

Abstract
Although the tricyclic antidepressant amitriptyline (ATL) is widely used in the clinic, the mechanism underlying its high therapeutic efficacy against neuropathic pain remains unclear. NMDA receptors (NMDARs) represent a target for ATL and are involved in sensitization of neuropathic pain. Here we describe two actions of ATL on NMDARs: 1) enhancement of Ca2+-dependent desensitization and 2) trapping channel block. Inhibition of NMDARs by ATL was found to be dependent upon external Ca2+ concentration ([Ca2+]) in a voltage-independent manner, with an IC50 of 0.72 μM in 4 mM [Ca2+]. The ATL IC50 value increased exponentially with decreasing [Ca2+], with an e-fold change observed per 0.69 mM decrease in [Ca2+]. Loading neurons with BAPTA abolished Ca2+-dependent inhibition, suggesting that Ca2+ affects NMDARs from the cytosol. Since there is one known Ca2+-dependent process in gating of NMDARs, we conclude that ATL most likely promotes Ca2+-dependent desensitization. We also found ATL to be a trapping open-channel blocker of NMDARs with an IC50 of 220 µM at 0 mV. An e-fold change in ATL IC50 was observed to occur with a voltage shift of 50 mV in 0.25 mM [Ca2+]. Thus, we disclose here a robust dependence of ATL potency on extracellular [Ca2+], and demonstrate that ATL bound in the NMDAR pore can be trapped by closure of the channel.
Subject terms: Ion transport, Ion channels in the nervous system, Cellular neuroscience
Introduction
More than half of patients with chronic neuropathic pain also suffer from depression1. Use of antidepressants as an adjuvant therapy for ameliorating chronic pain is a promising treatment strategy for patients displaying both neuropathic pain and depression2. However, the mechanisms underlying analgesic effects of many psychotropic drugs are currently unknown. Antidepressants represent a wide variety of compounds with diverse molecular structures, making it difficult to attribute their analgesic properties to action on specific targets. Among antidepressants with analgesic properties, amitriptyline (ATL) has long been successfully used in treatment of pain3.
ATL, a tricyclic antidepressant, demonstrates a broad profile of pharmacological actions. ATL effects considered to be relevant for antidepressant therapy include: 1) inhibition of monoamine uptake such as norepinephrine, dopamine, serotonin etc., 2) considerable M-cholinolytic, antihistamine and α-adrenolytic activities, and 3) block of Na+, K+ and Ca2+ channels [for review see4]. Additionally, ATL inhibits Ca2+ transport by Na+/Ca2+-exchanger (NCX) in synaptosomes and also demonstrates voltage- and Mg2+-dependent open-channel block of NMDA receptors (NMDARs)5,6. ATL is metabolized by the liver to nortriptyline, which further expands its wide set of molecular targets7,8.
It is well established that excitatory glutamatergic synaptic transmission is involved in sensitization of neuropathic pain. Nerve injury potentiates NMDAR activity at both pre- and postsynaptic sites9,10. In particular, injury of the dorsal root ganglion and spinal superficial dorsal horn neurons increases expression of α2δ-111, a voltage-activated Ca2+ channel subunit that also interacts with NMDARs, promoting synaptic and surface expression of α2δ-1-NMDAR complexes and sensitization of neuropathic pain12. We recently described that NMDARs are upregulated by NCX in physiological conditions and that inhibition of Ca2+ export from neurons by the NCX inhibitor (2-[4-[(4-nitrophenyl)methoxy]phenyl]ethyl ester, methanesulfonate, KB-R7943) or lithium enhances Ca2+-dependent desensitization of NMDARs13,14. Modulation of NMDAR activity by ATL may potentially contribute to its efficacy in treating neuropathic pain. Indeed, other drugs acting directly on NMDARs (e.g., ketamine15 and memantine16) or indirectly modulating NMDAR activity (e.g., gabapentinoids, which interact with α2δ-1 and prevent synaptic expression of α2δ-1-NMDAR complexes9) possess analgesic potential17.
Despite our knowledge of the molecular targets of ATL, the high therapeutic efficacy of ATL against neuropathic pain remains a puzzle. While ATL blocks open channels of NMDARs, the precise mechanism of the blockade is still unclear. In general, compounds satisfying the “sequential” model of open-channel block18,19 inhibit integral NMDA-activated currents less effectively than trapping channel blockers20–22. Relatively large ATL concentrations (15–60 µM) are required for the NMDAR channel block5. However, this somewhat contradicts clinical observations that neuropathic pain relief by ATL treatment can be rapidly achieved with dosages that are lower than those required for treatment of depression23.
To gain insights into the mechanism of ATL action on NMDARs, we used patch-clamp electrophysiology to study the effects of ATL on whole-cell NMDAR currents in primary cultures of rat cortical neurons, which express GluN1, GluN2A, and GluN2B subunits24 forming diheteromeric and triheteromeric NMDARs25,26. We also utilize electrophysiological recordings and structural modeling of NMDARs to investigate the mechanism of ATL binding to the NMDAR channel. We disclose here a novel mode of pharmacological action of ATL, a robust dependence of ATL potency on extracellular Ca2+ concentration, and demonstrate that ATL causes trapping open-channel block of NMDARs, binding to a site similar to MK-801 and memantine binding sites within the NMDAR channel pore.
Results
Contradictions in phenomenology of NMDAR inhibition by amitriptyline
We first examined general properties of ATL inhibition of whole-cell currents activated by NMDA in cortical neurons that suggest ATL has multiple mechanisms of action on NMDARs. 100 μM ATL, with 1 mM Ca2+ in the external solution, caused block of NMDAR currents that depended on membrane voltage. Fractions of blocked currents (see ‘Methods‘, ‘Analysis of membrane currents‘) were considerably larger at −70 mV than at −30 mV (Fig. 1a, Supplementary Table 1). As voltage dependence represents a key feature characterizing an open-channel blocker, the above observation suggests that ATL is an open-channel blocker of NMDARs. However, when the effect of 10 μM ATL was tested under the same conditions, voltage dependence was not observed. Fractions of blocked currents were similar both at −70 mV and at −30 mV (Fig. 1b). Furthermore, we did not find any voltage dependence in a wide membrane voltage range from −40 mV to +40 mV when NMDAR currents were inhibited by 20 μM ATL in an external solution containing 2 mM Ca2+ (Fig. 1c). This observation could not be easily explained within the framework of the open-channel block mechanism, unless the blocking molecule binds to a site that does not sense the plasma membrane electric field in the NMDAR external vestibule.
Figure 1.

Characterization of amitriptyline (ATL) effects on NMDA-activated currents. (a) An overlay of normalized currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV and −30 mV in the presence of 1 mM Ca2+ in the bathing solution (left). 100 μM ATL was applied at steady state. The agonist and drug applications are shown above the traces as bars. The histogram (right) depicts the fraction of blocked current (1 − Ib/Ic) obtained in each of experiments (circles) at −70 mV and −30 mV. Mean values ± S.E.M. are compared. ****Data are significantly different (p = 0.0001, Student’s two-tailed t-tests). (b) An overlay of normalized currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV and −30 mV in the presence of 1 mM Ca2+ in the bathing solution (on the left). 10 μM ATL was applied at the steady state. In the histogram (right), fractions of blocked current (1 − Ib/Ic) obtained in each of experiments (circles) at −70 mV and −30 mV mean values ± S.E.M are depicted. Data are not significantly different (p = 0.12, Student’s two-tailed t-tests). (c) Currents activated by 100 μM NMDA + 30 μM Gly recorded at −40 mV and + 40 mV in the presence of 2 mM Ca2+ in the bathing solution (on the left). 20 μM ATL was applied as shown above the traces by bars. Plot (right) depicts fractions of blocked current (1 − Ib/Ic) obtained in each experiment (circles) at −40 mV and +40 mV as well as mean values ± S.E.M. Data are not significantly different (p = 0.19, Student’s two-tailed t-tests). (d) An overlay of currents activated by 100 μM NMDA + 30 μM Gly normalized to the steady-state amplitude recorded at −70 mV in the presence of 0.25 mM and 2 mM Ca2+ in the bathing solution (left). 10 μM ATL was applied as shown by bars above the traces. The histogram (right) depicts the fraction of blocked current (1 − Ib/Ic) obtained at −70 mV in the presence of 0.25 mM and 2 mM Ca2+ in the bathing solution in each of experiments (circles) as well as mean values ± S.E.M. ***Data are significantly different (p = 0.0004, Student’s two-tailed t-tests). (e) An overlay of currents activated by 100 μM NMDA + 30 μM Gly normalized to the steady-state amplitude recorded at −70 mV in the presence of 0.25 mM and 2 mM Ca2+ in the bathing solution (left). 10 μM IEM-1754 was applied as shown by bars above the traces. Histogram (right) depicts fractions of blocked current (1 − Ib/Ic) obtained at −70 mV in the presence of 0.25 mM and 2 mM Ca2+ in the bathing solution in each of experiments (circles) as well as mean values ± S.E.M. Data are not significantly different (p = 0.82, Student’s two-tailed t-tests).
In the above experiments, we noticed that inhibition by lower (10 μM, 20 μM) ATL concentrations appeared to be facilitated in 2 mM Ca2+ (Fig. 1c) compared to 1 mM Ca2+ (Fig. 1b), suggesting possible involvement of extracellular Ca2+ in NMDAR block by ATL. We therefore examined ATL inhibition of NMDAR currents as a function of external Ca2+ concentration ([Ca2+]). Indeed, 10 μM ATL inhibited NMDAR currents in a Ca2+-dependent manner, with inhibition by ATL being far more pronounced in 2 mM Ca2+- than in 0.25 Ca2+- containing media (Fig. 1d).
In general, it cannot be excluded that Ca2+, as a permeant ion of NMDAR channels, could affect open-channel block. It is well established that the parameters of channel block of NMDARs by a blocking molecule27 and/or Mg2+28,29 are strongly affected by permeant ions. In addition, several binding sites for Ca2+ were found in the ion pathway of NMDARs30,31 that also likely interact with blocking molecules within the ion pore of NMDARs. A lack of information on this subject forced us to perform experiments with N-(tricyclo[3.3.1.13,7]dec-1-ylmethyl)-1,5-pentanediamine dihydrobromide (IEM-175432), a “pure” open-channel blocker that can bind to a shallow binding site in the NMDAR channel, preventing closure of the channel, as well as a deeper binding site, where it can be trapped in the channel19,27. Effects of 10 μM IEM-1754 on NMDA-activated currents were examined in the presence of 2 mM Ca2+ and 0.25 mM Ca2+ in the external solution. Figure 1e demonstrates that IEM-1754 caused block of NMDA-activated currents to a similar extent in both 2 mM Ca2+ and 0.25 mM Ca2+, suggesting that parameters of general open-channel block are not affected by extracellular Ca2+ within this [Ca2+] range. This later observation raises a question about the origin of the discrepancy we observed between the effects of low (10 μM, 20 μM) and high (100 μM) ATL concentrations on NMDARs - how could Ca2+ be contributing to inhibition by ATL?
Dependence of amitriptyline inhibition of NMDARs on external Ca2+
To quantify the effect of external [Ca2+] on inhibition by ATL, we studied concentration-inhibition relationships to estimate the concentration that causes half-maximal inhibition (IC50) in the presence of different [Ca2+]s in the bathing solution. In these experiments, increasing concentrations of ATL were sequentially applied during NMDA-activated currents at steady state (Fig. 2a). Amplitudes of currents were plotted as a function of ATL concentration and the IC50 values were measured by fitting the data to the Hill equation (Eq. 1). Increasing external [Ca2+] strongly decreased the IC50 value of NMDAR inhibition by ATL (Fig. 2b). The IC50 value measured in 4 mM Ca2+, 0.72 µM, increased over 100-fold in 0.25 mM Ca2+. The Ca2+-dependence of IC50 for the inhibition of NMDA-activated currents by ATL could be fit well with an exponential function (Fig. 2c, Supplementary Table 2). This dependence is rather sharp, with an e-fold change of the IC50 value achieved with a [Ca2+] shift of 0.63 mM. The single exponential approximation to the data (Fig. 2c) yielded the equation as follows: = 91 × exp (−[Ca2+]/0.63), where = 91 μM and 0.63 mM is the e-fold change value.
Figure 2.

Ca2+-dependent inhibition of NMDAR currents by amitriptyline (ATL). (a) Currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV in the presence of 0.25 mM and 2 mM Ca2+ in the bathing solution in the absence of ATL (0) and in the presence of rising ATL concentrations ([ATL]) indicated by numbers at corresponding level of currents (in μM). (b) Concentration-inhibition curves for ATL for currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV in the presence of different [Ca2+]s in the bathing solution. Symbols show mean values ± S.E.M of the relative amplitudes of currents (Ib/Ic) in the presence (Ib) and absence (Ic) of different ATL concentrations ([ATL]) from 5–9 measurements. Solid lines are fits to the data with the Hill equation (Eq. 1). (c) Dependence of IC50 (a concentration that causes a half-maximal inhibition) for ATL inhibition of NMDAR currents on external [Ca2+] obtained from experiments illustrated in panels a and b. Data from each experiment (symbols) and mean values ± S.E.M. are shown. The IC50 values are 63.0 ± 9.3 μM, h = 1.4 ± 0.1 (n = 8); 37.6 ± 7.7, h = 1.5 ± 0.1 (n = 7); 21.6 ± 8.7 μM, h = 1.6 ± 0.5 (n = 5); 4.9 ± 1.0 μM, h = 1.2 ± 0.1 (n = 9); and 0.72 ± 0.12 μM, h = 1.5 ± 0.2 (n = 10) in the presence of 0.25 mM, 0.5 mM, 1 mM, 2 mM and 4 mM Ca2+ in the bathing solution, respectively. The dotted line is an approximation of the data by a single exponential function. An e-fold change in IC50 is achieved by a shift of [Ca2+] of 0.63 mM.
In principle, external Ca2+ interactions with the Ca2+ binding sites found in the external vestibule of recombinant GluN1/GluN2A and GluN1/GluN2B receptors33–35 could contribute to the effects described above. However, Ca2+ influx through activated NMDAR channels could act from inside by promoting Ca2+-dependent desensitization. To determine whether Ca2+ affects NMDARs externally or internally, we performed experiments on neurons loaded with BAPTA, which chelates intracellular Ca2+. Figure 3a shows that in 1 mM external Ca2+ neurons loaded with BAPTA display far weaker desensitization of NMDARs, presumably due to ablation of Ca2+-dependent desensitization. In addition, a considerable increase of ATL concentration was required to inhibit currents activated by NMDA. This resulted in a shift of the concentration-inhibition relationship for ATL toward larger concentrations (Fig. 3b), so that the ATL IC50 in the presence of 1 mM external Ca2+ significantly elevated from 21.6 ± 8.7 µM (n = 5) to 105.8 ± 9.5 µM with BAPTA (n = 10, p = 0.0001, Student’s two-tailed t-tests, Fig. 3c). These data are consistent with our ideas that Ca2+ acts via the cytosol and that Ca2+-dependent desensitization of NMDARs contributes to the Ca2+ dependence of ATL effects on NMDARs.
Figure 3.

Loading neurons with BAPTA to chelate intracellular Ca2+ reduces ATL inhibition of NMDAR currents. (a) Currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV in the presence of 1 mM Ca2+ in the bathing solution in the absence of ATL (0) and in the presence of rising ATL concentrations ([ATL]) indicated by numbers at corresponding level of currents (in μM). The blue trace shown was recorded from intact neuron and the orange trace - from BAPTA-loaded neuron. (b) Concentration-inhibition curve for ATL obtained from currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV in the presence of 1 mM [Ca2+] in the bathing solution. Orange symbols represent mean values ± S.E.M of the relative amplitudes of currents (Ib/Ic) in the presence (Ib) and absence (Ic) of different ATL concentrations ([ATL]) measured from neurons loaded with BAPTA (n = 10). Solid orange line fits the data with the Hill equation (Eq. 1). The blue, gray and green curves are replotted from Fig. 2b to display the difference of results between control and BAPTA-loaded neurons. (c) Comparison of ATL IC50 values obtained in 1 mM [Ca2+] for control (21.6 ± 8.7 μM, n = 5) and BAPTA-loaded (105.8 ± 9.5 µM, n = 10) neurons. Data from each experiment (symbols) and mean values ± S.E.M. are shown. ****Data are significantly different (p = 0.0001, Student’s two-tailed t-tests).
We also tested whether ATL affects closed NMDARs. Regardless of [Ca2+] (0.25 mM or 2 mM), ATL applied prior to NMDA did not affect NMDA-activated currents. Simultaneous application of both ATL and NMDA was required for blockade. This suggests that activation of NMDARs and Ca2+ entry through the channels are indispensable conditions for the effects of ATL on NMDARs (Supplementary Fig. S1, Table 3).
It became clear that this pharmacological action of ATL does not fit the traditional open-channel block model and has not been described so far. Because Ca2+-dependent desensitization is the only known Ca2+-dependent process in NMDAR activation kinetics, we conclude that ATL can somehow enhance Ca2+-dependent NMDAR desensitization.
Open-channel block of NMDARs by amitriptyline
To study open-channel block of NMDARs by ATL, we performed experiments using 0.25 mM Ca2+-containing bathing solution to avoid complications that could be caused by Ca2+-dependent processes. Under these conditions, we tested whether ATL-induced block manifestations correspond well to basic predictions of open-channel block. ATL applied at steady state of NMDAR currents induced a decrease in steady-state current. The degree of block was dependent on ATL concentration (Fig. 4a). The block onset (τon) and offset (τoff) were well fit by a single exponential function (Eq. 2). The τon value decreased with a concentration increase whereas the τoff value did not depend on ATL concentration (Fig. 4b). These data allow an estimation of ATL binding parameters with NMDAR channels at −70 mV: 0.0016 ± 0.00009 s−1 µM−1 (n = 7) for the rate constant of binding (kon, Eq. 3), 0.204 ± 0.005 s−1 (n = 7) for the rate constant of dissociation (koff, Eq. 4) and 127.5 ± 7.8 µM (n = 7) for the apparent equilibrium dissociation constant (Kd, Eq. 5).
Figure 4.

Characteristics of open-channel block of NMDARs by amitriptyline (ATL). (a) An overlay of normalized currents activated by 100 μM NMDA + 30 μM Gly recorded at −70 mV in the presence of 0.25 mM Ca2+. 10 μM or 100 μM ATL was applied at steady state. Applications of agonist and drug are shown above the traces as bars. The onset and offset of ATL block of currents were well fitted by a single exponential function (smooth lines through corresponding currents), yielding time constants for block (τon) and unblock (τoff). (b) Histogram representing a comparison of rates of block onset (1/τon) and offset (1/τoff) obtained for 10 μM and 100 μM ATL at −70 mV. Values (circles) from each experiment, illustrated in panel a, as well as mean values ± S.E.M. are compared. ***Data are significantly different (p = 0.0003, Student’s two-tailed t-tests). The Kd value for ATL, calculated using Eqs. 3, 4 and 5, is 127.5 μM. (c) Currents activated by 100 μM NMDA + 30 μM Gly recorded in 0.25 mM Ca2+ at −100 mV, −70 mV and −30 mV in the absence of ATL (0) and in the presence of rising ATL concentrations ([ATL]) indicated by numbers at corresponding level of current (in μM). (d) Concentration-inhibition curves for ATL for currents activated by 100 μM NMDA + 30 μM Gly recorded at −100 mV, −70 mV and −30 mV in the presence of 0.25 mM Ca2+. Symbols depict mean values ± S.E.M of relative amplitudes of currents (Ib/Ic) measured in the presence (Ib) and absence (Ic) of different ATL concentrations from 5–10 experiments. Solid lines are fits to the data with the Hill equation (Eq. 1). (e) Voltage dependence of the IC50 value for ATL inhibition of NMDA-activated currents. Values (circles) from each experiment and mean values ± S.E.M. are shown. Dotted line depicts fit to the data with Eq. 6, which yielded IC50 (0) = 220 μM and a e-fold change of the IC50 of 50 mV.
An important feature of open-channel block is voltage-dependence. We therefore studied the dependence of concentration-inhibition relationships for ATL on holding potential. In 0.25 mM Ca2+-containing bathing solution, we sequentially applied increasing concentrations of ATL during NMDA-activated currents at steady state while holding the cell at −30 mV, −70 mV and −100 mV. To achieve a similar extent of block with depolarization, larger ATL concentrations were required (Fig. 4c), which is readily observed by comparing the concentration-inhibition curves obtained at different holding potentials (Fig. 4d). Therefore, the IC50 value for ATL block of NMDAR channels is voltage-dependent and could be well fit by an exponential (Fig. 4e, Supplementary Table 2). The dependence is rather weak - an e-fold change of the IC50 value could be achieved with a holding potential shift of 50 mV.
The widely accepted interpretation of the voltage-dependence of open-channel block is based on the Woodhull model36, which states that since a blocking molecule binding site is located within the ionic pathway, a blocker must traverse some fraction of the membrane electric field (δ) to reach its binding site. The δ value, therefore, reflects the depth of a blocker’s binding site in the voltage field (i.e. depth in the channel) and can be estimated using Eq. 6. We estimated δ and IC50 (0 mV), the two important parameters that characterize voltage dependence of NMDAR blockade, for ATL to be 0.51 and 220 μM, respectively, yielding the following equation: IC50Vm = 220 × exp (Vm • 0.021). The fact that the open-channel block of NMDARs by ATL is well measurable on macroscopic currents provides some clues for understanding whether a blocking molecule interacts with channel gating during open-channel block.
Trapping channel block of NMDARs by amitriptyline
Chemically diverse organic compounds can bind within the ionic pore after channel opening and inhibit current flow through NMDARs. Channel blockers differ with respect to their interaction with channel gaiting and an ability to prevent channel closure. Blockers that satisfy the sequential scheme of open-channel block (a so-called “foot-in-the-door” mechanism) interact with gaiting and prevent channel closure. While at a single-channel level this manifests in the prolongation of burst durations18,19, at a whole-cell level, macroscopic currents exhibit “tails” upon the simultaneous removal of agonists and a blocker37–40. An appearance of “tail currents” also suggests that NMDAR agonists are locked in their binding sites as long as a blocking molecule has not left the ionic pore39,40. The sequential model, in addition, predicts that a blocker Kd measured from the single-channel study should be much smaller than a blocker IC50 estimated from macroscopic currents41. Because ATL did not cause tail currents, we examined whether ATL can be trapped within the ionic pore of NMDARs by channel closure. Many other NMDAR channel blockers exhibit trapping, including Mg2+ 42, MK-80120, phencyclidine, ketamine, amantadine, memantine21,22, and argiotoxin-63643, a spider neurotoxin that blocks ionotropic glutamate gated channels44,45.
To address this point, we performed experiments in 0.25 mM Ca2+-containing bathing solution to minimize Ca2+-dependent desensitization of NMDARs using the protocol described in previous studies20,22,43. Application of NMDA elicited an inward current that rose rapidly to a peak and then decayed to a steady state. When 200 µM ATL was added to NMDA, the current was progressively blocked to a small residual component (Ib, Fig. 5a). After washout of agonist and ATL with a bathing solution containing 50 µM AP5 ((2 R)-amino-5-phosphonovaleric acid, a competitive antagonist of NMDAR glutamate binding sites used to prevent random opening of NMDARs by contaminating NMDA) (tAP5, Fig. 5a), a test application of NMDA was applied. This process was repeated multiple times on the same neuron, with the duration of ATL and agonist washout time (tAP5) increasing with each run.
Figure 5.

Trapping channel block of NMDARs by amitriptyline (ATL). (a) An example of whole-cell currents activated by 100 μM NMDA + 30 μM Gly recorded in a neuron at −70 mV in the presence of 0.25 mM Ca2+. When the current reached steady state, 200 μM ATL was added for 10 s, which caused a decrease of the current. After a washout of agonists and ATL of duration (tAP5) 0.5 s, 1 s, 2 s or 4 s with a bathing solution containing 50 μM AP5, a testing application of 100 μM NMDA + 30 μM Gly was given. Applications are shown as black bars above the trace. The rise time of the current activated by the testing application was well fitted by a double exponential function (Eq. 7, dotted line) that yielded amplitudes of fast (If) and slow (Is) components, and their time constants. (τf and τs). (b) Currents from a neuron activated by a testing application (100 μM NMDA + 30 μM Gly) recorded at −70 mV in 0.25 mM Ca2+ after washout durations of 0.5 s, 1 s and 4 s. Amplitudes of fast component (If) of the rise times are indicated by arrows. (c) The dependence of If contribution on the amplitude of testing current measured as If/(If + Is) on a delay duration (tAP5). Circles are single experimental measurements, from which mean values ± S.E.M. were calculated.
From the representative current trace shown on Fig. 5a, it is clear that washout of NMDA and ATL does not evoke a generation of “tail current”, suggesting that ATL is trapped inside the NMDAR channel by channel closure. The testing NMDA pulse applied after a 0.5 s washout caused a slowly rising current reflecting ATL unblock from the channels (Fig. 5b) which, if excluding a small fraction of non-blocked NMDARs (Ib), was well fit by a single-exponential function. When washout was prolonged to 1 s, 2 s and 4 s, the rise time of currents activated by the testing NMDA pulse became biphasic, with a fast component (τf) that resembled the rise time of NMDA-activated currents recorded under the control conditions (Fig. 5a) and a slow component (τs) that corresponded well to ATL unblock kinetics. Fitting these data with a double-exponential function yielded the rise time constants and amplitudes (If and Is) for the fast and slow components. Prolongation of the washout was accompanied by a progressive increase of If and a corresponding decrease of Is (Fig. 5c), while τf and τs did not depend on the washout duration (Fig. 5b). The τs values were 11 ± 3.2 s (n = 10), 13 ± 5.7 s (n = 12), 13.2 ± 4.0 s (n = 10) and 15 ± 2.9 s (n = 8) for 0.5 s, 1 s, 2 s, and 4 s delays, respectively, and did not differ significantly (Fig. 5a,b; p > 0.91 ANOVA). The increase of If with the longer washout durations could suggest that the blocking ATL molecules can somehow escape the closed channels, resulting in the fraction of unblocked channels increasing during longer washouts despite the absence of agonists and the presence of AP5 (Fig. 5a). The observation of the residual current (Ib) in the presence of large ATL concentration (200 µM) and the relatively fast escape of ATL molecules from closed channels could presumably reflect some slow permeation of ATL and the ability to dissociate when trapped. Thus, we conclude that ATL is an NMDAR open channel blocker that displays “partial trapping”, a profile shared with other clinically relevant NMDAR channel blockers such as memantine and amantadine22.
Modeling of amitriptyline binding site within NMDAR channel
Recently, it has been shown that NMDARs containing GluN2B subunits transfer the majority (about 80%) of NMDA-activated whole-cell currents in cortical neurons in primary culture46–48. In order to determine the structure of the ATL binding site in the ionic pore of NMDARs, we performed 3D structural modeling of a diheteromeric GluN1/GluN2B NMDAR followed by molecular docking of ATL into the NMDAR channel.
Docking simulations predict that the ATL molecule generally exists in a single, energetically preferred binding pose. In this pose, the tricyclic group is stabilized in a V-like conformation which consists of two centralized aromatic rings, the hydrophobic part of the molecule, situated 120 degrees apart. The amine group of ATL is located distantly at the end of an aliphatic chain of 5 Å in length consisting of 3 carbon atoms (Fig. 6a). Calculations of pKa of ATL revealed that 99% of molecules exist in the protonated form at the physiological pH range (7.2–7.4) with the positive charge distributed around the molecule (Fig. 6a).
Figure 6.

Molecular modeling of amitriptyline (ATL) interactions with the NMDAR ionic pore. (a) Charges of ATL atoms in the CHARMM27 force field. The carbon, hydrogen and nitrogen atoms are shown by blue, white and pink, respectively. The fractions of charge of individual atoms are indicated by numbers. Charge fraction of the positively charged hydrogen atom responsible for hydrophilic interaction with the NMDAR selectivity filter is printed in pink. (b) NMDA receptor lacking the amino terminal domain (pdb code 5UN159). Chains А and C (GluN1 subunits) are presented in orange; chains B and D (GluN2B subunits) are presented in blue. Ligand binding domains (LBD) and transmembrane domains (TMD) are denoted. Green square indicates the region of TMD external vestibule presented at higher space resolution in panels b and c. (c) ATL molecule binding site in of NMDAR channel predicted by molecular docking and molecular dynamics optimization. The key amino acids are shown (presented in thin sticks): N614, V642 and T646 of Chains A and C (GluN1); N612, L640 and T644 of Chains B and D (GluN2B). The distance between the hydrogen atom of amino group of ATL and the oxygen atom of N612 (Chain D, GluN2B) is shown by a dotted line. The estimated distance between these atoms is 1.7 Ǻ. (d) ATL molecule binding site in the NMDAR channel rotated 90° from projection shown in (c).
The NMDAR transmembrane domain (TMD) consists of M1–M4 helices of GluN1 and GluN2 subunits (Fig. 6b). The upper portion of the permeation pathway is a wide vestibule formed by residues in the M1, M3, and M4 helices. This vestibule is located superficially to a thin portion of the channel (the selectivity filter), which is formed by p-loops formed by the M2 segments of the transmembrane domains. Docking simulations predicted an energetically favorable binding pose for ATL in which the V-like group associates with the upper, wide portion of the vestibule while the amine group associates with the selectivity filter. The docked binding pose was found to be highly consistent with the NMDAR-ATL complex obtained by molecular dynamics (MD) simulations (Fig. 6c,d). MD simulations demonstrated that the hydrogen atom of the amine of ATL interacts with the oxygen atom of pore-loop asparagine residue N612 of one of two GluN2B subunits at a distance of 1.7 Å (Fig. 6c), a distance consistent with mutual atomic locations that allow the formation of a hydrogen bond. The V-like group of the ATL molecule is stabilized within the channel by hydrophobic interactions with surrounding and uncharged aliphatic amino acids in the M3 helices of all four subunits. M3 residues found to interact with the V-like group are GluN1(V642),GluN1(T646), GluN2B(L640), and GluN2B(T644).
Discussion
To clarify the mechanism of ATL effects on NMDARs, here we studied the dependence of ATL inhibition of NMDARs on membrane voltage and extracellular [Ca2+]. We observed a peculiarity in the discrepancy between the effects of low (10 μM) and high (100 μM) ATL concentrations. Notably, 10 μM ATL in the presence of 1 mM Ca2+ in the bathing solution inhibited NMDAR currents in a voltage-independent manner, and changes of extracellular [Ca2+] in any direction considerably altered magnitude of inhibition by ATL. In contrast, blockade of NMDAR currents by 100 μM ATL was voltage-dependent, which pointed toward ATL acting as an open-channel blocker, in agreement with a previous study5. To address whether these contradictory observations could be explained in terms of the open-channel block, we tested how external [Ca2+] could influence NMDAR inhibition by IEM-1754, a “pure” open-channel blocker without any other known mechanism of action19 on NMDARs. Blockade of NMDAR currents by IEM-1754 was resistant to a 4-fold increase of [Ca2+] in the external solution, suggesting a lack of Ca2+ effects on blockade. Similarly, the NMDAR channel blocker ketamine21 shows no sensitivity to changes in external [Ca2+] while memantine22 slows the rate of recovery from desensitization of GluN1/GluN2A NMDARs (but not GluN1/GluN2B receptors) in a Ca2+-dependent manner, suggesting that memantine stabilizes a Ca2+-dependent desensitized state of GluN1/GluN2A receptors49. In general, NMDAR open-channel block by a variety of structurally different compounds is not affected by external [Ca2+] to the extent displayed by ATL. Considering the strong dependence of the IC50 value of ATL on external [Ca2+], with a 1 mM increase of external [Ca2+] inducing a roughly 5-fold decrease of the IC50 for ATL inhibition of NMDAR currents, we conclude that Ca2+-dependent inhibition of NMDARs by ATL represents a mode of action that differs from canonical open-channel block. Since Ca2+-dependent desensitization is currently the only known Ca2+-dependent characteristic of NMDAR kinetics, we suggest that ATL promotes the Ca2+-dependent desensitization of NMDARs and this particular mechanism of action dominates at low ATL concentrations.
Ca2+-dependent desensitization of NMDARs
The mechanism by which ATL influences Ca2+-dependent desensitization of NMDARs is still unclear. Ca2+-dependent desensitization of NMDARs is trigged by Ca2+ entry to the cytosol during NMDAR activation, in binding of four Ca2+ ions to calmodulin, and subsequent binding of Ca2+-bound calmodulin to the C0 motif of the GluN1 subunit intracellular C-terminal domain50. There are a number of potential explanations of how ATL could influence Ca2+-dependent desensitization. For instance, we cannot exclude the possibility that NMDARs may have additional allosteric ATL binding sites beyond the binding site in the channel that could allow ATL to stabilize NMDARs in the Ca2+-dependent desensitized state. In addition, interactions with the Ca2+ binding sites found recently in the external vestibule of recombinant NMDARs33–35 could contribute to our observations. However, the lack of ATL effects on closed NMDARs argues against these assumptions (Supplementary Fig. S1). Alternatively, ATL could change the local Ca2+ concentration around NMDAR GluN1 C-terminal domains or inhibit Ca2+ export from neurons by acting on NCX, as shown in synaptosomes6. Inhibition of NCXs promotes Ca2+-dependent desensitization of NMDARs due to the tight molecular localization of NMDARs and NCXs in lipid rafts13,14, [for review see51. Besides, the presence of 10 mM EGTA in the pipette solution to control bulk intracellular Ca2+ NMDAR currents, recorded in our experiments, exhibited pronounced Ca2+-dependent desensitization. This is consistent to the previous observation, that even 40 mM EGTA could not affect Ca2+-dependent desensitization of NMDARs52 because this chelator binds Ca2+ much slower than calmodulin. By loading neurons with BAPTA, we were able to abolish the Ca2+-dependent component of ATL action in the presence of 1 mM Ca2+, suggesting that Ca2+ influx through NMDARs is required for augmentation of ATL block. Whereas this interpretation is consistent with potential action on NCXs, further investigation is required to gain insight into how Ca2+ modulates NMDAR inhibition by ATL.
Trapping channel block of NMDARs
To obtain “proper” parameters of open-channel block by ATL, we performed experiments in 0.25 mM Ca2+-containing external solution. In general, the characteristics of the blockade were: block onset (τon) depended on ATL concentration; recovery from block (τoff) did not depend on ATL concentration; ATL IC50 depended exponentially on membrane voltage that yielded IC50(0 mV) was 220 μM (and an e-fold change of the IC50 per 50 mV). These characteristics are consistent with open-channel block. The Woodhull model36 states that the voltage dependence of a blocker reflects a fraction of the membrane electric field that a blocking molecule must traverse to reach its binding site (δ) within the ionic pore. For ATL, δ equals 0.51, which is slightly smaller than values for ketamine (δ = 0.5553,), memantine (δ = 0.65–0.91) by different estimates22,54–57 and amantadine (δ = 0.7222,). This discrepancy could be due to the distributed charge density of the ATL molecule, a feature not shared by memantine or amantadine, or possible contribution of the voltage-independent inhibition to this estimate. An important feature of ATL block is that the NMDAR channel can close with ATL still bound, trapping the ATL molecule inside the channel. However, when agonists and ATL were simultaneously removed from the external solution we observed that the fraction of unblocked channels in a testing agonist application increased with longer washouts durations. This strongly suggests that ATL escaped from many of the blocked channels in the absence of agonists. This suggests that, like memantine and amantadine22, ATL exhibits “partial trapping”, open-channel block of NMDARs (Supplementary Fig. S2).
Structural modeling of amitriptyline binding site in ionic pore
The crystal structure of diheteromeric GluN1/GluN2B NMDAR accompanied with molecular dynamics simulations revealed that MK-801 and memantine bind within the vestibule of the ion channel, promote closure of the ion channel gate, and lodge between the M3-helix-bundle crossing and the M2-pore loops. The apex of the reentrant M2 is referred to as the Q/R/N site and is formed by asparagines N614 on GluN1 and N612 on GluN2B. These aspargines, together with the N + 1 site asparagines, N613 (GluN2B), are required for channel block by MK-80158,59. Both MK-80159 and memantine57 bind in the same channel vestibule with the positively charged amino group facing the Q/R/N sites. In silico modeling of ATL binding site within the GluN1/GluN2B ion channel demonstrated that the hydrophilic amine of ATL interacts with the pore-loop asparagine residue N612 of one of two GluN2B subunits. ATL is a tertiary amine and its amino group contains one hydrogen atom that can form one hydrogen bond at a time. The N-site asparagines N612 on GluN2B subunit also contribute to channel block by MK-801 and memantine58,59. In contrast, the secondary amine MK-801 and the primary amine memantine being protonated at physiological pH, have two hydrogen atoms covalently bound to their amino group. These hydrogens form stable hydrogen bonds with the two pore-loop asparagine residues N614 (GluN1) and N612 (GluN2B). The V-like hydrophobic part of the ATL molecule is stabilized by aliphatic amino acids V642 and T646 on M3 helixes of both GluN1 subunits. The V642 residue also contributes to stabilize the MK-801 molecule inside of the NMDAR channel59 interacting with its aromatic V-like group60. It is worth noting that MK-801 in animal models is effective against neuropathic pain, but it is not used in the clinic because of numerous side effects21,61–63.
Thus, our molecular docking simulations exhibited that ATL associates with a site in the channel vestibule with the positively charged amino group facing the Q/R/N site, a binding pose similar to both MK-80158 and memantine56,63. These molecules sharing an overlapping binding site is consistent with their experimentally verified similar action of trapping open-channel block of NMDARs.
In conclusion, here we have described two mechanisms of ATL action on NMDARs. ATL is widely used in medical practice to treat a number of mental disorders and performs as a reliable drug to treat neuropathic pain. It is well established that the therapeutic window for ATL concentrations in the blood plasma is within the range of 0.3–1 µM, but in brain tissue can reach 10 µM64. Whereas ATL shows voltage-dependent open-channel block of NMDARs at concentrations >70 µM, at lower concentrations and in physiological levels of extracellular [Ca2+] (2–4 mM), ATL seems to enhance Ca2+-dependent desensitization of NMDARs, displaying much more potent IC50 values ranging from 0.7–7 µM. This concentration range is within the therapeutic window for treatment of both depression as well as neuropathic pain. Considering that open-channel block and Ca2+-dependent inhibition of NMDARs by ATL represent two independent processes, the experimentally measured NMDAR block is determined by additive contributions of both mechanisms. Within the physiological membrane voltages and extracellular [Ca2+]s, the experimentally measured value () is predicted by 3-dimensional distribution (Fig. 7), calculated using equations for IC50Vm and (see Results) as = IC50Vm × /= 0.25 mM, which could be presented in the final form: = 317 × exp (Vm × a − [Ca2+]/b). The a value, which is zδF/RT, and the b value estimated from experiments were 0.021 and 0.63, respectively. When these parameters were optimized by fitting all bunch of the data (Supplimentary Table 2) with this equation, the best coincidence with the experimental data was achieved for a = 0.018 and b = 0.69 (Supplementary Table 4). This correction adjusted the depth of the ATL binding site in the channel to δ = 0.75, which became similar to memantine22,54–57 and amantadine22. The 3-dimensional distribution drawn from this equation illustrates that an enhancement of Ca2+-dependent desensitization of NMDARs represents a mechanism of ATL action that predominantly contributes to its therapeutic usage against neuropathic pain (Supplementary Fig. S3) sensitized by a hyperfunction of NMDARs9,10. It is widely accepted, in addition, that calmodulin- and Ca2+-dependent desensitization represents a common mechanism for the receptor and channel modulation and was described for NMDARs, TRPV-channels, L-type voltage-gated channels, SK-channels, adenosine receptors and many others [for review see51,65]. Because ATL may promote the Ca2+-dependent desensitization of NMDARs, we cannot exclude that similar ATL effects on the receptors and channels mentioned above could also occur. Regardless, this mode of ATL action requires more detailed investigations.
Figure 7.
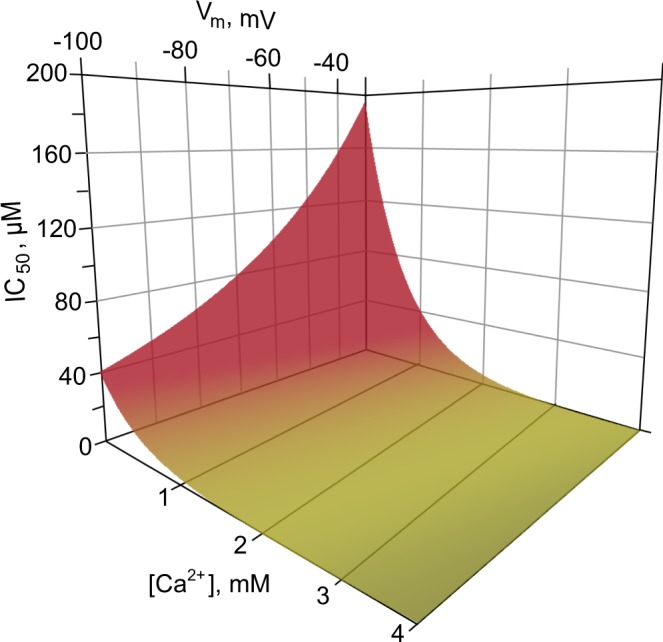
Predicted ATL IC50 values for NMDAR inhibition as a function of membrane voltage (Vm) and external [Ca2+]. The ATL IC50 was calculated as = IC50Vm × /, where equals 64 µM. The final form of the equation is: = 317 × exp (Vm × 0.018 − [Ca2+]/0.69). Green color corresponds to the region of ATL therapeutic concentrations in the blood serum.
Methods
Primary culture of cortical neurons
All manipulations on animals were performed in accordance with the guide of the Federation for Laboratory Animal Science Associations and were approved by the Animal Care and Use Committees of Sechenov Institute. Briefly, 16 days pregnant Wistar rats (14 female rats used in this study, supplied by the Sechenov Institute Animal Facility) were placed in a plastic box connected to a CO2 tank by a tube and then sacrificed by 1 min CO2 inhalation. Fetuses were removed and then primary cultures of rat cortical neurons were prepared using conventional procedures as described earlier27,66. Neurons were grown in Neurobasal culture medium supplemented with B-27 (Gibco-Invitrogen, UK) on glass coverslips coated with poly-D-lysine and were used for experiments after 10–15 days in culture66,67.
Patch clamp recordings
Whole-cell currents were recorded from cultured rat cortical neurons using a MultiClamp 700B patch-clamp amplifier with Digidata 1440 A controlled by pClamp v10.2 software (Molecular Devices). Recordings were 8-order low-pass filtered at 200 Hz and the acquisition rate was 20000 samples per second. Solution exchange was performed by means of a fast solution application system as described earlier13. Unless otherwise specified, the external bathing solution contained (in mM): 140 NaCl; 2.8 KCl; 1.0 CaCl2; 10 HEPES, at pH 7.2–7.4, osmolarity 310 mOsm. The intrapipette solution contained (in mM): 120 CsF, 10 CsCl, 10 EGTA, and 10 HEPES, osmolarity 300 mOsm14, with pH adjusted to 7.4 with CsOH. Patch pipettes of 2–4 MΩ were pulled from Sutter BF150-89-10 borosilicate capillaries. Experiments were performed at 23–25 °C. Under control conditions, neurons were voltage clamped at −70 mV. Data are reported without corrections for liquid junction potential, which was measured as −11 mV. To activate NMDARs, 100 µM NMDA and 30 µM glycine were co-applied. Some experiments were performed on neurons loaded with BAPTA. To load BAPTA, cortical neuron cultures were incubated in bathing solution containing 10 µM BAPTA-AM for 2 hours.
Drugs
Compounds were acquired from Sigma-Aldrich, St. Louis, MO, USA. The adamantane derivative IEM-1754 [N-(Tricyclo[3.3.1.13,7]dec-1-ylmethyl)-1,5-pentanediamine dihydrobromide] was synthesized by Dr. V. E. Gmiro at the Institute of Experimental Medicine, Russian Academy of Medical Sciences, St. Petersburg.
Analysis of membrane currents
Whole-cell membrane currents activated by NMDA were measured at steady state. The fraction of blocked current was calculated as 1 − Ib/Ic, where Ic is the amplitude of current activated by agonists and Ib is the amplitude of current activated by agonists in the presence of blocker.
To determine amitriptyline (ATL) blocking potency, NMDA elicited currents were measured in the absence and presence of different ATL concentrations ([B]). Amplitudes of currents measured in the presence of blocker were normalized to maximal current response in control (Ic). The IC50, a concentration of ATL causing 50% inhibition, and the Hill coefficient (h) were estimated by fitting concentration-inhibition curves with the Hill equation:
| 1 |
NMDAR current relaxations during block or unblock by ATL were fit with the single exponential equation:
| 2 |
where Imax and Imin are maximal and minimal amplitudes of currents during relaxations and τ represents the time constant of the exponential component during block onset (τon) or offset (unblock, τoff). From these measurements, an estimation of rate constants of block (kon) and unblock (koff) was estimated. kon was obtained from τon measured at 10 μM and 100 μM ATL (C1 and C2 respectively) using the equation:
| 3 |
The koff the reciprocal to measured τoff:
| 4 |
The equilibrium dissociation constant (Kd) for ATL was calculated as follows:
| 5 |
To obtain the voltage dependence of NMDAR block by ATL, its concentration-response curves were generated at three membrane holding potentials Vm = −100, −70 and −30 mV. The obtained values were fitted with the Woodhull equation36:
| 6 |
where IC50(0 mV) and δ were set as free parameters. IC50(0 mV) is the half-maximal inhibition concentration at Vm = 0 mV, δ is the fraction of the membrane voltage field exerting force on ATL at its binding site, z = 1 is an ATL molecule electric charge. For Faraday constant, gas constant and room temperature of 25 °C the RT/F ≈ 25.7 mV.
The rise time of currents activated by the testing NMDA + Gly application in trapping channel block experiments was fit in ClampFit (pClamp, Axon Instruments) using the double exponential function:
| 7 |
where I1 and I2 are amplitudes of the exponential and τ1 and τ2 are time constants of the exponential and C equals I1 + I2 + Ib.
Statistical analysis
Data are presented as representative measurements as well as mean values ± standard error of the mean (S.E.M.). Sample number (n) refers to the number of recorded cells. Groups were compared using ANOVA with Bonferroni correction and Student’s two-tailed t-test. Statistical significance is reported in the figures according to the following symbols *** and ****, which indicate p values below (<) 0.001 and 0.0001, respectively. Curve fitting was performed using OriginPro software (OriginLab Corp.).
Preparation of 3D-models
The three-dimensional model of ATL was generated from pdb entry 3APV68. Acid dissociation constants (pKa) of the compound and the percentage of different protonation states at neutral pH were calculated by MarvinView program 17.3.13.0 (ChemAxon Ltd., https://www.chemaxon.com/products/marvin/marvinview). There are several three-dimensional models of NMDAR in Protein Data Bank69. For our molecular modeling experiments, we used a model of NMDAR of African frog Xenopus laevis, with a molecule of dizocilpine (MK-801) bound inside the receptor channel. The code of the structure is 5UN170.
The chosen receptor model 5UN1 consists of two molecules of NMDAR in the crystalized unit containing the following polypeptide chains: chains A,C,E,G (GluN1 subunits) and chains B,D,F,H (GluN2B subunits). Subunits were expressed lacking the amino-terminal domains (∆ATD receptor) to facilitate structural analysis and crystallized with glycine, glutamate and MK-801. Its three-dimensional structure was determined by diffraction method with the resolution of 3.5 Å59.
For the molecular modeling experiments, we used one of the two NMDAR molecules of 5UN1 structure: chains A, C (GluN1) and B, D (GluN2B). Only the moieties of the receptor subunits that form the channel pore were used for simulation: amino acids 600–653 of GluN1 subunits and amino acids 598–653 of GluN2B subunits. Hereinafter, the numbering corresponds to the primary sequence of the receptor according to UniProt database:70 code for A0A1L8F5J9 GluN1 and code A7XY94 for GluN2B subunits. We removed all other amino acids and ligands from the structure. Missing atoms were added using the VMD software71 and the structure was optimized by energy minimization using NAMD software72.
Molecular docking procedure
Molecular docking of ATL into the NMDAR channel was performed using the Autodock Vina 1.1.2 software package73. The coordinates of the nitrogen atom of MK-801 in 5UN1 was used as the center of the Autodock search area. The search area size was set to 15 × 15 × 40 Å3, which covers the entire channel. The “exhaustiveness” parameter, which represents the amount of computational effort, was set to 24. The parameter “energy range”, characterizing the maximum scatter of energy values of conformations in the output file, was set to 3 kcal/mol. The number of the most optimal conformations in the output file (num_modes) was set to 10. The conformation with the best value of the binding energy was chosen for further optimization by molecular dynamics (MD) simulations.
Molecular dynamics simulations
The structure obtained by molecular docking was optimized with a 2 ns MD simulation with the help of NAMD software72 using CHARMM27 force filed73. Partial charges of atoms in the ATL molecule used in the CHARMM force field are presented in Fig. 6a. The complex was placed into a 1-palmitoyl-2-oleoyl-sn-glycero- 3-phosphatidylcholine (POPC) lipid bilayer with the help of the “Membrane” plug-in included in the VMD software package. The system was then solvated via the “Solvate” plug-in of the VMD suite74. 0.15 mol/L of NaCl was added to make the system neutral. The resulting structure had a total of 103 POPC lipid molecules, 20 sodium ions, 21 chloride ions and 7153 water molecules.
The MD simulation was preceded by a three-stage relaxation of the system. At the first stage, all atoms were fixed except the tails of the POPC molecules, which were relaxed by 1000 steps of conjugate gradient energy minimization followed by 500 ps of equilibration. At the second stage, NMDAR and ATL molecules were constrained, the energy of the system was minimized, and the system was equilibrated for 500 ps. At the final stage of the relaxation procedure, all molecules were released, and the system was equilibrated for 500 ps. Following the preparation stages, a 2 ns MD simulation was run with a time step of 2 fs. Temperature (310 K) and pressure (1 bar) were kept constant using a Langevin thermostat and piston methods. The particle-mesh Ewald method (PME)75 with nonbonded cutoff of 12 Å was applied to compute long-range electrostatic interactions. Periodic boundary conditions were used.
Supplementary information
Acknowledgements
We would like to thank Matthew B. Phillips from Department of Neuroscience of the University of Pittsburgh for reading, correcting, and helpful comments on the manuscript. This work is supported by RFBR grant 18-015-00023 and Russian government state program AAAA-A18-118012290427-7.
Author contributions
Y.D.S. and S.I.B. performed electrophysiological experiments. D.A.S. designed experiments, analyzed the data, prepared the figures, wrote the draft of the manuscript. P.A.A. prepared cell cultures and figures. D.A.B., N.N.S. and N.P.V. performed receptor 3D-molecular modeling, molecular docking and corresponding computational analysis. S.M.A. designed experiments, supervised the study, wrote the final version of the manuscript. All authors contributed to editing of the manuscript and approved the manuscript.
Data availability
The dataset generated and analyzed in this study is available upon request to the corresponding author.
Code availability
All code used in this study is available upon request to the corresponding author.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-56072-z.
References
- 1.Surah A, Baranidharan G, Morley S. Chronic pain and depression. BJA Education. 2014;14:85–89. [Google Scholar]
- 2.Dharmshaktu P, Tayal V, Kalra BS. Efficacy of antidepressants as analgesics: a review. J. Clin. Pharmacol. 2012;52:6–17. doi: 10.1177/0091270010394852. [DOI] [PubMed] [Google Scholar]
- 3.Bryson HM, Wilde MI. Amitriptyline. A review of its pharmacological properties and therapeutic use in chronic pain states. Drugs Aging. 1996;8:459–476. doi: 10.2165/00002512-199608060-00008. [DOI] [PubMed] [Google Scholar]
- 4.Lawson Kim. A Brief Review of the Pharmacology of Amitriptyline and Clinical Outcomes in Treating Fibromyalgia. Biomedicines. 2017;5(4):24. doi: 10.3390/biomedicines5020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barygin OI, et al. Inhibition of the NMDA and AMPA receptor channels by antidepressants and antipsychotics. Brain Res. 2017;1660:58–66. doi: 10.1016/j.brainres.2017.01.028. [DOI] [PubMed] [Google Scholar]
- 6.Lavoie PA, Beauchamp G, Elie R. Tricyclic antidepressants inhibit voltage-dependent calcium channels and Na+-Ca2+ exchange in rat brain cortex synaptosomes. Can. J. Physiol. Pharmacol. 1990;68:1414–1418. doi: 10.1139/y90-215. [DOI] [PubMed] [Google Scholar]
- 7.Olesen OV, Linnet K. Metabolism of the tricyclic antidepressant amitriptyline by cDNA-expressed human cytochrome P450 enzymes. Pharmacology. 1997;55:235–243. doi: 10.1159/000139533. [DOI] [PubMed] [Google Scholar]
- 8.Gillman PK. Tricyclic antidepressant pharmacology and therapeutic drug interactions updated. Br. J. Pharmacol. 2007;151:737–748. doi: 10.1038/sj.bjp.0707253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen SR, Zhou HY, Byun HS, Chen H, Pan HL. Casein kinase II regulates N-methyl-D-aspartate receptor activity in spinal cords and pain hypersensitivity induced by nerve injury. J. Pharmacol. Exp. Ther. 2014;350:301–312. doi: 10.1124/jpet.114.215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, et al. Chloride homeostasis critically regulates synaptic NMDA receptor activity in neuropathic pain. Cell Rep. 2016;15:1376–1383. doi: 10.1016/j.celrep.2016.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole RL, et al. Differential distribution of voltage-gated calcium channel alpha-2 delta (alpha2delta) subunit mRNA-containing cells in the rat central nervous system and the dorsal root ganglia. J. Comp. Neurol. 2005;491:246–269. doi: 10.1002/cne.20693. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, et al. The α2δ-1-NMDA receptor complex is critically involved in neuropathic pain development and gabapentin therapeutic actions. Cell Rep. 2018;22:2307–2321. doi: 10.1016/j.celrep.2018.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sibarov DA, Abushik PA, Poguzhelskaya EE, Bolshakov KV, Antonov SM. Inhibition of plasma membrane Na/Ca-exchanger by KB-R7943 or lithium reveals its role in Ca-dependent N-methyl-D-aspartate receptor inactivation. J. Pharmacol. Exp. Ther. 2015;355:484–495. doi: 10.1124/jpet.115.227173. [DOI] [PubMed] [Google Scholar]
- 14.Sibarov DA, Poguzhelskaya EE, Antonov SM. Downregulation of calcium-dependent NMDA receptor desensitization by sodium-calcium exchangers: a role of membrane cholesterol. BMC Neurosci. 2018;19:73. doi: 10.1186/s12868-018-0475-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 16.Ditzler K. Efficacy and tolerability of memantine in patients with dementia syndrome. A double-blind, placebo controlled trial. Arzneimittelforschung. 1991;41:773–780. [PubMed] [Google Scholar]
- 17.McEntire DM, et al. Pain transduction: a pharmacologic perspective. Expert. Rev. Clin. Pharmacol. 2016;9:1069–8010. doi: 10.1080/17512433.2016.1183481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neher E, Steinbach JH. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J. Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonov SM, Johnson JW. Voltage-dependent interaction of open-channel blocking molecules with gating of NMDA receptors in rat cortical neurons. J. Physiol. 1996;493:425–445. doi: 10.1113/jphysiol.1996.sp021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huettner JE, Bean BP. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc. Natl. Acad. Sci. USA. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacDonald JF, et al. Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J. Physiol. 1991;432:483–508. doi: 10.1113/jphysiol.1991.sp018396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. J. Neurophysiol. 1997;77:309–23. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- 23.Kamble SV, Motlekar SA, D’souza LL, Kudrigikar VN, Rao SE. Low doses of amitriptyline, pregabalin, and gabapentin are preferred for management of neuropathic pain in India: is there a need for revisiting dosing recommendations. Korean J. Pain. 2017;30:183–191. doi: 10.3344/kjp.2017.30.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong J, Russell SL, Pritchett DB, Molinoff PB, Williams K. Expression of mRNAs encoding subunits of the N-methyl-D-aspartate receptor in cultured cortical neurons. Mol. Pharmacol. 1994;45:846–853. [PubMed] [Google Scholar]
- 25.Li JH, et al. Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur. J. Neurosci. 1998;10:1704–1715. doi: 10.1046/j.1460-9568.1998.00169.x. [DOI] [PubMed] [Google Scholar]
- 26.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 27.Antonov SM, Gmiro VE, Johnson JW. Binding sites for permeant ions in the channel of NMDA receptors and their effects on channel block. Nat. Neurosci. 1998;1:451–61. doi: 10.1038/2167. [DOI] [PubMed] [Google Scholar]
- 28.Antonov SM, Johnson JW. Permeant ion regulation of N-methyl-D-aspartate receptor channel block by Mg2+ Proc. Natl. Acad. Sci. USA. 1999;96:14571–14576. doi: 10.1073/pnas.96.25.14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu Y, Auerbach A. Na+ occupancy and Mg2+ block of the N-methyl-D-aspartate receptor channel. J. Gen. Physiol. 2001;117:275–286. doi: 10.1085/jgp.117.3.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Premkumar LS, Auerbach A. Identification of a high affinity divalent cation binding site near the entrance of the NMDA receptor channel. Neuron. 1996;16:869–880. doi: 10.1016/S0896-6273(00)80107-5. [DOI] [PubMed] [Google Scholar]
- 31.Sharma G, Stevens CF. Interactions between two divalent ion binding sites in N-methyl-D-aspartate receptor channels. Proc. Natl. Acad. Sci. USA. 1996;93:14170–14175. doi: 10.1073/pnas.93.24.14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antonov SM, et al. Novel adamantane derivatives act as blockers of open ligand-gated channels and as anticonvulsants. Mol. Pharmacol. 1995;47:558–567. [PubMed] [Google Scholar]
- 33.Maki BA, Popescu GK. Extracellular Ca2+ ions reduce NMDA receptor conductance and gating. J. Gen. Physiol. 2014;144:379–392. doi: 10.1085/jgp.201411244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tu YC, Yang YC, Kuo CC. Modulation of NMDA channel gating by Ca2+ and Cd2+ binding to the external pore mouth. Sci. Rep. 2016;6:1–16. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chopra DA, Monaghan DT, Dravid SM. Bidirectional effect of pregnenolone sulfate on GluN1/GluN2A N-Methyl-D-aspartate receptor gating depending on extracellular calcium and intracellular milieu. Mol. Pharmacol. 2015;88:650–659. doi: 10.1124/mol.115.100396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Woodhull AM. Ionic blockade of sodium channel in nerve. J. Gen. Physiol. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costa AC, Albuquerque EX. Dynamics of the actions of tetrahydro-9-aminoacridine and 9-aminoacridine on glutamatergic currents: concentration-jump studies in cultured rat hippocampal neurons. J. Pharmacol. Exp. Ther. 1994;268:503–514. [PubMed] [Google Scholar]
- 38.Vorobjev VS, Sharonova IN. Tetrahydroaminoacridine blocks and prolongs NMDA receptor-mediated responses in a voltage-dependent manner. Eur. J. Pharmacol. 1994;253:1–8. doi: 10.1016/0014-2999(94)90750-1. [DOI] [PubMed] [Google Scholar]
- 39.Benveniste M, Mayer ML. Trapping of glutamate and glycine during open channel block of rat hippocampal neuron NMDA receptors by 9-aminoacridine. J. Physiol. 1995;483:367–84. doi: 10.1113/jphysiol.1995.sp020591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sobolevsky AI, Koshelev SG, Khodorov BI. Probing of NMDA channels with fast blockers. J. Neurosci. 1999;19:10611–10626. doi: 10.1523/JNEUROSCI.19-24-10611.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wright JM, Nowak LM. Effects of low doses of bicuculline on N-methyl-D-aspartate single-channel kinetics are not evident in whole-cell currents. Mol. Pharmacol. 1992;41:900–907. [PubMed] [Google Scholar]
- 42.Ascher P, Nowak L. The role of divalent cations in the N-methyl-D-aspartate responses of mouse central neurones in culture. J. Physiol. 1988;399:247–66. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poulsen MH, et al. Binding of ArgTX-636 in the NMDA receptor ion channel. J. Mol. Biol. 2014;427:176–189. doi: 10.1016/j.jmb.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grishin EV, et al. Structural-functional characteristics of argiopine - an ion channel blockers from the spider Argiope lobata venom. Bioorg. Khim. 1986;12:1121–1124. [PubMed] [Google Scholar]
- 45.Antonov SM, Dudel J, Franke C, Hatt H. Argiopine blocks glutamate-activated single-channel currents on crayfish muscle by two mechanisms. J. Physiol. 1989;419:569–87. doi: 10.1113/jphysiol.1989.sp017887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edman S, et al. TCN 201 selectively blocks GluN2A-containing NMDARs in a GluN1 co-agonist dependent but non-competitive manner. Neuropharmacology. 2012;63:441–449. doi: 10.1016/j.neuropharm.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volkmann RA, et al. MPX-004 and MPX-007: New pharmacological tools to study the physiology of NMDA receptors containing the GluN2A Subunit. PLoS One. 2016;11:1–20. doi: 10.1371/journal.pone.0148129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sibarov DA, Giniatullin R, Antonov SM. High sensitivity of cerebellar neurons to homocysteine is determined by expression of GluN2C and GluN2D subunits of NMDA receptors. Biochem. Biophys. Res. Commun. 2018;506:648–652. doi: 10.1016/j.bbrc.2018.10.140. [DOI] [PubMed] [Google Scholar]
- 49.Glasgow NG, Povysheva NV, Azofeifa AM, Johnson JW. Memantine and ketamine differentially alter NMDA receptor desensitization. J. Neurosci. 2017;37:9686–9704. doi: 10.1523/JNEUROSCI.1173-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–575. doi: 10.1016/S0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 51.Sibarov DA, Antonov SM. Calcium-dependent desensitization of NMDA receptors. Biochemistry (Mosc.) 2018;83:1173–1183. doi: 10.1134/S0006297918100036. [DOI] [PubMed] [Google Scholar]
- 52.Tong G, Jahr CE. Regulation of glycine-insensitive desensitization of the NMDA receptor in outside-out patches. J. Neurophysiol. 1994;72:754–61. doi: 10.1152/jn.1994.72.2.754. [DOI] [PubMed] [Google Scholar]
- 53.MacDonald JF, Miljkovic Z, Pennefather P. Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J. Neurophysiol. 1987;58:251–266. doi: 10.1152/jn.1987.58.2.251. [DOI] [PubMed] [Google Scholar]
- 54.Bresink I, et al. Effects of memantine on recombinant rat NMDA receptors expressed in HEK 293 cells. Br. J. Pharmacol. 1996;119:195–204. doi: 10.1111/j.1476-5381.1996.tb15971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frankiewicz T, Potier B, Bashir ZI, Collingridge GL, Parsons CG. Effects of memantine and MK-801 on NMDA-induced currents in cultured neurones and on synaptic transmission and LTP in area CA1 of rat hippocampal slices. Br. J. Pharmacol. 1996;117:689–697. doi: 10.1111/j.1476-5381.1996.tb15245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leiva R, et al. Pharmacological and electrophysiological characterization of novel NMDA receptor antagonists. ACS Chem. Neurosci. 2018;9:2722–2730. doi: 10.1021/acschemneuro.8b00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fedele L, et al. Disease-associated missense mutations in GluN2B subunit alter NMDA receptor ligand binding and ion channel properties. Nat. Commun. 2018;9:957. doi: 10.1038/s41467-018-02927-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wollmuth LP, Kuner T, Seeburg PH, Sakmann B. Differential contribution of the NR1- and NR2A-subunits to the selectivity filter of recombinant NMDA receptor channels. J. Physiol. 1996;491:779–797. doi: 10.1113/jphysiol.1996.sp021257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song X, et al. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature. 2018;556:515–519. doi: 10.1038/s41586-018-0039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kroemer RT, et al. Quantitative analysis of the structural requirements for blockade of the N-methyl-D-aspartate receptor at the phencyclidine binding site. J. Med. Chem. 1998;41:393–400. doi: 10.1021/jm9704412. [DOI] [PubMed] [Google Scholar]
- 61.Mao J, Price DD, Mayer DJ, Lu J, Hayes RL. Intrathecal MK-801 and local nerve anesthesia synergistically reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res. 1992;576:254–262. doi: 10.1016/0006-8993(92)90688-6. [DOI] [PubMed] [Google Scholar]
- 62.Medvedev IO, et al. Effects of low-affinity NMDA receptor channel blockers in two rat models of chronic pain. Neuropharmacology. 2004;47:175–183. doi: 10.1016/j.neuropharm.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 63.Limapichat W, Yu WY, Branigan E, Lester HA, Dougherty DA. Key binding interactions for memantine in the NMDA receptor. ACS Chem. Neurosci. 2013;4:255–260. doi: 10.1021/cn300180a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coudore F, Besson A, Eschalier A, Lavarenne J, Fialip J. Plasma and brain pharmacokinetics of amitriptyline and its demethylated and hydroxylated metabolites after one and six half-life repeated administrations to rats. Gen. Pharmacol. 1996;27:215–219. doi: 10.1016/0306-3623(95)02008-X. [DOI] [PubMed] [Google Scholar]
- 65.Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 66.Mironova EV, Evstratova AA, Antonov SM. A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture. J. Neurosci. Methods. 2007;163:1–8. doi: 10.1016/j.jneumeth.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 67.Han EB, Stevens CF. Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc. Natl. Acad. Sci. USA. 2009;106:10817–10822. doi: 10.1073/pnas.0903603106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nishi K, et al. Structural insights into differences in drug-binding selectivity between two forms of human alpha1-acid glycoprotein genetic variants, the A and F1*S forms. J. Biol. Chem. 2011;286:14427–14434. doi: 10.1074/jbc.M110.208926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berman HM, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.The UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research. 2019;47:D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Humphrey W, Dalke A, Schulten K. VMD - Visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 72.Phillips JC, et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu X, Lopes PE, Mackerell AD., Jr. Recent developments and applications of the CHARMM force fields. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012;2:167–185. doi: 10.1002/wcms.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Darden T, York D, Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset generated and analyzed in this study is available upon request to the corresponding author.
All code used in this study is available upon request to the corresponding author.