

NeuroLNC, a previously unknown long ncRNA, regulates neuronal activity by interacting with the neurodegeneration protein TDP-43.
Abstract
The cellular and the molecular mechanisms by which long noncoding RNAs (lncRNAs) may regulate presynaptic function and neuronal activity are largely unexplored. Here, we established an integrated screening strategy to discover lncRNAs implicated in neurotransmitter and synaptic vesicle release. With this approach, we identified neuroLNC, a neuron-specific nuclear lncRNA conserved from rodents to humans. NeuroLNC is tuned by synaptic activity and influences several other essential aspects of neuronal development including calcium influx, neuritogenesis, and neuronal migration in vivo. We defined the molecular interactors of neuroLNC in detail using chromatin isolation by RNA purification, RNA interactome analysis, and protein mass spectrometry. We found that the effects of neuroLNC on synaptic vesicle release require interaction with the RNA-binding protein TDP-43 (TAR DNA binding protein-43) and the selective stabilization of mRNAs encoding for presynaptic proteins. These results provide the first proof of an lncRNA that orchestrates neuronal excitability by influencing presynaptic function.
INTRODUCTION
Long noncoding RNAs (lncRNAs) are defined as RNA transcripts longer than 200 nucleotides that show no evidence of protein coding potential (1). While initially considered a “noisy” by-product of pervasive eukaryotic genome transcription (2), novel roles of lncRNAs have begun to emerge, and their investigation is rapidly gaining momentum in multiple fields of biology (3). Their significance is especially salient in the field of cancer biology, where lncRNAs have been found to be excellent biomarkers for predicting phenotypic changes in malignancies (4).
In the brain, the high abundance and strict developmental regulation of lncRNAs suggest that these molecules have bona fide regulatory functions in neuronal differentiation and possibly play a role in the establishment of disease (5, 6). Since lncRNAs can be rapidly turned over (7), their function might be of particular importance to the swift fine-tuning of synaptic activity during the delicate establishment of neuronal circuits and during continuous structural plasticity in the brain (8–10). Thus, it is imperative to understand the precise biological roles of these molecules in brain development and neuronal activation.
Notwithstanding, the importance of lncRNAs in the brain is often underestimated because of the inherent difficulty in designing coherent experimental strategies. A major confounding factor in these studies is the nonstringent transcription of neighboring genes, which often leads to false-positive correlations for lncRNAs that do not have a “true” molecular function. As a result, the number of candidates that need to be screened in these experiments is disproportionately high, and they provide relatively few functional lncRNAs. Moreover, the heterogeneous nature of the lncRNAs’ molecular functions means that their discovery necessitates the use of highly complex and often unestablished analysis techniques (11). These factors have inarguably hindered the study of lncRNAs, despite the biological significance of these molecules.
To gain more insight into the role of lncRNAs in the regulation of neuronal activity, here, we have developed a two-step screening strategy that lessens the confounding effect of pervasive transcription. Our approach takes advantage of a bioinformatic preselection, which screens for conserved correlation across species, followed by an assay for measuring synaptic vesicle (SV) exo-endocytosis, where the lncRNAs of interest are overexpressed. Because of the initial overexpression, we were able to discard all candidates that do not have a “true” molecular function early on in our experimental workflow. Our approach has allowed us to identify an lncRNA that we named neuroLNC, which is highly neurospecific and developmentally regulated and controls several crucial aspects of neuronal physiology. We also examined the molecular interactions of neuroLNC in detail with omics strategies and found that it interacts with the RNA-binding protein TDP-43 (TAR DNA binding protein-43) and promotes the selective stabilization of mRNAs encoding for presynaptic proteins. We propose that neuroLNC orchestrates neuronal excitability and influences the posttranscriptional regulation of a set of transcripts by serving as a nuclear organizing hub, implicating TDP-43 in the coordination of neurotransmitter release. More extensively, neuroLNC may be essential for ensuring efficient structural plasticity, and due to its interaction with TDP-43, might play a role in the modulation of disease states.
RESULTS
A screening strategy for lncRNAs involved in SV release identifies neuroLNC as a critical regulator of neurotransmitter release
While the roles of lncRNAs during neurogenesis and in the early steps of neuronal development have been more intensively studied (12), their importance during terminal differentiation, and in particular, in SV exo-endocytosis, is virtually unknown. We thus decided to specifically study this process, which arises relatively late during neuronal differentiation (13). To this aim, we developed a two-step screening strategy for identifying lncRNAs that regulate neurotransmitter release in primary neurons. Our strategy combines an initial “guilt by association” bioinformatic approach [global microarray meta-analysis (GAMMA)] based on correlation networks constructed using public transcriptional data (14) and a subsequent assay for measuring SV release based on a genetically encoded reporter (Fig. 1A) (15). When applying this approach, our GAMMA query yielded a total of 79 lncRNA candidates. We filtered these further to include only those transcripts that are distant from protein-coding genes conserved in rats, mice, and humans (data S1A). The resulting six candidates, detailed in data S1B, were cloned and further tested in an SV release assay. This allowed us to identify an lncRNA, “uc003wst.1,” which upon overexpression drastically increases the release of SVs in primary rat hippocampal neurons [351 and 268% increases following 60 and 600 action potentials (APs), respectively; Fig. 1, B and C, and fig. S1A], without affecting the total vesicle pool size (fig. S1B). This lncRNA only partially overlaps with the previously annotated LINC00599, depending on the gene assembly and the species analyzed (data S1B). Hence, to avoid confusion, we named it “neuroLNC.” Down-regulation of endogenous neuroLNC by short hairpin RNAs (shRNAs) resulted in decreased SV release (Fig. 1, D and E, and fig. S1, C to G), showing that its expression levels are directly related to the extent of neurotransmission.
Fig. 1. An integrated screening strategy identifies NeuroLNC, a conserved lncRNA that regulates SV release.
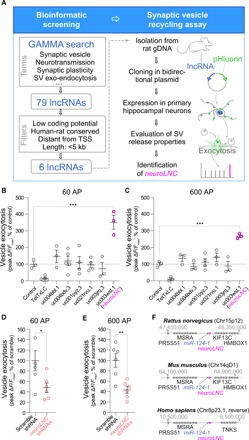
(A) Scheme depicting the experimental steps of the bioinformatic strategy and of the SV assay. gDNA, genomic DNA. (B) Quantification of the pHluorin experiments upon the pBI-driven overexpression of the lncRNAs following 60-AP field stimulation at 20 Hz. Mean values are plotted (± SEM). The tetanus toxin light chain (TetTxLC) was used as a negative control. Statistical test, one-way analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test versus control (***P < 0.001). (C) Quantification and statistics as in (B), following a 600-AP field stimulation at 20 Hz. (D and E) Quantification of the SV exo-endocytosis experiments following either a 60- or 600-AP 20-Hz field stimulation, respectively, upon neuroLNC down-regulation. Statistical test, unpaired two-tailed Student’s t test (*P < 0.05 and **P < 0.01). (F) Schematic representation of the genomic locus of neuroLNC for rat, mouse, and human.
NeuroLNC is conserved in rodents and humans, and its genomic locus is close to miR-124-1
Since lncRNAs can exert their function by influencing the transcripts in cis in their close proximity, we characterized the neuroLNC locus and its genomic organization in detail. NeuroLNC is highly conserved between rat and mouse, showing an overall 90.4% similarity and 87.7% identity. Orthologous analysis in rat, mouse, and human shows that for all three organisms its sequences have very low coding potential (fig. S2A) and that they share two conserved regions (CR1 and CR2) with ~80% homology, in agreement with what is observed for other notable lncRNAs (fig. S2B) (16). The genomic localization and the expression of neuroLNC are conserved among rodents and humans. While the gene itself is located on different chromosomes for different species, it always shares synteny with miR-124-1, the most abundant microRNA (miRNA) expressed in neurons (Fig. 1F and fig. S2, C to E) (17). Proximity to an miRNA is expected since regions harboring lncRNAs can be organized in complex polycistronic clusters together with several miRNAs (18). Yet, despite the fact that there is a correlation between neuroLNC and miR-124-1 expression (Fig. 2A), we established that neuroLNC is a separate entity from this miRNA (Fig. 2, B and C). For this, we isolated total RNA and genomic DNA [as a polymerase chain reaction (PCR) control] from rat. The RNA was retrotranscribed with random priming and PCR-amplified to reveal the presence of different RNA species. If upon retrotranscription the PCR was positive for a specific couple of primers, then it indicated that there are RNA molecules spanning the region. On the contrary, the absence of amplification suggested that there are no RNAs spanning the entire region. In total, eight different couples of primers were designed for the region between miR-124-1 and neuroLNC, and 13 different amplification reactions were tested. While the presence of several isoforms of neuroLNC is likely, there is, however, no continuity between miR-124-1 and neuroLNC. Moreover, neuroLNC does not interfere in cis with either miR-124-1 expression levels or its processing, measured by comparing relative levels of its 3p and 5p forms using miRNA specific quantitative polymerase chain reaction (miQPCR) (Fig. 2, D to F) (19). For the sake of completeness, we also characterized the distribution of the transcriptional starting sites (TSS) in the miR-124-1/neuroLNC locus. The region harbors at least 30 TSS, which give rise to several transcripts in both sense and antisense directions with respect to both miR-124-1 and neuroLNC, confirming our PCR results (Fig. 2G and data S1C). This observation is also in line with what has been previously observed, since several lncRNAs exhibit multiple splicing isoforms and TSS, which may contribute to cell type–specific or developmental stage–specific events (20). Although this complex picture suggests the presence of several neuroLNC isoforms, here, we focused on the original neuroLNC as was defined in our bioinformatic screening (detailed in data S1B).
Fig. 2. NeuroLNC and miRNA 124-1 are two separate entities with distinct TSS.
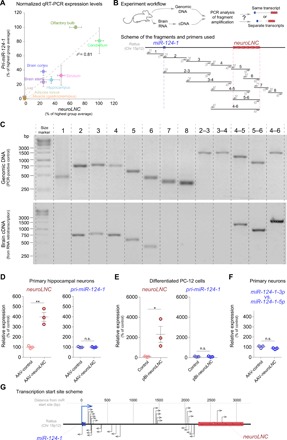
(A) Scatter plot showing the expression of neuroLNC and pri-miR-124-1. Since the two RNAs are transcribed from nearby genomic regions, we tested the correlation of the expression of the two RNAs (r2 = 0.81). (B) Experimental strategy used to test the presence of different RNA transcripts in the region between pri-miR-124-1 and neuroLNC. In the lower panel, the couple of primers used (light gray) and the fragments amplified (black) are represented to scale. (C) Results of the experiment exemplified in (B). The upper panel corresponds to the control PCR reaction on genomic DNA, and the lower panel shows the amplification of the retrotranscribed brain RNA. The lack of continuity between neuroLNC and mirR-124-1 (regions 2 and 3 and regions 3 and 4) indicates separate entities. For details concerning the primer sequences, refer to data S1J. bp, base pair; cDNA, complementary DNA. (D and E) Overexpression of neuroLNC in either primary hippocampal neurons (D) or differentiated PC-12 cells (E) does not influence the expression of pri-miR-124-1. The expression level values plotted are expressed as a percentage of the AAV-control or the control plasmid. NeuroLNC is significantly overexpressed, but there is no effect on the levels of pri-miR-124-1. Statistical tests, two-tailed Student’s t test (*P < 0.05 and **P < 0.01); n.s., not significant. (F) NeuroLNC overexpression does not influence the processing of miR-124-1 and the relative levels of its 3p and 5p forms. (G) TSS analysis. The scheme summarizes the observed starting sites, detailed in data S1C.
NeuroLNC is highly neurospecific, developmentally regulated, and activity dependent and influences calcium dynamics
We found that neuroLNC expression is highly restricted to the brain (Figs. 2A and 3A) and more specifically to the nucleus of neuronal cells (Fig. 3, B and C, and fig. S3A), while it is virtually undetectable in glial cells and cell lines used for modeling neuronal cells (fig. S3B). NeuroLNC is developmentally regulated in vitro since its levels initially peak during early development and then even out with aging (fig. S3C). In the rat brain in vivo, its expression is highest at P7 and is reduced to ~30% of its maximum in adults (fig. S3D). NeuroLNC expression in adults can still be detected in human brains as well (fig. S3E), suggesting a function that goes beyond differentiation and is kept during the entire life span of neurons.
Fig. 3. NeuroLNC is neuron specific and nuclear, regulates presynaptic Ca2+ influx, and is modulated by neuronal activity.
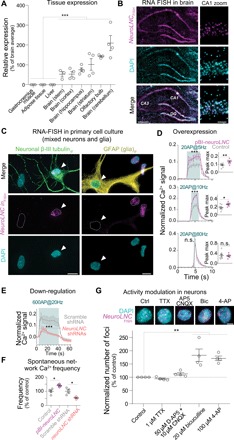
(A) Relative expression of neuroLNC in the rat central nervous system (CNS) and other tissues, as measured by quantitative real-time PCR (qRT-PCR). Statistical test, one-way ANOVA followed by Bonferroni’s multiple comparison test versus all non-CNS samples (***P < 0.001). (B) RNA fluorescence in situ hybridization (RNA-FISH) of neuroLNC on coronal brain slice of rat (P14). Note the localization to the nuclei of CA1 neurons in the zoom. Scale bars, 0.5 mm (left); 50 μm (right). DAPI, 4′,6-diamidino-2-phenylindole. (C) Combined RNA-FISH and immunofluorescence in primary mixed neuroglial cultures. Note that β-III tubulin–positive neurons are neuroLNC positive (left, white arrowhead), while cells positive for the glial marker glial fibrillary acidic protein (GFAP) are neuroLNC negative (right, white arrowhead). Scale bars, 10 μm. (D) Quantification of Ca2+ influx in primary hippocampal neurons upon neuroLNC overexpression following a 20-AP field stimulation at different frequencies. Mean values are plotted (± SEM). Insets indicate the difference in fluorescence measured at the maximum peak for each experiment. Statistical test, unpaired two-tailed Student’s t test (*P < 0.05; **P < 0.01; and ***P < 0.001). See fig. S5 for details. (E) Quantification of Ca2+ influx as in (D), following shRNA-mediated neuroLNC down-regulation with a 600-AP stimulation at 20 Hz. Upon neuroLNC down-regulation, Ca2+ influx is significantly decreased. (F) Physiological (unstimulated) network activity measured in neurons following overexpression or down-regulation of neuroLNC. Overexpression increases spontaneous activity, while down-regulation has the opposite effect. Unpaired two-tailed Student’s t test (*P < 0.05). (G) The number of neuroLNC foci following activity modulation of neurons. Blocking the activity of neurons with TTX or AP5 + CNQX has no effect. The number of neuroLNC foci is promoted by increasing neuronal activity by applying bicuculline or 4-AP. In the upper panels, representative images are shown. Scale bar, 5 μm. Normalized averages are shown. One-way ANOVA followed by Bonferroni’s multiple comparison test versus control (**P < 0.01).
Having confirmed the neurospecificity of neuroLNC and its involvement in terminal neuronal differentiation, we tested whether it could also affect presynaptic Ca2+ dynamics. Primary rat hippocampal neurons infected with an adeno-associated virus (AAV) overexpressing neuroLNC showed a significantly enhanced Ca2+ influx following a stimulation of 20 AP at 5 and 10 Hz but not at 80 Hz (Fig. 3D and fig. S4, A to C). On the contrary, neuroLNC down-regulation decreases the influx of Ca2+ during a prolonged 600-AP stimulation at 20 Hz (Fig. 3E). NeuroLNC levels also affect the frequencies of endogenous calcium bursts that can be observed in hippocampal cultures (Fig. 3F and fig. S4D) (21). Having demonstrated that neuroLNC is linked to neuronal activity, we turned to investigating whether the regulation of neuronal activity can influence the endogenous expression of neuroLNC. We tested this in terminally differentiated neurons [>14 days in vitro (DIV)], using single-molecule fluorescence in situ hybridization (FISH), and we evaluated whether a modulation of neuronal activity has an effect on the number of neuroLNC foci. While blocking neuronal activity with either tetrodotoxin (TTX) or 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX) had no direct effect on neuroLNC foci numbers, pharmacological intensification of neuronal activity with bicuculline or 4-aminopyridine (4-AP) significantly increased the number of foci 5 hours after treatment (Fig. 3G).
NeuroLNC regulates neurite elongation in vitro and neuronal migration in vivo
Given the role of lncRNAs and Ca2+ dynamics in neuronal development (12, 22) and the maximal expression of neuroLNC in the young brain (fig. S3D), it appeared likely that neuroLNC could also influence other developmental aspects in neurons. We therefore studied the role of neuroLNC in neurite growth in an in vitro assay that is used as a paradigm to study early neuronal development. Down-regulation of neuroLNC in rat primary hippocampal neurons decreased both neurite length and branching (Fig. 4, A and B), while its overexpression had the opposite effect (Fig. 4, C and D). As neurite growth is instrumental for neuronal polarization and migration (23), we tested whether neuroLNC could also influence mouse cortical neuron migration in vivo. Following in utero electroporation of shRNA constructs under a pol III promoter (U6), the electroporated precursor cells in the ventricular zone (VZ) terminally differentiate and become neurons. Neurons move to reach their final position in the cortical plate (CP), migrating through the subventricular zone (SVZ) and intermediate zone (IZ), eventually establishing connections with the marginal zone (MZ). NeuroLNC down-regulation decreases the migration of neurons in vivo from the VZ to the CP (Fig. 4, E and F). NeuroLNC overexpression has the opposite effect, since neurons show a more efficient migration pattern from the SVZ with respect to the control (Fig. 4, G and H). Together, the results presented so far suggest that neuroLNC exerts regulatory effects on the biology of neurons at different developmental stages and likely participates in the fine-tuning of neurotransmission.
Fig. 4. NeuroLNC influences neurite elongation and branching in vitro and neuronal migration in vivo.
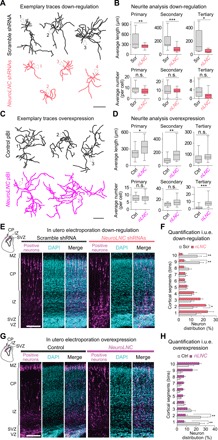
(A) Exemplary traces of reconstructed primary hippocampal neurons transfected with either scramble shRNAs or a pool of three shRNAs against neuroLNC. White arrowheads indicate position of the cell body, and numbers designate different cells. Scale bar, 250 μm. (B) Summary of morphometric analysis on reconstructed neurons upon neuroLNC down-regulation. Box plots summarize the data distribution of ~700 neurites as explained in the methods. Unpaired two-tailed Student’s t test (*P < 0.05; **P < 0.01; and ***P < 0.001). (C) Exemplary traces of reconstructed neurons as in (A), but upon neuroLNC overexpression. Scale bar, 250 μm. (D) Summary of the morphometric analysis of reconstructed neurons. Tracing and analysis of ~1000 neurites as in (B). (E) Exemplary confocal images of neuronal cells in E17.5 mouse cortices following E14.5 in utero electroporation (i.u.e.) with either scramble shRNAs or the shRNA pool against neuroLNC. Upper-left inset represents the portion of the coronal section imaged. Green fluorescent protein (GFP) was included for visualization of electroporated neurons (positive neurons are color-coded in magenta). Scale bar, 100 μm. (F) Frequency distribution and quantification of positive neurons upon neuroLNC down-regulation from the VZ (1st cortical segment) to the CP (10th cortical segment). Equal bins are used and data indicate mean ± SEM (experimental n = 5 in control and 7 in neuroLNC down-regulation). Two-way ANOVA with Bonferroni multiple comparison test for each single bin (*P < 0.05 and **P < 0.01). (G) Exemplary confocal images showing neuroLNC overexpression in an i.u.e. designed as in (E). A GFP version of the same plasmid was used as a negative control and coelectroporated for identifying positive neurons (color-coded in magenta). Scale bar, 100 μm. (H) Frequency distribution and quantification of positive neurons upon neuroLNC overexpression performed as in (F). Experimental n = 6 in both control neuroLNC overexpression. Two-way ANOVA with Bonferroni multiple comparison test for single bin pairs (**P < 0.01).
NeuroLNC interacts with DNA regions related to SV release and synapse organization
In neurons, lncRNAs may act as scaffolding adaptors in macromolecular complexes that finely regulate the execution of gene expression programs at different steps of mRNA and protein biogenesis (12). This could explain how a single molecular species can influence in trans several unrelated mechanisms such as neuritogenesis, calcium signaling, and neurotransmitter release. To test this hypothesis, we performed a series of experiments aimed at defining the molecular interactors of neuroLNC in vivo at the DNA, RNA, and protein level (Fig. 5A). Since neuroLNC is strictly nuclear (Fig. 3C), we first applied chromatin isolation by RNA purification (ChIRP) (24) from the rat brain cortex to identify the genome regions where neuroLNC may perform its regulatory function(s).
Fig. 5. NeuroLNC interacts in vivo with DNA and mRNA sequences of genes implicated in synaptic transmission, binds TDP-43, and requires its presence for modulating SV exocytosis.
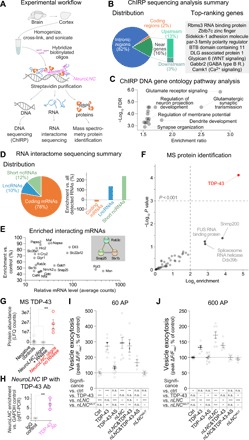
(A) Experimental workflow. For details and controls, see Materials and Methods and fig. S6. RBP, RNA binding protein. (B) Summary of DNA interactors of neuroLNC. All the hits are in data S1D. Among genes, preferentially bound top-ranking are shown in the gray inset. (C) Scatter plot of the false discovery rate (FDR, expressed as −log10) and enrichment ratio of the significant GO terms. GO analysis confirms the propensity of neuroLNC to be in proximity of genes related to chemical transmission, regulation of membrane potential, and synapse development. For an extensive list of GOs, see data S1E. (D) Summary of RNA targets found in the RNA interactome. (E) Summary of most abundant significantly enriched mRNA interactors. All mRNAs are significantly enriched versus the controls (P < 0.01), and >15 counts are represented in plot. For a comprehensive list, see data S1F. Among the most enriched mRNAs, three are also significantly enriched in STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) analysis (gray inset, FDR < 0.01). (F) Scatter plot of the MS identification of the protein interactors of neuroLNC. TDP-43 is the only highly significant hit (P < 0.001). For MS data, refer to data S1H. (G) Enrichment of TDP-43 in MS experiments, detailing the negative controls (oligos against LacZ and no probe) and RNA purification performed with neuroLNC oligos upon ribonuclease (RNase) treatment. (H) Reverse experiment, where upon immunoprecipitating TDP-43, the presence of neuroLNC was tested by qRT-PCR, and the enrichment versus the housekeeping mRNA glyceraldehyde phosphate dehydrogenase (GAPDH) was calculated. Ab, antibody; IgG, immunoglobulin G. (I and J) Quantification of the SV exo-endocytosis experiments upon pBI-driven overexpression of either TDP-43, its antisense RNA (to decrease its levels), neuroLNC, a mutated form of neuroLNC that does not contain UG-repeats and does not bind TDP-43 (neuroLNCMUT), or several combinations of these constructs. SV exo-endocytosis as in Fig. 1 (B and C), following either a 60- or 600-AP field stimulation at 20 Hz. Down-regulation of TDP-43 blocks the effects of neuroLNC at both 60 and 600 AP. The neuroLNCMUT shows no effect on SV exo-endocytosis. In both schemes below, graph significances are summarized. One-way ANOVA followed by Bonferroni’s multiple comparison test (*P < 0.05; **P < 0.01; and ***P < 0.001). LFQ, label-free quantification.
After confirming the specificity and the reliability of the experimental approach (fig. S5A), we found that neuroLNC interacts with the DNA regions of 3327 genes (Fig. 5B and data S1D). neuroLNC preferentially interacts with intronic regions (82%) and regions in the close vicinity of genes (16%), with a preference for upstream regions (Fig. 5B). This localization may suggest a function of neuroLNC in the transcription and/or splicing of these genes. Gene ontology (GO) analysis of the target genes shows that neuroLNC is significantly enriched in regions related to glutamate receptor signaling, regulation of membrane potential, and synapse organization (Fig. 5C and data S1E). These results are in line with the biological phenotype that we observe via neuroLNC modulation in neurons.
NeuroLNC binds a subset of mRNAs coding for SV proteins
To define the RNA molecules interacting with neuroLNC, we also characterized the RNAs that were coprecipitating during ChIRP in an RNA interactome experiment (Fig. 5, D and E, and data S1F). NeuroLNC mostly interacts with coding mRNAs (~78% of hits), although, the fraction of interacting ncRNAs is higher than expected from pure chance (twofold higher, χ2 statistic, 43.5; P < 0.0001; Fig. 5D). Among the most abundant mRNAs interacting with neuroLNC, there are two solute carriers (Slc35a1 and Slc22a12), the delta-like canonical Notch ligand 3 (Dll3), and three crucial mRNAs of proteins involved in neurotransmitter release (SNAP25, Stx1b, and Rab3c; Fig. 5E). The Notch signaling pathways have also been previously shown to be a target of LncND, an lncRNA that is enriched in radial glial cells in the VZ of the developing human brain (25). The fact that the overlap between the DNA-ChIRP and the RNA interactome analysis is limited (only 34 mRNAs, including Rab3c and Dll3 as detailed in data S1G) might reflect the heterogeneity of the complexes in which neuroLNC is involved.
NeuroLNC interacts with TDP-43, and the block of this interaction abolishes the effects of neuroLNC on neurotransmitter release
To define the protein interactors of neuroLNC, we also performed protein mass spectrometry (MS) upon neuroLNC enrichment, as previously described (Fig. 5, F and G; fig. S5B; and data S1H) (26). The sole highly significant enriched interactor of neuroLNC is the RNA-binding protein TDP-43, a central player in the pathogenesis of neurodegenerative disorders (27) such as frontotemporal lobar degeneration (FTLD; also known as frontotemporal dementia) and amyotrophic lateral sclerosis (ALS). We also found another important enriched player in ALS, the RNA-binding protein found in sarcoma (FUS), just below significance levels (enriched approximately seven times, P = 0.058).
Because of the importance of TDP-43, we studied its functional interactions with neuroLNC in more detail. We performed RNA immunoprecipitation (RIP) from the rat cortex with an antibody against TDP-43, and we confirmed that the endogenous TDP-43 in the brain is bound to neuroLNC (Fig. 5H and fig. S5C). Since TDP-43, which is mainly nuclear, can be delocalized to the cytosol during pathological modifications, we tested whether the overexpression of neuroLNC can delocalize it from the nucleus, but that was not the case (fig. S5D). Last, we tested whether there is any functional interaction between neuroLNC and TDP-43 in the same SV assay used in our initial SV screening (Fig. 5, I and J). To this purpose, TDP-43 was overexpressed or down-regulated (fig. S5E) in combination with neuroLNC overexpression. Since TDP-43 is known to bind a consensus sequence with uridine-guanine (UG)–repeats (28), we also included a mutated neuroLNC, where the UG-repeats were substituted (neuroLNCMUT; data S1I). Transient overexpression of either neuroLNC or TDP-43 in rat neurons increased SV release, as expected from our results, and in line with previous results obtained with neurospecific TDP-43 transgenic mice (29). The effect of neuroLNC was blocked by the down-regulation of TDP-43 or by the expression of neuroLNCMUT (Fig. 5, I and J). This indicates that at least some of the functions of neuroLNC are facilitated by its interaction with TDP-43.
NeuroLNC acts by increasing the levels of mRNA coding for SV proteins
Which is the molecular mechanism that allows neuroLNC to modulate presynaptic activity? It could be speculated that neuroLNC contributes to two distinct regulatory events, one at the level of chromatin, which regulates gene expression, and one specifically related to RNA biogenesis, splicing, or stabilization, which requires the corecruitment of TDP-43 to take place. Both of these events might result in the specific change of mRNA levels for key proteins in neuronal function. We tested this hypothesis by quantitative real-time PCR (qRT-PCR), where we examined the synaptic mRNAs that were mostly affected by AAV-mediated neuroLNC overexpression. When taken singularly, only SNAP25 and Syntaxin-1a were significantly, although modestly, increased (Fig. 6). As a side observation, because of the role of SNAP25 in the control of voltage-gated calcium channels (30), at least some of the observed calcium effects might be directly influenced by the differential expression of SNAP25. The class of mRNAs coding for SV proteins and SV protein–associated proteins was, as a whole, significantly increased upon neuroLNC overexpression (Fig. 6), while all other classes of synaptic and activity-related mRNAs tested were indistinguishable from the control (fig. S6).
Fig. 6. NeuroLNC overexpression increases the levels of SV protein and SV protein–associated mRNAs.
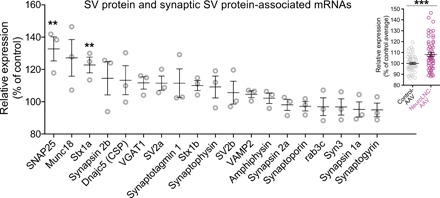
Results of the qRT-PCR following overexpression of neuroLNC in primary hippocampal neurons with an AAV. Data were GAPDH-normalized and expressed as relative increase with respect to the control cultures (infected with a control AAV). Considering individual mRNA species, only SNAP25 and Stx1a are significantly increased across all tested SV and SV-associated mRNAs. Statistical test, one-way ANOVA followed by Bonferroni’s multiple comparison test versus controls (**P < 0.01). An overall analysis of the relative expression of all the SV and SV-associated mRNAs against their expression in the controls shows a highly significant increase of the whole class (8.2 ± 1.8% increase), suggesting that the effect on all these mRNAs has a limited amplitude but is overall consistent, and single significant differences are probably too small to emerge because of the modest sampling depth. Statistical test, two-tailed Student’s t test (***P < 0.001).
DISCUSSION
The complex developmental programs of neurons rely on finely tuned regulatory mechanisms, which shape the architecture of brain circuits during synapse formation and stabilization. The establishment of neurotransmission, exemplified by SV exo-endocytosis and neurotransmitter release, has a direct influence on these regulatory mechanisms (31). Previous studies have revealed that hundreds of lncRNAs are dynamically regulated during neuronal development, brain activity, and synapse plasticity (5, 32, 33). Nevertheless, these observations often lack a causal relationship, and a direct link between lncRNA expression and SV exo-endocytosis has never been previously elucidated. To study this issue in detail, here we designed a strategy comprising a bioinformatic approach and a subsequent biological assay that quantifies SV release. Our strategy allowed us to identify neuroLNC, a nuclear lncRNA that not only regulates SV release but also influences other key aspects of neuronal physiology such as Ca2+ dynamics, neurite growth, and neuronal migration.
The initial bioinformatic strategy that we used enabled us to narrow down the most promising candidates that we then studied in detail with more time-consuming techniques on primary neurons. An obvious downside of our approach is that, although we specifically searched for keywords related to neurotransmission and brain plasticity, there is no direct correspondence between the broad terms used in the literature and the phenotypic behaviors of cells. This might explain why at the completion of our screening only one of the candidates turned out to influence the SV recycling assay that we used. Some of the search terms that are used for the bioinformatic screening may encompass different molecular mechanisms. Thus, we cannot exclude that some of the lncRNAs that we initially selected can still influence aspects of neuronal physiology that were not revealed by our biological validation. An obvious improvement for similar bioinformatic strategies would be to sample literature terms directly linked to more technical experimental aspects. At the same time, since we lack highly standardized experimental procedures for the moment, this might not improve the discovery success rate of the approach. Despite these limitations, the dual strategy that we describe here can be adapted to individual experimental needs and has the potential of serving as a blueprint for several future lncRNA studies.
Our screening led to the identification of neuroLNC, an activity-dependent lncRNA, which is developmentally regulated and localizes to the nucleus of neurons. One of the criteria that we used for the refinement of the initial bioinformatic results was a certain degree of conservation between humans and rodents, which would suggest that neuroLNC function is also conserved across species. An important observation is that, in contrast to several other developmentally regulated lncRNAs, its expression persists throughout adulthood, as observed in rat and human. This indicates that its role might be maintained in adult neurons, where it may contribute to activity regulation and structural plasticity. While this initial work focused on the developmental aspects of neuroLNC, future experiments will be necessary to elucidate its role in synapse maintenance and presynaptic plasticity.
Because of the large structural heterogeneity of lncRNAs, their mechanisms of action are extremely diverse (11). Natural antisense transcripts (NATs) are one of the most characterized subclasses of lncRNAs since they target protein-coding genes and directly destabilize their respective mRNAs through an antisense mechanism. In the brain, notable examples include the mRNA of the brain-derived neurotrophic factor (BDNF) and of the potassium voltage-gated channel A2 (KCNA2), which are targeted by two NATs (BDNF-AS and KCNA2-AS) and influence neuronal activity through a direct control of their respective mRNAs (9, 34). We excluded the possibility that neuroLNC acts as a NAT by characterizing in detail the genomic region from which it originates.
Our data show that neuroLNC is harbored in the vicinity of miR-124-1, but it is transcribed from an independent TSS and does not influence the stability or the processing of the neighboring miRNA (Fig. 2). For these reasons, neuroLNC should be defined as a large/long intergenic or intervening ncRNA. It is possible to speculate that the regulation of neuroLNC itself might take advantage of an antisense mechanism, since we also found some transcripts arising from the opposite direction in the neuroLNC locus. This remains for the moment hypothetical, although it is particularly interesting since it might be the basis of the feedback mechanism regulating its level during neuronal hyperactivation (Fig. 3G).
For lncRNAs that do not overlap as an antisense with a protein-coding mRNA in cis, defining the molecular mechanism of action remains extremely challenging (35), as also evidenced by the fact that the function of most lncRNAs remains unknown. One of the first steps to define the molecular role of lncRNAs is to identify their molecular interactors (11). This task is simplified by the subcellular localization of lncRNAs. NeuroLNC is strictly nuclear, and its most likely interactors include DNA, RNA, and proteins. We embarked in three systematic and unbiased screenings to describe the possible interactome of neuroLNC. Our screenings reveal that in vivo, the endogenous neuroLNC binds the DNA and the RNA of several neuronal genes implicated in neurotransmitter release, synapse organization, glutamatergic signaling, and regulation of neuritogenesis. While this does not narrow down the number of possible mechanisms of action, our findings on the physiology of neurons suggest that neuroLNC promotes the transcription of these genes and/or the stabilization of the mRNAs that are bound. We are aware that the validation of all these possible molecular interactions largely exceeds the purpose of this work; thus, we concentrated on the pathways that could explain the effects of neuroLNC on neurotransmission. We found that some key mRNAs regulating vesicle release such as SNAP25 and Syntaxin 1 are increased by overexpression (Fig. 6) and are also found interacting with neuroLNC (Fig. 5E). This is critical since SNAP25 and Syntaxin 1 are the target SNAREs (soluble N-ethylmaleimide–sensitive factor attachment protein receptors) promoting SV fusion.
Very few other lncRNAs have been directly linked to both neuronal excitability and neuronal growth and development. The most notable example is GM12371 (10), which shares several similarities with neuroLNC, including activity dependency, nuclear localization, and ability to modulate neurite complexity. At the same time, the two lncRNAs seem to have completely different mechanisms of actions, especially because the effects of GM12371 seem to be linked mostly to a postsynaptic phenotype, in contrast with the presynaptic modulation induced by neuroLNC.
An important finding is that neuroLNC binds the neurodegeneration-associated protein TDP-43 and that neuroLNCMUT, where the UG-repeats were substituted, is unable to potentiate SV release. This indicates that TDP-43 binding is essential for the activity of neuroLNC, as is also reinforced by the observation that the down-regulation of TDP-43 abolishes the effects of neuroLNC overexpression. Which is the precise molecular pathway that is regulated by the interaction between neuroLNC and TDP-43? This question is particularly difficult to address, also in light of the fact that TDP-43 modulates multiple RNA-related processes in the nucleus. These include the regulation of transcription, splicing, and RNA stability, as well as ncRNA processing (36). As an example, the activity-dependent lncRNA Gomafu might work in a similar manner, since it seems to serve as a scaffold that retains splicing factors that are released upon transient stimulation (32). Also in the case of Gomafu, the exact mechanism(s) of its function are not yet known. Moreover, TDP-43 participates in liquid-liquid and liquid-solid phase separation processes, influencing the structure of nuclear paraspeckles and of nuclear organization (37). NeuroLNC may influence any of these processes and possibly function as a scaffold for the assembly of macromolecular complexes that define the specificity of TDP-43 for presynaptic mRNAs, which result in their selective translation and/or stabilization. All these possibilities remain hypothetical and will require future experiments.
Aside from the precise details of the molecular pathway(s) that are regulated by the interaction between neuroLNC and TDP-43, one can speculate about the possible clinical significance of neuroLNC with respect to diseases such as FTLD and ALS. Future studies in humans should aim at determining whether neuroLNC expression is modified in the early stages of these diseases. Moreover, neuroLNC may be included in the detailed panel of DNA sequences analyzed in patients with FTLD and ALS. Similarly, it may be worthwhile assessing whether overexpression or down-regulation of neuroLNC in animal models modulates the development of TDP-43 proteinopathies and possibly influences the solubility of TDP-43 and/or modifies its interactome. Although promising, clinical applications related to neuroLNC should be carefully evaluated and will require future validations.
In conclusion, our results suggest that neuroLNC is involved in a feed-forward mechanism that enhances neurotransmission through the regulation of a subset of synaptic RNAs (summarized in fig. S7). More broadly, our work supports the idea that lncRNAs are pertinent for the regulation of neurotransmitter release and that their study may shed new light on the principles that govern neuronal physiology and degeneration.
MATERIALS AND METHODS
Bioinformatic screening
GAMMA is a previously developed algorithm (14) to identify correlated transcripts across microarray experiments. After the most correlated transcripts are identified, an entity-based literature-mining approach (38) is used to analyze their literature commonalities and predict ncRNA function. We considered the top 40 most highly correlated coding transcripts with the ncRNAs under scrutiny, and we analyzed their literature commonalities. These commonalities include terms such as diseases, chemicals, phenotypes, other genes, cellular functions, cellular structures, and biological processes, similar to GO associations. Thus, when function is not known, these transcriptional correlation networks can be used to infer function, tissue specificity, cellular localization, and phenotype and help prioritize experimentation. The use of GAMMA to successfully predict mRNA function has been previously reported in several other instances, but this is the first successful test of the approach to predict ncRNA functions. GAMMA was used to search for uncharacterized noncoding transcripts associated with the literature annotations: “Brain development,” “Synaptic vesicle,” “Synaptic vesicle exocytosis,” “Synaptic vesicle endocytosis,” “Regulation of synaptic plasticity,” “Synaptogenesis,” “Neurotransmission,” “Dendritic spine,” and “Neuronal protein.” These terms were used to define a “Guilt by association score” (data S1A) that was used during the first selection round. The filters used for scoring the transcripts were (i) low coding potential (as previously defined) (39); (ii) conservation across species, defined as percentage homology; (iii) distance from the TSS of a known annotated protein-coding gene; and (iv) size (sequences longer than 5 kb were excluded for obvious practical constraints).
Animal and human samples
All animal experiments were approved by the local authority, the Lower Saxony State Office for Consumer Protection and Food Safety (Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit) and the institutional ethical boards of the Katholieke Universiteit Leuven. Mixed neuroglial cultures were prepared from P2 Wistar rats. For neuroLNC, cloning and mRNA isolation samples were also collected from Wistar rats at various ages (as indicated in the respective experiments). For in utero electroporation, Swiss mice were used. All human midbrain samples were obtained from the Parkinson’s U.K. Brain Bank (Imperial College London, London, England). Snap-frozen tissue blocks were transported and stored under controlled temperature conditions (−80°C). The samples were conceded to the Department of Neurology of the University Medical Center Göttingen (Göttingen, Germany), and ethical approval was given by the Multicentre Research Ethics Committee (07/MRE09/72).
Cell culture and transfections
Mixed hippocampal neuroglial cultures and pure glia cultures were prepared from P2 rats as previously described (21). Neurons (between 10 and 14 DIVs) were transfected with the calcium-phosphate ProFection transfection kit (Promega, Fitchburg, WI, USA) as previously described (21). The use of this kit increased the reproducibility over time. Briefly, cells were incubated for 30 min in 37°C fresh Dulbecco’s modified Eagle’s medium (DMEM complemented with 10 mM MgCl2 and 5 mM Hepes at pH 7.5). For each coverslip, 50 μl of transfection mixture containing 2.5 μg of plasmid DNA, 200 mM CaCl2, and 1× Hepes-buffered solution was applied. The cultured neurons were incubated with the transfection mixture for 20 min at 37°C and 5% CO2 followed by three washes with fresh DMEM. Coverslips were placed back into their original culture medium and were cultured for 3 days at 37°C and 5% CO2 to allow transgene expression or down-regulation of the targets before further analyses. Human embryonic kidney (HEK) 293T, C6, and PC-12 cell lines were purchased from the German National Resource Center for Biological Material (DSMZ) and cultured following the recommended conditions. For transfection, PC-12 and C6 cells were detached by trypsin treatment and transfected using the SF cell line 4D-Nucleofector Kit (Lonza) following the instructions of the manufacturer. Following electroporation, transfected cells were seeded and incubated for 3 days at 37°C. The efficiency of down-regulation through TDP-43 antisense (TDP-43-AS) was tested in C6 cells followed by qRT-PCR for TDP-43. Differentiation of PC-12 cells was induced 1 day after transfection with plates coated with collagen (2 μg/ml; Roche) by adding 0.1% horse serum and nerve growth factor RPMI medium (50 ng/ml). To induce differentiation, PC-12 cells were cultured in this medium for 7 days, and the medium was changed every second day. Differentiation was assessed by morphological changes (neurite formation).
Plasmids, shRNAs, and DNA cloning
For all the oligonucleotide primers used during cloning, please refer to data S1J. Note that all plasmids were confirmed by sequencing and, upon reasonable request, they are available from the authors. The bidirectional pBI-CMV1 vector was purchased from Clontech (CA, USA) and modified with the QuikChange II Site-directed mutagenesis kit (Agilent, CA, USA) to delete the Bgl II restriction site within the multiple cloning site no. 1 (MCS1). The VAMP-2 pHluorin sequence was designed in silico on the basis of a previous publication (15), and it was ordered at GenScript (USA) and inserted in the MCS1 upon digestion with Kpn I and Not I. The plasmid encoding for the tetanus toxin light chain (pGEMTEZ-TetTxLC) was a gift from R. Jahn. TetTxLC or the six lncRNAs (data S1B) were inserted in the MCS2 of the pBI-CMV1-pHluorin plasmid with a PCR strategy upon either pGEMTEZ-TetTxLC or Rat genomic DNA amplification. The cloning primers used in the PCR strategy for insertion into the MCS2 added the following couples of restriction sites during amplification: Bcl I/Spe I (for TetTxLC and uc003wst.1), Bgl II/Xba I (uc021rvu.1, uc004bbl.1 and uc003jrq.3), Bgl II/Spe I (uc001pyz.3), or Bam HI/Xba I (uc004bdv.3). The X1 variant of rat TDP-43 (NCBI Reference Sequence: XM_006239382.3) was PCR-amplified from rat complementary DNA (cDNA) to add the Xba I and Sac II restriction sites and was inserted into the pBI-CMV1-pHluorin plasmid construct. For cloning of TDP-43-AS, the PCR fragment was inserted in antisense orientation using Bsu36 I and Sac II restrictions. The plasmid ensured an efficient reduction in TDP-43 expression levels with respect to control-transfected C6 cells as tested by qRT-PCR (fig. S5E). For in utero overexpression, neuroLNC was subcloned into the pCAGEN vector (obtained from Addgene) with a PCR strategy adding the Xho I and the Not I restriction sites. For viral overexpression, neuroLNC was cloned into an AAV backbone that was previously described (40) allowing the specific expression in neurons (through the human synapsin-1 promoter). For cloning in the viral vector, we used a PCR strategy allowing the addition of the restriction sites Asc I (also known as Sgs I) and Sbf I (also known as Sda I). Positive viral clones were always tested for the integrity of their inverted terminal repeats. For down-regulation, shRNAs against rat and mouse sequences were designed using the Biosettia tool (available at http://biosettia.com/support/shrna-designer/, Biosettia, CA, USA) and cloned following the manufacturer’s instructions into the pRNA1-M6-green plasmid [Biosettia; expressing green fluorescent protein (GFP) concomitantly with the shRNA]. As a negative control, the scramble vector provided by Biosettia was used. Note that among several tested down-regulation methods only the use of the mouse U6 pol-III promoter for the expression of shRNAs was efficiently down-regulating neuroLNC. The neuroLNCMUT sequence was designed in silico (as specified in data S1I) and was ordered at GenScript (USA). For all subclonings, we relied on commercial chemically competent bacteria (α-select bronze competent cells, Bioline or SURE bacteria from Agilent in case of adeno-associated viral constructs). For transformation, 50 μl of bacteria was heat-shocked at 42°C for 35 s and cooled briefly before addition of 400 μl of Super Optimal broth with Catabolite repression (SOC) medium (Sigma-Aldrich). After incubation for 45 min at 37°C and shaking at 300 rpm, cells were centrifuged and plated on agar plates with the appropriate antibiotic selection. Colonies were picked on the following morning and grown in LB. Midi preps were prepared using the Macherey-Nagel NucleoBond Xtra Midi EF Kit according to the low copy number protocol. DNA was eluted in double-distilled ultrapure water (ddH2O). Quality and amount of DNA were assessed by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
Virus preparation
Recombinant AAV particles were generated as described before (41). Briefly, vectors were propagated in HEK293T cells using the pDP6 helper plasmid (avoiding adenoviral contamination). Viral particles were purified by iodixanol step gradient centrifugation followed by heparin affinity chromatography. Fast protein liquid chromatography–purified/concentrated vectors were desalted by dialysis against phosphate-buffered saline (PBS). Viral titer was adjusted by serial dilution on primary hippocampal neurons by monitoring GFP expression levels (always expressed in a second cassette of the construct, driven by the human synapsin-1 promoter).
SV exo-endocytosis evaluation and calcium imaging
SV exo-endocytosis assays were performed as previously described (21, 42). Neurons were stimulated with an electric field generated in custom-made chambers housing 18-mm coverslips with platinum electrodes (8-mm distance between electrodes). For the generation of the electrical impulses, we used a Stimulus Isolator combined with an A310 Accupulser Stimulator (both from World Precision Instruments, USA) with an initial nominal output of 100 mA. Stimulation was performed either on neurons expressing the bidirectional series of pBI-CMV1-pHluorin plasmids (VAMP-2 based) or coexpressing the orange mOr2-Synaptophysin (mOr2-SypHy) indicator of SV recycling (21) together with the Biosettia plasmids for down-regulation. Coexpression was always confirmed by imaging both the enchanced GFP and the mOr2 channel. To test the efficiency of the pBI-CMV1-pHluorin plasmids, we included, in the initial screening, a version encoding the TetTxLC, which cleaves VAMP-2 and blocks neurotransmitter release. Unless otherwise stated, live experiments were performed in Tyrode’s solution [124 mM NaCl, 5 mM KCl, 30 mM glucose, 25 mM Hepes, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4)] supplemented with 10 μM CNQX (Tocris Bioscience, Cambridge, UK) and 50 μM 2-amino-5-phosphonopentanoic acid (D-AP5; Tocris Bioscience, Cambridge, UK) to avoid spontaneous network activation. At the end of each experiment, 50 mM NH4Cl was applied to evaluate total SV content. Live imaging was performed with an inverted Nikon Ti epifluorescence microscope (Nikon, Tokyo, Japan) equipped with a Plan Apochromat 60× 1.4 numerical aperture oil immersion objective, an HBO-100W lamp, an IXON X3897 Andor camera (Northern Ireland, UK), and an Okolab cage incubator system (Okolab, Ottaviano, Italy) to maintain a constant temperature of 37°C. The system was operated through the NIS-Elements AR software (version 4.20; Nikon). ND2 Images were imported using the Bio-Formats (Open Microscopy Environment) plug-in. For quantifications, the difference in fluorescence intensity before and at the peak of the electrical stimulation (ΔF) was normalized to the maximum of fluorescence (Fmax) measured upon NH4Cl application. For simplicity, all ΔF/Fmax values are expressed as a percentage of the signal measured in control cells (either expressing pBI-CMV1-pHluorin or the scramble shRNA vector from Biosettia). Each experiment (N) corresponds to a different neuronal preparation. For each neuronal preparation, the ΔF/Fmax values were collected and averaged from ~500 synapses from three or more coverslips. Image analysis was performed by identifying the boutons in the exocytotic hotspots with a custom-made National Institutes of Health ImageJ macro based on the “Time Series Analyzer” ImageJ plugin (available at http://rsbweb.nih.gov/ij/plugins/time-series.html). For visualization of calcium dynamics, neurons were loaded in their own medium for 30 min with the acetoxymethyl ester version of Cal-590 (Cal-590-AM; AAT Bioquest), a fluorescein-based Ca2+ indicator with single-photon excitation and emission peak wavelengths of 570 and 590 nm, respectively. After a 10-min reequilibration in their culture medium without dye, coverslips were quickly washed in Tyrode’s solution. Neurons were imaged and stimulated in the same settings as for the SV exo-endocytosis assay. Image analysis measuring signal intensities was performed using custom ImageJ macros. Briefly, circular regions of interest were placed on transfected cell bodies (identified by the GFP signal), and fluorescence was background-corrected. For each condition more than eight fields from three different preparations were measured. For the analysis of spontaneous activity, cells were imaged in the absence of CNQX and D-AP5, peaks surpassing baseline noise by at least twofold were recorded, and the frequency was determined. For stimulated activity (in the presence of CNQX and D-AP5), ΔF was calculated in a 10-s time frame around the stimulation maximum. ΔF was then normalized to the baseline signal measured just before the beginning of the stimulation (F0). For all experiments with neuroLNC overexpression, we also performed the analysis of the fluorescence at the peak maximum (which, depending on the timing of the imaging and of the stimulation, might be slightly shifted in different experiments).
Pharmacology
For the pharmacological modulation of neuronal activity, we used 1 μM TTX (Na+ Channel blocker; Cayman Chemical, USA), 20 μM bicuculline methobromide (GABAA receptor antagonist; Tocris Bioscience, Cambridge, UK), 100 μM 4-AP (K+ channel blocker; Tocris Bioscience, Cambridge, UK), 10 μM CNQX (AMPA/kainate receptor antagonist; Tocris Bioscience, Cambridge, UK), and 50 μM D-AP5 (N-methyl-d-aspartate receptor antagonist; Tocris Bioscience, Cambridge, UK). A total of >200 neurons from three different cultures were blindly analyzed for each condition.
Neurite elongation assay
Primary hippocampal neuron cultures were transfected at 10 DIVs with either a pool of three shRNAs or the pBI construct to down-regulate or overexpress neuroLNC, respectively. A total of >20 neurons per condition were analyzed following 4 days of expression, from three independent cultures. Neurons were manually traced in a blind experimental design and analyzed with the ImageJ plugin NeuronJ. The aspects considered were the average length and the number of primary, secondary, and tertiary neurites (arising respectively from the cell body, from a primary neurite, or from a secondary neurite).
In utero electroporation
Pregnant Swiss mice (E14.5) were anaesthetized by intramuscular injections of solution containing 117 μg of ketamine (Anesketin, Eurovet) and 0.7 μg of medetomidine hydrochloride (Domitor, Pfizer) per gram of body weight. Uterine horns were exposed, and a mixture of the required plasmids (1 to 2 μg/μl) was microinjected with the “Fast Green” dye (Sigma-Aldrich) in the lateral ventricles of embryos. Five current pulses (50-ms pulse/950-ms interval) were delivered across the head of the embryos (36 V), targeting the dorsal-medial part of the cortex. After 3 days of expression, the mothers were euthanized, the embryos were dissected, and the brains were quickly perfused with PBS and 4% paraformaldehyde (PFA). After dissection, the brains were post-fixed for 6 to 10 hours in 4% PFA at 4°C. Vibratome sections (100 m) were collected and prepared for imaging. Analysis of cell migration was performed as previously described (23) by dividing the cortical regions in 10 equal bins and by counting the number of positive neuronal nuclei in each bin. Note that also for neurons showed in Fig. 4 (E versus G), there are differences in the migration of neurons in the controls between experiments, as often observed when different control plasmids are used.
Single-molecule RNA in situ hybridization and immunofluorescence
FISH was performed with minor modifications from the method described in the QuantiGene ViewRNA ISH Cell Assay kit (Affymetrix). Briefly, adherent neuronal cultures were washed in Tyrode’s solution before being fixed in 4% PFA (Sigma) in PBS. Samples were further washed in PBS, and the detergent solution was provided by the manufacturer. Specific neuroLNC or glyceraldehyde phosphate dehydrogenase (GAPDH) oligonucleotide probes were designed and ordered from Affymetrix, diluted in probe diluent solution, and used for the hybridization. Coverslips were incubated upside down on the probe mix solution in a humidified chamber for 3 hours at 40°C. Alternating with washes in SSC buffer (UltraPure SSC, Thermo Fisher Scientific), incubation with the preamplifier, amplifier, and label mix (1:50 in respective diluent; 30 min at 40°C each) followed. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole), and coverslips were embedded on object slides in Mowiol (Calbiochem, Merck).
Quantitative real-time PCR
For RNA isolation from cells or rodent tissues, the miRNeasy Mini Kit (Qiagen) was used. First-strand cDNA was synthesized using SuperScript IV (Life Technologies) according to the manufacturer’s protocol. For qRT-PCR analyses, the LightCycler 480 SYBR Green I Master kit (Roche) was used on a LightCycler 480 Instrument II (Roche). All amounts were normalized to their GAPDH controls. miRNA expression level was assessed using the miQPCR method as previously described (19). For human samples, midbrain blocks were transferred shortly to a cryostat for the sake of having controlled temperature conditions during sampling (−20°C). Tissue punches were extracted from the frozen blocks using a 20-G Quincke Spinal Needle (Becton Dickinson), and the sampled material (around 20 mg) was stored into ribonuclease (RNAse)/deoxyribonuclease-free tubes. Total RNA was isolated from the samples using TRI Reagent (Sigma-Aldrich) following the manufacturer’s protocol. RNA precipitates were reconstituted in 15 μl of nuclease-free water, and the samples were cleaned and concentrated using the column-based RNA Clean & Concentrator-5 KIT (Zymo Research). RNA (1 μg) from each patient sample was reverse-transcribed using the QuantiTect Reverse Transcription Kit (Qiagen). cDNA samples were diluted 1:3 in nuclease-free water and kept at −20°C until further use. For human GAPDH, we used the primers “Hs_GAPDH_1_SG” provided in the QuantiTect Primer Assay (QT00079247). Unless otherwise stated, all other primers used in this study are summarized in data S1J.
Library preparation and sequencing
Library preparation for mRNA sequencing was performed as previously described (43). Library preparation for ChIRP sequencing was performed according to the NEBNext Ultra DNA Library Prep Kit for Illumina (New England BioLabs). Libraries were tested for quality and quantified using a NanoDrop 2000 (Thermo Fisher Scientific) and an Agilent 2100 Bioanalyzer (Agilent Technologies). Single-end 50–base pair (bp) sequencing data were generated on an Illumina HiSeq2000 using Illumina TruSeq SBS kits.
RNA-sequencing analysis
Quality assessment was based on the raw reads using the FASTQC (v0.10.1). The sequence reads (single-end 50 bp) were aligned to the rat reference genome (Rnor_6.0) with Bowtie2 (v2.0.2) using RSEM (v1.2.29) with default parameters. First, the rat reference genome was indexed using the Ensembl annotations (v83.6) with rsem-prepare-reference from RSEM software. Next, rsem-calculate-expression was used to align the reads and quantify the gene and isoform abundance. The output of rsem-calculate-expression separately gives the read count and transcripts per million value for each gene and isoform. The enrichment was at the end calculated for each biological experiment separately as summarized in data S1F. Briefly, the enrichment was defined for the odd and the even set separately, and an average enrichment was calculated across all enriched conditions.
ChIRP DNA analysis
Data were analyzed as previously described (44). Briefly, reads were aligned to the rat reference genome (Rnor_6.0) using Bowtie (v2.0.2) with default parameters allowing for two mismatches using seed alignment. Subsequently, aligned reads were filtered for high-quality, uniquely, and multimapped reads (MAPQ ! = [0, 2, 3, 4]). High-quality BAM files were merged for each condition using samtools (v0.1.18). Peak calling was performed using MACS2 (v2.1.2) with a minimum false discovery rate of 0.05. Peaks were assigned to the genes using PAVIS if they were located within 5 kb upstream of the TSS or in the transcribed region or within 1 kb downstream of the transcription end site.
GO and STRING analyses
GO analysis was conducted using WebGestalt using as a reference list all mapped entrez gene IDs from the selected (rnorvegicus) genome. For significance, we used an adjusted P value < 0.05 using the Benjamini-Hochberg method for controlling the false discovery rates during GO terms in biological processes. For details, please refer to data S1E. STRING analysis was conducted as previously described (43). Also in this case, for testing significance, we relied on an adjusted P value < 0.05 using the Benjamini-Hochberg method for controlling the false discovery rates.
5′-RACE PCR
For rapid amplification of 5′-cDNA ends, the GeneRacer Kit with SuperScript III RT and the TOPO TA Cloning Kit for Sequencing (Invitrogen) were used. The gene-specific primers (data S1J) were designed for the 5′ end amplification, and nested PCR products were cloned and sequenced for analyses as reported in data S1C.
ChIRP and RNA interactome analysis
ChIRP and RNA interactome experiments were performed as previously described (24, 26). Antisense oligo probes with 3′ biotin-triethyleneglycol modification (TEG) were designed using the online probe designer on the ChIRP Probe Designer (version 4.2, LGC Biosearch Technologies, Middlesex, UK) and synthesized by Sigma-Aldrich (data S1J). Note that the designed primers were blasted against whole rat genome and do not specifically bind other targets than neuroLNC. The 10 designed probes for neuroLNC were divided into two tiling pools (arbitrarily defined odd and even pools). We also designed 10 LacZ probes, which served as negative controls. We performed the neuroLNC enrichment from the brain of 2-day-old (P2) rats where neuroLNC expression is high. For ChIRP, cerebral cortices were homogenized with a micropestle in 500 μl of ice-cold low-sucrose buffer [0.32 M sucrose, 5 mM CaCl2, 5 mM Mg(Ac)2, 0.1 mM EDTA, 50 mM Hepes (pH 8), 1 mM dithiothreitol (DTT), and 0.1% Triton X-100]. Subsequently, the cortex homogenate was cross-linked with either 3% formaldehyde or 1% glutaraldehyde and sonicated for either 30 or 120 min for either ChIRP MS or ChIRP DNA/RNA, respectively. Enrichment of neuroLNC was tested by qRT-PCR for ChIRP RNA samples. ChIRP-derived RNA and DNA were sequenced, and eluted proteins were analyzed by MS proteomics as previously described (43).
MS identification of protein interactors following ChIRP/RNA interactome analysis
For proteomic analysis, we initially reversed the cross-linking by boiling the beads in a final concentration of 1× NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) at 95°C for 30 min on a thermomixer (at 750 rpm). After boiling, the samples were loaded on precast NuPAGE 4 to 12% Bis-Tris Protein Gels (Thermo Fisher Scientific) and separated for 20 min at constant voltage (200 V). The gels were stained overnight with Coomassie G250, and for each lane, five to six pieces were cut. In-gel digestion was performed overnight using trypsin. Samples were dried, and peptides were resuspended in 5% acetonitrile and 0.05% trifluoroacetic acid. Samples were further processed for liquid chromatography–MS in an online UltiMate 3000 RSLCnano high-performance liquid chromatography system (Thermo Fisher Scientific) coupled online to an Orbitrap Fusion Tribrid MS instrument (Thermo Fisher Scientific). Peptides were desalted on a reversed-phase C18 precolumn (3 cm long; inner diameter, 100 μm; outer diameter, 360 μm) for 3 min. After 3 min, the precolumn was switched online with the analytical column (~30 cm long; inner diameter, 75 μm) prepared in-house using ReproSil-Pur C18 AQ 1.9-μm reversed-phase resin. The peptides were separated with a linear gradient of 5 to 50% buffer B (80% acetonitrile and 0.1% formic acid) at a flow rate of 10 nl/min over 58-min gradient time. The temperature of the precolumn and the column was set to 50°C during chromatography. The MS data were acquired by scanning the precursors in a mass range from 380 to 1500 Da at a resolution of 120 K at a mass/charge ratio of 200. The acquired RAW data were analyzed using MaxQuant software on the basis of the Andromeda search engine. The updated rat UniProt database was used for identifying proteins (visit the PRIDE dataset identifier PXD013434 for details).
RNA immunoprecipitation
For confirming the interaction between neuroLNC and TDP-43, we used a RIP protocol. For this purpose, the cortices of P2 rats were homogenized by a micropestle in low-sucrose buffer [0.32 M sucrose, 5 mM CaCl2, 5 mM Mg(Ac)2, 0.1 mM EDTA, 50 mM Hepes (pH 8), 1 mM DTT, and 0.1% Triton X-100] on ice. The lysate was cross-linked by 1% formaldehyde and neutralized with 1.25 M glycine. In the following steps, the lysate was washed two times with 1 ml of cold Nelson-Jameson buffer (150 mM NaCl, 20 mM EDTA, 50 mM tris, 0.5% NP-40, and 1% Triton-X-100) and incubated in lysis buffer [100 mM tris-HCl (pH 8), 20 mM EDTA, and 2% SDS] for 10 min at 4°C. The lysate was precleared with Dynabeads protein A (Invitrogen) and then incubated with rabbit anti-rat TDP-43 antibody (Proteintech, no. 10782-2-AP) or Syntaxin 1A (SYnaptic SYstem, no. 110 302) as a negative control overnight and with beads for additional 1.5 hours at 4°C. After several washing steps, RNA was eluted from beads and purified by an miRNeasy Mini Kit (Qiagen). To achieve high-quality samples, RNAse inhibitor (SUPERase.In, Invitrogen) and protease inhibitors (phenylmethylsulfonyl fluoride and Roche cOmplete EDTA-free protease inhibitor cocktail) were added to all solutions. The purified RNA was either sequenced or reverse-transcribed for qRT-PCR analyses.
Western blot
Beads derived from ChIP were resuspended in 11 μl of Laemmli Buffer (4×) and boiled for 30 min at 95°C before dilution with water. Western blot for TDP-43 protein was performed using the mouse anti-rat TDP-43 (Abcam, no. 104223) and donkey anti-mouse IgG IRDye 800CW (Licor).
Immunocytochemistry
Primary neuron coverslips were washed with Tyrode’s solution, fixed with 4% PFA for 40 min, and quenched with glycine (1 M) for 20 min. Afterward, neurons were permeabilized with PBS containing 0.1% Triton and stained with primary and secondary antibodies in PBS containing 1.5% bovine serum albumin and 0.1% Triton for 1 hour. Mouse anti-rat TDP-43 (Abcam, no. 104223) and donkey anti-mouse cyanine3 (Dianova) were used as primary and secondary antibodies, respectively. Imaging was performed using an epifluorescence Nikon Eclipse Ti-E microscope equipped with an HBO-100W Lamp and an IXON X3897 Andor Camera. Capture settings were adjusted with the Nikon Imaging Software (NIS Elements).
Statistical analysis and graph construction
Depending on the data, we used Student’s t test or analyses of variance (ANOVAs; one or two ways) followed by Bonferroni’s multiple comparison post test (as indicated in each figure). Data distribution was assumed to be normal. Equal variances were formally tested and showed no difference between the groups. No statistical methods were used to predetermine sample sizes, and data were collected and processed randomly. Data analysis was performed blind to the condition when possible, and most of the times experiments were independently performed by two or more authors. Box-plot graphs were constructed with the box portion of the plot defined by the 25th and the 75th percentile, and the line dividing the box corresponds to the median. Whiskers indicate the 90th and the 10th percentile.
Supplementary Material
Acknowledgments
We thank M. Helm and T. Dankovich for carefully reading and commenting the original draft of this manuscript. We thank S. Sadman for comments on the final version of the manuscript. Funding: E.F.F. during this work was, in part, supported by an EMBO Long-Term Fellowship and a HFSP Fellowship (EMBO_LT_797_2012 and HFSP_LT000830/2013). The work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG,1967/7-1 and SFB-1286/B02) (to S.O.R.), from the SFB-1286/Z02 (to S.B.), and from the SFB-1286/10 (to H.U.). Author contributions: Conceptualization: E.F.F., S.O.R., S.B., and J.D.W. Methodology: S.K., V.K., S.M., V.B., R.R., S.B., A.G., S.K., J.D.W., E.F., L.C.G., and E.F.F. Formal analysis: S.K., V.K., E.F., V.B., R.R., S.B., J.D.W., and E.F.F. Resources and funds acquisition: S.O.R., H.U., and S.B. Writing of initial draft: E.F.F., S.K., and V.K. Writing of final draft, review, and editing: S.K., V.K., J.D.W., S.O.R., and E.F.F. Supervision: E.F.F. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials and/or in the deposited datasets. The ChIRP DNA and RNA interactome sequencing results have been deposited to the Gene Expression Omnibus (GEO) with the identifier GSE131103. The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD013434. All other data are available in data S1 or available from authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaay2670/DC1
Fig. S1. Details of the screening strategy used for studying the role of lncRNAs in neuronal activity and down-regulation in culture.
Fig. S2. NeuroLNC coding potential, conservation, and locus organization.
Fig. S3. Specificity of the RNA-FISH approach and expression of neuroLNC in rodent cultures and in adult human brain.
Fig. S4. Controls for the calcium dynamics experiments.
Fig. S5. Controls and additional information concerning Fig. 5.
Fig. S6. NeuroLNC overexpression does not affect the expression of the mRNAs that encode for calcium-related pathways, neuronal activity, or synaptic scaffolds.
Fig. S7. Schematic representation summarizing the function of NeuroLNC in the nucleus of neurons.
Data S1. Summary of data used here, see first sheet for details.
REFERENCES AND NOTES
- 1.Kapranov P., Cheng J., Dike S., Nix D. A., Duttagupta R., Willingham A. T., Stadler P. F., Hertel J., Hackermüller J., Hofacker I. L., Bell I., Cheung E., Drenkow J., Dumais E., Patel S., Helt G., Ganesh M., Ghosh S., Piccolboni A., Sementchenko V., Tammana H., Gingeras T. R., RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Struhl K., Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat. Struct. Mol. Biol. 14, 103–105 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Miao Y., Xu S.-Y., Chen L.-S., Liang G.-Y., Pu Y.-P., Yin L.-H., Trends of long noncoding RNA research from 2007 to 2016: A bibliometric analysis. Oncotarget 8, 83114–83127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanchez Calle A., Kawamura Y., Yamamoto Y., Takeshita F., Ochiya T., Emerging roles of long non-coding RNA in cancer. Cancer Sci. 109, 2093–2100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Briggs J. A., Wolvetang E. J., Mattick J. S., Rinn J. L., Barry G., Mechanisms of long Non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 88, 861–877 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Barry G., Briggs J. A., Hwang D. W., Nayler S. P., Fortuna P. R. J., Jonkhout N., Dachet F., Maag J. L. V., Mestdagh P., Singh E. M., Avesson L., Kaczorowski D. C., Ozturk E., Jones N. C., Vetter I., Arriola-Martinez L., Hu J., Franco G. R., Warn V. M., Gong A., Dinger M. E., Rigo F., Lipovich L., Morris M. J., O’Brien T. J., Lee D. S., Loeb J. A., Blackshaw S., Mattick J. S., Wolvetang E. J., The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci. Rep. 7, 40127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ayupe A. C., Tahira A. C., Camargo L., Beckedorff F. C., Verjovski-Almeida S., Reis E. M., Global analysis of biogenesis, stability and sub-cellular localization of lncRNAs mapping to intragenic regions of the human genome. RNA Biol. 12, 877–892 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernard D., Prasanth K. V., Tripathi V., Colasse S., Nakamura T., Xuan Z., Zhang M. Q., Sedel F., Jourdren L., Coulpier F., Triller A., Spector D. L., Bessis A., A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 29, 3082–3093 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modarresi F., Faghihi M. A., Lopez-Toledano M. A., Fatemi R. P., Magistri M., Brothers S. P., van der Brug M. P., Wahlestedt C., Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 30, 453–459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raveendra B. L., Swarnkar S., Avchalumov Y., Liu X.-A., Grinman E., Badal K., Reich A., Pascal B. D., Puthanveettil S. V., Long noncoding RNA GM12371 acts as a transcriptional regulator of synapse function. Proc. Natl. Acad. Sci. U.S.A. 115, E10197–E10205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao H., Wahlestedt C., Kapranov P., Strategies to annotate and characterize long noncoding RNAs: Advantages and pitfalls. Trends Genet. 34, 704–721 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Clark B. S., Blackshaw S., Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front. Genet. 5, 164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaech S., Banker G., Culturing hippocampal neurons. Nat. Protoc. 1, 2406–2415 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Wren J. D., A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics 25, 1694–1701 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sankaranarayanan S., De Angelis D., Rothman J. E., Ryan T. A., The use of pHluorins for optical measurements of presynaptic activity. Biophys. J. 79, 2199–2208 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnsson P., Lipovich L., Grandér D., Morris K. V., Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 1840, 1063–1071 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T., Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12, 735–739 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Dill T. L., Naya F. J., A hearty dose of noncoding RNAs: The imprinted DLK1-DIO3 locus in cardiac development and disease. J. Cardiovasc. Dev. Dis. 5, 37 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benes V., Collier P., Kordes C., Stolte J., Rausch T., Muckentaler M. U., Häussinger D., Castoldi M., Identification of cytokine-induced modulation of microRNA expression and secretion as measured by a novel microRNA specific qPCR assay. Sci. Rep. 5, 11590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knauss J. L., Sun T., Regulatory mechanisms of long noncoding RNAs in vertebrate central nervous system development and function. Neuroscience 235, 200–214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Truckenbrodt S., Viplav A., Jähne S., Vogts A., Denker A., Wildhagen H., Fornasiero E. F., Rizzoli S. O., Newly produced synaptic vesicle proteins are preferentially used in synaptic transmission. EMBO J. 37, e98044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenberg S. S., Spitzer N. C., Calcium signaling in neuronal development. Cold Spring Harb. Perspect. Biol. 3, a004259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gärtner A., Fornasiero E. F., Munck S., Vennekens K., Seuntjens E., Huttner W. B., Valtorta F., Dotti C. G., N-cadherin specifies first asymmetry in developing neurons. EMBO J. 31, 1893–1903 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu C., Quinn J., Chang H. Y., Chromatin isolation by RNA purification (ChIRP). J. Vis. Exp. 61, 3912 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rani N., Nowakowski T. J., Zhou H., Godshalk S. E., Lisi V., Kriegstein A. R., Kosik K. S., A Primate lncRNA mediates notch signaling during neuronal development by sequestering miRNA. Neuron 90, 1174–1188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chu C., Zhang Q. C., da Rocha S. T., Flynn R. A., Bharadwaj M., Calabrese J. M., Magnuson T., Heard E., Chang H. Y., Systematic discovery of Xist RNA binding proteins. Cell 161, 404–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee E. B., Lee V. M.-Y., Trojanowski J. Q., Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 13, 38–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhardwaj A., Myers M. P., Buratti E., Baralle F. E., Characterizing TDP-43 interaction with its RNA targets. Nucleic Acids Res. 41, 5062–5074 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsai K.-J., Yang C.-H., Fang Y.-H., Cho K.-H., Chien W.-L., Wang W.-T., Wu T.-W., Lin C.-P., Fu W.-M., Shen C.-K. J., Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med. 207, 1661–1673 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pozzi D., Corradini I., Matteoli M., The control of neuronal calcium homeostasis by SNAP-25 and its impact on neurotransmitter release. Neuroscience, (2018) [Online ahead of print]. [DOI] [PubMed] [Google Scholar]
- 31.Andreae L. C., Burrone J., The role of neuronal activity and transmitter release on synapse formation. Curr. Opin. Neurobiol. 27, 47–52 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barry G., Briggs J. A., Vanichkina D. P., Poth E. M., Beveridge N. J., Ratnu V. S., Nayler S. P., Nones K., Hu J., Bredy T. W., Nakagawa S., Rigo F., Taft R. J., Cairns M. J., Blackshaw S., Wolvetang E. J., Mattick J. S., The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 19, 486–494 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Lipovich L., Dachet F., Cai J., Bagla S., Balan K., Jia H., Loeb J. A., Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics 192, 1133–1148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao X., Tang Z., Zhang H., Atianjoh F. E., Zhao J.-Y., Liang L., Wang W., Guan X., Kao S.-C., Tiwari V., Gao Y.-J., Hoffman P. N., Cui H., Li M., Dong X., Tao Y.-X., A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat. Neurosci. 16, 1024–1031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andersen R. E., Lim D. A., Forging our understanding of lncRNAs in the brain. Cell Tissue Res. 371, 55–71 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Prasad A., Bharathi V., Sivalingam V., Girdhar A., Patel B. K., Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 12, 25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maharana S., Wang J., Papadopoulos D. K., Richter D., Pozniakovsky A., Poser I., Bickle M., Rizk S., Guillén-Boixet J., Franzmann T. M., Jahnel M., Marrone L., Chang Y.-T., Sterneckert J., Tomancak P., Hyman A. A., Alberti S., RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360, 918–921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wren J. D., Bekeredjian R., Stewart J. A., Shohet R. V., Garner H. R., Knowledge discovery by automated identification and ranking of implicit relationships. Bioinformatics 20, 389–398 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Kang Y.-J., Yang D.-C., Kong L., Hou M., Meng Y.-Q., Wei L., Gao G., CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 45, W12–W16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sousa-Ferreira L., Garrido M., Nascimento-Ferreira I., Nobrega C., Santos-Carvalho A., Álvaro A. R., Rosmaninho-Salgado J., Kaster M., Kügler S., Pereira de Almeida L., Cavadas C., Moderate long-term modulation of neuropeptide Y in hypothalamic arcuate nucleus induces energy balance alterations in adult rats. PLOS ONE 6, e22333 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Malik J. M. I., Shevtsova Z., Bähr M., Kügler S., Long-term in vivo inhibition of CNS neurodegeneration by Bcl-XL gene transfer. Mol. Ther. 11, 373–381 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Fornasiero E. F., Raimondi A., Guarnieri F. C., Orlando M., Fesce R., Benfenati F., Valtorta F., Synapsins contribute to the dynamic spatial organization of synaptic vesicles in an activity-dependent manner. J. Neurosci. 32, 12214–12227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fornasiero E. F., Mandad S., Wildhagen H., Alevra M., Rammner B., Keihani S., Opazo F., Urban I., Ischebeck T., Sakib M. S., Fard M. K., Kirli K., Centeno T. P., Vidal R. O., Rahman R.-U., Benito E., Fischer A., Dennerlein S., Rehling P., Feussner I., Bonn S., Simons M., Urlaub H., Rizzoli S. O., Precisely measured protein lifetimes in the mouse brain reveal differences across tissues and subcellular fractions. Nat. Commun. 9, 4230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halder R., Hennion M., Vidal R. O., Shomroni O., Rahman R.-U., Rajput A., Centeno T. P., van Bebber F., Capece V., Vizcaino J. C. G., Schuetz A.-L., Burkhardt S., Benito E., Sala M. N., Javan S. B., Haass C., Schmid B., Fischer A., Bonn S., DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 19, 102–110 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/12/eaay2670/DC1
Fig. S1. Details of the screening strategy used for studying the role of lncRNAs in neuronal activity and down-regulation in culture.
Fig. S2. NeuroLNC coding potential, conservation, and locus organization.
Fig. S3. Specificity of the RNA-FISH approach and expression of neuroLNC in rodent cultures and in adult human brain.
Fig. S4. Controls for the calcium dynamics experiments.
Fig. S5. Controls and additional information concerning Fig. 5.
Fig. S6. NeuroLNC overexpression does not affect the expression of the mRNAs that encode for calcium-related pathways, neuronal activity, or synaptic scaffolds.
Fig. S7. Schematic representation summarizing the function of NeuroLNC in the nucleus of neurons.
Data S1. Summary of data used here, see first sheet for details.