

Abstract
Objective
Mutations in optineurin (OPTN) have been identified in familial and sporadic amyotrophic lateral sclerosis (ALS). We screened a cohort of Chinese patients for mutations in optineurin. We also performed an extensive literatures review of all mutations in optineurin identified previously to detect genotype–phenotype associations.
Methods
All 16 exons of the OPTN gene in a cohort of 15 familial ALS indexes and 275 sporadic ALS patients of Chinese origin were sequenced by targeted next generation sequencing.
Results
Two known heterozygous missense mutations in the OPTN, c.1481T> G (p.L494W), and c.1546G> C (p.E516Q), as well as one novel heterozygous missense mutation c.1690G> C (p.D564H) were each detected in one sporadic ALS patient. The patient carrying the p.E516Q mutation developed clinical features of ALS‐frontotemporal dementia (FTD) and the patient carrying the p.D564H mutation showed a phenotype of ALS. They both had an aggressive course, with a survival of 18 and 14 months respectively. Literature review showed that the clinical phenotypes in OPTN mutated ALS were not homogeneous, although some individuals showed a relatively slow progression and a long duration, some mutations carriers developed an aggressive progression and a short survival.
Interpretation
OPTN mutations contribute to ALS in Chinese population and account for 0.8% of sporadic ALS patients and 1.5% of familial ALS in the pooled Chinese ALS cohorts. Mutations in optineurin can cause aggressive ALS+/−FTD.
Introduction
Amyotrophic lateral sclerosis (ALS) is a syndrome characterized by degeneration of neurons in the brain stem and spinal cord, leading to progressive muscle weakness and atrophy. Typically, death due to respiratory paralysis occurs in 3 to 5 years. Approximately 10% of ALS patients are familial ALS (FALS), the other 90% of cases are sporadic ALS (SALS).1 Mutations in more than 20 genes have been linked to ALS.2 In 2010 Maruyama et al. first identified mutations of optineurin (OPTN) in both FALS and SALS in Japanese population.3 Interestingly, some ALS patients carrying OPTN mutations demonstrated a relatively slow progression and a long duration.3 Here we screened a cohort of Chinese patients for mutations in optineurin and identified two OPTN mutations presenting aggressive ALS+/‐frontotemporal dementia (FTD). We also performed an extensive literatures review of all mutations in optineurin identified previously to detect genotype–phenotype associations.
Patients and Methods
Study population
Fifteen FALS probands and 275 SALS patients (173 males, 117 females, mean age of onset ± SD 55.3 ± 11.6 years) referred to the Neurology Department, Fujian Medical Union Hospital and Department of Neurology, Henan Provincial People's Hospital between January 2017 and December 2018 were enrolled in the study. A diagnosis of definite, probable, or laboratory supported probable ALS was established using the revised El Escorial criteria.4 Familial ALS was diagnosed if one or more first‐ or second‐ degree relatives developed ALS or FTD.5 A patient was classified as having ALS + FTD if they presented with major cognitive or behavioral change at any stage during the course. Ethical approval was obtained from the ethics committee of Fujian Medical Union Hospital and Henan Provincial People's Hospital. Blood samples were collected after the individuals had signed an informed consent document.
Genetic studies
Genomic DNA was extracted from peripheral blood by standard methods from ethylenediaminetetraacetic acid (EDTA) blood samples collected from a peripheral vein, using an Axygen DNA Blood Genomic Minikit (Axygen, NY, USA). Targeted NGS was used (full methods are available upon request). The targeted regions were designed to include all 16 exons with intronic 50 bp flanking sites and 3' and 5' untranslated regions (UTRs) of the OPTN gene (NM_001008212.2). The identified variants were Sanger sequenced for confirmation. Patients carrying OPTN variants were also screened for mutations in other known ALS related genes SOD1, FUS, TARDBP, KIF5A, ANXA11, TIA1, CCNF, NEK1, CHCHD10, TUBA4A,TBK1, SQSTM1, CHCHD10, MATR3, hnRNPA1, hnRNPA2B1, VAPB, SPG11, VCP, PFN1, ANG, ALS2, DAO, UBQLN2, SIGMAR1, SETX, FIG4, DCTN1, as well as the presence of the GGGGCC expansions in the C9orf72 gene.
Bioinformatic analysis
Variant frequencies were initially determined in dbSNP, the 1000 Genomes Project, and Exome Aggregation Consortium (ExAC) to remove common single nucleotide polymorphisms (SNPs). Only nonsynonymous, splicing and frameshift variants with minor allele frequency (MAF) <1% or absent in population databases were selected for further assessment. Protein sequence alignment was performed using UniProt (https://www.uniprot.org/align/). The effect of the detected OPTN missense variants on protein structure or function was analyzed in silico with three prediction programs: SIFT (https://sift.bii.a-star.edu.sg), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2), and Mutation Taster (http://www. mutationtaster.org). PyMOL software (Delano Scientifi c LLC) was used for structure analysis of variants.
Results
Genetic analysis
Two known heterozygous missense mutations in the OPTN gene, c.1481T> G (p.L494W), and c.1546G> C (p.E516Q) were each identified in one SALS patient (Fig. 1A and 1). Another heterozygous missense variant in the OPTN, c.1690G> C (p.D564H) was detected in one SALS case (Fig. 1C). The p.D564H variant is absent from the public population database gnomAD and have a frequency of 0.00003295 in ExAC. It is absent from 600 ethnicity matched controls and CMDB (Chinese Millionome Database, https://db.cngb.org/cmdb/). The p.D564H variant lies in a highly conserved zinc finger domain (Fig. 1D) and predicted to have a deleterious effect by the SIFT, PolyPhen‐2, and Mutation Taster (Table 1). Structure analysis based on the atomic NMR model of human Optineurin (PDB code 2LO4) showed that Asp564 is located at the transition point from coil to helix in the zinc finger domain of optineurin. The side chain delta 1 and delta 2 oxygen atoms of Asp564 form tightly coupled hydrogen bonds with the Thr567. The replacement of Asp564 by a bulky Histidine residue would very likely to introduce sterechemical clashes, thus affect zinc finger domain stability (Fig. 1E).
Figure 1.
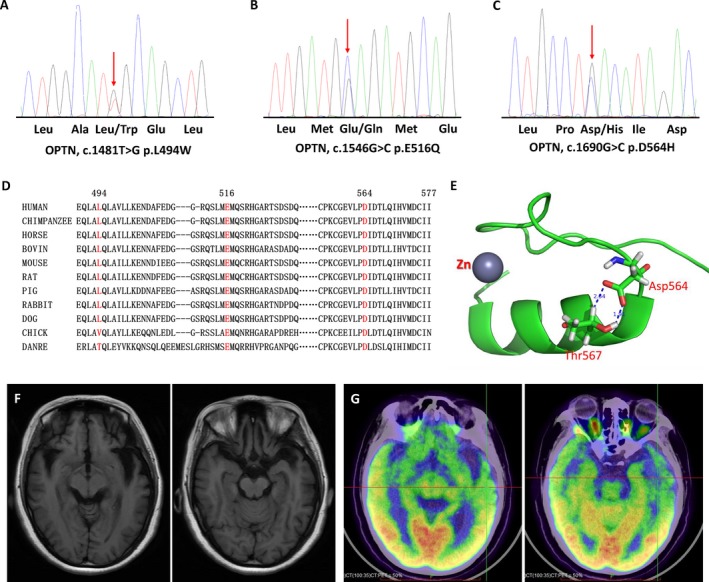
Sequencing chromatograms and pathogenicity analysis of the OPTN mutations. Sequencing chromatograms of the OPTN p.L494W (A), p.E516Q (B) and p.D564H (C) mutation. (D) Evolutionary conservation of the p.L494W, p.E516Q and p.D564H mutation in OPTN. L494, E516, and D564 are highly conserved across species. (E) Structure analysis of p.D564H mutation. The structure analysis is based on the atomic NMR model of human Optineurin (PDB code 2LO4). The amino acids position was renumbered according to the full length of OPTN protein (NM_021980). This segment of the OPTN protein is the zinc finger domain. The Asp564 is located at the transition point from coil to helix. The side chain delta 1 and delta 2 oxygen atoms of Asp564 form tightly coupled hydrogen bonds with the Thr567, as represented by the dash line the labeled distance 2.34Åand 1.91Å. The replacement of Asp564 by a bulky Histidine residue would very likely introduce sterechemical clashes, thus affect zinc finger domain stability. (F) Brain MRI of the patient with OPTN p.E516Q mutation showed brain atrophy especially in bilateral frontal and temporal lobes. (G) PET of the patient with OPTN p.E516Q mutation revealed hypo‐metabolic changes in several brain regions, especially in bilateral frontal and temporal lobes.
Table 1.
Genetic profile of OPTN mutations identified in our study.
| Exon | Nucleoid changes |
Amino acid changes |
1000genome | ExAC | Evolutionary conservation | SIFT | Polyphen2 | Mutation Taster | ACMG |
|---|---|---|---|---|---|---|---|---|---|
| 14 | c.1481T> G | p.L494W | 0 | 0.000008237 | Yes | Damage | Damage | Neutral | Likely pathogenic |
| 14 | c.1546G> C | p.E516Q | 0 | 0.00002472 | Yes | Tolerent | Damage | Damage | Likely pathogenic |
| 16 | c.1690G> C | p.D564H | 0 | 0.00003295 | Yes | Damage | Damage | Damage |
Variant of uncertain significance |
Phenotype of patients with OPTN mutations
The patient with OPTN p.L494W mutation was a 46‐year‐old man presented with left hand weakness. The symptom progressed gradually and he had some difficulties when lift heavy things. Eight months after symptom onset he began to develop weakness of right hand. Three months later the symptoms involve left lower limb. Neurological examination revealed obvious atrophy and fasciculations in both upper limbs. The deep tendon reflexes were decreased in upper limbs but hyperactive in lower limbs. Babinski sign was negative bilaterally. He scored 42/48 on ALS Functional Rating Scale Revised (ALSFRS‐R) and 28/30 on Montreal Cognitive Assessment testing. Electromyography demonstrated acute and chronic neurogenic changes in all limbs, and thoracic and sternocleidomastoid muscles. Nineteen months after symptoms onset his left upper limb was totally paralyzed, but he still could lift light things with right hand and walked slowly independently. No bulbar symptoms were observed.
The OPTN p.E516Q mutation carrier was a woman presented with dysarthria at the age of 38, followed within two months by language impairment, such as slowed word‐finding, repeating the same sentences. Emotional incontinence with exaggerated crying was also seen. Ten months after onset she began to exhibit drinking bucking and dysphagia, she could hardly speak due to severe dysarthria. Memory impairment was also noticed. One month later she developed weakness of upper limbs and had difficulties in lifting things and combing. Neurologic examination at one year after onset revealed obvious atrophy and fasciculations in tongue and upper limbs. The deep tendon reflexes were hyperactive in all limbs with bilateral positive palmomental reflex and Babinski sign. Edinburgh Cognitive and Behavioral ALS Screen Chinese version6 identified cognitive impairment in verbal fluency, language, and executive functioning, and memory, with a score of 75/136. ALSFRS‐R at initial assessment was 30. Electromyography demonstrated acute denervation and chronic reinnervation in upper limbs, thoracic muscles, and sternocleidomastoid muscles. Brain MRI showed brain atrophy especially in bilateral frontal and temporal lobes (Fig. 1F). PET revealed hypo‐metabolic changes in several brain regions, especially in bilateral frontal and temporal lobes (Fig. 1G). The patient developed weakness of lower limbs 14 months after onset. Two month later she could hardly eat and had obvious weight loss. She developed aspiration pneumonia and died 18 months after onset due to respiratory failure. Both her parents were still alive and had no symptoms of muscle weakness, dementia, or eye pain.
The patient carrying the OPTN p.D564H mutation was a 54‐year‐old male with a history of diabetes who presented with weakness of left lower limb. The symptoms spread quickly to involve right lower limb. He felt unsteady when walking and fell down several times. Two months after onset he began to develop dysarthria. One month later the weakness progressed to upper limbs. He had some difficulties in lift things. On neurologic examination, obvious atrophy and fasciculations were observed in tongue and all limbs, with split hand sign. The deep tendon reflexes were hyperactive in all limbs. The palmomental reflex and Babinski sign were positive bilaterally. The sensory was normal. Electromyography demonstrated acute denervation in all limbs and thoracic muscles, as well as chronic reinnervation in all limbs and sternocleidomastoid muscles. ALSFRS‐R at initial assessment was 37 with an estimated progression rate since symptom onset of 3.6 points/month. Scores of Montreal Cognitive Assessment testing was 27/30. He became bedridden 8 months after onset. He could neither speak nor eat 10 months after onset and resulted in a weight loss of more than 10Kg. Therefore, he began nasogastric tube feeding. The patient died 14 months after symptom onset due to respiratory failure. His father died of stroke. His mother and his brother and sister were alive and had no history of dementia or glaucoma.
Discussion
In this study, we screened for OPTN mutations by targeted NGS in a Chinese cohort comprising 15 unrelated FALS probands and 275 SALS patients. Two known heterozygous missense mutations, c.1481T>G (p.L494W) and c.1546G>C (p.E516Q), as well as one novel heterozygous missense variant, c.1690G>C (p.D564H), were each identified in one SALS patient. No OPTN mutations were identified in the FALS indexes.
The pathogenicity of p.D564H variant is supported by several lines of evidences. The mutation is absent from public population database gnomAD, Chinese Millionome Database, and have a very low frequency (<0.0001) in ExAC, and absent from inhouse healthy controls. The mutant position is highly conserved across species, implicating functional importance of Asp564. Furthermore, different in silico prediction programs demonstrate the variant to be damage. Structure analysis show that replacement of Asp564 by a bulky Histidine residue would very likely introduce sterechemical clashes, thus affect zinc finger domain stability (Fig. 1E).
OPTN mutations were first linked to ALS in Japanese population, accounting for 3.3% of FALS and 0.2% of SALS patients.3 Subsequent studies showed that the frequency of OPTN mutations in Caucasian populations ranged from 0–4.6% in FALS patients and 0–2.7% in SALS cases. Taking together, the frequency of OPTN mutations is 0.5% (15/2,789) in FALS and 0.4% (25/7,018) in SALS patients in Caucasian populations (Table S1). Soong et al detected one OPTN mutation in one out of 131 Chinese SALS patients while no mutations were found in 30 FALS probands, the frequency of OPTN mutations in SALS was 0.8% in the study.7 Li et al identified four OPTN mutations in four out of 511 Chinese SALS patients. Liu et al screened for OPTN mutations in a cohort of Chinese ALS patients and the frequency was 5.0% (1/20) in FALS and 1.3% (3/234) in SALS patients.8 Recently Zhang et al detected one OPTN mutation in one out of 311 Chinese SALS cases, leading a frequency of 0.3% (1/311).9 The frequency of OPTN mutations in SALS is 1.1% (3/275) in our study. Taking together, the frequency of OPTN mutations is 0.8% (12/1,462) in SALS and 1.5% (1/65) in FALS patients of Chinese origin. Our review showed that the frequency of OPTN mutations is 2.5% (8/320) in FALS and 0.7% (25/3,645) in SALS patients in Asian populations. Therefore, optineurin mutations seem to be more frequent in Japanese and Chinese ALS patients than in Caucasian ALS cases.
Totally 61 mutations in OPTN have been reported to date, including 36 missense mutations, three delete mutations, eight nonsense mutations, nine frameshift mutations, and five splicing mutations (Fig. 2). Most mutations are heterozygous except seven homozygous mutation p.D127Rfs*21,10 p.G291fs*, p.K359fs*,11 p.E135X,12 p.S174X,13 p.Q398X, and Ex5del.3 There are four compound heterozygous mutation, p.D220Mfs*12 and p.Q165X,14 p.L430Rfs*16 and p.S262X.15 Twenty‐eight OPTN mutation carriers had a definite family history of ALS/FTD. Nineteen families demonstrate an AD hereditary and nine families had an AR hereditary pattern (Table S2). Optineurin consists of five domains: two coiled coil domains, LC3‐interacting region, ubiquitin‐binding domain, and the zinc finger domain. The p.L494W mutation lies in C‐terminal coiled coil domain, where more than half of the ALS related OPTN mutations (31 mutations) are localized. The p.D564H mutation lies in C‐terminal zinc finger domain. To date, only three OPTN mutations have been found in the zinc finger domain.8, 16, 17
Figure 2.
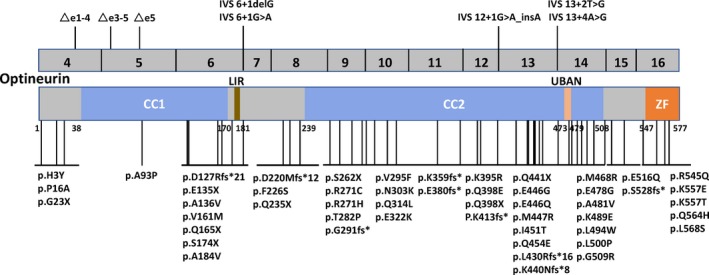
Schematic graph of the optineurin protein and overview of the OPTN mutations linked to ALS. CC, coiled coil domain; LIR, LC3‐interacting region; UBAD, ubiquitin‐binding domain; ZF, zinc finger domain.
OPTN p.L494W mutation has been reported in one SALS by Soong et al. The patient demonstrated the phenotype of flail arm syndrome at age 48 and had no bulbar involvement 9 years after symptoms onset.7 The p.L494W mutation carrier in our study presented with left hand weakness at age 46 and showed the phenotype of classic ALS. OPTN p.E516Q mutation has been detected in one SALS presented with weakness of upper limbs at age 48 and died of respiratory failure 14 months following disease onset.9 The p.E516Q mutation carrier in our study was a 38‐year‐old female who presented with dysarthria and demonstrated typical clinical, electromyographic and imaging features of ALS‐FTD and an aggressive progression, with a survival of 18 months. In accordance with our result, three familial ALS cases with OPTN mutation demonstrated a phenotype of ALS‐FTD have been reported (Table S2),15, 18, 19 with a survival of 18–22 months. In addition, four patients who had a familial history and carried OPTN mutations initially presented with features of FTD and then progressed to ALS have also been reported, with a survival between 16–54 months.10, 18 The patient with OPTN p.D564H mutation in our study developed classic ALS phenotype and an aggressive course at age 54 with a survival of only 14 months. Interestingly, an Italian FALS patient carrying p.K557T mutation in the zinc finger domain of optineurin also demonstrated severe phenotype of ALS and survived only 9 months.16
The clinical characters of ALS cases with OPTN mutations reported to date were summarized in Table S2. Fifty‐one OPTN mutated ALS patients with detailed clinical features were identified, including 34 men and 17 women (male:female = 2:1). The age of onset was 51.9 years (range 23–83 years). Nine patients had bulbar symptoms at onset, 38 cases had spinal onset, and four patients presented with cognitive impairment. The clinical phenotypes in OPTN mutated ALS were not homogeneous, although some individuals showed a relatively slow progression and a long duration7, 16, 20, 21 as reported by Maruyama et al,3 some mutations carriers developed an aggressive progression and a short survival.3, 8, 9, 10, 14, 15, 16, 18, 19, 22, 23 Interestingly, among the OPTN mutant ALS cases with an aggressive course, one harbored OPTN p.A136V and SOD1 p.S69P mutation,8 two harbored OPTN p.E468R mutation and C9orf72 expansions, the other one carried OPTN p.E468R mutation, C9orf72 and AXTN expansions,18 suggesting variability in phenotype of OPTN mutant ALS may partly be due to the oligogenic basis of ALS.2 Both the patient carrying OPTN p.E516Q mutation and the patient with p.D564H mutation had a rapid progression. However, further sequencing did not detect mutations in other known ALS related genes including C9orf72.
ALS‐associated OPTN mutations include deletions, missense, frameshift, and nonsense mutations (Fig. 2). Functional studies identified three major neuroprotective mechanisms of optineurin: regulation of autophagy, mitigation of inflammatory signaling, and blockade of necroptosis.24 Deletions, nonsense and frameshift mutations in OPTN were found in either homozygous or heterozygous state, suggesting that complete loss of function or haploinsufficiency of optineurin is enough to cause or contribute to ALS.25 Most ALS‐linked missense optineurin mutations are disproportionally enriched in the C‐terminus coiled coil domain, ubiquitin‐binding domain, and zinc finger domain. Indeed, the strongest evidences for pathogenicity exists for the heterozygous missense mutation p.E478G in the ubiquitin‐binding domain of optineurin, suggesting that it causes disease by gain‐of‐function or haploinsufficiency.25 The aggressive course of the patients carrying OPTN p.E516Q mutation and p.D564H mutation in our study suggest severe functional impairment of optineurin for the two mutations. However, further function studies are needed to explicit the mechanism in neurodegeneration of OPTN p.E516Q and p.D564H mutation.
In conclusions, mutations in OPTN contribute to ALS in Chinese population and account for about 0.8% of SALS and 1.5% of FALS patients in the pooled Chinese ALS cohorts. Mutations in optineurin can cause aggressive ALS +/− FTD.
Author Contributions
Z‐Y. Z, S‐M. F, and H‐P. H were involved in conception and design of the study, acquisition and analysis of data and manuscript drafting and completion. All remaining authors were involved in acquisition and analysis of data and manuscript review.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Table S1. Summary of genetic studies screening for OPTN mutations.
Table S2. Summary of clinical features of ALS patients with OPTN mutations.
Acknowledgments
The authors thank the patients and their families for their cooperation in this study, Dr. Victor W. Zhang from Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, and AmCare Genomics Lab , Guangzhou, China, for kind helps in structure analysis of the variant, as well as the anonymous reviewers for their helpful suggestions on the quality improvement of our report. This study was supported by grants from the National Natural Science Foundation of China [grant number 81671271], Joint Funds for the innovation of science and Technology, Fujian province [grant number 2017Y91010015] and Program for New Century Excellent Talents in Fujian Province University [grant number 2017B014].
Funding Information
This study was supported by grants from the National Natural Science Foundation of China [grant number 81671271], Joint Funds for the innovation of science and Technology, Fujian province [grant number 2017Y91010015] and Program for New Century Excellent Talents in Fujian Province University [grant number 2017B014].
Funding Statement
This work was funded by National Natural Science Foundation of China grant 81671271; Program for New Century Excellent Talents in Fujian Province University grant 2017B014; Joint Funds for the innovation of science and Technology, Fujian province grant 2017Y91010015.
Contributor Information
Hua‐Pin Huang, Email: hh-p@163.com.
Zhang‐Yu Zou, Email: fmuzzy@fjmu.edu.cn.
References
- 1. Brown RH, Al‐Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med 2017;377:162–172. [DOI] [PubMed] [Google Scholar]
- 2. Volk AE, Weishaupt JH, Andersen PM, et al. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet 2018;30:252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010;465:223–226. [DOI] [PubMed] [Google Scholar]
- 4. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 5. Ludolph A, Drory V, Hardiman O, et al. A revision of the El Escorial criteria ‐ 2015. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:291–292. [DOI] [PubMed] [Google Scholar]
- 6. Ye S, Ji Y, Li C, et al. The edinburgh cognitive and behavioural ALS screen in a chinese amyotrophic lateral sclerosis population. PLoS ONE 2016;11:e0155496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soong BW, Lin KP, Guo YC, et al. Extensive molecular genetic survey of Taiwanese patients with amyotrophic lateral sclerosis. Neurobiol Aging 2014;35:e2421–2426. [DOI] [PubMed] [Google Scholar]
- 8. Liu Q, Liu F, Cui B, et al. Mutation spectrum of Chinese patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2016;87:1272–1274. [DOI] [PubMed] [Google Scholar]
- 9. Li C, Ji Y, Tang L, et al. Optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in China. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:485–489. [DOI] [PubMed] [Google Scholar]
- 10. Goldstein O, Nayshool O, Nefussy B, et al. OPTN 691_692insAG is a founder mutation causing recessive ALS and increased risk in heterozygotes. Neurology 2016;86:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ozoguz A, Uyan O, Birdal G, et al. The distinct genetic pattern of ALS in Turkey and novel mutations. Neurobiol Aging 2015;36:e1769–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muller K, Brenner D, Weydt P, et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry 2018;89:817–827. [DOI] [PubMed] [Google Scholar]
- 13. Gotkine M, De Majo M, Wong CH, et al. A novel optineurin truncation mutation identified in a consanguineous Palestinian family with Amyotrophic Lateral Sclerosis confirms loss of function as a disease mechanism. F1000Research 2016;5:2754(poster). [Google Scholar]
- 14. Beeldman E, van der Kooi AJ, de Visser M, et al. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:410–411. [DOI] [PubMed] [Google Scholar]
- 15. Pottier C, Rampersaud E, Baker M, et al. Identification of compound heterozygous variants in OPTN in an ALS‐FTD patient from the CReATe consortium: a case report. Amyotroph Lateral Scler Frontotemporal Degener 2018;19:469–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Del Bo R, Tiloca C, Pensato V, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2011;82:1239–1243. [DOI] [PubMed] [Google Scholar]
- 17. Lattante S, Conte A, Zollino M, et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012;79:66–72. [DOI] [PubMed] [Google Scholar]
- 18. Farhan SMK, Gendron TF, Petrucelli LOPTN, et al. p.Met468Arg and ATXN2 intermediate length polyQ extension in families with C9orf72 mediated amyotrophic lateral sclerosis and frontotemporal dementia. Am J Med Genet B Neuropsychiatr Genet 2018;177:75–85. [DOI] [PubMed] [Google Scholar]
- 19. Nishiyama A, Niihori T, Warita H, et al. Comprehensive targeted next‐generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol Aging 2017;53:194.e1‐194.e8. [DOI] [PubMed] [Google Scholar]
- 20. Naruse H, Takahashi Y, Kihira T, et al. Mutational analysis of familial and sporadic amyotrophic lateral sclerosis with OPTN mutations in Japanese population. Amyotroph Lateral Scler 2012;13:562–566. [DOI] [PubMed] [Google Scholar]
- 21. Tumer Z, Bertelsen B, Gredal O, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging 2012;33:e201–205. [DOI] [PubMed] [Google Scholar]
- 22. van Blitterswijk M, van Vught PW, van Es MA, et al. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging 2012;33:e1011–1017. [DOI] [PubMed] [Google Scholar]
- 23. Weishaupt JH, Waibel S, Birve A, et al. A novel optineurin truncating mutation and three glaucoma‐associated missense variants in patients with familial amyotrophic lateral sclerosis in Germany. Neurobiol Aging 2013;34:e1519–1515. [DOI] [PubMed] [Google Scholar]
- 24. Bansal M, Swarup G, Balasubramanian D. Functional analysis of optineurin and some of its disease‐associated mutants. IUBMB Life 2015;67:120–128. [DOI] [PubMed] [Google Scholar]
- 25. Markovinovic A, Cimbro R, Ljutic T, et al. Optineurin in amyotrophic lateral sclerosis: Multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurogibol 2017;154:1–20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of genetic studies screening for OPTN mutations.
Table S2. Summary of clinical features of ALS patients with OPTN mutations.