

Abstract
Traumatic brain injury (TBI) is a progressive and complex pathological condition that results in multiple adverse consequences, including impaired learning and memory. Transplantation of mesenchymal stem cells (MSCs) has produced limited benefits in experimental TBI models. Fibroblast growth factor 21 (FGF21) is a novel metabolic regulator that has neuroprotective effects, promotes remyelination, enhances angiogenesis, and elongates astrocytic processes. In this study, MSCs were genetically engineered to overexpress FGF21 in order to improve their efficacy in TBI. MSCs overexpressing FGF21 (MSC-FGF21) were transplanted to mouse brain by intracerebroventricular injection 24 h after TBI was induced by controlled cortical impact (CCI). Hippocampus-dependent spatial learning and memory, assessed by the Morris water maze test, was markedly decreased 3–4 weeks after TBI, a deficit that was robustly recovered by treatment with MSC-FGF21, but not MSC-mCherry control. Hippocampus-independent learning and memory, assessed by the novel object recognition test, was also impaired; these effects were blocked by treatment with both MSC-FGF21 and MSC-mCherry control. FGF21 protein levels in the ipsilateral hippocampus were drastically reduced 4 weeks post-TBI, a loss that was restored by treatment with MSC-FGF21, but not MSC-mCherry. MSC-FGF21 treatment also partially restored TBI-induced deficits in neurogenesis and maturation of immature hippocampal neurons, whereas MSC-mCherry was less effective. Finally, MSC-FGF21 treatment also normalized TBI-induced impairments in dendritic arborization of hippocampal neurons. Taken together, the results indicate that MSC-FGF21 treatment significantly improved TBI-induced spatial memory deficits, impaired hippocampal neurogenesis, and abnormal dendritic morphology. Future clinical investigations using MSC-FGF21 to improve post-TBI outcomes are warranted.
Keywords: fibroblast growth factor 21, mesenchymal stem cells, neurogenesis, spatial learning and memory, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is the most common form of brain trauma and estimated to result in death or hospital admission for more than 10 million people annually worldwide.1,2 TBI can be divided into mild, moderate, and severe TBI. Even mild TBI causes an array of symptoms that may persist for up to 3 years post-injury, including headache, anxiety, depression, and irritability.3 Cognitive and memory impairments are also frequently associated with TBI.4 Deficits in learning and memory have been reported in several cognitive domains, including executive function, attention, working memory, episodic memory, verbal learning, and processing speed.5,6 In both animal models and human studies, cognitive impairment after TBI correlated with neural atrophy and progressive cell death in the hippocampus and frontal cortex.7,8
Although the pathophysiology of TBI has been extensively studied, no effective therapies presently exist, and clinical intervention relies largely on pathological stabilization immediately after injury and subsequent long-term rehabilitative care. Thus, an urgent need exists to explore novel treatment options to improve both immediate and long-term outcomes in TBI. In this context, stem cell therapy has emerged as a potential therapeutic strategy for treating central nervous system diseases, including brain trauma. Because mesenchymal stem cells (MSCs) secrete trophic factors that activate endogenous neurorestorative processes within the injured brain, their transplantation has been investigated in experimental TBI studies.9–11 In particular, harnessing the secretome of MSCs may be a potentially effective strategy because chemokines, cytokines, growth factors, gene products, and immunomodulatory peptides act as signaling molecules to induce functional recovery by promoting neurogenesis, angiogenesis, and synaptogenesis.12,13 In the context of TBI, MSC-mediated cell survival, neurogenesis, angiogenesis, and synaptogenesis in the hippocampus, the most vulnerable structure affected by TBI, may promote functional recovery of the hippocampus-associated learning and memory impairments that are frequent hallmarks of brain trauma.14
MSC transplantation is a promising therapeutic strategy given the accessibility, ease of handling, and isolation of these cells. Nevertheless, the severe and complex nature of the injured brain tissue microenvironment in TBI which may, in turn, affect MSC survival has cast doubt on whether this method will be truly useful for treating TBI.15 However, some studies found that pre-conditioning stem cells with pharmacological agents or hypoxia before transplantation significantly improved their therapeutic profile against brain- and cardiac-tissue injuries.14,16–18 Moreover, pre-conditioning stem cells was found to improve survival of the transplanted cells and augment the benefits of cell transplantation. For instance, cotreatment of MSCs with the mood stabilizers, lithium and valproate, both of which are known to have neuroprotective properties, improved their therapeutic efficacy and markedly altered gene expression after transplantation into experimental ischemic stroke rats and transgenic Huntington's disease mice.19,20 Specifically, priming MSCs with lithium improved their migration and homing abilities by inducing matrix metalloproteinase (a matrix metallopeptidase) and C-X-C chemokine receptor type 4 (a chemokine receptor), respectively, in vitro.21 In ischemic rats, post-insult MSC injection enhanced angiogenesis in the ischemic brain and improved behavioral performance.19 In N171-82Q Huntington's disease mice, pre-conditioning MSCs with lithium and valproate induced robust changes in the expression of genes involved in trophic effects and also enhanced the survival of MSCs in the brain after intranasal delivery, producing more significant improvements in motor performance than MSCs not subjected to pre-conditioning.20
One of the most prominent trophic proteins induced by lithium and valproate cotreatment in neurons and MSCs is fibroblast growth factor-21 (FGF21).20,22 FGF21, which facilitates the metabolism of glucose and lipids, has traditionally been thought to be produced only in peripheral tissues.23 However, recent studies found that FGF21 can be made in brain neurons and that it exhibits robust neuroprotective properties against glutamate excitotoxicity in primary brain neurons through v-akt murine thymoma viral oncogene homolog 1 (Akt-1)-dependent mechanisms.22 FGF21 also promoted the elongation of neurite-like processes in primary astrocytes as well as in an astrocytic cell line.24 FGF21 was also found to be dramatically downregulated in the cortex and striatum after cerebral ischemia; pharmacological manipulation of its levels partially restored function by reducing the size of brain infarction and improved functional outcomes.25
In light of the promising neuroprotective and neurotrophic features of FGF21, the present study genetically engineered MSCs to overexpress FGF21 and then assessed the beneficial effects of intracerebroventricular (ICV) injection of MSCs overexpressing FGF21 (MSC-FGF21) in a mouse model of TBI. The effects of MSC-FGF21 on hippocampus-dependent and -independent spatial learning and memory, hippocampal neurogenesis, and dendritic morphology were assessed and compared to sham-operated control mice, vehicle-treated TBI mice, and TBI mice treated with MSC-mCherry control.
Methods
Cell culture
Cryopreserved Gibco® MSCs isolated from the bone marrow of C57BL/6 mice were purchased from Invitrogen Life Technologies (Carlsbad, CA). MSCs were isolated at ≤8 weeks of gestation through mechanical and enzymatic digestion and cultured as previously reported.20 Cells had been stained positive for the cell-surface protein markers, CD29, CD34, CD44, and stem cells antigen-1 (>70%), and negative for CD117 (<5%) in flow cytometry assays performed by the manufacturer.
Lentiviral transduction of mesenchymal stem cells
MSCs were transduced with lentivirus particles consisting of a bicistronic control vector with an mCherry reporter gene (control) or with the open reading frame of Mus musculus FGF21 containing mCherry reporter gene (GenCopoeia, Rockville, MD) under the control of the human elongation factor 1-α promoter. Briefly, MSCs were seeded at a density of 5000 cells/well in a 12-well plate. On the following day, MSCs were transduced with the lentiviral transduction particles at a multiplicity of infection of 30; media were replenished with standard MSC growth media 20 h later. The mCherry control and FGF21-mCherry MSCs underwent three rounds of fluorescence-activated cell sorting to isolate a highly enriched population of MSCs expressing mCherry reporter gene.
Experimental animals
Wild-type C57B/L male mice (total number = 56) were obtained from the National Laboratory Animal Centre in Taipei, Taiwan. Animals were housed in cages under a standard 12-h light/dark cycle with food and water available ad libitum, and their body weights were measured biweekly. All procedures were approved by the Institutional Animal Care and Use Committee and with the approval of the Ethics Committee for animal use in Taipei Medical University (Taipei, Taiwan).
Controlled cortical impact–induced traumatic brain injury
TBI was induced in 8- to 9-week-old male mice using a controlled cortical impact (CCI) device. The unilateral, moderate CCI injury was induced as described previously.26 Briefly, mice were anesthetized by an intraperitoneal injection of zolazepam (50 mg/kg) and rompun (20 mg/kg) and positioned in a stereotaxic frame (Stoelting, Wood Dale, IL) before TBI. Under sterile conditions, the skin was retracted, and a 4-mm craniotomy was performed on the right cerebral hemisphere (2.0 mm posterior to the bregma and 2.0 mm lateral to the sagittal suture). To expose the intact dura, the skullcap was carefully removed. To induce a mild-to-moderate TBI, an impacting tip with a diameter of 3 mm driven by pneumatic piston was used to impact the exposed cortical surface at a velocity of 5 m/s, a depth of 1 mm, and a dwell time of 250 ms. After the CCI injury, the scalp was sutured and the mouse was placed on a heating pad to maintain a body temperature of 36–37°C throughout the recovery phase. Sham (non-injured) mice underwent craniotomy, but not CCI injury (14 mice).
Transplantation of mesenchymal stem cells
Twenty-four hours after TBI induction, MSCs were collected from the culture flasks and resuspended in phosphate-buffered saline (PBS; pH 7.4). Cell viability and number were examined by the Trypan Blue exclusion assay, and cell concentration was then adjusted to 1.5 × 104 cells/5 μL of PBS. Anesthetized mice were positioned on a stereotaxic frame, and a hole was drilled on the contralateral side of the injury hemisphere at coordinates of 0 mm caudal to bregma and 1 mm lateral to the midline. Using an automatic syringe pump system (Singa, Taoyuan, Taiwan) and a Hamilton syringe (27 gauges), 5 μL of cell suspension was ICV-infused at a depth of 3 mm beneath the dura mater. The cell suspension was infused over 5 min at a rate of 1 μL/min, and the needle was left in place for another 5 min. TBI animals were infused with either vehicle (in this case, PBS; n = 14), mCherry control MSCs (n = 14), or FGF21-MSCs (n = 14). The scalp was then sutured. Animals were kept at 37°C during the recovery phase and closely monitored. No animals died after transplantation.
Behavioral assessments
Morris water maze test
To evaluate spatial learning and memory impairments in mice, the Morris water maze (MWM) test was performed at 7, 14, 21, and 28 days after TBI. The experimental procedure is outlined in Figure 1A. The experimental procedure was similar to those described previously, with some modifications.27–30 The apparatus included a circular pool (150 cm in diameter and 60 cm in depth) filled with water (30 cm in depth) and a Plexiglas platform (10 cm in diameter), which was surrounded by visual cues visible to the mice. The pool was subdivided into four equal quadrants, and the platform was placed 1 cm beneath the surface of the water located in one of the four quadrants. Animals were trained for 4 consecutive days with five trials daily before the CCI injury, with an intertrial interval of 10 min. During the training trials, animals were gently placed into the tank in one of the four quadrants and facing the target quadrant; they were allowed 2 min to locate the hidden platform. The location of the platform was kept constant in the acquisition trials. If an animal failed to find the platform within 120 sec, it was gently guided toward it. Once on the platform, the animal was allowed to remain there for 15 sec.
FIG. 1.

MSC-FGF21 treatment suppressed learning and memory deficits in mice with traumatic brain injury (TBI) induced by controlled cortical impact (CCI). Twenty-four hours after TBI, mice were treated with MSC-mCherry control or MSC-FGF21 by intracerebroventricular (ICV) infusion. Learning and memory performance was assessed by the Morris Water Maze (MWM) test (A) at the indicated time points (B and C) and by the novel object recognition (NOR) test on day 14 (E). The characteristic path traced by each group in the MWM test to find the hidden platform (the red dot represents the starting point, and the green dot represents the end point) at day 28 post-injury is shown in (D). Data are presented as mean ± SEM (n = 6–8 per group). *p < 0.05; **p < 0.01; ***p < 0.001 between indicated groups using one-way ANOVA. ANOVA, analysis of variance; FGF21, fibroblast growth factor 21; MSC, mesenchymal stem cell; PBS, phosphate-buffered saline; SEM, standard error of the mean.
After each trial, animals were towel dried and returned to their cages. We exclude only the mice that failed to swim in five attempts with a 15-min intrainterval in the first day of training to reduce the pre-injury performing bias that could confound with the testing results as previously reported.31 After TBI induction, animals were tested on days 7, 14, 21, and 28 (sham, n = 6; TBI + PBS, n = 7; TBI + MSC-mCherry, n = 8; TBI + MSC-FGF21, n = 8). The time that animals spent reaching the underwater platform within 2 min was defined as the escape latency, which was used as an indicator of spatial memory retention. All animals were monitored and tracked by computerized video tracking system software (FG34PATH; HaSoTec, Rostock, Germany). Escape latencies for five trials with a 15-min intertrial interval were recorded.
Novel object recognition test
The novel object recognition (NOR) test, which evaluates the tendency of rodents to discriminate between new and familiar objects, was conducted to assess the extent of impaired memory in mice at day 14 after TBI (sham, n = 8; TBI + PBS, n = 8; TBI + MSC-mCherry, n = 8; TBI + MSC-FGF21, n = 8). Two days before the test, animals were individually habituated for 10 min every day in an open field box (60 × 60 cm) made of dark Plexiglas. On the day of the experiment, animals were habituated in the box for 10 min and then transferred to their home cage. After 2 min, animals were placed back into the same box for 10 min after two objects of the same material and shape, that were placed in a symmetrical position, had been added. After 1 h (acquisition phase), one of the objects was replaced with a novel object and each mouse's exploratory behavior was analyzed for 10 min. Exploration was defined as reaching the object, sniffing it from a distance of <2 cm, and/or touching it with the nose. After each session, the objects were cleaned with 75% ethanol to prevent odor recognition.
Data were analyzed and expressed as the time spent at each object and as a discrimination index (DI), which is the amount of time spent exploring the novel object compared with the familiar one. Specifically, the DI was defined as (time spent at the novel object – time spent at the familiar object) / (time spent at the novel object + time spent at the familiar object) × 100.
Western blot analysis
After the final behavioral assessments on day 28, animals were anesthetized with a mixture of zolazepam/rompun and then decapitated (n = 6 for each group). Brains were then carefully removed, and the ipsilateral hippocampi were dissected and collected in vials and rapidly frozen in liquid nitrogen. Samples were then homogenized in modified radioimmunoprecipitation assay lysis buffer containing protease inhibitor (Thermo Fisher Scientific, Rockford, IL). Lysates were chilled on ice and then centrifuged at 130,000g for 5 min at 4°C. Supernatants were collected and the protein concentrations were determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Irvine, CA).
Protein samples from tissue lysates were denatured at 95°C for 5 min after a proper dilution with 5 × sodium dodecyl sulfate buffer. Protein samples (30 μg) were resolved using electrophoresis in a 10% polyacrylamide gel and then transferred onto polyvinylidene difluoride membranes. After the transfer, membranes were blocked in 3% bovine serum albumin/1% Tween 20 in Tris-buffered saline for 1 h at room temperature and then incubated overnight with the specific primary antibodies (anti-β-actin, 1:2,000; Abeomics, San Diego, CA; and anti-FGF21, 1:1,000; Aviscera Bioscience, Santa Clara, CA) at 4°C. After washing with Tris-buffered saline/Tween solution, membranes were incubated with appropriate horseradish peroxidase–conjugated anti-rabbit immunoglobulin G antibody (1:5,000) for 1 h at room temperature. Blots were developed and enhanced with a chemiluminescence plus detection kit (Amersham Life Sciences, Piscataway, NJ). Protein bands were then detected by enhanced chemiluminescence western blot detection reagents (Millipore, Billerica, MA). All western blot images were acquired using the ChemiDoc Touch System; signal intensity of the bands was quantified by Image Lab (Bio-Rad Laboratories) and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) band signals.
Tissue collection and processing
Animals anesthetized with a mixture of zolazepam/rompun underwent transcardiac perfusion with PBS followed by chilled 4% paraformaldehyde in PBS (n = 6 for each group). Brains were then carefully removed from the skull and post-fixed for 1 day at 4°C overnight. After post-fixation, brains were dehydrated in 20%, and then 30%, sucrose for cryoprotection. Brains were embedded in optimal cutting temperature compound (Sakura Finetek, Torrance, CA) and frozen on a dry ice/ethanol bath for cryosectioning. Coronal sections were cut at a thickness of 14 microns using a sliding microtome (CM3050S; Leica, Tokyo, Japan) and stored at −20°C.
Immunohistochemistry and immunofluorescence
After a series of PBS washes, sections were incubated in 0.2% Triton X-100 in PBS for 15 min followed by blocking with 3% normal goat serum and 3% bovine serum albumin for 1 h. Sections were then incubated overnight at 4°C with diluted primary antibodies in blocking buffer that included rabbit anti–neuronal nuclei (ABN78, 1:250; EMD Millipore, Burlington, MA), guinea pig anti-doublecortin (DCX; AB2253, 1:200; EMD Millipore), and rabbit anti-mCherry (GTX128508, 1:100; GeneTex, Irvine, CA). After primary antibody incubation, sections were incubated with the appropriate Alexa Fluor 488–, 555–, or 647–conjugated secondary antibodies (1:1,000; Invitrogen, Carlsbad, CA) for 1 h, then washed with cold PBS and cover-slipped with Fluoroshield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; GeneTex). Microscope analyses were performed using a wide-field Olympus microscope (BX43) equipped with bright field and fluorescence microscopy. Digital image documentation was performed with a DP80 camera (Olympus, Tokyo, Japan). Quantification of immuno-positive cells was accomplished using the Particle Analysis command in ImageJ (National Institutes of Health [NIH], Bethesda, MD).
To analyze dendritic morphology of immature neurons in vivo, projection images were traced using the plug-in, “Simple Neurite Tracer and Sholl analysis” from Fiji (ImageJ; NIH). Dentate granule cells with dendritic trees were analyzed for total dendritic length. Dendritic complexity was measured using Sholl analysis plugged into NIH ImageJ by counting the number of dendrites that crossed a series of concentric circles from the cell soma. For each group (n = 6), six neurons per animal were drawn from randomly selected sections and analyzed for neuronal morphology.
Statistical analysis
Experimental results are expressed as mean ± standard error of the mean (SEM) and were analyzed using analysis of variance (ANOVA; one-way or two-way; GraphPad Prism; GraphPad Software Inc., San Diego, CA), as appropriate. Post-hoc analyses were performed using Bonferroni tests. Statistical significance was defined as p < 0.05.
Results
Mesenchymal stem cell/fibroblast growth factor 21 transplantation attenuated traumatic brain injury–induced spatial learning and memory deficits assessed by the Morris water maze test
The MWM was used to assess the treatment effects of MSC-FGF21 or MSC-mCherry ICV infusion on hippocampus-dependent spatial learning and memory. The experimental procedure is outlined in Figure 1A. Compared to sham-treated control animals, it took vehicle (PBS)-treated TBI mice longer to find the hidden platform on days 21 and 28 post-injury (p < 0.005; Fig. 1B–D). However, in TBI mice treated with MSC-FGF21, escape latencies were reduced to the levels observed for sham-operated control mice on days 21 and 28 post-TBI. This reduction was also statistically significant between the MSC-FGF21– and MSC-mCherry–treated TBI groups (p < 0.05; Fig. 1B–D). Interestingly, MSC-mCherry treatment did not reduce escape latencies, which remained elevated in this group compared to the sham-operated group (p < 0.01 on days 21 and 28). In addition, the reduction in escape latencies observed in the MSC-FGF21 group was time dependent, given that escape latencies remained significantly higher in the MSC-FGF21 group compared to the sham group on day 7 post-TBI (p < 0.01; Supplementary Fig. S1A), but began to decrease on day 14 (Supplementary Fig. S1B).
Mesenchymal stem cell treatment ameliorated learning and memory dysfunction in the novel object recognition test
On day 14 post-injury, animals were assessed by the NOR task (Fig. 1E). Compared to sham-operated animals, vehicle-treated mice exhibited drastic memory impairments, as indicated by total DI blockade (p < 0.05); these results are consistent with previous studies indicating that malfunctions in the perirhinal cortex and hippocampus can affect memory and reduce the ability to discriminate novel from familiar objects in the NOR test.32,33 In contrast, treatment with either MSC-mCherry or MSC-FGF21 robustly improved NOR performance compared to vehicle-treated mice (p < 0.05).
Localization and survival of mesenchymal stem cells transplant in the injured brain hemisphere
Presence of transplanted MSCs in the injured brain hemisphere was determined by mCherry immunoreactivity at 28 days post-injury (n = 3). Expression of mCherry by transplanted MSC-mCherry (Fig. 2A) and MSC-FGF21 (Fig. 2B) was used as a cellular label to localize the presence of transplanted cells. Transplanted MSCs were localized in the ipsilateral hemisphere primarily in the injured cortex and hippocampus. Both treated group showed a pronounced mCherry immunoreactivity. Thus, these results suggest that the transplanted MSCs survive at least until 27 days post-transplantation.
FIG. 2.
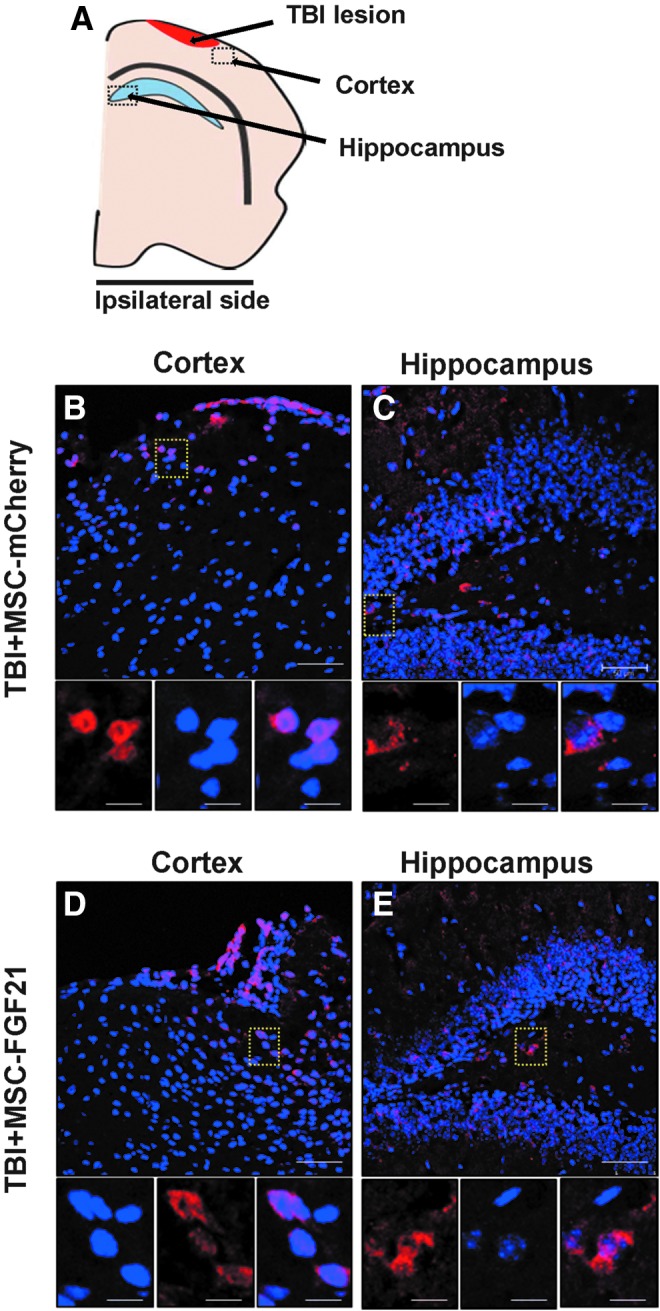
Presence of mCherry-labeled MSCs in the ipsilateral brain hemisphere. (A) Schematic illustration of the location of the detected mCherry-labeled MSCs in the ipsilateral brain hemisphere. Representative images of mCherry immunereactivity in coronal sections of ipsilateral hemisphere of injured brains at 28 days post-injury showing the presence and selectivity of MSC-mCherry (B) and MSC-FGF21 (C) after MSCs were injected into the contralateral lateral ventricle. MSC-mCherry and MSC-FGF21 cells were stably overexpressing the mCherry protein and used as cellular label to localize the transplanted MSCs. MSC-mCherry and MSC-FGF21 were located mainly in the ipsilateral cortex and hippocampus (dashed boxes). Scale bar, 100 μm, unless stated otherwise. FGF21, fibroblast growth factor 21; MSCs, mesenchymal stem cells; TBI, traumatic brain injury.
Hippocampal fibroblast growth factor 21 protein levels were decreased by traumatic brain injury and restored by treatment with mesenchymal stem cell/fibroblast growth factor 21
FGF21 protein levels in lysates of the ipsilateral hippocampus were evaluated in all animal groups in order to assess whether FGF21 expression levels changed as a result of TBI and/or treatment with MSC-mCherry or MSC-FGF21. Western blot results demonstrated that FGF21, which appeared as a protein band around 30 kDa, was decreased by approximately 50% in the ipsilateral hippocampal lysates on day 28 post-TBI (Fig. 3A,B) in vehicle-treated animals. Treatment with MSC-FGF21 completely restored the loss of hippocampal FGF21, whereas treatment with MSC-mCherry had no effect, suggesting that MSC-FGF21 treatment may release FGF21 in the ipsilateral hippocampus, thus contributing to the beneficial effects of MSC-FGF21 in TBI mice.
FIG. 3.

Treatment with MSC-FGF21 restored FGF21 protein levels in the ipsilateral hippocampus. On day 28 post-injury, ipsilateral hippocampal tissues were removed and lysates were prepared, followed by western blot analysis to determine FGF21 protein levels. (A) Representative western blot results from four groups of animals using GAPDH as the loading control. (B) Quantified FGF21 western blot results from hippocampal tissue lysate (n = 6 per group). Data are presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 between indicated groups using one-way ANOVA. ANOVA, analysis of variance; FGF21, fibroblast growth factor 21; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; MSC, mesenchymal stem cell; PBS, phosphate-buffered saline; SEM, standard error of the mean; TBI, traumatic brain injury.
Mesenchymal stem cell/fibroblast growth factor 21 treatment enhanced the neurogenesis and maturation of newborn neurons in the ipsilateral hippocampus at the chronic stage after traumatic brain injury
To identify the effects of TBI insult at the chronic stage, 28 days after TBI, on generating newborn neurons in the ipsilateral hippocampus, brain sections were labeled with immature neuron marker DCX. The density of the DCX-positive cells in the granule cell layer of the ipsilateral hippocampal dentate gyrus (DG) was counted and analyzed.
In vehicle-treated mice, the density of DCX-labeled cells in the DG was decreased by more than 70% on day 28 post-TBI compared to the sham-operated animals, indicating impaired neurogenesis at the chronic stage after TBI (p < 0.005; Fig. 4A,B). This impairment in neurogenesis was robustly suppressed in MSC-FGF21–treated mice (p < 0.05), for whom values approached those observed in sham-operated control mice. There was a trend toward increased DCX density in the DG of MSC-mCherry–treated TBI mice, but this increase did not reach statistical significance.
FIG. 4.

Treatment with MSC-FGF21 increased the density and maturation of immature neurons in the dentate gyrus (DG) of the ipsilateral hippocampus in traumatic brain injury (TBI) model mice. On day 28 post-TBI, immunohistochemical staining with doublecortin (DCX) and DAPI was performed in the DG of the ipsilateral hippocampus. (A) Representative results of immunostaining of DCX (red), DAPI (blue), and their overlaid images in the granule cell layer of the DG of the ipsilateral hippocampus from four groups of mice. The horizontal bar represents 50 um. (B) Quantified results of the density of DCX-labeled immature neurons in the DG from four groups of mice (n = 6–8 animals per group). (C) Typical morphology of three types of DCX-labeled immature neurons in the DG showing the expression of long and branched processes, short and unbranched processes, or no processes. The horizontal bar represents 10 um. (D) Quantified results showing the percentages of DCX-positive immature neurons with no processes, short unbranched processes, or long branched processes from the ipsilateral DG granule cell layer of the four groups of animals. Quantified results shown are mean ± SEM (n = 6). *p < 0.05; **p < 0.01; ***p < 0.001 between the indicated groups using one-way ANOVA. ANOVA, analysis of variance; DAPI, 4′,6-diamidino-2-phenylindole; FGF21, fibroblast growth factor 21; MSC, mesenchymal stem cell; PBS, phosphate-buffered saline; SEM, standard error of the mean.
Potential differences in maturation of the newly generated immature neurons were also examined on day 28 after TBI. The DCX-labeled neurons in the hippocampal DG exhibited three types of morphological features: cells with no processes; cells with short, unbranched processes; and cells with long and/or branched processes (Fig. 4C). Sham-operated mice predominantly expressed the cell type with long, branched processes, whereas the vehicle-treated group expressed all three cell types in approximately equal proportions (Fig. 4D). Notably, TBI mice treated with MSC-FGF21 showed greater percentages of the cell type with long, branched processes (50.5%) than cell types with no processes (24%) or with short, unbranched processes (25.5%). Percentages of cell types expressing long processes were highest in MSC-FGF21–treated TBI mice (55.5%), followed by MSC-mCherry TBI mice (41%), and then finally by vehicle-treated TBI mice (34%), whose values were significantly lower than that of sham control animals (59%).
Mesenchymal stem cell/fibroblast growth factor 21 treatment rescued the dendritic morphology defects of immature newborn neurons in the ipsilateral hippocampus
Because TBI induced by CCI has been reported to increase the heterogeneity of dendritic branch morphology in cells born after injury,37 we sought to categorize DCX-labeled immature neurons in the ipsilateral DG on day 28 into three classes: “typical” Y-shaped neurons with one apical primary dendrite; Y-shaped neurons with one short apical primary dendrite; and V-shaped neurons with more than one primary dendrite (Fig. 5A). Compared with the sham control group, CCI-induced TBI reduced the percentage of Y-shaped neurons with one apical primary dendrite from 59% to 34%, but increased the percentage of V-shaped neurons with more than one primary dendrite from 11% to 33% in the ipsilateral hippocampal DG (Fig. 5B). MSC-FGF21 treatment largely blocked these two TBI-induced parameters, with changes of 43% in Y-shaped neurons with one apical primary dendrite and 13% V-shaped neurons with more than one primary dendrite. MSC-mCherry treatment produced similar, but less potent, effects (40% were Y-shaped neurons with one apical primary dendrite and 25% were V-shaped neurons with more than one primary dendrite) than MSC-FGF21.
FIG. 5.

MSC-FGF21 treatment blocked dendritic morphology defects in immature newborn hippocampal neurons. On day 28 after TBI, doublecortin (DCX)-labeled immature hippocampal neurons were examined for dendritic morphology. (A) Representative dendritic morphology of immature hippocampal neurons, which were divided into three classes according to their dendritic phenotypes. (B) Distribution percentages of these three classes of neurons with distinct dendritic morphology in four groups of animals. Scholl analysis was performed to measure total dendritic length (C), number of dendritic branches (D), and distance from the first branch to the soma (E) of DCX-positive immature hippocampal neurons in four groups of animals. Quantified data are mean ± SEM (n = 6 animals per group). *p < 0.05; **p < 0.01; ***p < 0.001 using one-way ANOVA. ANOVA, analysis of variance; FGF21, fibroblast growth factor 21; MSC, mesenchymal stem cell; PBS, phosphate-buffered saline; SEM, standard error of the mean; TBI, traumatic brain injury.
Because neuronal insults can cause dendritic morphology defects in adult newborn neurons,38,39 we further analyzed the arborization of immature neurons in the DG of the ipsilateral hippocampus on day 28 after TBI (Fig. 5C–E). Dendritic arborization of DCX-labeled immature neurons in vehicle-treated TBI mice was significantly impaired compared to sham-operated animals, with decreased total dendritic length (p < 0.01), fewer branches (p < 0.01), and shorter distances between the first proximal branch and cell soma (p < 0.05). Compared to the vehicle-treated TBI and sham control groups, MSC-FGF21 treatment almost completely blocked the long-term effects of TBI on loss of total dendritic length (p < 0.01) and reduced the number of branches (p < 0.05), though it had no effect on distance of the first branch to the cell soma. MSC-mCherry treatment significantly protected against the decrease in total dendritic length compared to the vehicle-treated group (p < 0.05), but had no other significant effects.
Discussion
This pre-clinical study is the first to demonstrate that MSC-FGF21 treatment leads to a number of long-lasting improvements in TBI-induced spatial memory deficits, impaired hippocampal neurogenesis, and abnormal dendritic morphology at the chronic stage after TBI. First, treatment with MSCs overexpressing FGF21 was found to suppress TBI-induced learning and memory deficits. Second, endogenous FGF21 levels were found to be significantly decreased determined by western blotting in the ipsilateral hippocampus for weeks after TBI, and treatment with MSC-FGF21 rescued these decreases. Third, MSC-FGF21 treatment enhanced hippocampal neurogenesis at the chronic stage after TBI injury. Finally, FGF21 overexpression enhanced the ability of MSCs to normalize the long-term TBI-induced defects in the dendritic morphology of immature newborn neurons in the hippocampus. Taken together, these findings shed new light on potential novel therapies for TBI-induced memory impairments.
Memory deficits are particularly common in human TBI patients, either as a consequence of direct effects on memory encoding or through secondary effects on learning, concentration, and attention.40–43 Using a spatially challenging behavioral test, one study found that immature DCX-expressing neurons were required for the successful acquisition of spatial learning.44 Consistent with these reports, the present study found that impaired performance in the MWM and NOR tests correlated with drastic reductions in the generation of newborn immature DCX-expressing neurons observed in the DG of TBI mice at the chronic stage after TBI. Notably, MSC-FGF21 treatment effectively reduced spatial learning memory deficits, as indicated by shortened escape latencies in the MWM test and increased time spent in novel object exploration in the NOR test. Further, treatment with MSC-FGF21, but not MSC-mCherry, control enhanced the generation of newborn immature DCX-expressing neurons in the DG of TBI animals at the chronic stage after TBI. Although our data do not provide comprehensive proof that the enhanced cognitive performance observed after MSC-FGF21 treatment was the direct consequence of enhanced neurogenesis, the robust improvement in the number of immature neurons in the DG provides compelling evidence that these events are related.
The vulnerability of immature DCX-expressing neurons in the DG of the hippocampus to TBI impact at the acute stage has been well established.33–35 However, the long-term effects of TBI on the hippocampal neurogenesis and generating new neurons are underestimated at the chronic stage after TBI. Although the reason for the observed decline in neurogenesis demonstrated by robust reduction of DCX-positive immature neurons at the chronic stage after TBI is unclear, it could be related to the exhaustion and depletion of neural stem and progenitor cells pool attributed to the extensive proliferation of neural stem cells at the acute stage. The depletion of neural stem cells attributed to accelerated proliferation has been reported in an induced neural degeneration mouse model, suggesting that the neuroregenerative capacity of neural stem cells is limited.45 Our results indicating long-term effects of TBI on hippocampal neurogenesis and ability of generating immature neurons at the chronic stage after TBI are demonstrated by the significant reduction in the DCX-positive immature neurons in the DG 28 days post-injury.
It should be noted that the learning and memory deficits detected by the MWM test were rescued by treatment with MSC-FGF21, but not MSC-mCherry, whereas the deficits detected by the NOR test were alleviated by both MSC-FGF21 and MSC-mCherry. There was no significant difference between the observed improvement in MSC-FGF21 and MSC-mCherry. These results suggest that the observed improvements in the NOR test by MSC-FGF21 treatment was not attributed to FGF21 secretion, but rather attributed to MSCs themselves. Our results are in agreement with previous reports that have demonstrated significant enhancement in the NOR test after transplantation of MSC in murine models of brain injury models.46,47 One possible explanation for beneficial effects of MSC treatment in the NOR test is attributed to the numerous trophic and growth factors that were produced by MSCs to stimulate the host cells and ameliorate inflammation post-injury.
MWM is a measure of hippocampus-dependent spatial memory, whereas the NOR involves incidental encoding and does not typically require hippocampal involvement.33 In this respect, the results that were obtained from the two experiments are not contradictive. Accumulating studies suggest that hippocampal or fornix lesions do not affect object recognition, whereas lesions in the perirhinal cortex severely disrupt object recognition.48–53 Although these brain regions may form a functional network for higher-order cognitive functions (such as ordering objects or planning appropriate response strategies), they have been found to function independently when processing object familiarity or spatial information.33 Taken together, our data suggest that TBI-induced spatial and learning memory deficits are improved by MSC-FGF21 treatment attributed to enhanced hippocampal neurogenesis, and that MSC-FGF21 and MSC-mCherry are equally effective in improving object recognition memory that involves hippocampus-independent mechanisms.
FGF21 is a metabolic regulator that plays a role in controlling glucose homeostasis, insulin sensitivity, and ketogenesis.54,55 It is known for its peripheral actions and is produced mainly in the liver, pancreas, and adipose tissue; however, FGF21 protein can also be found in some brain regions, including the cortex, hippocampus, and striatum,54,56,57 and is involved in some prominent neurophysiological functions such as neuroendocrine control of ovulation.58 A previous study found that FGF21 expression can be induced in the brain and primary neurons, and that it exhibits robust neuroprotective properties against glutamate excitotoxicity through mechanisms involving Akt-1 activation and glycogen synthase kinase 3 inhibition.22 FGF21 also induces elongation of neurite-like processes in primary astrocytes and an astrocytic cell line.24 These properties suggest that FGF21 may be a promising therapeutic tool for promoting neuronal survival and enhancing neuron-glia interactions after TBI.
Transplanted MSCs were localized at the injured hemisphere after 27 days post-transplantation (28 days post-injury), suggesting that MSC-mCherry and MSC-FGF21 are able to migrate successfully toward the injured hemisphere (primarily in cortex and hippocampus) and survive for at least 27 days. Whereas previous published reports have evaluated expression of neuronal, oligodendrocyte, microglial, or astrocytic markers by MSCs after transplantation to determine the cells' fate, such an evaluation was not the main aim of the current study.59,60 FGF21 secretion by transduced MSCs was the main therapeutic effect of MSC-FGF21 transplantation, by providing stable and sustained secretion of FGF21 into ipsilateral cortex and hippocampus. This is evidenced by the significant increase of FGF21 levels in the ipsilateral hippocampus and the enhanced hippocampal-dependent cognitive functions in the animals that were treated with MSC-FGF21, compared to MSC-mCherry–treated animals.
In this study, CCI insult drastically reduced FGF21 levels in the ipsilateral hippocampus on day 28 after TBI, a decrease that correlated with impaired neurogenesis in the ipsilateral hippocampus at the chronic stage after TBI. However, MSC-mCherry–treated TBI mice exhibited less neurogenesis than MSC-FGF21 TBI mice. This suggests that the improved neurogenesis observed in the ipsilateral hippocampus in mice treated with MSC-FGF21 is most likely attributable to increased FGF21 levels rather than activity of the MSC secretome. It is possible that FGF21 improved the generation of newborn neurons and neurogenesis in hippocampus, thus restoring the drastic decline of neurogenesis and newborn immature neurons over an extended period.
TBI is known to impair multiple aspects of neurogenesis, including neural stem cell proliferation, immature neuron survival, dendrite development and arborization, and functional integration into the hippocampal neuronal network.33,61–64 In particular, impaired dendrite development, arborization, and morphology of immature neurons significantly affect functional synaptic integration and disrupt the signaling transduction needed to produce action potential in neurons, ultimately contributing to post-traumatic deficits in learning and memory.65 In this study, we found that dendritic length and complexity were dramatically reduced in immature hippocampal neurons in vehicle-treated TBI mice, but that MSC-FGF21 treatment alleviated these impairments in dendritic morphology, including in the number of branches and total dendritic length. Interestingly, MSC-mCherry treatment suppressed dendritic length impairments, but not the number of branches, resulting in less morphologically complex immature hippocampal neurons. These results indicate that MSC-FGF21 treatment generated immature neurons at the chronic stage post-TBI that were morphologically more similar to those observed in the sham-operated control group versus those in either vehicle-treated or MSC-mCherry–treated mice.
Importantly, the improved neurogenesis and normalization of dendritic morphology in response to MSC-FGF21 treatment could affect the functional integration and plasticity of immature neurons in the hippocampus, and contribute to recovery from memory deficits after TBI. Figure 6 is a schematic illustration of the effects of treatment with MSC-FGF21 or MSC-mCherry on TBI-induced deficits in hippocampal FGF21 protein levels, neurogenesis, morphology of immature neurons, and learning/memory.
FIG. 6.

Schematic illustration of the effects of treatment with MSC-FGF21 or MSC-mCherry on traumatic brain injury (TBI)-induced deficits in hippocampal FGF21 levels, neurogenesis, immature neuronal morphology, and spatial learning and memory. Graphical representation of immature neurons 28 days post-injury in sham-operated controls and TBI mice treated with vehicle, MSC-mCherry, or MSC-FGF21. Hippocampus-dependent spatial learning and memory were severely impaired in vehicle-treated TBI mice compared to sham-operated controls. This impairment was associated with decreased hippocampal FGF21 levels, deficits in neurogenesis and maturation of immature neurons, as well as abnormal dendritic morphology in the hippocampal dentate gyrus. Although MSC-mCherry treatment suppressed some aspects of the morphological deficits observed in immature neurons, decreases in the number of immature neurons and in hippocampal FGF21 levels were still observed, as were impairments in spatial learning and memory. MSC-FGF21 treatment after TBI reversed all injury-induced deficits in hippocampal FGF21, neurogenesis, neuronal morphology, and spatial learning and memory. CCI, controlled cortical impact; FGF21, fibroblast growth factor 21; MSC, mesenchymal stem cell; PBS, phosphate-buffered saline.
From a treatment perspective, systemic administration of recombinant human FGF21 has been shown to reduce brain edema, protect against blood–brain barrier disruptions, and improve sensorimotor deficits in mice 24 h after TBI induction.56 Further, peripherally derived FGF21 was shown to improve remyelination by interacting with the FGF21 coreceptor, β-klotho, in a mouse model of toxin-induced demyelination.66 FGF21 turns over rapidly, with a half-life of only 1–2 h and can be readily cleared from the brain by circulation.67 In this study, injected MSCs were genetically engineered to continuously overexpress FGF21, thus maintaining high, steady levels of brain FGF21 for up to 28 days post-TBI. As a result, we were able to demonstrate, for the first time, that MSC-FGF21 treatment leads to long-lasting improvements in TBI-induced spatial memory deficits, impaired hippocampal neurogenesis, and abnormal dendritic morphology.
It should be noted that previous studies found that few MSCs differentiate into other cell types after transplantation to the brain and, further, that the beneficial effects of MSCs in the injured brain mainly are largely attributed to their trophic and neuroprotective effects.19,20 Given that MSCs can be derived from homologous sources to minimize immune response and have low potential of inducing tumorigenesis, these preliminary, pre-clinical findings suggest that the current strategy could be applied to patients who suffer from moderate and severe brain injuries. The present study also demonstrated that MSC-FGF21 was significantly more effective than MSC-mCherry in TBI mice. Collectively, our results suggest that future clinical investigations of MSC-FGF21 strategies in human cases of TBI are warranted.
Supplementary Material
Acknowledgments
This study was supported by Ministry of Science and Technology Grants (MOST 104-2923-B-038-004 -MY2, MOST 107-2314-B-038-063, and MOST 107-2314-B-038-042) and Taipei Medical University (TMU 106-5400-004-400 and TMU 106-5310-001-400). We thank the partial support from the Intramural Research Program of NIMH, NIH in the USA and the Translational Laboratory, Department of Medical Research, Taipei Medical University Hospital for generously lending facility support. We also thank Ms. Ioline Henter (NIMH-NIH, USA) for invaluable editorial assistance.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
References
- 1. Murray C.J., and Lopez A.D. (1997). Mortality by cause for eight regions of the world: Global Burden of Disease Study. Lancet 349, 1269–1276 [DOI] [PubMed] [Google Scholar]
- 2. Hyder A.A., Wunderlich C.A., Puvanachandra P., Gururaj G., and Kobusingye O.C. (2007). The impact of traumatic brain injuries: a global perspective. NeuroRehabilitation 22, 341–353 [PubMed] [Google Scholar]
- 3. Waljas M., Iverson G.L., Lange R.T., Hakulinen U., Dastidar P., Huhtala H., Liimatainen S., Hartikainen K., and Ohman J. (2015). A prospective biopsychosocial study of the persistent post-concussion symptoms following mild traumatic brain injury. J. Neurotrauma 32, 534–547 [DOI] [PubMed] [Google Scholar]
- 4. Rabinowitz A.R. and Levin H.S. (2014). Cognitive sequelae of traumatic brain injury. Psychiatr. Clin. North Am. 37, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McAllister T.W., Flashman L.A., McDonald B.C., and Saykin A.J. (2006). Mechanisms of working memory dysfunction after mild and moderate TBI: evidence from functional MRI and neurogenetics. J. Neurotrauma 23, 1450–1467 [DOI] [PubMed] [Google Scholar]
- 6. Johansson B., Berglund P., and Ronnback L. (2009). Mental fatigue and impaired information processing after mild and moderate traumatic brain injury. Brain Inj. 23, 1027–1040 [DOI] [PubMed] [Google Scholar]
- 7. Henninger N., Dutzmann S., Sicard K.M., Kollmar R., Bardutzky J., and Schwab S. (2005). Impaired spatial learning in a novel rat model of mild cerebral concussion injury. Exp. Neurol. 195, 447–457 [DOI] [PubMed] [Google Scholar]
- 8. Palacios E.M., Sala-Llonch R., Junque C., Fernandez-Espejo D., Roig T., Tormos J.M., Bargallo N., and Vendrell P. (2013). Long-term declarative memory deficits in diffuse TBI: correlations with cortical thickness, white matter integrity and hippocampal volume. Cortex 49, 646–657 [DOI] [PubMed] [Google Scholar]
- 9. Li Y., and Chopp M. (2009). Marrow stromal cell transplantation in stroke and traumatic brain injury. Neurosci. Lett. 456, 120–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aertker B.M., Bedi S., and Cox C.S. Jr (2016). Strategies for CNS repair following TBI. Exp. Neurol. 275, Pt. 3, 411–426 [DOI] [PubMed] [Google Scholar]
- 11. Mahmood A., Lu D., and Chopp M. (2004). Intravenous administration of marrow stromal cells (MSCs) increases the expression of growth factors in rat brain after traumatic brain injury. J. Neurotrauma 21, 33–39 [DOI] [PubMed] [Google Scholar]
- 12. Xiong Y., Mahmood A., and Chopp M. (2010). Angiogenesis, neurogenesis and brain recovery of function following injury. Curr. Opin. Investig. Drugs 11, 298–308 [PMC free article] [PubMed] [Google Scholar]
- 13. Walker P.A., Shah S.K., Harting M.T., and Cox C.S. Jr (2009). Progenitor cell therapies for traumatic brain injury: barriers and opportunities in translation. Dis. Model Mech. 2, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei L., Fraser J.L., Lu Z.Y., Hu X., and Yu S.P. (2012). Transplantation of hypoxia preconditioned bone marrow mesenchymal stem cells enhances angiogenesis and neurogenesis after cerebral ischemia in rats. Neurobiol. Dis. 46, 635–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gold E.M., Su D., Lopez-Velazquez L., Haus D.L., Perez H., Lacuesta G.A., Anderson A.J., and Cummings B.J. (2013). Functional assessment of long-term deficits in rodent models of traumatic brain injury. Regen. Med. 8, 483–516 [DOI] [PubMed] [Google Scholar]
- 16. Xu Y., Hu X., Wang L., Jiang Z., Liu X., Yu H., Zhang Z., Chen H., Chen H., Steinhoff G., Li J., and Jian'an W. (2013). Preconditioning via angiotensin type 2 receptor activation improves therapeutic efficacy of bone marrow mononuclear cells for cardiac repair. PLoS One 8, e82997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sakata H., Niizuma K., Yoshioka H., Kim G.S., Jung J.E., Katsu M., Narasimhan P., Maier C.M., Nishiyama Y., and Chan P.H. (2012). Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J. Neurosci. 32, 3462–3473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sun X., Fang B., Zhao X., Zhang G., and Ma H. (2014). Preconditioning of mesenchymal stem cells by sevoflurane to improve their therapeutic potential. PLoS One 9, e90667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai L.K., Wang Z., Munasinghe J., Leng Y., Leeds P., and Chuang D.M. (2011). Mesenchymal stem cells primed with valproate and lithium robustly migrate to infarcted regions and facilitate recovery in a stroke model. Stroke 42, 2932–2939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Linares G.R., Chiu C.T., Scheuing L., Leng Y., Liao H.M., Maric D., and Chuang D.M. (2016). Preconditioning mesenchymal stem cells with the mood stabilizers lithium and valproic acid enhances therapeutic efficacy in a mouse model of Huntington's disease. Exp. Neurol. 281, 81–92 [DOI] [PubMed] [Google Scholar]
- 21. Tsai L.K., Leng Y., Wang Z., Leeds P., and Chuang D.M. (2010). The mood stabilizers valproic acid and lithium enhance mesenchymal stem cell migration via distinct mechanisms. Neuropsychopharmacology 35, 2225–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leng Y., Wang Z., Tsai L.K., Leeds P., Fessler E.B., Wang J., and Chuang D.M. (2015). FGF-21, a novel metabolic regulator, has a robust neuroprotective role and is markedly elevated in neurons by mood stabilizers. Mol. Psychiatry 20, 215–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inagaki T., Lin V.Y., Goetz R., Mohammadi M., Mangelsdorf D.J., and Kliewer S.A. (2008). Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 8, 77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Leng Y., Wang J., Wang Z., Liao H.M., Wei M., Leeds P., and Chuang D.M. (2016). Valproic acid and other HDAC inhibitors upregulate FGF21 gene expression and promote process elongation in glia by inhibiting HDAC2 and 3. Int. J. Neuropsychopharmacol. 19, pyw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z., Leng Y., Wang J., Liao H.M., Bergman J., Leeds P., Kozikowski A., and Chuang D.M. (2016). Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of alpha-tubulin acetylation and FGF-21 up-regulation. Sci. Rep. 6, 19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu F., Wang Z., Tchantchou F., Chiu C.T., Zhang Y., and Chuang D.M. (2012). Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 29, 362–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morris R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Methods 11, 47–60 [DOI] [PubMed] [Google Scholar]
- 28. Lai J.H., Karlsson T.E., Wu J.C., Huang C.Z., Chen Y.H., Kang S.J., Brodin A.T.S., Hoffer B.J., Olson L., Chiang Y.H., and Chen K.Y. (2019). Role of Nogo receptor-1 for recovery of balance, cognition, and emotion after mild traumatic brain injury in mice. J. Neurotrauma 36, 1054–1059 [DOI] [PubMed] [Google Scholar]
- 29. Czurko A., Czeh B., Seress L., Nadel L., and Bures J. (1997). Severe spatial navigation deficit in the Morris water maze after single high dose of neonatal x-ray irradiation in the rat. Proc. Natl. Acad. Sci. U. S. A. 94, 2766–2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scherbel U., Raghupathi R., Nakamura M., Saatman K.E., Trojanowski J.Q., Neugebauer E., Marino M.W., and McIntosh T.K. (1999). Differential acute and chronic responses of tumor necrosis factor-deficient mice to experimental brain injury. Proc. Natl. Acad. Sci. U. S. A. 96, 8721–8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vorhees C.V., and Williams M.T. (2006). Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 1, 848–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barker G.R., Bird F., Alexander V., and Warburton E.C. (2007). Recognition memory for objects, place, and temporal order: a disconnection analysis of the role of the medial prefrontal cortex and perirhinal cortex. J. Neurosci. 27, 2948–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Barker G.R., and Warburton E.C. (2011). When is the hippocampus involved in recognition memory? J. Neurosci. 31, 10721–10731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grady M.S., Charleston J.S., Maris D., Witgen B.M., and Lifshitz J. (2003). Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: analysis by stereological estimation. J. Neurotrauma 20, 929–941 [DOI] [PubMed] [Google Scholar]
- 35. Gao X., Deng-Bryant Y., Cho W., Carrico K.M., Hall E.D., and Chen J. (2008). Selective death of newborn neurons in hippocampal dentate gyrus following moderate experimental traumatic brain injury. J. Neurosci. Res. 86, 2258–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brown J.P., Couillard-Despres S., Cooper-Kuhn C.M., Winkler J., Aigner L., and Kuhn H.G. (2003). Transient expression of doublecortin during adult neurogenesis. J. Comp. Neurol. 467, 1–10 [DOI] [PubMed] [Google Scholar]
- 37. Villasana L.E., Kim K.N., Westbrook G.L., and Schnell E. (2015). Functional integration of adult-born hippocampal neurons after traumatic brain injury(1,2,3). eNeuro 2, ENEURO..0056-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Niv F., Keiner S., Krishna, Witte O.W., Lie D.C., and Redecker C. (2012). Aberrant neurogenesis after stroke: a retroviral cell labeling study. Stroke 43, 2468–2475 [DOI] [PubMed] [Google Scholar]
- 39. Jessberger S., Zhao C., Toni N., Clemenson G.D. Jr., Li Y., and Gage F.H. (2007). Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J. Neurosci. 27, 9400–9407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Whyte J., Polansky M., Cavallucci C., Fleming M., Lhulier J., and Coslett H.B. (1996). Inattentive behavior after traumatic brain injury. J. Int. Neuropsychol. Soc. 2, 274–281 [DOI] [PubMed] [Google Scholar]
- 41. McAllister T.W., Flashman L.A., Sparling M.B., and Saykin A.J. (2004). Working memory deficits after traumatic brain injury: catecholaminergic mechanisms and prospects for treatment—a review. Brain Inj. 18, 331–350 [DOI] [PubMed] [Google Scholar]
- 42. Mathias J.L., and Mansfield K.M. (2005). Prospective and declarative memory problems following moderate and severe traumatic brain injury. Brain Inj. 19, 271–282 [DOI] [PubMed] [Google Scholar]
- 43. Serra-Grabulosa J.M., Junque C., Verger K., Salgado-Pineda P., Maneru C., and Mercader J.M. (2005). Cerebral correlates of declarative memory dysfunctions in early traumatic brain injury. J. Neurol. Neurosurg. Psychiatry 76, 129–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vukovic J., Borlikova G.G., Ruitenberg M.J., Robinson G.J., Sullivan R.K., Walker T.L., and Bartlett P.F. (2013). Immature doublecortin-positive hippocampal neurons are important for learning but not for remembering. J. Neurosci. 33, 6603–6613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Child K.M., Herrick D.B., Schwob J.E., Holbrook E.H., and Jang W. (2018). The neuroregenerative capacity of olfactory stem cells is not limitless: implications for aging. J. Neurosci. 38, 6806–6824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Donega V., Nijboer C.H., van Velthoven C.T., Youssef S.A., de Bruin A., van Bel F., Kavelaars A., and Heijnen C.J. (2015). Assessment of long-term safety and efficacy of intranasal mesenchymal stem cell treatment for neonatal brain injury in the mouse. Pediatr. Res. 78, 520–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mishra S.K., Rana P., Khushu S., and Gangenahalli G. (2017). Therapeutic prospective of infused allogenic cultured mesenchymal stem cells in traumatic brain injury mice: a longitudinal proton magnetic resonance spectroscopy assessment. Stem Cells Transl. Med. 6, 316–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Good M.A., Barnes P., Staal V., McGregor A., and Honey R.C. (2007). Context- but not familiarity-dependent forms of object recognition are impaired following excitotoxic hippocampal lesions in rats. Behav. Neurosci. 121, 218–223 [DOI] [PubMed] [Google Scholar]
- 49. Langston R.F., and Wood E.R. (2010). Associative recognition and the hippocampus: differential effects of hippocampal lesions on object-place, object-context and object-place-context memory. Hippocampus 20, 1139–1153 [DOI] [PubMed] [Google Scholar]
- 50. Mumby D.G., and Pinel J.P. (1994). Rhinal cortex lesions and object recognition in rats. Behav. Neurosci. 108, 11–18 [DOI] [PubMed] [Google Scholar]
- 51. Ennaceur A., Neave N., and Aggleton J.P. (1996). Neurotoxic lesions of the perirhinal cortex do not mimic the behavioural effects of fornix transection in the rat. Behav. Brain Res. 80, 9–25 [DOI] [PubMed] [Google Scholar]
- 52. Bussey T.J., Muir J.L., and Aggleton J.P. (1999). Functionally dissociating aspects of event memory: the effects of combined perirhinal and postrhinal cortex lesions on object and place memory in the rat. J. Neurosci. 19, 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Norman G., and Eacott M.J. (2004). Impaired object recognition with increasing levels of feature ambiguity in rats with perirhinal cortex lesions. Behav. Brain Res. 148, 79–91 [DOI] [PubMed] [Google Scholar]
- 54. Badman M.K., Pissios P., Kennedy A.R., Koukos G., Flier J.S., and Maratos-Flier E. (2007). Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]
- 55. Galman C., Lundasen T., Kharitonenkov A., Bina H.A., Eriksson M., Hafstrom I., Dahlin M., Amark P., Angelin B., and Rudling M. (2008). The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 8, 169–174 [DOI] [PubMed] [Google Scholar]
- 56. Makela J., Tselykh T.V., Maiorana F., Eriksson O., Do H.T., Mudo G., Korhonen L.T., Belluardo N., and Lindholm D. (2014). Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1alpha in human dopaminergic neurons via Sirtuin-1. Springerplus 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Inagaki T., Dutchak P., Zhao G., Ding X., Gautron L., Parameswara V., Li Y., Goetz R., Mohammadi M., Esser V., Elmquist J.K., Gerard R.D., Burgess S.C., Hammer R.E., Mangelsdorf D.J., and Kliewer S.A. (2007). Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 5, 415–425 [DOI] [PubMed] [Google Scholar]
- 58. Owen B.M., Bookout A.L., Ding X., Lin V.Y., Atkin S.D., Gautron L., Kliewer S.A., and Mangelsdorf D.J. (2013). FGF21 contributes to neuroendocrine control of female reproduction. Nat. Med. 19, 1153–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kopen G.C., Prockop D.J., and Phinney D.G. (1999). Marrow stromal cells migrate throughout forebrain and cerebellum, and they differentiate into astrocytes after injection into neonatal mouse brains. Proc. Natl. Acad. Sci. U. S. A. 96, 10711–10716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang S., Kan Q., Sun Y., Han R., Zhang G., Peng T., and Jia Y. (2013). Caveolin-1 regulates neural differentiation of rat bone mesenchymal stem cells into neurons by modulating Notch signaling. Int. J. Dev. Neurosci. 31, 30–35 [DOI] [PubMed] [Google Scholar]
- 61. Chirumamilla S., Sun D., Bullock M.R., and Colello R.J. (2002). Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J. Neurotrauma 19, 693–703 [DOI] [PubMed] [Google Scholar]
- 62. Rice A.C., Khaldi A., Harvey H.B., Salman N.J., White F., Fillmore H., and Bullock M.R. (2003). Proliferation and neuronal differentiation of mitotically active cells following traumatic brain injury. Exp. Neurol. 183, 406–417 [DOI] [PubMed] [Google Scholar]
- 63. Ramaswamy S., Goings G.E., Soderstrom K.E., Szele F.G., and Kozlowski D.A. (2005). Cellular proliferation and migration following a controlled cortical impact in the mouse. Brain Res. 1053, 38–53 [DOI] [PubMed] [Google Scholar]
- 64. Ibrahim S., Hu W., Wang X., Gao X., He C., and Chen J. (2016). Traumatic brain injury causes aberrant migration of adult-born neurons in the hippocampus. Sci. Rep. 6, 21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen J., Hu J., Liu H., Xiong Y., Zou Y., Huang W., Shao M., Wu J., Yu L., Wang X., Wang X., and Lin L. (2018). FGF21 protects the blood-brain barrier by upregulating PPARgamma via FGFR1/beta-klotho after traumatic brain injury. J. Neurotrauma 35, 2091–2103 [DOI] [PubMed] [Google Scholar]
- 66. Kuroda M., Muramatsu R., Maedera N., Koyama Y., Hamaguchi M., Fujimura H., Yoshida M., Konishi M., Itoh N., Mochizuki H., and Yamashita T. (2017). Peripherally derived FGF21 promotes remyelination in the central nervous system. J. Clin. Invest. 127, 3496–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Xu J., Stanislaus S., Chinookoswong N., Lau Y.Y., Hager T., Patel J., Ge H., Weiszmann J., Lu S.C., Graham M., Busby J., Hecht R., Li Y.S., Li Y., Lindberg R., and Veniant M.M. (2009). Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models—association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab. 297, E1105–E1114 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.