

Abstract
The number and spacing of B-cell epitopes on antigens have a profound impact on the activation of B cells and elicitation of antibody responses, the quantitative aspects of which may be utilized for rational design of vaccines. Ni-chelating liposomes have been widely used as protein carriers in experimental studies of vaccine delivery, owing to the convenience and versatility of this conjugation chemistry. However, the epitope number per particle as well as the stability of protein conjugation on liposomes remain far less characterized. Here we have developed quantitative methods to measure the average spatial density of proteins on liposomes using both ensemble and single-molecule techniques and demonstrated their utility using liposomes conjugated with native proteins of two different sizes. These studies revealed that the initial density of protein conjugation on Ni-chelating liposomes can be finely controlled, but the density can decrease over time upon dilution due to the noncovalent nature of Ni-chelation chemistry. These results indicate that an alternative method other than the Ni-chelation chemistry is needed for stable conjugation of epitopes onto liposomes and also suggest a general strategy that can be used to precisely regulate the epitope density on liposomes for B-cell antigen delivery.
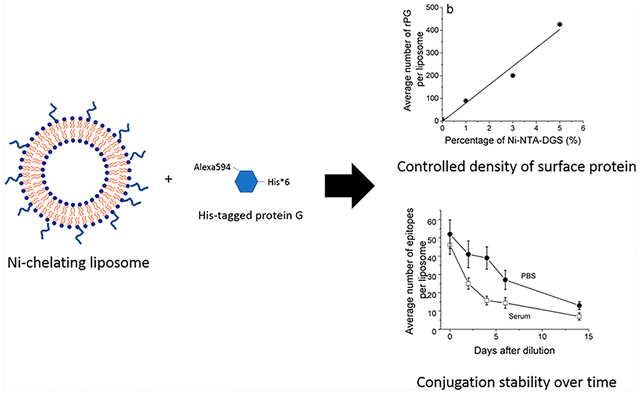
Graphical Abstract
INTRODUCTION
Vaccination is a concept that can be broadly applied to the prevention and treatment of human diseases, including but not limited to infectious diseases, cancer, and neurodegenerative diseases. Currently, there are more than 90 vaccine products that are licensed for immunization and distribution in the US. The mechanisms of protection in the majority of these licensed vaccines are correlated with antibodies.1 As a result, how to optimally activate antigen-specific B cells for the production of memory B cells is likely to be the key to the success of these vaccines.
There are extensive and well controlled studies in the literature that document the impact of epitope density on B-cell responses. Quantitative differences in epitope density can easily produce quantitative or qualitative differences in antibody responses in mouse models of immunization.2–5 Indeed, the variation of the spatial density of a protein on a particulate antigen, e.g., vaccine candidate, has a direct impact on the antibody response in animals.6,7 These studies revealed that the spatial density of epitopes on particulate antigens is a critical feature that one needs to consider for the delivery of B-cell based vaccines. However, application of this fundamental principle to vaccine delivery remains a challenge for two reasons: (i) a robust antigen-delivery system is yet to be developed that allows the delivery of antigens, especially protein antigens with well-defined spatial densities, and (ii) the techniques that are necessary to precisely quantitate the spatial density of epitopes on particulate antigens continue to lag behind.
Varieties of nanomaterials have been developed over the past decade for experimental delivery of various immunogens.8,9 A simple yet versatile approach is the protein-conjugated liposomes. Liposomes are well documented drug delivery systems with a safe record of clinical applications.10,11 Liposome and liposome-derived nanovesicles have become important platforms for carrying antigens and immune-stimulatory molecules for vaccine development.12 A variety of different parameters including the lipid composition, size, charge, and antigen conjugation can be manipulated for various applications.13 At least two lipid-related vaccine products have been approved, Inflexal V14 and Epaxal,15 targeting influenza and hepatitis A, respectively; and more liposome-based vaccines are at different stages of clinical trials.
A potential advantage of liposomes for vaccine delivery is that a purified protein can be attached to the particle surface through chemical conjugation, during which the density of the protein averaged over a single particle may be controlled. This strategy, if it works, may allow a fine control over the spatial density of epitopes per particle to optimize the B-cell antibody response. For anchoring epitopes of interest on the surface of liposomes, a variety of conjugation methods have been developed.16 Among them, Ni-NTA-his-tag conjugation has been a widely accepted choice due to the convenience of its operation. Chelator-lipids were first synthesized and characterized for immobilization of engineered proteins at a self-assembled lipid interface in 1994.17 Since then, many groups have used liposomes containing metal-ion-chelating lipid to attach a histidine-tagged protein of interest, which led to an elevated level of immune response compared to traditional forms of antigen.18–21 However, the spatial density of the proteins attached on these liposomes was often not reported in these studies.
In this paper, we explore the feasibility to finely control the surface density of epitopes attached to unilamellar liposomes, and develop quantitative methods to characterize the spatial density of epitopes averaged over a single particle. It is worth noting that proteins attached onto the outer surface of liposomes, either covalently or noncovalently, may still be capable of two-dimensional diffusion on the liposome surface, which may allow for the formation of protein clusters with high local spatial density. Whether this phenomenon may occur during B-cell recognition of the particulate antigens is a subject of future research. Specifically, herein, the term “spatial density” is referred to as the spatial density of proteins that is averaged over a single particle. Our results revealed that the initial density of proteins conjugated on liposomes via Ni-chelation chemistry can be finely controlled, but the density decreases over time upon dilution. These results thus indicate that Ni-chelation chemistry is not stable for conjugation of B-cell epitopes onto liposomes and also suggest a strategy to regulate the average spatial density of proteins on liposomes for optimization of antibody responses.
RESULTS
Characterization of the Spatial Density of rPG on Liposomal Surface.
To facilitate the quantitation of the spatial density of proteins conjugated onto liposomes, we have chosen to use a fluorescent labeled B1 domain of the streptococcal protein G, herein referred to as rPG, for attachment onto liposomes via the Ni-NTA coordination chemistry. The single and site-specific labeling of Alexa-594 fluorophore on this protein allows us to quantitate the spatial density of protein attached on liposomes using both ensemble and single-molecule fluorescence approaches. We prepared liposomes by the lipid thin film hydration method, followed by membrane extrusion.22–24 For conjugation of proteins onto the liposome surface, a minor percentage of Ni-NTA-DGS lipid was doped into DMPC during liposome preparation. Extrusion through the polycarbonate membrane of 100 nm pore size allowed liposome formation with a relatively uniform size. As shown in Table S1, these synthesized Ni-chelating liposomes had an average diameter of 97 ± 1 nm (mean ± standard error) with a mean polydispersity index of 0.04 ± 0.02 (mean ± standard error). The average diameter of these liposomes does not change significantly with the increase in the molar percentage of Ni-NTA-DGS lipids in the range of 0–20% that we have investigated.
To quantitate the spatial density of rPG on liposomes, we started out by focusing on liposomes that carried 1% of Ni-NTA-DGS. After liposome synthesis, a designated amount of rPG was incubated with liposomes for 1 h at 37 °C, followed by purification through gel filtration. This experiment was then repeated using the same amount of liposomes (0.44 μmol DMPC) but with increasing concentrations of rPG. As shown in Table S2, these liposomes after rPG association had a measurable increase in their diameters compared to that of the liposome alone before rPG binding, consistent with protein binding on the surface of the liposomes. To determine the average number of rPG molecules per liposome, we first used a fluorescence plate reader to measure the fluorescence intensity from the purified liposome samples. Comparison with a standard curve that was constructed using free rPG protein at various concentrations in PBS buffer under the same instrument conditions (Figure S1) allowed us to estimate the concentration of rPG in liposome samples. Control experiments showed that incubation of liposomes with rPG did not cause any change in the fluorescence signal for Alexa-594, suggesting that the quantum yield of Alexa-594 was not significantly perturbed by rPG attachment onto liposomes. Based on the molarity of liposomes calculated using eq 1, we can thus calculate the average number of rPG molecules per liposome. This result is shown in Figure 1a. As the quantity of rPG increased from 2.5 to 80 μg, the average number of rPG per liposome also increased gradually and showed a trend to reaching a plateau, resembling a typical binding isotherm. It is worth noting that at 80 μg rPG, the molar ratio between Ni and rPG is 1:1.6, suggesting that the trend of the approaching plateau is a result of saturation of all the available Ni-NTA binding sites on liposomes, instead of surface crowding, i.e., saturation as a result of the crowding of proteins on liposome surface.
Figure 1.

Characterization of rPG-conjugated liposomes using the ensemble approach. (a) Average number of rPG per liposome as a function of the initial input quantity of rPG during incubation with liposomes that contain 1% Ni-NTA-DGS. The error bars are standard deviations from three independent repeats of the same experiment. The solid line serves as a guide for the eye only. (b) Average number of rPG per liposome as a function of the percentage of the Ni-NTADGS incorporated into the liposomes (a representative of two). The solid line is a result of linear regression for the experimental data points.
To provide further proof for this point, we have thus compared liposomes containing 1%, 3%, and 5% Ni-NTA-DGS (all in molar percentage) for their protein surface densities, as shown in Figure 1b. These liposomes were prepared by incubating the same moles of liposomes (0.44 μmol DMPC for liposomes with 1% Ni-NTA-DGS) with 30 μg rPG, followed by gel filtration. The average number of rPG molecules per liposome increased in a linear fashion with the percentage of Ni-NTA-DGS in the liposomes, as indicated by the solid line from a linear regression analysis. The average number of rPG per liposome observed from liposomes containing 5% Ni-NTA is 3-fold higher than the plateau in Figure 1a, which clearly demonstrates that the trend of saturation observed in Figure 1a is not due to surface crowding. Rather, it is due to the limited number of Ni-NTA sites per liposome. These results also suggest that one potential strategy to regulate the spatial densities of proteins on these liposome surface is to adjust the percentage of Ni-NTA-DGS lipids incorporated into liposomes.
The above approach to estimate the average number of proteins on liposomes is an ensemble-based measurement, where we assume each liposome has an average number of lipid molecules, based on which to calculate liposome molarity and thus the number of rPG molecules per liposome. Moreover, for the above approach to work, the fluorescence intensity of Alexa-594 fluorophore should not change significantly upon conjugation onto liposomes through protein G, i.e., rPG free in solution and rPG conjugated on the surface of a liposome should have similar fluorescence intensity; otherwise, any change in fluorophore quantum yield has to be taken into account when doing estimations using this approach, which is not trivial. To provide an alternative and yet direct measurement for the spatial densities of rPG on individual liposomes, we have resorted to single-molecule fluorescence approach.
As shown in Figure 2a, we deposited purified rPG-conjugated liposomes on poly(L-lysine) coated coverslips, and use epifluorescence imaging to directly visualize individual liposomes through excitation of the Alexa-594 fluorophore by a 592 nm laser. Individual liposomes showed up in a dark background as bright spots. These liposomes were prepared by incubating 2.5 μg rPG with liposomes containing 1% Ni-NTA-DGS (0.44 μmol DMPC), followed by gel filtration. Neither liposome samples alone nor free rPG alone at the same concentrations yielded these bright spots under the microscope. Moreover, no fluorescent spots were visible when rPG was incubated with liposomes without Ni-NTA-DGS, suggesting that these fluorescent spots were liposomes conjugated with rPG through Ni-NTA but not protein aggregates. In order to determine the number of rPG molecules per liposome, we selected to work at a laser power of 100 mW. This illumination condition allowed us to collect the initial fluorescence intensity from individual spots, and the constant illumination of the sample under this condition also allowed us to observe the photobleaching of individual fluorophores with time. As shown in Figure 2b, these photobleaching events display a hallmark feature of “steps”, where the fluorescence persists for a finite time followed by a sudden decrease in fluorescence intensity. These steps correspond to the photobleaching of individual molecules. We have used a step-detection algorithm that we developed previously25 to identify steps from these real-time fluorescence traces. Measurement of this “step” size yields the fluorescence intensity of a single Alexa-594 fluorophore, which can be used as an internal reference to convert the initial fluorescence intensity of individual liposomes to the number of Alexa-594 fluorophores, as we have done recently for individual HIV-1 virions using the same quantitation methodology.26 As shown in Figure 2c, the histogram of the individual photobleaching step sizes for Alexa-594 molecules (N = 163) could be well described by the sum of two Gaussians (black curve), one centered at 649 ± 136 analog-to-digital units (a.u., mean ± standard deviation) and the other centered at 1164 ± 235 a.u. Under a statistical significance level of 5%, Pearson’s Chi-square test27 selected the double-Gaussian distribution (P-value = 0.84) and rejected the single-Gaussian distribution (P-value = 0.02) as the model to describe the data. The difference in peak values is close to 2-fold. As we have observed previously, the secondary peak may result from the photobleaching of two Alexa-594 molecules that occurred almost simultaneously, which could not be resolved by either the finite camera exposure time or the step-finding algorithm.28,29 The fluorescence intensity of a single Alexa-594 fluorophore can then be used to calculate the total number of Alexa-594 molecules per liposome based on a ratio comparison30,31 with the initial fluorescence intensity of Alexa-594 associated with individual liposomes. Figure 2d shows the distribution for the number of rPG molecules per liposome gathered from this analysis. It is very clear that the individual liposomes are heterogeneous in the number of rPG molecules per liposome, ranging from a value below 20 to a value above 100. Under a typical statistical significance level of 5%, Pearson’s Chi-square test27 selected a single-Gaussian distribution (P-value = 0.2249, shown in black curve) and strongly rejected Poisson distribution (P-value <0.0001) as the model to describe the data. The arithmetic mean value is 53, which is very close to the value of 57 ± 3 rPG molecules per liposome estimated using the ensemble approach as described above (Figure 1a). Therefore, the results from the single-molecule fluorescence measurements further support the use of the ensemble approach to estimate the average spatial density of proteins under our current experimental conditions.
Figure 2.

Characterization of rPG-conjugated liposomes using a single-molecule fluorescence approach. These liposomes were prepared by incubating 2.5 μg rPG with liposomes containing 1% Ni-NTA-DGS (0.44 μmol DMPC), followed by gel filtration. Immediately before imaging, the liposome sample was diluted 1000-fold in PBS and deposited onto a coverslip. (a) Snapshot of the epi-fluorescence image of rPG-conjugated liposomes deposited on poly(l-lysine) coated coverslip. The scale bar represents 10 μm. (b) Representative time trajectories of single-molecule photobleaching recorded from individual rPG-conjugated liposomes. (c) Histogram of the individual photobleaching step sizes for Alexa-594 molecules identified from the fluorescence time courses (b) using a step-finding algorithm (n = 163), which could be described by the sum of two Gaussians (black curve). (d) Histogram for the number of rPG molecules per liposome based on the initial fluorescence intensity of individual liposomes normalized by the average fluorescence intensity of a single Alexa-594 fluorophore (N = 45). The solid line is a fit to a Gaussian probability density function.
Characterization of the Average Spatial Density of mTFP on Liposomal Surface.
The B1 domain of the streptococcal protein G is a very small protein: even after the site-specific labeling of the Alexa-594 fluorophore, the molecular weight is still less than 9 kD. To test if the above results for protein G may also be relevant to larger protein antigens, we have overexpressed and purified a green fluorescent protein variant, mTFP,32 which carries a hexahistidine tag for attachment onto liposomes through Ni-NTA coordination chemistry. This protein adopts a “β-can”32 structure that is typical of green fluorescent proteins and the hexahistidine-tagged version of the protein has a molecular weight of 28 kDa.
We prepared liposomes containing varied percentages of Ni-NTA-DGS, and incubated the purified mTFP proteins with various liposomes followed by gel filtration to remove free proteins. To develop an approach that is more applicable to generic protein antigens, we also decided to use the silver staining method instead of fluorescence to quantitate the amount of proteins present in the purified liposome samples. This method entails a standard curve based on band intensities quantitated from a silver-stained polyacrylamide gel using the proteins of interest at various quantities. The linear dependence between the band intensity and the quantity of mTFP protein (Figure S2) allows us to estimate the amount of mTFP proteins present in the purified liposome samples, by running the purified liposome sample on the same gel.
As shown in Figure 3a, the average number of mTFP molecules per liposome determined in this way shows a linear dependence on the percentage of Ni-NTA-DGS contained in the liposomes, consistent with the trend observed for rPG on liposomes (Figure 1b). With 20% Ni-NTA-DGS, on average, over 600 molecules of mTFP can be conjugated per liposome. It is worth noting that for 20% Ni-NTA-DGS in a liposome of 100 nm diameter, theoretically there are more than 17 000 Ni-NTA groups per liposome available for binding with hexahistidine-tagged proteins. Therefore, 600 molecules may not have reached the limit that one can conjugate on individual liposomes. To obtain more insight on the capacity of individual liposomes for conjugation of mTFP, we have thus decided to carry out the following experiment: we prepared liposomes containing 20% Ni-NTA-DGS lipids, and then incubated these liposomes with increasing quantities of mTFP proteins. After gel filtration to remove excess free proteins, we carried out three separate measurements on these purified liposomes: (i) run polyacrylamide gel to quantitate the amount of mTFP proteins based on silver staining (Figure S2); and (ii) using Stewart assay to determine phospholipid content and thus derive the moles of liposome based on the average diameter measured from dynamic light scattering; and (iii) using inductively coupled plasma optical emission spectrometry (ICP-OES) to directly measure Ni content for each purified liposome sample in order to verify the amount of Ni-NTA-DGS.
Figure 3.

Characterization of mTFP-conjugated liposomes. (a) Average number of mTFP per liposome as a function of the percentage of the Ni-NTA-DGS incorporated into the liposomes. The various liposomes were prepared by incubation of the same amount of moles of liposomes (0.07 μmol DMPC for liposomes with 5% Ni-NTA-DGS) with 40 μg of mTFP, followed by gel filtration to remove free mTFP proteins. (b) Left y-axis: the average number of mTFP molecules per liposome as a function of the initial mTFP input during incubation with liposomes containing 20% Ni-NTA-DGS. The initial amounts of liposomes were all identical among different samples (0.18 μmol DMPC). Right y-axis: the percentage of Ni-NTA-DGS incorporated into liposomes measured by ICP-OES. The error bars are standard deviations from three independent repeats of the same experiment.
As shown in Figure 3b, the amount of mTFP conjugated on liposomes increased with increasing quantities of mTFP initially incubated with liposomes (filled circles), showing a trend of reaching a plateau. Meanwhile, the percentage of Ni-NTA-DGS that we experimentally measured for these purified samples using ICP-OES (open circles) yielded an excellent agreement with theoretical expectations, with a mean and standard deviation of 20.3 ± 0.7% among these samples. It is worth noting that under these conditions, even for the highest quantities of mTFP we tried, the amount of Ni-NTA was still in excess, with a molar ratio of 3:1 between Ni-NTA-DGS and the mTFP protein. This result thus indicates that there were more Ni-NTA binding sites on liposomes than the amount of free mTFP available, and this trend of plateau is unlikely to be a result of limiting the Ni-NTA-DGS quantity in liposomes. More likely, it is due to the crowding of mTFP molecules on the liposomal surface. The radius of gyration for a globular protein of 28 kDa is close to 2 nm.33 For a liposome of 100 nm diameter, the upper limit on the number of mTFP molecules per liposome is thus estimated to be 2500 with the assumption that the entire surface of the liposome can be fully occupied by arrays of mTFP molecules without significant vacancies in between. This estimation thus supports our interpretation on the mechanism behind the trend of plateau observed in Figure 3b (filled circles).
To summarize, the above results with rPG and mTFP revealed two limiting conditions of protein conjugation on Ni-NTA liposomes: (i) when the percentage of Ni-NTA-DGS lipids is low, the average number of proteins conjugated per liposome will be limited by the percentage of Ni-NTA-DGS lipids, which we showed using a small protein rPG; (ii) when the percentage of Ni-NTA-DGS is high, the average number of proteins conjugated per liposome can also be limited by the crowding of proteins on liposomal surface, which we showed using mTFP. Overall, these results indicate that the epitope density on these liposomes can be quantitatively adjusted and controlled.
Stability of the Ni-NTA Noncovalent Conjugation Chemistry.
Although the use of Ni-NTA coordination chemistry is of great convenience considering the varieties of recombinant proteins that can be purified using hexahistidine technology, the noncovalent nature of this coordination chemistry may give rise to undesired drawbacks. The binding affinity between hexahistidine-tagged proteins and Ni-NTA-modified surface has been reported to be 0.1–1 μM.34,35 In particular, Groves and co-workers have shown that there is a great degree of heterogeneity in the Ni-NTA coordination complex formed between the hexahistidine tag and the nickel ions, which depends on the initial binding conditions and results in different off rates observed for proteins attached to supported lipid bilayer through this noncovalent chemistry and a changing protein density with time.36
To examine the stability of proteins conjugated onto liposome surface through this noncovalent chemistry, we have thus measured the dissociation of proteins from liposomes by direct observation using fluorescence microscopy. This experiment was done by using the rPG-conjugated liposomes, the same preparation of liposomes that we used in Figure 2 for quantitation of the number of rPG molecules per liposome using the single-molecule fluorescence approach. The starting sample has 53 rPG molecules per liposome on average (Figure 2c). To monitor the kinetics of dissociation, the liposome sample was diluted 1000-fold into PBS buffer on Day 0. The diluted sample was stored in the dark in a polypropylene test tube for a designated time at 22 °C and then aliquots were taken out for measurement of the protein density using the single-molecule fluorescence approach as illustrated above, and also the size using dynamic light scattering. Although the size of liposomes does not change over a two-week period (Figure S3), as shown in Figure 4, the average number of rPG molecules per liposome dropped with increasing days after the initial dilution (black circles). The average number of rPG per liposome decreased from 53 ± 4 (Mean ± standard error) on Day 0 to 27 ± 3 after a week, and 13 ± 1 after 2 weeks. In contrast, the liposome sample without dilution still had similar numbers of rPG molecules per liposome bound after 2 weeks (data not shown). To assay the stability of the liposomes in more physiologically relevant media, we also conducted the same experiments with every condition identical but in 100% fetal bovine serum (ThermoFisher CAT#10082), and used the same epi-fluorescence imaging approach to quantitate the number of fluorophores per liposome as a function of time. As shown in Figure 4, the average number of rPG molecules per liposome dropped with increasing days after the initial dilution (open squares). The average number of rPG per liposome decreased from 46 ± 3 (Mean ± standard error) on Day 0 to 14 ± 2 after a week, and 7 ± 1 after 2 weeks. Compared to PBS, this trend suggest that proteins conjugated on liposomes may undergo even faster dissociation upon entering biological milieu, and this instability of protein conjugation is an issue intrinsic to the Ni-NTA chelation chemistry. Qualitatively, this dissociation of proteins from liposomes upon dilution in serum is also consistent with Platt et al., who demonstrated that proteins bound on liposomes via Ni-chelation chemistry will dissociate rapidly in the presence of serum.37
Figure 4.

Kinetics of epitope dissociation from individual liposomes. The average number of rPG molecules per liposome measured from single-molecule fluorescence approach as a function of the days after the initial dilution of the liposome samples in either PBS (black circles) or 100% fetal bovine serum (open squares). For each data point, 45 individual liposomes were randomly selected from fluorescence images for each sample and analyzed for the average number of fluorescent epitopes per liposome. The error bars represent 1.96 standard error of the mean.
DISCUSSION
In this study, we have tested the feasibility to adjust the average number of protein molecules conjugated on liposomal surface and established quantitative methods to measure the average spatial density of proteins on the surface of liposomes after conjugation. Our method of ensemble estimation is supported by direct single-molecule measurement of protein densities on individual liposomes (Figure 2). Proximity of fluorophores is less of an issue for our current quantitation using fluorescence, because even for the highest spatial density we have in this case, 500 rPG per liposome (Figure 1b), the average distance between rPG molecules is estimated to be 8 nm, which is still large enough to prohibit quenching through energy transfer or complex formation. Based on our studies, when the percentage of Ni-NTA-DGS is low, the spatial density of a protein that one can attach onto the liposome surface is limited by the percentage of Ni-NTA-DGS under saturating protein conditions (Figure 1a). On the other hand, as the percentage of Ni-NTA-DGS increases, the spatial density of proteins will be eventually limited by the surface crowding effect (Figure 3b), i.e., the available liposomal surface will be fully occupied by the attached proteins. These results thus suggest that the spatial density of proteins on liposomes may be quantitatively controlled.
The results from rPG conjugation on the liposome surface suggest a potential strategy for regulating the spatial density of epitopes on liposomes as particulate antigens, which is an important parameter for B cell antibody response and vaccination.1 This strategy entails the following important steps: (i) purification of target proteins containing specific tags or active groups for chemical conjugation; (ii) liposome synthesis using a mixture of lipids, in which a small percentage of lipids carry specific and reactive head groups; (iii) after liposome synthesis, use excess protein for conjugation to the outer surface of the liposome via the specific conjugation chemistry; (iv) gel filtration to purify protein-conjugated liposomes away from excess proteins. Compared to other alternative procedures, this strategy has the following advantages: (i) conjugation of protein post-liposome synthesis helps to keep proteins in their native states, and avoid their exposure to organic solvents used for liposome synthesis; (ii) because of excess proteins used for conjugation, variation of the percentage of the specific lipid head-groups will thus allow the regulation of protein density. Systematic enumeration of the specific lipid percentage may allow formation of liposomes with surface epitopes at discrete densities, which would be very important tools for antibody-based vaccine development.
For the above strategy, there are currently at least two different methods available for epitope conjugation onto the outer surface of liposomes: Ni-chelation chemistry and Michael addition of a thiol group to a maleimide. As we showed for Ni-NTA coordination chemistry, this conjugation method is amenable to the fine control of epitope densities on the surface. However, due to its noncovalent nature, the spatial density of proteins is not stable but decreases upon dilution (Figure 4). This is not desired for the long-term storage of this formulation nor in vivo vaccination, because the time-dependent change of epitope density will confound the interpretation of the immunogenicity data based on liposome particles that use this noncovalent chemistry. Thus, the alternative approach such as maleimide–thiol reaction to conjugate epitopes onto the liposome surface is worth pursuing in the future, and the covalent chemistry may yield a spatial density that is stable over time. Moreover, from the perspective of future applications, the maleimide–thiol conjugation is less likely to be a regulatory concern because several FDA approved drug products contain maleimide–thiol conjugates.38–40 Also, this chemistry has been widely used in the conjugation of drugs to macromolecules. There is wide interest in the pharmaceutical and biotech industry to further optimize the chemistry of this conjugation to improve drug products.41,42 We thus expect synergistic efforts toward the use of this conjugation chemistry. In contrast, substantial studies in the literature have documented the adverse biological effects of nickel ions in vivo, which include tissue inflammation that is correlated with their distributions,43 allergy,44 toxicity, and carcinogenicity.45–47 Moreover, the hexahistidine tags might also be immunogenic in vivo, which may elicit antibodies targeting histidine repeats and thus represent an off-target effect, whose safety profile remains to be demonstrated. As a result, Ni-chelating liposomes may encounter significant safety challenges toward a final clinical product.
Based on the stability comparison for the protein conjugation (Figure 4), and the safety issues that we have outlined above, it is likely that the Michael addition of thiol to maleimide is a superior strategy than Ni-NTA coordination chemistry to conjugate epitopes onto liposomes for elicitation of antibody responses. Future experiments should explore maleimide–thiol reaction chemistry to conjugate epitopes at regulated spatial density on liposome surface, and test these liposomal particles in animals for optimal B-cell antibody response.
Finally, as we mentioned in the Introduction, our current quantitation of protein density is an average over a single particle. These proteins attached onto liposome surface may still undergo two-dimensional diffusion, which may allow formation of protein clusters and a high local surface density. Whether this may occur as a result of B cell recognition remains to be investigated in the future.
MATERIALS AND METHODS
Preparation of Ni-Chelating Liposomes.
All liposomes used in this study were prepared using oil-in-water micro-emulsion precursor followed by membrane extrusion as originally described in the literature.22–24 Two different lipids at designated molar ratios were used in the synthesis of liposomes: DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) and Ni-NTA-DGS (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl] (nickel salt)) (Avanti Lipids) following previous literature.20,21 Briefly, various amount of Ni-NTA-DGS lipid was added into 5 mg DMPC in a round-bottom bottle to reach the indicated molar percentage for the Ni-NTA-DGS lipids. The lipid mixture in chloroform was then vacuum desiccated to allow thin film formation at the bottom of the bottle. 1 mL PBS buffer was then added to the bottle to hydrate the lipid film through gentle rocking at 37 °C overnight. The next morning, the content was retrieved and extruded through polycarbonate membrane of 1000 and 100 nm sequentially, ten times each. The resulting liposomes were then stored at 4 °C for all experiments.
Purification of Hexahistidine-Tagged Proteins.
We have used two recombinant proteins of different sizes in this study: the B1 domain of streptococcal protein G and the monomeric teal fluorescent protein (mTFP).32 The B1 domain of protein G has a molecular weight of 7.7 kDa while the mTFP has a molecular weight of 28 kDa, which provides a reasonable size difference for testing conjugation onto liposomes. Both proteins carry a hexahistidine tag at the C-terminus of the respective proteins for attachment onto liposomes through Ni-NTA coordination chemistry. Both proteins were house expressed in E. coli BL21(DE3) pLysS strain and purified to greater than 95% purity using nickel-nitrilotriacetic acid (Ni-NTA) agarose technology, as judged by intensity comparisons on an acrylamide gel loaded with different amounts of final purified proteins. The native B1 domain of protein G does not carry any cysteine residues.48 To site-specifically label a single Alexa-594 fluorophore onto the protein for single-molecule studies, we have engineered a single cysteine at position 2, after the N-terminal methionine. We have developed a protocol to label the single cysteine on the B1 domain with high efficiency. Briefly, the purified B1 domain was treated with 5-fold molar excess of TCEP at 4 °C for 18 h. The excess TCEP was removed by passing the protein through a desalting column. The Alexa-594 maleimide (ThermoFisher) freshly dissolved in water was then added to the protein at a 10-fold molar excess. The cross-linking reaction was incubated at 20 °C for 8 h. The reaction was stopped by adding 2-mercaptoethanol to a final concentration of 20 mM. After additional 30 min incubation at 20 °C, the labeled protein was extensively dialyzed at 4 °C using a 3 kD molecular weight cutoff membrane against the storage buffer containing 50 mM HEPES, 150 mM NaCl, pH 7.0, at 22 °C in order to remove excess dye molecules. The concentration of the labeled protein was measured by absorbance at 280 nm (corrected for dye absorbance at this wavelength) using an extinction coefficient of 9530 M−1 cm−1, and the concentration of the conjugated Alexa-594 was quantified simultaneously by absorbance at 590 nm with an extinction coefficient of 9.00 × 104 M−1 cm−1. The efficiency of dye labeling was thus calculated to be 96%. The status of single fluorophore labeling was also confirmed by positive ion electrospray mass spectrometry. This fluorescently labeled B1 domain of the streptococcal protein G is referred to as rPG throughout this manuscript.
Attachment of Proteins to Liposomes and Size Exclusion Chromatography.
For attachment of purified proteins onto the liposome surface, the purified proteins were incubated with synthesized liposomes at 37 °C for 1 h at the indicated molar ratio between the proteins and the Ni-NTA group, which is sufficient to reach equilibrium as indicated by a comparison with the result from an overnight incubation at the same temperature. The mixture at 400 μL volume was then directly applied to the top of a 10 mL Sepharose CL-4B (GE Life Sciences) gel filtration column that was already equilibrated in PBS in order to remove excess free proteins not bound to liposomes. The column was run by gravity at 4 °C and again PBS was used as the elution buffer. Twenty fractions were collected after the sample application and each fraction contained 0.5 mL eluate. To identify fractions containing liposomes, dynamic light scattering measurement was conducted for samples from each fraction and the fractions with most of the liposomes were pooled and further stored at 4 °C. For each coupling of proteins to liposomes reported in this work, the coupling efficiencies of proteins were calculated as the ratio between the amount of proteins on liposomes after the size-exclusion column and the initial input protein and reported as percentages in Table S3. The yield of lipid recovery was also calculated as the ratio between the amount of lipids present in liposomes after the size-exclusion column and the initial input lipids and reported as percentages in Table S3.
Stewart Assay to Measure Lipid Content.
We have used the established Stewart assay49 to determine the phospholipid content in the purified liposomes, based on which the molarity of the liposomes can be further estimated. It is worth noting, however, that under Stewart assay conditions, Ni-NTA-DGS alone can also give rise to non-negligible absorbance at 470 nm that is in linear proportion to the molarity of this specific lipid. Thus, throughout our studies, the phospholipid content measured has been corrected based on the percentage of Ni-NTA-DGS in the liposome sample, although this correction was minor for most cases. Purified liposomes bound with fluorescence labeled B1 domain of the streptococcal protein G or mTFP were both examined using Stewart assay for phospholipid quantitation. Briefly, 100 μL liposome samples were added to 1 mL chloroform in a clean glass tube, and then 1 mL ferrothiocynate solution containing 0.1 M ferric chloride hexahydrate and 0.4 M ammonium thiocyanate was added. The mixture was vigorously vortexed for 1 min and further kept at 22 °C in the dark for at least 1 h or until its full separation into two layers. The lower aqueous layer was then taken for absorption measurement at 470 nm. The phospholipid content was then calculated based on the comparison with a standard curve of DMPC lipids measured under identical conditions.
Estimation of Liposome Molarity Based on Geometric Considerations.
To further estimate the molarity of the liposome based on the phospholipid content, we used geometric considerations of liposomes. Briefly, we calculated the number of lipid molecules N per liposome using the following equation:
| (1) |
where D is the diameter of the liposome, h is the thickness of the bilayer, and a is the area of the lipid molecule headgroup. In our calculation for liposomes prepared using DMPC as the major lipids, the bilayer thickness h was taken as 4.6 nm based on previous experimental measurement on DMPC bilayers.50 The area of the DMPC lipid headgroup was taken as 0.657 nm2 based on X-ray scattering measurement on DMPC lipid vesicles.51 The average diameters of the liposomes were measured using dynamic light scattering for each liposomal sample using Malvern Zetasizer Nano ZS at 20 °C. The molarity of the liposomes was then calculated based on the measured phospholipid content.
Fluorescence Plate Reader to Measure rPG Content in Liposomes.
To measure the rPG protein content in the purified liposome samples, we employed fluorescence plate reader (Synergy HT, Biotek). Briefly, the fluorescence of Alexa-594 was recorded using an excitation filter of 590 nm with 20 nm bandwidth, and an emission filter of 645 nm with 40 nm bandwidth. Comparison with a standard curve prepared and measured using known quantities of corresponding rPG free in PBS solution (0 to 3 ng/μL) yields an estimate of protein content in liposome samples. The corresponding liposome samples without proteins contributed negligibly to the fluorescence reading at the above settings.
Epifluorescence Imaging of Liposome Samples.
To use single-molecule fluorescence to quantitate the spatial density of rPG on individual liposomes, we have employed an epifluorescence imaging approach. Liposome samples after gel filtration were diluted 1000-fold and immediately applied onto poly(l-lysine) coated coverslips for epifluorescence imaging under an IX71 Olympus Advanced Research Fluorescence microscope. Alexa-594 was excited using a 592 nm fiber laser and its emission monitored using a 630 nm filter with 40 nm bandwidth. To record fluorescence with single-molecule sensitivity, a laser power of 100 mW, EM gain of 100, and an exposure time of 20 ms were used throughout so that the recorded fluorescence intensity can be directly compared across experiments. All the imaging experiments were conducted at 20 °C. The irradiance of the incident laser on the sample was estimated to be 87 mW/cm2 or less, and the heating of the sample by laser irradiation was negligible under these conditions.
Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES).
We have used ICP-OES (Optima 2000 DV, PerkinElmer) to quantitatively measure the amount of nickel ions in mTFP-conjugated liposomes collected through a gel filtration column. The atomic emission of Ni was detected at both wavelengths of 221 and 231 nm. To quantitate the amount of Ni in the liposome samples, a series of reference Ni samples from 100 to 1000 ppb were prepared and standard curves were constructed. The final concentration of Ni in each sample was determined based on the standard curve and the average values from both 221 and 231 nm wavelengths was used as the final concentration of the Ni. Throughout our studies, reference yttrium samples served as internal controls for the instrument by measuring their emissions at 371 nm.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the MCubed project #805 to WC and JM by the University of Michigan, the Upjohn Research Award to WC and the NIH Director’s New Innovator Award 1DP2OD008693-01 to WC. ZC was partially supported by a Summer Award from the Rackham Graduate School at the University of Michigan. We thank Dr. Robert E. Campbell for providing the E. coli plasmid encoding mTFP. The plasmid for the B1 domain of the streptococcal protein G was obtained from Dr. John Louis through Addgene. We thank Dr. KD Lee and Dr. Allison Matyas for assistance in size exclusion column chromatography. We thank Dr. Jin H. Kim for purification of mTFP and rPG protein. We thank Dr. Michael DeSantis and Richard Schutzman for assistance with epifluorescence microscopy and image analysis. We thank James Wang for help in preparation of protein conjugated liposomes and Cheng Lab members for helpful discussions.
ABBREVIATIONS
- mTFP
monomeric teal fluorescent protein
- rPG
recombinant protein G site-specifically labeled with Alexa-594
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- Ni-NTA-DGS
1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt)
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.8b00033.
Size and characterization of Ni-chelating liposomes; coupling efficiency of proteins onto liposomes together with the yield of lipid recovery; Standard curves for rPG and mTFP for quantitation of proteins on liposomes; Size of rPG-conjugated liposomes as a function of time after dilution (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Cheng W (2016) The Density Code for the Development of a Vaccine? J. Pharm. Sci 105 (11), 3223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Dintzis HM, Dintzis RZ, and Vogelstein B (1976) Molecular determinants of immunogenicity: the immunon model of immune response. Proc. Natl. Acad. Sci. U. S. A 73 (10), 3671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bachmann MF, Rohrer UH, Kundig TM, Burki K, Hengartner H, and Zinkernagel RM (1993) The influence of antigen organization on B cell responsiveness. Science 262 (5138), 1448–51. [DOI] [PubMed] [Google Scholar]
- (4).Chackerian B, Lowy DR, and Schiller JT (1999) Induction of autoantibodies to mouse CCR5 with recombinant papillomavirus particles. Proc. Natl. Acad. Sci. U. S. A 96 (5), 2373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Feldmann M, and Easten A (1971) The relationship between antigenic structure and the requirement for thymus-derived cells in the immune response. J. Exp. Med 134 (1), 103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hanson MC, Abraham W, Crespo MP, Chen SH, Liu H, Szeto GL, Kim M, Reinherz EL, and Irvine DJ (2015) Liposomal vaccines incorporating molecular adjuvants and intra-structural T-cell help promote the immunogenicity of HIV membrane-proximal external region peptides. Vaccine 33 (7), 861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ingale J, Stano A, Guenaga J, Sharma SK, Nemazee D, Zwick MB, and Wyatt RT (2016) High-Density Array of Well-Ordered HIV-1 Spikes on Synthetic Liposomal Nanoparticles Efficiently Activate B Cells. Cell Rep 15 (9), 1986–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Irvine DJ, Hanson MC, Rakhra K, and Tokatlian T (2015) Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev 115 (19), 11109–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hartwell BL, Antunez L, Sullivan BP, Thati S, Sestak JO, and Berkland C (2015) Multivalent nanomaterials: learning from vaccines and progressing to antigen-specific immunotherapies. J. Pharm. Sci 104 (2), 346–61. [DOI] [PubMed] [Google Scholar]
- (10).Allen TM, and Cullis PR (2013) Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Delivery Rev 65 (1), 36–48. [DOI] [PubMed] [Google Scholar]
- (11).Allen TM, and Cullis PR (2004) Drug delivery systems: entering the mainstream. Science 303 (5665), 1818–22. [DOI] [PubMed] [Google Scholar]
- (12).Watson DS, Endsley AN, and Huang L (2012) Design considerations for liposomal vaccines: influence of formulation parameters on antibody and cell-mediated immune responses to liposome associated antigens. Vaccine 30 (13), 2256–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schwendener RA (2014) Liposomes as vaccine delivery systems: a review of the recent advances. Ther. Adv. Vaccines 2 (6), 159–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Herzog C, Hartmann K, Kunzi V, Kursteiner O, Mischler R, Lazar H, and Gluck R (2009) Eleven years of Inflexal V-a virosomal adjuvanted influenza vaccine. Vaccine 27 (33), 4381–7. [DOI] [PubMed] [Google Scholar]
- (15).Bovier PA (2008) Epaxal: a virosomal vaccine to prevent hepatitis A infection. Expert Rev. Vaccines 7 (8), 1141–50. [DOI] [PubMed] [Google Scholar]
- (16).Marques-Gallego P, and de Kroon AI (2014) Ligation strategies for targeting liposomal nanocarriers. BioMed Res. Int 2014, 129458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schmitt L, Dietrich C, and Tampe R (1994) Synthesis and Characterization of Chelator-Lipids for Reversible Immobilization of Engineered Proteins at Self-Assembled Lipid Interfaces. J. Am. Chem. Soc 116 (19), 8485–8491. [Google Scholar]
- (18).van Broekhoven CL, Parish CR, Demangel C, Britton WJ, and Altin JG (2004) Targeting dendritic cells with antigen-containing liposomes: a highly effective procedure for induction of antitumor immunity and for tumor immunotherapy. Cancer Res. 64 (12), 4357–65. [DOI] [PubMed] [Google Scholar]
- (19).Patel JD, O’Carra R, Jones J, Woodward JG, and Mumper RJ (2007) Preparation and characterization of nickel nanoparticles for binding to his-tag proteins and antigens. Pharm. Res 24 (2), 343–52. [DOI] [PubMed] [Google Scholar]
- (20).Watson DS, Platt VM, Cao L, Venditto VJ, and Szoka FC Jr. (2011) Antibody response to polyhistidine-tagged peptide and protein antigens attached to liposomes via lipid-linked nitrilotriacetic acid in mice. Clin. Vaccine Immunol 18 (2), 289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pejawar-Gaddy S, Kovacs JM, Barouch DH, Chen B, and Irvine DJ (2014) Design of lipid nanocapsule delivery vehicles for multivalent display of recombinant Env trimers in HIV vaccination. Bioconjugate Chem 25 (8), 1470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Hope MJ, Bally MB, Webb G, and Cullis PR (1985) Production of large unilamellar vesicles by a rapid extrusion procedure: characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta, Biomembr 812 (1), 55–65. [DOI] [PubMed] [Google Scholar]
- (23).Mayer LD, Hope MJ, and Cullis PR (1986) Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta, Biomembr 858 (1), 161–8. [DOI] [PubMed] [Google Scholar]
- (24).Olson F, Hunt CA, Szoka FC, Vail WJ, and Papahadjopoulos D (1979) Preparation of liposomes of defined size distribution by extrusion through polycarbonate membranes. Biochim. Biophys. Acta, Biomembr 557 (1), 9–23. [DOI] [PubMed] [Google Scholar]
- (25).Arunajadai SG, and Cheng W (2013) Step detection in single-molecule real time trajectories embedded in correlated noise. PLoS One 8 (3), e59279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).DeSantis MC, Kim JH, Song H, Klasse PJ, and Cheng W (2016) Quantitative Correlation between Infectivity and Gp120 Density on HIV-1 Virions Revealed by Optical Trapping Virometry. J. Biol. Chem 291 (25), 13088–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Evans RD The Atomic Nucleus, McGraw-Hill, 1969, Chapter 27. [Google Scholar]
- (28).Pang Y, Song H, Kim JH, Hou X, and Cheng W (2014) Optical trapping of individual human immunodeficiency viruses in culture fluid reveals heterogeneity with single-molecule resolution. Nat. Nanotechnol 9 (8), 624–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hou X, and Cheng W (2011) Single-molecule detection using continuous wave excitation of two-photon fluorescence. Opt. Lett 36 (16), 3185–7. [DOI] [PubMed] [Google Scholar]
- (30).Coffman VC, and Wu JQ (2012) Counting protein molecules using quantitative fluorescence microscopy. Trends Biochem. Sci 37 (11), 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lawrimore J, Bloom KS, and Salmon ED (2011) Point centromeres contain more than a single centromere-specific Cse4 (CENP-A) nucleosome. J. Cell Biol 195 (4), 573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ai HW, Henderson JN, Remington SJ, and Campbell RE (2006) Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochem. J 400 (3), 531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Garcia De La Torre J, Huertas ML, and Carrasco B (2000) Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J 78 (2), 719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nieba L, Nieba-Axmann SE, Persson A, Hamalainen M, Edebratt F, Hansson A, Lidholm J, Magnusson K, Karlsson AF, and Pluckthun A (1997) BIACORE analysis of histidine-tagged proteins using a chelating NTA sensor chip. Anal. Biochem 252 (2), 217–28. [DOI] [PubMed] [Google Scholar]
- (35).Dorn IT, Neumaier KR, and Tampe R (1998) Molecular recognition of histidine-tagged molecules by metal-chelating lipids monitored by fluorescence energy transfer and correlation spectroscopy. J. Am. Chem. Soc 120 (12), 2753–2763. [Google Scholar]
- (36).Nye JA, and Groves JT (2008) Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir 24 (8), 4145–9. [DOI] [PubMed] [Google Scholar]
- (37).Platt V, Huang Z, Cao L, Tiffany M, Riviere K, and Szoka FC Jr. (2010) Influence of multivalent nitrilotriacetic acid lipidligand affinity on the circulation half-life in mice of a liposome-attached His6-protein. Bioconjugate Chem 21 (5), 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Senter PD, and Sievers EL (2012) The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol 30 (7), 631–7. [DOI] [PubMed] [Google Scholar]
- (39).Peddi PF, and Hurvitz SA (2013) Trastuzumab emtansine: the first targeted chemotherapy for treatment of breast cancer. Future Oncol. 9 (3), 319–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lang L (2008) FDA approves Cimzia to treat Crohn’s disease. Gastroenterology 134 (7), 1819. [DOI] [PubMed] [Google Scholar]
- (41).Fontaine SD, Reid R, Robinson L, Ashley GW, and Santi DV (2015) Long-term stabilization of maleimide-thiol conjugates. Bioconjugate Chem 26 (1), 145–52. [DOI] [PubMed] [Google Scholar]
- (42).Lyon RP, Setter JR, Bovee TD, Doronina SO, Hunter JH, Anderson ME, Balasubramanian CL, Duniho SM, Leiske CI, Li F, and Senter PD (2014) Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol 32 (10), 1059–62. [DOI] [PubMed] [Google Scholar]
- (43).Wataha JC, O’Dell NL, Singh BB, Ghazi M, Whitford GM, and Lockwood PE (2001) Relating nickel-induced tissue inflammation to nickel release in vivo. J. Biomed. Mater. Res 58 (5), 537–44. [DOI] [PubMed] [Google Scholar]
- (44).Saito M, Arakaki R, Yamada A, Tsunematsu T, Kudo Y, and Ishimaru N (2016) Molecular Mechanisms of Nickel Allergy. Int. J. Mol. Sci 17, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Magaye R, and Zhao J (2012) Recent progress in studies of metallic nickel and nickel-based nanoparticles’ genotoxicity and carcinogenicity. Environ. Toxicol. Pharmacol 34 (3), 644–50. [DOI] [PubMed] [Google Scholar]
- (46).Marchetti C (2014) Interaction of metal ions with neurotransmitter receptors and potential role in neurodiseases. BioMetals 27 (6), 1097–113. [DOI] [PubMed] [Google Scholar]
- (47).Kim HS, Kim YJ, and Seo YR (2015) An Overview of Carcinogenic Heavy Metal: Molecular Toxicity Mechanism and Prevention. J. Cancer Prev 20 (4), 232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, and Clore GM (1991) A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science 253 (5020), 657–61. [DOI] [PubMed] [Google Scholar]
- (49).Stewart JC (1980) Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem 104 (1), 10–4. [DOI] [PubMed] [Google Scholar]
- (50).Johnson SJ, Bayerl TM, McDermott DC, Adam GW, Rennie AR, Thomas RK, and Sackmann E (1991) Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys. J 59 (2), 289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lewis BA, and Engelman DM (1983) Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol 166 (2), 211–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.