

Abstract

A novel carbon material made of porous graphene-like nanosheets was synthesized from biomass resources by a simple catalytic graphitization process using nickel as a catalyst for applications in electrodes for energy storage devices. A recycled fiberboard precursor was impregnated with saturated nickel nitrate followed by high-temperature pyrolysis. The highly exothermic combustion of in situ formed nitrocellulose produces the expansion of the cellulose fibers and the reorganization of the carbon structure into a three-dimensional (3D) porous assembly of thin carbon nanosheets. After acid washing, nickel particles are fully removed, leaving nanosized holes in the wrinkled graphene-like sheets. These nanoholes confer the resulting carbon material with ≈75% capacitance retention, when applied as a supercapacitor electrode in aqueous media at a specific current of 100 A·g–1 compared to the capacitance reached at 20 mA·g–1, and ≈35% capacity retention, when applied as a negative electrode for lithium-ion battery cells at a specific current of 3720 mA·g–1 compared to the specific capacity at 37.2 mA·g–1. These findings suggest a novel way for synthesizing 3D nanocarbon networks from a cellulosic precursor requiring low temperatures and being amenable to large-scale production while using a sustainable starting precursor such as recycled fiberwood.
1. Introduction
The industrial interest of carbon materials in any of their allotropic forms has considerably grown in the past century due to their interesting intrinsic physicochemical properties such as good electronic conductivity, high chemical stability, tailorable surface properties, ease of processing, and nonhazardous nature.1 Carbon materials find applications in fields as diverse as thermal2 and energy storage systems,3 removal of metal contaminants,4 and organics in water purification,5 catalyst supports,6 gas adsorption and storage,7 or biosensing.8 In the field of energy storage, carbon materials dominate the composition of commercial electrodes in electrochemical double-layer capacitors (EDLCs or “supercapacitors”)9−13 and of anodes for lithium-ion (LIBs)14−16 and sodium-ion batteries (SIBs).17,18
The working principle of EDLCs relies on physical charge separation,19 which allows the devices to withstand thousands of cycles without remarkable capacitance loss20,21 while delivering high power densities. For this application, mesoporous materials of different compositions,22,23 metal–organic framework (MOF)-derived nanoporous materials,24,25 and two-dimensional (2D) layered materials26 have shown promising electrochemical properties as electrodes for supercapacitor applications. Notwithstanding that, carbon-based electrodes are still receiving the most attention and dominate the commercial market. Since the capacitance is predominantly related to the surface area accessible to the electrolyte, intensive research efforts have been devoted to design advanced carbon materials with tailored pore characteristics.27,28 However, aside from either increasing the specific surface area and storage active sites by chemical and physical activation processes29 or attempting to control the final surface properties by template methods,30 there is currently a new focus toward the development of three-dimensional (3D) nanostructured materials with hierarchical and interconnected porosity due to their improved storage performance at high charge/discharge rates.31 This interest has aroused not only for EDLC applications but also for the application of carbonaceous materials in LIBs32 and SIBs.33
In the case of LIBs, one of the essential and desired requirements for their implementation in automotive applications is a higher power density, which enables charging the device in a short period of time without compromising energy density. However, state-of-the-art graphite anodes deliver extremely poor rate capabilities that cannot satisfy such a demand whilst also arising safety concerns related to lithium-metal plating on the anode surface.34 As for EDLCs, nanostructured amorphous carbon anodes have shown promise in supplying better rate capabilities35−37 when compared to graphite anodes due to shorter lithium diffusion length paths and smaller charge transfer resistance.
Dimensionality has proven to have a decisive effect on electronic and ionic transport properties of carbon materials,38 positioning layered carbon structures amongst the most promising electrode materials.39 Nevertheless, the main limitation of sheet-like morphologies when evaluated for EDLC and LIB applications is that isolated sheets suffer from agglomeration during electrode preparation as a result of strong van der Waals intersheet interaction, limiting the area accessible by the electrolyte only to the electrode edges rather than across the whole volume.40 For instance, in high-loaded anodes for LIBs, the diffusion of lithium ions across the electrode is extremely poor at high charging rates due to the high ion path tortuosity.41
This fact usually leads to a reduction in the achievable capacitance (EDLCs)/capacity (LIBs) compared to what is expected in relation to the material properties. One of the main approaches to overcome the above shortcomings and endow such materials with better electrochemical performance is the introduction of porosity or holes onto the sheets in order to facilitate the ionic access to the whole electrode area.42 A variety of procedures have been addressed to produce porous nanosheets: high-energy bombardment with electrons or ions, nanolithography and etching, liquid-phase oxidation, acid etching with nitric acid (HNO3), guided etching with catalysts or reactive nanoparticles,42 or chemical activation43 and oxidation.44
Another novel strategy for shortening ionic diffusion lengths is the development of graphene-like nanosheets arranged into a porous hierarchical 3D network.45−47 Some attempts already reported to design such microstructures include template-directed deposition or assembly of graphene,48 development of graphene aerogels by self-assembly in freeze-drying processes,49,50 and taking advantage of the naturally developed hierarchical structure of some biomass precursors.44
The use of biomass resources as sustainable starting precursors to obtain carbon materials is currently of high interest. Most biomass, especially that coming from agricultural waste, can be seen as a renewable resource, and thus there is interest in exploring ways to turn a waste biomass into a high-value product.51 Porous carbon nanosheet networks have been previously obtained from various biomass precursors, including, for example, from cellulose via ball milling treatment followed by KOH activation,52 from peanut skins and rice husk via a sulfuric acid-assisted hydrothermal process followed by activation,53,54 from sugarcane bagasse pith/chitosan by carbonization and activation with KOH treatments,43 and from soybean milk using NaCl as a template.36
Herein, a one-step process for the synthesis of a three-dimensional porous graphene-like carbon material is presented. A recycled cellulose precursor was first impregnated with a saturated nickel nitrate solution and then carbonized in a nitrogen atmosphere. Upon heat treatment, a fast thermal decomposition causes the breakage of the original cell wall structure of the biomass source, while nickel acts as catalyst promoting graphitization. A subsequent acid etching process removes the nickel particles and introduces pores into the structure. The resulting microstructure of the carbon material, consisting of holey graphene-like sheets, would be interesting for electrochemical energy storage applications. The thermal behavior during pyrolysis and the resulting microstructural features and surface properties are evaluated with a view to their electrochemical behavior as electrodes for supercapacitor devices and as a negative electrode (anode) for LIBs.
2. Results and Discussion
2.1. Synthesis and Microstructural Characterization
Thermogravimetric (TGA) and differential scanning calorimetry (DSC) analysis were performed to study the thermal behavior of samples while heating under an inert atmosphere. Figure 1 shows the TGA and DSC analysis under nitrogen of raw MDF precursor and MDF impregnated with an aqueous nickel nitrate solution in comparison with that of MDF impregnated with the nickel nitrate isopropanol-based solution previously reported in ref (55). In that paper, focused on the graphitization of MDF wood by means of a nickel catalyst, the authors decided to use isopropanol as a solvent instead of water to avoid the swelling of the wood fibers and ensure the consistency of the monolithic carbon scaffold.
Figure 1.
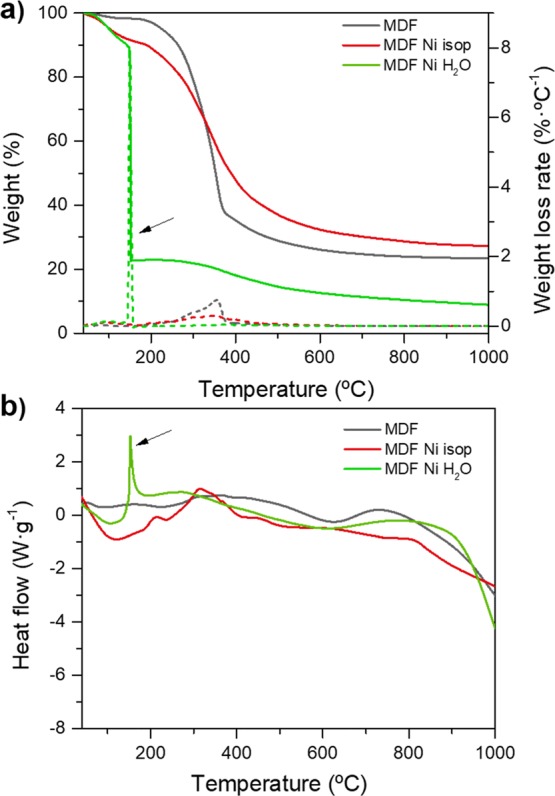
TGA/DSC analysis using a heating rate of 10 °C·min–1 from room temperature to 1000 °C under a constant nitrogen flow rate of 100 mL·min–1. Comparison between the thermal behavior of raw MDF, MDF impregnated with Ni(NO3)2 isopropanol-based solution55 and MDF impregnated with Ni(NO3)2 water-based solution: (a) Weight curve (left; solid lines) and derivative weight loss (right; dashed lines) versus temperature during pyrolysis; (b) heat flow curve versus temperature (black arrows point to the exothermic reactions).
The raw MDF precursor exhibits a smooth and continuous weight loss at temperatures between 200 and 500 °C attributed to the decomposition of the main polysaccharide chains of cellulosic precursors and breakdown of C–O, C–C, and C–H bonds, leaving a solid carbon template with ≈20–25% of the initial weight at temperatures above 600 °C (Figure 1a). No further weight loss is observed for untreated samples, whereas samples impregnated with an isopropanol-based nickel nitrate solution55 exhibit a fairly similar behavior except for a more progressive weight loss arising from the decomposition of a nickel nitrate component (TGA/DSC analysis of a hexahydrate nickel nitrate powder in an inert atmosphere shown in Figure S1, Supporting Information).
In contrast, samples impregnated with an aqueous nickel nitrate solution undergo an abrupt thermal decomposition with a large weight loss of ≈67% at 150 °C (pointed out by arrows in Figure 1). At this temperature, the sample weight abruptly decays from 89 to 22%. A highly exothermic reaction can be confirmed by the DSC analysis (enthalpy of ≈360 J·g–1) shown in Figure 1b at the same temperature. From then on, no significant changes are detected, apart from a slight weight reduction up to ≈9% at 1000 °C. However, this remaining carbon weight content after pyrolysis may not be representative since part of the powdered sample was released from the alumina crucible during the exothermic reaction. It is evident that, due to the large amount of released energy, the actual sample temperature should be much higher than the externally applied temperature as measured in the furnace’s thermocouple.
The main morphological and microstructural features were investigated by scanning (SEM) and transmission electron (TEM) microscopy. Figure 2 shows a summary of representative SEM micrographs of MDF-derived carbon obtained by conventional pyrolysis and carbon obtained after impregnation with the catalyst solution followed by pyrolysis. The typical microstructure of MDF-derived carbon without a catalyst consists of an agglomerate of compacted hollow cellulosic fibers with an average outer diameter of ≈10–15 μm, as seen in Figure 2a,b. In contrast, for samples impregnated with the catalyst, the morphology of the carbonized fibers is severely modified after heating to as little as 300 °C, suggesting a large inner volume expansion (SEM micrographs shown in Figure 2c,d). Instead of a dense fibrous structure, the carbonization process transforms the original structure to an open and highly complex three-dimensional cross-linked framework. Inner porosity is originated by the randomly oriented stacking of crumpled graphene-like nanosheets assembled along the fiber axis. When the pyrolysis temperature is increased up to 1000 °C (Figure 2e), no significant morphological changes occur, but it becomes more apparent that the sheets’ surfaces are covered by globular nanosized particles.
Figure 2.

Representative SEM micrographs: (a, b) Front and side views of a representative fiber from conventional, nontreated MDF carbon carbonized at 300 °C; (c, d) front and side views of a representative fiber of MDF Ni H2O 300 °C sample; (e) MDF Ni H2O 1000 °C sample where globular nickel nanoparticles are appreciable under light contrast before acid washing, and (f) MDF Ni H2O 1000 °C after acid washing with HCl.
The interconnected sheet-like morphology is confirmed in representative TEM images shown in Figure 3. The microstructure obtained at a processing temperature of 300 °C consists of a network of thin platelets stacked on top of each other. These isolated sheets exhibit a very thin flake morphology, indicated by the low and almost homogeneous contrast under the electron beam, along with a large number of crumples and ripples on their surface (edges observable by lighter contrast lines in the dark-field (DF) image shown in Figure 3a). Due to the presence of these wrinkles, it was difficult to establish an average value for the sheet thickness.
Figure 3.

Representative TEM micrographs: (a) Dark-field (DF) STEM image of MDF Ni H2O 300 °C sample; (b) bright-field TEM image of the same sample showing the homogeneous distribution of Ni nanoparticles (particle size distribution shown in the inset of the figure); and (c) combined elemental mapping and STEM images of MDF Ni H2O 1000 °C. The green-colored area corresponds to the nickel phase, while the red one corresponds to carbon. (d–f) Representative TEM micrographs of MDF Ni H2O 1000 °C after acid washing (the letter G represents the hollow ordered graphitic structures, and the arrows show the edges and corrugations on the sheets’ surface).
A close-up TEM view shown in Figure 3b reveals a homogeneous distribution of nickel nanoparticles (dark areas) with an average diameter of ≈4 nm at a pyrolysis temperature of 300 °C. Combined STEM-HAADF and EDX analysis (compositional map shown in Figure 3c) confirm the composition of the globular nickel particles over the thin carbon matrix. Furthermore, since nickel particles are well known to induce the graphitization of amorphous carbon materials in situ during pyrolysis from temperatures below 1000 °C,55,56 they are surrounded by well-ordered graphitic layers formed by a solution-precipitation mechanism as reported previously by Gutiérrez-Pardo et al.55 The coarsening of nickel nanoparticles at these comparatively low temperatures can be explained by a decrease in their melting point when compared to the bulk eutectic melting temperature (1455 °C) due to size effects and the solution of carbon into the catalytic nanoparticles.57
When etched with hydrochloric acid (SEM image in Figure 2f and TEM images in Figure 3d–f), nickel particles were fully removed from the material, leaving many in-plane holes and pores in the carbon sheets with either spherical or faceted shapes (further details in the next section). The catalyst concentration in the carbon scaffold after impregnation and carbonization up to 1000 °C (as representative sample) was ≈34.9 wt %. After acid etching, the catalyst amount fell to ≈1.6 wt % according to ICP-OES analysis. The microstructure of the MDF Ni H2O 1000 °C sample after acid washing is similar to that reported for few-layer graphene and graphene oxide materials56,58,59 and comparable to graphene fibers synthesized in previous works by a wet-spinning technique,60−62 with the difference of using a biomass resource as a starting precursor and avoiding arduous synthesis procedures. Furthermore, we can observe microporosity arising from nickel removal, resulting in microstructures similar to those recently shown by holey graphene materials.42
SEM analysis of the fibers after impregnation but before pyrolysis (shown in Figure S2, Supporting Information) confirms that the impregnation with a water-based nitrate solution enhances the swelling and debonding of the fibers in MDF wood; nonetheless, individual fibers maintain their shape and structure, so it is expected that the rearrangement of the microstructure takes place upon heat treatment and it is not only a direct effect of the impregnation.
It is then clear that impregnation with an aqueous nickel nitrate solution is not only depositing a nickel precursor onto the fiber’s surface that later induces graphitization but also affecting the chemical state of the wood precursor itself. Deeper insight into these changes was obtained from Fourier transform infrared (FTIR-ATR) spectroscopy analysis. As shown in Figure 4, the infrared spectra of as-received MDF, prior to any impregnation or pyrolysis, exhibit similar characteristic peaks than those of pure cellulose.63 The absorption bands located at ≈3330 and 1700–1500 cm–1 indicate O–H group’s stretching and scissoring vibrations, respectively. Bands located at wavenumbers of ≈2920 and ≈1350 cm–1 are ascribed to C–H stretching and bending vibration modes in the glucose units. Moreover, absorption peaks at ≈1030 and 900 cm–1 correspond to C–O stretching in secondary and primary alcohols or aliphatic ethers and to the β-glycosidic linkages between glucose units in cellulose.64 When comparing MDF after impregnation with that of raw MDF wood spectra, besides an increased intensity of the O–H vibration arising from adsorbed water, significant changes are noticeable in the region between 1750 and 750 cm–1. Three new distinctive absorption peaks appeared within this range (at ≈800, ≈1270, and ≈1640 cm–1), related to the different vibrations of the nitrate groups and hence confirming the formation of N–O stretching vibrations.
Figure 4.
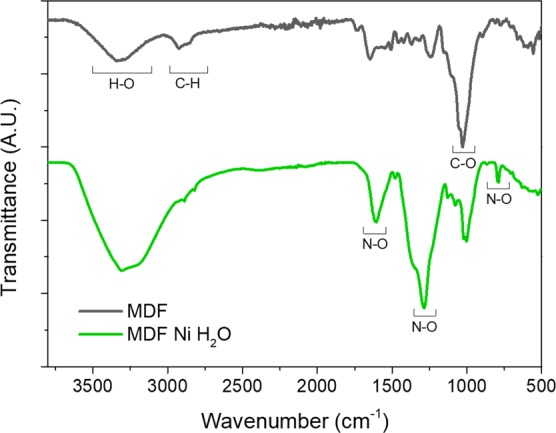
FTIR spectra of raw MDF and MDF impregnated with a water-based Ni(NO3)2 solution.
The hierarchical and anisotropic wood cellular wall structure is characterized by the stacking of parallel elongated hollow cells aligned along the tree growth direction. Such a three-dimensional fiber wall structure is composed of different layers, as extensively reported elsewhere,65 but the main components of the wood are typically 40–50% of semicrystalline cellulose fibers covered by hemicellulose, which represents 20–30% by weight. Both components are bound together by an amorphous lignin matrix that connects the wood cells and fills the space between them, providing the wood with a structural support and mechanical strength.66 A single cellulose fiber (10–50 μm in diameter) is itself composed of an arrangement of elementary structural units called nanofibrils (2.5–3.5 nm in diameter for single fibrils and 20–50 nm for aggregates67), which are bound together by van der Waals forces and intra- and interchain hydrogen bonding.68
As we have demonstrated, the highly exothermic reaction at temperatures below 200 °C results in both a fast thermal decomposition and the breakage and rearrangement of the original cell wall fibers, whereas when samples are heated in air, they burn explosively at temperatures below 100 °C (results not shown here). Nitro-containing compounds are well known for their potential explosiveness, and so, for comparison purposes, we synthesized nitrocellulose from cotton by the usual method of esterification of hydroxyl groups with a nitric/sulfuric acid mixture (weight ratio of 1:3)69 and studied its FTIR spectrum (Figure S3, Supporting Information). In this sample, the most distinctive feature is a decrease in the intensity of the O–H stretching mode and the formation of three intense bands at ≈1660 cm–1 (asymmetric NO2 stretching), ≈1280 cm–1 (symmetric NO2 stretching), and ≈840 cm–1 (NO stretching) similar to those shown by MDF impregnated with a water-based nickel nitrate solution (Figure 4).70,71
TGA results under nitrogen support the hypothesis that the microstructural expansion can be ascribed to the rapid and massive capillary release of volatile matter and gases during thermal decomposition. This would indeed lead to a sudden pressure build up inside the cellulose fiber and the rupture of the links between cellulose chains, causing the restacking of the cell wall to form a network of interconnected and stacked carbon nanosheets. Thus, the explosive reaction of nitrocellulose provides enough energy for the rearrangement of the cellulose units and their carbonization at such low temperatures.
Similar approaches using massive capillary release of volatile species have been carried out to synthetize 3D porous graphene architectures72 and isolate cellulose nanofibrils through a steam explosion process.73 A previous work reported a morphological change of fibrous cotton to interconnected carbon nanosheets after impregnation with magnesium nitrate, attributing the change to the oxidation caused by release of NO2 and CO2 compounds. In that work, much lower solution concentrations were considered and the thermal behavior was not studied.74
2.2. Bulk Structure, Surface Chemical Composition and Textural Properties
Nitrogen adsorption–desorption measurements at 77 K were performed to determine surface properties and pore size distributions. Figure 5a shows type IV adsorption–desorption isotherms according to the IUPAC classification, suggesting a mesoporous structure with a small hysteresis loop closing at relative pressures close to 0.4. The BET specific surface area (Table 1) at 300 and 1000 °C is ≈391 and 333 m2·g–1, respectively. These values are up to three times higher than the value shown for conventional nickel nitrate-treated MDF carbons using isopropanol as a solvent at 1000 °C (≈122 m2·g–1) since the resulting carbon structure has a strongly increased accessible porosity. The pore size distribution (shown in the inset of Figure 5a) is in good agreement with the pores observed by TEM analysis (Figure 5b,c). At 300 °C, most of the pore volume lies in the range of ≈2–4 nm, while, due to the coarsening effect of nickel particles, the pore distribution at 1000 °C becomes broader and bimodal. Despite the fact that, at 1000 °C, most nickel nanoparticles have already formed agglomerates (Figure 3c), after acid washing, pores with a much lower diameter can also be seen. These pores may have been originated as a result of the fast thermal decomposition and activation shown at 150 °C by nitrogen and oxygen species gases. The increase in pressure can also cause nickel particles to leave holes into the thin carbon sheets while gases are released.
Figure 5.

(a) N2 adsorption/desorption isotherms and pore size distributions (inset of the figure) calculated by applying the BJH method to the desorption data of MDF Ni H2O 300 and 1000 °C samples. (b, c) High-resolution TEM images showing the porous structure. (d) X-ray diffraction patterns of MDF Ni H2O 300 and 1000 °C. (e, f) Raman spectra of MDF Ni H2O 300 and 1000 °C. Spectra were deconvoluted into relevant carbonaceous bands as described in the main text, using a least-squares method and pseudo-Voigt line shapes. The green line corresponds to the residual, while the red one represents the fitted data.
Table 1. Summary of Relevant Microstructural and Textural Parameters of MDF Samples Impregnated with Nickel Nitrate Solutions from Nitrogen Adsorption/Desorption Measurements and Raman Fitting of Relevant Carbonaceous Bandsa.
| parameters | MDF Ni H2O 300 °C | MDF Ni H2O 1000 °C | MDF Ni isop 1000 °C |
|---|---|---|---|
| SBET (m2·g–1) | 391 | 333 | 122 |
| IG/ID1 | 0.84 | 1.56 | 0.50 |
| α | 0.45 | 0.60 | 0.33 |
| I2D/IG | 0.19 | 0.89 | 0.33 |
| GFWHM (cm–1) | 50 ± 6 | 15 ± 1 | 31 ± 2 |
| 2DFWHM (cm–1) | 200 ± 20 | 30 ± 1 | 60 ± 7 |
Data for MDF Ni isop is from ref (55).
XRD and Raman spectroscopy measurements were performed to gain insights into the degree of crystallinity of the resulting carbon material. Figure 5d shows XRD patterns of MDF Ni H2O at 300 and 1000 °C. The XRD pattern at 300 °C exhibits two broad reflections centered at 2θ ≈ 26 and 44° similar to those of (002) and (100) lattice planes of graphite (pdf 26-1079) with a lack of ordered stacked regions. As the temperature increases up to 1000 °C, the intensity of the (002) reflection peak of graphite increases, while the full width at half-maximum decreases due to the precipitation of graphitic structures surrounding nickel particles. Notwithstanding that, a broad background and a shoulder are still observable at values around 26° associated to the presence of an amorphous fraction. No significant peaks from remaining nickel were observed.
Raman spectra shown in Figure 5e,f exhibit three prominent bands within the 1000 to 3000 cm–1 wavenumber range. The so-called D1 band (located at 1350 cm–1, A1g symmetry) reflects the vibration of graphene layer edges in amorphous carbons and is almost absent in defect-free graphite and graphene. The G band (located at 1580 cm–1, E2g symmetry) originates from the in-plane stretching vibration of the C=C double bond (sp2-bonded carbon), while the 2D peak (located at 2690 cm–1) area, intensity, and shape are indicative of the stacking level of graphene layers.75,76 Other defect bands indicating a disordered graphitic lattice are the D2 band (1620 cm–1) arising from the vibration of surface graphene layers, the D3 band (1500 cm–1) related to the vibration of amorphous regions, and the D4 band (located at 1150 cm–1) showing the local disorder by the presence of ionic impurities.77 The intensity ratio between the D1 and G bands is indicative of the degree of graphitization, whereas the G/2D intensity ratio accounts for the stacking level. Since these ratios are often used to characterize structural properties, Raman spectra were fitted using a least-squares method and pseudo-Voigt line shapes, and main results are summarized in Table 1. Raman results reported by Gutiérrez-Pardo et al.55 for nickel-treated MDF carbon using isopropanol as a solvent at 1000 °C are also included in the table for comparison purposes.
A clear enhancement in crystallinity is obtained when increasing the carbonization temperature from 300 to 1000 °C, noted by an increase in the G peak intensity relative to the D1 band, along with the separation of both bands and the narrowing of the FWHM of the G band (data shown in Table 1). The continuous solution of amorphous carbon into metal particles and precipitation of graphite crystals causes an increase in the degree of graphitization with increasing treatment temperature. The degree of graphitization α, as obtained from deconvolution of Raman spectra, increased from 0.45 to 0.6 at 300 and 1000 °C, respectively. These values are much higher than usual for nickel-graphitized samples at such temperatures (at 1000 °C usually α ≈ 0.3355). Interestingly, at 300 °C, we can already observe the precipitation of graphitic regions by TEM analysis (Figure S4, Supporting Information), while it is well known that such an effect takes place at temperatures above 800 °C.55,78 We attribute this early beginning of the graphitization to the self-propagating exothermic reaction taking place at 150 °C, which may have increased the temperature locally.
In addition, the intensity ratio between the 2D and G bands (I2D/IG) is about 0.19 and 0.89 at 300 and 1000 °C, respectively. These features along with the maximum peak position suggest a few-layer graphene structure following Ferrari et al.,76 which is hardly differentiable from that of graphite. At 300 °C, the characteristic 2D peak is almost absent and increases in intensity with increasing pyrolysis temperature. In a previous work, there was considerable debate regarding the Raman spectra of graphene-related materials.79 The Raman spectra of ideal monolayer graphene materials shows a strong 2D band contribution along with a low-intensity G peak,80 whereas the D1 peak is usually not observable in defect-free graphene. However, the presence of defects, including wrinkles, remnant functional groups, and porosity/holes can result in a deviation from this general trend, when compared to mechanically exfoliated or CVD-grown graphene. Some works showed equally intense D and G bands with no appreciable 2D band, while others reported a broad 2D peak similar to that of amorphous carbon.81−83 Nevertheless, SEM and TEM analyses confirm a microstructure similar to that of graphene-related materials.
X-ray photoelectron spectroscopy (XPS) measurements were performed to investigate the surface chemical composition and sp3 to sp2 carbon contribution ratio. Main results are listed in Table 2 (spectra shown in Supporting Information, Figure S5). Samples carbonized at 300 °C show a large oxygen content (≈29 atom %) with a C1s/O1s atomic ratio close to 2, decreasing to ≈11 atom % at 1000 °C (C1s/O1s ratio close to 8). Likewise, the N atomic concentration decreases gradually from 7 atom % (300 °C) to 3 atom % (1000 °C). These reductions confirm the almost complete removal of functional groups as the pyrolysis temperature is increased. To gain further insights into the structure, the C1s peak was deconvoluted into five contributions using Gaussian–Lorentzian functions: sp2-hybridized carbon (centered at ≈284.6 eV), sp3-bonded carbon (≈285.3 eV), C–O (≈286.5 eV), epoxy and hydroxyl groups, C=O carbonyl groups (centered at ≈287.7 eV), O—C=O carboxyl groups (∼288.7 eV), and π–π* shake-up line (≈291 eV).84 Results of these fits are shown in Supporting Information (Figure S5). From this data, the ratio of sp3 to sp2 contributions was estimated to decrease from 0.36 at 300 °C to 0.26 at 1000 °C due to the nickel-assisted graphitization, in agreement with our interpretation of the Raman spectra.
Table 2. XPS Elemental Analysisa.
| parameter | MDF Ni H2O 300 °C | MDF Ni H2O 1000 °C | MDF Ni isop 1000 °C |
|---|---|---|---|
| carbon (%) | 63 | 86 | 86 |
| oxygen (%) | 29 | 11 | 11 |
| nickel (%) | 1 | 1 | |
| nitrogen (%) | 7 | 3 | 2 |
| C/O ratio | 2.2 | 7.8 | 7.8 |
| sp3/sp2 ratio | 0.36 | 0.26 | 0.41 |
Atomic content of C, O, Ni, and N, estimation of C/O and sp3 to sp2 ratio from peak deconvolution.
2.3. Electrochemical Characterization
The electrochemical performance of MDF Ni H2O 1000 °C as a supercapacitor electrode was evaluated in a symmetrical two-electrode setup. Figure 6 summarizes the main results. CV curves (Figure 6a) at different sweep rates exhibit a roughly rectangular shape with small deviations at ≈0.8–1 V, suggesting a dominant double-layer formation mechanism. Slight differences in CV curves in terms of curve shape and area were observed when increasing the sweep rate from 5 to 200 mV·s–1. Consistent with these results, the galvanostatic potential profiles (Figure 6b) also show an almost linear charging/discharging behavior, indicating little pseudocapacitive effects.
Figure 6.

Summary of relevant electrochemical measurements in a symmetrical two-electrode cell setup of MDF Ni H2O 1000 °C. (a) Representative CV curves at different sweep rates ranging from 5 to 200 mV·s–1. (b) Typical galvanostatic charge/discharge curves at different specific currents. (c) Variation of specific capacitance as a function of specific current from 20 mA·g–1 to 100 A·g–1. (d) Variation of specific capacitance as a function of cycle number, measured up to 10,000 cycles at a rate of 2 A·g–1.
Figure 6c summarizes the specific capacitance calculated from the GCD curves as a function of the specific currents, ranging between 0.02 and 100 A·g–1. At the lowest studied specific current, 20 mA·g–1, a specific capacitance of 72 F·g–1 was achieved. Interestingly, the sample still shows a capacitance of ≈55 F·g–1 at a current density of 100 A·g–1, meaning a retention of ≈75% of their maximum capacitance even at such a high current. The small increase in capacitance at high current densities can be attributed to slight overshooting of the potentiostat. This can result in hydrogen evolution that can be stored in the pores and introduce a small amount of pseudocapacitance, overestimating the cell capacitance.85,86 More than 96% of the original capacitance was retained after long-term cycling experiments of 10,000 charge/discharge GCD cycles (Figure 6d), indicating good cycling stability.
Although the experimental conditions (either in material preparation, heat-treatment temperature, or electrochemical studies) are difficult to compare, Table S1 (Supporting Information) gives a comparison of previous electrochemical studies on biomass-derived carbon electrodes for EDLC applications. For a fair comparison, data selection of this table was based on carbon materials with similar surface areas (below 1000 m2·g–1). The obtained capacitance values in this work at low current densities are not impressive compared to other biomass-derived carbon materials due to our limited surface area (≈333 m2·g–1). However, MDF Ni H2O shows good rate capability and long-term cycling stability on account of the hierarchical open and porous carbon structure obtained from the explosion-assisted activation strategy.
To further investigate electrochemical properties, the synthesized porous nanosheets were also evaluated as an anode for LIBs, focusing on the rate capability compared to that of commercial graphite anodes. The electrochemical performance was assessed in a half-cell setup (three-electrode configuration) by galvanostatic charge/discharge experiments at a specific current between 37.2 and 3720 mA g–1 using lithium metal as a CE and RE. Figure 7a shows the delivered specific capacities of MDF Ni H2O 1000 °C carbon at different specific currents. The rate performance of a commercial graphite material for LIBs was also measured for comparison purposes. Figure 7b shows the capacity retention of these anode materials (considering that the maximum capacity (100%) is the one achieved at a specific current of 37.2 mA·g–1) as a function of specific current to better compare the rate capability.
Figure 7.

(a) Discharge capacities of MDF Ni H2O 1000 °C carbon electrodes compared to one commercial graphite (SMG A4) during cycling at different specific currents. (b) Capacity retention as a function of specific charge/discharge current. Rate performance investigations: Cycles 1–3: 37.2 mA·g–1; Cycles 4–30: 372 mA·g–1; Cycles 31–70: specific currents of 37.2, 74.4, 186, 372, 744, 1166, 1860, and 3720 mA·g–1 for each step (five cycles); Cycle 70 onward: 372 mA·g–1.
As can be seen, the commercial graphite exhibits the highest achievable capacity at a specific current of 37.2 mA·g–1 (capacity, Q ≈ 338 mAh·g–1), lower than the theoretical capacity of the maximum intercalation stage LiC6 (≈372 mAh·g–187). The MDF Ni H2O 1000 °C sample only reaches a maximum capacity of ≈204 mAh·g–1, related to the less graphitic structure that decreases the storage capacity.16 However, the commercial graphite anode shows a very poor rate capability: increasing the current up to 744 mA·g–1 leads to a significant capacity drop (≈137 mAh·g–1), and the capacity retention amounts to only 40%. Furthermore, the maximum achievable capacity at the maximum studied current of 3720 mA·g–1 is only ≈14 mAh·g–1 (4%). Such strong capacity fading has also been reported previously by Cheng et al.88 for other commercial graphites due to the limited and rather slow diffusion as Li+ ions intercalate via the prismatic plane of graphite and move between adjacent graphene sheets. In contrast, the capacity of MDF Ni H2O 1000 °C only drops to 154 ± 4 and 72 ± 2 mAh·g–1 when increasing the specific current up to 744 and 3720 mA·g–1, retaining 35% of the maximum capacity at the highest specific current. This good rate capability is attributed to the presence of an interconnected and open porous structure, which allows faster Li+ insertion/de-insertion during charge/discharge processes. Apart from good rate capability, electrodes also show good long-term cycling stability (Figure S6): after 200 cycles at a charge/discharge current of 372 mA·g–1, a capacity retention of 97% was maintained, while commercial graphite only retains 75% of the capacity achieved at the beginning of the test. However, it has to be kept in mind that reactions at the Li metal CE might also influence the cycling performance in this cell setup. To confirm the long-term stability, it would be necessary to assemble LIB full cells using state-of-the-art layered oxide cathode materials.
Although MDF Ni H2O 1000 °C exhibits good rate capability compared to a commercial graphite and good long-term stability as anode for LIBs, its main drawback is related to its high specific surface area and presumably also surface heterogeneity (cf. ref (89)) that results in a low first cycle Coulombic efficiency (CEff90,91) of only 27 ± 1%, while the graphite electrode exhibits an efficiency of 85% (Figure S7).
Based on these results, it is clear that, as-synthesized, these materials have some limitations for their application in both supercapacitors and LIBs. However, this work serves as a preliminary study to assess its electrochemical performance in two applications that require very different material characteristics. From this point on, strategies required to improve the performance in one application would result in worse properties for the other one.
On the one hand, for supercapacitor applications, the low surface area (≈333 m2·g–1) limits the capacitance value. It is expected that the larger the accessible surface to the electrolyte, the higher the reachable capacitance. However, our materials exhibited a limited specific surface because our main goal was to study the effect of carrying out a simple process of catalytic graphitization and subsequent acid washing. To improve the surface properties, we propose a simultaneous catalytic graphitization and chemical activation by impregnation with both a graphitization catalyst and an activation agent, such as zinc chloride, potassium chloride, or potassium hydroxide.92−95 This combination could allow to achieve much higher specific capacitances compared to what was obtained by each method separately. It is important to note that, although the increase in the specific surface would be beneficial for supercapacitors, it would also be detrimental for LIBs, as it will be discussed below.
On the other hand, the low CEff obtained in LIB experiments hinders the practical use of these anodes in an LIB full-cell configuration (where the amount of lithium is limited by the positive electrode material) due to the irreversible consumption of Li ions when the SEI is formed. Viable and low-cost prelithiation strategies (chemical, electrochemical, or prelithiation in direct contact with metallic lithium15) could be used in the future to compensate the active lithium losses in the first charge/discharge cycles, therefore achieving a higher CEff. The simplest strategy to simultaneously improve the specific capacity while decreasing the surface area would be to increase the starting catalyst concentration. Numerous papers have confirmed that the extent of graphitization within the carbon material can be enhanced through an increase in the starting catalyst concentration.96−99 Furthermore, the higher the starting catalyst loading, the lower the surface area after acid etching due to a coarsening effect of catalyst particles upon heating, thus allowing an increased first cycle Ceff.96,98 An increase in the degree of crystallinity of the samples could directly lead to an enhanced capacity due to the intercalation of the ions into more ordered, stacked graphene sheets. Nevertheless, it is also possible that the higher degree of graphitization would worsen the rate capability due to the slower kinetics of the lithium intercalation reaction.
3. Conclusions
A graphene-like porous carbon framework was synthesized from a recycled biomass resource by a novel, simple, and cost-effective strategy: a modified catalytic graphitization process using a saturated nickel nitrate water-based solution to impregnate the raw precursor. After pyrolysis, the microstructure of the original material undergoes a morphological transformation, resulting in randomly stacked, wrinkled graphene-like nanosheets covered by nickel nanoparticles. By acid etching with HCl, nickel particles were fully removed, leaving in-plane pores into the sheets. The thermal analysis of the pyrolysis process revealed that the raw impregnated precursor undergoes a self-propagating exothermic reaction, which causes a fast thermal decomposition at only 150 °C. We propose that the fast thermal decomposition at this temperature is promoted by the high content of nitrate groups after impregnation of the cellulosic precursor. The massive release of the volatile matter and nitrogen functional species gases upon thermal decomposition leads to a sudden increase in pressure, which causes the rearrangement of the structure. In addition, nickel particles act as a catalyst to induce graphitization at lower than usual temperatures, resulting in highly crystalline sheets at only 1000 °C, as confirmed by both Raman and XPS studies.
The electrochemical properties of the carbon material as an electrode for supercapacitors and as an anode for LIBs were investigated. The MDF Ni H2O 1000 °C sample showed interesting rate capability as a supercapacitor electrode in aqueous media, retaining ∼75% of the capacitance at a specific current of 100 A·g–1 when compared to the capacitance at 20 mA·g–1. In addition, a capacity retention of ≈35% was reported when applied as an anode for LIB cells at the highest specific current of 3720 mA·g–1 compared to the capacity achieved at 37.2 mA·g–1.
Although the electrochemical properties are not impressive, it is important to highlight the simplicity of the synthesis method. Findings of this work suggest a novel and promising way for developing three-dimensional graphene-like scaffolds but using a simpler, cheaper, and eco-friendlier process than other previously reported synthesis routes. This method requires comparatively low temperatures and is amenable to large-scale production while using a sustainable and inexpensive starting raw material such as recycled fiberwood. However, some strategies should be addressed to improve the electrochemical properties of these materials for energy storage applications, depending on the targeted device. For its application in supercapacitors, the specific surface area should be increased to increase the cell capacitance. However, for application in lithium batteries, the specific surface area must be reduced to improve the Coulombic efficiency in the first cycles.
4. Experimental Section
4.1. Materials
An industrially manufactured wood precursor, cellulose-based medium-density fiberboard (MDF), was chosen as the starting material.14 Prior to pyrolysis, as-received wood pieces were first cut and then dried for 48 h at 80 °C and later impregnated with a 3.0 M nickel nitrate solution in deionized water (Ni(NO3)2·6H2O; purity: 99.99%; Panreac) under reduced pressure to evacuate air and ensure full impregnation. Afterward, the samples were dried at 40 °C for 1 week. The impregnation step helped disaggregate the cellulose fibers so that the MDF lost its integrity. The mass gain after impregnation was ≈25 ± 6%.
Pyrolysis was subsequently carried out in flowing nitrogen under a heating ramp of 5 °C·min–1 up to the desired peak temperature followed by a holding time of 30 min and then cooling down to room temperature. Peak temperatures used in this study were 300 and 1000 °C; however, as it was shown, a highly exothermic reaction takes place between 150 and 200 °C, meaning that for samples heated to a furnace temperature of 300 °C, actual temperatures can be much higher. This exothermic reaction also helped to further disaggregate the fibers, resulting in a loose powder as the final product.
For the development of porosity, remaining nickel particles were removed by ultrasonic stirring in hydrochloric acid (HCl; 37%, Panreac) followed by intensive washing in deionized water until neutral pH. These samples are labeled as “MDF Ni H2O” followed by a number that describes the pyrolysis temperature. MDF-derived samples prepared through the same route but using isopropanol as the solvent (labeled as “MDF Ni isop”) and carbon samples prepared without a catalyst (labeled as “MDF”) were also synthesized for comparison purposes.
4.2. Characterization
Thermal behavior during heating in an inert atmosphere was studied by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) experiments (SDT-600, TA Instruments) using a nitrogen flow rate of 100 mL·min–1 and a heating rate of 10 °C·min–1 up to 1000 °C. Calibration tests were periodically performed using a standard sapphire sample. Fourier transform infrared (FTIR) spectroscopy measurements (Two spectrophotometer, Perkin-Elmer) were performed using the attenuated total reflection (ATR) technique, which allows direct measurement of the as-received material without any further preparation.
Morphological and structural characteristics of the carbon samples were studied by scanning electron microscopy (SEM; Hitachi S5200) and transmission electron microscopy (TEM; Talos F2000S, FEI). Compositional analysis distribution was analyzed by combining high-angle annular dark field (HAADF) and energy-dispersive X-ray spectroscopy (EDX) in the scanning transmission electron microscopy (STEM) mode. For sample preparation, carbon materials were first ground with a mortar and pestle and then dispersed in ethanol by ultrasonic stirring. Then, a drop of the resulting dispersion was deposited on a copper grid with a reticulated amorphous carbon film.
The mass loading of the nickel catalyst in the carbon precursor before and after acid etching (representative carbonization temperature of 1000 °C) was determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES; SpectroBlue, Spectro).
The structural order was characterized by Raman spectroscopy measurements (LabRam Jobin Yvon, Horiba) using a green excitation wavelength of 532 nm. To quantify the extent of the graphitization, Raman spectra were fitted to pseudo-Voigt line shapes, with special interest in the integrated intensities and widths (FWHM) of the D1 (≈1350 cm–1), G (≈1580 cm–1), and 2D (≈2700 cm–1) bands. The degree of graphitization (α) was calculated from the integrated intensity area of the G and D1 bands as follows:
| 1 |
The degree of graphitization ranges in absolute values from 0 to 1. For an ideal graphite structure, only the G band should be detected, and thus α should be 1.
X-ray diffraction (XRD; Bruker D8I-90 diffractometer) measurements with Cu Kα (λ = 0.154 nm) radiation within a scanning angle (2θ) range 10–70° were used to further assess the crystallinity of the samples. Surface properties were determined by nitrogen adsorption/desorption measurements at 77 K using a Micromeritics ASAP 2420 analyzer. Specific surface area and pore size distributions were determined by applying Brunauer–Emmett–Teller (BET) and the Barrett–Joyner–Halenda (BJH) models to the adsorption/desorption isotherms.
X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Leybold-Heraeus LHS10/20 spectrometer (Al Kα1 monochromatic radiation) to study the elemental surface composition. The binding energy was calibrated against the 1s line of carbon (284.6 eV), and after deconvolution of the C1s spectra, the sp3 to sp2 bonding ratios were determined. Curve fitting was performed using CasaXPS software.
4.3. Electrochemical Measurements
4.3.1. Supercapacitor Cells
The working electrodes (WEs) were obtained by preparing a slurry of 80 wt % carbon as an active material, 10 wt % carbon black as a conductive agent (Super C65, Imerys Graphite & Carbon), and 10 wt % of polytetrafluoroethylene (PTFE) as a binder (60 wt % dispersion in H2O, Sigma-Aldrich). The above electrode paste was first homogeneously coated on a nickel foam current collector (12 mm diameter, bulk density 0.45 g·cm–3, porosity: 95%, Goodfellow) and then dried at 80 °C overnight. The active mass loading for each electrode was ≈5 mg.
Symmetric supercapacitors were assembled and studied in a two-electrode setup using Swagelok-type laboratory cells, that is, two equal carbon electrodes and a fiberglass filter (Whatman) as a separator. Total active mass in the device was ≈10 mg, following best practices in the field.100 An aqueous 6 M KOH solution (85% pellets, Panreac) was chosen as an electrolyte. In order to ensure a constant pressure between both electrodes, a stainless steel spring was incorporated into the cell. All experiments were carried out on a Solartron 1287A potentiostat/galvanostat with a 1260A frequency response analyzer. Cyclic voltammetry (CV) experiments were carried out at scanning rates from 1 to 200 mV·s–1 within a voltage range between 0.0 and 1.0 V. Galvanostatic charge/discharge (GCD) experiments were performed at specific currents from 2 × 10–2 to 100 A·g–1 in a voltage window between 0 and 1 V. Long-term cycling stability was evaluated by 10,000 GCD cycles at a specific current of 2 A·g–1. The specific capacitance was calculated following the equation: C = 2·I·Δt·(ΔV·m)−1, where C (F·g–1) is the specific capacitance, I is the applied current, Δt is the discharge time, ΔV is the voltage window, and m is the active mass of the WE.
4.3.2. Lithium-Ion Battery Cells
The carbon sample graphitized using nickel as acatalyst at 1000 °C was also studied as an anode material in a half-cell configuration for LIB cells in Swagelok-type cells. The electrode composition was 90 wt % active material, 5 wt % Super C65 (Imerys Graphite & Carbon) as a conductive agent, and 5 wt % sodium carboxymethyl cellulose (Na-CMC; Walocel CRT 2000 PPA 12, Dow Wolff Cellulosics) dissolved in water as a binder. The electrode paste was cast on dendritic Cu foil by means of a laboratory-scale doctor-blade technique. The mass loading of the active material in each electrode was ≈1.7–1.9 mg·cm–2.
Three-electrode cells were assembled using high-purity lithium metal foil of 12 and 6 mm in diameter (Albemarle Corporation) as the counter (CE) and reference (RE) electrodes, respectively, and the carbonaceous electrodes as WE. GCD experiments were carried out in a potential range between 0.02 and 1.5 V versus Li/Li+ at specific currents between 37.2 and 3720 mA·g–1: first three formation cycles at 37.2 mA·g–1 followed by 20 cycles at 372 mA·g–1 and each five cycles at specific currents of 37.2, 74.4, 186, 372, 744, 1116, 1860, and 3720 mA·g–1, and finally, long-term cycling performance was evaluated over 200 cycles at 372 mA·g–1. Lithium hexafluorophosphate (LiPF6) dissolved in a mixture of ethylene carbonate/ethyl methyl carbonate (1.0 M LiPF6 in EC/EMC, weight ratio of 3:7; supplied by BASF SE; purity: battery grade) solvents was chosen as the electrolyte plus 2 wt % vinylene carbonate (VC) as a solid-electrolyte interphase (SEI) forming additive.101 At least three cells were evaluated for each sample to ensure a high reproducibility of the results, and the associated standard deviation is represented as error bars in the figures.
For comparison in terms of rate capability, a commercial anode graphite (SMG A4; active mass loading of 2.3 mg·cm–2, Hitachi) with the same electrode composition was also considered for LIB cell investigations. The BET surface area of this graphite (data not shown here) was assessed by N2 adsorption isotherms to be 1.58 m2·g–1. Particle size measurements were performed by laser scattering (Cilas 1064, Quantachrome) with a powder dispersion in water. Four repetitions were carried out, yielding values of d0.1 (μm) = 1.78 ± 0.01; d0.5 (μm) = 4.26 ± 0.02; and d0.9 (μm) = 6.81 ± 0.07; average value of 4.32 ± 0.04 μm.
Acknowledgments
This work was supported by the Spanish Government Agency Ministerio de Economía y Competitividad (MINECO) (grant number MAT2016-76526-R). Microstructural and thermal characterization as well as X-ray diffraction were performed at the CITIUS central services of the University of Seville. A.G.-M. is grateful to the German Academic Exchange Service (DAAD) for funding her time at MEET under the “Short-term research grants 2018” program. The authors from the University of Münster thank the MWIDE NRW for funding this work in the project “GrEEn” (funding code: 313-W044A).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03142.
Thermogravimetric analysis of nickel nitrate hexahydrate under nitrogen, SEM micrographs of impregnated biomass before pyrolysis, FTIR spectra of nitrocellulose, TEM micrographs of carbon pyrolyzed at 300 °C, XPS survey spectra and deconvolution of the C1s peak, Comparison of electrochemical properties with other biomass derived carbons, Cycling performance of a commercial SMG A4 graphite and MDF Ni H2O derived carbon anode materials, and first charge–discharge potential profile of MDF Ni H2O derived carbon anode (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Leitner K.; Lerf A.; Winter M.; Besenhard J. O.; Villar-Rodil S.; Suárez-García F.; Martínez-Alonso A.; Tascón J. M. D. Nomex-derived activated carbon fibers as electrode materials in carbon based supercapacitors. J. Power Sources 2006, 153, 419–423. 10.1016/j.jpowsour.2005.05.078. [DOI] [Google Scholar]
- Badenhorst H. A review of the application of carbon materials in solar thermal energy storage. Solar Energy 2019, 192, 35–68. 10.1016/j.solener.2018.01.062. [DOI] [Google Scholar]
- Titirici M.-M.; White R. J.; Brun N.; Budarin V. L.; Su D. S.; del Monte F.; Clark J. H.; MacLachlan M. J. Sustainable carbon materials. Chem. Soc. Rev. 2015, 44, 250–290. 10.1039/C4CS00232F. [DOI] [PubMed] [Google Scholar]
- Xu J.; Cao Z.; Zhang Y.; Yuan Z.; Lou Z.; Xu X.; Wang X. A review of functionalized carbon nanotubes and graphene for heavy metal adsorption from water: Preparation, application, and mechanism. Chemosphere 2018, 195, 351–364. 10.1016/j.chemosphere.2017.12.061. [DOI] [PubMed] [Google Scholar]
- Satayeva A. R.; Howell C. A.; Korobeinyk A. V.; Jandosov J.; Inglezakis V. J.; Mansurov Z. A.; Mikhalovsky S. V. Investigation of rice husk derived activated carbon for removal of nitrate contamination from water. Sci. Total Environ. 2018, 630, 1237–1245. 10.1016/j.scitotenv.2018.02.329. [DOI] [PubMed] [Google Scholar]
- Samad S.; Loh K. S.; Wong W. Y.; Lee T. K.; Sunarso J.; Chong S. T.; Wan Daud W. R. Carbon and non-carbon support materials for platinum-based catalysts in fuel cells. Int. J. Hydrogen Energy 2018, 43, 7823–7854. 10.1016/j.ijhydene.2018.02.154. [DOI] [Google Scholar]
- Zhang C.; Kong R.; Wang X.; Xu Y.; Wang F.; Ren W.; Wang Y.; Su F.; Jiang J.-X. Porous carbons derived from hypercrosslinked porous polymers for gas adsorption and energy storage. Carbon 2017, 114, 608–618. 10.1016/j.carbon.2016.12.064. [DOI] [Google Scholar]
- Alshehri R.; Ilyas A. M.; Hasan A.; Arnaout A.; Ahmed F.; Memic A. Carbon Nanotubes in Biomedical Applications: Factors, Mechanisms, and Remedies of Toxicity. J. Med. Chem. 2016, 59, 8149–8167. 10.1021/acs.jmedchem.5b01770. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Tetard L.; Zhai L.; Thomas J. Supercapacitor electrode materials: nanostructures from 0 to 3 dimensions. Energy Environ. Sci. 2015, 8, 702–730. 10.1039/C4EE03229B. [DOI] [Google Scholar]
- Song W.-L.; Li X.; Fan L.-Z. Biomass derivative/graphene aerogels for binder-free supercapacitors. Energy Storage Mater. 2016, 3, 113–122. 10.1016/j.ensm.2016.01.010. [DOI] [Google Scholar]
- Beguin F.; Frackowiak E.. Carbons for electrochemical energy storage and conversion systems; Crc Press: 2009. [Google Scholar]
- Balducci A. Electrolytes for high voltage electrochemical double layer capacitors: A perspective article. J. Power Sources 2016, 326, 534–540. 10.1016/j.jpowsour.2016.05.029. [DOI] [Google Scholar]
- Schütter C.; Pohlmann S.; Balducci A. Industrial Requirements of Materials for Electrical Double Layer Capacitors: Impact on Current and Future Applications. Adv. Energy Mater. 2019, 1900334. 10.1002/aenm.201900334. [DOI] [Google Scholar]
- Gomez-Martin A.; Martinez-Fernandez J.; Ruttert M.; Heckmann A.; Winter M.; Placke T.; Ramirez-Rico J. Iron-Catalyzed Graphitic Carbon Materials from Biomass Resources as Anodes for Lithium-Ion Batteries. ChemSusChem 2018, 11, 2776–2787. 10.1002/cssc.201800831. [DOI] [PubMed] [Google Scholar]
- Holtstiege F.; Bärmann P.; Nölle R.; Winter M.; Placke T. Pre-Lithiation Strategies for Rechargeable Energy Storage Technologies: Concepts, Promises and Challenges. Batteries 2018, 4, 4. 10.3390/batteries4010004. [DOI] [Google Scholar]
- Winter M.; Besenhard J. O. Lithiated carbons. Handb. Battery Mater. 1998, 383–418. 10.1002/9783527611676.ch17. [DOI] [Google Scholar]
- Irisarri E.; Ponrouch A.; Palacin M. R. Review—Hard Carbon Negative Electrode Materials for Sodium-Ion Batteries. J. Electrochem. Soc. 2015, 162, A2476–A2482. 10.1149/2.0091514jes. [DOI] [Google Scholar]
- Simone V.; Boulineau A.; de Geyer A.; Rouchon D.; Simonin L.; Martinet S. Hard carbon derived from cellulose as anode for sodium ion batteries: Dependence of electrochemical properties on structure. J. Energy Chem. 2016, 25, 761–768. 10.1016/j.jechem.2016.04.016. [DOI] [Google Scholar]
- Zhang L. L.; Zhao X. S. Carbon-based materials as supercapacitor electrodes. Chem. Soc. Rev. 2009, 38, 2520–2531. 10.1039/b813846j. [DOI] [PubMed] [Google Scholar]
- Vlad A.; Singh N.; Galande C.; Ajayan P. M. Design Considerations for Unconventional Electrochemical Energy Storage Architectures. Adv. Energy Mater. 2015, 5, 1402115. 10.1002/aenm.201402115. [DOI] [Google Scholar]
- Lin Z.; Goikolea E.; Balducci A.; Naoi K.; Taberna P.-L.; Salanne M.; Yushin G.; Simon P. Materials for supercapacitors: When Li-ion battery power is not enough. Mater. Today 2018, 21, 419–436. 10.1016/j.mattod.2018.01.035. [DOI] [Google Scholar]
- Huang H.-S.; Chang K.-H.; Suzuki N.; Yamauchi Y.; Hu C.-C.; Wu K. C.-W. Evaporation-Induced Coating of Hydrous Ruthenium Oxide on Mesoporous Silica Nanoparticles to Develop High-Performance Supercapacitors. Small 2013, 9, 2520–2526. 10.1002/smll.201202786. [DOI] [PubMed] [Google Scholar]
- Bastakoti B. P.; Huang H.-S.; Chen L.-C.; Wu K. C. W.; Yamauchi Y. Block copolymer assisted synthesis of porous α-Ni(OH)2 microflowers with high surface areas as electrochemical pseudocapacitor materials. Chem. Commun. 2012, 48, 9150–9152. 10.1039/c2cc32945j. [DOI] [PubMed] [Google Scholar]
- Liu B.; Shioyama H.; Jiang H.; Zhang X.; Xu Q. Metal–organic framework (MOF) as a template for syntheses of nanoporous carbons as electrode materials for supercapacitor. Carbon 2010, 48, 456–463. 10.1016/j.carbon.2009.09.061. [DOI] [Google Scholar]
- Yang J.; Ma Z.; Gao W.; Wei M. Layered Structural Co-Based MOF with Conductive Network Frames as a New Supercapacitor Electrode. Chem. – Eur. J. 2017, 23, 631–636. 10.1002/chem.201604071. [DOI] [PubMed] [Google Scholar]
- Acerce M.; Voiry D.; Chhowalla M. Metallic 1T phase MoS2 nanosheets as supercapacitor electrode materials. Nat. Nanotechnol. 2015, 10, 313. 10.1038/nnano.2015.40. [DOI] [PubMed] [Google Scholar]
- Salunkhe R. R.; Kaneti Y. V.; Yamauchi Y. Metal–Organic Framework-Derived Nanoporous Metal Oxides toward Supercapacitor Applications: Progress and Prospects. ACS Nano 2017, 11, 5293–5308. 10.1021/acsnano.7b02796. [DOI] [PubMed] [Google Scholar]
- Panja T.; Bhattacharjya D.; Yu J.-S. Nitrogen and phosphorus co-doped cubic ordered mesoporous carbon as a supercapacitor electrode material with extraordinary cyclic stability. J. Mater. Chem. A 2015, 3, 18001–18009. 10.1039/C5TA04169D. [DOI] [Google Scholar]
- Wang J.; Kaskel S. KOH activation of carbon-based materials for energy storage. J. Mater. Chem. 2012, 22, 23710–23725. 10.1039/c2jm34066f. [DOI] [Google Scholar]
- Singh S. B.; De M. Alumina based doped templated carbons: A comparative study with zeolite and silica gel templates. Microporous Mesoporous Mater. 2018, 257, 241–252. 10.1016/j.micromeso.2017.08.047. [DOI] [Google Scholar]
- Li X.; Wei B. Supercapacitors based on nanostructured carbon. Nano Energy 2013, 2, 159–173. 10.1016/j.nanoen.2012.09.008. [DOI] [Google Scholar]
- Yang J.; Zhou X.-y.; Zou Y.-l.; Tang J.-j. A hierarchical porous carbon material for high power, lithium ion batteries. Electrochim. Acta 2011, 56, 8576–8581. 10.1016/j.electacta.2011.07.047. [DOI] [Google Scholar]
- Tang H.; Wang M.; Lu T.; Pan L. Porous carbon spheres as anode materials for sodium-ion batteries with high capacity and long cycling life. Ceram. Int. 2017, 43, 4475–4482. 10.1016/j.ceramint.2016.12.098. [DOI] [Google Scholar]
- Waldmann T.; Hogg B.-I.; Wohlfahrt-Mehrens M. Li plating as unwanted side reaction in commercial Li-ion cells – A review. J. Power Sources 2018, 384, 107–124. 10.1016/j.jpowsour.2018.02.063. [DOI] [Google Scholar]
- Holtstiege F.; Koç T.; Hundehege T.; Siozios V.; Winter M.; Placke T. Toward High Power Batteries: Pre-lithiated Carbon Nanospheres as High Rate Anode Material for Lithium Ion Batteries. ACS Appl. Energy Mater. 2018, 1, 4321–4331. 10.1021/acsaem.8b00945. [DOI] [Google Scholar]
- Guo S.; Chen Y.; Shi L.; Dong Y.; Ma J.; Chen X.; Song H. Nitrogen-doped biomass-based ultra-thin carbon nanosheets with interconnected framework for High-Performance Lithium-Ion Batteries. Appl. Surf. Sci. 2018, 437, 136–143. 10.1016/j.apsusc.2017.12.144. [DOI] [Google Scholar]
- Yi Z.; Liang Y.; Lei X.; Wang C.; Sun J. Low-temperature synthesis of nanosized disordered carbon spheres as an anode material for lithium ion batteries. Mater. Lett. 2007, 61, 4199–4203. 10.1016/j.matlet.2007.01.054. [DOI] [Google Scholar]
- Chen L.; Feng Y.; Lang H.-W.; Wu Z.-Y.; Yu S.-H. Macroscopic-Scale Three-Dimensional Carbon Nanofiber Architectures for Electrochemical Energy Storage Devices. Adv. Energy Mater. 2017, 7, 1700826. 10.1002/aenm.201700826. [DOI] [Google Scholar]
- Liu Y.; Peng X. Recent advances of supercapacitors based on two-dimensional materials. Appl. Mater. Today 2017, 8, 104–115. 10.1016/j.apmt.2017.05.002. [DOI] [Google Scholar]
- Jiang Z.-j.; Jiang Z.; Chen W. The role of holes in improving the performance of nitrogen-doped holey graphene as an active electrode material for supercapacitor and oxygen reduction reaction. J. Power Sources 2014, 251, 55–65. 10.1016/j.jpowsour.2013.11.031. [DOI] [Google Scholar]
- Billaud J.; Bouville F.; Magrini T.; Villevieille C.; Studart A. R. Magnetically aligned graphite electrodes for high-rate performance Li-ion batteries. Nat. Energy 2016, 1, 16097. 10.1038/nenergy.2016.97. [DOI] [Google Scholar]
- Lin Y.; Liao Y.; Chen Z.; Connell J. W. Holey graphene: a unique structural derivative of graphene. Mater. Res. Lett. 2017, 5, 209–234. 10.1080/21663831.2016.1271047. [DOI] [Google Scholar]
- Niu Q.; Gao K.; Tang Q.; Wang L.; Han L.; Fang H.; Zhang Y.; Wang S.; Wang L. Large-size graphene-like porous carbon nanosheets with controllable N-doped surface derived from sugarcane bagasse pith/chitosan for high performance supercapacitors. Carbon 2017, 123, 290–298. 10.1016/j.carbon.2017.07.078. [DOI] [Google Scholar]
- Ding J.; Wang H.; Li Z.; Cui K.; Karpuzov D.; Tan X.; Kohandehghan A.; Mitlin D. Peanut shell hybrid sodium ion capacitor with extreme energy-power rivals lithium ion capacitors. Energy Environ. Sci. 2015, 8, 941–955. 10.1039/C4EE02986K. [DOI] [Google Scholar]
- Seo D. H.; Rider A. E.; Han Z. J.; Kumar S.; Ostrikov K. Plasma Break-Down and Re-Build: Same Functional Vertical Graphenes from Diverse Natural Precursors. Adv. Mater. 2013, 25, 5638–5642. 10.1002/adma201301510. [DOI] [PubMed] [Google Scholar]
- Fan Z.-J.; Yan J.; Wei T.; Ning G.-Q.; Zhi L.-J.; Liu J.-C.; Cao D.-X.; Wang G.-L.; Wei F. Nanographene-Constructed Carbon Nanofibers Grown on Graphene Sheets by Chemical Vapor Deposition: High-Performance Anode Materials for Lithium Ion Batteries. ACS Nano 2011, 5, 2787–2794. 10.1021/nn200195k. [DOI] [PubMed] [Google Scholar]
- Guo D.; Lai L.; Cao A.; Liu H.; Dou S.; Ma J. Nanoarrays: design, preparation and supercapacitor applications. RSC Adv. 2015, 5, 55856–55869. 10.1039/C5RA09453D. [DOI] [Google Scholar]
- Huang T.; Zheng B.; Kou L.; Gopalsamy K.; Xu Z.; Gao C.; Meng Y.; Wei Z. Flexible high performance wet-spun graphene fiber supercapacitors. RSC Adv. 2013, 3, 23957–23962. 10.1039/c3ra44935a. [DOI] [Google Scholar]
- Zhao M.-Q.; Zhang Q.; Huang J.-Q.; Tian G.-L.; Nie J.-Q.; Peng H.-J.; Wei F. Unstacked double-layer templated graphene for high-rate lithium–sulphur batteries. Nat. Commun. 2014, 5, 3410. 10.1038/ncomms4410. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Liu F.; Casano G.; Bhavsar R.; Kinloch I. A.; Derby B. Pristine Graphene Aerogels by Room-Temperature Freeze Gelation. Adv. Mater. 2016, 28, 7993–8000. 10.1002/adma.201602393. [DOI] [PubMed] [Google Scholar]
- Fic K.; Platek A.; Piwek J.; Frackowiak E. Sustainable materials for electrochemical capacitors. Mater. Today 2018, 21, 437–454. 10.1016/j.mattod.2018.03.005. [DOI] [Google Scholar]
- Mo R.-J.; Zhao Y.; Zhao M.-M.; Wu M.; Wang C.; Li J.-P.; Kuga S.; Huang Y. Graphene-like porous carbon from sheet cellulose as electrodes for supercapacitors. Chem. Eng. J. 2018, 346, 104–112. 10.1016/j.cej.2018.04.010. [DOI] [Google Scholar]
- Li J.; Qi H.; Wang Q.; Xu Z.; Liu Y.; Li Q.; Kong X.; Huang J. Constructing graphene-like nanosheets on porous carbon framework for promoted rate performance of Li-ion and Na-ion storage. Electrochim. Acta 2018, 271, 92–102. 10.1016/j.electacta.2018.02.147. [DOI] [Google Scholar]
- Horax K. M.; Bao S.; Wang M.; Li Y. Analysis of graphene-like activated carbon derived from rice straw for application in supercapacitor. Chin. Chem. Lett. 2017, 28, 2290–2294. 10.1016/j.cclet.2017.11.004. [DOI] [Google Scholar]
- Gutiérrez-Pardo A.; Ramírez-Rico J.; de Arellano-López A. R.; Martínez-Fernández J. Characterization of porous graphitic monoliths from pyrolyzed wood. J. Mater. Sci. 2014, 49, 7688–7696. 10.1007/s10853-014-8477-8. [DOI] [Google Scholar]
- Thambiliyagodage C. J.; Ulrich S.; Araujo P. T.; Bakker M. G. Catalytic graphitization in nanocast carbon monoliths by iron, cobalt and nickel nanoparticles. Carbon 2018, 134, 452–463. 10.1016/j.carbon.2018.04.002. [DOI] [Google Scholar]
- Magnin Y.; Zappelli A.; Amara H.; Ducastelle F.; Bichara C. Size Dependent Phase Diagrams of Nickel-Carbon Nanoparticles. Phys. Rev. Lett. 2015, 115, 205502. 10.1103/PhysRevLett.115.205502. [DOI] [PubMed] [Google Scholar]
- Deng S.; Berry V. Wrinkled, rippled and crumpled graphene: an overview of formation mechanism, electronic properties, and applications. Mater. Today 2016, 19, 197–212. 10.1016/j.mattod.2015.10.002. [DOI] [Google Scholar]
- Li Z.; Zhang W.; Guo J.; Yang B.; Yuan J. Improved synthesis of fluffy and wrinkled reduced graphene oxide for energy storage application. Vacuum 2015, 117, 35–39. 10.1016/j.vacuum.2015.03.032. [DOI] [Google Scholar]
- Chen L.; He Y.; Chai S.; Qiang H.; Chen F.; Fu Q. Toward high performance graphene fibers. Nanoscale 2013, 5, 5809–5815. 10.1039/c3nr01083j. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Gao C. Graphene fiber: a new trend in carbon fibers. Mater. Today 2015, 18, 480–492. 10.1016/j.mattod.2015.06.009. [DOI] [Google Scholar]
- Xu Z.; Zhang Y.; Li P.; Gao C. Strong, Conductive, Lightweight, Neat Graphene Aerogel Fibers with Aligned Pores. ACS Nano 2012, 6, 7103–7113. 10.1021/nn3021772. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yan R.; Chen H.; Lee D. H.; Zheng C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. 10.1016/j.fuel.2006.12.013. [DOI] [Google Scholar]
- Aslan D. I.; Özoğul B.; Ceylan S.; Geyikçi F. Thermokinetic analysis and product characterization of Medium Density Fiberboard pyrolysis. Bioresour. Technol. 2018, 258, 105–110. 10.1016/j.biortech.2018.02.126. [DOI] [PubMed] [Google Scholar]
- Zhang N.; Li S.; Xiong L.; Hong Y.; Chen Y. Cellulose-hemicellulose interaction in wood secondary cell-wall. Modell. Simul. Mater. Sci. Eng. 2015, 23, 085010 10.1088/0965-0393/23/8/085010. [DOI] [Google Scholar]
- Mathews S. L.; Pawlak J.; Grunden A. M. Bacterial biodegradation and bioconversion of industrial lignocellulosic streams. Appl. Microbiol. Biotechnol. 2015, 99, 2939–2954. 10.1007/s00253-015-6471-y. [DOI] [PubMed] [Google Scholar]
- Cabane E.; Keplinger T.; Künniger T.; Merk V.; Burgert I. Functional lignocellulosic materials prepared by ATRP from a wood scaffold. Sci. Rep. 2016, 6, 31287. 10.1038/srep31287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim Z.; Afrin S. Nanocellulose as Novel Supportive Functional Material for Growth and Development of Cells. Cell Dev. Biol. 2015, 04, 2. 10.4172/2168-9296.1000154. [DOI] [Google Scholar]
- Saunders C. W.; Taylor L. T. A review of the synthesis, chemistry and analysis of nitrocellulose. J. Energ. Mater. 1990, 8, 149–203. 10.1080/07370659008012572. [DOI] [Google Scholar]
- López-López M.; de la Ossa M. Á. F.; Galindo J. S.; Ferrando J. L.; Vega A.; Torre M.; García-Ruiz C. New protocol for the isolation of nitrocellulose from gunpowders: Utility in their identification. Talanta 2010, 81, 1742–1749. 10.1016/j.talanta.2010.03.033. [DOI] [PubMed] [Google Scholar]
- Costa M. N.; Veigas B.; Jacob J. M.; Santos D. S.; Gomes J.; Baptista P. V.; Martins R.; Inácio J.; Fortunato E. A low cost, safe, disposable, rapid and self-sustainable paper-based platform for diagnostic testing: lab-on-paper. Nanotechnology 2014, 25, 094006 10.1088/0957-4484/25/9/094006. [DOI] [PubMed] [Google Scholar]
- Amiinu I. S.; Zhang J.; Kou Z.; Liu X.; Asare O. K.; Zhou H.; Cheng K.; Zhang H.; Mai L.; Pan M.; Mu S. Self-Organized 3D Porous Graphene Dual-Doped with Biomass-Sponsored Nitrogen and Sulfur for Oxygen Reduction and Evolution. ACS Appl. Mater. Interfaces 2016, 8, 29408–29418. 10.1021/acsami.6b08719. [DOI] [PubMed] [Google Scholar]
- Kaushik A.; Singh M. Isolation and characterization of cellulose nanofibrils from wheat straw using steam explosion coupled with high shear homogenization. Carbohydr. Res. 2011, 346, 76–85. 10.1016/j.carres.2010.10.020. [DOI] [PubMed] [Google Scholar]
- Yang C.; Xiong J.; Ou X.; Wu C.-F.; Xiong X.; Wang J.-H.; Huang K.; Liu M. A renewable natural cotton derived and nitrogen/sulfur co-doped carbon as a high-performance sodium ion battery anode. Mater. Today Energy 2018, 8, 37–44. 10.1016/j.mtener.2018.02.001. [DOI] [Google Scholar]
- Ferrari A. C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. 10.1016/j.ssc.2007.03.052. [DOI] [Google Scholar]
- Ferrari A. C.; Meyer J. C.; Scardaci V.; Casiraghi C.; Lazzeri M.; Mauri F.; Piscanec S.; Jiang D.; Novoselov K. S.; Roth S.; Geim A. K. Raman Spectrum of Graphene and Graphene Layers. Phys. Rev. Lett. 2006, 97, 187401. 10.1103/PhysRevLett.97.187401. [DOI] [PubMed] [Google Scholar]
- Sadezky A.; Muckenhuber H.; Grothe H.; Niessner R.; Pöschl U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. 10.1016/j.carbon.2005.02.018. [DOI] [Google Scholar]
- Hoekstra J.; Beale A. M.; Soulimani F.; Versluijs-Helder M.; Geus J. W.; Jenneskens L. W. Base Metal Catalyzed Graphitization of Cellulose: A Combined Raman Spectroscopy, Temperature-Dependent X-ray Diffraction and High-Resolution Transmission Electron Microscopy Study. J. Phys. Chem. C 2015, 119, 10653–10661. 10.1021/acs.jpcc.5b00477. [DOI] [Google Scholar]
- Kaniyoor A.; Ramaprabhu S. A Raman spectroscopic investigation of graphite oxide derived graphene. AIP Advances 2012, 2, 032183 10.1063/1.4756995. [DOI] [Google Scholar]
- Lin Z.; Ye X.; Han J.; Chen Q.; Fan P.; Zhang H.; Xie D.; Zhu H.; Zhong M. Precise Control of the Number of Layers of Graphene by Picosecond Laser Thinning. Sci. Rep. 2015, 5, 11662. 10.1038/srep11662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johra F. T.; Lee J.-W.; Jung W.-G. Facile and safe graphene preparation on solution based platform. J. Ind. Eng. Chem. 2014, 20, 2883–2887. 10.1016/j.jiec.2013.11.022. [DOI] [Google Scholar]
- Yang Z.; Zhao W.; Niu Y.; Zhang Y.; Wang L.; Zhang W.; Xiang X.; Li Q. Direct spinning of high-performance graphene fiber supercapacitor with a three-ply core-sheath structure. Carbon 2018, 132, 241–248. 10.1016/j.carbon.2018.02.041. [DOI] [Google Scholar]
- Ma W.; Liu Y.; Yan S.; Miao T.; Shi S.; Yang M.; Zhang X.; Gao C. Systematic characterization of transport and thermoelectric properties of a macroscopic graphene fiber. Nano Res. 2016, 9, 3536–3546. 10.1007/s12274-016-1231-6. [DOI] [Google Scholar]
- Hou D.; Liu Q.; Cheng H.; Zhang H.; Wang S. Green reduction of graphene oxide via Lycium barbarum extract. J. Solid State Chem. 2017, 246, 351–356. 10.1016/j.jssc.2016.12.008. [DOI] [Google Scholar]
- Fic K.; Meller M.; Frackowiak E. Interfacial Redox Phenomena for Enhanced Aqueous Supercapacitors. J. Electrochem. Soc. 2015, 162, A5140–A5147. 10.1149/2.0251505jes. [DOI] [Google Scholar]
- Vix-Guterl C.; Frackowiak E.; Jurewicz K.; Friebe M.; Parmentier J.; Béguin F. Electrochemical energy storage in ordered porous carbon materials. Carbon 2005, 43, 1293–1302. 10.1016/j.carbon.2004.12.028. [DOI] [Google Scholar]
- Winter M.; Moeller K.-C.; Besenhard J.. Carbonaceous and graphitic anodes. In Lithium Batteries; Springer: 2009, 145–194. [Google Scholar]
- Cheng Q.; Yuge R.; Nakahara K.; Tamura N.; Miyamoto S. KOH etched graphite for fast chargeable lithium-ion batteries. J. Power Sources 2015, 284, 258–263. 10.1016/j.jpowsour.2015.03.036. [DOI] [Google Scholar]
- Placke T.; Siozios V.; Schmitz R.; Lux S. F.; Bieker P.; Colle C.; Meyer H.-W.; Passerini M. S.; Winter M. Influence of graphite surface modifications on the ratio of basal plane to “non-basal plane” surface area and on the anode performance in lithium ion batteries. J. Power Sources 2012, 200, 83–91. 10.1016/j.jpowsour.2011.10.085. [DOI] [Google Scholar]
- Meister P.; Jia H.; Li J.; Kloepsch R.; Winter M.; Placke T. Best practice: performance and cost evaluation of lithium ion battery active materials with special emphasis on energy efficiency. Chem. Mater. 2016, 28, 7203–7217. 10.1021/acs.chemmater.6b02895. [DOI] [Google Scholar]
- Placke T.; Siozios V.; Rothermel S.; Meister P.; Colle C.; Winter M. Assessment of surface heterogeneity: A route to correlate and quantify the 1st cycle irreversible capacity caused by SEI formation to the various surfaces of graphite anodes for lithium ion cells. Z. Phys. Chem. 2015, 229, 1451–1469. 10.1515/zpch-2015-0584. [DOI] [Google Scholar]
- Peng H.; Ma G.; Sun K.; Mu J.; Zhang Z.; Lei Z. Formation of Carbon Nanosheets via Simultaneous Activation and Catalytic Carbonization of Macroporous Anion-Exchange Resin for Supercapacitors Application. ACS Appl. Mater. Interfaces 2014, 6, 20795–20803. 10.1021/am505066v. [DOI] [PubMed] [Google Scholar]
- Sun L.; Tian C.; Li M.; Meng X.; Wang L.; Wang R.; Yin J.; Fu H. From coconut shell to porous graphene-like nanosheets for high-power supercapacitors. J. Mater. Chem. A 2013, 1, 6462–6470. 10.1039/c3ta10897j. [DOI] [Google Scholar]
- Zhang X.; Zhang K.; Li H.; Wang Q.; Jin L. E.; Cao Q. Synthesis of porous graphitic carbon from biomass by one-step method And its role in the electrode for supercapacitor. J. Appl. Electrochem. 2018, 48, 415–426. 10.1007/s10800-018-1170-x. [DOI] [Google Scholar]
- Qin J.; Zhu S.; Feng C.; Zhao N.; Shi C.; Liu E.-Z.; He F.; Ma L.; Li J.; He C. In-situ space-confined catalysis for fabricating 3D mesoporous graphene and their capacitive properties. Appl. Surf. Sci. 2018, 433, 568–574. 10.1016/j.apsusc.2017.10.035. [DOI] [Google Scholar]
- Thompson E.; Danks A. E.; Bourgeois L.; Schnepp Z. Iron-catalyzed graphitization of biomass. Green Chem. 2015, 17, 551–556. 10.1039/C4GC01673D. [DOI] [Google Scholar]
- Bitencourt C. S.; Luz A. P.; Pagliosa C.; Pandolfelli V. C. Role of catalytic agents and processing parameters in the graphitization process of a carbon-based refractory binder. Ceram. Int. 2015, 41, 13320–13330. 10.1016/j.ceramint.2015.07.115. [DOI] [Google Scholar]
- Yan Q.; Li J.; Zhang X.; Zhang J.; Cai Z. In situ formation of graphene-encapsulated iron nanoparticles in carbon frames through catalytic graphitization of kraft lignin. Nanomater. Nanotechnol. 2018, 8, 1847980418818955. 10.1177/1847980418818955. [DOI] [Google Scholar]
- Kubo S.; Uraki Y.; Sano Y. Catalytic graphitization of hardwood acetic acid lignin with nickel acetate. J. Wood Sci. 2003, 49, 188–192. 10.1007/s100860300030. [DOI] [Google Scholar]
- Stoller M. D.; Ruoff R. S. Best practice methods for determining an electrode material’s performance for ultracapacitors. Energy Environ. Sci. 2010, 3, 1294–1301. 10.1039/c0ee00074d. [DOI] [Google Scholar]
- Nie M.; Demeaux J.; Young B. T.; Heskett D. R.; Chen Y.; Bose A.; Woicik J. C.; Lucht B. L. Effect of Vinylene Carbonate and Fluoroethylene Carbonate on SEI Formation on Graphitic Anodes in Li-Ion Batteries. J. Electrochem. Soc. 2015, 162, A7008–A7014. 10.1149/2.0021513jes. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.