

Abstract
IL-17 cytokines, in particular IL-17A, are critical effectors in psoriasis. Antibodies blocking IL-17A are highly efficacious in treating psoriasis. Likewise, disruption of IL-17 cytokines signaling, such as via loss of the adaptor CIKS/Act1, ameliorates inflammation in mouse models of psoriasis. IL-17A promotes a cascade of effects, including robust production of IL-19 in both humans and mice. IL-19, along with IL-20 and IL-24 signal via IL-20 receptors and comprise a subgroup within the IL-10 cytokine family. The role of these three cytokines in psoriasis is unsettled. They have been linked to inflammatory processes, including psoriatic pathology, but these cytokines have also been reported to suppress inflammation in other contexts. Here we demonstrate that signaling via IL-20 receptors, including in response to IL-19, delimited aspects of imiquimod-induced psoriatic inflammation. IL-20 receptor-signaling suppressed dermal production of the CCL2 chemokine and thereby reduced CCL-2-driven infiltration of inflammatory cells into the dermis, including IL-17A-producing γδT cells. This constitutes a negative feedback, since IL-17A strongly induces IL-19 in keratinocytes. The effects of IL-17 cytokines in this inflammatory setting are dynamic, they are central to development of both dermal and epidermal hallmarks of psoriasis, but also initiate a path to mitigate inflammatory damage.
INTRODUCTION
Psoriasis is a common chronic inflammatory skin disease characterized by thickening of the epidermis and scaling, caused by abnormal proliferation and differentiation of keratinocytes, as well as by microvascular changes and infiltration of leukocytes into the epidermis and especially the dermis. Although underlying mechanisms are still incompletely understood, the IL-23/IL-17A pathway plays a key role in the pathogenesis (Boehncke and Schon, 2015)). Importantly, monoclonal antibodies targeting IL-17A or its receptor are highly efficacious in treating psoriasis (Patel and Kuchroo, 2015). IL-17A (a.k.a. IL-17) is the signature cytokine of the IL-17 family (IL-17A–F). Increased expression of IL-17A has been linked not only to psoriasis, but also other inflammatory diseases (Miossec and Kolls, 2012). Dermal γδT cells are the main producers of IL-17A in several acute mouse models of psoriasis (Cai et al., 2011, Ha et al., 2014), while dermal tissue-resident memory CD4+ T cells are the primary source of IL-17A in the chronic disease in humans (Matos et al., 2017). However, numbers of dermal γδT cells are also increased in human psoriatic lesions and may be critical during initiation of the disease (Cai et al., 2011, Papotto et al., 2018). Regardless of the cellular source, IL-17A is the critical cytokine driving psoriatic inflammation in humans and in the IMQ mouse model. In addition to IL-17A, IL-17C and IL-17E may also contribute to human psoriasis and mouse models. They function downstream of IL-17A, are produced by and act on keratinocytes, with IL-17C in particular targeting genes largely overlapping with those of IL-17A (Ramirez-Carrozzi et al., 2011, Xu et al., 2018).
All members of IL-17 family signal via heteromeric receptors composed of members of the IL-17 receptor family (RA–RE) (Monin and Gaffen, 2018). Upon ligand engagement, the adaptor CIKS (a.k.a. Act1, Traf3ip2) is recruited for signal transmission; consequently, CIKS is critical for IL-17 cytokines-induced pathology in mouse models psoriasis, collagen-induced arthritis, lupus, and asthma (Claudio et al., 2009, Ha et al., 2014, Pisitkun et al., 2010, Pisitkun et al., 2012).
IL-19, IL-20 and IL-24 comprise a subgroup of cytokines within the IL-10 superfamily (Ouyang et al., 2011). All three cytokines signal via a receptor composed of IL-20RB and IL-20RA (IL-20R type1), while IL-20 and IL-24 can additionally signal via IL-20R type2 (IL-20RB and IL-22RA1). IL-19, IL-20 and IL-24 are produced mainly by epithelial, but also other cell populations, while their receptors are found primarily, but not exclusively on non-hematopoietic cells. All three cytokines have been linked to psoriasis. Expression of IL-19 is highly up regulated in human psoriatic skin lesion and has been associated with pathology in this context. This notion is largely based on in vitro studies in which IL-19, having negligible effects on its own, enhanced some effects of IL-17 on keratinocyte cultures, but did not alter proliferation, differentiation or migration of these cells (Bissonnette et al., 2017, Witte et al., 2014). It remains to be established whether IL-19/IL-20 receptor signaling is part of the pathogenic sequelae of IL-17 in vivo. Transgenic mice overexpressing IL-20 or IL-24 display some epidermal hyperplasia, although those overexpressing IL-19 do not (He and Liang, 2010). Interestingly, all three cytokines have been associated with both pro- and anti-inflammatory functions, depending on context/study (Autieri, 2018, Canto et al., 2014, Caparros and Frances, 2018, Fujimoto et al., 2017, Gough et al., 2017, Horiuchi et al., 2015, Kako et al., 2016, Kragstrup, 2016, Kumar et al., 2018, Matsuo et al., 2015, Myles et al., 2013, Niess et al., 2018, Steinert et al., 2017, Xie et al., 2016). The in vivo roles of IL-19, IL-20 and IL-24 in psoriatic inflammation warrant further investigations.
Here we demonstrate that contrary to prevailing theory, loss of IL-19 did not ameliorate IMQ-induced psoriatic inflammation. Instead, mice lacking IL-20 receptors (IL-20RB-deficient), and thus unable to transmit signals from IL-19, IL-20 and IL-24, exhibited notably increased psoriatic pathology, and in particular, increased numbers of IL-17A-producing γδT cells. Injection of IL-19 reduced excessive dermal infiltration of these cells in an IMQ-treated mutant mouse impaired in IL-19 production by keratinocytes. IL-19/IL-20 receptor signaling capped IL-17A production, comprising a negative feedback. Signaling via IL-20 receptors did so, at least in part, by inhibiting CCL2 chemokine-dependent recruitment of inflammatory cells into the dermis. The inflammatory IL-17 cytokines thus initiate a path to prevent excessive inflammation.
RESULTS
Loss of IL-19 does not ameliorate IMQ-induced psoriatic inflammation
We confirmed that IL-19 was highly induced in IMQ-induced psoriatic inflammation and that its expression was largely dependent on IL-17 cytokines-signaling in keratinocytes. Strongly increased levels of IL-19 were noted in the epidermal ear layer (Figure 1a; left panel) of wild type (WT), but not mutant mice specifically lacking CIKS in keratinocytes (CIKS Δ KC) (Ha et al., 2014); IL-24 was less prominently induced than IL-19, and trended slightly lower in the CIKS DKC mutant mice, while expression of IL-20 was barely detectable (Figure 1a; middle, right panels). Similar results were obtained with dorsal whole skin samples (Supplementary Figure S1a). Prior data in human patients suggested keratinocytes to be the primary source of IL-19 (Romer et al., 2003). We found that IL-17A directly induced IL-19, as seen with primary keratinocyte cultures, the human keratinocyte cell line, HaCaT and with mouse ear skin tissue explants (Figure 1b; left, middle and right panels, respectively).
Figure 1. IL-17 induces IL-19 in keratinocytes.

(a) Relative mRNA expression of IL-20 family cytokines in epidermis of IMQ- or control-treated ears from wild-type (WT) and CIKS DKC [K5-cre;Traf3ip2−/flx] mice. (b) Relative mRNA expression of IL-19 in primary keratinocytes from WT and CIKS KO mice, HaCaT cells and WT ear explants stimulated with IL-17A for 6h (left, middle and right panels, respectively). (*p < 0.05; mean ± SEM; n =5–8, except n=3 for primary CIKS KO keratinocytes).
To investigate the role IL-19 in the IMQ model we made use of IL-19-deficient (KO) mice. Following IMQ treatment, both WT and IL-19 KO mice developed psoriasis-like pathology, including epidermal thickening and infiltration of immune cells into the skin; KO phenotypes tended to be slightly worse (Figure 2a). We noted a mild, albeit not significant increase in the numbers of dermal γδT and monocytic cells in the skin of IMQ-treated IL-19 KO relative to WT mice (Figure 2b). IL-19 was therefore not required to execute pathologic effects of IL-17 in this model, and may have had a mildly protective effect instead. IL-24 expression trended higher in IL-19 KO vs WT mice, which may have had a compensatory effect (Supplementary Figure S1b); this prompted us to investigate the combined action of these cytokines.
Figure 2. IMQ-induced psoriatic inflammation in IL-19 KO mice.

(a) Representative H&E-stained dorsal sections of IMQ-treated WT and IL-19 KO mice. Left graph: Epidermal thickening quantitated via measurements of epidermal area from sections. Scale bar 100 mm. Right graph: Numbers of CD45+ cells in dorsal skin sections. (b) Numbers of dermal gdT cells (CD45+, TCRγδintermediate), IL-17A+ γδT cells and monocytic cells (CD45+ Ly6C+, CD11b+, excluding MHCII−, CD64− cells) based on flow cytometric analyses of IMQ-treated dorsal skin cells from WT and IL-19 KO mice. (for a and b: mean ± SEM; n = 6 mice per group).
Exacerbated IMQ-induced psoriatic inflammation in IL-20 receptors-deficient mice
To delineate the combined contributions of IL-19, IL-20 and IL-24, we made use of IL-20RB-deficient mice (IL-20RB KO), which lack IL-20 receptors. Visual inspection of IMQ-treated WT and IL-20RB KO mice did not show obvious gross differences in psoriasis-like dorsal skin pathologies, but analysis of sections revealed that IL-20RB KO mice exhibited a notably increased thickening of the epidermis (with acanthosis and hyperkeratosis) (Figure 3a; consistent with increased proliferation in basal keratinocytes (Supplementary Figure S2a)). IL-20RB receptor-signaling thus partially ameliorated the epidermal psoriatic phenotype.
Figure 3. Exacerbated psoriatic inflammation in IL-20RB KO mice, including a rise in IL-17A+ γδTcells.
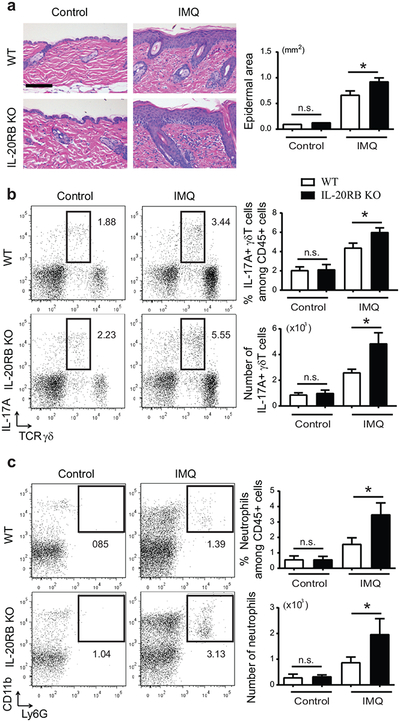
(a) Representative H&E-stained dorsal sections of IMQ- or control-treated WT and IL-20RB KO mice. Scale bar 100 μm. Epidermal thickening quantitated via measurement of epidermal area from sections. (b, c) Representative flow cytometric analyses of IMQ- or control-treated dorsal skin cells from WT and IL-20RB KO mice analyzed for expression of markers as shown within the CD45+ gate. (b) Numbers and percentages of IL-17A+ γδT cells and (c) neutrophils generated from flow cytometric analyses. (for a-c: *p < 0.05; mean ± SEM; n = 8–12 mice per group).
IMQ-treated IL-20RB KO mice showed increased dermal infiltration compared to WT mice (Figure 3a) and we detected a significant increase in the percentage and especially total number of IL-17A+ γδT cells in dorsal skin (Figure 3b). Similarly, we observed more neutrophils (Figure 3c), consistent with increased IL-17A. In addition to IL-17A, IMQ-induced expression of IL-1b mRNA was also augmented in dorsal skin (but not skin draining lymph nodes (sDLNs)) of IL-20RB KO mice (Supplementary Figures S2b,c). IL-β drives IL-17A expression by γδT cells. There were no significant changes in expression of IL-23a or IL-12b in lesional skin of IL-20RB KO compared to WT mice, although IL-12b trended higher (not shown). IL-17A mRNAs and IL-17A+ γδT cells as well as neutrophils were also increased in sDLNs of IL-20RB KO compared to WT mice (Supplementary Figures S2d,e).
IL-19 limits recruitment of IL-17A-producing dermal γδT cells
To assess whether IL-19/IL-20RB signaling could cap accumulation of dermal IL-17A+ γδT cells, we intradermally injected IL-19 into IMQ-treated ears of CIKS Δ KC mice. As shown, IMQ-treatment of CIKS ΔKCs largely failed to generate IL-19, as IL-17 signaling into keratinocytes was blocked; furthermore, these mutant mice exhibited increased cellular infiltration, in particular of IL-17A+ γδT cells (Ha et al., 2014). Injection of CIKS ΔKC mice with IL-19 significantly reduced total numbers of IL-17A+ γδT cells in skin, along with other immune cell infiltrates (Figure 4a). Remarkably, IL-19 injection of the mutants led to an increase of in particular IL-17A+ γδT cells in ear sDLNs (Figure 4b).
Figure 4. IL-19 limits accumulation of IL-17+ dermal γδT cells.

(a, b) Representative flow cytometric analyses for markers shown of cells obtained from IMQ-treated ears (a) and earsDLNs (b) of CIKS ΔKC mice i.d. injected with PBS or IL-19. Numbers and percentages of IL-17A+ γδT cells generated from flow cytometric analyses. (**p < 0.01; mean ± SEM; n = 6–8 mice per group). (c, d) Representative flow cytometric analyses for markers shown of cells obtained from IMQ-treated dorsal skin (c) and sDLNs (d) of WT mice injected i.p. with PBS or FTY720. Numbers and percentages of IL-17A+ γδT cells generated from flow cytometric analyses. (**p < 0.01; mean ± SEM; n = 6–9 mice per group).
This suggested that IL-19 curtailed recruitment of IL-17A+ γδT cells from DLNs into skin following IMQ treatments. To confirm, we administered FTY720, which blocks egress from lymph nodes. In agreement with a prior report (Ramirez-Valle et al., 2015), FTY720 interfered with the IMQ-induced rise in IL-17A+ γδT cells in dorsal skin (Figure 4c), while at the same time retaining and increasing the numbers of these cells in sDLNs, along with other cells (Figure 4d). Ramirez-Valle et al additionally posited that IMQ-induced migration of IL-17A+ γδT cells from sDLNs to skin was dependent on the chemokine receptor CCR2. We confirmed that CCR2 was highly expressed on dermal γδT cells and that IMQ failed to induce accumulation of IL-17A+ γδT cells in mice deficient in CCR2 (Supplementary Figure S3a,b).
IL-19/IL-20RB limits accumulation of IL-17A+ γδT cells via reduction of CCL2
To elucidate mechanisms by which IL-19/IL-20RB signaling caps accumulation of IL-17A+ γδT cells, we focused on CCL2, the primary CCR2 ligand. CCL2 mRNA expression was significantly higher in dorsal skin of IMQ-treated CIKS ΔKC and IL-20RB KO compared to WT mice and trended higher in IL-19 KO mice (Figure 5a). Nearly all CCL2 expression in IL-20RB KO mice occurred in the dermal layer of ear skin (Figure 5b).
Figure 5. IL-19/IL-20 receptor signaling reduces CCL2 and thereby limits accumulation of IL-17A+ dermal γδT.

(a) Relative mRNA expression for CCL2 in IMQ-treated dorsal skin of CIKS ΔKC, IL-20RB KO, IL-19 KO and WT mice (*p < 0.05; mean ± SEM; n = 6–15 mice per group). (b) Relative mRNA expression for CCL2 in epidermis (Epi) and dermis (Derm) of IMQ-treated ears of IL-20RB KO mice. (**p < 0.01; mean ± SEM; n =7). (c, d) Representative flow cytometric analyses of IMQ-treated ear skin cells (c) and ear sDLNs (d) of CIKS KC mice i.d. injected with α-CCL2 or control antibodies, analyzed for expression of markers shown. Numbers and percentages of IL-17A+ γδT cells generated from flow cytometric analyses. (*p < 0.05, **p< 0.01; mean ± SEM; n = 6–8).
To confirm that CCL2 was essential for increased accumulation of dermal IL-17A+ γδT cells in IMQ-treated CIKS DKC mice, we administered neutralizing antibodies to CCL2. Intradermal α-CCL2-injections in ears of CIKS ΔKCs led to significantly lower cellular infiltration into skin, including IL-17A+ dermal γδT cells (Figure 5c). At the same time, α-CCL2 injections increased numbers of IL-17A+ γδT cells in ear sDLNs, similar to IL-19 injections (Figure 5d). Thus IL-19/IL-20RB dampened the IMQ-induced influx of IL-17A+ γδT from sDLNs into skin, at least in part by reducing CCL2 expression.
We investigated CCL2 and IL-19 expression during the course of the IMQ-treatments of WT mice. CCL2 mRNA was strongly induced in dorsal skin by 24h after the first IMQ application, but expression began to return to baseline with subsequent applications. By contrast, IL-19 mRNA induction was delayed, peaking at day 3 of treatment; it gradually fell thereafter, remaining above pretreatment levels at the end of IMQ treatments. These results are consistent with the notion that IL-17-induced IL-19 dampened induction of CCL2 after its rapid initial rise (Figure 6a). Measurements for CCL2 protein in dorsal skin confirmed the early rise and subsequent decline (Figure 6b). CCL2 was primarily produced in the dermis, measured at peak times (Figure 6c). As noted above, expression of CCL2 persisted in IL-20RB KO mice, and originated primarily in the dermis, assessed at the end of treatments.
Figure 6. IL-19 downregulates expression of CCL2 in dermal fibroblast.

(a,b) Relative mRNA expression of CCL2 and IL-19 (a) and CCL2 protein levels (b) in dorsal skin of WT mice during the course of 5 successive IMQ-treatments. (c) Protein levels of CCL2 in one-time IMQ-or control-treated epidermal and dermal layers of ears of WT mice (**p < 0.01; mean ± SEM; n = 6–10 per group). (d) Relative mRNA expression of CCL2 in primary dermal fibroblast cultures of WT and IL-20RB KO mice stimulated (or not) with IFNγ and TNFα in the presence/absence of IL-19 or IL-24, as shown (**p < 0.01; mean ± SEM; n = 10 per group).
Based on these findings we examined for effects of IL-19 and IL-24 on expression of CCL2 in primary dermal fibroblasts, the main cells in the dermis. CCL2 was induced upon stimulation with IFNɣ and TNFα, inflammatory cytokines also present in psoriatic inflammation; addition of IL-19 or IL-24 significantly reduced this induction, dependent on the presence of IL-20RB (Figure 6d). This suggests one mechanism by which IL-19 and IL-24 may impair CCL2 expression.
DISCUSSION
The present study revealed that IMQ-induced psoriatic inflammation was not ameliorated in IL-19-deficient mice and, instead, slightly worsened it. This finding calls into question the oft surmised role of IL-19 as a critical downstream executioner of IL-17A-induced psoriatic pathologies. The most direct evidence for such a role stemmed from in vitro stimulations of keratinocyte cultures and explants, in which IL-19, though largely unable to elicit responses by itself, augmented some effects of IL-17A (Witte et al., 2014). However, whether the augmentation of IL-17A-mediated induction of e.g. anti-microbial proteins by IL-19 noted in the in vitro cultures translates to a pathologic role in psoriatic inflammation in vivo was not addressed. Our data indicate that IL-19 is likely to have a much more nuanced function in psoriatic inflammation, as we demonstrated an anti-inflammatory function. Injection of this cytokine dampened the excessive infiltration of leukocytes - including IL-17A+ γδT- into skin seen in CIKS ΔKC mice. IL-17 cytokines cannot signal into keratinocytes in these mutant mice, and in consequence, IMQ-induced IL-19 production is largely blunted. As shown previously, epidermal pathology was notably ameliorated in these mutants, but not so infiltration of leukocytes into the dermis, which instead was exacerbated compared to WT mice (Ha et al., 2014). As documented here, intradermal injection of these CIKS ΔKC mice with IL-19 largely reversed excessive cellular infiltration, thereby delimiting production of IL-17A by infiltrating dermal γδT cells. Since IL-19 is produced by keratinocytes in response to IL-17A, IL-19 functioned as a negative feedback regulator of IL-17A. Furthermore, the excessive leukocyte infiltration and IL-17A production observed in IMQ-treated CIKS ΔKC mice was thus due, at least in part, to the severe drop in IL-19 in these mutant mice.
Based on these findings, it is not apparent why IL-19 deficient mice did not exhibit exacerbated dermal infiltration in the IMQ model, including increased IL-17A production. This contrasts with CIKS ΔKC mice, in which loss of IL-17-induced IL-19 exacerbated and i.d. injection of IL-19 reversed these pathologic phenotypes. Possibly, mice lacking IL-19 may have partially compensated the loss via IL-24 (IL-20 was barely detectable). This notion is supported by exacerbated psoriatic pathology observed in mice deficient in IL-20 receptors (IL-20RB KO), the receptors required for signaling by all three cytokines. In these mutants, IMQ treatments resulted not only in increased leukocyte infiltration, including IL-17A-producing γδT cells and neutrophils, but also increased epidermal thickening. Furthermore, IL-24 was induced and trended higher in IMQ-treated IL-19 KO compared to WT mice; it also suppressed CCL2 expression, similar to IL-19 (see below). Therefore, the combined action of these IL-20RB ligands exerted an overall anti-inflammatory effect, ameliorating several pathologic phenotypes associated with psoriatic inflammation, in distinction with the view that these cytokines help drive inflammation. It is important to note though that signaling via IL-20 receptors, including response to the highly induced IL-19, did not prevent IMQ-induced pathology, but instead delimited the extent of inflammation. Future research will need to address which receptors and cell types mediate the effects of the IL-20RB ligands and whether they cross-regulate each other.
Our findings demonstrate that IL-19/IL20RB signaling restrained cellular infiltration, at least in part by suppressing CCL2 in skin. CCL2 is the primary ligand for CCR2. It attracts monocytes, neutrophils and is critical for dermal γδT cell recruitment into skin from sDLNs under inflammatory conditions. This stands in contrast with homeostatic conditions, where the CCL20/CCR6 pathway is primarily responsible for recruitment (McKenzie et al., 2017, Ramirez-Valle et al., 2015). (CCL20 expression was not altered in IMQ-treated CIKS ΔKC vs WT [not shown]). Importantly, CCL2 expression was notably elevated in CIKS ΔKC and IL-20RB KO mice, and trended higher in IL-19 KO mice. Intra-dermal administration of IL-19 or CCL2-neutralizing antibodies reduced overall leukocyte infiltration in CIKS DKC mice, including IL-17A+ γδT cells. FTY720 more specifically prevented infiltration of the latter cells in WT mice; they are stored in lymph nodes, unable to egress upon FTY treatment, while monocytes and neutrophils can readily enter from the circulation.
Additional lines of investigation support the view that IL-19/IL-20RB can limit production of CCL2. CCL2 was rapidly induced in WT mice upon initial exposure to IMQ, but expression waned with subsequent exposures, coincident with the delayed rise in IL-19; expression of IL-19 fell only modestly thereafter and remained above baseline. By contrast, CCL2 expression was sustained in IL-20RB KO mice at the end of the IMQ treatments, consistent with loss of IL-19 signaling. We also noted that early IMQ-induced CCL2 production in WT mice was particularly evident in the dermis, as was sustained expression in IL-20RB KO mice. (Dermal CCL2 expression originated almost exclusively from CD45− stromal cells [per cell basis] at the early peak in IMQ-treated WT mice; at the end of treatments, CD45+ cells also contributed, although they constitute only a small fraction of total dermal cells [not shown]). Fibroblasts are the most abundant CD45− cell type in the dermis and we found that IL-19 and IL-24 notably diminished expression of CCL2 induced by TNFα plus IFNγ in primary dermal fibroblast cultures. These inflammatory cytokines are also present in psoriatic inflammation and this may be one mechanism by which IL-19 (and IL-24) signaling via IL20-RB restricts expression of CCL2, although additional mechanisms may exist.
Our findings show that IL-19/IL-20RB signaling does not function as a mere mediator of pathologic consequences to IL-17A in skin, but has more nuanced roles, including specific anti-inflammatory effects, imposing a cap on IL-17A production in the IMQ model. The role of IL-20 receptors-signaling in skin is thus more in line with constraining, rather than eliminating psoriatic inflammation, and helping to promote barrier defenses. Whether such a role could also have long-term detrimental consequences in chronic psoriasis remains an open question. IL-20 receptors-mediated signals are integral to the communication between epidermal, stromal and immune cells in skin.
MATERIALS & METHODS
Mice
Mice strains used: [Il20rb−/−] (Zheng et al., 2008), [Il19−/−] (gifts from Genentech, S. San Francisco, CA), [Traf3ip2flx/flx] and [Traf3ip2−/−] (Pisitkun et al., 2012), [K5-cre; Traf3ip2−/flx] (CIKSΔKC) (Ha et al., 2014), and [Ccr2−/−] (Jackson Laboratories, Bar Harbor, ME) (all C57BL/6). 8–10 weeks-old mice with littermate controls were used. All mice were bred and housed in a NIAID facility, and all experiments were performed with the approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Experimentally induced psoriatic inflammation
Aldara cream containing 5% Imiquimod was applied to shaved dorsal skin or to ears for 5 consecutive days, as described (Ha et al., 2014, van der Fits et al., 2009). Mice were harvested on day 6. In some experiments mice were intra-dermally injected with IL-19 (1ug/ml) (or PBS) or with α-CCL2 (10ug/ml) (or control IgG) on days 3 to 5 just prior to IMQ applications (reagents from R&D Systems, Minneapolis MN). FTY720 (Cayman Chemical, Ann Arbor, MI; 1mg/kg) was injected i.p. every other day.
Cellular analysis
Lymph nodes were mechanically dissociated to obtain single-cell suspensions (Pisitkun et al., 2010). For separation of dermal and epidermal layers, ears were split into dorsal and ventral halves, cartilage and fat was removed and halves were floated dermal side down in a 0.5 M ammonium thiocyanate solution (Sigma, St. Louis MO), incubated at 37C for 20 min, washed in PBS, then layers were separated with forceps. Single-cell suspensions from dorsal skin were prepared as described (Ha et al., 2014) and stained with Aqua (Invitrogen, Carlsbad, CA) and antibodies against one or more of the following proteins: IL-17A, Ly6C(AL 21) (BD Biosciences, San Jose, CA); TCRγδ(UC7–13D5), IL-17A(eBio17B7), and IL-17F(eBio18F10) (eBioscience, San Diego, CA); Ly6G (IA8), CD45.2(104), TCRγδ(GL3), CD11b(M1/70), TCRvγ4(UC3–10A6), MHCII(M5/114.15.2) and CD64(X54–5/7.1) (Biolegend, San Diego, CA); CCR2 (R&D Systems, Minneapolis, MN). For intracellular staining, cells were treated with cell stimulation cocktail (plus protein transport inhibitors) (eBioscience, San Diego, CA) for 4 hr. Data were collected with FACSCanto and FACSCelesta (BD Biosciences, San Jose, CA) and analyzed using FlowJo (Tree Star, Ashland OR).
Histology, tissue analysis
Mouse dorsal skin tissues were fixed in 4% formaldehyde, stained with H&E and visualized with an Olympus BX50. Epidermal areas were quantitated on H&E stained slides from multiple mice with ImageJ software (NIH). Sections were prepared for immunofluorescence as described (Ha et al., 2014) and stained with primary antibodies against K5 (1:100; Lifespan Biosciences) and Ki67 (1:100; BD Pharmingen). Secondary antibodies were labeled with guinea pig IgG (Alexa Fluor 546) and mouse IgG (Alexa Fluor 488) (1:1000; Molecular Probes, Eugene OR). Slides were mounted with Vectashield without DAPI (Vector Labs) and visualized with a Leica AF6000LX fluorescence microscope. To prepare protein extracts from skin, frozen tissue sections were homogenized with a protease inhibitor cocktail (Roche, Basel Switzerland) in PBS; extracts were analyzed for CCL2 with Elisa assay kit (R&D Systems, Minneapolis, MN).
In vitro cultures
Isolation of keratinocytes and fibroblasts from neonatal mice and their culture conditions were as reported (Ha et al., 2014). HaCaT cells were obtained from Dr. Maria Morasso (NIH). For ear explants, ears were washed with betadine solution, split to remove cartilage and fat and then tissue was placed dermal side down in complete culture media in the presence of absence of cytokine overnight. Cultures were stimulated as indicated with one or more of the following cytokines: TNFα (20 ng/mL), IFNγ (10 ng/mL) (PeproTech, Rocky Hill NJ); IL-17A (100 ng/mL), IL-19 (100 ng/mL) (R&D Systems, Minneapolis MN).
Quantitative real-time PCR
RNA was purified using TRIzol (Invitrogen) and RNeasy kit (Qiagen); cDNA was generated with cDNA synthesis kit (Qiagen), and quantitative real-time PCR was performed (Taqman protocol). The mouse primers for Gapdh, Il19, Il20, Il24, Ccl2, Il1a, Il1b, Il17a, Il17f, Il22, and Cxcl1 were obtained from Applied Biosystems (Foster City CA). All values were normalized to Gapdh.
Statistical analyses
All data are presented as the mean ± SEM. Student’s t test (two-tailed) was used to evaluate significance; p values <0.05 were considered to be statistically significant, and values <0.01 highly significant.
Supplementary Material
ACKNOWLEGEMENTS
We greatly appreciate the constructive inputs provided by members of the Siebenlist laboratory. This research was supported by the Intramural Research Programs of NIAID, NIH.
Abbreviations:
- sDLN
skin-draining lymph node
- IMQ
Imiquimod
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA AVAILABILITY STATEMENT
No datasets were generated or analyzed during the current study.
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Autieri MV. IL-19 and Other IL-20 Family Member Cytokines in Vascular Inflammatory Diseases. Front Immunol 2018;9:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonnette R, Fuentes-Duculan J, Mashiko S, Li X, Bonifacio KM, Cueto I, et al. Palmoplantar pustular psoriasis (PPPP) is characterized by activation of the IL-17A pathway. J Dermatol Sci 2017;85(1):20–6. [DOI] [PubMed] [Google Scholar]
- Boehncke WH, Schon MP. Psoriasis. Lancet 2015;386(9997):983–94. [DOI] [PubMed] [Google Scholar]
- Cai Y, Shen X, Ding C, Qi C, Li K, Li X, et al. Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 2011;35(4):596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto E, Garcia Planella E, Zamora-Atenza C, Nieto JC, Gordillo J, Ortiz MA, et al. Interleukin-19 impairment in active Crohn’s disease patients. PLoS One 2014;9(4):e93910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparros E, Frances R. The Interleukin-20 Cytokine Family in Liver Disease. Front Immunol 2018;9:1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol 2009;182(3):1617–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto Y, Azuma YT, Matsuo Y, Kuwamura M, Kuramoto N, Miki M, et al. Interleukin-19 contributes as a protective factor in experimental Th2-mediated colitis. Naunyn Schmiedebergs Arch Pharmacol 2017;390(3):261–8. [DOI] [PubMed] [Google Scholar]
- Gough P, Ganesan S, Datta SK. IL-20 Signaling in Activated Human Neutrophils Inhibits Neutrophil Migration and Function. J Immunol 2017;198(11):4373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha HL, Wang H, Pisitkun P, Kim JC, Tassi I, Tang W, et al. IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc Natl Acad Sci U S A 2014;111(33):E3422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Liang P. IL-24 transgenic mice: in vivo evidence of overlapping functions for IL-20, IL-22, and IL-24 in the epidermis. J Immunol 2010;184(4):1793–8. [DOI] [PubMed] [Google Scholar]
- Horiuchi H, Parajuli B, Wang Y, Azuma YT, Mizuno T, Takeuchi H, et al. Interleukin-19 acts as a negative autocrine regulator of activated microglia. PLoS One 2015;10(3):e0118640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kako F, Gabunia K, Ray M, Kelemen SE, England RN, Kako B, et al. Interleukin-19 induces angiogenesis in the absence of hypoxia by direct and indirect immune mechanisms. Am J Physiol Cell Physiol 2016;310(11):C931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kragstrup TW. The IL-20 receptor axis in immune-mediated inflammatory arthritis: novel links between innate immune recognition and bone homeostasis. Scand J Rheumatol 2016;45(sup128):53–7. [DOI] [PubMed] [Google Scholar]
- Kumar NP, Moideen K, Banurekha VV, Nair D, Babu S. Modulation of Th1/Tc1 and Th17/Tc17 responses in pulmonary tuberculosis by IL-20 subfamily of cytokines. Cytokine 2018;108:190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos TR, O’Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. The Journal of clinical investigation 2017;127(11):4031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Azuma YT, Kuwamura M, Kuramoto N, Nishiyama K, Yoshida N, et al. Interleukin 19 reduces inflammation in chemically induced experimental colitis. Int Immunopharmacol 2015;29(2):468–75. [DOI] [PubMed] [Google Scholar]
- McKenzie DR, Kara EE, Bastow CR, Tyllis TS, Fenix KA, Gregor CE, et al. IL-17-producing gammadelta T cells switch migratory patterns between resting and activated states. Nat Commun 2017;8:15632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 2012;11(10):763–76. [DOI] [PubMed] [Google Scholar]
- Monin L, Gaffen SL. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb Perspect Biol 2018;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles IA, Fontecilla NM, Valdez PA, Vithayathil PJ, Naik S, Belkaid Y, et al. Signaling via the IL-20 receptor inhibits cutaneous production of IL-1beta and IL-17A to promote infection with methicillin-resistant Staphylococcus aureus. Nat Immunol 2013;14(8):804–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niess JH, Hruz P, Kaymak T. The Interleukin-20 Cytokines in Intestinal Diseases. Front Immunol 2018;9:1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Rutz S, Crellin NK, Valdez PA, Hymowitz SG. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol 2011;29:71–109. [DOI] [PubMed] [Google Scholar]
- Papotto PH, Reinhardt A, Prinz I, Silva-Santos B. Innately versatile: gammadelta17 T cells in inflammatory and autoimmune diseases. J Autoimmun 2018;87:26–37. [DOI] [PubMed] [Google Scholar]
- Patel DD, Kuchroo VK. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015;43(6):1040–51. [DOI] [PubMed] [Google Scholar]
- Pisitkun P, Claudio E, Ren N, Wang H, Siebenlist U. The adaptor protein CIKS/ACT1 is necessary for collagen-induced arthritis, and it contributes to the production of collagen specific antibody. Arthritis Rheum 2010;62(11):3334–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun P, Ha HL, Wang H, Claudio E, Tivy CC, Zhou H, et al. Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 2012;37(6):1104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 2011;12(12):1159–66. [DOI] [PubMed] [Google Scholar]
- Ramirez-Valle F, Gray EE, Cyster JG. Inflammation induces dermal Vgamma4+ gammadeltaT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc Natl Acad Sci U S A 2015;112(26):8046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer J, Hasselager E, Norby PL, Steiniche T, Thorn Clausen J, Kragballe K. Epidermal overexpression of interleukin-19 and −20 mRNA in psoriatic skin disappears after short term treatment with cyclosporine a or calcipotriol. J Invest Dermatol 2003;121(6):1306–11. [DOI] [PubMed] [Google Scholar]
- Steinert A, Linas I, Kaya B, Ibrahim M, Schlitzer A, Hruz P, et al. The Stimulation of Macrophages with TLR Ligands Supports Increased IL-19 Expression in Inflammatory Bowel Disease Patients and in Colitis Models. J Immunol 2017;199(7):2570–84. [DOI] [PubMed] [Google Scholar]
- van der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 2009;182(9):5836–45. [DOI] [PubMed] [Google Scholar]
- Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, et al. IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol 2014;134(11):2757–67. [DOI] [PubMed] [Google Scholar]
- Xie W, Fang L, Gan S, Xuan H. Interleukin-19 alleviates brain injury by anti-inflammatory effects in a mice model of focal cerebral ischemia. Brain Res 2016;1650:172–7. [DOI] [PubMed] [Google Scholar]
- Xu M, Lu H, Lee YH, Wu Y, Liu K, Shi Y, et al. An Interleukin-25-Mediated Autoregulatory Circuit in Keratinocytes Plays a Pivotal Role in Psoriatic Skin Inflammation. Immunity 2018;48(4):787–98 e4. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 2008;14(3):282–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.