination and proteasomal degradation. However, under reduced oxygen conditions, HIF α-subunits are not degraded and so are able to form a dimer with the β-subunit and thus regulate specific gene responses.68
Activated HIF binds to upstream promoters of pro-fibrogenic genes and induces the expression of matrix modifying factors including matrix metallopeptidase 2, tissue inhibitor of metalloproteinase 1, collagen I and CTGF leading to accumulation of extracellular matrix in the tubulointerstitum.68,69 Modulation of tubular epithelial cells to mesenchymal phenotype (EMT) may be mediated via HIF-1α dependent signaling pathways.70,71 In line with this notion, genetic ablation of HIF-1α in proximal tubular epithelial cells was shown to ameliorate the development of tubulointertitial fibrosis and inflammation in mice with UUO.71 In addition, there was a reduction in the number of S100A4-positive fibroblasts, possibly derived from epithelial to mesenchymal transition.71,72 In the endothelium, sustained HIF-1α expression, stimulated via targeted EC specific knockdown of PHD-2, resulted in EndoMT and development of renal fibrosis.73 Thus, sustained HIF signaling under hypoxic conditions might play an important role in the development of renal fibrosis.
Additional signaling pathways including TGF-β, nuclear factor-κB (NF-κB), PI3K/Akt, angiotensin II /reactive oxygen species and the Notch pathway can be activated in response to renal hypoxia, which subsequently may stimulate myofibroblasts to produce extracellular proteins in the renal intertitium.74 Inhibition of several of these pathways (angiotensin II, TGF-β) have been shown to contribute to the AKI-to-CKD transition.75,76
Effects of hypoxia on inflammatory responses
It is now appreciated that renal hypoxia is associated with inflammation in models of progressive kidney disease. Yamaguchi et al. demonstrated that renal tissue hypoxia and inflammation co-exist and are reciprocally modulated in both acute and chronic kidney diseases.77 Inflammation may be modulated by overexpression of the CCAAT/enhancer-binding protein δ (CEBPD), an inflammatory response gene expressed in proximal tubular cells in both acute and chronic hypoxic conditions. Data from both in vitro studies and in vivo models suggest that the effects of hypoxia on CEBPD expression are mediated through NF-κB-dependent pathways.77 CEBPD binds to the promotor of HIF-1α and subsequently increases HIF-1α expression in tubular epithelial cells, which in turn stimulates infiltration of inflammatory cells and production of inflammatory cytokines.77
Infiltration of inflammatory cells in the renal interstitium contributes to development of renal fibrosis and plays a vital role in transition of AKI to CKD.1,78,79 Renal inflammation, characterized by infiltration of macrophages, neutrophils and lymphocytes is evident in experimental models of kidney disease80 and in human renal biopsies.81,82 Moreover, the number of interstitial macrophages positively correlates with interstitial fibrosis and negatively correlates with vascular density.65
The role for inflammation in the AKI to CKD transition has received significant attention.83 Inflammatory cells are thought to initiate phagocytosis and release inflammatory and profibrotic cytokines such as TGF-β. TGF-β, mainly secreted by macrophages,84 but also by tubules 85 promotes renal scar formation by activating myofibroblast proliferation and reducing the production of extracellular matrix-degrading proteinases.86,87 Thalidomide, an immunomodulatory drug, attenuated infiltration of macrophages and reduced the development of interstitial fibrosis in the kidney.80 Specific ablation of the TGF-βRII from macrophages protected against the development of interstitial fibrosis following severe renal I/R injury.88 Interleukin-1 receptor-associated kinase -M is, a kinase enzyme, associated with switching macrophages from an M1 inflammatory to an M2 pro-repair phenotype. Deletion of this kinase resulted in sustained M1 activation and increased severity of fibrosis following I/R injury.89
In addition, increased IL17 expression from either T-cells or NK (natural killer) cells has been shown to contribute renal fibrosis in an AKI-to-CKD model.90
An assessment of hypoxia in the AKI to CKD transition
Evaluation of the chronic hypoxia hypothesis in the AKI-to-CKD transition necessitates addressing fundamental features directly related to the hypothesis such as: a) Is renal hypoxia present following AKI? and b) Does alteration of renal oxygen tension alter progression of renal disease? In our opinion, available evidence in animal models is supportive but not definitive regarding the role of chronic hypoxia in the setting of AKI-to-CKD. Moreover, because of the lack of studies investigating renal hypoxia in patients, few clinical studies have helped to provide significant insight into potential roles of hypoxia in the AKI-to-CKD transition. Below we summarize some of the experimental evidence addressing these questions.
a) Is renal hypoxia present following AKI?
To date, multiple techniques have been used to detect renal tissue hypoxia in the pathogenesis of kidney disease. These techniques include pimonidazole adduct immunohistochemistry (PIM), assessment of expression of HIFs, Clark-type micro electrodes and blood oximetry, oxygen telemeter, and blood oxygen level dependent-magnetic resonance imaging (BOLD-MRI).91 Excellent reviews are available summarizing these methods in depth and illustrate advantages and limitations.91 Below, we categorize and summarize both qualitative and quantitative approaches that have been used and highlight where these have been applied in AKI and transition to CKD (Table 2).
Table 2.
Methods for assessment of renal tissue pO2 in the setting of AKI and AKI-to-CKD transition
| Methods | In vivo/processed tissue | Data type | Tissue pO2 in AKI/ AKI-to-CKD transition | References |
|---|---|---|---|---|
| Pimonidazole adduct immune histochemistry | paraffin-embedded sections | Qualitative | ~5 weeks following recovery from I/R injury, positive pimonidizole staining was observed in the renal outer medulla of rats, positive pimonidizole staining was largely absent in kidney tissue at both 24 h and 5 days following I/R in rats | (Basile et al)60 (Ow et al 2018)110 |
| HIF immune histochemistry | paraffin-embedded sections/frozen sections | Qualitative | Increased expression of HIF-1α in renal tubules 28 days following sever I/R injury in mice | (Li et al 2019)101 |
| Clark-type micro electrodes | Anesthetized rat | Quantitative | Evidence of marked outer medullary hypoxia 20 minutes after injection of indomethacin or 10 minutes after injection of sodium iothalamate in rat, Renal tissue pO2 was consistently greater in rats subjected to ischemia than sham ischemia after 24 h post I/R | (Heyman et al 1991)107 (Ow et al 2018)110 |
| Radio-telemeter based oxygen sensor | Conscious rat | Quantitative | Inner medullary tissue pO2 did not differ significantly between sham rats and ischemic rats across 5 days after I/R | (Ow et al 2018)110 |
| BOLD-MRI | Human, rat, mice | Qualitative | Increased R2* values observed 24 hours post I/R in mice and in renal medulla 20 minutes after administration of iodinated contrast media in human | (Oostendorp et al 2011)130 (Hofmann et al 2006)131 |
| Urinary oxygen tension | Human, sheep | Quantitative | Urinary pO2 decreased during 24 hours period of development of sepsis in sheep and in human urinary pO2 decreased across the course of the cardiac surgery | (Lankadeva et al 2016)126 (Zhu et al 2018)125 |
Abbreviation: AKI, Acute kidney injury; CKD, Chronic kidney disease; I/R, Ischemia reprfusion; HIF,_Hypoxia inducible factor; BOLD-MRI, Blood oxygen level dependent-magnetic resonance imaging; I/R, Ischemia reperfusion.
Qualitative methods
PIM:
Pimonidazole is oxidized by molecular oxygen and reduced to a hydroxylamine intermediate in cells with oxygen tension lower than ~10 mmHg.92,93 These hydroxylamine intermediates bind to thiol containing proteins forming adducts. For in vivo assessment of hypoxia, pimonidazole is administered to animals prior to tissue harvest and the protein adducts are detected using immunohistochemistry, which provides excellent spatial resolution of hypoxic regions.94 Positive staining for PIM adduct formation has been observed in the medulla and cortico-medullary region in kidneys of healthy rats, consistent with the notion that the mammalian renal medulla is hypoxic.95 In a rat model of adenine-induced CKD, pimonidazole-adducts were confined to the tubular epithelium in a close spatial relationship with the interstitial fibrosis in both the cortex and medulla.95,96 This technique has also been used to detect cellular hypoxia in experimental models of diabetic nephropathy97 and a rat model of glomerulonephritis.64
Enhanced PIM-staining was found in post ischemic rat kidney 5 weeks following I/R despite the recovery of serum creatinine and a return of total renal blood flow.60 Unilateral nephrectomy (UNX), a maneuver designed to increase tubular workload and thereby increased oxygen consumption, in combination with 1-kidney I/R injury prominently enhanced PIM-staining when compared to kidneys following bilateral I/R. In contrast, PIM staining was reduced by a maneuver which increased renal blood flow after induction of AKI, i.e., oral supplementation with L-arginine.60 The enhancement of hypoxia by UNX hastened the development of CKD as illustrated by fibrosis and proteinuria, while reduction of hypoxia by L-arginine sharply attenuated CKD progression.60
The major limitation of this method is that it provides poor temporal resolution of hypoxia as an assessment can only be performed in processed tissue i.e., paraffin-embedded sections or in frozen sections at a discrete time point.91 Caution should also be taken to ensure minimal false positive staining, particularly in cellular debris and casts.96 In addition, hypoxia may be introduced during tissue harvest leading to elevated signal 94 (and D. P. Basile, unpublished observations).
Hypoxia inducible factors (HIFs):
As described earlier, cellular adaptation to hypoxia is mediated by cellular accumulation of HIFs and therefore, increased HIF or HIF-target gene expression could be interpreted as evidence of local tissue hypoxia. Both in vivo 98 and in vitro 99,100 studies have demonstrated increased cellular levels of HIFs under hypoxic conditions. Increased expression of HIF-1α and HIF-2α were reported in various renal cell populations when rats were exposed to systemic hypoxia induced by either bleeding anemia, functional anemia (0.1% carbon monoxide), renal ischemia, or cobalt chloride (which is known to mimic hypoxia). HIF expression co-localized with HIF target-genes such as HO-1, EPO and GLUT-1.98 Recently, Li et al. reported sustained hypoxia up to 28 days following severe I/R injury in mice using PIM and a corresponding expression of HIF-1α in renal tubules. The chronic induction of FoxO3, an anti-fibrotic factor, following I/R was dependent on the activity of HIF-1α fibrosis following AKI.101
One advantage of this approach is that it provides potential insight into activation of hypoxic signaling in renal biopsies. For example, Rosenberger et al. detected tubular HIF1α 2 weeks following renal transplant, with higher levels being associated with longer cold-ischemic times.102 At 3 months post-transplant, HIF-1α was not detectable in normal functioning grafts, but was evident in grafts with evidence of clinical or subclinical rejection.102 In another study, HIF-1α expression was found in infiltrating cells and the number of HIF-1α-positive cells was associated with poor graft survival.103
Quantitative methods
These aforementioned approaches provide no quantitative information regarding actual pO2 levels in the kidney, making evaluation of the hypoxia hypothesis challenging. The Clark-type microelectrode is an oxygen sensor that measures soluble oxygen concentrations in a liquid medium or in tissues based on an oxidation-reduction reaction.91 The Clark-type microelectrode has been used to determine renal tissue pO2 at various depths below the kidney surface, so can provide some degree of spatial resolution of renal tissue pO2.104,105 In 1997, Liss and colleagues introduced a modified Clark electrode with an outer tip diameter of 5.5±1.9 μm and measured renal tissue pO2 in the cortex was 45±2 mmHg (meant standard error of mean), outer medulla (32 ±2 mmHg) and inner medulla (25±2 mmHg) of rats.106 The approach has been used to demonstrate reductions in medullary oxygen tension in response to radiographic contrast agents or indomethacin in rats.106‘107 Renal medullary hypoxia has also been detected in kidneys of diabetic rats, which was improved by administration of anti-oxidants such as α-tocopherol.108 In addition, this approach has been used to evaluate reductions in pig renal medulla in response to graded levels of renal artery occlusion, a model associated with progressive fibrosis.109 However, in a model of I/R induced AKI in rats, renal pO2 levels measured with the Clark-electrodes were similar 5 days following reperfusion when compared to sham-operated control rats.110 A similar observation was also found in rats instrumented with radio-telemeter in renal medulla or post mortem assessment of cellular hypoxia by PIM.110 In that study, the marked deficit in renal oxygen consumption (49%) was observed in rats 5 days after reperfusion than after sham-ischemia which likely provided some protection against development of renal hypoxia in a rat model of I/R injury during subacute phase.
Recently, Koeners et al. developed a radio-telemeter based oxygen sensor for continuous monitoring of renal tissue pO2 in freely moving rats.111 This method has been shown to generate stable recordings of medullary or cortical tissue pO2 for up to 3 weeks.111,112 In a model of angiotensin II induced hypertension and CKD, Emans et al. used this approach to detect cortical tissue hypoxia, within 15 hours of renin-angiotensin-aldosterone system activation.112 These investigators also used this approach to detect renal medullary hypoxia in rats treated for 2 weeks with N-ω-nitro-L-arginine (a nitric oxide synthase inhibitor).113
Techniques with potential application in clinical practice
While oxygen sensing micro-electrodes methods provide quantitative information regarding renal tissue pO2, their application in human subjects is limited due to their invasive nature. The following represent less invasive approaches which are required to evaluate pO2 in patients.
BOLD-MRI:
BOLD-MRI utilizes the paramagnetic properties of deoxyhemoglobin to generate images that detect changes in renal blood oxygenation and does not require administration of contrast agents for image acquisition.114 BOLD-MRI measures R2* values, defined as the time required for the MRI signal to decay in the transverse plane. This value is proportional to the blood content of deoxyhemoglobin. R2* values assigned in 3D voxels provides spatial information on regional oxygenation.114
Experimental evidence demonstrates a linear relationship between blood R2* values measured by BOLD-MRI and tissue pO2 (ranging from 0.2 to 10 kPa).115 Studies by Prasad et al. using healthy young volunteer subjects, have shown a decrease in R2*, reflective of an increase in oxygenation in the renal medulla following furosemide administration.116 This observation is consistent with those of previous studies in which medullary tissue pO2, measured with Clark-electrodes was increased in anesthetized rats when furosemide was administered to inhibit transport in the thick ascending limb.117
Until recently BOLD-MRI has been extensively used to detect changes in renal blood oxygenation in both experimental animals and human subjects in response to diuretics, diabetic nephropathy, radiocontrast nephropathy and renal artery stenosis.118 However, recently, Prujim et al. conducted a three-year follow up study of patients with CKD, hypertensive patients without CKD, and normotensive controls. These investigators reported that low cortical oxygenation predicted a decline in yearly estimated glomerular filtration rate in patients with CKD.119 Although promising, a potential concern regarding BOLD-MRI is that it provides information on blood pO2, rather than tissue pO2 directly.91 In addition, multiple external and internal factors including hydration status,120 low pH,121 body temperature,121 dietary sodium intake,122 vascular volume and vessel geometry123 may influence BOLD-MRI signal thus R2* values.
Recently, Kodama and colleagues have employed a new method that utilizes a combination of electron paramagnetic resonance imaging and dynamic nuclear polarization-MRI technique in living animals in order to quantify renal oxygen tension in mmHg.124 This double-resonance technique allows co-registration of tissue oxygen tension and renal structure non-invasively by using paramagnetic contrast agents such as oxygen-sensitive triarylmethyl radical 0X63.124 0X63, which is administered intravenously, interacts with dissolved paramagnetic oxygen and broadens the spectral line width of 0X63 and thereby provides quantitative information related to tissue oxygen tension. Kodama et al. have shown that renal oxygen tensions estimated by using this dynamic nuclear polarization-MRI technique were similar to those estimated with the “gold standard” microelectrode method in experimental mice models of type 1 and type 2 diabetes.124
Urinary PO2
Measurement of urinary pO2 as a “physiological biomarker” for risk of AKI has received increasing attention for its potential usefulness in clinical settings.125 Evidence demonstrates that urinary pO2 reflects medullary tissue pO2.126-128 In 1965, Leonhardt et al. performed a preliminary study for measurement of pO2 in human kidney by using oxygen micro-electrode and showed excellent agreement between pelvic urinary pO2 and medullary pO2 in human patients under anesthetized conditions.128 Therefore urinary pO2 could represent a potential biomarker for risk of AKI.
Recently, Lankadeva et al. for the first time utilized fiber-optic probes for measurement of urinary pO2 in an ovine model of acute kidney injury.126 In that study, changes in medullary pO2 closely paralleled changes in urinary pO2 during development of AKI in conscious sheep. The fiber-optic probe, which works on the principle of fluorescence lifetime oximetry,129 was inserted into a standard bladder catheter without blocking urine flow. Zhu et al. studied patients at high risk of AKI associated with cardiac surgery requiring cardiopulmonary bypass.125 These investigators demonstrated that urinary pO2 decreased progressively during cardiac surgery requiring cardiopulmonary bypass.125 Strikingly, urinary pO2 was reduced in patients who developed AKI relative to patients who did not develop AKI (18.7±9.0 mmHg vs 26.7±17.2, P=0.03) during post bypass epoch, and urinary hypoxia was an independent predictor for the development of postoperative AKI.125
However, at the current time, no studies have sought to evaluate the degree to which urinary pO2 may be persistent following an episode of AKI or whether this may correlate with the future development of CKD.
b) Does alteration of renal oxygen tension alter progression of renal disease?
Over the last two decades, an increasing amount of experimental evidence suggests the presence of renal tissue hypoxia in multiple forms of CKD, including diabetic nephropathy,132-134 the remnant kidney model,135 hypertension,136 polycystic kidney disease, 137,138 CKD after recovery from I/R injury.139 In many cases, such as in I/R injury60 or in STZ induced diabetes,133 evidence of hypoxia precedes evidence of progressive disease suggesting hypoxia may be a cause rather than an effect of the subsequent development of interstitial fibrosis.
However, whether hypoxia drives progression is not firmly established. As described, progression of CKD relies on multiple interacting pathways in the setting of renal injury (Figure 1). Much of the evidence regarding the role of hypoxia is largely indirect. It should be recognized that experimental interventions with the potential to influence renal hypoxia might also affect other pathophysiological pathways making it difficult to firmly establish a role for hypoxia per se. For example, studies from our group have shown that preservation of peritubular capillaries by VEGF-121 attenuates CKD progression following AKI.33 While these results may be attributable to improved oxygen delivery to tissue, VEGF-121 prevents initial endothelial damage 33 and therefore this treatment may also attenuate the activation of pericytes to myofibroblasts to reduce the population of this fibrotic cell type. Recent studies also have shown that when modified VEGF-121 is administered to pigs with renal artery stenosis, there is a shift from pro-inflammatory M1 macrophages to pro-repair M2 macrophages with sustained VEGF expression.140
In addition, losartan was shown to increase renal blood flow and attenuate renal fibrosis in post-ischemic rats. 141 This result is consistent with a potential role for Ang II blockade to improve oxygenation as a means of suppressing progression. However, blockade of Ang II signaling is also known to affect glomerular hydrostatic pressure, influence macrophage and lymphocyte activity, as well as reduce tubular expression of pro-fibrotic growth factors such as TGF-β. Finally, as described above, L-arginine increased RBF and attenuated hypoxia and progression to CKD in post AKI rats,60 but the putative activation of NO-related events could easily be thought to influence inflammatory processes.
Other investigators have attempted to evaluate whether exacerbation of hypoxia might hasten the development of CKD. For example, reduced renal mass results in a greater degree of renal hypoxia and accelerates the AKI-to-CKD progression.24 This approach would also tend to support a role for hypoxia in CKD progression. However, other studies have shown that reduction in renal mass results in a greater percentage of tubules which fail to redifferentiate completely.16 As described earlier, undifferentiated tubules may provide a strong driving force for the development of fibrosis. Taken together, while a number of studies seem to support a likely role for hypoxia in progression, experimental maneuvers designed to increase or decrease hypoxia may affect other processes making firm conclusions difficult.
Despite these complications, an interesting study by Friedrich-Persson and colleagues attempted to evaluate if renal tissue hypoxia per se may be sufficient to initiate the development of nephropathy, even in the absence of other confounding factors like hyperglycemia, hypertension, oxidative stress or prior tissue damage.142 In their study, normoglycemic rats received 2, 4-dinitrophenol, a mitochondrial uncoupler that increases oxygen consumption, for 30 consecutive days by oral gavage. This resulted in increased oxygen consumption and a significant reduction of oxygen tension in both the renal cortex and medulla. This treatment resulted in increased proteinuria, infiltration of inflammatory cells and tubular injury as indicated by increased expression of vimentin positive cells. In contrast, renal markers of oxidative stress, mean arterial pressure, renal blood flow, glomerular filtration rate and urinary excretion of Na+ remained unaffected in rats that received chronic dinitrophenol treatment.142 These studies suggest that renal tissue hypoxia, by itself, has the potential to act as a pathogenic factor in the kidney.
A developing area of interest that may impinge on this conversation may come from information of patients residing in high altitudes. As reviewed by Luks et al. reductions in inspired pO2 are associated with reduced renal pO2.143 There are limited studies demonstrating that Navajo Indians living at high altitude have higher rates of ESRD compared with other Native Americans living at low altitudes.144 Another study compared patients with type 2 diabetes living near sea level or above 1700 meter.145 Despite a similar level of glycemic control, it was reported that patients at high altitude had increased levels of proteinuria and serum creatinine versus patient near sea level.145
Future Directions
The last 20 years has seen a dramatic increase in the understanding of the pathophysiology of the AKI to CKD transition and the identification of several molecular pathways mediated by hypoxia. However, it remains to be determined whether hypoxia represents an important mediator of the AKI-to-CKD transition. Few longitudinal studies have been conducted in an effort to evaluate patients with AKI at risk for progression of CKD. We propose that long-term follow up studies should consider evaluation of renal hypoxia as one of the potential risk factors which may predict future progression. Given the improvement in non-invasive methods for measurement of renal hypoxia such as a newly developed dynamic nuclear polarization-MRI technique would be useful for understanding the role of hypoxia in pathogenesis of AKI-CKD transition. This information might therefore be helpful in identifying whether hypoxia in patients with AKI represents potential risk factor for progression of CKD, and perhaps such approaches could identify which patients with AKI that may require long term follow up care following discharge.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2:1303–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lameire NH, Bagga A, Cruz D, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382:170–79. [DOI] [PubMed] [Google Scholar]
- 3.Benedetto U, Luciani R, Goracci M, et al. Miniaturized cardiopulmonary bypass and acute kidney injury in coronary artery bypass graft surgery. Ann Thorac Surg. 2009;88:529–35. [DOI] [PubMed] [Google Scholar]
- 4.Uchino S, Kellum JA, Bellomo R, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. Jama. 2005;294:813–18. [DOI] [PubMed] [Google Scholar]
- 5.Fahling M, Seeliger E, Patzak A, Persson PB. Understanding and preventing contrast-induced acute kidney injury. Nat Rev Nephrol. 2017;13:169–80. [DOI] [PubMed] [Google Scholar]
- 6.Moore PK, Hsu RK, Liu KD. Management of acute kidney injury: core curriculum 2018. Am J Kidney Dis. 2018;72:136–48. [DOI] [PubMed] [Google Scholar]
- 7.Rifkin DE, Coca SG, Kalantar-Zadeh K. Does AKI truly lead to CKD? J Am Soc Nephrol. 2012;23:979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishani A, Xue JL, Himmelfarb J, et al. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20:223–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amdur RL, Chawla LS, Amodeo S, Kimmel PL, Palant CE. Outcomes following diagnosis of acute renal failure in U.S. veterans: focus on acute tubular necrosis. Kidney Int. 2009;76:1089–97. [DOI] [PubMed] [Google Scholar]
- 10.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81:442–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basile DP, Bonventre JV, Mehta R, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27:687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basile DP. Rarefaction of peritubular capillaries following ischemic acute renal failure: a potential factor predisposing to progressive nephropathy. Curr Opin Nephrol Hypertens. 2004;13:1–7. [DOI] [PubMed] [Google Scholar]
- 13.Nony PA, Schnellmann RG. Mechanisms of renal cell repair and regeneration after acute renal failure. J Pharmacol Exp Ther. 2003;304:905–12. [DOI] [PubMed] [Google Scholar]
- 14.Dong Y, Zhang Q, Wen J, et al. Ischemic duration and frequency determines AKI-to-CKD progression monitored by dynamic changes of tubular biomarkers in IRI mice. Front Physiol. 2019;10:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nowak G, Schnellmann RG. Renal cell regeneration following oxidant exposure: inhibition by TGF-beta1 and stimulation by ascorbic acid. Toxicol Appl Pharmacol. 1997;145:175–83. [DOI] [PubMed] [Google Scholar]
- 16.Polichnowski AJ, Lan R, Geng H, Griffin KA, Venkatachalam MA, Bidani AK. Severe renal mass reduction impairs recovery and promotes fibrosis after AKI. J Am Soc Nephrol. 2014;25:1496–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lan R, Geng H, Polichnowski AJ, et al. PTEN loss defines a TGF-beta-induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol. 2012;302:1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26:1765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nature medicine. 2010;16:535–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geng H, Lan R, Wang G, et al. Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol. 2009;174:1291–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Borst MH, Prakash J, Sandovici M, et al. c-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J Pharmacol Exp Ther. 2009;331:896–05. [DOI] [PubMed] [Google Scholar]
- 22.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki T, Kimura M, Asano M, Fujigaki Y, Hishida A. Role of atrophic tubules in development of interstitial fibrosis in microembolism-induced renal failure in rat. Am J Pathol. 2001;158:75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281:887–99. [DOI] [PubMed] [Google Scholar]
- 25.Kramann R, Tanaka M, Humphreys BD. Fluorescence microangiography for quantitative assessment of peritubular capillary changes after AKI in mice. J Am Soc Nephrol. 2014;25:1924–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Basile DP, Friedrich JL, Spahic J, et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300:721–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Babickova J, Klinkhammer BM, Buhl EM, et al. Regardless of etiology, progressive renal disease causes ultrastructural and functional alterations of peritubular capillaries. Kidney Int. 2017;91:70–85. [DOI] [PubMed] [Google Scholar]
- 28.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwon O, Hong SM, Sutton TA, Temm CJ. Preservation of peritubular capillary endothelial integrity and increasing pericytes may be critical to recovery from postischemic acute kidney injury. Am J Physiol Renal Physiol. 2008;295:351–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steegh FMEG, Gelens MACJ, Nieman FHM, et al. Early loss of peritubular capillaries after kidney transplantation. J Am Soc Nephrol. 2011;22:1024–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw SK, Perkins BN, Lim YC, et al. Reduced expression of junctional adhesion molecule and platelet/endothelial cell adhesion molecule-1 (CD31) at human vascular endothelial junctions by cytokines tumor necrosis factor-alpha plus interferon-gamma does not reduce leukocyte transmigration under flow. Am J Pathol. 2001;159:2281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ehling J, Bábíčková J, Gremse F, et al. Quantitative micro-computed tomography imaging of vascular dysfunction in progressive kidney diseases. J Am Soc Nephrol. 2016;27:520–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leonard EC, Friedrich JL, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol. 2008;295:1648–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jung YJ, Kim DH, Lee AS, et al. Peritubular capillary preservation with COMP-angiopoietin-1 decreases ischemia-reperfusion-induced acute kidney injury. Am J Physiol Renal Physiol. 2009;297:952–60. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014;306:970–80. [DOI] [PubMed] [Google Scholar]
- 36.Cantaluppi V, Gatti S, Medica D, et al. Microvesicles derived from endothelial progenitor cells protect the kidney from ischemia-reperfusion injury by microRNA-dependent reprogramming of resident renal cells. Kidney Int. 2012;82:412–27. [DOI] [PubMed] [Google Scholar]
- 37.Lee SY, Hörbelt M, Mang HE, et al. MMP-9 gene deletion mitigates microvascular loss in a model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2011;301:101–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xing L, Cui R, Peng L, et al. Mesenchymal stem cells, not conditioned medium, contribute to kidney repair after ischemia-reperfusion injury. Stem Cell Res Ther. 2014;5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee D, Shenoy S, Nigatu Y, Plotkin M. Id proteins regulate capillary repair and perivascular cell proliferation following ischemia-reperfusion injury. PLoS One. 2014;9:96–06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82:172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horbelt M, Lee SY, Mang HE, et al. Acute and chronic microvascular alterations in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2007;293:688–95. [DOI] [PubMed] [Google Scholar]
- 42.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72:151–56. [DOI] [PubMed] [Google Scholar]
- 43.Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol. 2015;26:1027–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol. 2008;294:928–36. [DOI] [PubMed] [Google Scholar]
- 45.Lindenmeyer MT, Kretzler M, Boucherot A, et al. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol. 2007;18:1765–76. [DOI] [PubMed] [Google Scholar]
- 46.Barylski M, Kowalczyk E, Banach M, Ciecwierz J, Pawlicki L, Kowalski J. Plasma total antioxidant activity in comparison with plasma NO and VEGF levels in patients with metabolic syndrome. Angiology. 2009;60:87–92. [DOI] [PubMed] [Google Scholar]
- 47.Smith SW, Chand S, Savage CO. Biology of the renal pericyte. Nephrol Dial Transplant. 2012;27:2149–55. [DOI] [PubMed] [Google Scholar]
- 48.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol. 2008;173:1617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin SL, Chang FC, Schrimpf C, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178:911–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Humphreys BD, Lin SL, Kobayashi A, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 2010;176:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen YT, Chang FC, Wu CF, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–81. [DOI] [PubMed] [Google Scholar]
- 52.Kida Y, Duffield JS. Pivotal role of pericytes in kidney fibrosis. Clin Exp Pharmacol Physiol. 2011;38:417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrimpf C, Xin C, Campanholle G, et al. Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol 2012;23:868–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kramann R, Wongboonsin J, Chang-Panesso M, Machado FG, Humphreys BD. Gli1+ pericyte loss induces capillary rarefaction and proximal tubular injury. J Am Soc Nephrol 2017;28:776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansell P, Welch WJ, Blantz RC, Palm F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin Exp Pharmacol Physiol 2013;40:123–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brezis M, Rosen S. Hypoxia of the renal medulla — its implications for disease. N Engl J Med. 1995;332:647–55. [DOI] [PubMed] [Google Scholar]
- 57.Molema G, Aird WC. Vascular heterogeneity in the kidney. Semin Nephrol. 2012;32:145–55. [DOI] [PubMed] [Google Scholar]
- 58.Evans RG, Ince C, Joles JA, et al. Haemodynamic influences on kidney oxygenation: clinical implications of integrative physiology. Clin Exp Pharmacol Physiol. 2013;40:106–22. [DOI] [PubMed] [Google Scholar]
- 59.Evans RG, Eppel GA, Michaels S, et al. Multiple mechanisms act to maintain kidney oxygenation during renal ischemia in anesthetized rabbits. Am J Physiol Renal Physiol. 2010;298:1235–43. [DOI] [PubMed] [Google Scholar]
- 60.Basile DP, Donohoe DL, Roethe K, Mattson DL. Chronic renal hypoxia after acute ischemic injury: effects of L-arginine on hypoxia and secondary damage. Am J Physiol Renal Physiol. 2003;284:338–48. [DOI] [PubMed] [Google Scholar]
- 61.Fine LG, Bandyopadhay D, Norman JT. Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia Kidney Int. 2000;57:22–26. [PubMed] [Google Scholar]
- 62.Fine LG, Norman JT. Chronic hypoxia as a mechanism of progression of chronic kidney diseases: from hypothesis to novel therapeutics Kidney Int. 2008;74:867–72. [DOI] [PubMed] [Google Scholar]
- 63.Fine LG, Orphanides C, Norman JT. Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int Suppl. 1998;65:74–78. [PubMed] [Google Scholar]
- 64.Matsumoto M, Tanaka T, Yamamoto T, et al. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol. 2004;15:1574–81. [DOI] [PubMed] [Google Scholar]
- 65.Eardley KS, Kubal C, Zehnder D, et al. The role of capillary density, macrophage infiltration and interstitial scarring in the pathogenesis of human chronic kidney disease. Kidney Int. 2008;74:495–04. [DOI] [PubMed] [Google Scholar]
- 66.Matsumoto M, Makino Y, Tanaka T, et al. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol. 2003;14:1825–32. [DOI] [PubMed] [Google Scholar]
- 67.Nordquist L, Friederich-Persson M, Fasching A, et al. Activation of hypoxia-inducible factors prevents diabetic nephropathy. J Am Soc Nephrol. 2015;26:328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol. 2006;291:271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nangaku M, Eckardt KU. Hypoxia and the HIF system in kidney disease J Mol Med. 2007;85:1325–30. [DOI] [PubMed] [Google Scholar]
- 70.Sun S, Ning X, Zhang Y, et al. Hypoxia-inducible factor-1alpha induces twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 2009;75:1278–87. [DOI] [PubMed] [Google Scholar]
- 71.Higgins DF, Kimura K, Bernhardt WM, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang S, Zeng H, Chen ST, et al. Ablation of endothelial prolyl hydroxylase domain protein-2 promotes renal vascular remodelling and fibrosis in mice. J Cell Mol Med. 2017;21:1967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu M, Ning X, Li R, et al. Signalling pathways involved in hypoxia-induced renal fibrosis. J Cell Mol Med. 2017;21:1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng S- Y, Chou Y- H, Liao F- L, et al. Losartan reduces ensuing chronic kidney disease and mortality after acute kidney injury. Sci Rep. 2016;6:34265–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gewin LS. Transforming growth factor-beta in the acute kidney injury to chronic kidney disease transition. Nephron. 2019:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamaguchi J, Tanaka T, Eto N, Nangaku M. Inflammation and hypoxia linked to renal injury by CCAAT/enhancer-binding protein delta. Kidney Int. 2015;88:262–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ascon M, Ascon DB, Liu M, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75:526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Burne-Taney MJ, Yokota N, Rabb H. Persistent renal and extrarenal immune changes after severe ischemic injury. Kidney Int. 2005;67:1002–09. [DOI] [PubMed] [Google Scholar]
- 80.Santana AC, Degaspari S, Catanozi S, et al. Thalidomide suppresses inflammation in adenine-induced CKD with uraemia in mice. Nephrol Dial Transplant. 2013;28:1140–49. [DOI] [PubMed] [Google Scholar]
- 81.Kincaid-Smith P, Nicholls K, Birchall I. Polymorphs infiltrate glomeruli in mesangial IgA glomerulonephritis. Kidney Int. 1989;36:1108–11. [DOI] [PubMed] [Google Scholar]
- 82.Rastaldi MP, Ferrario F, Crippa A, et al. Glomerular monocyte-macrophage features in ANCA-positive renal vasculitis and cryoglobulinemic nephritis. J Am Soc Nephrol. 2000;11:2036–43. [DOI] [PubMed] [Google Scholar]
- 83.Kinsey GR. Macrophage dynamics in AKI to CKD progression. J Am Soc Nephrol. 2014;25:209–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nacu N, Luzina IG, Highsmith K, et al. Macrophages produce TGF-β-induced (β-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts. J Immunol. 2008;180:5036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ito Y, Goldschmeding R, Kasuga H, et al. Expression patterns of connective tissue growth factor and of TGF-beta isoforms during glomerular injury recapitulate glomerulogenesis. Am J Physiol Renal Physiol. 2010;299:545–58. [DOI] [PubMed] [Google Scholar]
- 86.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li J, Ren J, Liu X, et al. Rictor/mTORC2 signaling mediates TGFbeta1 - induced fibroblast activation and kidney fibrosis. Kidney Int. 2015;88:515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chung S, Overstreet JM, Li Y, et al. TGF-beta promotes fibrosis after severe acute kidney injury by enhancing renal macrophage infiltration. JCI Insight. 2018;3:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lech M, Grobmayr R, Ryu M, et al. Macrophage phenotype controls long-term AKI outcomes--kidney regeneration versus atrophy. J Am Soc Nephrol. 2014;25:292–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mehrotra P, Collett JA, McKinney SD, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: compensatory role of natural killer cells in athymic rats. Am J Physiol Renal Physiol. 2017;312:385–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Evans RG, Gardiner BS, Smith DW, O’Connor PM. Methods for studying the physiology of kidney oxygenation Clin Exp Pharmacol Physiol. 2008;35:1405–12. [DOI] [PubMed] [Google Scholar]
- 92.Arteel G, Thurman R, Yates J, Raleigh J. Evidence that hypoxia markers detect oxygen gradients in liver: pimonidazole and retrograde perfusion of rat liver. Br J Cancer. 1995;72:889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arteel G, Thurman R, Raleigh J. Reductive metabolism of the hypoxia marker pimonidazole is regulated by oxygen tension independent of the pyridine nucleotide redox state. Eur J Biochem. 1998;253:743–50. [DOI] [PubMed] [Google Scholar]
- 94.Rosenberger C, Rosen S, Paliege A, Heyman SN. Pimonidazole adduct immunohistochemistry in the rat kidney: detection of tissue hypoxia. Methods Mol Biol 2009;466:161–74. [DOI] [PubMed] [Google Scholar]
- 95.Fong D, Ullah MM, Lai JG, et al. Renal cellular hypoxia in adenine-induced chronic kidney disease. Clin Exp Pharmacol Physiol. 2016;43:896–05. [DOI] [PubMed] [Google Scholar]
- 96.Ow CPC, Ullah MM, Ngo JP, Sayakkarage A, Evans RG. Detection of cellular hypoxia by pimonidazole adduct immunohistochemistry in kidney disease: Methodological pitfalls and their solution. Am J Physiol Renal Physiol. 2019;317:322–32. [DOI] [PubMed] [Google Scholar]
- 97.Rosenberger C, Khamaisi M, Abassi Z, et al. Adaptation to hypoxia in the diabetic rat kidney. Kidney Int. 2008;73:34–42. [DOI] [PubMed] [Google Scholar]
- 98.Rosenberger C, Mandriota S, Jürgensen JS, et al. Expression of hypoxia-inducible factor-1α and −2α in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13:1721–32. [DOI] [PubMed] [Google Scholar]
- 99.Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996;271:1172–80. [DOI] [PubMed] [Google Scholar]
- 100.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li L, Kang H, Zhang Q, D’Agati VD, Al-Awqati Q, Lin L. Lox03 activation in hypoxic tubules prevents chronic kidney disease. J Clin Invest. 2019;129:2374–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rosenberger C, Pratschke J, Rudolph B, et al. Immunohistochemical detection of hypoxia-inducible factor-1 alpha in human renal allograft biopsies. J Am Soc Nephrol. 2007;18:343–51. [DOI] [PubMed] [Google Scholar]
- 103.Yu TM, Wen MC, Li CY, et al. Expression of hypoxia-inducible factor-1 alpha (HIP-1 alpha) in infiltrating inflammatory cells is associated with chronic allograft dysfunction and predicts long-term graft survival. Nephrol Dial Transplant. 2013;28:659–70. [DOI] [PubMed] [Google Scholar]
- 104.Aukland K, Krog J. Renal oxygen tension. Nature. 1960; 188:671. [DOI] [PubMed] [Google Scholar]
- 105.Baumgartl H, Leichtweiss HP, Lubbers DW, Weiss C, Huland H. The oxygen supply of the dog kidney: measurements of intrarenal pO2. Microvasc Res. 1972;4:247–57. [DOI] [PubMed] [Google Scholar]
- 106.Liss P, Nygren A, Revsbech NP, Ulfendahl HR. Intrarenal oxygen tension measured by a modified clark electrode at normal and low blood pressure and after injection of x-ray contrast media. Pflugers Arch. 1997;434:705–11. [DOI] [PubMed] [Google Scholar]
- 107.Heyman SN, Brezis M, Epstein FH, Spokes K, Silva P, Rosen S. Early renal medullary hypoxic injury from radiocontrast and indomethacin. Kidney Int. 1991;40:632–42. [DOI] [PubMed] [Google Scholar]
- 108.Palm F, Cederberg J, Hansell P, Liss P, Carlsson PO. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46:1153–60. [DOI] [PubMed] [Google Scholar]
- 109.Warner L, Gomez SI, Bolterman R, et al. Regional decreases in renal oxygenation during graded acute renal arterial stenosis: a case for renal ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ow CPC, Ngo JP, Ullah MM, et al. Absence of renal hypoxia in the subacute phase of severe renal ischemia reperfusion injury. Am J Physiol Renal Physiol. 2018;315:1358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Koeners MP, Ow CP, Russell DM, et al. Telemetry-based oxygen sensor for continuous monitoring of kidney oxygenation in conscious rats. Am J Physiol Renal Physiol. 2013;304:1471–80. [DOI] [PubMed] [Google Scholar]
- 112.Emans TW, Janssen BJ, Pinkham MI, et al. Exogenous and endogenous angiotensin-II decrease renal cortical oxygen tension in conscious rats by limiting renal blood flow. J Physiol. 2016;594:6287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Emans TW, Janssen BJ, Joles JA, Krediet CTP. Nitric oxide synthase inhibition induces renal medullary hypoxia in conscious rats. J Am Heart Assoc. 2018;7:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Prasad PV. Evaluation of intra-renal oxygenation by BOLD MRI. Nephron Clin Pract. 2006;103:58–65. [DOI] [PubMed] [Google Scholar]
- 115.Pedersen M, Dissing TH, Morkenborg J, et al. Validation of quantitative BOLD MRI measurements in kidney: application to unilateral ureteral obstruction. Kidney Int. 2005;67:2305–12. [DOI] [PubMed] [Google Scholar]
- 116.Prasad PV, Edelman RR, Epstein FH. Noninvasive evaluation of intrarenal oxygenation with BOLD MRI. Circulation. 1996;94:3271–75. [DOI] [PubMed] [Google Scholar]
- 117.Brezis M, Agmon Y, Epstein FH. Determinants of intrarenal oxygenation. I. effects of diuretics. Am J Physiol. 1994;267:1059–62. [DOI] [PubMed] [Google Scholar]
- 118.Li L- P, Halter S, Prasad PV. Blood oxygen level-dependent MR imaging of the kidneys. Magn Reson Imaging Clin N Am. 2008;16:613–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pruijm M, Milani B, Pivin E, et al. Reduced cortical oxygenation predicts a progressive decline of renal function in patients with chronic kidney disease. Kidney Int. 2018;93:932–40. [DOI] [PubMed] [Google Scholar]
- 120.Prasad PV, Epstein FH. Changes in renal medullary pO2 during water diuresis as evaluated by blood oxygenation level-dependent magnetic resonance imaging: effects of aging and cyclooxygenase inhibition. Kidney international. 1999;55:294–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pruijm M, Mendichovszky IA, Liss P, et al. Renal blood oxygenation level-dependent magnetic resonance imaging to measure renal tissue oxygenation: a statement paper and systematic review. Nephrol Dial Transplant. 2018;33:22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pruijm M, Hofmann L, Maillard M, et al. Effect of sodium loading/depletion on renal oxygenation in young normotensive and hypertensive men. Hypertension. 2010;55:1116–22. [DOI] [PubMed] [Google Scholar]
- 123.Pohlmann A, Arakelyan K, Hentschel J, et al. Detailing the relation between renal T2* and renal tissue pO2 using an integrated approach of parametric magnetic resonance imaging and invasive physiological measurements. Invest Radiol. 2014;49:547–60. [DOI] [PubMed] [Google Scholar]
- 124.Kodama Y, Hyodo F, Yamato M, et al. Dynamic nuclear polarization magnetic resonance imaging and the oxygen-sensitive paramagnetic agent OX63 provide a noninvasive quantitative evaluation of kidney hypoxia in diabetic mice. Kidney Int. 2019;96:787–92. [DOI] [PubMed] [Google Scholar]
- 125.Zhu MZL, Martin A, Cochrane AD, et al. Urinary hypoxia: an intraoperative marker of risk of cardiac surgery-associated acute kidney injury. Nephrol Dial Transplant. 2018;33:2191–01. [DOI] [PubMed] [Google Scholar]
- 126.Lankadeva YR, Kosaka J, Evans RG, Bailey SR, Bellomo R, May CN. Intrarenal and urinary oxygenation during norepinephrine resuscitation in ovine septic acute kidney injury. Kidney Int. 2016;90:100–08. [DOI] [PubMed] [Google Scholar]
- 127.Lankadeva YR, Kosaka J, Evans RG, Bellomo R, May CN. Urinary oxygenation as a surrogate measure of medullary oxygenation during angiotensin II therapy in septic acute kidney injury. Crit Care Med. 2018;46:41–48. [DOI] [PubMed] [Google Scholar]
- 128.Leonhardt KO, Landes RR, McCauley RT. Anatomy and physiology of intrarenal oxygen tension: priliminary study of the effects of anesthetics. Anesthesiology. 1965;26:648–58. [DOI] [PubMed] [Google Scholar]
- 129.O’Hara JA, Hou H, Demidenko E, Springett RJ, Khan N, Swartz HM. Simultaneous measurement of rat brain cortex PtO2 using EPR oximetry and a fluorescence fiber-optic sensor during normoxia and hyperoxia. Physiol Meas. 2005;26:203–13. [DOI] [PubMed] [Google Scholar]
- 130.Oostendorp M, de Vries EE, Slenter JM, et al. MRI of renal oxygenation and function after normothermic ischemia-reperfusion injury. NMR Biomed. 2011;24:194–00. [DOI] [PubMed] [Google Scholar]
- 131.Hofmann L, Simon-Zoula S, Nowak A, et al. BOLD-MRI for the assessment of renal oxygenation in humans: acute effect of nephrotoxic xenobiotics. Kidney Int. 2006;70:144–50. [DOI] [PubMed] [Google Scholar]
- 132.Palm F, Ortsater H, Hansell P, Liss P, Carlsson PO. Differentiating between effects of streptozotocin per se and subsequent hyperglycemia on renal function and metabolism in the streptozotocin-diabetic rat model. Diabetes Metab Res Rev. 2004;20:452–59. [DOI] [PubMed] [Google Scholar]
- 133.Franzen S, Pihl L, Khan N, Gustafsson H, Palm F. Pronounced kidney hypoxia precedes albuminuria in type 1 diabetic mice. Am J Physiol Renal Physiol. 2016;310:807–09. [DOI] [PubMed] [Google Scholar]
- 134.Ries M, Basseau F, Tyndal B, et al. Renal diffusion and BOLD MRI in experimental diabetic nephropathy. Blood oxygen level-dependent. J Magn Reson Imaging. 2003;17:104–13. [DOI] [PubMed] [Google Scholar]
- 135.Manotham K, Tanaka T, Matsumoto M, et al. Evidence of tubular hypoxia in the early phase in the remnant kidney model. J Am Soc Nephrol. 2004;15:1277–88. [DOI] [PubMed] [Google Scholar]
- 136.Welch WJ, Baumgartl H, Lubbers D, Wilcox CS. Renal oxygenation defects in the spontaneously hypertensive rat: role of AT1 receptors. Kidney Int. 2003;63:202–08. [DOI] [PubMed] [Google Scholar]
- 137.Ding A, Kalaignanasundaram P, Ricardo SD, et al. Chronic treatment with tempol does not significantly ameliorate renal tissue hypoxia or disease progression in a rodent model of polycystic kidney disease. Clin Exp Pharmacol Physiol. 2012;39:917–29. [DOI] [PubMed] [Google Scholar]
- 138.Ow CP, Abdelkader A, Hilliard LM, Phillips JK, Evans RG. Determinants of renal tissue hypoxia in a rat model of polycystic kidney disease. Am J Physiol Regul Integr Comp Physiol. 2014;307:1207–15. [DOI] [PubMed] [Google Scholar]
- 139.Papazova DA, Friederich-Persson M, Joles JA, Verhaar MC. Renal transplantation induces mitochondrial uncoupling, increased kidney oxygen consumption, and decreased kidney oxygen tension. Am J Physiol Renal Physiol. 2015;308:22–28. [DOI] [PubMed] [Google Scholar]
- 140.Engel J, Guise E, Williams M, Bidwell G, Chade AR. Targeted VEGF therapy induces long-term renal recovery in chronic kidney disease via macrophage polarization. Hypertension. 2019;(In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. 2015;88:776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Friederich-Persson M, Thorn E, Hansell P, Nangaku M, Levin M, Palm F. Kidney hypoxia, attributable to increased oxygen consumption, induces nephropathy independently of hyperglycemia and oxidative stress. Hypertension. 2013;62:914–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Luks AM, Johnson RJ, Swenson ER. Chronic kidney disease at high altitude. J Am Soc Nephrol. 2008;19:2262–71. [DOI] [PubMed] [Google Scholar]
- 144.Hochman ME, Watt JP, Reid R, O’Brien KL. The prevalence and incidence of end-stage renal disease in Native American adults on the Navajo reservation. Kidney Int. 2007;71:931–37. [DOI] [PubMed] [Google Scholar]
- 145.Sayarlioglu H, Erkoc R, Dogan E, et al. Nephropathy and retinopathy in type 2 diabetic patients living at moderately high altitude and sea level. Ren Fail. 2005;27:67–71. [PubMed] [Google Scholar]