

Abstract
The mankind relies on the use of antibiotics for a healthy life. The epidemic-like emergence of drug-resistant bacterial strains is increasingly becoming one of the leading causes of morbidity and mortality, which gives rise to design a potential antimicrobial peptide (AMP). Here, we have designed the potential AMP using the extensive dynamics simulation since protein–peptide interactions are linked to large conformational changes. Therefore, we have employed the advanced computational avenue CABS molecular docking method that enabled the flexible peptide-protein molecular docking with a large-scale rearrangement of the protein. Lead AMP was investigated against the wild-type (WT) and mutant-PBP5 (MT-PBP5) proteins (antiresistance property). AMP20 showed strong interactions with wtPBP5 and mtPBP5 and involvement of a large number of elements in interactions determined through an atomic model study. Full flexibility analysis showed the stable interaction of AMP20 with both the wild-type and mutant form of PBP5 with root-mean-square deviation (RMSD) values of ∼4.51 and 4.85 Å, respectively. Moreover, peptide dynamics showed involvement of all residues of AMP20 through contact map analysis, and extensive simulation confirmed the stable interaction of AMP20, with lower values of RMSD, radius of gyration, and root-mean-square fluctuation. This study paves the way for a potential approach to design the AMP with amino acid walking and large-scale conformational rearrangements of amino acids.
Introduction
The humanity relies on the use of antibiotics for a healthy life. However, the excessive dependency and blind use and abuse of one of the discoveries in the field of medicine have led to a situation where the disease has mastered its antidote.1 From the “golden age of antibacterials”, we are now heading for a “post-antibiotic era”. The global crisis of antibiotic resistance is projected to cause a staggering death toll of one person every 3 s by 2050.2 Among these “superbugs”, Enterobacteriaceae (a family of Gram-negative bacteria) poses a major concern3 since these pathogens are a natural inhabitant of our microbiome. Unfortunately, in the past two years, only two classes of antibiotics have reached the market, cyclic lipopeptides and oxazolidinones, and both of these drugs are inadequate as they only target Gram-positive bacteria.4Escherichia coli, a Gram-negative bacteria, is the most predominant facultative anaerobic species in the gastrointestinal tract of mammals.5 It is usually a harmless microbe, but by acquiring specific virulent genes, it can cause a number of significant illnesses. Antimicrobial-resistant E. coli strains have been reported worldwide.6 Consequently, the development of antibacterial agents with novel modes of action has become indispensable to combat drug-resistant E. coli. Antimicrobial peptides as novel therapeutic agents represent a solution to ward off evolving pathogens. Hence, we aim to design the potential antimicrobial peptide (AMP) that can inhibit the bacterial cell wall synthesis by targeting the penicillin-binding protein (PBP). PBP is majorly responsible for cell wall biosynthesis by its trans-glycosylation and trans-peptidation.7 Among the different PBPs, PBP-5 is reported to be conserved with a high-resolution crystal structure (X-ray diffraction resolution of 1.8 Å).7
With the advancement of structural biology and computational approaches, new and innovative methods have been used to derive the potential peptide, which is rapidly expanding the field of peptide therapeutics and rational drug design. With the rapid success of peptide-based therapies and notable advantages over small drug-like specificity, higher selectivity, low toxicity, and minimal adverse side effects,8−10 antigenic peptides are capable of generating effective immunity by administration of the antigens along with adjuvants. For an effective treatment, peptidic antigens or drugs with sustainable delivery system have shown significant results in medical science.11,12 To develop and design the potential approach to AMP, we have made a large library of antimicrobial therapeutics with large variations and diversity using ranalexin (a natural antimicrobial peptide), which reduces the toxicity and side effects with its nature.13 We have modified the AMPs with (KPCI) amino acids, reported to be potential residues that enhance the binding specificity of AMP targeting the antibacterial proteins by replacing (N, W, V, L, M, F, H, Y, N, E, L, F) and (A, Y, N).14 During the molecular interaction study of protein-peptide, high flexibility of protein targets and the phenomenon of large-scale conformational rearrangements of the receptor protein and peptide ligands occur, which are needed to analyze to study the stability of an interacting complex.15 Conventionally, molecular docking is used for the interaction study of peptide sequences. The commonly used algorithms for molecular docking of protein-peptide are restricted to the rearrangement and flexibility analysis of peptide only and ablating the flexibility of the receptor protein.16−20 Hence, we have used the CABS-dock coarse-grained-based approach, which has been found to be a potential tool for large-scale protein rearrangements while explicating the peptide docking.21 Molecular docking is an approach to study the binding of the receptor and ligand molecules based on various computational theoretical algorithms. The molecular docking approach is widely employed in many applications including binding studies of target receptors with ligand, peptide interactions, drug interactions, nanoparticle interaction, binding mechanism analysis, etc.
In order to design the potential antibacterial peptide, we have targeted the PBP5 protein. PBP plays a key role in bacterial wall biosynthesis.22 It is well known that substitution in PBP can lead to antibiotic-resistant strains of pathogenic bacteria.23 Gramicidin A, an antimicrobial peptide, is already a successful antibiotic.24 This area of research has gained considerable interest.25 Hence, we aimed to design a novel antimicrobial peptide that binds better with wtPBP and mutated PBP as well. We have also discussed binding mechanisms with the wild-type and mutant form of PBP5 antibacterial target. To our best knowledge, the previous studies of designing of AMP with both forms of PBP5 (wild-type and mutant form) were limited to regular simulations of the complex and excluding the extensive dynamics simulation analysis linked to large conformational changes. Therefore, in this work, we have performed the flexible peptide-protein molecular docking with a large-scale rearrangement of the protein chains. Again to our knowledge, amino acid walking (by changing the amino acids in a large sequence sequentially) has not been pursued. As we present here, a novel approach that involves the amino acid walking and their analysis by molecular docking and followed by extensive simulation of the AMP-PBP5 complex in an explicit docking method were performed. This particular approach paves the way for the development of novel antimicrobial peptides against drug-resistant pathogens.
Results and Discussion
Structure Retrieval of Penicillin-Binding Protein 5 and Assessment
PBP5 is the most abundant type of PBP in E. coli and catalyzes the major d-alanine carboxypeptidase activity in vivo.26 We have retrieved the crystal structure of wtPBP5 of E. coli (strain K12) from the Protein Data Bank (PDB ID: 1NZO). A three-dimensional (3D) structure was determined by X-ray diffraction study and reported at a resolution of 1.85 Å. PBP5 was made up of only one chain (Chain-A) with 363 amino acids. Chain-A of PBP5 was found complexed with the small molecule named β-mercaptoethanol (for structural stabilization). The PBP5 structure forms a covalent acyl-enzyme complex with β-lactam antibiotics.27 The 3D structure was evaluated for its stereochemical and physiochemical properties. Stereochemical properties were analyzed through Ramachandran plot assessment using the procheck server. The Ramachandran plot showed that 91.5% of residues were in most favored regions, 8.2% of residues were in additional allowed regions, and 0.3% of residues were in generously allowed regions with no residues in disallowed regions of the protein structure.
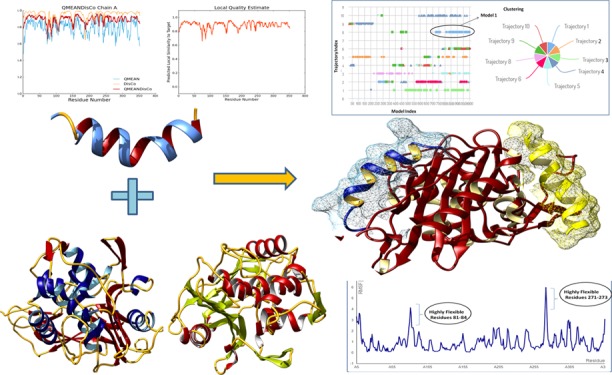
Physiochemical properties were determined by the Swiss server, and the QMEAN score was calculated to be 0.53, representing the overall absolute quality of the wtPBP5 structure for the various properties (solvation energy, torsion angle energy, solvent accessibility, and atom pairwise energy) (Figure 1). The scores for these particular parameters came in the region light blue to blue of the QMEAN plot of the wtPBP5 protein and indicated the good values of the physicochemical properties. Positive QMEAN values (blue region) indicate that the model scores higher than experimental structures on average. The white area (light blue) in the bar plots (numerical values close to zero) indicates that the property is similar to what one would expect from experimental structures of a similar size. The accuracy of the assessment was enhanced by using QMEANDisco. This is consistent with the observation of interatomic distance in the PBP5 structure with ensembling information from the experimentally identified protein structures, which were homologs to the query structure sequence. The local quality plot showed the similarity of the native structures and reflected to possess a high quality with a score of more than 0.6, a value that is expected for high-quality structures. In addition, Errat and Verify3D web interfaces also confirmed the good quality of the structures. Errat showed that the structural residues lie under the range error or warning region of the plot derived from the NMR/X-ray-derived 3D structure database (Figure 2). The observed structure was found to be very similar to the expected structures of databases, as shown in the plot. Verify3D showed that more than 99% of residues have a score of more than 0.2 in the 1D-2D profile of NMR/X-ray-derived 3D structures.
Figure 1.
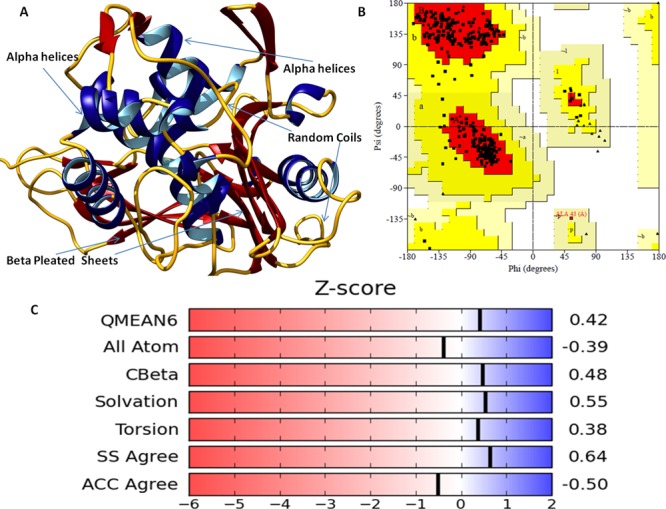
(A) Diagram depicting the three-dimensional structure of the PBP5 protein in newcartoon view showing the α-helices in dark blue color with its interior in cyan, pleated sheets in dark red color, and random coils in dark yellow color. (B) Ramachandran plot of the PBP5 protein. (C) Various physiochemical properties of the PBP5 crystal structure (all atoms, solvation energy, torsion, and solvent accessibility) lying in the acceptable region (light blue to blue).
Figure 2.

(A) Diagram depicting the accuracy of assessment of the crystal structure of PBP5 by QMEANDisco. QMEAN values (blue region) indicate that the PBP5 model scores higher than experimental structures on average by QMEANDisco. (B) Local quality plot showing the similarity of the native structures and high quality with a score of more than 0.6, expected for high-quality structures. (C) Verify3D web interfaces also confirming the good quality of the structures with minimal deviations in the acceptable range. (D) Structural residues lying under the range of the error-prone or warning region of the plot derived from the Errat protein analysis program.
Three-Dimensional Structure Retrieval of Mutant Penicillin-Binding Protein 5 and Assessment
Gly105-Asp mutation in PBP5 markedly impairs deacylation with only minor effects on acylation and abolishes CPase activity. We have used the Gly105-Asp mtPBP5 structure for mutant resistance study. The crystal structure of a mutant of penicillin-binding protein 5 of E. coli (strain K12) was retrieved from the Protein Data Bank (PDB ID: 1NJ4). The mutant-type PBP5 structure was determined by X-ray diffraction study and reported at a 1.9 Å resolution.27 The mutation causes conformational changes in the structure. Hence, the structure was evaluated using the Ramachandran plot and Verify3D servers. Ramachandran plot assessment showed that 92.6% of residues were in most favored regions, 7.1% of residues were in additional allowed regions, and 0.3% of residues were in generously allowed regions with no disallowed regions of the mutant-type PBP5 protein structure. The mtPBP5 structure was also found similar to the expected structure, defined on the basis of NMR-based known structures (Figure S1). Verify3D showed that more than 99% of residues of the mtPBP5 structure have a score of more than 0.2 in the 1D-2D profile of NMR/X-ray-derived 3D structures, also shown in Figure S2.
Molecular Docking Analyses of the AMP Library with PBP5
The designed library was docked with the target protein PBP5 using the ClusPro molecular docking interface. The prepared target receptor PBP5 protein was uploaded, and its unstructured terminal residues were removed, and then ligands (designed AMP library) were uploaded. The ClusPro server works using a piper rigid body molecular docking algorithm and helps in ligand rotation with 70,000 rotations at all grid points (X, Y, and Z) about the target protein with a spacing width of 1.0 Å. Then, using the clustering techniques to find near-native conformations along with eliminating the non-native clusters, the 1000 best energy conformations with the lowest score were clustered, and among them, the 30 largest clusters were refined by minimizing the Charmm energy of the complexes. The clustering of the poses starts with the lowest energy pose and grouping all poses within 9 Å. The results showed that AMP20 has a minimum free energy with a weighted lowest energy score of −845.2 kcal/mol both in the center and to the whole structure, among the whole library. AMP20 was found to form strong interactions with PBP5 with hydrogen bond of 3.02 Å bond length between Cys14 (position SG) of AMP20 and Asp175 (position OD2) of the PBP5 target protein and strong hydrophobic interactions (Figure 3). Fifteen residues of PBP5 were found to be involved in strong hydrophobic interactions with AMP20 in three regions. AMP20 binding regions at PBP5 were as follows: first region (26–41) with involved residues Asn26, Ser27, Gly28, Lys29, and Gly41; second region (140–174) with involved residues Leu140, Gln170, and Arg174; and third region (297–345) with involved residues Tyr297, Val328, and Pro345 (Figure 4). Furthermore, to validate our results, comparative analyses were performed on lead AMP20 with a known antimicrobial peptide, ranalexin (as discussed in the Introduction). ClusPro resulted in docking scores of −719.1 and −732.4 kcal/mol for the center and lowest binding energy scores for RN, respectively, which are lower scores than AMP20. These results confirmed the potency of our designed AMP20 with higher binding with the target protein PBP5 (Table 1).
Figure 3.
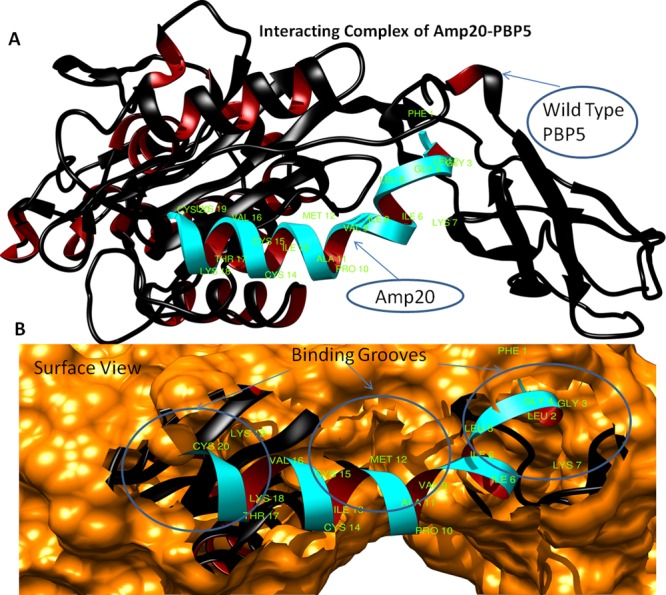
(A) Structural model of PBP5 with ligand AMP20 (cyan color) interaction and binding sites (green color), newcartoon view. (B) Surface view of binding site pocket residues with best fit confirmation and superimposition of AMP20 (cyan color) to the PBP5 binding grooves.
Figure 4.
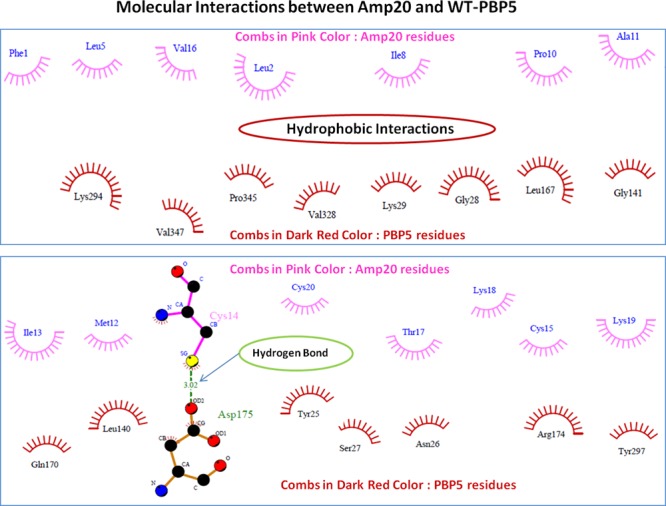
Depiction of molecular interactions of AMP20 with the PBP5 protein. Involved hydrophobic interactions are shown in the combs in pink color (AMP20) and dark red (PBP5 structure), and hydrogen bonds are shown in green color with a bond length of 3.02 Å.
Table 1. Molecular Docking Scores of Top-Ranked AMPs with Wild-Type PBP5.
| S. No. | AMP code | top-ranked peptide | center energy values (kcal/mol) | lowest energy values (kcal/mol) |
|---|---|---|---|---|
| 1 | AMP1(RN) | FLGGLIKIVPAMICAVTKKC | –719.1 | –732.4 |
| 2 | AMP6 | FLGGLIKIVPAMICKVTKKC | –822.6 | –822.6 |
| 3 | AMP19 | FLGGLIKIVPACICAVTKKC | –769.2 | –816.4 |
| 4 | AMP20 | FLGGLIKIVPAMICCVTKKC | –845.2 | –845.2 |
| 5 | AMP21 | FLGGLIKIVPAMICACTKKC | –775.4 | –811.7 |
| 6 | AMP23 | ILGGLIKIVPAMICAVTKKC | –754.2 | –754.2 |
| 7 | AMP25 | FLGGLIKIIPAMICAVTKKC | –827.6 | –827.6 |
| 8 | AMP30 | FLGGLIKIVPAMICAVRKKC | –832.1 | –853.3 |
| 9 | AMP35 | FLGGLIKKWPWWPWRR | –763.0 | –857.6 |
| 10 | AMP38 | FLGGLIKWWPWRR | –763.3 | –839.8 |
| 11 | AMP47 | FLGGLIKKPCIMICAVTKKC | –734.5 | –872.3 |
| 12 | AMP48 | FLGGLIKIVKPCIICAVTKKC | –724.8 | –830.0 |
| 13 | AMP53 | FLGGLIKIVPAMICAKPCIC | –726.2 | –856.6 |
Bacterial Resistance Study of AMP20 with Mutant PBP5 Protein
Bacterial resistance study of the lead peptide (AMP20) was performed by molecular docking with mutant PBP5. A mutant form of the PBP5 protein is reported to have bacterial resistance. AMP20 was docked using the ClusPro program, with the same protocol given above. Interaction analyses of AMP20 showed that it has a very strong interaction with the mutant form of PBP5 as well, with a binding energy score of −759 kcal/mol. It has a strong interaction with mtPBP5 with three hydrogen bonds of less than 3 Å bond length and strong hydrophobic interactions. Hydrogen bonds were found between Cys20 (oxygen atom) of AMP20 and Arg198 (amino position NH1) of 2.78 Å bond length, Ile13 (oxygen atom) of AMP20 with Asn112 (position ND2) of 2.65 Å bond length, and Gly30 (oxygen atom) of AMP20 with His151 (position NE2) of 2.87 Å bond length. A total of 14 residues of PBP5 were found to be involved in hydrophobic interactions with AMP20. It was found to bind at three regions: first region (73–113) with involved residues Asn73, Met89, Phe90, Gly111, Asp113, and Gln126; second region (150−162) with involved residues Leu153 and Asp154; and third region (197–216) with involved residues Asn197, Gly200, His216, and Arg248 (Figure S3). To compare the results, we have also docked the known antimicrobial peptide ranalexin (RN) with mtPBP5, which resulted in a lower binding score (−585.5 kcal/mol) than AMP20 (Table 2 and Figure 5) and weak hydrophobic interactions with only two hydrogen bonds between Ala15 of AMP20 and Arg174 (amino position NE) of 3.01 Å bond length and Lys18 of AMP20 and Arg174 (amino position NH2) of 2.67 Å bond length (Figure S4).
Table 2. Molecular Docking Results of Top-Ranked AMPs with Mutant PBP5 (mtPBP5).
| S. No. | code | peptide | center energy (kcal/mol) | lowest energy (kcal/mol) |
|---|---|---|---|---|
| 1 | AMP1 | FLGGLIKIVPAMICAVTKKC | –585.5 | –747.5 |
| 2 | AMP6 | FLGGLIKIVPAMICKVTKKC | –619.2 | –727.1 |
| 3 | AMP20 | FLGGLIKIIPAMICAVTKKC | –759.0 | –759.0 |
| 4 | AMP25 | FLGGLIKIIPAMICAVTKKC | –610.5 | 769.2 |
| 5 | AMP30 | FLGGLIKIVPAMICAVRKKC | –738.4 | –789.5 |
Figure 5.
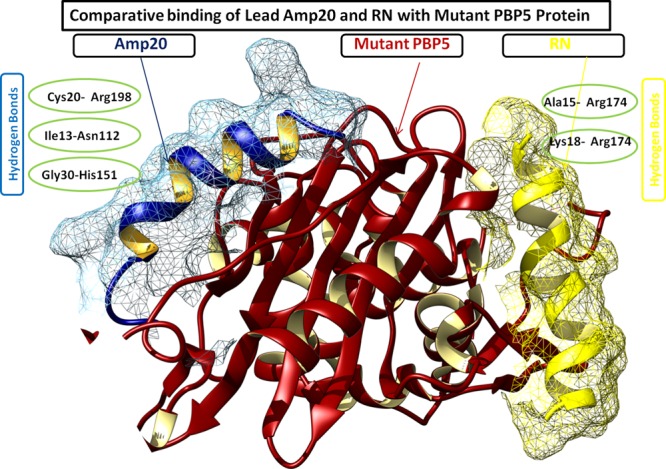
Comparative binding depiction of AMP20 and ranalexin peptide (RN) at different binding regions of mtPBP5. Notably, AMP20 has strong interactions with three hydrogen bonds, whereas RN binds with two hydrogen bonds.
Full Flexibility Analysis of the AMP20-PBP5 Complex
The CABS-dock program was employed to perform the molecular dynamics simulation to analyze the interaction sites, allowing the full flexibility of the AMP20 peptide to the target protein backbone (Figure 6). The 3D structures of the PBP5 receptor and the peptide sequence (AMP20) were given as input files for CABS simulation. The CABS-dock server followed the multistage protocol that consists of multiple programs and associating scripts with the CABS model. The whole procedure involved the flexible docking by the CABS algorithm and the initial filtering of probable solutions from all generated models and a further selection of representative models by the clustering protocol and reconstruction to the all-atom representation of the AMP20-PBP5 complex and further local optimization of final models.
Figure 6.

CABS-dock energy-based top-ranked models obtained for AMP20-PBP5 in the CABS-dock energy graph for top 10 models (shown in different colors). The marker (Model 1) indicates the best model produced in the simulation studies, where the peptide binds close to the PBP5 protein receptor in an open flexible conformation.
Conformation flexibility analyses of interactions were the key properties of protein interaction systems for biological function. Flexibility analysis of the AMP20-PBP5 system was performed through near-native dynamics for 10 ns MD simulations (all-atom, explicit water, for all protein metafolds using the four most popular force fields) using the CABS model. The AMP20 peptide was studied for its rearrangement and flexibility analyses for 50 Monte Carlo simulation cycles. CABS used the well-established coarse-grained protein simulation. The CABS force field included the knowledge-based statistical potentials (sequence-dependent short-range conformational preferences, context-dependent potential of pairwise interactions of side chains, and model of the main chain hydrogen bonds) accounting for the solvent effect in an implicit fashion. The complex system was simulated by a random series of small local moves (controlled by a Monte Carlo scheme) whose long-term evolution describes the protein dynamics. The resulting 10,000 models, during the simulation, up to 1000 models were selected for further steps through all unbound states, where the interaction energy between the peptide and the receptor was zero, and from the remaining 100 models, top 10 trajectories were picked by the lowest interaction energy of AMP20-PBP5 models.
Among the top models, cluster 1 resulted in specific and flexible interactions with a large number of elements (number of models grouped in a cluster) of 127 and an average RMSD (root–mean-square deviation) (average pairwise Cα RMSD value between models grouped in a cluster) of 4.51 Å with a cluster density (the number of elements divided by average cluster RMSD) of 28.115. During the docking simulation, residues Lys294, Val347, Pro345, Val328, Lys29, Gly28, Leu167, Gly141, Gln170, Leu140, Ser27, Asn26, Arg174, Tyr297, and Tyr25 were found to bind AMP20 with the significant flexibility of the PBP5 protein. Moreover, four clusters of AMP20 were found to bind in the proximity of the binding grooves of PBP5 with strong interactions, in the range of less than 3 Å, with a buried area of the complex of 2047.9 Å2. The critical binding analyses of these clusters elucidate the binding sites of PBP5 by reconstructing the complex system and showed that all residues of AMP20 (cluster 1) are involved in the interaction with PBP5.
Thereafter, we have analyzed the AMP20 flexibility binding analysis with mtPBP5. The top model resulted in specific and flexible interactions with a large number of elements of 103 and an average RMSD of 4.80 Å with a cluster density of 21.453. All top-ranked clusters showed the strong interaction of AMP20 to the closed vicinity of PBP5, except for cluster 5 and cluster 8. Notably, the mtPBP5 protein resulted in conformational structural changes; hence, AMP20 binds to the slightly different region to PBP5, particularly at the center for residues Arg248, Asn197, Gly200, His216, Gln126, Lys84, Asn73, Gln111, Asp113, Met89, Phe90, Ser87, Leu153, and Asp154 of PBP5. Here, we found the efficient binding of AMP20 and that this approach provides a potential method to define the binding site of the target protein through flexibility rearrangement simulation studies. Our results showed the flexibility of peptide, with binding to target, the phenomenon is known as conformational selection for molecular recognition of peptide.
Contact Map Analysis
The molecular interactions of AMP20 with PBP5 were analyzed using the contact map analysis using the CABS simulation program. We determined the intermolecular protein-peptide contacts focusing on the dynamics of the PBP5 protein to define the part of the protein, most prominent for interaction with AMP20. The contact map showed that the different regions of PBP5, that is, 26–32, 52–60, 136–140, 174–175, 295–299, 327–330, and 341–344, were involved in the interaction in close proximity with AMP20 (bond length of 4.5 Å). Observed contact maps are presented in Figure 7. Similarly, contact map analysis was performed for mtPBP5 interaction with lead AMP20. For the mtPBP5 protein, regions 84–90, 110–116, 151–154, 198–216, and 248–249 were found in close contact with AMP20, with a bond length of 4.5 Å. Notably, residues Ala139 and Gln56 of wtPBP5 and residues Asn112 and Arg198 of mtPBP5 were found to be the most conserved residues involved in the interaction of a complex in a bond length of 3.0 Å.
Figure 7.

Protein-peptide contact maps for the AMP20-PBP5 complex system. (A) Most frequent contacts formed between AMP20 peptide residues with wtPBP5. (B) Contacts formed between AMP20 peptide residues with mtPBP5.
Peptide Dynamics
We have used the intraprotein contact maps and root-mean-square fluctuation (RMSF) average values over simulation trajectories to qualitatively analyze the dynamics of the flexible AMP20-PBP5 complex system. The RMSF plot showed the very mild fluctuations in amino acid side chains, which reflect the uninterrupted interaction between the ligand and receptor, whereas a small region (81–84 and 271–273 residues of PBP5) in the plot indicated the highly flexible regions in the complex (Figure 8). Higher peaks at the terminal regions of PBP5 showed that it undergoes through significant conformational changes with high fluctuations. Overall, RMSF values lying in the range of 1–3 Å indicated the stable binding of the AMP20. AMP20 with mild fluctuations formed the different arrangements in the proximity of the binding site of the protein PBP5. A similar conclusion to simulation studies can be drawn from the contact map obtained.
Figure 8.

AMP20-PBP5 complex system RMSF (root-mean-square fluctuation) averaged values over the trajectory from the CABS-dock simulation (blue line). Fluctuation run shows the consistent flexible interaction, with a small flexible region at residues 81–84 and 271–273.
AMP20-PBP5 Complex System Dynamics
In addition to the flexibility analysis of the complex system using CABS and AMP20 peptide dynamics study, we have confirmed our results with regular molecular dynamics simulation using an NAMD full setup program through the MDWeb interface. After the simulation run of 100 ps, a stable trajectory complex system was observed. The whole process of simulation included the cleaning of a complex structure, fixing of side chains, the addition of hydrogen atoms, neutralization, the addition of a solvent box, and minimization and equilibration of the system to finally achieve the structure prepared by simulation. The equilibration of the system also included heating the solvent to 300 K and reducing the restraints to just the protein backbone. These steps were carried out to provide the structure with the necessary conditions to study its dynamics along a span of time. Once the structure was prepared, the water molecules in it were removed to reduce the size of the system to achieve the dry trajectory for plotting various graphs for the analysis of the simulated complex structure. The RMSD values were assessed and showed the low deviation within a range of ∼0.15 to ∼0.0601.2 Å in a very similar pattern for the peptide bound to the whole PBP5 sequence during the molecular dynamics simulation of 100 ps time length, which indicates the stability of the AMP20-wtPBP5 complex. All-atom interactions were used to identify the protein complex conformational changes due to isotopic crystallographic B factors. RMSD analysis showed the stable and strong interaction between the AMP20 and the target PBP5 protein as the quality assessment criteria are based on ligand RMSD between the predicted model and the experimental peptide structure after superimposition of the receptor molecules. Moreover, the atomic fluctuation (B factor) and movement of the residues of the complex system were assessed. Atomic fluctuation measures the mobility of the backbone atoms of the system in the dynamics of the complex. We observed the minimal variations to the corresponding binding region of the AMP20 to PBP5, as shown in Figure 9. The atomic fluctuation profile for the binding region was found to be in the range of ∼1 to ∼8 Å, except at two regions (198–201 and 290–296) of PBP5. This indicated the lesser fluctuation per residues of the protein complex contributing to the protein stability of the complex.
Figure 9.

Molecular dynamics simulation plot of the AMP20-PBP5 complex. (A) Plot depicting the RMSD structural deviation per residue of the complex. RMSD values show a minimal deviation in the interacting complex. (B) Atomic fluctuation per residue of the target protein with lower deviation. (C) Radius of gyration plot investigation showing the compactness of the protein complex till 10 snapshots. The compactness of protein receptor increased till nine snapshots during the simulation.
The compactness of the protein complex was also calculated by the radius of gyration (Rg) plot. Rg analysis showed that the complex had a slightly fluctuating Rg in the range of 23.42–23.47 Å and then it gets stabilized with an average Rg score of 23.4689 Å, lower values than the trajectory analysis of wtPBP5 (23.5338 Å). These conformational rearrangements of compactness of the AMP20-PBP5 complex system indicate the flexible interaction with the most favored conformation of AMP20 to the binding grove of PBP5. Furthermore, we have analyzed the interaction stability of AMP20 with mtPBP5 and showed the stable binding of the AMP20-mtPBP5 system with minimal deviation in values of RMSD and RMSF (Supporting Information). These outcomes evidently showed the dynamic nature of the AMP20 ensemble.
Physicochemical Analysis of AMP Constructs
Various physicochemical properties of the lead antibacterial peptide (AMP20) were calculated using the ProtParam server. It uses an amino acid sequence of the peptide construct for analysis. The molecular weight of the construct was calculated to be 2.137 kDa and, the number of atoms was 315, which shows that the construct has good antigenic properties. Further analysis showed that AMP20 has a pI (isoelectric point) value of 9.11, which indicates that the peptide construct is slightly basic in nature with a charge of +3. AMP20 fulfills the criteria for the potential antimicrobial peptide since it possesses the characteristic properties reported for effective antimicrobial peptides (short length, 12–50 amino acid residues long; positively charged, net charge of +2 to +9; and amphipathic nature).28 The estimated half-life for AMP20 was computed to be 1.1 h in mammalian reticulocyte, and the instability index was 14.18, which signified the stability of the peptide. The estimated aliphatic index was 131.50, which indicated that the protein is thermostable, and proteins with a high value of aliphatic index represent thermostability. Moreover, the grand average of hydropathicity (GRAVY) was calculated to be 1.435; a positive value indicated that the protein is hydrophobic in nature and it can cross the cell membrane and also be passed through the hydrophobic layers.
Secondary Structure Analysis
The secondary structure of the lead AMP20 construct was predicted from the online server SOPMA. It predicts the secondary structure on the basis of the amino acid sequence of the 511 proteins in the databases. Overall, the secondary structure prediction result showed that the α-helices contents were 65.00%, extended strands (β sheets) were 20.00%, and random coils were 15.00% (Figure 10).
Figure 10.

(A) 3D crystal structure of the potential antibacterial peptide (AMP20), newcartoon view. (B) Ramachandran plot showing the existence of the AMP20 structure within the favored (94.1% of residues) and allowed (5.9% of residues) regions with no residues in the outlier region. (C) Secondary structure analysis of the peptide.
Peptide Tertiary Structure Refinement
The predicted tertiary (3D) structure of antimicrobial construct (AMP20) was further processed using the GalaxyRefine server for model refinement. GalaxyRefine generated five models after refinement, out of which Model 1 was selected with its stable structural properties. Also, GDT-HA was calculated to be 0.9625, while RMSD, MolProbity, clash score, and poor rotamers were calculated to be 0.0663, 1.346, 6.3, and 0.0, respectively. Model 1 was found to be the best-refined model after comparison of different scores of it with remaining models (Table S1). Moreover, the Ramachandran plot was also evaluated to assess the phi and psi angle conformations, where we found that 94.1% of residues are in the favored region and 5.9% of residues are in the allowed region of the three-dimensional structure.
Allergenicity Analysis of AMP
Allergenicity is an important characteristic property of an effective antimicrobial peptide. An allergen molecule can induce the overreaction to the immune system. Hence, AMP20 was evaluated and found as a non-allergen. Non-alleregenicty of AMP20 was identified on the basis of several descriptors including the size, hydrophobicity, helix contents, and β-strand forming propensities. The AMP20 peptide string was transformed into uniform vectors by cross-covariance with a machine learning method and classified as a non-allergen.29
Conclusions
Herein, we have designed the AMP targeting PBP5 (a potential antibacterial target protein), both the wild-type and mutant form are responsible for bacterial resistance, using the CABS-dock methods. Amino acid walking and extensive simulation dynamics studies were performed on the AMP-PBP5 complex for large-scale conformational rearrangements. For potential AMP determination, a large library was designed. In order to enhance the diversity and specificity of the AMP library, we have incorporated the potential amino acids (KPCI) and followed the amino acid walking approach. The obtained large library was evaluated by using the molecular docking, which showed that the AMP20 has the highest binding free energy score of −845.2 kcal/mol. Lead AMP20 was found to possess a strong interaction with potential hydrogen bonds and hydrophobic forces. In one of the recent computational studies, where AMP was designed, however, they have performed only molecular docking and did not involve molecular simulation analysis,30 overcoming the issues we have employing the extensive molecular dynamics simulation studies with refining the approach for designing the potential AMP with the larger flexibility of the protein. Many reports suggested the use of molecular dynamics simulation studies to design the potent AMP with stable and consistent interaction with the target protein. CABS-dock, a coarse-grained simulation method, was employed for large-scale PBP5 protein motion during an explicit designed AMP study. During the interaction study, full flexibility of ligand peptide structures and large-scale flexibility of the AMP-receptor protein structure with involvement of elements were analyzed.
The large-scale conformational rearrangement analysis of the AMP20-PBP5 complex showed that the accuracy of the best model is 4.51 Å with the involvement of 127 elements in the interactions and resulted in the improved model quality using the all-atom refinement. Moreover, the molecular dynamics simulation analyses confirmed the importance of various amino acids in the binding of AMP20 with the target PBP5. To our best knowledge, AMP identification with close binding to PBP5 has not been reported with large conformation flexibility studies and contact map analysis. The full flexibility analysis resulted in the stable binding of the lead peptide (AMP20) with wtPBP5 and mtPBP5 proteins with lower RMSD values of ∼4.51 and 4.85 Å, respectively, and suggested the high potency of the AMP against the target antibacterial protein. Contact maps were employed to study the peptide dynamics, and it showed the involvement of all amino acids of AMP20 in the interaction with the target PBP5. Moreover, our results were supported by the optimal physicochemical properties with the non-allergen characteristic of the AMP20. In conclusion, our approach helped in designing the potential AMP by studying its binding dynamics and side-chain fluctuation to the large-scale flexibility of the target wild-type and mutant PBP receptor protein. The designed AMP with molecular modeling studies, including molecular docking, dynamics simulation, and large-scale rearrangement/flexibility studies, confirmed the target specificity, which will restrict the nonspecific binding and result in reduced side effects. The present study provides an effective computational approach to design the AMP with amino acid walking and large-scale rearrangements of the protein using the CABS-dock.
Materials and Methods
Structure Retrieval and Assessment of PBP5 (Wild-Type and Mutant Form)
PBP5 of E. coli functions as d-alanine carboxypeptidase (CPase) activity, cleaving d-alanine from the C terminus of the cell wall of peptides. Like all PBPs, PBP5 forms a covalent acyl-enzyme complex with a β-lactam antibiotic. Importantly, PBP5 is distinguished by its high rate of deacylation of the acyl-enzyme complex (t(1/2) of approximately 10 min).27 We have retrieved and assessed the crystal structures of the wtPBP5 and mtPBP5 of E. coli from the Protein Data Bank.
Designing of AMP Library by the Amino Acid Walking Approach
AMPs are essential components of the innate immune system and produced by all classes of life from prokaryotes to mammalians to protect themselves against invasion by microbial pathogens.31,32 Hence, a large library of AMPs was designed by using the computational approaches that have attributed to potential avenues for therapeutic AMP development with high diversity. We have taken naturally occurring AMP ranalexin (FLGGLIKIVPAMICAVTKKC) to modify and design the large library of AMPs. It is reported that one of the potential strategies to develop new antimicrobial peptides is to modify the template sequence of naturally occurring peptides and enhancing their activity.13 We have modified the ranaxelin antimicrobial peptide by inserting the potential amino acids (K, P, C, and I) at different positions. Amino acids (K, P, C, and I) have been reported to enhance the binding specificity of AMP and possess the compositional importance for designing the potential AMPs. We have replaced the (N, W, V, L, M, F, H, Y, N, E, L, F) and (A, Y, N) amino acids with (KPCI) since these were reported to be less important in designing the antibacterial peptides.14 We have employed the peptide walking approach to the target peptide by incorporating K, P, C, and I amino acids to the ranaxelin peptide to sequentially form the 5′ to 3′ order at every position to make the library diverse and specific to the target protein PBP5. The designed AMP library was analyzed through molecular docking and molecular dynamics simulation and advanced peptide approaches including CABS.
Molecular Docking Analyses
Molecular docking analysis of the designed library was performed using the ClusPro protein-peptide docking program.33 The ClusPro suite works on the basis of fast Fourier transformation to determine the lowest binding energy conformation by analyzing the semidefinite program for the protein receptor–ligand complex and also refined the stable interaction cluster by using the Monte Carlo simulations.34 The designed library of peptides was studied against both wild-type and mutant PBP5 in order to optimize the efficiency of lead AMP for bacterial resistance. The docked complex of PBP5-lead AMP with the lowest binding energy was selected and studied for involved molecular interaction using the Ligplot+ (v.1.4.5) module. Ligplot determines the hydrogen bonds and hydrophobic interactions of the complex system.35
Full Flexibility Analysis of Lead AMP with Both Wild-Type and Mutant PBP5
CABS-dock (http://biocomp.chem.uw.edu.pl/CABSdock/), a coarse-grained simulation method, was employed for large-scale PBP5 protein motion during an explicit designed AMP study since large structural changes in a protein target in a molecular docking approach are the requisite parameters for classical modeling servers.15,36−38 During the interaction study, the CABS method involves the full flexibility of ligand peptide structures and large-scale flexibility of the protein structure to the binding site. CABS has been used extensively in wide applications, including the elucidation of binding mechanism, peptide dynamics, and folding assessment of the proteins and structure conformational studies.39−46
Peptide Dynamics and Molecular Dynamics Studies of the Complex
Molecular dynamics and stability of AMP20 with wtPBP5 and mtPBP5 were studied using the MDWeb program. MDWeb determines the root-mean-square deviation and atomic fluctuations of the interacting complex by scrutinizing the stable binding trajectories.47 An NAMD full molecular dynamics setup was employed for carrying out the atomic fluctuation analyses, and the resulting dry trajectory was analyzed to determine the RMSD and atomic fluctuations.
Physicochemical Properties Analysis of Lead AMP Constructs
Shortlisted AMP from the large library through CABS and molecular dynamics study was studied for its physicochemical properties using the ProtParam server.48 The structural stability of the lead AMP was analyzed by studying its secondary and tertiary structure conformations. The 3D structure of the construct was also analyzed using the GalaxyRefine server49 for model refinement and also evaluated its Ramachandran plot to assess the stereochemical properties and favorable conformations. In addition to stability, allergenicity was also analyzed for the lead AMP since an allergen molecule can induce the overreaction to the immune system.
Acknowledgments
We are grateful to the University of Delhi and SRM-IST for providing support and the necessary facilities to carry out the research work. N.K. thanks the CSIR-RA and DBT-SRF for providing the fellowship and support for the research. D.S. particularly thanks the UGC-SRF for providing the fellowship for the ongoing research.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03035.
Three-dimensional structure of mutant penicillin-binding protein 5 and assessment (Figures S1 and S2), molecular interactions of AMP20 with mutant PBP5 (Figure S3) and ranalexin with mutant PBP5 protein (Figure S4), studies of dynamic stability of AMP with mutant PBP5 protein, and peptide tertiary structure refinement data (PDF)
Author Contributions
§ N.K. and D.S. contributed equally as co-first authors.
Author Contributions
N.K., D.S., and R.C. designed and performed the experimental studies. N.K. and D.S. carried out the in silico experiments. The manuscript was written by N.K., D.S., and R.T.
The authors declare no competing financial interest.
Supplementary Material
References
- Prestinaci F.; Pezzotti P.; Pantosti A. Antimicrobial resistance: a global multifaceted phenomenon. Pathog. Global Health 2015, 109, 309–318. 10.1179/2047773215Y.0000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J. I. M. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 1–16. [Google Scholar]
- Iredell J.; Brown J.; Tagg K. Antibiotic resistance in Enterobacteriaceae: mechanisms and clinical implications. BMJ 2016, 352, h6420. 10.1136/bmj.h6420. [DOI] [PubMed] [Google Scholar]
- Coates A. R. M.; Halls G.; Hu Y. Novel classes of antibiotics or more of the same?. Br. J. Pharmacol. 2011, 163, 184–194. 10.1111/j.1476-5381.2011.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasheed M. U.; Thajuddin N.; Ahamed P.; Teklemariam Z.; Jamil K. Antimicrobial drug resistance in strains of Escherichia coli isolated from food sources. Rev. Inst. Med. Trop. Sao Paulo 2014, 56, 341–346. 10.1590/S0036-46652014000400012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . Global antimicrobial resistance surveillance system (GLASS) report Early implementation 2016-2017. ISBN: 978-92-4-151344-9 (2018). https://apps.who.int/iris/bitstream/handle/10665/259744/9789241513449-eng.pdf?sequence=1
- Macheboeuf P.; Contreras-Martel C.; Job V.; Dideberg O.; Dessen A. Penicillin Binding Proteins: key players in bacterial cell cycle and drug resistance processes. FEMS Microbiol. Rev. 2006, 30, 673–691. 10.1111/j.1574-6976.2006.00024.x. [DOI] [PubMed] [Google Scholar]
- Fosgerau K.; Hoffmann T. Peptide therapeutics: Current status and future directions. Drug Discovery Today 2015, 20, 122–128. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Singh V. K.; Kumar N.; Kalsan M.; Saini A.; Chandra R. A Novel Peptide Thrombopoietin Mimetic Designing and Optimization Using Computational Approach. Front. Bioeng. Biotechnol. 2016, 4, 69. 10.3389/fbioe.2016.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N.; Tomar R.; Pandey A.; Tomar V.; Singh V. K.; Chandra R. Preclinical evaluation and molecular docking of 1, 3-benzodioxole propargyl ether derivatives as novel inhibitor for combating the histone deacetylase enzyme in cancer. Artif. Cells Nanomed. Biotechnol. 2017, 46, 1288–1299. 10.1080/21691401.2017.1369423. [DOI] [PubMed] [Google Scholar]
- Kaurav M.; Madan J.; Sudheesh M. S.; Pandey R. S. Combined adjuvant-delivery system for new generation vaccine antigens: alliance has its own advantage. Artif. Cells, Nanomed., Biotechnol. 2018, 46, S818–S831. 10.1080/21691401.2018.1513941. [DOI] [PubMed] [Google Scholar]
- Sharma A.; Sharma D.; Baldi A.; Jyoti K.; Chandra R.; Madan J. Imiquimod-oleic acid prodrug loaded cream reduced drug crystallinity and induced indistinguishable cytotoxicity and apoptosis in mice melanoma tumour. J. Microencapsulation 2019, 1–16. 10.1080/02652048.2019.1677796. [DOI] [PubMed] [Google Scholar]
- Duval E.; Zatylny C.; Laurencin M.; Baudy-Floc’h M.; Henry J. KKKKPLFGLFFGLF: a cationic peptide designed to exert antibacterial activity. Peptides 2009, 30, 1608–1612. 10.1016/j.peptides.2009.06.022. [DOI] [PubMed] [Google Scholar]
- Meher P. K.; Sahu T. K.; Saini V.; Rao A. R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. 10.1038/srep42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes D. A.; Devaurs D.; Kavraki L. E. Understanding the challenges of protein flexibility in drug design. Expert Opin. Drug Discovery 2015, 10, 1301–1313. 10.1517/17460441.2015.1094458. [DOI] [PubMed] [Google Scholar]
- London N.; Raveh B.; Cohen E.; Fathi G.; Schueler-Furman O. Rosetta FlexPepDock web server - High resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. 10.1093/nar/gkr431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trellet M.; Melquiond A. S. J.; Bonvin A. M. A Unified Conformational Selection and Induced Fit Approach to Protein-Peptide Docking. PLoS One 2013, 8, e58769 10.1371/journal.pone.0058769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antes I. DynaDock: A now molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins: Struct., Funct., Bioinf. 2010, 78, 1084–1104. 10.1002/prot.22629. [DOI] [PubMed] [Google Scholar]
- Schindler C. E. M.; De Vries S. J.; Zacharias M. Fully Blind Peptide-Protein Docking with pepATTRACT. Structure 2015, 23, 1507–1515. 10.1016/j.str.2015.05.021. [DOI] [PubMed] [Google Scholar]
- Lee H.; Heo L.; Lee M. S.; Seok C. GalaxyPepDock: a protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. 10.1093/nar/gkv495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurcinski M.; Jamroz M.; Blaszczyk M.; Kolinski A.; Kmiecik S. CABS-dock web server for the flexible docking of peptides to proteins without prior knowledge of the binding site. Nucleic Acids Res. 2015, 43, W419–W424. 10.1093/nar/gkv456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvage E.; Kerff F.; Terrak M.; Ayala J. A.; Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 2008, 32, 234–258. 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- Calvez P.; Breukink E.; Roper D. I.; Dib M.; Contreras-Martel C.; Zapun A. Substitutions in PBP2b from β-Lactam-resistant Streptococcus pneumoniae Have Different Effects on Enzymatic Activity and Drug Reactivity. J. Biol. Chem. 2017, 292, 2854–2865. 10.1074/jbc.M116.764696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J. W.; Hung Y. J.; Yang C. H.; Chen Y. C. The Antimicrobial Activity of Gramicidin A Is Associated with Hydroxyl Radical Formation. PLoS One 2015, 10, e0117065 10.1371/journal.pone.0117065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenssen H.; Hamill P.; Hancock R. E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuhashi M.; Tamaki S.; Curtis S. J.; Strominger J. L. Mutational evidence for identity of penicillin-binding protein 5 in Escherichia coli with the major D-alanine carboxypeptidase IA activity. J. Bacteriol. 1979, 137, 644–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas R. A.; Krings S.; Tomberg J.; Nicola G.; Davies C. Crystal structure of wild-type penicillin-binding protein 5 from Escherichia coli: implications for deacylation of the acyl-enzyme complex. J. Biol. Chem. 2003, 278, 52826–52833. 10.1074/jbc.M310177200. [DOI] [PubMed] [Google Scholar]
- Mahlapuu M.; Håkansson J.; Ringstad L.; Björn C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. 10.3389/fcimb.2016.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrov I.; Naneva L.; Doytchinova I.; Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 2014, 30, 846–851. 10.1093/bioinformatics/btt619. [DOI] [PubMed] [Google Scholar]
- Nagra S.; Kumar D.; Bhattacharyya R.; Banerjee D.; Mukherjee T. Designing of a penta-peptide against drug resistant E. coli. Bioinformation 2017, 13, 192–195. 10.6026/97320630013192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badosa E.; et al. A library of linear undecapeptides with bactericidal activity against phytopathogenic bacteria. Peptides 2007, 28, 2276–2285. 10.1016/j.peptides.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Hara S.; Mukae H.; Sakamoto N.; Ishimoto H.; Amenomori M.; Fujita H.; Ishimatsu Y.; Yanagihara K.; Kohno S. Plectasin has antibacterial activity and no affect on cell viability or IL-8 production. Biochem. Biophys. Res. Commun. 2008, 374, 709–713. 10.1016/j.bbrc.2008.07.093. [DOI] [PubMed] [Google Scholar]
- Kozakov D.; Hall D. R.; Xia B.; Porter K. A.; Padhorny D.; Yueh C.; Beglov D.; Vajda S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. 10.1038/nprot.2016.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D.; Hall D. R.; Beglov D.; Brenke R.; Comeau S. R.; Shen Y.; Li K.; Zheng J.; Vakili P.; Paschalidis I. C.; Vajda S. Achieving reliability and high accuracy in automated protein docking: ClusPro, PIPER, SDU, and stability analysis in CAPRI rounds. Proteins: Struct., Funct., Bioinf. 2010, 78, 3124–3130. 10.1002/prot.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace A. C.; Laskowski R. A.; Thornton J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- Blaszczyk M.; Kurcinski M.; Kouza M.; Wieteska L.; Debinski A.; Kolinski A.; Kmiecik S. Modeling of protein-peptide interactions using the CABS-dock web server for binding site search and flexible docking. Methods 2016, 93, 72–83. 10.1016/j.ymeth.2015.07.004. [DOI] [PubMed] [Google Scholar]
- Li H.; Lu L.; Chen R.; Quan L.; Xia X.; Lü Q. PaFlexPepDock: Parallel Ab-initio docking of peptides onto their receptors with full flexibility based on Rosetta. PLoS One 2014, 9, e105715. 10.1371/journal.pone.0094769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wabik J.; Kurcinski M.; Kolinski A. Coarse-Grained Modeling of Peptide Docking Associated with Large Conformation Transitions of the Binding Protein: Troponin I Fragment–Troponin C System. Molecules 2015, 20, 10763–10780. 10.3390/molecules200610763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurcinski M.; Kolinski A.; Kmiecik S. Mechanism of folding and binding of an intrinsically disordered protein as revealed by ab initio simulations. J. Chem. Theory Comput. 2014, 10, 2224–2231. 10.1021/ct500287c. [DOI] [PubMed] [Google Scholar]
- Kmiecik S.; Gront D.; Kouza M.; Kolinski A. From coarse-grained to atomic-level characterization of protein dynamics: Transition state for the folding of B domain of protein A. J. Phys. Chem. B. 2012, 116, 7026–7032. 10.1021/jp301720w. [DOI] [PubMed] [Google Scholar]
- Kmiecik S.; Kolinski A. Characterization of protein-folding pathways by reduced-space modeling. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 12330–12335. 10.1073/pnas.0702265104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik S.; Kolinski A. Simulation of chaperonin effect on protein folding: A shift from nucleation - Condensation to framework mechanism. J. Am. Chem. Soc. 2011, 133, 10283–10289. 10.1021/ja203275f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz M.; Kolinski A.; Kmiecik S. CABS-flex: Server for fast simulation of protein structure fluctuations. Nucleic Acids Res. 2013, 41, W427–W431. 10.1093/nar/gkt332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamroz M.; Kolinski A.; Kmiecik S. CABS-flex predictions of protein flexibility compared with NMR ensembles. Bioinformatics 2014, 30, 2150–2154. 10.1093/bioinformatics/btu184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk M.; Jamroz M.; Kmiecik S.; Kolinski A. CABS-fold: Server for the de novo and consensus-based prediction of protein structure. Nucleic Acids Res. 2013, 41, W406–W411. 10.1093/nar/gkt462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kmiecik S.; Jamroz M.; Kolinski M. Structure prediction of the second extracellular loop in G-protein-coupled receptors. Biophys. J. 2014, 106, 2408–2416. 10.1016/j.bpj.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospital A.; Andrio P.; Fenollosa C.; Cicin-Sain D.; Orozco M.; Gelpí J. L. MDWeb and MDMoby: an integrated web-based platform for molecular dynamics simulations. Bioinformatics 2012, 28, 1278–1279. 10.1093/bioinformatics/bts139. [DOI] [PubMed] [Google Scholar]
- Gasteiger E.; Hoogland C.; Gattiker A.; Duvaud S.; Wilkins M. R.; Appel R. D.; Bairoch A.. Protein Identification and Analysis Tools on the ExPASy Server; Walker J. M., Ed.; The Proteomics Protocols Handbook; Humana Press, 2005; pp 571–607. [Google Scholar]
- Heo L.; Park H.; Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. 10.1093/nar/gkt458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.