

Abstract
Efficient site-directed insertion of heterologous DNA into a genome remains an outstanding challenge. Recombinases that can integrate kilobase-sized DNA constructs are difficult to reprogram to user-defined loci, while genomic insertion using CRISPR-Cas methods relies on inefficient host DNA repair machinery. Here, we describe a Cas-Transposon (CasTn) system for genomic insertions that uses a Himar1 transposase fused to a catalytically dead dCas9 nuclease to mediate programmable, site-directed transposition. Using cell-free in vitro assays, we demonstrated that the Himar–dCas9 fusion protein increased the frequency of transposon insertion at a single targeted TA dinucleotide by >300-fold compared to a random transposase, and that site-directed transposition is dependent on target choice while robust to log-fold variations in protein and DNA concentrations. We also showed that Himar–dCas9 mediates directed transposition into plasmids in Escherichia coli. This work highlights CasTn as a new modality for host-independent, programmable, site-directed DNA insertions.
Introduction
Genome engineering relies on molecular tools for targeted and specific modification of a genome to introduce insertions, deletions, and substitutions. While numerous advances have emerged over the last decade to enable programmable editing and deletion of bacterial and eukaryotic genomes, targeted genomic insertion remains an outstanding challenge.1 Integration of desired heterologous DNA into the genome needs to be precise, programmable, and efficient—three key parameters of any genome integration methodology. Currently available genome integration tools are limited by one or more of these factors. Recombinases such as Flp2 and Cre3 that mediate recombination at defined recognition sequences to integrate heterologous DNA have limited programmability.4,5 Site-specific nucleases such as CRISPR-associated (Cas) nucleases,6,7 zinc-finger nucleases (ZFNs),8 and transcription activator-like effector nucleases (TALENs)9 can be programmed to generate double-strand DNA breaks that are then repaired to incorporate a template DNA. However, this process relies on host homology-directed repair machinery, which is variable and often inefficient, especially as the size of the DNA insertion increases.10
Transposable elements are selfish genetic systems capable of integrating large pieces of DNA into both prokaryotic and eukaryotic genomes. Among various known transposable elements,11,12 the Himar1 transposon from the horn fly Haematobia irritans13 has been co-opted as a popular tool for insertional mutagenesis. The Himar1 transposon is mobilized by the Himar1 transposase, which like other Tc1/mariner-family transposases, functions as a homodimer to bind the transposon DNA at the flanking inverted repeats, excise the transposon, and paste it into a random TA dinucleotide on a target DNA.13–16 Himar1 requires no host factors for transposition and functions in vitro,13 in bacteria,17 and in mammalian cells,18 and is capable of inserting transposons >7 kb in size.19 A hyperactive mutant of the transposase, Himar1C9, which contains two amino acid substitutions and increases transposition efficiency by 50-fold,20 has enabled the generation of transposon insertion mutant libraries for genetic screens in diverse microbes.21–23 However, because Himar1 transposons are inserted randomly into TA dinucleotides, their utility in targeted genome insertion applications has thus far been limited.
There has been great interest in harnessing the integration capabilities of transposases for genome editing. Synthetic approaches to increase the specificity of random transposon insertions aim to increase the affinity of the transposon or the transposase to specific DNA motifs. IS608, which is directed by base-pairing interactions between a transposon end and target DNA to insert 3′ to a tetranucleotide sequence, was shown to be targeted more specifically by increasing the length of the guide sequence in the transposon end.24 However, altering transposon flanking end sequences affects the physical structure and biochemical activity of the transposon, limiting the range of viable sequence alterations that can be made. Several studies have described fusing transposases to DNA-binding protein (DBP) domains to direct transposon insertions to specific loci. Fusing the Gal4 DNA-binding protein to Mos1 (a Tc1/mariner family member) and piggyBac transposases increased the frequency of integration sites near Gal4 recognition sites.25 Fusion of DNA-binding zinc-finger or transcription activator-like (TAL) effector proteins to piggyBac enabled integration into specified genomic loci in human cells.26–28 ISY100 transposase (also a Tc1/mariner family member) has been fused to a Zif268 Zinc-finger domain to increase specificity of transposon insertions to DNA adjacent to Zif268 binding sites.29
More recently, researchers have begun uniting the powerful integration abilities of transposases with precision targeting by RNA-guided Cas nucleases to achieve targeted transposon integration. In nature, CRISPR-associated Tn7-like transposases have been discovered in cyanobacteria30 and in Vibrio cholerae.31 In each of these studies, a Tn7-like transposase was found to be genetically encoded in close association with a CRISPR-Cas system. The RNA-guided Cas-effector complex was deficient in DNA cleavage but recruited the Tn7-like transposase protein subunits to insert transposons locally near its binding site, thereby enabling programmable insertions of transposons both in vitro and in vivo in Escherichia coli genomes. Other studies draw upon synthetic biology research showing that Cas nucleases can be repurposed as RNA-guided DNA-binding protein domains for manipulation of DNA sequences and gene expression at user-defined loci, in applications such as CRISPR interference (CRISPRi),32,33 CRISPR activation (CRISPRa),33,34 FokI-dCas9 dimeric nucleases,35,36 base editors,37,38 dCas9-targeted Gin serine recombinase,39 and targeted histone modifiers.40,41 Likewise, transposases that naturally insert transposons randomly can be fused to catalytically dead Cas9 (dCas9) for targeted transposition. A recent study showed that a synthetic Hsmar1 transposase–dCas9 fusion protein enabled directed transposition in cell-free reactions.42
In this study, we developed a novel synthetic system, Cas-Transposon (CasTn), which unites the DNA integration capability of the Himar1 transposase and the programmable genome targeting capability of dCas9 to enable site-directed transposon insertions at user-defined genetic loci. This gRNA-targeted Himar1–dCas9 fusion protein integrates mini-transposons carrying synthetic DNA payloads into specific loci with nucleotide precision (Fig. 1A), which we demonstrated in both cell-free in vitro reactions and in a plasmid assay in E. coli. With further improvements to the system, CasTn can potentially function in a variety of organisms because the Himar1–dCas9 protein requires no host factors to function. An optimized CasTn platform may allow integration of a synthetic module of genes into a target locus, expanding the toolbox available to genome engineers in metabolic engineering43 and emergent gene drive applications.44
FIG. 1.

Schematics of the in vitro Cas-Transposon (CasTn) test system. (A) Overview of Himar1–dCas9 protein function. The Himar1–dCas9 fusion protein is guided to the target insertion site by a gRNA, where it is tethered by the dCas9 domain. The Himar1 domain dimerizes with that of another fusion protein to cut-and-paste a Himar1 transposon into the target gene, which is knocked out in the same step. (B) Implementation of the CasTn system in vitro. Transposon donor and target plasmids were mixed with purified protein and gRNA. Following purification of transposition reactions, a mix of donor, target, and transposition product plasmids was obtained and analyzed by several assays. cmR, chloramphenicol resistance; GFP, green fluorescent protein; carbR, carbenicillin resistance; oriR, origin of replication. (C) Sodium dodecyl sulfate polyacrylamide gel electrophoresis of purified Himar–dCas9 protein. (D) Schematic of target plasmid–transposon junction polymerase chain reaction (PCR) assay. The PCR was performed using primer 1, which binds the transposon, and primer 2, which binds the target plasmid. Site-specific transposition results in an enrichment for a PCR product corresponding with the expected transposition product. PCR amplicons for transposition reactions containing gRNA-guided transposases and random, unguided transposases were analyzed by next-generation sequencing. (E) Schematic of transformation assay. In vitro reaction products were transformed into electrocompetent Escherichia coli to isolate single transposition events from individual colonies containing a transposition product, and to calculate the efficiency of transposition (fraction of all target plasmids bearing a transposon conferring chloramphenicol resistance).
Methods
Strains, media, and growth conditions
All E. coli strains were grown aerobically in LB Lennox broth at 37°C with shaking, with antibiotics added at the following concentrations: carbenicillin (carb) 50 μg/mL, kanamycin (kan) 50 μg/mL, chloramphenicol (chlor) 20–34 μg/mL, and spectinomycin (spec) 240 μg/mL for S17 derivative strains and 60 μg/mL for non-S17 derivative strains. Supplements were added at the following concentrations: diaminopimelic acid (DAP) 50 μM, anhydrotetracycline (aTc) 1–100 ng/mL, and magnesium chloride (MgCl2) 20 mM.
Buffer compositions
Buffers used in the study were as follows. Protein resuspension buffer (PRB): 20 mM Tris-HCl pH 8.0, 10 mM imidazole, 300 mM NaCl, 10% v/v glycerol. One tablet of cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail (Roche) was dissolved in 10 mL buffer immediately before use. Protein wash buffer (PWB): 20 mM Tris-HCl pH 8.0, 30 mM imidazole, 500 mM NaCl, 10% v/v glycerol. Protein elution buffer (PEB): 20 mM Tris-HCl pH 8.0, 500 mM imidazole, 500 mM NaCl, 10% v/v glycerol. Dialysis buffer 1 (DB1): 25 mM Tris-HCl pH 7.6, 200 mM KCl, 10 mM MgCl2, 2 mM DTT, 10% v/v glycerol. Dialysis buffer 2 (DB2): 25 mM Tris-HCl pH 7.6, 200 mM KCl, 10 mM MgCl2, 0.5 mM DTT, 10% v/v glycerol. 10 × Annealing buffer: 100 mM Tris-HCl pH 8.0, 1 M NaCl, 10 mM EDTA (pH 8.1).
Design and construction of the Himar–dCas9 transposase
The gene encoding fusion protein Himar1C9–XTEN–dCas9 (Himar–dCas9) was constructed from the hyperactive Himar1C9 transposase gene on plasmid pSAM-BT21 and the dCas9 gene from pdCas9-bacteria (Addgene plasmid #44249). Flexible peptide linker sequence XTEN35 was synthesized as a gBlock® (Integrated DNA Technologies). DNA sequences were polymerase chain reaction (PCR) amplified using Kapa Hifi Master Mix (Kapa Biosystems) and cloned into expression vectors using NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs). Himar–dCas9 and Himar1C9 genes were cloned into a C-terminal 6 × His-tagged T7 expression vector (yielding plasmids pET–Himar–dCas9 and pET–Himar) for protein production and purification. Himar–dCas9, dCas9, and Himar1C9 genes were cloned into tet-inducible bacterial expression vectors (yielding plasmids pHdCas9, pdCas9–carb, and pHimar1C9, respectively) to assess protein function in vivo. Tet-inducible bacterial expression vectors for Himar–dCas9 that additionally feature constitutive gRNA expression cassettes were constructed to evaluate site-specificity of Himar–dCas9 in vivo: pHdCas9–gRNA1, pHdCas9–gRNA4, pHdCas9–gRNA5, pHdCas9–gRNA5–gRNA16 containing gRNA_1, gRNA_4, gRNA_5, and both gRNA_5 and gRNA_16, respectively. Himar–dCas9 was cloned into a mammalian expression vector with an N-terminal 3 × FLAG tag and SV40 nuclear localization signal (pHdCas9-mammalian), and this mammalian variant of the Himar–dCas9 protein was purified from C-terminal 6 × His-tagged expression vector pET–Himar–dCas9-mammalian. Plasmids used in this study are described in Table 1. All gRNAs used in this study are described in Table 2.
Table 1.
Plasmids used in this study
| Plasmid | Origin of replication | Size (bp) | Selection | Features | Purpose |
|---|---|---|---|---|---|
| pET–Himar–dCas9 |
ROP |
10,864 |
carb |
6 × His tag, IPTG-inducible T7 promoter |
Himar–dCas9 protein purification |
| pET–Himar | ROP | 6,712 | carb | 6 × His tag, IPTG-inducible T7 promoter | Himar1C9 protein purification |
| pET–Himar–dCas9-mammalian | ROP | 10,969 | carb | 6 × His tag, IPTG-inducible T7 promoter | Protein purification of Himar–dCas9 with 3 × FLAG, SV40 NLS |
| pGT-B1 | pBBR1 | 6,235 | carb | Constitutive sfGFP gene | Target plasmid for in vitro assays |
| pHimar6 | R6K | 3,394 | kan | Himar transposon with chlor resistance cassette, RP4 oriT | Himar transposon donor plasmid for in vitro and Escherichia coli in vivo assays |
| pTarget | ColE1 | 3,237 | spec | Constitutive sfGFP gene | Target plasmid for E. coli in vivo assays |
| pZE41-eGFP | ColE1 | 3,154 | spec | eGFP on pL-tetO promoter | Target plasmid for in vitro testing of mammalian gRNAs |
| pHimar1C9 | p15A | 3,846 | carb | Himar1C9 on tet-inducible promoter | Bacterial expression vector for Himar1C9 |
| pHdCas9–gRNA1 | p15A | 8,200 | carb | Himar–dCas9 on tet-inducible promoter, constitutively expressed gRNA_1 | Bacterial expression vector for Himar–dCas9 and gRNA_1 |
| pHdCas9–gRNA4 | p15A | 8,200 | carb | Himar–dCas9 on tet-inducible promoter, constitutively expressed gRNA_4 | Bacterial expression vector for Himar–dCas9 and gRNA_4 |
| pHdCas9–gRNA5 | p15A | 8,200 | carb | Himar–dCas9 on tet-inducible promoter, constitutively expressed gRNA_5 | Bacterial expression vector for Himar–dCas9 and gRNA_5 |
| pHdCas9 | p15A | 7,738 | carb | Himar–dCas9 on tet-inducible promoter | Bacterial expression vector for Himar–dCas9 |
| pdCas9-carb | p15A | 6,847 | carb | dCas9 on tet-inducible promoter | Bacterial expression vector for Himar–dCas9 |
| pHdCas9–gRNA5–gRNA16 | p15A | 8,191 | chlor | Himar–dCas9 on tet-inducible promoter, constitutively expressed gRNA_5 and gRNA_16 | Bacterial expression vector for Himar–dCas9, gRNA_5, gRNA_16 |
| pdCas9–gRNA5–gRNA16 | p15A | 7,099 | chlor | dCas9 on tet-inducible promoter, constitutively expressed gRNA_5 and gRNA_16 | Bacterial expression vector for dCas9, gRNA_5, gRNA_16 |
| pHP–M1 | pBR322 | 4,161 | carb | Himar transposon with promoterless puroR-mCherry cassette; gRNA M1 expressed from U6 promoter | Himar transposon donor plasmid for mammalian cells; expression vector for gRNA M1 |
| pHP–M2 | pBR322 | 4,161 | carb | Himar transposon with promoterless puroR-mCherry cassette; gRNA M2 expressed from U6 promoter | Himar transposon donor plasmid for mammalian cells; expression vector for gRNA M2 |
| pHP–M1–M2 | pBR322 | 4,540 | carb | Himar transposon with promoterless puroR-mCherry cassette; gRNAs M1 and M2 expressed from U6 promoter | Himar transposon donor plasmid for mammalian cells; expression vector for gRNAs M1 and M2 |
| pHP–M3 | pBR322 | 4,159 | carb | Himar transposon with promoterless puroR-mCherry cassette; Esp3I/BsmBI cloning site for gRNAs can also be expressed as a gRNA from U6 promoter | Himar transposon donor plasmid for mammalian cells; expression vector for non-targeting gRNA M3; Golden Gate cloning vector for gRNAs |
| pHP | pBR322 | 3,742 | carb | Himar transposon with promoterless puroR-mCherry cassette | Himar transposon donor plasmid for mammalian cells |
| pHP-on | pBR322 | 5,340 | carb | Himar transposon with puroR-mCherry cassette on CMV promoter; gRNAs M1 and M2 expressed from U6 promoter | mCherry expression plasmid for transfection positive control |
| pHdCas9-mammalian | pBR322 | 8,727 | carb | Himar–dCas9 gene with N-terminal 3 × FLAG, N-terminal SV40 NLS, and C-terminal 6 × His, expressed from CMV promoter | Himar–dCas9 expression vector for mammalian cells |
| pcDNA5/FRT/Hyg-eGFP | pBR322 | 4,795 | carb | FRT site with downstream hygromycin resistance-eGFP cassette | Insert HygroR-eGFP into FLP-in cell genome to create transposon insertion target site |
| pcDNA5/FRT/Hyg-Himar | pBR322 | 6,601 | carb | FRT site with downstream hygromycin resistance-eGFP cassette; eGFP is interrupted by puroR-mCherry transposon | Create positive control FLP-in cell line containing site-specific integration of Himar transposon |
IPTG, isopropyl β-d-1-thiogalactopyranoside; GFP, green fluorescent protein.
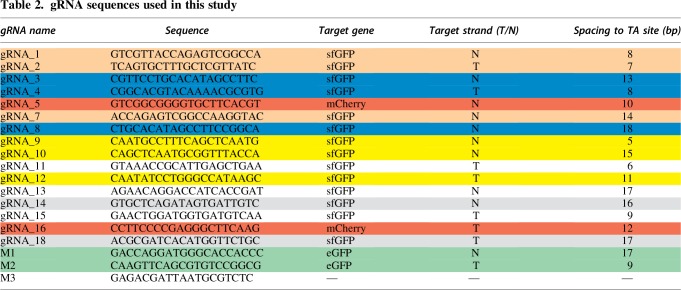
T indicates that the gRNA is complementary to the Template strand of the gene, while N indicates that the gRNA complements the Non-template strand. gRNAs that target the same TA insertion site are labeled with the same color. gRNAs 11, 13, and 15 all target different sites uniquely. M3 is a non-targeting gRNA.
Measurement of Himar–dCas9 gene expression knockdown in E. coli
We measured expression knockdown of mCherry in E. coli strain EcSC83 (MG1655 galK::mCherry-specR). Tet-inducible expression vectors pHdCas9–gRNA5–gRNA16 and pdCas9–gRNA5–gRNA16 were used to produce either Himar–dCas9 or dCas9 (a positive control) in each strain along with two gRNAs targeting mCherry. We measured expression knockdown of green fluorescent protein (GFP) encoded on the pTarget plasmid in the E. coli S17 strain. Tet-inducible expression vectors (pHdCas9–gRNA1, pHdCas9–gRNA4, pHdCas9–gRNA5, pHdCas9 for negative control) were used to express Himar–dCas9 along with a GFP-targeting gRNA in S17 with pTarget.
Saturated overnight E. coli cultures were diluted 1:40 into fresh LB media containing aTc to induce Himar–dCas9 or dCas9 expression. Aliquots of induced cultures (200 μL) were grown with shaking on 96-well plates at 37°C on a BioTek plate reader. Measurements of OD600 and mCherry (excitation 580 nm, emission 610 nm) and GFP (excitation 485 nm, emission 528 nm) fluorescence were taken 12 h post induction.
Measurement of Himar–dCas9 transposase activity in E. coli
Himar–dCas9 and Himar1C9 proteins were expressed in MG1655 E. coli from tet-inducible expression vectors pHdCas9 and pHimar1C9, respectively. These strains were conjugated with DAP-auxotrophic donor strain EcGT2 (S17 asd::mCherry-specR)45 containing transposon donor plasmid pHimar6, which has a 1.4 kb Himar1 mini-transposon containing a chlor resistance cassette and the R6K origin of replication, which does not replicate in MG1655.
Donor and recipient cultures were grown overnight at 37°C; donors were grown in LB with DAP and kan, and recipients were grown in LB with carb. Donor culture (100 μL) was diluted in 4 mL fresh media. Recipient culture (100 μL) was diluted in 4 mL fresh media with 1 ng/mL aTc to induce transposase expression. Both cultures were grown for 5 h at 37°C. Donor and recipient cultures were centrifuged and re-suspended twice in phosphate-buffered saline (PBS) to wash the cells. Donor (109) and recipient (109) cells were mixed, pelleted, re-suspended in 20 μL PBS, and dropped onto LB agar with 1 ng/mL aTc. The cell droplets were dried at room temperature and then incubated for 2 h at 37°C. After conjugation, cells were scraped off, re-suspended in PBS, and plated ± chlor (20 μg/mL) to select for recipient cells with an integrated transposon. Transposition rates were measured as the ratio of chlor-resistant colony-forming units (CFUs) to total CFUs.
Purification of Himar–dCas9 protein
His-tagged Himar–dCas9 was purified by nickel affinity chromatography from Rosetta2 cells (Novagen) bearing plasmid pET–Himar–dCas9 or pET–Himar–dCas9-mammalian. Saturated overnight culture (1 mL) grown in LB with chlor (34 μg/mL) and carb was diluted in 100 mL fresh media and grown to OD0.6–0.8 at 37°C with shaking. Isopropyl β-d-1-thiogalactopyranoside (IPTG; 0.2 mM) was added to induce protein expression, and the flask was incubated for 16 h at 18°C with shaking. The cells were pelleted by centrifugation at 7,197 g for 5 min at 4°C and then re-suspended in 5 mL ice-cold PRB. Cells were lysed in an ice water bath using a Qsonica sonicator at 40% power for a total of 120 s in 20 s on/off intervals. The cell suspension was mixed by pipetting, and the sonication step was repeated. The lysate was centrifuged at 7,197 g for 10 min at 4°C to pellet cell debris, and the cleared cell lysate was collected.
All subsequent steps were performed at 4°C. Ni-NTA agarose (1 mL; Qiagen) was added to a 15 mL polypropylene gravity flow column (Qiagen) and equilibrated with 5 mL of PRB. Cleared cell lysate was added to the column and incubated on a rotating platform for 30 min. The lysate was flowed through, and the nickel resin was washed with 50 mL PWB. The protein was eluted with PEB in five fractions of 0.5 mL each. Each elution fraction was analyzed by running an sodium dodecyl sulfate polyacrylamide gel electrophoresis. Elution fractions 2–4 were combined and dialyzed overnight in 500 mL DB1 using 10K MWCO Slide-A-Lyzer™ Dialysis Cassettes (Thermo Fisher Scientific). The protein was dialyzed again in 500 mL DB2 for 6 h. The dialyzed protein was quantified with the Qubit Protein Assay Kit (Thermo Fisher Scientific) and divided into single-use aliquots that were snap frozen in dry ice and ethanol and stored at −80°C. SDS-PAGE of purified Himar–dCas9 is shown in Figure 1C.
Purification of Himar1C9 protein
C-terminal 6 × His-tagged Himar1C9 was purified by nickel affinity chromatography from Rosetta2 cells (Novagen) bearing plasmid pET–Himar. Saturated overnight culture (1 mL) grown in LB with chlor (34 μg/mL) and carb was diluted in 100 mL fresh media and grown to OD0.9 at 37°C with shaking. IPTG (0.5 mM) was added to induce protein expression, and the flask was incubated at 37°C with shaking for 1 h. The cells were pelleted as described above, and the protein was purified using the His-Spin Protein Miniprep Kit (Zymo Research) according to the manufacturer's instructions, using the denaturing buffer protocol. The purified protein was dialyzed, frozen, and stored as described above. Purified Himar1C9 was used in control in vitro reactions along with commercially available purified dCas9 (Alt-R® S.p. dCas9 Protein V3; Integrated DNA Technologies).
In vitro transposition reaction setup
We characterized the specificity and efficiency of transposition by purified Himar–dCas9 within in vitro reactions (Fig. 1B). Each reaction was performed in a buffer consisting of 10% glycerol, 2 mM dithiothreitol (DTT), 250 μg/mL bovine serum albumin (BSA), 25 mM HEPES (pH 7.9), 100 mM NaCl, and 10 mM MgCl2. Plasmid DNA was purified using the ZymoPureII midiprep kit (Zymo Research). Background E. coli genomic DNA was purified using the MasterPure Gram Positive DNA Purification Kit (Epicentre). All DNAs were purified again using the Zymo Clean and Concentrator-25 Kit (Zymo Research) to remove all traces of RNAse. gRNAs were synthesized using the GeneArt™ Precision gRNA Synthesis Kit (Invitrogen). Concentrations of DNAs and gRNAs were measured using a Qubit 4 fluorometer (Invitrogen).
To set up in vitro reactions, frozen aliquots of Himar–dCas9 protein and gRNAs were thawed on ice. The protein was diluted to a 20 × final concentration in DB2 buffer, and gRNAs were diluted to the same molarity in nuclease-free water. The diluted protein and gRNA were mixed in equal volumes and incubated at room temperature for 15 min. Transposon donor DNA, target plasmid DNA, and background DNA (if applicable) were mixed on ice with 10 μL 2 × transposition buffer master mix and water to reach a volume of 18 μL. The protein/gRNA mixture (2 μL) was added last to the reaction. In reactions where the transposase/gRNA complex was preloaded onto the target plasmid, the target plasmid was mixed with protein and gRNA and incubated at 30°C for 10 min, and donor DNA was added last. Transposition reactions were incubated for 3–72 h at 30–37°C and then heat inactivated at 75°C for 20 min. Transposition products were purified using magnetic beads46 and eluted in 45 μL nuclease-free water.
Quantitative PCR assay for site-specific insertions in transposition reactions
One method used to evaluate the specificity and efficiency of Himar–dCas9 within in vitro transposition reactions was a series of quantitative PCRs (qPCRs; Fig. 1D). For each reaction, two qPCRs were performed to obtain the measure of relative Cq: one PCR amplifying transposon–target plasmid junctions, and another PCR amplifying the target plasmid backbone to normalize for template DNA input across samples. Relative Cq values shown in this study are the differences between the two Cq values.
For in vitro transposition into pGT-B1 (target plasmid used in in vitro experiments), primers p433 and p415 were used for junction PCRs, and primers p828 and p829 were used for control PCRs. For in vitro transposition into pTarget (target plasmid used for in vivo bacteria experiments) or pZE41-eGFP (target plasmid used to test mammalian CasTn components in vitro), primers p898 and p415 were used for junction PCRs, and primers p899 and p900 were used for control PCRs. All qPCR primers used in this study are listed in Table 3.
Table 3.
Oligonucleotides used in this study
| Name | Sequence (5′→3′) | Target | Temperature (°C) | Function |
|---|---|---|---|---|
| p433 |
CGCTTACAATTTCCATTCGCCATTC |
pGT-B1 |
67 |
qPCR for Himar transposon–pGT-B1 junction |
| p415 | CCCTGCAAAGCCCCTCTTTACG | pHimar6 transposon | 71 | qPCR for Himar–transposon junctions |
| p771 | GCGTAAAGGCGAAGAGC | pGT-B1 | 64 | PCR for Himar–transposon–pGT-B1 junction |
| p828 | CTGCGCAACCCAAGTGCTAC | pGT-B1 | 70 | Control qPCR for pGT-B1 |
| p829 | CAGTCCAGAGAAATCGGCATTCA | pGT-B1 | 67 | Control qPCR for pGT-B1 |
| p923 | Biotin/GCCATAAACTGCCAGGCATCAA | pHimar6 transposon | 68 | In vitro transposon sequencing library preparation |
| p922 | CCTTCTTGCGCATCTCACG | pGT-B1 | 67 | In vitro transposon sequencing library preparation |
| Adapter_T | Phosphate/AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | Anneal to make Y-shaped adapter for Tn-seq library prep | ||
| Adapter_B | GTCTCGTGGGCTCGGGCTCTTCCGATCT*N*N | Anneal to make Y-shaped adapter for Tn-seq library prep | ||
| p790 | AATGATACGGCGACCACCGAGATCTACAC TAGATCGCCGCCAGACCGGGGACTTATCATCCAACCTGT |
Himar transposon IR | 73 | Add barcode and P5 sequence to Himar transposon ends for Illumina sequencing |
| p791 | AATGATACGGCGACCACCGAGATCTACAC CTCTCTATCGCCAGACCGGGGACTTATCATCCAACCTGT |
Himar transposon IR | 73 | Add barcode and P5 sequence to Himar transposon ends for Illumina sequencing |
| p792 | AATGATACGGCGACCACCGAGATCTACAC TATCCTCTCGCCAGACCGGGGACTTATCATCCAACCTGT |
Himar transposon IR | 73 | Add barcode and P5 sequence to Himar transposon ends for Illumina sequencing |
| p793 | AATGATACGGCGACCACCGAGATCTACAC AGAGTAGACGCCAGACCGGGGACTTATCATCCAACCTGT |
Himar transposon IR | 73 | Add barcode and P5 sequence to Himar transposon ends for Illumina sequencing |
| p794 | AATGATACGGCGACCACCGAGATCTACAC GTAAGGAGCGCCAGACCGGGGACTTATCATCCAACCTGT |
Himar transposon IR | 73 | Add barcode and P5 sequence to Himar transposon ends for Illumina sequencing |
| p795 | AATGATACGGCGACCACCGAGATCTACAC ACTGCATACGCCAGACCGGGGACTTATCATCCAACCTGT |
Himar transposon IR | 73 | Add barcode and P5 sequence to Himar transposon ends for Illumina sequencing |
| p712 | CGCCAGACCGGGGACTTATCATCCAACCTGT | Himar transposon IR | 67 | Read 1 primer for Illumina sequencing |
| p713 | CGGAAGAGCCCGAGCCCACGAGAC | Himar sequencing library | 67 | Index 1 primer for Illumina sequencing |
| p898 | TTTGAGTGAGCTGATACCGCTC | ColE1 oriR | 67 | qPCR for Himar transposon–plasmid junctions in pTarget and pZE41-eGFP |
| p899 | GAGCGGTATCAGCTCACTCAAA | ColE1 oriR | 67 | Control qPCR for pTarget and pZE41-eGFP |
| p900 | TCCCTTAACGTGAGTTTTCGTTCC | ColE1 oriR | 67 | Control qPCR for pTarget and pZE41-eGFP |
| p931 | GGATAACATGGCCATCATCAAGGAG | mCherry | 67 | Control qPCR for Himar transposons in mammalian gDNA |
| p932 | CCTCGTTGTGGGAGGTGATG | mCherry | 68 | Control qPCR for Himar transposons in mammalian gDNA |
| p933 | AGGTCCTGTGAGCAAGGG | HygroR/eGFP cassette | 67 | qPCR for Himar transposon–eGFP junctions in mammalian gDNA |
| p946 | AGAGTTCTTGCAGCTCGGTG | mammalian Himar transposon | 68 | qPCR for Himar transposon–eGFP junctions in mammalian gDNA |
IR, inverted repeat; oriR, origin of replication.
PCR reactions contained 1 μL each of 10 μM forward and reverse primers, 1 μL purified transposition products as template DNA, 7 μL water, and 10 μL Q5 2 × Master Mix (NEB) + SYBR Green. Reactions were thermocycled using a Bio-Rad C1000 touch qPCR machine for 1 min at 98°C, followed by 35 cycles of 98°C denaturation for 10 s, 68°C annealing for 15 s, and 72°C extension for 2 min. PCR products were qualitatively analyzed for specificity on agarose DNA gels.
Transposon sequencing library preparation
To survey the distribution of transposition events performed by Himar–dCas9, we performed transposon sequencing on in vitro reaction products (Supplementary Fig. S2). Transposon junctions were PCR amplified from transposition reactions using primer sets p923/p433 and p923/p922 with Q5 HiFi 2 × Master Mix (NEB) + SYBR Green. Primer p923 binds the Himar1 transposon from pHimar6, while p433 and p922 bind to target plasmid pGT-B1. PCR reactions were performed on a Bio-Rad C1000 touch qPCR machine with the same thermocycling conditions described in the qPCR protocol, but were stopped in the exponential phase to avoid oversaturation of PCR products. PCR products were purified using magnetic beads,46 and 100–200 ng DNA per sample was digested with MmeI (NEB) for 1 h in a reaction volume of 40 μL. The digestion products were purified using Dynabeads M-270 streptavidin beads (Thermo Fisher Scientific) according to the manufacturer's instructions. The digested transposon ends, bound to magnetic Dynabeads, were mixed with 1 μg sequencing adapter DNA (see next section), 1 μL T4 DNA ligase, and T4 DNA ligase buffer in a total reaction volume of 50 μL. The ligations were incubated at room temperature (∼23°C) for 1 h, and then the beads were washed according to the manufacturer's instructions and re-suspended in 40 μL water.
Dynabeads (2 μL) were used as a template for the final PCR using barcoded P5 and P7 primers and Q5 HiFi 2 × × Master Mix (NEB) + SYBR Green. Reactions were thermocycled using a Bio-Rad C1000 touch qPCR machine for 1 min at 98°C, followed by cycles of 98°C denaturation for 10 s, 67°C annealing for 15 s, and 72°C extension for 20 s until the exponential phase. Equal amounts of DNA from all PCR reactions were combined into one sequencing library, which was purified and size selected for 145 bp products using the Select-a-Size Clean and Concentrator Kit (Zymo). The library was quantified with the Qubit dsDNA HS Assay Kit (Invitrogen) and combined at a ratio of 7:3 with PhiX sequencing control DNA. The library was sequenced using a MiSeq V2 50 Cycle Kit (Illumina) with custom read 1 and index 1 primers spiked into the standard read 1 and index 1 wells. Reads were mapped to the pGT-B1 plasmid using Bowtie 2.47
Construction of sequencing adapter
Oligonucleotides Adapter_T and Adapter_B were diluted to 100 μM in nuclease-free water. Ten microliters of each oligo was mixed with 2.5 μL water and 2.5 μL 10 × annealing buffer. The mixture was heated to 95°C and cooled at 0.1°C/s to 4°C to yield 25 μL of 40 μM sequencing adapter, which was stored at −20°C.
Transformation assay for in vitro transposition reaction products
Another method used to measure transposition specificity and efficiency was transformation of the reaction product DNA into competent E. coli and analyzing transposon inserts in individual transformants (Fig. 1E). Purified DNA (5 μL) from an in vitro transposition reaction was mixed with 45 μL distilled water and chilled on ice. Thawed MegaX electrocompetent E. coli (10 μL; Invitrogen) was added and mixed by pipetting gently. The mixture was transferred to an ice-cold 0.1 cm gap electroporation cuvette (Bio-Rad) and electroporated at 1.8 kV. Cells were recovered in 1 mL SOC and incubated with shaking at 37°C for 90 min. The cells were plated on LB + chlor (34 μg/mL) to select for target plasmids (pGT-B1) containing transposons, and on LB + carb to measure the electroporation efficiency of pGT-B1. The efficiency of transposition was measured as the ratio of chlor-resistant transformants to carb-resistant transformants. To assess specificity of inserted transposons, we performed colony PCR on transformants using the primer set p433/p415 with KAPA2G Robust HotStart ReadyMix (Kapa Biosystems) to amplify junctions between the Himar1 transposon from pHimar6 and the pGT-B1 target plasmid, which were analyzed by Sanger sequencing. Although this primer set was expected to amplify only the junctions arising from transposon insertions in a single orientation (not the reverse orientation), due to recombination and inversion of the transposon in some MegaX cells after transformation, this PCR was sensitive enough to detect the location of the transposon insertion into pGT-B1 in all colonies, but not the direction of the transposon.
To assess the direction of transposon insertion into pGT-B1 plasmids, ElectroMAX™ Stbl4™ electrocompetent E. coli, which have lower rates of recombination, were transformed with DNA from in vitro transposition reactions as described above. We performed colony PCR on transformants using primer sets p771/p415 (amplifying “forward” transposon–target junctions) and p433/p415 (amplifying “reverse” junctions) to assess for directionality (Supplementary Fig. S6).
In vivo assays for transposition into a target plasmid
S17 E. coli were sequentially electroporated with plasmid pTarget as a target plasmid and then one of several pHdCas9–gRNA plasmids (pHdCas9–gRNA1, pHdCas9–gRNA4, pHdCas9–gRNA5, or pHdCas9), which are bacterial expression vectors for Himar–dCas9 and a gRNA (Fig. 4A and Table 1). Transformants were selected on LB with carb and spec (240 μg/mL). Transformants were grown from a single colony to mid-log phase in liquid selective media, electroporated with 130 ng pHimar6 transposon donor plasmid DNA, and recovered in 1 mL LB for 1 h at 37°C with shaking post electroporation. One hundred microliters of a 10–3 dilution of the transformation was plated on LB agar plates with spec (240 μg/mL), carb, chlor (20 μg/mL), MgCl2 (20 mM), and aTc (0–2 ng/mL). Plates were grown at 37°C for 16 h. Between 103 and 104 colonies were scraped off each plate into 2 mL PBS and homogenized by pipetting. The cells (500 μL) were miniprepped using the QIAprep kit (Qiagen).
FIG. 4.

Himar–dCas9 performs site-directed transposition into plasmids in E. coli. (A) Three plasmids were transformed into S17 E. coli to create a testbed for Himar–dCas9 transposition specificity in vivo. Post-transposition plasmids were extracted from the bacteria and analyzed by PCR and by transformation into competent E. coli with Sanger sequencing of plasmids from individual colonies. (B) Himar–dCas9 knocks down green fluorescent protein (GFP) expression from the pTarget plasmid in vivo in E. coli with gRNA_1, which targets the non-template strand (N) of the GFP gene. Himar–dCas9 does not knock down GFP fluorescence when expressed with a gRNA complementing the template strand (T) or with a non-targeting gRNA (NT) or no gRNA. These cells did not contain transposon donor DNA. n = 2 per gRNA and ATC concentration; error bars indicate standard deviation. (C) PCR assay of in vitro transposition reactions using donor plasmid pHimar6 and recipient plasmid pTarget. Donor and recipient plasmids (2.27 nM each) along with 30 nM Himar–dCas9/gRNA complex were incubated for 3 h at 30°C. Expected PCR products of targeted insertions are shown with red arrows. (D) PCR analysis of pTarget–transposon junctions resulting from in vivo transposition in bacteria. Three out of five gRNA_1 PCR products show enrichment for the targeted insertion product. Transpositions A, B, C, and D with gRNA_1 were also analyzed by transformation and colony analysis. (E) Plasmid pools from four independent in vivo transposition experiments using gRNA_1 were transformed into E. coli, and the resultant colonies were analyzed by PCR and Sanger sequencing. The pie charts show the number of colonies containing on- and off-target transposition products from each plasmid pool, with the chart area proportional to the total number of colonies.
Minipreps from each transformation were evaluated by qPCR for junctions between the transposon from pHimar6 and the pTarget plasmid and by a transformation assay. qPCR assays for transposon–target plasmid junctions were performed as described above, using primers p898 and p415 and 10 ng miniprep DNA as PCR template. The control PCR to normalize for pTarget DNA input was performed with primers p899 and p900. In transformations, 150 ng plasmid DNA was electroporated into 10 μL MegaX electrocompetent cells diluted in 50 μL ice-cold distilled water. Cells were immediately recovered in 1 mL LB and incubated with shaking at 37°C for 90 min. The cells were plated on LB agar with chlor (20 μg/mL) and spec (60 μg/mL) to select for pTarget plasmids containing a transposon from pHimar6. We performed colony PCR using the primer set p898/p415 with KAPA2G Robust HotStart ReadyMix (Kapa Biosystems) to amplify transposon–pTarget junctions, which were analyzed by Sanger sequencing.
Generation of Chinese hamster ovary cell lines for transposition assays
Chinese hamster ovary (CHO) cells were cultured in Ham's F-12K (Kaighn's) Medium (Thermo Fisher Scientific) with 10% fetal bovine serum and 1% penicillin-streptomycin. The eGFP+ CHO cell line was generated by transfection of plasmids pcDNA5/FRT/Hyg-eGFP and pOG44 into the Flp-In™-CHO cell line (Thermo Fisher Scientific) followed by selection in media with hygromycin (500 μg/mL). An eGFP–, mCherry+, puromycin-resistant site-specific transposition positive control cell line was generated by transfection of plasmids pcDNA5/FRT/Hyg-Himar and pOG44 into the Flp-In™-CHO cell line followed by selection in media with puromycin (10 μg/mL). Transfections were performed on cells at 70% confluence on six-well plates using 12 μL of Lipofectamine 2000 and 1,000 ng of each plasmid. Antibiotic selection was initiated 48 h after transfection. Polyclonal transfected cells were trypsinized and passaged for use in subsequent experiments.
In vivo transposition assays in mammalian cells
The eGFP+ CHO cell line was transfected with a pHP plasmid (transposon donor and gRNA expression vector) and the pHdCas9-mammalian expression plasmid. Transfections were performed on cells at 70% confluence on six-well plates using 12 μL of Lipofectamine 2000 and 1,250 ng of each plasmid. In the transposition negative control, the pHP–M1–M2 plasmid was transfected without the pHdCas9-mammalian plasmid. Transfection efficiencies were 40–70% based on flow cytometry measurements of mCherry expression in cells 24 h post transfection of control plasmid pHP-on. Antibiotic selection with puromycin (10 μg/mL) was initiated 48 h after transfection. Cells from each transfection were trypsinized after 9 days of selection, and the whole volume was transferred into a single well of a 12-well plate and grown for four more days in puromycin media. During 13 days of antibiotic selection, the medium was changed every 24 h. Post-selection cells were trypsinized and diluted 1:5 in fresh media and analyzed on a Guava easyCyte flow cytometer (Millipore). Gates for mCherry and GFP fluorescence were set using mCherry–/eGFP– CHO cells, mCherry–/eGFP+ CHO cells, and mCherry+/eGFP– transposition positive control CHO cells.
Genomic DNA from trypsinized cells was extracted using the Wizard Genomic DNA Purification Kit (Promega) for PCR analysis. qPCR for transposon–gDNA junctions was performed as described above using primers p933 and p946. The control PCR to normalize for DNA input was performed using primers p931 and p932. Purified gDNA (10 ng per sample) was used as PCR template.
Results
Design of an engineered programmable, site-directed transposase protein
The design of the CasTn system leverages key insights from previous studies on Himar1 transposases and dCas9 fusion variants.7,20,29,32,34–36 The dCas9 protein is a well-characterized catalytically inactive Cas9 nuclease from Streptococcus pyogenes that contains the D10A and H841A amino acid substitutions7,32 and has been used as an RNA-guided DNA-binding protein for transcriptional modulation.32–34 Himar1C9 is a hyperactive Himar1 transposase variant that efficiently catalyzes transposition in diverse species and in vitro,20 highlighting its robust ability to integrate without host factors in a variety of cellular environments. The C-terminus of Himar1C9 was fused to the N-terminus of dCas9 using flexible protein linker XTEN35 (N-SGSETPGTSESATPES-C), as previous studies have described fusing other proteins to the N-terminus of dCas9 and to the C-terminus of mariner-family transposases.29,35,36
Because Himar1C9–dCas9 (Himar–dCas9) is a novel synthetic protein, we verified that both the Himar1 and dCas9 components remained functional. To check that Himar–dCas9 was capable of binding a DNA target specified by a gRNA, we expressed Himar–dCas9 in an E. coli strain with a genomically integrated mCherry gene, along with two gRNAs targeting mCherry (gRNA_5 and gRNA_16 in Table 2). We observed knockdown of mCherry expression, indicating that the DNA binding functionality of Himar–dCas9 was intact (Supplementary Fig. S1A). To verify Himar–dCas9 transposition activity, we conjugated a Himar1 mini-transposon with a chloramphenicol resistance gene (on plasmid pHimar6) from EcGT2 donor E. coli into MG1655 E. coli expressing Himar–dCas9 or Himar1C9 transposase. We measured the transposition rate as the proportion of recipient cells that acquired a genomically integrated transposon (Supplementary Fig. S1B). Himar–dCas9 mediates transposition events in E. coli, although at a lower rate (about 2 log-fold) compared with Himar1C9, which may be associated with lower expression of Himar–dCas9, which is a much larger and metabolically costly protein to produce, or with altered DNA affinity by dCas9, even in the absence of gRNA.48
An in vitro reporter system to assess site-directed transpositions by Himar–dCas9
To establish and optimize parameters for site-directed transposition, we next developed an in vitro reporter system to explore the transposition activity of Himar–dCas9. Purified Himar–dCas9 protein was mixed with transposon donor plasmid pHimar6 (containing a Himar1 mini-transposon with a chlor resistance gene), a transposon target pGT-B1 plasmid (containing a GFP gene), and one or more gRNAs targeted to various loci along GFP (Fig. 1B and Tables 1 and 2). We analyzed transposon insertion events into the pGT-B1 plasmid by several assays. First, quantitative PCR (qPCR) of target plasmid–transposon junctions, using one primer designed to anneal to a part of the transposon DNA and one primer designed to anneal to a part of pGT-B1, enabled qualitative assessment of transposition specificity based on enrichment of qPCR products of the expected amplicon size, as well as quantitative estimation of transposition rate (Fig. 1D and Table 3). For every transposon–target junction qPCR, we also performed a control qPCR that amplifies the target plasmid's backbone to control for variations in DNA input between samples. Relative Cq measurements, an estimation of transposition efficiency, were taken as the difference between the Cq values from the junction and control qPCR reactions. Next-generation transposon sequencing (Tn-seq) further enabled measurement of the distribution of inserted transposons within the target plasmid (Fig. 1D and Supplementary Fig. S2). Finally, transposition reaction products were transformed into competent E. coli to probe the specificity of transposition insertion sites further (Fig. 1E). Because the donor pHimar6 plasmid has a R6K origin of replication that is unable to replicate in E. coli without the pir replication gene, we selected for transformants containing the target pGT-B1 plasmid with an integrated transposon. Transposition efficiency was determined by dividing the number of chloramphenicol-resistant transformants (CFUs with a target plasmid carrying a transposon) by the number of carbenicillin-resistant transformants (total CFUs with a target plasmid). Sanger sequencing of the target plasmid from chloramphenicol-resistant transformants revealed the site of integration and the transposition specificity.
Efficiency and site-specificity of Himar–dCas9 transposon insertions is gRNA dependent
Using the in vitro reporter system, we first assessed how the orientation of the gRNA relative to the target TA dinucleotide affects the site specificity of transposition. We tested gRNAs spaced 5–18 bp from a TA site, targeting either the template or non-template strand of GFP (Fig. 2A and Table 2). Using the qPCR assay, we found that a single gRNA is sufficient to effect site-directed transposition by Himar–dCas9, but not by unfused Himar1C9 and dCas9, indicating that Himar–dCas9 bound to a target site mediates transposition locally (Fig. 2B and Supplementary Fig. S3). The site-specificity of these insertions is dependent on the gRNA spacing to the target TA site. All gRNA-directed insertion events occurred at the nearest TA distal to the 5′ end of the gRNA, as evidenced by gel purification and Sanger sequencing of enriched PCR bands (Fig. 2B) and by transposon sequencing of reaction products (Supplementary Fig. S4). Site-directed transposition was robust in reactions using gRNAs with 7–9 bp and 16–18 bp spacings, but did not occur at all at short spacings (5–6 bp), likely due to steric hindrance by Himar–dCas9 at short distances. At spacings of 11–13 bp, there was a very faint expected PCR band, indicating that site-directed transposition at those sites was relatively poor. Slightly stronger bands at 14–15 bp spacings indicate intermediate performance of Himar–dCas9 in site-directed transposition. These findings are consistent with the previously observed spacing dependence for FokI–dCas9 proteins that use the same XTEN peptide linker.35 The bimodal distribution of robustly targeting gRNA spacings may be due to the DNA double helix providing steric hindrance, since optimal spacings are approximately one helix turn (∼10 bp) apart.
FIG. 2.

Himar–dCas9 specificity is dependent on gRNA spacing and target site. (A) Illustration of gRNA strand orientation and spacings to TA insertion site. (B) PCR analysis of transposon–target junctions from in vitro reactions containing 30 nM Himar–dCas9/gRNA complex, 2.27 nM transposon donor DNA, and 2.27 nM target DNA. Reactions (n = 3) were run using gRNAs with spacings between 5 and 18 bp from the TA insertion site. Non-targeting gRNA (gRNA_5), no gRNA, and no transposase controls were also performed. Red arrows indicate expected site-specific PCR products for each gRNA. Error bars indicate standard deviation. (C) Transposon sequencing results for reactions with no gRNAs (left, gray, n = 4) or with gRNA_4 (blue, n = 3), gRNA_8 (orange, n = 3), gRNA_12 (green, n = 3), or gRNA_5 (black, n = 3). The baseline random distribution of transposons along the recipient plasmid in each panel with a gRNA is shown in light gray.
To assess the distribution of transposon insertions around the target pGT-B1 plasmid, we performed transposon sequencing on transposition products resulting from three GFP-targeting gRNAs (gRNA_4, gRNA_8, and gRNA_12), a non-targeting gRNA, and no gRNA (Fig. 2C and Supplementary Fig. S4). Although these distributions may not represent the true abundance of transposition events at each location, since sequencing was performed on size-biased PCR amplicons of transposon–target junctions, transposon distributions could be compared across reactions. The baseline distribution of random transposon insertions was generated from reactions with no gRNA. Random insertions were present throughout the 6.2 kb pGT-B1 plasmid, with a spike in transposition abundance at position 5999, a TA site in the middle of a 12 bp stretch of T/A nucleotides. This result is consistent with the observation that Himar1 transposase preferentially inserts transposons into flexible, T/A-rich DNA.49 In contrast, gRNA-directed insertions were less likely to be inserted into position 5,999 and were enriched at their respective gRNA-adjacent TA sites compared with baseline (Fig. 2C). gRNA_4, with an optimal spacing of 8 bp from the target TA site, produced the best-targeted insertions, with 42% of sequenced transposon insertions being exactly at the target site, a 342-fold enrichment over baseline. Comparison of targeted insertion fold-enrichment across different gRNAs suggests that the specific target site and flanking DNA play a role in the specificity of transposon integration. For instance, gRNA_12 had a higher fold-enrichment of insertions at its target site than gRNA_8, but a lower fraction of measured insertions, suggesting that the target site of gRNA_12 may be intrinsically disfavored for transposition. Together, these results further show that Himar–dCas9 mediates directed transposon insertion to an intended integration site with the help of an optimally spaced gRNA.
Given that mariner transposases dimerize in solution in the absence of DNA,50 we hypothesized that Himar–dCas9 dimerizes spontaneously, and the active Himar1 dimer is guided to a gRNA-specific target locus by one of the dCas9 domains in the Himar–dCas9 dimer (Fig. 1A). This mechanism is consistent with the observation that one gRNA is sufficient to direct targeted transposition. Further support for this hypothesis comes from in vitro reactions containing pairs of gRNAs targeting the same TA site but complementing opposite strands (Supplementary Fig. S5). If Himar1 subunits did not spontaneously dimerize, then dimerization of Himar–dCas9 would be enhanced by loading two monomers onto the same target plasmid in close proximity. We set up reactions in which target DNA was first preloaded with either paired or single gRNA/Himar–dCas9 complexes and then mixed with transposon donor DNA (Supplementary Fig. S5A). In these experiments, the final reaction contained 5 nM Himar–dCas9, 5 nM donor DNA, 5 nM target DNA, and 2.5 nM each of two gRNAs. We observed no difference in transposition rate or specificity between the gRNA/Himar–dCas9 complexes preloaded as pairs or as singletons (Supplementary Fig. S5B and C). The observation that preloading pairs of Himar–dCas9 complexes does not improve transposition is consistent with the hypothesis that transposase dimers formed before one of the gRNA/dCas9 domains targeted the dimer to its final location.
Site-directed transposition by Himar–dCas9 is robust across a range of protein and DNA concentrations in vitro
To assess the robustness of Himar–dCas9 to various experimental conditions and to determine the optimal parameters for site-directed transposition, we explored different concentrations of (1) protein–gRNA complexes, (2) transposon donor plasmid (pHimar6) DNA, (3) target plasmid (pGT-B1) DNA, and (4) background off-target DNA within in vitro transposition reactions containing a single gRNA (gRNA_4). We also performed in vitro reactions over different temperatures and reaction times.
Varying concentrations of Himar–dCas9/gRNA complexes, we detected site-directed transposition by PCR in in vitro reactions with at least 3 nM of Himar–dCas9/gRNA complexes, using 5 nM donor and 5 nM target plasmids (Fig. 3A). Increasing the Himar–dCas9/gRNA concentration increased the yield of targeted transposition events. The trend of higher transposition rates at higher transposase concentrations was confirmed by the transformation assay (Fig. 3B), which also enabled precise analysis of transposition specificity from individual transformants. At 30 nM Himar–dCas9/gRNA complex, the specificity of transposon insertion into the targeted TA site was 44% (11/25 colonies). The specificity of insertion at 100 nM of the complex remained stable at 47.5% (19/40 colonies). The directionality of transposons inserted into the GFP gene was split approximately 50/50 based on screens of transformants (Supplementary Fig. S6), supporting the hypothesis that insertion of transposons in a cell-free reaction is not directionally biased.
FIG. 3.

Himar–dCas9-mediated site-directed transposition is robust to changes in ribonucleoprotein complex and DNA concentration. Target plasmids were pGT-B1 and donor plasmids were pHimar6. (A) PCR analysis of transposition reactions (n = 3) using varying levels of Himar–dCas9/gRNA_4 complexes. Reactions were performed for 3 h at 30°C with 5 nM donor and recipient plasmid DNA. (B) Transformation assay to measure transposition rates in reactions using varying levels of Himar–dCas9/gRNA_4 complexes (n = 5). Reactions were performed for 3 h at 30°C with 5 nM of donor and recipient plasmid DNA. (C) PCR analysis of transposition reactions (n = 3) using varying levels of donor plasmid DNA. Reactions were performed for 3 h at 30°C with 5 nM of recipient plasmid DNA and 30 nM Himar–dCas9/gRNA_4 complex. (D) PCR analysis of transposition reactions (n = 3) using varying levels of recipient plasmid DNA. Reactions were performed for 3 h at 30°C with 0.5 nM of donor plasmid DNA and 30 nM Himar–dCas9/gRNA_4 complex. (E) PCR analysis of transposition reactions (n = 3) performed for different lengths of time in the presence or absence of background nonspecific DNA. Reactions were performed at 37°C with 1 nM recipient plasmid DNA, 1 nM donor plasmid DNA, and 100 nM Himar–dCas9/gRNA_4 complex. Background E. coli genomic DNA was present at 10 × the mass of recipient plasmid DNA. (F) Quantitative PCR measurement of transposition efficiency in reactions shown in panel (E). n = 3 for each reaction condition. In all panels, red arrows indicate the expected targeted transposition PCR product for gRNA_4, and error bars indicate standard deviation. Cq measurements correspond to log-scale differences in transposase activity.
Next, we explored whether site-directed transposition was affected by DNA concentrations of the donor or target plasmids. Using 5 nM target plasmid DNA, transposition activity was robust across 0.05–5 nM of donor plasmid DNA, with greater rates of transposition at higher donor DNA concentrations (Fig. 3C). Similarly, using 0.5 nM of donor plasmid DNA, site-directed transposition occurred across target plasmid concentrations of 0.25–10 nM (Fig. 3D). While the absolute rate of transposition (as assessed by Cq of the transposon–target junction qPCR) was higher at higher target DNA concentrations, the relative Cq remained relatively stable across target DNA concentrations, indicating that a similar proportion of target plasmids received a transposon in each reaction.
We also tested whether the gRNA-guided Himar–dCas9 could efficiently transpose into a targeted site in the presence of background DNA and whether the amount of transposition changed over longer reaction times. We added up to 10 × (by mass) more background E. coli genomic DNA than target plasmid DNA to in vitro transposition reactions. Across different ratios of target-to-background DNA concentrations tested, Himar–dCas9 was able to locate the gRNA-targeted site and insert transposons with no observed loss of specificity or efficiency (Supplementary Fig. S7A). When we performed similar reactions containing 10 × background DNA at 37°C and over longer time courses instead of the standard protocol of 30°C for 3 h, to mimic conditions in living cells, we observed similar results (Supplementary Fig. S7B and C and Fig. 3E and F). The relative Cq and PCR band intensity of transposon–target junctions increased slightly between 3 and 16 h, suggesting that gRNA-guided transposases are faster at locating the target site than catalyzing transposition and that the increase in site-specific transposon insertions over time is performed by gRNA–dCas9 bound transposases. After 16 h, site-specific transposition events reached a plateau; the loss of specific transposon–target junctions observed at 72 h by PCR is likely due to degradation of reaction components (Supplementary Fig. S7B and Fig. 3E).
Together, these results highlight that Himar–dCas9/gRNA mediates site-directed transposon insertions across a range of experimental conditions, including physiologically relevant temperatures and reactant concentrations. In bacteria, 1 nM corresponds to approximately one molecule per cell, while in eukaryotic cells, 1 nM corresponds to approximately 1,000 molecules per cell.51 Targeted transposition was observed to occur at protein concentrations of 1–100 nM (1–100 molecules of protein per bacterium) and DNA concentrations of <1 to 10 nM (1–10 DNA copies per bacterium). In bacteria, these concentrations are physiologically achievable with low protein expression and with transposon donor/target DNA present as a single chromosomal copy or on a low/medium copy number plasmid. Notably, we did not experimentally find an upper limit of protein/DNA concentrations for effective site-directed transposition beyond the loss of specific targeting due to increased background transpositions. Nevertheless, the CasTn system can be used with different plasmid expression systems to modulate copy numbers of both protein and DNA.
Himar–dCas9 mediates site-directed transposon insertions into plasmids in vivo in E. coli
Since Himar–dCas9 robustly facilitated site-directed transposon integration in vitro, we tested the ability of HdCas9 to mediate site-specific transposition in two in vivo systems in E. coli and in mammalian cells. In the first system, we transformed a set of three plasmids into S17 E. coli: pTarget, which contains a GFP target gene; pHimar6, the transposon donor plasmid; and a tet-inducible expression vector for Himar–dCas9 and a gRNA (Fig. 4A). We grew these cells on selective agar plates with MgCl2 and anhydrotetracycline (aTc) to enable transposition and then extracted all plasmids. Transposition specificity was determined by two methods: PCR of transposon–target plasmid junctions, and transformation of plasmids into competent cells and analysis of transposon insertions in transformants.
We first verified that the Himar–dCas9 system components functioned in vivo. By measuring transcriptional repression of GFP in E. coli containing pTarget and one of several Himar–dCas9/gRNA expression vectors, we confirmed that gRNAs targeted Himar–dCas9 to the pTarget plasmid and determined the optimal concentration of aTc for inducing Himar–dCas9 expression (Fig. 4B). Consistent with previously reported results, gRNA_1, which targets the non-template strand of GFP, caused knockdown of GFP expression, but gRNA_4, which targets the template strand and does not sterically hinder RNA polymerase, did not cause GFP knockdown.32 Himar–dCas9 concentrations reached saturation at aTc induction levels of 2 ng/mL, as further increasing the concentration of aTc did not result in further knockdown of GFP by gRNA_1. We also validated that purified Himar–dCas9 protein with gRNA_1 or gRNA_4 mediated targeted transposition into the GFP gene of pTarget in vitro (Fig. 4C).
In the in vivo assay, S17 E. coli containing pTarget, a Himar–dCas9/gRNA expression, and pHimar6 were grown on agar plates containing a saturating concentration of MgCl2 and 1 ng/mL aTc to induce expression of Himar–dCas9 while avoiding overproduction inhibition of Himar1C9.52 After 16 h of growth at 37°C, we analyzed the pooled plasmids from all colonies for site-specific transposon insertions. PCR for transposon–target plasmid junctions showed that gRNA_1 produced detectable site-specific transposon insertions into pTarget in three out of five independent replicates (Fig. 4D). gRNA_4, however, did not produce an enrichment of PCR products corresponding to its target site.
We further evaluated the site specificity of transposition by transforming the plasmid pools into E. coli and analyzing individual transformants by colony PCR and Sanger sequencing in order to confirm that Himar–dCas9 with gRNA_1 mediated precisely targeted transposon insertions into pTarget. In three out of four independent replicates with gRNA_1, transformations produced colonies with mostly or all site-specific transposition products (Fig. 4E). In transformations of four plasmid pools from cells without a gRNA, we did not obtain any transformants with a transposon integrated into pTarget. Taken together, these results demonstrate in vivo directed transposition by an engineered Himar–dCas9 system for the first time.
In a second in vivo test system, we tested the ability of Himar–dCas9 to mediate site-specific transposition into a genomic locus in CHO cells. We transfected CHO cells containing a single-copy constitutively expressed genomic eGFP gene with two plasmids: one containing a Himar transposon and gRNA expression operons, and the other being a Himar–dCas9 expression vector (Supplementary Fig. S8A). The mammalian Himar–dCas9 was fused to an N-terminal 3 × -FLAG tag and SV40 nuclear localization signal (NLS) and a C-terminal 6 × -His tag. Two gRNAs were designed to target the eGFP gene at the same TA insertion site, complementing opposite strands. We tested these gRNAs individually and as a pair, along with a non-targeting gRNA and no gRNA. In vitro experiments demonstrated that the two gRNAs individually mediated site-specific transposition by the purified 3 × -FLAG-NLS-HdCas9-6 × -His protein (Supplementary Fig. S8B).
The Himar transposon contained a promoterless puromycin resistance gene and mCherry gene, both of which would be inserted in-frame into the eGFP locus and expressed if targeted by Himar–dCas9 in the correct orientation (Supplementary Fig. S8A). Because the transposon genes would only be expressed if the transposon were integrated downstream of a genomic promoter, puromycin selection for transposon mutants was stringent against false-positive clones resulting from plasmid integration into the genome. We verified that transposon insertions into the target locus resulted in successful expression of puromycin resistance and mCherry by constructing a positive control cell line with the transposon cloned into that locus (Supplementary Fig. S8C).
Following transfection, we selected for cells with an integrated transposon using puromycin. From each transfection of approximately 106 cells, we obtained about 20 colonies representing independent transposition events. Negative controls for transposition, which were transfected with only the transposon donor plasmid, did not produce viable cells, indicating clean selection against background plasmid integration events. All colonies from each transfection were pooled for analysis by flow cytometry and PCR for transposon–target junctions. Transfections with no gRNA resulted in few eGFP– cells, while some transfections with at least one gRNA (including the non-targeting gRNA) produced eGFP– cells (Supplementary Fig. S8C and D). However, PCR for the expected eGFP–transposon junction in genomic DNA showed no evidence of targeted transposition in any of the transfections, suggesting that the eGFP– cells had lost eGFP expression by another mechanism (Supplementary Fig. S8E). Although we did not observe targeted transposition by HdCas9 into a genomic locus here, an optimized mammalian testbed may enable screening for site-specific transposition events among larger samples of transposon insertions and shed light on the determinants of site-specific transposition in mammalian cells.
Discussion
In this study, we demonstrated the successful engineering of a programmable transposase for site-directed transposon insertion in cell-free reactions and into plasmids in E. coli. The Himar–dCas9 fusion protein mediates site-directed transposition into a user-specified locus. We characterized the activity of Himar–dCas9 in vitro across various reaction conditions, demonstrating that the site-directed transposition activity is robust. We further showed that transposition is dependent on the gRNA orientation relative to the target TA insertion site and the surrounding target DNA sequence. In in vivo studies of Himar–dCas9 in E. coli, we showed that gRNA_1 successfully directed transposition to a single locus on a medium copy-number target plasmid in >80% of detected transposition events.
Comparing the results of the in vitro and in vivo studies, there were persistent off-target transposition events in both types of studies, while gRNA strand complementarity appears to affect site-specific transposition in vivo but not in vitro. We attribute the off-target transposition to constitutive activity of Himar1C9 domains within the Himar–dCas9 fusion protein, even in transposase dimers that are not bound to the gRNA-targeted site. The difference in transposition targeting by gRNA_1 and gRNA_4, which both resulted in site-directed transposition in vitro, may be explained by the presence of other DNA-interacting elements in vivo. As discussed above, gRNA_1-guided Himar–dCas9 bound the non-template strand of the GFP gene, sterically hindering RNA polymerase from traversing downstream to its target TA site, while gRNA_4-guided Himar–dCas9 did not hinder RNA polymerase from accessing its upstream TA site, unwinding the target DNA, and blocking transposition. This difference in relative accessibility of the targeted TA site by Himar–dCas9 versus RNA polymerase may explain the strand-dependence of in vivo transposition targeting by gRNAs.
While site-directed insertion was demonstrated here for a medium copy-number target plasmid in E. coli, additional optimizations may be necessary to improve Himar–dCas9 targeting to a genomic locus and detection of targeted genomic insertion events in bacteria. Assays for targeted genomic transposition using counter-selectable genomic markers such as galK, tolC, or sacB as targets may be useful for detecting site-specific transposon insertions.53–55 The use of inducible Himar–dCas9 can improve the differentiation between gene knockout (via targeted transposition) and knockdown (via transcriptional blockade by Himar–dCas9) to improve the sensitivity and selectivity of insertion events. To reduce the leakiness of inducible expression systems that may yield unintended off-target transpositions, further improvements could be made to express the Himar–dCas9/gRNA complex transiently through acquisition of a non-replicative vector (e.g., a R6K plasmid) in the target cell by conjugation or transformation. Transient presence of the transposon donor vector would also be a useful feature in genomic targeting studies so that the pool of integrated transposons could be isolated and analyzed by Tn-seq.
The CasTn system requires significant further optimization to enable targeted genomic transposon insertions in mammalian cells. We introduced the system components into CHO cells and obtained antibiotic-resistant clones from transfections involving both the Himar–dCas9 expression vector and the transposon donor vector, but not control transfections of only the transposon donor, indicating that the resistant cells had undergone transposon insertion into the genome. However, it is unclear how well the HdCas9 protein and gRNAs were expressed in the nucleus to enable transposition. Alternative methods of delivering these components into cells, such as electroporation of nucleoprotein complexes, may be required to achieve optimal levels inside the nucleus. More sensitive screens for transposon insertions among larger pools of transposon insertions are also required to interrogate Himar–dCas9 targeting in mammalian cells.
Protein engineering of Himar–dCas9 could improve efficiency of transposition and reduce the frequency of off-target transposition across all cellular contexts. Because the Himar1 transposase functions as a dimer, mutating the dimerization interface of this protein may result in less off-target activity. Two dimerization-deficient Himar–dCas9 molecules could be directed as a pair, using two gRNAs, to a target site to increase the rate of dimerization locally while minimizing off-target insertions. Alternatively, two dimerization-deficient Himar1 monomers could be fused to one dCas9 subunit to improve targeting of dimers to a desired insertion site, similar to DNA base editors that utilize a pair of linked deaminase monomers fused to a Cas9 variant.37 Additional modification of the Himar1 protein by rational mutagenesis may further improve efficiency. Previous studies have shown that Himar1, like other mariner transposases, is rate limited by the synapsis of transposon ends to the protein dimer.52,56 Himar1 mutants with single amino-acid substitutions in the conserved WVPHEL motif have higher transposition rates than the Himar1C9 mutant because the allosteric inhibition of transposon synapsis is disrupted.52 Thus, a hyperactive WVPHEL mutation, which removes the natural rate-limiting step of transposon synapsis, combined with a mutation in the dimerization interface to slow down dimerization of Himar1 (normally a fast step), may result in a Himar1 protein that dimerizes more specifically at dCas9-targeted locations and then efficiently catalyzes transposition at those sites. Finally, CasTn systems constructed with alternative protein components can be explored, including other transposases (e.g., Tn5), DNA-binding domains (e.g., Cas homologs), and peptide linkers.
Another consideration in the improvement of the CasTn platform is that the inserted transposons introduce unwanted flanking end DNA along with a desired genetic construct. In our system, the transposon flanking ends were symmetric, enabling recombination and inversion of the inserted construct, which may be undesirable in genome engineering applications where stability is important. To minimize recombination, it would be possible to mutate non-conserved residues in each transposon flanking end to eliminate symmetry without compromising transposase activity.21,22 To eliminate unwanted flanking end DNA, one may use CasTn in conjunction with other methods for genome modification, such CRISPR-Cas genome editing, recombineering, or MAGE, to delete the transposon flanking ends after insertion of an operon into the genome. Optimized CasTn technologies may thus expand and complement the set of currently available tools for targeted genome editing.
Supplementary Material
Acknowledgments
We thank Saeed Tavazoie, Alexandra Ketcham, and Anupama Khare for guidance on transposon sequencing. Eric Greene, Justin Steinfeld, and Chu Jian (Frank) Ma provided materials and guidance on in vitro protein expression and purification. We thank members of the Wang lab for helpful scientific discussions and for assistance with experiments.
Author Disclosure Statement
A provisional patent application has been filed by The Trustees of Columbia University in the City of New York based on this work. The authors declare no additional competing financial interests.
Funding Information
This work was supported by funding from the Office of Naval Research (N00014-15-1-2704 to H.H.W.); Defense Advanced Research Projects Agency (W911NF-15-2-0065 to H.H.W.); National Institutes of Health (1DP5OD009172 to H.H.W., F30DK111145 to S.P.C., and T32GM007367 to S.P.C.); and Burroughs Wellcome Fund (1016691 to H.H.W.).
Supplementary Material
References
- 1. Esvelt KM, Wang HH. Genome-scale engineering for systems and synthetic biology. Mol Syst Biol 2013;9:641 DOI: 10.1038/msb.2012.66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andrews BJ, Proteau GA, Beatty LG, et al. The FLP recombinase of the 2 micron circle DNA of yeast: interaction with its target sequences. Cell 1985;40:795–803. DOI: 10.1016/0092-8674(85)90339-3 [DOI] [PubMed] [Google Scholar]
- 3. Abremski K, Hoess R. Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. J Biol Chem 1984;259:1509–1514 [PubMed] [Google Scholar]
- 4. Bolusani S, Ma CH, Paek A, et al. Evolution of variants of yeast site-specific recombinase Flp that utilize native genomic sequences as recombination target sites. Nucleic Acids Res 2006;34:5259–5269. DOI: 10.1093/nar/gkl548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buchholz F, Stewart AF. Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat Biotechnol 2001;19:1047–1052. DOI: 10.1038/nbt1101-1047 [DOI] [PubMed] [Google Scholar]
- 6. Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–823. DOI: 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012;337:816–821. DOI: 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Urnov FD, Rebar EJ, Holmes MC, et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 2010;11:636–646. DOI: 10.1038/nrg2842 [DOI] [PubMed] [Google Scholar]
- 9. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 2013;14:49–55. DOI: 10.1038/nrm3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kowalczykowski SC. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb Perspect Biol 2015;7 DOI: 10.1101/cshperspect.a016410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munoz-Lopez M, Garcia-Perez JL. DNA transposons: nature and applications in genomics. Curr Genomics 2010;11:115–128. DOI: 10.2174/138920210790886871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Curcio MJ, Derbyshire KM. The outs and ins of transposition: from mu to kangaroo. Nat Rev Mol Cell Biol 2003;4:865–877. DOI: 10.1038/nrm1241 [DOI] [PubMed] [Google Scholar]
- 13. Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J 1996;15:5470–5479. DOI: 10.1002/j.1460-2075.1996.tb00930.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richardson JM, Dawson A, O'Hagan N, et al. Mechanism of Mos1 transposition: insights from structural analysis. EMBO J 2006;25:1324–1334. DOI: 10.1038/sj.emboj.7601018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Richardson JM, Colloms SD, Finnegan DJ, et al. Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell 2009;138:1096–1108. DOI: 10.1016/j.cell.2009.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Claeys Bouuaert C, Lipkow K, Andrews SS, et al. The autoregulation of a eukaryotic DNA transposon. eLife 2013;2:e00668 DOI: 10.7554/eLife.00668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nat Rev Microbiol 2013;11:435–442. DOI: 10.1038/nrmicro3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Sankar U, Lampe DJ, et al. The Himar1 mariner transposase cloned in a recombinant adenovirus vector is functional in mammalian cells. Nucleic Acids Res 1998;26:3687–3693. DOI: 10.1093/nar/26.16.3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lampe DJ, Grant TE, Robertson HM. Factors affecting transposition of the Himar1 mariner transposon in vitro. Genetics 1998;149:179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lampe DJ, Akerley BJ, Rubin EJ, et al. Hyperactive transposase mutants of the Himar1 mariner transposon. Proc Natl Acad Sci U S A 1999;96:11428–11433. DOI: 10.1073/pnas.96.20.11428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goodman AL, McNulty NP, Zhao Y, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 2009;6:279–289. DOI: 10.1016/j.chom.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods 2009;6:767–772. DOI: 10.1038/nmeth.1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang JK, Pritchett MA, Lampe DJ, et al. In vivo transposon mutagenesis of the methanogenic archaeon Methanosarcina acetivorans C2A using a modified version of the insect mariner-family transposable element Himar1. Proc Natl Acad Sci U S A 2000;97:9665–9670. DOI: 10.1073/pnas.160272597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Morero NR, Zuliani C, Kumar B, et al. Targeting IS608 transposon integration to highly specific sequences by structure-based transposon engineering. Nucleic Acids Res 2018;46:4152–4163. DOI: 10.1093/nar/gky235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maragathavally KJ, Kaminski JM, Coates CJ. Chimeric Mos1 and piggyBac transposases result in site-directed integration. FASEB J 2006;20:1880–1882. DOI: 10.1096/fj.05-5485fje [DOI] [PubMed] [Google Scholar]
- 26. Owens JB, Urschitz J, Stoytchev I, et al. Chimeric piggyBac transposases for genomic targeting in human cells. Nucleic Acids Res 2012;40:6978–6991. DOI: 10.1093/nar/gks309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Owens JB, Mauro D, Stoytchev I, et al. Transcription activator like effector (TALE)-directed piggyBac transposition in human cells. Nucleic Acids Res 2013;41:9197–9207. DOI: 10.1093/nar/gkt677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo W, Galvan DL, Woodard LE, et al. Comparative analysis of chimeric ZFP-, TALE- and Cas9-piggyBac transposases for integration into a single locus in human cells. Nucleic Acids Res 2017;45:8411–8422. DOI: 10.1093/nar/gkx572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Feng X, Bednarz AL, Colloms SD. Precise targeted integration by a chimaeric transposase zinc-finger fusion protein. Nucleic Acids Res 2010;38:1204–1216. DOI: 10.1093/nar/gkp1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Strecker J, Ladha A, Gardner Z, et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 2019;365:48–53. DOI: 10.1126/science.aax9181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Klompe SE, Vo PLH, Halpin-Healy TS, et al. Transposon-encoded CRISPR-Cas systems direct RNA-guided DNA integration. Nature 2019;571:219–225. DOI: 10.1038/s41586-019-1323-z [DOI] [PubMed] [Google Scholar]
- 32. Qi LS, Larson MH, Gilbert LA, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013;152:1173–1183. DOI: 10.1016/j.cell.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bikard D, Jiang W, Samai P, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res 2013;41:7429–7437. DOI: 10.1093/nar/gkt520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gilbert LA, Larson MH, Morsut L, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013;154:442–451. DOI: 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 2014;32:577–582. DOI: 10.1038/nbt.2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsai SQ, Wyvekens N, Khayter C, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 2014;32:569–576. DOI: 10.1038/nbt.2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gaudelli NM, Komor AC, Rees HA, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017;551:464–471. DOI: 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Komor AC, Kim YB, Packer MS, et al. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533:420–424. DOI: 10.1038/nature17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chaikind B, Bessen JL, Thompson DB, et al. A programmable Cas9-serine recombinase fusion protein that operates on DNA sequences in mammalian cells. Nucleic Acids Res 2016;44:9758–9770. DOI: 10.1093/nar/gkw707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kearns NA, Pham H, Tabak B, et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 2015;12:401–403. DOI: 10.1038/nmeth.3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hilton IB, D'Ippolito AM, Vockley CM, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 2015;33:510–517. DOI: 10.1038/nbt.3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bhatt S, Chalmers R. Targeted DNA transposition in vitro using a dCas9-transposase fusion protein. Nucleic Acids Res 2019;47:8126–8135. DOI: 10.1093/nar/gkz552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pickens LB, Tang Y, Chooi YH. Metabolic engineering for the production of natural products. Annu Rev Chem Biomol Eng 2011;2:211–236. DOI: 10.1146/annurev-chembioeng-061010-114209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Esvelt KM, Smidler AL, Catteruccia F, et al. Concerning RNA-guided gene drives for the alteration of wild populations. eLife 2014;3 DOI: 10.7554/eLife.03401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ronda C, Chen SP, Cabral V, et al. Metagenomic engineering of the mammalian gut microbiome in situ. Nat Methods 2019;16:167–170. DOI: 10.1038/s41592-018-0301-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 2012;22:939–946. DOI: 10.1101/gr.128124.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–359. DOI: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sundaresan R, Parameshwaran HP, Yogesha SD, et al. RNA-independent DNA cleavage activities of Cas9 and Cas12a. Cell Rep 2017;21:3728–3739. DOI: 10.1016/j.celrep.2017.11.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vigdal TJ, Kaufman CD, Izsvak Z, et al. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J Mol Biol 2002;323:441–452. DOI: 10.1016/s0022-2836(02)00991-9 [DOI] [PubMed] [Google Scholar]
- 50. Trubitsyna M, Morris ER, Finnegan DJ, et al. Biochemical characterization and comparison of two closely related active mariner transposases. Biochemistry 2014;53:682–689. DOI: 10.1021/bi401193w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Milo R, Jorgensen P, Moran U, et al. BioNumbers—the database of key numbers in molecular and cell biology. Nucleic Acids Res 2010;38:D750–753. DOI: 10.1093/nar/gkp889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lampe DJ. Bacterial genetic methods to explore the biology of mariner transposons. Genetica 2010;138:499–508. DOI: 10.1007/s10709-009-9401-z [DOI] [PubMed] [Google Scholar]
- 53. Warming S, Costantino N, Court DL, et al. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 2005;33:e36 DOI: 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li XT, Thomason LC, Sawitzke JA, et al. Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res 2013;41:e204 DOI: 10.1093/nar/gkt1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. DeVito JA. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res 2008;36:e4 DOI: 10.1093/nar/gkm1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu D, Chalmers R. Hyperactive mariner transposons are created by mutations that disrupt allosterism and increase the rate of transposon end synapsis. Nucleic Acids Res 2014;42:2637–2645. DOI: 10.1093/nar/gkt1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.