

Abstract
Exposure of biological molecules to oxidants is inevitable and therefore commonplace. Oxidative stress in cells arises from both external agents and endogenous processes that generate reactive species, either purposely (e.g. during pathogen killing or enzymatic reactions) or accidentally (e.g. exposure to radiation, pollutants, drugs, or chemicals). As proteins are highly abundant and react rapidly with many oxidants, they are highly susceptible to, and major targets of, oxidative damage. This can result in changes to protein structure, function, and turnover and to loss or (occasional) gain of activity. Accumulation of oxidatively-modified proteins, due to either increased generation or decreased removal, has been associated with both aging and multiple diseases. Different oxidants generate a broad, and sometimes characteristic, spectrum of post-translational modifications. The kinetics (rates) of damage formation also vary dramatically. There is a pressing need for reliable and robust methods that can detect, identify, and quantify the products formed on amino acids, peptides, and proteins, especially in complex systems. This review summarizes several advances in our understanding of this complex chemistry and highlights methods that are available to detect oxidative modifications—at the amino acid, peptide, or protein level—and their nature, quantity, and position within a peptide sequence. Although considerable progress has been made in the development and application of new techniques, it is clear that further development is required to fully assess the relative importance of protein oxidation and to determine whether an oxidation is a cause, or merely a consequence, of injurious processes.
Keywords: oxidative stress, oxygen radicals, post-translational modification, protein chemical modification, protein cross-linking, carbonyl, disulfide, hydroperoxide, protein chemical modification, reactive oxygen species
Introduction
Biological systems are exposed to a wide variety of oxidizing species—both free radicals and two-electron oxidants. These species are often termed “reactive oxygen species,” although this is a misleading term, as the reactivity of these species varies enormously (see below). Oxidants are generated both deliberately (e.g. to kill invading pathogens or as intermediates in enzymatic reactions) or unintentionally (e.g. via electron leakage from electron transport chains, metabolism of drugs, exposure to chemicals, pollutants, and radiation). These processes have been reviewed in Refs. 1, 2.
The formation of these oxidants and their reactions are limited by cellular and organismal defense systems, which include enzymes that remove oxidants or oxidant precursors (e.g. superoxide dismutases, peroxiredoxins, thioredoxin/thioredoxin reductase, GSH peroxidase isoforms, and catalases), and water- and lipid-soluble oxidant scavengers, including thiols (e.g. GSH and thioredoxin), ascorbic acid (vitamin C), urate, tocopherol isoforms (vitamin E), quinols (e.g. reduced coenzyme Q10), carotenoids, and polyphenols. Although these systems are efficient, and in many cases show redundancy (i.e. multiple processes remove the same species), they are not 100% effective, and a large body of data indicates that biological targets suffer resulting damage. These protective systems are therefore complemented by systems that either repair damage or remove the modified molecules (e.g. methionine sulfoxide reductases, disulfide reductases/isomerases, glutaredoxins, sulfiredoxins, proteasomes, lysosomes, proteases, phospholipases, and DNA repair enzymes) (1, 2).
Despite this battery of preventative and repair systems, many studies have reported increased damage, and accumulation of this, in human, animal, and microbial and plant systems exposed to stress conditions (reviewed in Refs. 1, 2). A higher level of damage may arise from increased oxidant generation, a decrease or failure of defense systems, or (most commonly) a mixture of both processes, as many defense systems are themselves subject to damage or show reduced activity due to co-factor depletion. This concept of an altered balance between formation and removal gave rise to the term “oxidative stress” (2), although it is now apparent that this is an oversimplification of a complex picture, as limited stress (“eustress”) can be beneficial in priming and protecting a system against greater damage (“distress”) (2). Increasing age is often associated with a decrease in enzyme levels or activity, and in some cases decreased levels of co-factors and essential trace elements, such that increased levels of oxidants and modified products are formed (1, 2). These changes can be accelerated by disease or environmental factors, despite the presence of feedback loops (e.g. antioxidant-response elements, including the Nrf-2 pathway; DNA damage–response element; OxyR; SoxRS) that up-regulate the synthesis of protective species (1, 2). In this study, we review the basic chemistry and biochemistry of protein modification by oxidants, with a focus on methods available for the detection, identification, and quantification of these changes.
Proteins as targets of oxidative damage
Proteins are major components of most biological systems and constitute ∼70% of the dry mass of cells and tissues. The rate of reaction of an oxidant with a biological target depends on the concentration of the target, multiplied by the rate constant for its reaction with the oxidant. Both of these factors result in proteins being major targets for damage as proteins are both present at high concentrations (up to 1–3 mm in plasma and 5–10 mm in cells) and have high rate constants for reaction with oxidants. Thus, oxidant damage in most biological systems is likely to be skewed toward protein modification (3, 4). This is clearly an oversimplification of a complex situation, as other factors are known to play an important role, including localization of the generating system relative to the target and particularly membrane barriers, micro-environments, binding, or association of the oxidant system to a target, the occurrence of secondary reactions, and intra- and intermolecular transfer reactions (3, 4). However, it is likely that proteins are major sites of damage in many situations, although it should also be noted that the extent of damage and its biological importance may be very different (3, 4). Thus, low levels of modification of a critical target may have much greater consequences than a high level of damage to noncritical materials.
Radicals (e.g. HO•, CO3˙̄, NO2·, ROO•, RO•, R•, and many others), two-electron oxidants (e.g. peroxides, 1O2, O3, ONOOH, HOCl, and related species), and metal–oxo complexes can all modify proteins, although the reactivity and selectivity of these oxidants are highly variable (3, 4). Reactions of secondary products (e.g. aldehydes, quinones, and dehydroalanine) are a further source of modifications (5, 6). Together, these generate a wide variety of post-translational modifications that alter amino acid and protein composition and structure, charge, hydrophobicity/hydrophilicity, folding, and function (3, 4, 7, 8).
Nature and reactivity of oxidant species
In the following section, a brief overview is presented on the formation and reactivity of a number of key oxidant species relevant to mammalian systems. Although each of these species is described as single entities, it should be noted that nearly all of these species undergo further interconversion reactions as illustrated in Fig. 1 (although this is situation-dependent) that result in complex mixtures in most reaction systems (3, 4).
Figure 1.
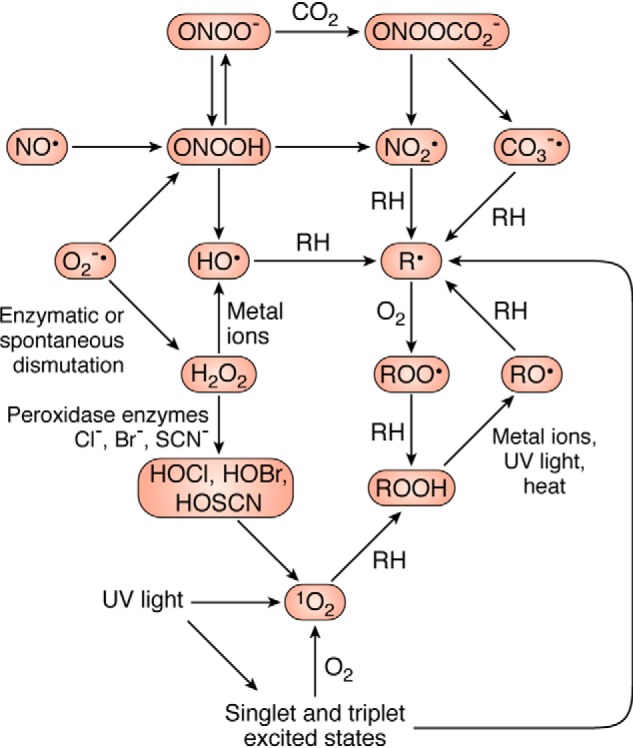
Overview of interconversion processes of common biological oxidants. The extent of these reactions depends on the circumstances and reaction conditions and is therefore only intended as guide to the complexity of examining oxidant reactions. Adapted from Ref. 4.
Superoxide radical-anions (O2˙̄)
Superoxide O2˙̄ radicals are generated continuously by most organisms as a result of the use of O2 as a terminal electron acceptor in some electron transport chains, such as those of mitochondria, the endoplasmic reticulum cytochrome P450 system, and the plasma membrane NADH/NADPH oxidase systems (1, 2). Incomplete coupling of electron transfer results in single electron leakage to O2 (estimated at 1–3%) (1). O2˙̄ is poorly reactive, with iron–sulfur complexes a significant target (9). O2˙̄ undergoes rapid spontaneous, and superoxide dismutase-catalyzed, disproportionation to give hydrogen peroxide (H2O2) and O2, with the former being a precursor of further oxidizing species, as well as being a reactive species in its own right (10). O2˙̄ is generated, at very high fluxes over short time periods, by membrane-associated NADPH oxidases (NOXs and DUOXs), at the expense of NADPH via an “oxidative burst” (11). O2˙̄ is also formed by heme proteins (cytochromes, oxyhemoglobin, and oxymyoglobin) (12), uncoupled nitric-oxide synthase, and xanthine oxidase, among others (1).
Hydrogen peroxide (H2O2)
In addition to formation from O2˙̄ by dismutation, H2O2 is also generated directly by a number of enzymes (e.g. NADPH oxidase-4, monoamine oxidases, hexose oxidases, amino acid oxidases, other oxidoreductases, protein-disulfide isomerases, and Ero1p during protein synthesis and folding (2, 13)). Direct oxidation of biological targets by H2O2 is both limited in extent and is usually slow. Thus, direct reaction is limited to Cys, selenocysteine, and Met residues with these typically occurring with very low rate constants (see also below), with the exception of reaction with some specialized enzymes (e.g. peroxiredoxins and GSH peroxidases), which have catalytic centers that facilitate rapid O–O bond cleavage. Such environments can elevate the rate constant for reaction by a million-fold (2, 13). H2O2 is a substrate for a large family of peroxidase enzymes, with these reactions used both synthetically (e.g. in the formation of thyroid hormones by thyroid peroxidase and in the generation of collagen matrices by peroxidasins) and to kill invading pathogens (2, 13).
Hypohalous acids and other reactive halogen species
Reaction of H2O2 with heme peroxidases, such as myeloperoxidase, eosinophil peroxidase, and lactoperoxidase, results in the formation of hypohalous acids (HOX, X = Cl, Br, I, or SCN) (14). These vary markedly in reactivity and oxidizing capacity, with hypochlorous acid (HOCl, familiar to many as the active component in household bleach) being the most reactive and powerful oxidant (15, 16). HOCl (which exists in equilibrium with Cl2 at low pH values) is a key component of the innate immune response against pathogens, with this generated at high concentrations in neutrophil phagolysosomes (17). Release of myeloperoxidase to the extracellular space (instead of into phagolysosomes) and subsequent reaction with H2O2 can, however, result in host tissue damage, with the level and activity of myeloperoxidase associated with tissue damage in acute and chronic inflammatory conditions (17). Comparison of HOCl and H2O2 illustrates a key point about oxidants and their reactivity: H2O2 is a strong oxidant (reduction potential 1.32 V) and a more powerful oxidant than HOCl (reduction potential 1.28 V), but this reacts very slowly when compared with HOCl (cf. the rate constant, k, for reaction of HOCl with the amino acid methionine is ∼108-fold higher than for H2O2 (4, 15)), so evolution has favored the use of HOCl over H2O2, for kinetic rather than thermodynamic reasons. The low reactivity of H2O2 is likely to be one of the major reasons why this species is used biologically as a messenger molecule: slow and selective reactivity allows for specificity in transmission of messages as only a limited number of select targets are likely to be activated. Highly-reactive species such as HOCl would not allow such specificity of transfer of information.
Nitric oxide (NO•)
NO• is a key vascular regulator and messenger molecule, with this generated from arginine by nitric-oxide synthase enzymes (NOSs)2 (18). The concentration generated by constitutive NOS isoforms is low (pico- to nanomolar), consistent with a regulatory function, but higher levels (up to micromolar) are formed by the inducible NOS isoform of macrophages, at sites of inflammation (19). NO• reacts slowly with most biological targets, consistent with its role as a signaling molecule (18, 20, 21), with fast reactions occurring primarily with transition metal ions (e.g. the iron center of heme proteins, including that of soluble guanylate cyclase, the major effector of NO• signaling) and with other radicals (18, 22). Reaction with other radicals can be protective (e.g. by terminating lipid peroxidation chain reactions) but also damaging when the product is a powerful oxidant, as is seen in the formation of peroxynitrous acid (19).
Peroxynitrous acid (ONOOH)
Diffusion-controlled (i.e. k 109–1010 m−1 s−1) reaction of NO• with O2˙̄ gives ONOO−(peroxynitrite). This species exists in equilibrium with the corresponding conjugate acid peroxynitrous acid (ONOOH), with the pKa (6.8) favoring the anion at most biological pH values (19). However, the acid (ONOOH) form is typically the more reactive species with protein targets. Reaction with CO2 and some boronic acid probes are exceptions, with the rate constant for these targets being higher with ONOO− (19). The reactivity of ONOOH is therefore modulated by CO2 (which is in equilibrium with HCO3−) as a result of the formation of ONOOCO2−, which decomposes to give CO3˙̄ and NO2· that can either diffuse out of the solvent cage or recombine (reviewed in Ref. 19). ONOOH is a potent oxidizing/nitrating species (19) that can give rise to both two-electron and one-electron oxidation products. The former arises from direct reactions of ONOOH, and the latter is formed from limited homolysis to give HO• and NO2·, and subsequent radical chemistry (19).
Hydroxyl (HO•) and other oxygen-centered radicals
Metal ion (primarily Fe and Cu)-catalyzed decomposition of H2O2 generates the highly-reactive and powerful oxidant HO• via Fenton and pseudo-Fenton reactions (23); this species is also formed directly from water by high-energy radiation (24). Metal ion–catalyzed decomposition of organic and lipid hydroperoxides gives alkoxyl radicals (RO•) that are less powerful oxidants than HO•, but more reactive than peroxyl radicals (ROO•) that are typically generated from rapid (typically diffusion-controlled) addition of O2 to carbon-centered radicals (R•) (4). The latter arise from hydrogen atom abstraction from biological targets by reactive radicals (24). R• and ROO• are key intermediates in lipid peroxidation chain reactions, i.e. typically initiated by hydrogen abstraction from methylene groups of polyunsaturated fatty acids. ROO• are the key chain carriers in lipid peroxidation (25), but ROO• also appear to play a role in (short) chain reactions on proteins (26).
UV light, singlet oxygen (1O2), and other photochemically-generated species
UV light with very short wavelengths (UVC) is strongly absorbed by atmospheric molecules, such as ozone, and hence does not give rise to significant effects at the earth's surface. In contrast, longer wavelength UV light, particularly UVB (λ 280–320 nm) and UVA (λ 320–400 nm) wavelengths, can oxidize molecules via light absorption by suitable chromophores and generation of either excited state species (i.e. species with an electron in a higher orbital, type 2 photoreactions) or radicals (type 1 photoreactions), as a result of photoejection of an electron (27–29). The excited state species (usually triplet species due to the short lifetime of singlet states) can either induce direct oxidation by, for example, electron or hydrogen atom abstraction or by energy transfer to molecular oxygen O2 to give singlet oxygen (1O2) (27–29). In addition to its formation by type 2 photoreactions, 1O2 is also formed by some enzyme-catalyzed reactions, via termination reactions of ROO•, and some metal ion-catalyzed processes; 1O2 is therefore an important intermediate in both light-induced and “dark” reactions (28, 29).
Negative and positive aspects of oxidative damage
Oxidant species can generate damage to all components of biological systems, including lipids, proteins, and DNA (1, 2, 4), and these modifications have been linked to a wide range of pathologies, including some cancers, neurological disorders (Alzheimer's and Parkinson's diseases and amyotrophic lateral sclerosis), sepsis, hypertension, cardiovascular diseases, including atherosclerosis, ischemia-reperfusion injury to multiple organs, renal and ocular damage, cataracts, chronic obstructive pulmonary disease, cystic fibrosis, asthma, rheumatoid and osteoarthritis, motor neuron disease, irritable bowel syndrome, pancreatitis, hepatitis, sunburn and photodamage, and many more (1, 2, 30). Although it is likely that some of these are only associations (i.e. oxidation is not causative but merely a consequence of other injurious processes), oxidative damage appears, at least in some cases, to be a contributing factor (i.e. causative). Methodologies that can detect and quantify oxidative damage are therefore of considerable importance, not least as potential biomarkers to assess therapeutic strategies. Oxidation can also be a valuable tool in the treatment of some diseases, with radiation and photodynamic therapy of tumors (31–33), the use of redox-cycling drugs (e.g. doxorubicin and nitroaromatics (34)), and photochemical tissue bonding in wound closure after surgery being good examples (35). The following section outlines current knowledge of the modifications arising from oxidant interactions with proteins, and subsequently how these can be detected and quantified.
Protein modifications induced by reactive oxidants
The majority of radical reactions with proteins occur via three major pathways— hydrogen atom abstraction from C–H, S–H, N–H, or O–H bonds, electron abstraction from electron-rich sites, and addition to electron-rich centers (aromatic rings and sulfur species) (36). The first of these reactions results in the formation of carbon-centered species (R•), thiyl radical (RS•), nitrogen-centered species (primarily indolyl radicals from Trp), and oxygen-centered radicals (primarily phenoxyl radicals from Tyr). Most R• radicals, including those generated from oxidation of aliphatic side chains (Leu, Ile, Val, and Pro, etc.) react rapidly with O2 to give ROO• at diffusion-controlled rates (k ∼109 m−1 s−1) (4). Although these reactions are fast, the O2 concentration is low in most biological samples (5–100 μm), which may limit ROO• formation and result in dimers (R–R) when the R• concentration is relatively high (Fig. 2) (37). In contrast, RS•, Tyr phenoxyl radicals, and Trp indolyl radicals react with O2 with much lower rate constants (k <107 m−1 s−1 for RS•, ≪105 m−1 s−1 for TrpN•, and <103 m−1 s−1 for TyrO• (38–40)), resulting in higher yields of cross-linked products, including disulfides (RSSR), Trp–Trp, and Tyr–Tyr (41). Crossed dimers (e.g. Tyr–Trp) are also known (41, 42). The chemical structures of some of the most abundant and commonly examined products are given in Fig. 3.
Figure 2.
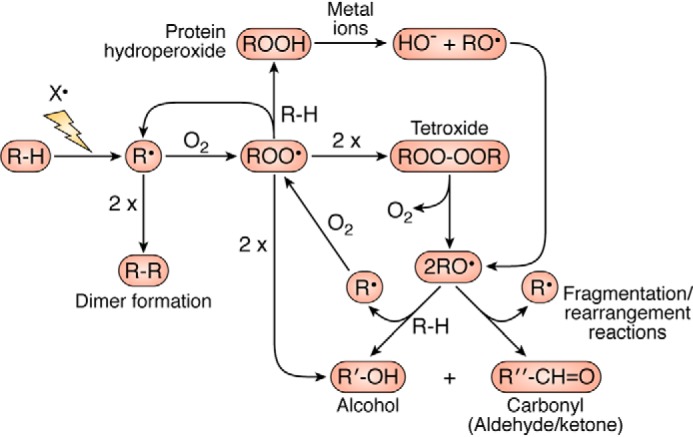
Summary of O2-dependent reactions of carbon, peroxyl, and alkoxyl radical reactions on proteins and the occurrence of short-chain reactions. In this scheme, R, R′, and R″ are used to designate carbon-centered species with different chemical structures.
Figure 3.

Chemical structures of some of the most abundant and/or commonly examined side-chain oxidation products.
For ROO• generated on aliphatic side chains, hydrogen-atom abstraction is a major reaction with other X–H bonds, with this resulting in hydroperoxide (ROOH) formation (Fig. 2). These are major products with multiple different attacking species (4, 43). Dimerization reactions of ROO• have also been characterized, but these appear to be of modest importance, particularly when the radical flux is low (4). When this does occur, and the ROO• is a tertiary species (e.g. on Val and Leu), a tetroxide is generated (ROOOOR) that can decompose to give two alkoxyl radicals (RO•) and O2 (Fig. 2) (44, 45). The RO• can subsequently undergo β-scission fragmentation reactions, to give a carbonyl compound and a further radical (Fig. 2) (46, 47). These processes may be partly responsible for the occurrence of (short) chain reactions on proteins and the generation of protein-bound (or released) carbonyls (46, 47). Alternatively, RO• may undergo hydrogen atom abstraction radicals to generate alcohols (Fig. 2). With primary or secondary ROO•, dimerization reactions yield 1 mol of alcohol, carbonyl, and O2 (Fig. 2) (44). The major products from radical-mediated oxidation of aliphatic side chains are therefore hydroperoxides, alcohols, carbonyls, and fragmentation products.
Highly-reactive radicals such as HO• react with little selectivity (48), except when these are generated in a site-specific manner, such as by bound metal ions, with this resulting in damage localized to the vicinity of the binding site (49). Other less-reactive radicals can show marked selectivity. As most biologically-important radicals are electrophilic, they react most rapidly, and to the greatest extent, with electron-rich sites (4) resulting in damage to a subset of amino acid side chains; these data are summarized in Table 1. These include the sulfur-containing amino acids Cys, Met, and cystine, and the aromatic residues, Trp, Tyr, Phe, and His. For both Cys and His, the reactions are pH-dependent, with the rate of oxidation occurring more rapidly at higher pH values (4). In the case of Cys, a wide range of products can be formed, including disulfides and oxyacids (50–52). With Met, the major product is usually the sulfoxide (53), and to a much lesser extent the sulfone and carbon-centered radical products (53). For the aromatic amino acids, hydroxylated and dimeric species predominate, although with both Trp and His multiple ring opened products are generated (36, 54). A (nonexhaustive) list of products arising from such reactions is given in Table 2.
Table 1.
Overview of selectivity of different oxidizing species for protein side chains
These data are approximations, as the reactivity of both the oxidant and the side chains are environment- and pH-dependent: reactions at Cys, His, and selenocysteine (Sec) are particularly pH-dependent, with greater reactivity observed with the ionized species. The pKa values of these residues are also environment-dependent and can vary significantly. These data are only an approximate indication of major sites of damage.
| Major targets | |
|---|---|
| Radical oxidants | |
| Hydroxyl radical: HO• | All, including peptide backbone |
| Alkoxyl radical: RO• | All, but slower reactions than with HO• |
| Peroxyl radical: ROO• | Cys, Met, Trp, Tyr |
| Superoxide radical anion: O2˙̄ | Fe-S clusters |
| Carbonate radical anion: CO3˙̄ | Cys, Met, Trp, Tyr, His |
| Nitrogen dioxide radical: NO3˙̄ | Cys, Tyr, Trp (and Tyr/Trp radicals) |
| Two-electron (non-radical) oxidants | |
| Peroxynitrous acid: ONOOH | Cys, cystine, Met, Trp, selenomethionine (Sec) |
| UVB light (λ 280–320 nm) | Trp, Tyr, cystine |
| UVA light (λ 320–400 nm) | No direct amino acid absorption |
| Singlet oxygen: 1O2 | Cys, Met, Trp, Tyr, His, Sec |
| Hypochlorous acid: HOCl | Cys, Met, cystine, His, α-amino, Lys, Trp, Tyr (slow), Sec, selenomethionine, Arg (slow) |
| Hypobromous acid: HOBr | Cys, Met, His, cystine, α-amino, Lys, Trp, Tyr, Sec |
| Hypothiocyanous acid: HOSCN | Cys, Sec |
| Hydrogen peroxide (H2O2) and other peroxides | Cys, Sec, Met (slow), selenomethionine |
| Quinones and aldehydes | Cys, Sec, Lys, Arg |
Table 2.
Selected major products (both stable and unstable) generated from oxidation of protein side chains
Products generated from the free amino acids and species formed from glycation/glycoxidation reactions are not included. This list is likely to be incomplete as the material balance (material lost versus products identified) is poor in most cases, indicating the probable presence of additional species. For details on the origins of these species and references, see the text. The chemical structures of some of the most abundant, or commonly examined species are given in Fig. 3.
| Amino acid side chain | Three-letter code | Single-letter code | Products |
|---|---|---|---|
| Alanine | Ala | A | Serine |
| Dehydroalanine | |||
| Lanthionine (dehydroalanine–Cys cross-link) | |||
| Lysinoalanine (dehydroalanine–Lys cross-link) | |||
| Hydroperoxide | |||
| Carbonyl (serine aldehyde, 2-oxo species) | |||
| Arginine | Arg | R | Hydroperoxides |
| Alcohols | |||
| Carbonyls (glutamic semi-aldehyde and others) | |||
| His–Arg cross-links | |||
| Asparagine | Asn | N | Hydroperoxides |
| Aspartic acid | Asp | D | Hydroperoxides |
| Carbonyl | |||
| Decarboxylated species (serine aldehyde) | |||
| Cysteine | Cys | C | Cystine (disulfide) |
| Sulfenic acid: RSOH | |||
| Sulfinic acid: RSO2H | |||
| Sulfonic acid: RSO3H | |||
| Sulfenamide (sulfenyl amide): RS-NHR′ | |||
| Sulfinamide (sulfinyl amide): RSO-NHR′ | |||
| Sulfonamide (sulfonyl amide): RSO2-NHR′ | |||
| Nitrosocysteine: RSNO | |||
| Nitrocysteine: RSNO2 | |||
| Sulfenylchloride: RSCl | |||
| Persulfides: R(S)nH | |||
| Multiple adducts to α-,β-unsaturated aldehydes, aldehydes, and quinones | |||
| Lanthionine (dehydroalanine-Cys cross-link) | |||
| His–Cys cross-link | |||
| Thioethers (addition products) | |||
| Glutamic acid | Glu | E | Hydroperoxides |
| Alcohols | |||
| Carbonyls (3-oxo species) | |||
| Decarboxylated species (aspartate semi-aldehyde) | |||
| Glutamine | Gln | Q | Hydroperoxides |
| Carbonyls (3-oxo species) | |||
| Glycine | Gly | G | Hydroperoxide |
| Histidine | His | H | Hydroperoxides and endoperoxides |
| Hydroxyhistidine | |||
| 2-Oxohistidine | |||
| Nitrohistidine | |||
| Aspartyl urea (ring-opened product) | |||
| Formyl asparagine (ring-opened product) | |||
| Asparagine (ring-opened product) | |||
| Aspartic acid (ring-opened product) | |||
| Di-histidine cross-link (His–His) | |||
| His–Cys cross-link | |||
| His–Lys cross-link | |||
| His–Arg cross-link | |||
| Isoleucine | Ile | I | Hydroperoxides |
| Alcohols (hydroxyisoleucines) | |||
| Carbonyls (3- and 4-oxo species) | |||
| Leucine | Leu | L | Hydroperoxides |
| Alcohols (hydroxyleucines) | |||
| Carbonyls (4-oxo species) | |||
| Lysine | Lys | K | Hydroperoxides |
| Alcohols (hydroxylysines) | |||
| Carbonyls (α-aminoadipic semi-aldehyde and others) | |||
| Chloramines (RNHCl) | |||
| Bromamines (RNHBr) | |||
| Nitriles | |||
| Lysinoalanine (dehydroalanine–Lys cross-link) | |||
| Methionine | Met | M | Methionine sulfoxide: RSOR′ |
| Methionine sulfone: RSO2R′ | |||
| Dehydromethionine | |||
| Carbonyls (aspartate 4-semialdehyde arising from loss of–SMe function) | |||
| Homocysteic acid: RSO3H (cleavage of S–CH3 bond) | |||
| Phenylalanine | Phe | F | 2-Hydroxyphenylalanine (ortho-Tyr) |
| 3-Hydroxyphenylalanine (meta-Tyr) | |||
| Tyr (4-hydroxyphenylalanine) | |||
| 2- or 4-nitrophenylalanine | |||
| Proline | Pro | P | Hydroperoxides |
| Alcohols (hydroxyprolines) | |||
| Carbonyls (2-pyrrolidinone and ring-opened species such as glutamic semi-aldehyde) | |||
| Selenocysteine | Sec | U | Mixed seleno-thiol cross-linked species (RSe-SR′) |
| Selenenic acid: RSeOH | |||
| Seleninic acid: RSeO2H | |||
| Selenonic acid: RSeO3H | |||
| Dehydroalanine | |||
| Selenomethionine | Selenomethionine selenoxide | ||
| Serine | Ser | S | Carbonyls (serine aldehyde, 2-oxo species) |
| Threonine | Thr | T | Carbonyls (2-amino-3-ketobutyric acid and others) |
| Tryptophan | Trp | W | Hydroperoxides and endoperoxides |
| N-Formylkynurenine | |||
| Hydroxy N-formylkynurenine | |||
| Di-hydroxy N-formylkynurenine | |||
| Kynurenine | |||
| Kynurenic acid | |||
| 3-Hydroxykynurenine (and downstream products, including xanthurenic acid, 3-hydroxyanthranilic acid, quinolinic acid, and picolinic acid) | |||
| Hydroxytryptophan (multiple isomers) | |||
| 5- and 6-nitrotryptophan | |||
| Chlorotryptophan | |||
| Hydropyrroloindole | |||
| 2-Oxindole species | |||
| Di-oxindole species | |||
| Hydroxytryptophandione | |||
| Di-tryptophan (multiple isomers with both C–C and C–N linkages) | |||
| Trp–Tyr cross-link species | |||
| Tyrosine | Tyr | Y | Hydroperoxides and endoperoxides |
| DOPA | |||
| DOPA quinone | |||
| Trihydroxyphenylalanine (TOPA) | |||
| 3-Nitrotyrosine | |||
| 3,5-Dinitrotyrosine | |||
| 3-Chlorotyrosine | |||
| 3,5-Dichlorotyrosine | |||
| 3-Bromotyrosine | |||
| 3,5-Dibromotyrosine | |||
| Di-tyrosine: Tyr–Tyr cross-link (both C–C and C–O linkages | |||
| Trp–Tyr cross-link species | |||
| Valine | Val | V | Hydroperoxides |
| Alcohols (3- and 4-hydroxyvalines) | |||
| Carbonyls (3-oxo species) |
Radical-mediated damage can also be detected on the peptide backbone (36, 55, 56), with this appearing to occur primarily via hydrogen-atom abstraction from the α-carbon (the side-chain attachment site) (56, 57). Subsequent reactions of the initial R• formed in this process result in fragmentation of the peptide backbone, with this occurring via two different pathways involving ROO• and RO• (Fig. 4) (36, 55, 56). These pathways have been reviewed recently (4). With species such as HO•, this can result in a large range of different cleavage sites along a protein backbone (often detected as a “smear” on protein gels), although some selectivity in fragmentation has been reported (58, 59), particularly at metal ion–binding sites (60). This may also be due to particular stabilizing factors, such as the capacity to form a planar intermediate that maximizes stabilization of the intermediate α-carbon species (61) or a high level of solvent exposure on the surface of a protein (e.g. at turns between helices).
Figure 4.

Overview of radical reactions resulting in cleavage of the protein backbone. This can arise from both direct reactions at backbone sites (principally at the α-carbon) and also indirectly via initial oxidation at side-chain sites with subsequent radical transfer to the backbone, either intra- or intermolecularly. For further details see main text and Refs. 3, 4, 51, 52.
Two-electron oxidants typically show markedly greater selectivity than most radicals, due to the higher-energy barriers for many of these reactions (Table 1) (4). For species such as ONOOH, direct two-electron processes can occur (19), including reaction with Cys, cystine, and Met residues to give oxygenated species (Table 1). Oxidation of metal-ion centers can also occur via two-electron pathways. However, these reactions occur in competition with homolysis of ONOOH to give radicals (and hence one-electron oxidation products), and reaction of the anion ONOO− with CO2 to give the corresponding carbonate adduct (although the identity of this species is disputed (62, 63)). The adduct has been reported to have a short life-time (a few nanoseconds (62)) and to decompose to give NO2· and CO3˙̄, and thereby generates radical-mediated products. As NO2· is formed from both ONOOH and ONOOCO2−, nitrated products are commonly detected, with these being mainly generated from Tyr and Trp residues via dimerization reactions of NO2· with the TyrO• and TrpN• species formed by HO• or CO3˙̄ (19). More limited modifications are detected from radical chemistry at Phe and His (64, 65), but oxidation of Cys and Met likely occurs via both one- and two-electron reactions (65).
With hypohalous acids (HOCl, etc.), reaction occurs most rapidly with the sulfur amino acids (Cys > Met > cystine) to give a mixture of species (Tables 1 and 2) (66). Reaction also occurs, albeit less rapidly, with nitrogen nucleophiles (i.e. His, the α-amino group, and Lys) to give short-lived chloramines (RNHCl) (15, 66, 67) that retain the oxidizing capacity of HOCl but react much less rapidly and with greater selectivity (67, 68). Reaction also occurs with Trp and Tyr, although with lower rate constants (15). With Trp, oxygenated (and possibly chlorinated) species are formed (66), and with Tyr, the major product is 3-chloro-Tyr (69), a well-established biomarker of this oxidant, even though this species is formed slowly and in low yield (Table 2) (15).
1O2 reacts primarily with sulfur (Cys, Met, and cystine) and aromatic residues (Trp, Tyr, and His) (27, 29), with reaction at the former species to give the dimer (cystine) and oxygenated products (Cys oxyacids, Met sulfoxide, and oxygenated disulfides) (27, 29). With Trp, Tyr, and His, the initial products are endoperoxides formed by cycloaddition reactions, with these subsequently undergoing ring opening to give hydroperoxides, oxygenated products, and further cyclized materials (Table 2) (27, 29). As with the radical chemistry of Trp and His, these reactions can result in ring opening reactions and a similar (complex) mixture of species. The products from these amino acids therefore do not allow the initial oxidant to be easily identified.
From the above discussion it is clear that different oxidants have very different chemical behaviors and reaction kinetics, and these differences can be magnified or decreased by a range of other factors that influence oxidant selectivity. This is discussed further in the following section.
Factors affecting oxidant selectivity
The extent of damage by a particular oxidant can be modulated by multiple factors, including the accessibility of the oxidant to the target residue (e.g. Trp residues are often buried within protein structures and have limited solvent accessibility), and also electrostatic interactions with residues on the protein surface (e.g. the presence of charge on oxidants such as O2˙̄ and CO3˙̄). Neutral species may induce greater damage than charged species, and also at more remote locations, due to the greater propensity of such species to traverse membranes and hence diffuse away from their site of generation. The neutral species may also be better electrophiles and provide better leaving groups. This is exemplified by the greater reactivity of HOCl over −OCl and ONOOH over ONOO− (15, 19). Ionization of side-chain residues on a protein increases their electron density, increasing their capacity to act as a nucleophile and be more readily oxidized; thus, the thiolate anion, RS−, is more reactive than the parent RSH (and similarly for the related selenium-containing amino acid, selenocysteine, Sec), and the Tyr phenolate anion is more readily oxidized than the neutral phenol (70, 71).
Readily oxidized residues (e.g. Cys, Trp, and Tyr) can undergo long-range electron transfer reactions and thereby function as radical “sinks” both within and between proteins (72–75). Such transfers can occur over long distances (e.g. in ribonucleotide reductase, DNA photolyase, and photosystem II), such that initial oxidation at one site can be rapidly transferred to another remote residue, with the electron transfer occurring through bonds or space (Fig. 5) (76, 77). Consequently, the initial site of oxidation may not be the final site of modification. As the one-electron reduction potentials of Trp and Tyr are similar, radical formation at one residue can result in equilibration between residues, assuming suitable electron transfer pathways are available (73). One consequence of this is that radical termination reactions (e.g. dimerization of two TyrO•, two TrpN•, or cross-reaction of these species with NO2· to give nitrated products) may occur via the most accessible, or reactive, TyrO• or TrpN• rather than at the site of initial radical generation. Thus, cross-link formation involving Tyr and Trp radicals, and formation of products such as 3-nitro-Tyr, may be determined by the accessibility and reactivity of a particular residue, rather than the extent of initial reaction at that site (Fig. 5).
Figure 5.

Initial oxidation at electron-rich sites (e.g. Tyr and Trp residues but also Met, His, and Cys) can result in rapid electron transfer both within, and between, protein molecules. This can result in subsequent reactions and products being formed at sites that were not the initial site of oxidation and at locations remote from the initial site.
In the light of these data, the next section summarizes commonly used methods to detect protein alterations, starting with modifications at the intact protein level (i.e. changes that markedly affect protein mass and structure: “gross changes”) and then progressing to techniques that identify and quantify changes at an amino acid level, and at specific sites within a protein sequence.
Detection and quantification of protein oxidation
Gross modification of parent proteins
Oxidation of proteins can generate both fragmentation and aggregation of proteins. The latter can involve both covalent cross-linking as well as noncovalent interactions. Separation methods based on mass or charge (e.g. one- or two-dimensional electrophoresis and column chromatography) with subsequent detection methods (e.g. silver staining or immunoblotting) can provide limited information about such changes. This works best with purified proteins or limited mixtures, but it has severe limitations with complex samples and also when comparing healthy versus diseased samples, as the protein pools may be very different in such cases, even when two-dimensional gels are used to enhance resolution. Immunoblotting with specific antibodies can provide high sensitivity and specificity detection, but this approach is severely limited with regard to both the quantification and identification of modifications. Artifactual proteolysis or aggregation is also a serious concern. Both oxidant-mediated fragmentation and aggregation can be investigated using these approaches, but as fragmentation is often nonspecific or poorly-specific, discrete bands or spots (from 2D gels) are rare, with “band smears” being the usual outcome.
Aggregation or cross-linking is more readily analyzed, as dimers (for example) generated by any pairing of residues are likely to provide bands/spots of similar mass. Care clearly needs to be taken as multiple proteins are typically present in each band or fraction. Reduced antibody recognition of a specific native epitope can be used as a method of assessing modification to that site, in either immunoblotting studies or ELISA. These approaches are limited by the availability of specific antibodies but have been used successfully in a large number of studies ranging from isolated proteins to tissues, and they have the advantage of very-high sensitivity. Increased information can be obtained if antibodies against both parent protein epitopes, and specific products (see below), are available (78–85). An example of this approach are the studies that have examined HOCl-mediated damage to the extracellular matrix underlying endothelial cells. Binding of three specific antibodies (anti-fibronectin, anti-laminin, and anti-thrombospondin) was decreased on treatment with HOCl, implicating damage to these proteins (86). However, analysis of such data can be complex, as damage may also enhance antibody binding by exposing cryptic epitopes. Thus, low doses of HOCl appear to increase the affinity of anti-fibronectin antibodies to plasma fibronectin, whereas high concentrations have the opposite effect (87). This has been rationalized in terms of the generation of an extended fibronectin conformation at low HOCl doses, and aggregation with high concentrations.
An absence of positive data from ELISA or immunoblotting studies does not preclude the presence of damage, as epitopes may become inaccessible on protein oligomerization or as a result of other structural changes. Quantification is also challenging as this is very dependent on the sensitivity of the antibody: strong signals may be detected for low-abundance material, whereas abundant species may give a weak (or no) signal with a poor antibody. Separation of modified species by HPLC, for example, has been employed successfully with oxidants that are highly selective and that induce damage at a limited set of residues. An example is the separation of modified apolipoproteins AI and AII (88, 89) after mild oxidation of high-density lipoproteins or plasma, with loss of the parent isoforms, and the formation of newly-oxidized species, as detected by HPLC (88). Subsequent MS analysis of the fractions identified the modifications as loss of parent Met and generation of the corresponding sulfoxide.
Other biophysical techniques (e.g. CD, light scattering, small-angle neutron scattering, small-angle X-ray scattering, turbidity methods, X-ray crystallography, and NMR spectroscopy) can also yield information on protein structure, particularly the generation of fragments or aggregates, as these methods are sensitive to changes in protein mass, the size of particles, modified secondary structure, and altered charge and solubility. X-ray crystallographic studies have provided evidence for increased electron density between residues in aggregated proteins supporting the presence and identification of particular cross-links and their nature (exact site of linkage and intra- versus inter-molecular; see, for example, data for oxidized peroxiredoxin 5, thioredoxin 2, and γS-crystallin (90–92)). However, with the exception of X-ray crystallography and NMR, these methods do not provide definitive information as to the sites and modifications, and these techniques are (currently) limited to homogeneous samples (often single proteins) with high modification levels.
Total amino acid analysis
This methodology can provide important quantitative data on the consumption of parent species, arising from all potential modification reactions, and for some species the yields of products can also be assessed (e.g. methionine sulfoxide, see below). Such data are important with regard to obtaining a material balance, something that has been difficult to achieve even with the simplest systems. An overview of this approach is provided in Fig. 6. Differences between loss and total product formation can provide vital information with regard to the generation of alternative (known or unknown) species (see also below). Typically, proteins are isolated (e.g. by precipitation from homogenates/lysates using TCA or organic solvents), cleaned up (e.g. delipidation), and subsequently subjected to hydrolysis to give the free amino acids and products (Fig. 6) (54). Processing is preferentially carried out in the presence of enzyme inhibitors and antioxidants to decrease artifacts (54). Proteolysis can be achieved using acid conditions (with this resulting in loss of Cys, cystine, and some Trp species, although this depends on the acid), alkaline conditions (which preserves Trp species, but results in loss of other species such as the product DOPA), or nonspecific proteases, such as Pronase (54, 80, 93). The free amino acids (and any products) are then separated (e.g. by HPLC/UPLC) and quantified by mass spectrometry (MS), fluorescence (either directly, for some aromatic species, or by pre-column fluorescent tagging of free amino groups using reagents such as o-phthaldialdehyde, OPA), UV absorption, or electrochemical methods (Fig. 6) (54, 80, 93). The combination of acid hydrolysis (using methane sulfonic acid) and OPA tagging allows data to be obtained for all common amino acids with the exception of Cys/cystine (which are acid-sensitive), Asn and Gln (which are converted to Asp and Glu, respectively), and Pro (which does not react with OPA, being an imine) (54). Enzymatic hydrolysis results in lower levels of artifactual oxidation due to the mild conditions (typically overnight incubation at 37 °C, pH 7.4) and hence preservation of acid/base-sensitive materials, but quantification can be problematic due to self-digestion of the protease resulting in release of additional amino acids although this is often limited (80).
Figure 6.

Workflow to assess protein amino acid composition and their associated modifications. Proteins isolated and purified before digestion or hydrolysis to their constituent amino acids. Free amino acids and/or related oxidation products are then separated by LC. For some applications, pre-column or post-column derivatization of the amino acids and related products is required before separation to enable detection and quantification using one or more detection methods, which typically include MS, fluorescence, UV or visible absorption, or electrochemical (EC) detection. Abbreviations used are as follows: MSA, methane sulfonic acid; NFK, N-formylkynurenine.
Quantitative data can be obtained by use of standard curves generated using amino acid mixtures, with heavy atom labeling (usually 2H, 13C, or 15N) in the case of MS analysis (54). Lys quantification can be problematic due to the second side-chain amino group, which results in multiple peaks if labeling (such as tagging with OPA or other amine-reactive tags) is incomplete. An internal standard (e.g. homo-Arg) allows sample recovery and derivatization efficiency to be assessed (54). The use of sacrificial oxidation targets (e.g. tryptamine), anoxic conditions, antioxidants, and other inhibitors are important to prevent significant losses, and data are typically normalized to nonmodified amino acids to compensate for any losses during processing (54). Comparison with expected amino acid compositions is recommended for studies using pure proteins, to ensure that materials are not lost (or the extent compensated for) during processing. Some modifications (e.g. Trp products during acid hydrolysis) are also known to be lost during processing (see below), which may result in an underestimation of the level of damage. MS analysis can also be readily used for studies on free amino acids, peptides, and for proteins. For the last of these, analysis is usually undertaken on loss of specific peptides after digestion using trypsin (or other enzymes) or at the intact protein level. Absolute quantification can, however, be tricky to achieve (see below).
The total amino acid analysis approach is limited in that it only provides overall levels and not data on the sites of modifications within a sequence, nor the data on what proteins they might be present on, when analyzing complex mixtures. However, these data are an important complement to other approaches, such as MS peptide mass mapping, where only a limited number of species are typically analyzed.
Loss of some amino acids can be quantified by alternative methods. Direct fluorescence (typically λex 280–285 nm and λem 340–345 nm) has been widely used to examine Trp residues (94), but this can be problematic, as Trp fluorescence is environment-sensitive (hence its use in examining protein unfolding (95)) and is difficult to use in the presence of other species that absorb or exhibit fluorescence at these wavelengths. This may include proteins with high concentrations of Tyr residues, heme proteins, species arising from Trp degradation, and some glycoxidation products. Lys and Arg can be quantified by reagents that give strongly fluorescent derivatives, such as fluorescamine (96) and 9,10-phenanthrenequinone (97) respectively. These methods are rapid, sensitive, and give limited artifacts during sample preparation, but neither reagent is entirely specific, and hence needs to be used with care.
A number of methods have been developed to allow quantification of the loss of the key redox-sensitive amino acid Cys on proteins. Detection and quantification of Cys products are covered below. Methods include spectrophotometric (e.g. using 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), Ellman's reagent (98, 99), or 4,4′-dithiodipyridine (100)), fluorometric (e.g. using ThioGlo 1, (10-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)-9-methoxy-3-oxo-3H-naphthol[2,1-b]pyran-2-carboxylic acid methyl ester), a naphthopyranone maleimide derivative, and related species (101), biotinylated reagents with various detection methods, MS (102), and click chemistry (103, 104). These approaches can be used both directly on complex samples to give an overall readout or after protein separation using 1D or 2D gels (105) and HPLC/UPLC (101, 102). With fluorescent or biotinylated thiol derivatization reagents, care should be taken to ensure specificity of the probe (54). The use of MS with isotope-coded tags is a powerful method for determining the sites and extent of Cys oxidation in complex biological mixtures (reviewed in Ref. 106).
The oxidation of the major low-molecular-mass cellular thiol, glutathione (GSH), is widely employed as an oxidative stress marker (e.g. Ref. 107), particularly when expressed relative to its major oxidation product, the disulfide GSSG, or the total of these species (e.g. Ref. 108). As GSSG formation can be reversed by GSH reductase (109, 110), this reaction can be exploited to determine levels of GSH and the total of GSSG and GSH spectrophotometrically. GSSG levels can then be determined by difference in the values. The GSH concentration can be determined as described above, and also via the consumption of NADPH, the co-factor for GSH reductase (109, 110). GSSG levels can also be determined using genetically-encoded probes, such as roGFP fused to glutaredoxin; these probes are not responsive to thioredoxin-mediated changes probably for steric reasons. These probes allow real-time imaging of redox changes at specific and defined locations within living cells, although they also have some significant caveats; in particular, these are not direct oxidant probes, for they only report on the redox status of the specific environment being examined (111, 112). Kits are available for such measurements. Direct measurements of both GSH and GSSG can be achieved after separation (e.g. by HPLC/UPLC) with various detection methods, including electrochemistry (e.g. Ref. 113) and fluorescent tagging (e.g. with dansyl chloride or monobromobimane (114)). The levels and ratio of Cys/cystine and other thiols present in plasma have also been used as oxidative stress indicators in plasma (115). The absolute amounts, and ratio, of protein-bound Cys and low-molecular-mass thiols can be individually assessed after separation of the high- and low-molecular-weight fractions (e.g. by use of spin filters or protein precipitation).
Reduced and oxidized thiols on proteins separated on 1D or 2D gels can be assessed by a number of methods (reviewed in Ref. 116), including the use of fluorescent-tagging (e.g. 5-iodoacetamidofluorescein, a fluorescent derivative of iodoacetamide) or biotin-tagging (e.g. Ref. 117). Reversible thiol modifications can be examined by use of derivatization reagents after oxidant treatment, reduction of the reversible modification, and use of a second orthogonal tag (also see below and Ref. 117).
Direct quantification of disulfides (e.g. cystine) is complex but can be achieved by mild protein digestion methods (e.g. using chemicals such as cyanogen bromide or enzymatic approaches), which cleave the polypeptide backbone between the half-cystinyl residues, under conditions that minimize thiol-disulfide exchange and disulfide reduction. Diagonal electrophoresis was used in early studies (reviewed in Ref. 118), but this is now often examined using MS partial digestion and LC separation (e.g. Ref. 119). Other MS methods have also been developed for cystine (115).
Detection of protein oxidation intermediates
Modification of proteins and peptides by oxidants can result in the generation of a number of intermediate species that have modest lifetimes and stabilities. As the assay of these materials requires specialized methods, these are discussed separately from the analysis of long-lived (“stable”) products, which are discussed later in this review. An overview of these methods is provided in Fig. 7.
Figure 7.

Approaches and experimental methods to detect reactive intermediates on proteins.
Radicals
Radicals, both initiating species and those formed on peptides and proteins, can be detected by a number of approaches including direct spectroscopic methods such as UV-visible, resonance Raman, conductivity, and electron paramagnetic resonance (EPR). Because of the short half-lives of most radicals, techniques with rapid response times are required, and of the above methods, only EPR is specific for radicals (120). Measurements using other techniques can be readily confounded by more abundant nonradical species, and hence they are only typically used with very clean systems coupled to rapid radical generation methods (e.g. pulse radiolysis, flash photolysis, and stopped flow) (120). These other methods, although limited in applicability, can provide valuable kinetic (rate constant) data.
The use of EPR spectroscopy to detect and identify amino acid, peptide, and protein radicals in both isolated and complex systems has been reviewed elsewhere (121–123). Although this is a very powerful analytical technique for identification of radicals (the “gold standard”), quantification is very challenging due to the short lifetime of these species. This can be (partially) overcome by use of ancillary techniques such as rapid-flow methods, in situ photolysis or radiolysis, freeze-quenching, and spin trapping (e.g. using nitrone compounds, such as 5,5-dimethyl-1-pyrroline N-oxide, DMPO), although each of these has advantages and disadvantages (reviewed in Refs. 121–123). Spin trapping is the most widely used as it allows studies on fluids, cells, tissues, and intact animals (e.g. mice and rats; see e.g. Refs. 120, 122, 123). In this technique, a compound (the spin trap, typically a nitroso or nitrone) is added, with the aim of generating more stable, detectable adducts (Fig. 7). Analysis of the resulting spectra can then yield information on the species present (121–123). The technique is artifact-prone and needs to be carried out with care. The resulting data can be definitive with regard to the species present, but also have a number of inherent caveats. In particular, the sensitivity of the method allows minor pathways to be detected and potentially misinterpreted, and the data do not provide information as to the absolute radical concentrations. Differences in the rates of trapping (or adduct decay) may make a minor species appear important when compared with a major pathway that yields very transient species. The long lifetimes of some protein–radical adducts have allowed this approach to be combined with other analytical methods, including MS, to provide detailed information (reviewed in Ref. 121).
A related method, immunospin trapping, has been developed to detect protein radicals (reviewed in Refs. 124, 125), with this method utilizing the decay of a nitroxide spin adduct to a nitrone species to generate an antigen that is recognized by an antibody. This then allows the sensitive and specific detection of the former radical species, on isolated proteins, in cells, and in animals (primarily rodents). Immunospin trapping system can also be combined with LC/MS/MS and with the anti-DMPO antibody used to screen separate fractions (e.g. from HPLC or size-exclusion columns) for the presence of adducted DMPO. This allows residues that previously contained a radical to be readily detected, as the mass addition arising from the presence of DMPO can be readily detected (124, 125).
Hydroperoxides
Hydroperoxides are major products of both radical- and 1O2-mediated damage to proteins in the presence of O2. These species are formed on many side chains in high yield by a wide range of insults, including oxygen-derived radicals, 1O2, activated white cells, ONOOH, and metal ion-catalyzed systems (Table 2) (reviewed in Ref. 4). Decomposition of these species, which have lifetimes of minutes to many hours, by metal ions or UV light can give further radicals (RO•, R•, and ROO•) that propagate damage, including to lipids, other proteins, DNA, and RNA (4, 126, 127). Two-electron reduction by both low-molecular-mass reductants (e.g. GSH) and some protein and enzyme systems (128) gives the corresponding (stable) alcohols as products. In contrast, reaction of ROOH/H2O2 with critical Cys residues present on proteins or enzymes can inhibit enzyme activity and exacerbate damage (51, 52, 129, 130). Alcohols consistent with the formation and subsequent decay of hydroperoxides have been detected in healthy and disease specimens (e.g. human lens cataracts (131) and atherosclerotic lesions (132)), consistent with the occurrence of this chemistry in vivo. Hydroperoxides can be quantified by multiple methods, including iodometric titration, the ferrous oxidation–xylenol orange (FOX) assay, and also by use of boronic acid probes (Fig. 7) (133–135). In the first of these, reaction of ROOH with iodide ions (I−) in the presence of acid generates triiodide (I3−) that can be quantified by its absorbance at 358 nm. This method is quantitative and has a well-defined 1:1 stoichiometry, but it needs be performed under strict anoxic conditions due to the sensitivity of acidified iodide solutions to O2; it is therefore technically demanding (133). The FOX (ferrous oxidation–xylenol orange) method assays hydroperoxide-mediated oxidation of a Fe(II)–xylenol orange complex to the Fe(III) form, with the latter quantified via its absorbance at 560 nm (134, 135). This assay is generic for all hydroperoxides, and also H2O2 (so samples are typically pre-treated with catalase to remove this species), and has been adapted to allow quantification of both protein- and lipid-derived hydroperoxides (134). This method has a low sensitivity to O2, but it is not compatible with some buffers and has a poorly-defined stoichiometry; consequently, data are typically reported as H2O2 equivalents obtained by use of a standard curve generated using this species (136). Boronic acid probes that give fluorescent products on reaction with hydroperoxides have also been introduced (137), and these can provide real-time data, although they may be limited to in vitro systems, as multiple other oxidants also react with these pro-fluorescent species (138, 139).
Chloramines/bromamines
Reaction of nucleophilic nitrogen centers (e.g. imidazole, amines, and amides), with hypohalous acids (HOCl and HOBr) generates N-chloro and N-bromo species (RNHX, where X = Cl, Br (14, 140)). These species can be formed on both the N-terminal amine and the side chains of Lys and His and to lesser extent Arg, Asn, Gln, and backbone amides (14). Quantification is possible by their UV absorption bands (Fig. 7) (λ ∼250 and ∼290 nm, for RNHCl and RNHBr respectively (141)), but these overlap with many other species in complex systems, so quantification is usually achieved by reaction with an added probe such as 5-thio-2-nitrobenzoic acid (TNB, which is oxidized to the corresponding dimer, DTNB (141)), with quantification achieved via the loss of absorbance from TNB at 412 nm. Oxidation of TNB to DTNB also occurs with many oxidants (e.g. HOCl, HOBr, HOSCN, H2O2, and other peroxides, ONOOH, 1O2, and many radicals), and hence it is not specific for any particular species. Iodometric titration can also be employed (see section on “Hydroperoxides”), but again this lacks specificity. Iodide ions have also been used to catalyze the oxidation of 3,3′,5,5′-tetramethylbenzidine and dihydrorhodamine, by chloramines, to optically absorbing or fluorescent products (Fig. 7) (142); this method is more specific but also has some drawbacks, including slow reaction (142). N-Chloro species have also been identified by LC/MS, but the instability of these species at elevated temperatures limits quantitative assessment (143).
Sulfenic acids and related species
Sulfenic acids (RSOH, with the formation of these species often termed S-sulfenylation), sulfenyl chlorides (RSCl), and S-nitrosated species (RSNO, S-nitrosylated) are major intermediates formed by two-electron oxidants with Cys residues and related species (50–52, 144). S-Nitrosylated and S-nitrated (RSNO2) species are also formed by one-electron mechanisms involving reaction of RS• with NO• and NO2·, respectively. RSOH and the corresponding sulfenyl amides RS-NHC(O)R′ (145) are key intermediates in the catalytic and regulatory processes of some proteins and enzymes (52), particularly as these are formed in a reversible manner and hence may provide protection against irreversible oxidation of critical Cys residues and provide a facile “on-off” switch for enzyme activity and act as redox switches (146). Most of these species (with the exception of RSNO) react rapidly with other thiols (and other nucleophiles and oxidants) to give disulfides, thiosulfinates, and sulfinic (RSO2H) or sulfonic acids (RSO3H) (147). The higher oxyacids are usually irreversible oxidation products, although RSO2H can be reduced by sulfiredoxins (148). As sulfenic acids are key redox switches, considerable effort has been expended in developing methods to quantify these intermediates (148, 149). Most methods for the detection of RSOH rely on chemical derivatization or trapping methods, with the prototypic species being 5,5-dimethyl-1,3-cyclohexanedione (dimedone), which reacts with RSOH (and other species (150)) to form a stable thioether adduct (50–52), which can be quantified by MS or by use of fluorescent or biotinylated tags (Fig. 7) (144, 151–153). Reduction of sulfenic acids by arsenite has been utilized to develop a “biotin-switch” method for labeling protein RSOH, with the reduced amino acid subsequently labeled with biotin-maleimide. The adducts can then be detected using immunoblotting with streptavidin-horseradish peroxidase or separated using streptavidin-agarose (154).
S-Nitrosated, sometimes (incorrectly) named as S-nitrosylated, Cys residues are also key signaling species and may act as a reservoir of NO• (reviewed in Refs. 155, 156). Protein S-nitrosation has also been implicated in multiple disease states, particularly those involving neurodegeneration and inflammation (reviewed in Refs. 157, 158). The classical “biotin-switch” technique that has been widely used quantifies these species (158–160). In its classical form, nonmodified thiols are first blocked, and following the removal of excess alkylating reagent, ascorbate is added to selectively reduce any S-nitrosothiols (but not other species) to free thiols that are then labeled and detected by immunoblotting or fluorescent tagging following separation. Multiple iterations and improvements of this method have been proposed to enhance its specificity and to allow more rigorous quantification of RSNO levels. Many drawbacks have been reported, and considerable care and appropriate controls need to be employed to minimize artifacts (161, 162).
Multiple studies have reported MS methods to detect RSNO species (163, 164). As with other unstable intermediates, considerable care needs to be taken to avoid artifactual changes in the levels and sites of modification, as it is well-established that some RSNO species undergo ready transnitrosation reactions (164–166).
In biological systems, nitrosation is readily reversed, and this appears to be primarily driven by enzymatic reactions with reduction of low-molecular-mass species, such as S-nitrosated GSH (GSNO), being catalyzed by the widespread enzyme S-nitrosoglutathione reductase (GSNOR) (165–167), whereas removal of protein species is catalyzed by members of the thioredoxin family (168).
Persulfidation (RSSH formation) is a relatively recently identified modification of proteins that can act as both a redox control mechanism and sensor of redox stress (169). These species are formed, at least in part, via downstream reactions of H2S, a relatively recent addition to the family of a gaseous signal transmitter family, the other members being NO• and CO. Conversion of Cys to Cys-SSH occurs through sulfuration or persulfidation processes and may involve oxidized H2S species and particularly polysulfides (H2Sn), as well as other pathways. H2S is generated from Cys and homocysteine by widely-expressed enzymes, including cystathionine β-synthase, cystathionine γ-lyase, cysteine aminotransferase, and 3-mercaptopyruvate sulfurtransferase. As these species are expressed in the vascular wall, H2S has been proposed as a regulator of vascular tone, neuronal health, the integrity of endothelial cells barriers, smooth muscle cell proliferation and survival, angiogenesis, and as a modulator of inflammation (169–171) Persulfides can be detected and quantified using a tag-switch method (172); this should not be confused with the biotin-switch method (173), by MS methods, and also by use of fluorescent dyes, although the last of these, like most fluorescent dye approaches (138, 174), is likely to be artifact-prone (175–178).
Detection and quantification of products
The above discussion of methods available to detect loss of parent materials and detection of intermediate species illustrates some of the positives and negatives of these approaches. In particular, it is hopefully clear that detecting minor losses of a parent amino acid against a large background of native species is challenging, as is the detection of transient reactive intermediates, which are often present at low concentrations. In contrast, detection of stable products can afford more compelling data and often yields higher-quality quantitative data (as detection of a small increase against a theoretical background of zero can be achieved more readily), although again none of the methods available are without significant pitfalls and caveats. A description of available methods together with their advantages and drawbacks is presented below.
Generic markers of protein oxidation
Carbonyls are generated on proteins by multiple pathways and on a wide variety of residues, although with very variable yields (reviewed in Refs. 8, 36, 179). They can also arise from glycation/glycoxidation reactions, which may confound use of these as a quantitative, and exclusive, marker of oxidation. These species are also not specific to particular oxidants (47). Carbonyls can be formed on most amino acids (Table 2), although some yield higher concentrations than others (reviewed in Ref. 36). Metal ion–catalyzed oxidation systems give relatively high yields on Arg, Pro, and Lys residues, but modification is not exclusive to these sites (180, 181). Recent advances in MS identification methods—and particularly enrichment techniques—has expanded knowledge of the amino acids that give these species their chemical identity and yields (182). Different oxidants give different patterns of carbonyls, and both protein-bound and low-molecular-mass fragments can be formed (46, 47). The low-molecular-mass species arise from fragmentation reactions of RO• (see above and Refs. 46, 47), and these can be significant contributors to the total yield, although they are infrequently quantified. Quantification solely of protein-bound species is therefore likely to underestimate the total extent of damage (47, 183). Carbonyl levels have been shown to increase with age as well as with multiple diseases (reviewed in Refs. 8, 179, 184, 185).
Carbonyls can be quantified via their reaction with 2,4-dinitrophenylhydrazine (and related species) to give the hydrazone, with these assayed by optical absorbance (at 370 nm) or by antibodies against standards (Fig. 7) (179, 186, 187). A number of commercial kits are available that use this technology in either ELISA or immunoblotting approaches after separation on 1- or 2-D gels. The former gives the total yield of protein carbonyls, whereas the latter provides qualitative data on the proteins on which these may be present. Similar separation methods have been employed with fluorescent tags (e.g. fluorescein 5-thiosemicarbazide (188)).
Similar chemistry underlies the use of biotin-hydrazine and related species, which react to give the corresponding hydrazones. The latter can be reduced using cyanoborohydride, and a biotin tag can be used for enrichment before MS analysis (Fig. 8) (182, 189). This can be undertaken at both the protein and peptide level (190–192), but in the former case the high abundance of native peptides after proteolysis can result in the carbonyl products being missed, due to ion suppression and the use of only a limited number of the most abundant ions (typically parent peptide species) for further investigation (191, 192). The high affinity of biotin for avidin/streptavidin can result in a poor release of enriched materials in some cases, but this problem now seems to have been resolved by the use of 95 °C water as the eluent (182). Carbonyls can also be reduced with tritiated borohydride with subsequent and radioactive counting (Fig. 8), but this assumes that carbonyls are the only reducible species, which may not always be correct (187). The methods available for detecting carbonyls have recently been reviewed (188).
Figure 8.

Overview of methods for the detection and analysis of carbonyls (both protein-bound and low-molecular mass) arising from protein oxidation.
Most Cys oxidation products, with the exception of sulfenyl chlorides (which are very transient), are formed by multiple different species and hence are not diagnostic of the initial species. Furthermore, the wide range of species (and particularly cystine) that can be generated from Cys do not typically allow these species to be easily used as markers of oxidation. As outlined above, sulfenic acids can be quantified, but their reactive nature usually prohibits accurate assessment of the absolute extent of Cys oxidation. The higher oxyacids are commonly detected in MS experiments, but they are often not quantified as they can be generated as artifacts during processing and because oxyacids can also be generated from oxidative cleavage of cystine (probably via intermediate thiosulfinate species) (193, 194). Sulfenic and sulfinic acids can also be generated enzymatically and hence are not exclusive markers of reactive, diffusible oxidants. Thus, cysteine dioxygenase enzymes, which are common in plants, have been characterized in mammalian tissues, and these nonheme iron-containing enzymes can oxidize N-terminal Cys residues to the sulfinic acid. Interestingly, these enzymes also contain an internal Cys–Tyr thioether cross-link, which markedly enhances the enzymatic activity (195). These enzymes are key regulators of hypoxia responses in both animals and plants (195). Furthermore, there is abundant evidence for enzymatic Cys oxidation occurring by way of redox relays involving proteins with highly-reactive Cys residues, such as peroxiredoxins and STAT3, with initial oxidation of one protein by oxidants such as H2O2 allowing subsequent oxidative transfer to target proteins in a controlled and specific manner. This process appears to be a key pathway in H2O2-mediated cell signaling (196–199).
Methionine sulfoxide, which exists as two stereoisomers (R- and S-), is readily formed by many oxidants, although the rate constants for its formation vary by ∼10 orders of magnitude (4). Recent studies have however shown that the sulfoxide can also be generated on proteins, and particularly the key cytoskeletal protein actin, by the enzymatic action of the MICAL-family of proteins (200). This oxidation appears to play a key role in the regulation of actin depolymerization and also potentially membrane trafficking (see Refs. 200 and Fremont et al. (201). The detection of methionine sulfoxide may therefore not always be a marker of diffusible reactive oxidant species. Although the sulfoxide can be oxidized further to the sulfone, this is typically a slow and minor process (202). Levels in diseased tissue samples are often elevated, but the species is also readily formed by artifactual oxidation (cf. its ready detection in many MS analyses (203, 204)); hence, the true in vivo values may be overestimated. Methods have been developed to circumvent this problem (203, 204), but these are often not readily applicable to complex samples as they require complex cleavage and derivatization procedures (204) or complete oxidation of all Met residues using H218O2 and the use of the +2-Da mass shift arising from 18O incorporation to differentiate artifactual oxidation from “real” oxidation (203). Although an improvement, this method still has problems with peptides containing multiple Met residues, but this may be resolved using a method employing theoretical isotope distributions (205). The use of significant levels of peroxide in this method may also induce other alterations. Biological levels may also be perturbed by the repair of this product by methionine sulfoxide reductases (206, 207). However, the detection of elevated (relative to control) levels can be a useful indicator of enhanced damage, and its ready and rapid oxidation indicate that this is a sensitive marker. A recent study has reported a genetically-encoded ratiometric fluorescent biosensor MetROx that allows quantification of the R stereoisomer (207).
Immunological detection of oxidation products
Antibodies have been raised against a number of both generic and specific products formed on peptides and proteins. The antibodies vary significantly with regard to their specificity and selectivity, with some commercial species having a very poor reputation. In other cases, sensitive antibodies appear to have been generated, but either the exact epitope is unknown or there is significant cross-reactivity with other materials. This approach therefore has to be used with extreme care, and (both positive and negative) data need to be validated using alternative methods. Some of the better antibodies show tremendous sensitivity (as good or better than the most sensitive MS machines), but quantification is a significant problem, irrespective of whether these are employed in immunoblotting or ELISA formats (reviewed in Ref. 208). Thus, the absence of a signal does not necessarily imply the absence of a product (e.g. this may be due to epitope inaccessibility), and a strong signal does not necessarily result from a high yield of product. Different oxidation sites on a protein, or on different proteins, may react with an antibody to a greater or lesser extent depending on the position of the epitope in the structure and the surrounding environment. This is compounded in the case of polyclonal species and by use of poorly-defined materials as the original antigen; the latter may result in the recognition of multiple species.
Accurate quantification requires authentic epitope standards, which are typically not available for peptides and proteins. Relative quantification (i.e. versus the parent materials) may be less problematic, but still prone to error. Nonetheless, immunoblot or ELISA data can be a useful method for determining relative changes and as a screening tool. Antibodies can also be of tremendous use in enriching or semi-purifying low concentration materials. Unfortunately, some peptide and protein oxidation products have proven to be difficult to generate antibodies against; these include some chlorinated and brominated species (e.g. 3-chloro-Tyr and 3-bromo-Tyr) and methionine sulfoxide. Good commercial antibodies are available for 3-nitro-Tyr, 6-nitro-Trp, and the cross-linked species Tyr–Tyr. An antibody 2D10G9 (HOP-1) raised against HOCl-modified proteins (209) also recognizes HOBr-induced damage (210), and although the exact epitope that is recognized is unknown, this antibody has nonetheless yielded useful data on HOCl-mediated damage (209, 211–213). Some commercial antibodies raised against advanced glycation end products (AGEs) also recognize multiple species (or even different products) as a result of the use of poor original antigens. Appropriate controls are therefore critical. Similarly, antibodies have also been raised against derivatized products, with a well-established example being those raised against the 2,4-dinitrophenylhydrazones formed from the reaction of carbonyls with 2,4-dinitrophenylhydrazine (see above and Ref. 214). These antibodies, which are widely available in commercial kits, can be used for both immunoblotting (on 1D and 2D gels (215, 216), with derivatization carried out after isoelectric focusing so as to not alter the pI of the proteins (217)) and in ELISA (214). Considerable efforts have been expended to develop standardized methods to allow data to be compared with confidence across different labs (218, 219).
Detection and quantification of specific oxidation products
Aromatic side-chain modification products are widely used to assess protein oxidation, as these are typically easily oxidized and give stable products, some of which can be readily quantified (Fig. 6). Some of these materials are also informative with regard to the generating oxidant, unlike the situation with carbonyls, alcohols, most Cys products, and Met sulfoxide. Thus, 3-chloro-Tyr, 3-bromo-Tyr, and 3-nitro-Tyr/6-nitro-Trp are established biomarkers of chlorinating, brominating, and nitrating oxidants, respectively (69, 220–222). Chlorination appears to be exclusive (in mammalian systems) for myeloperoxidase-derived HOCl (14), and bromination can arise from HOBr generated by both myeloperoxidase and eosinophil peroxidase (220), and nitrating species appear to be predominantly generated via reactions of NO2· that can arise from ONOOH, ONOOCO2−, and also peroxidase-catalyzed oxidation of NO2− (19). Other Tyr and Trp oxidation products are less informative, but are still valuable markers of damage, with DOPA and Tyr–Tyr generated by multiple species (223). Similarly, oxidation of Phe to give 2-hydroxytyrosine (o-Tyr) and 3-hydroxytyrosine (m-Tyr), conversion of Trp to hydroxylated and ring-opened species (e.g. N-formylkynurenine), and His to ring-opened species occur with multiple oxidants, including HO•, some ROO• and RO•, ONOOH, HOCl, and HOBr, and some metal–ion oxo complexes (36, 54, 93). A number of these species, although moderately long-lived, can undergo additional reactions (e.g. conversion of N-formylkynurenine to kynurenine and other species, DOPA to the quinone and cyclized products, and His-adducts to Asn and Asp (36, 54, 93)).
Multiple methods have been developed to quantify these products, including HPLC/UPLC with UV (all species), fluorescence (e.g. DOPA, o- and m-Tyr, and Tyr–Tyr), electrochemical (e.g. Tyr–Tyr, 3-chloro-, 3-bromo-, and 3-nitro-Tyr), and MS detection, GC/MS, and immunological methods (immunoblotting/ELISA) (Fig. 6) (42, 54, 93, 224). Most GC methods have been superseded by LC approaches due to an increased risk of artifacts during derivatization to make the species volatile (224). DOPA (and other catechol species) can also be detected (but not quantified) by redox staining methods after separation of the proteins by SDS-PAGE or column chromatography (225).
These methods can be employed at an amino acid level (i.e. after hydrolysis, see Fig. 6) to give free amino acids and products at the peptide level (e.g. by MS peptide mass mapping, Fig. 9) and in some cases at the intact protein level. The first of these has the advantage that this gives the total yield of a particular species, but results in the loss of positional information. In contrast, peptide mass mapping, gives precise positional data but is less quantitative (see below). Intact protein studies have the advantage that they do not require the extensive handling and processing of the other methods and can provide an overall picture of the extent of oxidation (as judged, e.g. by the presence of ions corresponding to the addition of single or multiple oxygen atoms (194)), but they do not typically provide positional data or quantitative data. Recent advances in column technology allow large numbers of parent and modified amino acids to be detected in short LC runs, potentially allowing a more complete picture to be obtained as to the extent of modification of multiple species in single runs. However, control for ionization efficiency and suppression effects typically require the use of heavy isotope (2H, 13C, and 15N) standards (226). Although these are readily available for the parent amino acids, the pool of isotopic homologues for modified amino acids is low.
Figure 9.

Workflow to assess protein modifications by peptide mass–mapping approaches. Proteins were isolated and purified before digestion to peptides with trypsin or other protease enzymes. Peptides analyzed by MS following cleanup and LC separation. Peptide mass fingerprinting analysis and/or peptide fragmentation analysis by MS-MS with appropriate database searches using fixed or variable modifications followed by validation to limit artifacts.
Amino acid, peptide, and intact protein MS methods are therefore complementary and ideally should be employed together with measurement of parent loss (see above), allowing a material balance to be obtained. A number of recent studies have illustrated the importance of such data, as it is clear that in many cases there is a significant missing mass, consistent with the presence of multiple unidentified products (42, 227).
The powerful and increasingly widely used technique of peptide mass mapping of both native proteins and post-translational modifications, including oxidation products, has been discussed extensively elsewhere (228–232), and hence only very brief details will be given here (Fig. 9). The protein is digested enzymatically (e.g. with trypsin, but increasingly with other species) to release peptides that are then analyzed by LC/MS with specific peptide ions, typically the most abundant, subject to further fragmentation to give the amino acid sequence from the series of ions (usually b and y) detected (231–233). A range of different fragmentation techniques can be employed to obtain good sequence coverage (231, 232). Complete coverage (both for the parent and also modified species) is highly desirable but often is not achieved due to the generation of either very short or very long peptides (which can be outside of the detectable mass range of a spectrometer, but this is machine-dependent) or other factors (231, 232). A reduction in sequence coverage is often also encountered with modified proteins, particularly if extensive cross-linking or alteration/blocking of cleavage sites occurs (e.g. modifications at Lys or Arg, which prevent cleavage by trypsin) (231, 232). This can sometimes be circumvented by use of alternative cleavage enzymes. Data analysis can be automated, with both fixed and variable modifications included in the analysis. The former allows processing artifacts (e.g. deamination) to be eliminated, but in some cases oxidation species are also included (e.g. Cys and Met sulfoxide), which prevent data being obtained on these species (228, 231, 232). Variable modifications are typically specified (e.g. +16 and +32 from addition of one or two oxygen atoms, etc.) at defined amino acid residues. Lists of known products have been compiled in both papers (see Refs. 49, 228) and in databases (e.g. NCBI database, www.ncbi.nlm.nih.gov), and multiple search engines are available, but increasing the number of modifications examined increases the computational power required in a significant manner.
Directed searches are not ideal, but often employed, as only a defined subset of modifications are detected, and this therefore provides a potentially misleading picture. Open and unbiased searches (i.e. with no pre-defined modifications) are preferable (49), but are very time- and resource-demanding. All automated analyses should be manually validated and checked for chemical reality, which is again demanding on resources. Both loss of the parent peptides and generation of the modified species should ideally be examined, as this can limit artifacts and provide data on the extent of conversion of a particular residue to products. The number of ions chosen for secondary fragmentation, and hence sequencing, can also be a limiting and significant factor, as modified species are typically present at low levels and may be lost in a sea of higher abundance species. However, fragmentation of a larger number of ions severely impacts the run times.
Sample treatment before analysis is often necessary and can limit the information obtained. Because of the high propensity for artifactual oxidation at Cys residues during processing, and also to enhance sequence coverage of proteins containing native disulfide bonds, reduction and alkylation is typically carried out (234). This therefore results in loss of most information about oxidant-mediated changes at both Cys and cystine, although the higher oxyacids (Cys–SO2H and Cys–SO3H) are still detected (193). Several recent studies have, however, shown that reduction and alkylation can also decrease the levels of other modifications (e.g. 3-chloro-Tyr (235, 236) and others (234, 237)), although the exact reasons for this and its quantitative importance remain to be determined. Sequencing without reduction and alkylation can markedly decrease sequence coverage with disulfide-rich proteins (235, 236), as the peptides will remain covalently linked via the disulfide, and this is a particular problem with disulfide-rich species.
Quantification of modifications from peptide mass mapping studies is important, but it can be complex and subject to a large number of limitations. Data are often presented as relative site occupancies with this defined as the percentage of a particular modification at a specific site, relative to the sum of all the detected peptides (native and modified) containing the site (238). Accurate quantification therefore requires knowledge, and detection, of all modifications that occur at the site, and this is rarely achieved. If the extent of missing modified peptide data is low and the percentage conversion of the parent to products is low, the error is low, but with high extents of modification (i.e. only low levels of parent remain), any missing modified peptides can have a major impact. Inherent in such analyses is the assumption that the native and modified peptides behave identically under the MS conditions (e.g. ionization), and this is clearly not always the case (e.g. where suppression of ionization occurs or the modified peptide has a different number of charges). In theory, these effects can be discounted by use of standards, but this is rarely done as specific modified peptides can be costly and tricky to prepare, especially for complex samples where many parent and modified species would need to be generated and examined. Instability of modifications, under the conditions used for MS sample preparation and analysis, is another potential confounding factor as this can perturb the extent of modification detected. Thus, although direct LC/MS can be used to detect hydroperoxides, sulfenic acids, chloramines/bromamines, and S-nitrosated species present on amino acids and peptides (see above), these materials are often too labile to survive digestion and processing when present on intact proteins. Information on some of these materials can be obtained by derivatization (see above), but in some cases this is not possible without loss of information (e.g. hydroperoxides are refractory to derivatization, and reduction of the alcohol eliminates knowledge whether the species came from a hydroperoxide or another pathway).
Detection, identification, and quantification of protein cross-links
The generation and subsequent accumulation of cross-linked or aggregated (noncross-linked) proteins have been linked to a number of human pathologies, and hence there is considerable interest in the detection, characterization, and quantification of these species (41). Tyr–Tyr is a well-established protein cross-link, but other cross-linked species have been identified, involving DOPA quinone (from further oxidation of DOPA), Trp–Trp, Trp–Tyr, His–His, His–Lys, His–Arg, thioethers (lanthionine; from reaction of dehydroalanine with Cys), lysinoalanine (from reaction of dehydroalanine with Lys), sulfonium species derived from Met, carbonyl-Lys, and Schiff base-related species (e.g. involving allysine), and some advanced glycation end products (reviewed in Ref. 41). Some of these are generated enzymatically and deliberately (e.g. allysine species formed by lysyl oxidase and related species (239)), and Met sulfonium cross-links by peroxidasin enzymes in extracellular matrix assembly (240, 241), and some quinone and Tyr–Tyr related cross-links in insect exoskeletons and glues (242, 243), but others appear accidental and driven by oxidation (reviewed in Ref. 41).
Multiple methods have been developed to analyze for these species (Fig. 10). Direct UV absorbance, and particularly fluorescence, detection have been used for Tyr–Tyr (λex ∼280 nm or 305–315 nm, λem ∼410 nm) (54), but this is problematic if other fluorophores are present (e.g. Trp and Trp-derived products, AGEs). Thus, prior isolation and separation are often required (see above). As with other modifications, data can be obtained at the free amino acid, peptide, and protein levels, with the workflow depending on sample, the species being analyzed, and the desired information (identity, quantity, and location).
Figure 10.

Overview of approaches to detect cross-links on proteins.
Both GC-MS and LC-MSn have been used to quantify in situ and released cross-linked species. Free Tyr–Tyr levels in urine and plasma have been quantified by both LC-MSn and GC-MS, with LC-MSn having a higher sensitivity (244–247), and also have been used with tissue samples such as eye lens proteins (248). Chemical hydrolysis followed by MS analysis has been employed to detect and quantify protein cross-links, with isotope-labeled standards added before acid or alkaline hydrolysis depending on the cross-link (249). For di-Trp cross-links, alkaline hydrolysis is typically used, due to the sensitivity of indoles to acid-mediated cleavage (250). Enzymatic hydrolysis has also been employed (250), although this is less efficient, and auto-digestion of the protease can confound data. These methods allow (approximate) quantification of the total cross-link yield, but not positional data. The low levels of Tyr–Tyr detected in some studies with high levels of oligomerization indicate that other species must also play a major role in the detected aggregation (252).
Analysis at the peptide level typically involves proteolytic digestion and subsequent LC-MSn analysis, but this is challenging as linear (noncross-linked) peptides usually constitute the majority of the peptides, and the cross-linked species are easily missed (41). A number of different strategies have therefore been applied to isolate or enrich cross-linked species, at the protein or peptide level, to reduce complexity and facilitate analysis. For oligomers containing intermolecular (but not intramolecular) cross-links, size-exclusion chromatography (SEC) (253, 254) or SDS-PAGE can be used to isolate cross-linked from noncross-linked species. SEC can be advantageous, as it avoids in-gel digestion that can be inefficient for cross-linked species. Strong cation-exchange and charged-base fractional diagonal chromatography can also be used for intermolecular cross-link enrichment (255, 256), as these species often have a larger number of protonatable sites (e.g. two N termini) than linear peptides. Multistep enrichment methods have also been reported (257).
The abundance limitation has also been partly overcome by use of 18O labeling with trypsin (or GluC, LysC, or pepsin (258–260)) as the cleavage enzyme. This utilizes the capacity of these enzymes to incorporate two 18O atoms from H218O into the C terminus of the generated peptides. The first 18O arises from the peptide–backbone cleavage reaction, and the second is from enzyme-mediated carbonyl–oxygen exchange (261). This results in cross-linked peptides with two C termini having four 18O atoms incorporated compared with two for linear peptides. Thus, proteins are digested separately in H216O and H218O, and then mixed in a 1:1 ratio prior to MS analysis (Fig. 11). As the isotope analogues co-elute, a mass difference of 4 Da is observed for the linear peptides from the two 18O atoms incorporated, but an 8-Da mass difference is detected for cross-linked peptides; this +8-Da shift is diagnostic for the cross-linked species (Fig. 11) (41, 262). This method has been used to identify His–His links in IgG (263), Trp–Trp links in superoxide dismutase (264), Tyr–Tyr cross-links in tropoelastin (82), and Tyr–Tyr, Tyr–Trp, Tyr–Lys, His–Arg, and His–Lys cross-links in glucose-6-phosphate dehydrogenase, RNase, and lysozyme (42, 227, 262). This approach has also been shown to work in protein extracts from Gram-positive bacterium with Tyr–Tyr, Tyr–Trp, and Trp–Trp cross-links detected (262). This approach is cost-effective, widely applicable, and able to be used on clinical materials, as the labeling is carried out after tissue sampling and occurs only in the protease substrates (41). The method does however have some challenges, including the difficulty in achieving complete 18O incorporation as the exchange reactions are sometimes slow (with this resulting in mixed isotopic isoforms, although this can be minimized with longer incubation times (262)), and protease-catalyzed back-exchange of 16O, which can occur after the H216O and H218O digests are mixed (41). The latter can be minimized by combining the samples immediately prior to analysis.
Figure 11.

Outline of isotope-labeling method to detect and characterize protein cross-links using MS.
MS/MS spectra of cross-linked peptides can be highly complex, due to the presence of fragment ions from both peptide chains, even though one is typically underrepresented in the fragment ion spectra (83). The reasons for this have been partly elucidated (peptide length, with longer peptides dissociating more effectively; fragmentation efficiency; and the presence of specific amino acids (251)). The fragmentation pattern can be further complicated by cross-linked cleavage (e.g. the C–N bonds seen in some Tyr–Trp cross-links and the N–S bonds of sulfilimines (240, 253, 264)) but not others (e.g. the C–C bonds of Tyr–Tyr). Electron-transfer dissociation and higher-energy collisional dissociation have been shown to generate abundant ions (262), with detection of ions from both peptides allowing the precise site of the cross-link to be determined, with some of the ions typically arising from species that retain the cross-link (42, 82, 227, 262). The detection and identification of cross-links are, however, still in its infancy, and there is likely to be significant scope for further development in this area and also with regard to the quantification of these species.
Conclusions
Over the last few years considerable progress has been made in the development of new and powerful techniques to detect, identify, and quantify modifications on proteins, but it is clear that there is still much to be done. There is a pressing need for reliable and robust methods for the absolute quantification of oxidation products formed from amino acids, peptides, and proteins, which can be applied to complex biological systems.
As many current methods are less than optimal, it is highly advisable to use multiple different analytical methods or approaches, as these are more likely to give an accurate representation. Data obtained using single methods can be misleading, even when this involves highly-sophisticated technologies such as MS. In addition, quantitative methods are preferable when compared with qualitative approaches, as the absolute magnitude of a change is much more compelling and informative than “fold differences,” when there is no absolute reference value available. Thus, a 10-fold change may be of little biological consequence when it corresponds to a change from (for example) 0.1 to 1% in intact protein levels, whereas a 10-fold change from 10 to 100% is likely to be of major significance. In either case, it is critical to try and address whether the observed modification(s) and their quantitative changes are consistent with the observed biological, structural, or functional effects and to therefore address the question of causality.
Only with the development of better and complementary methodologies, as well as more accurate methods to quantify absolute changes, will the relative importance of protein oxidation/modification become clear when compared with other targets (e.g. to lipids, carbohydrates, and DNA) and whether protein alteration is a cause, or merely a consequence, of injurious processes.
This work was supported by Novo Nordisk Foundation Grants NNF13OC0004294 (to M. J. D.) and NNF17OC0028990 (to C. L. H.) and Danish Council for Independent Research/Natural Sciences Grant DFF-7014-00047 (to M. J. D.). The authors declare that they have no conflicts of interest with the contents of this article.
- NOS
- nitric-oxide synthase
- OPA
- o-phthaldialdehyde
- DMPO
- ,5-dimethyl-1-pyrroline N-oxide
- TNB
- as 5-thio-2-nitrobenzoic acid
- DTNB
- 5,5′-dithiobis(2-nitrobenzoic acid
- AGE
- advanced glycation end product
- DOPA
- 3,4-dihydroxyphenylalanine
- SEC
- size-exclusion chromatography
- FOX
- ferrous oxidation-xylenol orange
- UPLC
- ultra-performance liquid chromatography.
References
- 1. Halliwell B., and Gutteridge J. M. (2015) Free Radicals in Biology & Medicine, 5th Ed., Oxford University Press, Oxford, UK [Google Scholar]
- 2. Sies H., Berndt C., and Jones D. P. (2017) Oxidative stress. Annu. Rev. Biochem. 86, 715–748 10.1146/annurev-biochem-061516-045037 [DOI] [PubMed] [Google Scholar]
- 3. Davies M. J. (2005) The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109 10.1016/j.bbapap.2004.08.007 [DOI] [PubMed] [Google Scholar]
- 4. Davies M. J. (2016) Protein oxidation and peroxidation. Biochem. J. 473, 805–825 10.1042/BJ20151227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grimsrud P. A., Xie H., Griffin T. J., and Bernlohr D. A. (2008) Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 283, 21837–21841 10.1074/jbc.R700019200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shu N., Lorentzen L. G., and Davies M. J. (2019) Reaction of quinones with proteins: kinetics of adduct formation, effects on enzymatic activity and protein structure, and potential reversibility of modifications. Free Radic. Biol. Med. 137, 169–180 10.1016/j.freeradbiomed.2019.04.026 [DOI] [PubMed] [Google Scholar]
- 7. Gianazza E., Crawford J., and Miller I. (2007) Detecting oxidative post-translational modifications in proteins. Amino Acids 33, 51–56 10.1007/s00726-006-0410-2 [DOI] [PubMed] [Google Scholar]
- 8. Dalle-Donne I., Aldini G., Carini M., Colombo R., Rossi R., and Milzani A. (2006) Protein carbonylation, cellular dysfunction, and disease progression. J. Cell. Mol. Med. 10, 389–406 10.1111/j.1582-4934.2006.tb00407.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hausladen A., and Fridovich I. (1994) Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 269, 29405–29408 [PubMed] [Google Scholar]
- 10. Fridovich I. (1975) Superoxide dismutases. Annu. Rev. Biochem. 44, 147–159 10.1146/annurev.bi.44.070175.001051 [DOI] [PubMed] [Google Scholar]
- 11. Babior B. M. (1987) The respiratory burst oxidase. Trends Biochem. Sci. 12, 241–243 10.1016/0968-0004(87)90118-6 [DOI] [Google Scholar]
- 12. Misra H. P., and Fridovich I. (1972) The generation of superoxide radical during the autoxidation of hemoglobin. J. Biol. Chem. 247, 6960–6962 [PubMed] [Google Scholar]
- 13. Sies H. (2017) Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 11, 613–619 10.1016/j.redox.2016.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Davies M. J., Hawkins C. L., Pattison D. I., and Rees M. D. (2008) Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid. Redox Signal. 10, 1199–1234 10.1089/ars.2007.1927 [DOI] [PubMed] [Google Scholar]
- 15. Pattison D. I., and Davies M. J. (2001) Absolute rate constants for the reaction of hypochlorous acid with protein side chains and peptide bonds. Chem. Res. Toxicol. 14, 1453–1464 10.1021/tx0155451 [DOI] [PubMed] [Google Scholar]
- 16. Pattison D. I., Davies M. J., and Hawkins C. L. (2012) Reactions and reactivity of myeloperoxidase-derived oxidants: differential biological effects of hypochlorous and hypothiocyanous acids. Free Radic. Res. 46, 975–995 10.3109/10715762.2012.667566 [DOI] [PubMed] [Google Scholar]
- 17. Klebanoff S. J., Kettle A. J., Rosen H., Winterbourn C. C., and Nauseef W. M. (2013) Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 93, 185–198 10.1189/jlb.0712349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moncada S., Palmer R. M., and Higgs E. A. (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 43, 109–142 [PubMed] [Google Scholar]
- 19. Ferrer-Sueta G., Campolo N., Trujillo M., Bartesaghi S., Carballal S., Romero N., Alvarez B., and Radi R. (2018) Biochemistry of peroxynitrite and protein tyrosine nitration. Chem. Rev. 118, 1338–1408 10.1021/acs.chemrev.7b00568 [DOI] [PubMed] [Google Scholar]
- 20. Förstermann U., and Sessa W. C. (2012) Nitric oxide synthases: regulation and function. Eur. Heart J. 33, 829–837, 837a–837d 10.1093/eurheartj/ehr304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davis K. L., Martin E., Turko I. V., and Murad F. (2001) Novel effects of nitric oxide. Annu. Rev. Pharmacol. Toxicol. 41, 203–236 10.1146/annurev.pharmtox.41.1.203 [DOI] [PubMed] [Google Scholar]
- 22. Cary S. P., Winger J. A., Derbyshire E. R., and Marletta M. A. (2006) Nitric oxide signaling: no longer simply on or off. Trends Biochem. Sci. 31, 231–239 10.1016/j.tibs.2006.02.003 [DOI] [PubMed] [Google Scholar]
- 23. Koppenol W. H. (1993) The centennial of the Fenton reaction. Free Radic. Biol. Med. 15, 645–651 10.1016/0891-5849(93)90168-T [DOI] [PubMed] [Google Scholar]
- 24. von Sonntag C. (1987) The Chemical Basis of Radiation Biology, Taylor and Francis, London [Google Scholar]
- 25. Yin H., and Porter N. A. (2005) New insights regarding the autoxidation of polyunsaturated fatty acids. Antioxid. Redox Signal. 7, 170–184 10.1089/ars.2005.7.170 [DOI] [PubMed] [Google Scholar]
- 26. Neuzil J., Gebicki J. M., and Stocker R. (1993) Radical-induced chain oxidation of proteins and its inhibition by chain-breaking antioxidants. Biochem. J. 293, 601–606 10.1042/bj2930601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pattison D. I., Rahmanto A. S., and Davies M. J. (2012) Photo-oxidation of proteins. Photochem. Photobiol. Sci. 11, 38–53 10.1039/C1PP05164D [DOI] [PubMed] [Google Scholar]
- 28. Davies M. J. (2004) Reactive species formed on proteins exposed to singlet oxygen. Photochem. Photobiol. Sci. 3, 17–25 10.1039/b307576c [DOI] [PubMed] [Google Scholar]
- 29. Davies M. J. (2003) Singlet oxygen-mediated damage to proteins and its consequences. Biochem. Biophys. Res. Commun. 305, 761–770 10.1016/S0006-291X(03)00817-9 [DOI] [PubMed] [Google Scholar]
- 30. Butterfield D. A., Perluigi M., Reed T., Muharib T., Hughes C. P., Robinson R. A., and Sultana R. (2012) Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid. Redox Signal. 17, 1610–1655 10.1089/ars.2011.4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Symonds P., Deehan C., Meredith C., and Mills J. (2012) Walter and Miller's Textbook of Radiotherapy: Radiation Physics, Therapy and Oncology, 7th Ed., Churchill Livingston, New York [Google Scholar]
- 32. Kwiatkowski S., Knap B., Przystupski D., Saczko J., Kȩdzierska E., Knap-Czop K., Kotlińska J., Michel O., Kotowski K., and Kulbacka J. (2018) Photodynamic therapy- mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 106, 1098–1107 10.1016/j.biopha.2018.07.049 [DOI] [PubMed] [Google Scholar]
- 33. Beharry A. A. (2018) Next-generation photodynamic therapy: new probes for cancer imaging and treatment. Biochemistry 57, 173–174 10.1021/acs.biochem.7b01037 [DOI] [PubMed] [Google Scholar]
- 34. Tacar O., Sriamornsak P., and Dass C. R. (2013) Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 65, 157–170 10.1111/j.2042-7158.2012.01567.x [DOI] [PubMed] [Google Scholar]
- 35. Xu N., Yao M., Farinelli W., Hajjarian Z., Wang Y., Redmond R. W., and Kochevar I. E. (2015) Light-activated sealing of skin wounds. Lasers Surg. Med. 47, 17–29 10.1002/lsm.22308 [DOI] [PubMed] [Google Scholar]
- 36. Hawkins C. L., and Davies M. J. (2001) Generation and propagation of radical reactions on proteins. Biochim. Biophys. Acta 1504, 196–219 10.1016/S0005-2728(00)00252-8 [DOI] [PubMed] [Google Scholar]
- 37. Dizdaroglu M., and Simic M. G. (1983) Isolation and characterization of radiation-induced aliphatic peptide dimers. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 44, 231–239 10.1080/09553008314551091 [DOI] [PubMed] [Google Scholar]
- 38. Schöneich C. (2016) Thiyl radicals and induction of protein degradation. Free Radic. Res. 50, 143–149 10.3109/10715762.2015.1077385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fang X., Jin F., Jin H., and von Sonntag C. (1998) Reaction of the superoxide radical with the N-centered radical derived from N-acetyltryptophan methyl ester. J. Chem. Soc. Perkin Trans. 2, 259–263 10.1039/A706979K [DOI] [Google Scholar]
- 40. Hunter E. P., Desrosiers M. F., and Simic M. G. (1989) The effect of oxygen, antioxidants, and superoxide radical on tyrosine phenoxyl radical dimerization. Free Radic. Biol. Med. 6, 581–585 10.1016/0891-5849(89)90064-6 [DOI] [PubMed] [Google Scholar]
- 41. Hägglund P., Mariotti M., and Davies M. J. (2018) Identification and characterization of protein cross-links induced by oxidative reactions. Expert Rev. Proteomics 15, 665–681 10.1080/14789450.2018.1509710 [DOI] [PubMed] [Google Scholar]
- 42. Leinisch F., Mariotti M., Rykaer M., Lopez-Alarcon C., Hägglund P., and Davies M. J. (2017) Peroxyl radical- and photo-oxidation of glucose-6-phosphate dehydrogenase generates cross-links and functional changes via oxidation of tyrosine and tryptophan residues. Free Radic. Biol. Med. 112, 240–252 10.1016/j.freeradbiomed.2017.07.025 [DOI] [PubMed] [Google Scholar]
- 43. Gebicki J. M. (1997) Protein hydroperoxides as new reactive oxygen species. Redox Rep. 3, 99–110 10.1080/13510002.1997.11747096 [DOI] [PubMed] [Google Scholar]
- 44. Russell G. A. (1957) Deuterium-isotope effects in the autoxidation of aralkyl hydrocarbons. Mechanism of the interaction of peroxy radicals. J. Am. Chem. Soc. 79, 3871–3877 10.1021/ja01571a068 [DOI] [Google Scholar]
- 45. Ando W. (ed) (1992) Organic Peroxides, pp. 1–845, John Wiley and Sons, Chichester, UK [Google Scholar]
- 46. Headlam H. A., Mortimer A., Easton C. J., and Davies M. J. (2000) β-Scission of C-3 (β-carbon) alkoxyl radicals on peptides and proteins: a novel pathway which results in the formation of α-carbon radicals and the loss of amino acid side chains. Chem. Res. Toxicol. 13, 1087–1095 10.1021/tx0001171 [DOI] [PubMed] [Google Scholar]
- 47. Headlam H. A., and Davies M. J. (2004) Markers of protein oxidation: different oxidants give rise to variable yields of bound and released carbonyl products. Free Radic. Biol. Med. 36, 1175–1184 10.1016/j.freeradbiomed.2004.02.017 [DOI] [PubMed] [Google Scholar]
- 48. Buxton G. V., Greenstock C. L., Helman W. P., and Ross A. B. (1988) Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms, and hydroxyl radicals (•OH/•O−) in aqueous solution. J. Phys. Chem. Ref. Data 17, 513–886 10.1063/1.555805 [DOI] [Google Scholar]
- 49. Rykaer M., Svensson B., Davies M. J., and Hägglund P. (2017) Unrestricted mass spectrometric data analysis for identification, localization and quantification of oxidative protein modifications. J. Proteome Res. 16, 3978–3988 10.1021/acs.jproteome.7b00330 [DOI] [PubMed] [Google Scholar]
- 50. Poole L. B. (2015) The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 80, 148–157 10.1016/j.freeradbiomed.2014.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Devarie-Baez N. O., Silva Lopez E. I., and Furdui C. M. (2016) Biological chemistry and functionality of protein sulfenic acids and related thiol modifications. Free Radic. Res. 50, 172–194 10.3109/10715762.2015.1090571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yang J., Carroll K. S., and Liebler D. C. (2016) The expanding landscape of the thiol redox proteome. Mol. Cell. Proteomics 15, 1–11 10.1074/mcp.O115.056051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schöneich C. (2005) Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer's disease. Biochim. Biophys. Acta 1703, 111–119 10.1016/j.bbapap.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 54. Hawkins C. L., Morgan P. E., and Davies M. J. (2009) Quantification of protein modification by oxidants. Free Radic. Biol. Med. 46, 965–988 10.1016/j.freeradbiomed.2009.01.007 [DOI] [PubMed] [Google Scholar]
- 55. Garrison W. M. (1968) in Current Topics in Radiation Research (Ebert M., and Howard A., eds) Vol. 4, pp. 43–94, North-Holland Publishing Co., Amsterdam, Netherlands, and Manchester, UK [Google Scholar]
- 56. Davies M. J. (1996) Protein and peptide alkoxyl radicals can give rise to C-terminal decarboxylation and backbone cleavage. Arch. Biochem. Biophys. 336, 163–172 10.1006/abbi.1996.0545 [DOI] [PubMed] [Google Scholar]
- 57. Morgan P. E., Pattison D. I., and Davies M. J. (2012) Quantification of hydroxyl radical-derived oxidation products in peptides containing glycine, alanine, valine, and proline. Free Radic. Biol. Med. 52, 328–339 10.1016/j.freeradbiomed.2011.10.448 [DOI] [PubMed] [Google Scholar]
- 58. Schuessler H., and Davies J. V. (1983) Radiation-induced reduction reactions with bovine serum albumin. Int. J. Radiat. Biol. 43, 291–301 10.1080/09553008314550331 [DOI] [PubMed] [Google Scholar]
- 59. Schuessler H., and Denkl P. (1972) X-ray inactivation of lactate dehydrogenase in dilute solution. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 21, 435–443 10.1080/09553007214550511 [DOI] [PubMed] [Google Scholar]
- 60. Chevion M. (1988) A site-specific mechanism for free radical induced biological damage: the essential role of redox-active transition metals. Free Radic. Biol. Med. 5, 27–37 10.1016/0891-5849(88)90059-7 [DOI] [PubMed] [Google Scholar]
- 61. Easton C. J., and Hay M. P. (1986) Preferential reactivity of glycine residues in free radical reactions of amino acid derivatives. J. Chem. Soc. Chem. Commun. 1986, 55–57 10.1039/C39860000055 [DOI] [Google Scholar]
- 62. Augusto O., Goldstein S., Hurst J. K., Lind J., Lymar S. V., Merenyi G., and Radi R. (2019) Carbon dioxide-catalyzed peroxynitrite reactivity–The resilience of the radical mechanism after two decades of research. Free Radic. Biol. Med. 135, 210–215 10.1016/j.freeradbiomed.2019.02.026 [DOI] [PubMed] [Google Scholar]
- 63. Serrano-Luginbuehl S., Kissner R., and Koppenol W. H. (2018) Reaction of CO2 with ONOO−: one molecule of CO2 is not enough. Chem. Res. Toxicol. 31, 721–730 10.1021/acs.chemrestox.8b00068 [DOI] [PubMed] [Google Scholar]
- 64. Ischiropoulos H., and al-Mehdi A. B. (1995) Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 364, 279–282 10.1016/0014-5793(95)00307-U [DOI] [PubMed] [Google Scholar]
- 65. Alvarez B., and Radi R. (2003) Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25, 295–311 10.1007/s00726-003-0018-8 [DOI] [PubMed] [Google Scholar]
- 66. Hawkins C. L., Pattison D. I., and Davies M. J. (2003) Hypochlorite-induced oxidation of amino acids, peptides and proteins. Amino Acids 25, 259–274 10.1007/s00726-003-0016-x [DOI] [PubMed] [Google Scholar]
- 67. Thomas E. L. (1979) Myeloperoxidase, hydrogen peroxide, chloride antimicrobial system: nitrogen-chlorine derivatives of bacterial components in bactericidal action against Escherichia coli. Infect. Immun. 23, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Peskin A. V., Midwinter R. G., Harwood D. T., and Winterbourn C. C. (2004) Chlorine transfer between glycine, taurine, and histamine: reaction rates and impact on cellular reactivity. Free Radic. Biol. Med. 37, 1622–1630 10.1016/j.freeradbiomed.2004.08.010 [DOI] [PubMed] [Google Scholar]
- 69. Kettle A. J. (1996) Neutrophils convert tyrosyl residues in albumin to chlorotyrosine. FEBS Lett. 379, 103–106 10.1016/0014-5793(95)01494-2 [DOI] [PubMed] [Google Scholar]
- 70. Ferrer-Sueta G., Manta B., Botti H., Radi R., Trujillo M., and Denicola A. (2011) Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 24, 434–450 10.1021/tx100413v [DOI] [PubMed] [Google Scholar]
- 71. Criado S., Soltermann A. T., Marioli J. M., and García N. A. (1998) Sensitized photooxidation of di- and tripeptides of tyrosine. Photochem. Photobiol. 68, 453–458 10.1111/j.1751-1097.1998.tb02499.x [DOI] [PubMed] [Google Scholar]
- 72. Butler J., Land E. J., Prutz W. A., and Swallow A. J. (1982) Charge transfer between tryptophan and tyrosine in proteins. Biochim. Biophys. Acta 705, 150–162 10.1016/0167-4838(82)90173-X [DOI] [Google Scholar]
- 73. Prütz W. A., Butler J., Land E. J., and Swallow A. J. (1989) The role of sulphur peptide functions in free radical transfer: a pulse radiolysis study. Int. J. Radiat. Biol. 55, 539–556 10.1080/09553008914550591 [DOI] [PubMed] [Google Scholar]
- 74. Prütz W. A., and Land E. J. (1979) Charge transfer in peptides. Pulse radiolysis investigation of one-electron reactions in dipeptides of tryptophan and tyrosine. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 36, 513–520 10.1080/09553007914551301 [DOI] [PubMed] [Google Scholar]
- 75. Prüutz W. A., Siebert F., Butler J., Land E. J., Menez A., and Montenay-Garestier T. (1982) Charge transfer in peptides. Intramolecular radical transformations involving methionine, tryptophan and tyrosine. Biochim. Biophys. Acta 705, 139–149 10.1016/0167-4838(82)90172-8 [DOI] [Google Scholar]
- 76. Aubert C., Vos M. H., Mathis P., Eker A. P., and Brettel K. (2000) Intraprotein radical transfer during photoactivation of DNA photolyase. Nature 405, 586–590 10.1038/35014644 [DOI] [PubMed] [Google Scholar]
- 77. Barry B. A., Chen J., Keough J., Jenson D., Offenbacher A., and Pagba C. (2012) Proton coupled electron transfer and redox active tyrosines: structure and function of the tyrosyl radicals in ribonucleotide reductase and photosystem II. J. Phys. Chem. Lett. 3, 543–554 10.1021/jz2014117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kato Y., Wu X., Naito M., Nomura H., Kitamoto N., and Osawa T. (2000) Immunochemical detection of protein dityrosine in atherosclerotic lesion of apo-E–deficient mice using a novel monoclonal antibody. Biochem. Biophys. Res. Commun. 275, 11–15 10.1006/bbrc.2000.3265 [DOI] [PubMed] [Google Scholar]
- 79. Al-Hilaly Y. K., Biasetti L., Blakeman B. J., Pollack S. J., Zibaee S., Abdul-Sada A., Thorpe J. R., Xue W.-F., Serpell L. C., Goedert M., Wright J. A., Wang X., Brown D. R., Celej M. S., Winner B., et al. (2016) The involvement of dityrosine crosslinking in α-synuclein assembly and deposition in Lewy bodies in Parkinson's disease. Sci. Rep. 6, 39171 10.1038/srep39171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Degendorfer G., Chuang C. Y., Hammer A., Malle E., and Davies M. J. (2015) Peroxynitrous acid induces structural and functional modifications to basement membranes and its key component, laminin. Free Radic. Biol. Med. 89, 721–733 10.1016/j.freeradbiomed.2015.09.018 [DOI] [PubMed] [Google Scholar]
- 81. Degendorfer G., Chuang C. Y., Kawasaki H., Hammer A., Malle E., Yamakura F., and Davies M. J. (2016) Peroxynitrite-mediated oxidation of plasma fibronectin. Free Radic. Biol. Med. 97, 602–615 10.1016/j.freeradbiomed.2016.06.013 [DOI] [PubMed] [Google Scholar]
- 82. Degendorfer G., Chuang C. Y., Mariotti M., Hammer A., Hoefler G., Hägglund P., Malle E., Wise S. G., and Davies M. J. (2018) Exposure of tropoelastin to peroxynitrous acid gives high yields of nitrated tyrosine residues, di-tyrosine cross-links and altered protein structure and function. Free Radic. Biol. Med. 115, 219–231 10.1016/j.freeradbiomed.2017.11.019 [DOI] [PubMed] [Google Scholar]
- 83. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A. L., and Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 10.1074/mcp.M113.034009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tiwari M. K., Leinisch F., Sahin C., Møller I. M., Otzen D. E., Davies M. J., and Bjerrum M. J. (2018) Early events in copper-ion catalyzed oxidation of α-synuclein. Free Radic. Biol. Med. 121, 38–50 10.1016/j.freeradbiomed.2018.04.559 [DOI] [PubMed] [Google Scholar]
- 85. Fuentes-Lemus E., Silva E., Leinisch F., Dorta E., Lorentzen L. G., Davies M. J., and López-Alarcon C. (2018) α- and β-casein aggregation induced by riboflavin-sensitized photo-oxidation occurs via di-tyrosine cross-links and is oxygen concentration dependent. Food Chem. 256, 119–128 10.1016/j.foodchem.2018.02.090 [DOI] [PubMed] [Google Scholar]
- 86. Vissers M. C., and Thomas C. (1997) Hypochlorous acid disrupts the adhesive properties of subendothelial matrix. Free Radic. Biol. Med. 23, 401–411 10.1016/S0891-5849(96)00619-3 [DOI] [PubMed] [Google Scholar]
- 87. Olszowski S., Olszowska E., Kusior D., Piwowarczyk M., and Stelmaszynska T. (2003) Hypochlorite action on plasma fibronectin promotes its extended conformation in complexes with antibodies. J. Protein Chem. 22, 449–456 10.1023/B:JOPC.0000005460.94172.1d [DOI] [PubMed] [Google Scholar]
- 88. Pankhurst G., Wang X. L., Wilcken D. E., Baernthaler G., Panzenböck U., Raftery M., and Stocker R. (2003) Characterization of specifically oxidized apolipoproteins in mildly oxidized high density lipoprotein. J. Lipid Res. 44, 349–355 10.1194/jlr.M200256-JLR200 [DOI] [PubMed] [Google Scholar]
- 89. Garner B., Witting P. K., Waldeck A. R., Christison J. K., Raftery M., and Stocker R. (1998) Oxidation of high density lipoproteins. I. Formation of methionine sulfoxide in apolipoproteins AI and AII is an early event that accompanies lipid peroxidation and can be enhanced by α-tocopherol. J. Biol. Chem. 273, 6080–6087 10.1074/jbc.273.11.6080 [DOI] [PubMed] [Google Scholar]
- 90. Evrard C., Capron A., Marchand C., Clippe A., Wattiez R., Soumillion P., Knoops B., and Declercq J. P. (2004) Crystal structure of a dimeric oxidized form of human peroxiredoxin 5. J. Mol. Biol. 337, 1079–1090 10.1016/j.jmb.2004.02.017 [DOI] [PubMed] [Google Scholar]
- 91. Smeets A., Evrard C., Landtmeters M., Marchand C., Knoops B., and Declercq J. P. (2005) Crystal structures of oxidized and reduced forms of human mitochondrial thioredoxin 2. Protein Sci. 14, 2610–2621 10.1110/ps.051632905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Thorn D. C., Grosas A. B., Mabbitt P. D., Ray N. J., Jackson C. J., and Carver J. A. (2019) The structure and stability of the disulfide-linked γS-crystallin dimer provide insight into oxidation products associated with lens cataract formation. J. Mol. Biol. 431, 483–497 10.1016/j.jmb.2018.12.005 [DOI] [PubMed] [Google Scholar]
- 93. Davies M. J., Fu S., Wang H., and Dean R. T. (1999) Stable markers of oxidant damage to proteins and their application in the study of human disease. Free Radic. Biol. Med. 27, 1151–1163 10.1016/S0891-5849(99)00206-3 [DOI] [PubMed] [Google Scholar]
- 94. Ehrenshaft M., Deterding L. J., and Mason R. P. (2015) Tripping up Trp: modification of protein tryptophan residues by reactive oxygen species, modes of detection, and biological consequences. Free Radic. Biol. Med. 89, 220–228 10.1016/j.freeradbiomed.2015.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Eftink M. R. (1998) The use of fluorescence methods to monitor unfolding transitions in proteins. Biochemistry 63, 276–284 [PubMed] [Google Scholar]
- 96. Weigele M., DeBarnardo S. L., Tengi J. P., and Leimgruber W. (1972) A novel reagent for the fluorometric assay of primary amines. J. Am. Chem. Soc. 94, 5927–5928 10.1021/ja00771a084 [DOI] [Google Scholar]
- 97. Smith R. E., and MacQuarrie R. (1978) A sensitive fluorometric method for the determination of arginine using 9,10-phenanthrenequinone. Anal. Biochem. 90, 246–255 10.1016/0003-2697(78)90029-5 [DOI] [PubMed] [Google Scholar]
- 98. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 10.1016/0003-9861(59)90090-6 [DOI] [PubMed] [Google Scholar]
- 99. Hu M. L. (1994) Measurement of protein thiol groups and glutathione in plasma. Methods Enzymol. 233, 380–385 10.1016/S0076-6879(94)33044-1 [DOI] [PubMed] [Google Scholar]
- 100. Hansen R. E., Østergaard H., Nørgaard P., and Winther J. R. (2007) Quantification of protein thiols and dithiols in the picomolar range using sodium borohydride and 4,4′-dithiodipyridine. Anal. Biochem. 363, 77–82 10.1016/j.ab.2007.01.002 [DOI] [PubMed] [Google Scholar]
- 101. Sharov V. S., Dremina E. S., Galeva N. A., Williams T. D., and Schöneich C. (2006) Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem. J. 394, 605–615 10.1042/BJ20051214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schöneich C., and Sharov V. S. (2006) Mass spectrometry of protein modifications by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 41, 1507–1520 10.1016/j.freeradbiomed.2006.08.013 [DOI] [PubMed] [Google Scholar]
- 103. Liu Y., Hou W., Sun H., Cui C., Zhang L., Jiang Y., Wu Y., Wang Y., Li J., Sumerlin B. S., Liu Q., and Tan W. (2017) Thiol-ene click chemistry: a biocompatible way for orthogonal bioconjugation of colloidal nanoparticles. Chem. Sci. 8, 6182–6187 10.1039/C7SC01447C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chen G., Feng H., Xi W., Xu J., Pan S., and Qian Z. (2019) Thiol-ene click reaction-induced fluorescence enhancement by altering the radiative rate for assaying butyrylcholinesterase activity. Analyst 144, 559–566 10.1039/C8AN01808A [DOI] [PubMed] [Google Scholar]
- 105. Baty J. W., Hampton M. B., and Winterbourn C. C. (2002) Detection of oxidant sensitive thiol proteins by fluorescence labeling and two-dimensional electrophoresis. Proteomics 2, 1261–1266 10.1002/1615-9861(200209)2:9<1261::AID-PROT1261>3.0.CO;2-Q [DOI] [PubMed] [Google Scholar]
- 106. Turecek F. (2002) Mass spectrometry in coupling with affinity capture-release and isotope-coded affinity tags for quantitative protein analysis. J. Mass Spectrom. 37, 1–14 10.1002/jms.275 [DOI] [PubMed] [Google Scholar]
- 107. Yang C. S., Chou S. T., Liu L., Tsai P. J., and Kuo J. S. (1995) Effect of ageing on human plasma glutathione concentrations as determined by high-performance liquid chromatography with fluorimetric detection. J. Chromatograph. B 674, 23–30 10.1016/0378-4347(95)00287-8 [DOI] [PubMed] [Google Scholar]
- 108. Moriarty-Craige S. E., and Jones D. P. (2004) Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 24, 481–509 10.1146/annurev.nutr.24.012003.132208 [DOI] [PubMed] [Google Scholar]
- 109. Meister A. (1988) Glutathione metabolism and its selective modification. J. Biol. Chem. 263, 17205–17208 [PubMed] [Google Scholar]
- 110. Deponte M. (2013) Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 1830, 3217–3266 10.1016/j.bbagen.2012.09.018 [DOI] [PubMed] [Google Scholar]
- 111. Albrecht S. C., Sobotta M. C., Bausewein D., Aller I., Hell R., Dick T. P., and Meyer A. J. (2014) Redesign of genetically encoded biosensors for monitoring mitochondrial redox status in a broad range of model eukaryotes. J. Biomol. Screen. 19, 379–386 10.1177/1087057113499634 [DOI] [PubMed] [Google Scholar]
- 112. Ezeriņa D., Morgan B., and Dick T. P. (2014) Imaging dynamic redox processes with genetically encoded probes. J. Mol. Cell Cardiol. 73, 43–49 10.1016/j.yjmcc.2013.12.023 [DOI] [PubMed] [Google Scholar]
- 113. Richie J. P. Jr., and Lang C. A. (1987) The determination of glutathione, cyst(e)ine, and other thiols and disulfides in biological samples using high-performance liquid chromatography with dual electrochemical detection. Anal. Biochem. 163, 9–15 10.1016/0003-2697(87)90085-6 [DOI] [PubMed] [Google Scholar]
- 114. Martin J., and White I. N. (1991) Fluorimetric determination of oxidised and reduced glutathione in cells and tissues by high-performance liquid chromatography following derivatization with dansyl chloride. J. Chromatogr. 568, 219–225 10.1016/0378-4347(91)80356-H [DOI] [PubMed] [Google Scholar]
- 115. Johnson J. M., Strobel F. H., Reed M., Pohl J., and Jones D. P. (2008) A rapid LC-FTMS method for the analysis of cysteine, cystine and cysteine/cystine steady-state redox potential in human plasma. Clin. Chim. Acta 396, 43–48 10.1016/j.cca.2008.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Eaton P. (2006) Protein thiol oxidation in health and disease: techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic. Biol. Med. 40, 1889–1899 10.1016/j.freeradbiomed.2005.12.037 [DOI] [PubMed] [Google Scholar]
- 117. Baty J. W., Hampton M. B., and Winterbourn C. C. (2005) Proteomic detection of hydrogen peroxide-sensitive thiol proteins in Jurkat cells. Biochem. J. 389, 785–795 10.1042/BJ20050337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Creighton T. E. (1984) Disulfide bond formation in proteins. Methods Enzymol. 107, 305–329 10.1016/0076-6879(84)07021-X [DOI] [PubMed] [Google Scholar]
- 119. Yazdanparast R., Andrews P., Smith D. L., and Dixon J. E. (1986) A new approach for detection and assignment of disulfide bonds in peptides. Anal. Biochem. 153, 348–353 10.1016/0003-2697(86)90102-8 [DOI] [PubMed] [Google Scholar]
- 120. Brustolon M., and Giamello E. (eds) (2008) Electron Paramagnetic Resonance: A Practitioner's Toolkit, pp. 1–608, John Wiley and Sons, Inc., Hoboken, NJ [Google Scholar]
- 121. Davies M. J., and Hawkins C. L. (2004) EPR spin trapping of protein radicals. Free Radic. Biol. Med. 36, 1072–1086 10.1016/j.freeradbiomed.2003.12.013 [DOI] [PubMed] [Google Scholar]
- 122. Hawkins C. L., and Davies M. J. (2014) Detection and characterisation of radicals in biological materials using EPR methodology. Biochim. Biophys. Acta 1840, 708–721 10.1016/j.bbagen.2013.03.034 [DOI] [PubMed] [Google Scholar]
- 123. Davies M. J. (2016) Detection and characterisation of radicals using electron paramagnetic resonance (EPR) spin trapping and related methods. Methods 109, 21–30 10.1016/j.ymeth.2016.05.013 [DOI] [PubMed] [Google Scholar]
- 124. Mason R. P., and Ganini D. (2019) Immuno-spin trapping of macromolecules free radicals in vitro and in vivo–One stop shopping for free radical detection. Free Radic. Biol. Med. 131, 318–331 10.1016/j.freeradbiomed.2018.11.009 [DOI] [PubMed] [Google Scholar]
- 125. Mason R. P. (2016) Imaging free radicals in organelles, cells, tissue, and in vivo with immuno-spin trapping. Redox Biol. 8, 422–429 10.1016/j.redox.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Luxford C., Morin B., Dean R. T., and Davies M. J. (1999) Histone H1- and other protein- and amino acid-hydroperoxides can give rise to free radicals which oxidize DNA. Biochem. J. 344, 125–134 10.1042/bj3440125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Luxford C., Dean R. T., and Davies M. J. (2000) Radicals derived from histone hydroperoxides damage nucleobases in RNA and DNA. Chem. Res. Toxicol. 13, 665–672 10.1021/tx000053u [DOI] [PubMed] [Google Scholar]
- 128. Morgan P. E., Dean R. T., and Davies M. J. (2004) Protective mechanisms against peptide and protein peroxides generated by singlet oxygen. Free Radic. Biol. Med. 36, 484–496 10.1016/j.freeradbiomed.2003.11.021 [DOI] [PubMed] [Google Scholar]
- 129. Morgan P. E., Dean R. T., and Davies M. J. (2002) Inhibition of glyceraldehyde-3-phosphate dehydrogenase by peptide and protein peroxides generated by singlet oxygen attack. Eur. J. Biochem. 269, 1916–1925 10.1046/j.1432-1033.2002.02845.x [DOI] [PubMed] [Google Scholar]
- 130. Dremina E. S., Sharov V. S., Davies M. J., and Schöneich C. (2007) Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem. Res. Toxicol. 20, 1462–1469 10.1021/tx700108w [DOI] [PubMed] [Google Scholar]
- 131. Fu S., Dean R., Southan M., and Truscott R. (1998) The hydroxyl radical in lens nuclear cataractogenesis. J. Biol. Chem. 273, 28603–28609 10.1074/jbc.273.44.28603 [DOI] [PubMed] [Google Scholar]
- 132. Fu S., Davies M. J., Stocker R., and Dean R. T. (1998) Evidence for roles of radicals in protein oxidation in advanced human atherosclerotic plaque. Biochem. J. 333, 519–525 10.1042/bj3330519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jessup W., Dean R. T., and Gebicki J. M. (1994) Iodometric determination of hydroperoxides in lipids and proteins. Methods Enzymol. 233, 289–303 10.1016/S0076-6879(94)33032-8 [DOI] [Google Scholar]
- 134. Gay C. A., and Gebicki J. M. (2003) Measurement of protein and lipid hydroperoxides in biological systems by the ferric-xylenol orange method. Anal. Biochem. 315, 29–35 10.1016/S0003-2697(02)00606-1 [DOI] [PubMed] [Google Scholar]
- 135. Wolff S. P. (1994) Ferrous ion oxidation in the presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 233, 182–189 10.1016/S0076-6879(94)33021-2 [DOI] [Google Scholar]
- 136. Bou R., Codony R., Tres A., Decker E. A., and Guardiola F. (2008) Determination of hydroperoxides in foods and biological samples by the ferrous oxidation-xylenol orange method: a review of the factors that influence the method's performance. Anal. Biochem. 377, 1–15 10.1016/j.ab.2008.02.029 [DOI] [PubMed] [Google Scholar]
- 137. Michalski R., Zielonka J., Gapys E., Marcinek A., Joseph J., and Kalyanaraman B. (2014) Real-time measurements of amino acid and protein hydroperoxides using coumarin boronic acid. J. Biol. Chem. 289, 22536–22553 10.1074/jbc.M114.553727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hardy M., Zielonka J., Karoui H., Sikora A., Michalski R., Podsiadły R., Lopez M., Vasquez-Vivar J., Kalyanaraman B., and Ouari O. (2018) Detection and characterization of reactive oxygen and nitrogen species in biological systems by monitoring species-specific products. Antioxid. Redox Signal. 28, 1416–1432 10.1089/ars.2017.7398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zielonka J., Sikora A., Hardy M., Joseph J., Dranka B. P., and Kalyanaraman B. (2012) Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem. Res. Toxicol. 25, 1793–1799 10.1021/tx300164j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Weiss S. J., Lampert M. B., and Test S. T. (1983) Long-lived oxidants generated by human neutrophils: characterization and bioactivity. Science 222, 625–628 10.1126/science.6635660 [DOI] [PubMed] [Google Scholar]
- 141. Thomas E. L., Grisham M. B., and Jefferson M. M. (1986) Preparation and characterization of chloramines. Methods Enzymol. 132, 569–585 10.1016/S0076-6879(86)32042-1 [DOI] [PubMed] [Google Scholar]
- 142. Dypbukt J. M., Bishop C., Brooks W. M., Thong B., Eriksson H., and Kettle A. J. (2005) A sensitive and selective assay for chloramine production by myeloperoxidase. Free Radic. Biol. Med. 39, 1468–1477 10.1016/j.freeradbiomed.2005.07.008 [DOI] [PubMed] [Google Scholar]
- 143. Raftery M. J. (2007) Detection and characterization of N-α-chloramines by electrospray tandem mass spectrometry. Anal. Biochem. 366, 218–227 10.1016/j.ab.2007.04.016 [DOI] [PubMed] [Google Scholar]
- 144. Yang J., Gupta V., Tallman K. A., Porter N. A., Carroll K. S., and Liebler D. C. (2015) Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 10, 1022–1037 10.1038/nprot.2015.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Takakura K., Beckman J. S., MacMillan-Crow L. A., and Crow J. P. (1999) Rapid and irreversible inactivation of protein tyrosine phosphatases PTP1b, CD45, and LAR by peroxynitrite. Arch. Biochem. Biophys. 369, 197–207 10.1006/abbi.1999.1374 [DOI] [PubMed] [Google Scholar]
- 146. Ross S. H., Lindsay Y., Safrany S. T., Lorenzo O., Villa F., Toth R., Clague M. J., Downes C. P., and Leslie N. R. (2007) Differential redox regulation within the PTP superfamily. Cell. Signal. 19, 1521–1530 10.1016/j.cellsig.2007.01.026 [DOI] [PubMed] [Google Scholar]
- 147. Hogg D. R. (1990) The Chemistry of Sulphenic Acids and Their Derivatives (Saul P. ed) pp. 361–402, John Wiley & Sons, New York [Google Scholar]
- 148. Akter S., Fu L., Jung Y., Conte M. L., Lawson J. R., Lowther W. T., Sun R., Liu K., Yang J., and Carroll K. S. (2018) Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 14, 995–1004 10.1038/s41589-018-0116-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Li R., and Kast J. (2017) Biotin switch assays for quantitation of reversible cysteine oxidation. Methods Enzymol. 585, 269–284 10.1016/bs.mie.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 150. Forman H. J., Davies M. J., Krämer A. C., Miotto G., Zaccarin M., Zhang H., and Ursini F. (2017) Protein cysteine oxidation in redox signaling: caveats on sulfenic acid detection and quantification. Arch. Biochem. Biophys. 617, 26–37 10.1016/j.abb.2016.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Furdui C. M., and Poole L. B. (2014) Chemical approaches to detect and analyze protein sulfenic acids. Mass Spectrom. Rev. 33, 126–146 10.1002/mas.21384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Gupta V., Paritala H., and Carroll K. S. (2016) Reactivity, selectivity, and stability in sulfenic acid detection: a comparative study of nucleophilic and electrophilic probes. Bioconjug. Chem. 27, 1411–1418 10.1021/acs.bioconjchem.6b00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Lo Conte M., Lin J., Wilson M. A., and Carroll K. S. (2015) A chemical approach for the detection of protein sulfinylation. ACS Chem. Biol. 10, 1825–1830 10.1021/acschembio.5b00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Saurin A. T., Neubert H., Brennan J. P., and Eaton P. (2004) Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U.S.A. 101, 17982–17987 10.1073/pnas.0404762101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Janssen-Heininger Y. M., Mossman B. T., Heintz N. H., Forman H. J., Kalyanaraman B., Finkel T., Stamler J. S., Rhee S. G., and van der Vliet A. (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17 10.1016/j.freeradbiomed.2008.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chiesa J. J., Baidanoff F. M., and Golombek D. A. (2018) Don't just say no: differential pathways and pharmacological responses to diverse nitric oxide donors. Biochem. Pharmacol. 156, 1–9 10.1016/j.bcp.2018.08.002 [DOI] [PubMed] [Google Scholar]
- 157. Nakamura T., and Lipton S. A. (2008) Emerging roles of S-nitrosylation in protein misfolding and neurodegenerative diseases. Antioxid. Redox Signal. 10, 87–101 10.1089/ars.2007.1858 [DOI] [PubMed] [Google Scholar]
- 158. Jaffrey S. R., Erdjument-Bromage H., Ferris C. D., Tempst P., and Snyder S. H. (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197 10.1038/35055104 [DOI] [PubMed] [Google Scholar]
- 159. Devarie-Baez N. O., Zhang D., Li S., Whorton A. R., and Xian M. (2013) Direct methods for detection of protein S-nitrosylation. Methods 62, 171–176 10.1016/j.ymeth.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Chiappetta G., Ndiaye S., Igbaria A., Kumar C., Vinh J., and Toledano M. B. (2010) Proteome screens for Cys residues oxidation: the redoxome. Methods Enzymol. 473, 199–216 10.1016/S0076-6879(10)73010-X [DOI] [PubMed] [Google Scholar]
- 161. Gladwin M. T., Wang X., and Hogg N. (2006) Methodological vexation about thiol oxidation versus S-nitrosation. Free Radic. Biol. Med. 41, 557–561 10.1016/j.freeradbiomed.2006.05.025 [DOI] [PubMed] [Google Scholar]
- 162. Forrester M. T., Foster M. W., and Stamler J. S. (2007) Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J. Biol. Chem. 282, 13977–13983 10.1074/jbc.M609684200 [DOI] [PubMed] [Google Scholar]
- 163. Beuve A., Wu C., Cui C., Liu T., Jain M. R., Huang C., Yan L., Kholodovych V., and Li H. (2016) Identification of novel S-nitrosation sites in soluble guanylyl cyclase, the nitric oxide receptor. J. Proteomics 138, 40–47 10.1016/j.jprot.2016.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wu C., Liu T., Wang Y., Yan L., Cui C., Beuve A., and Li H. (2018) Biotin switch processing and mass spectrometry analysis of S-nitrosated thioredoxin and its transnitrosation targets. Methods Mol. Biol. 1747, 253–266 10.1007/978-1-4939-7695-9_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lancaster J. R., Jr. (2017) How are nitrosothiols formed de novo in vivo? Arch. Biochem. Biophys. 617, 137–144 10.1016/j.abb.2016.10.015 [DOI] [PubMed] [Google Scholar]
- 166. Wynia-Smith S. L., and Smith B. C. (2017) Nitrosothiol formation and S-nitrosation signaling through nitric oxide synthases. Nitric Oxide 63, 52–60 10.1016/j.niox.2016.10.001 [DOI] [PubMed] [Google Scholar]
- 167. Liu L., Hausladen A., Zeng M., Que L., Heitman J., and Stamler J. S. (2001) A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 410, 490–494 10.1038/35068596 [DOI] [PubMed] [Google Scholar]
- 168. Benhar M., Forrester M. T., Hess D. T., and Stamler J. S. (2008) Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320, 1050–1054 10.1126/science.1158265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Filipovic M. R., Zivanovic J., Alvarez B., and Banerjee R. (2018) Chemical biology of H2S signaling through persulfidation. Chem. Rev. 118, 1253–1337 10.1021/acs.chemrev.7b00205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Bełtowski J. (2019) Synthesis, metabolism, and signaling mechanisms of hydrogen sulfide: an overview. Methods Mol. Biol. 2007, 1–8 10.1007/978-1-4939-9528-8_1 [DOI] [PubMed] [Google Scholar]
- 171. Paul B. D., and Snyder S. H. (2018) Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 149, 101–109 10.1016/j.bcp.2017.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Zhang D., Macinkovic I., Devarie-Baez N. O., Pan J., Park C. M., Carroll K. S., Filipovic M. R., and Xian M. (2014) Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 53, 575–581 10.1002/anie.201305876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., and Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal. 2, ra72 10.1126/scisignal.2000464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Kalyanaraman B., Darley-Usmar V., Davies K. J., Dennery P. A., Forman H. J., Grisham M. B., Mann G. E., Moore K., Roberts L. J. 2nd., and Ischiropoulos H. (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6 10.1016/j.freeradbiomed.2011.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kouroussis E., Adhikari B., Zivanovic J., and Filipovic M. R. (2019) Measurement of protein persulfidation: improved tag-switch method. Methods Mol. Biol. 2007, 37–50 10.1007/978-1-4939-9528-8_4 [DOI] [PubMed] [Google Scholar]
- 176. Meng W., Chen Y., Feng Y., Zhang H., Xu Q., Sun M., Shi W., Cen J., Zhao J., and Xiao K. (2018) An off-on fluorescent probe for the detection of mitochondria-specific protein persulfidation. Org. Biomol. Chem. 16, 6350–6357 10.1039/C8OB01608A [DOI] [PubMed] [Google Scholar]
- 177. Aroca A., Benito J. M., Gotor C., and Romero L. C. (2017) Persulfidation proteome reveals the regulation of protein function by hydrogen sulfide in diverse biological processes in Arabidopsis. J. Exp. Bot. 68, 4915–4927 10.1093/jxb/erx294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhang D., Du J., Tang C., Huang Y., and Jin H. (2017) H2S-induced sulfhydration: biological function and detection methodology. Front. Pharmacol. 8, 608 10.3389/fphar.2017.00608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Dalle- Donne I., Rossib R., Giustarinib D., Milzania A., and Colomboa R. (2003) Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. 329, 23–38 10.1016/S0009-8981(03)00003-2 [DOI] [PubMed] [Google Scholar]
- 180. Requena J. R., Levine R. L., and Stadtman E. R. (2003) Recent advances in the analysis of oxidized proteins. Amino Acids 25, 221–226 10.1007/s00726-003-0012-1 [DOI] [PubMed] [Google Scholar]
- 181. Requena J. R., Chao C. C., Levine R. L., and Stadtman E. R. (2001) Glutamic and aminoadipic semialdehydes are the main carbonyl products of metal-catalyzed oxidation of proteins. Proc. Natl. Acad. Sci. U.S.A. 98, 69–74 10.1073/pnas.98.1.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Havelund J. F., Wojdyla K., Davies M. J., Jensen O. N., Møller I. M., and Rogowska-Wrzesinska A. (2017) Improved enrichment strategy for novel carbonylated amino acids in proteins from human plasma. J. Proteomics 156, 40–51 10.1016/j.jprot.2016.12.019 [DOI] [PubMed] [Google Scholar]
- 183. Headlam H. A., and Davies M. J. (2002) β-Scission of side-chain alkoxyl radicals on peptides and proteins results in the loss of side chains as aldehydes and ketones. Free Radic. Biol. Med. 32, 1171–1184 10.1016/S0891-5849(02)00814-6 [DOI] [PubMed] [Google Scholar]
- 184. Beal M. F. (2002) Oxidatively modified proteins in aging and disease. Free Radic. Biol. Med. 32, 797–803 10.1016/S0891-5849(02)00780-3 [DOI] [PubMed] [Google Scholar]
- 185. Stadtman E. R., and Levine R. L. (2003) Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids 25, 207–218 10.1007/s00726-003-0011-2 [DOI] [PubMed] [Google Scholar]
- 186. Levine R. L., Wehr N., Williams J. A., Stadtman E. R., and Shacter E. (2000) Determination of carbonyl groups in oxidized proteins. Methods Mol. Biol. 99, 15–24 10.1385/1-59259-054-3:15 [DOI] [PubMed] [Google Scholar]
- 187. Levine R. L., Williams J. A., Stadtman E. R., and Shacter E. (1994) Carbonyl assays for determination of oxidatively modified proteins. Methods Enzymol. 233, 346–357 10.1016/S0076-6879(94)33040-9 [DOI] [PubMed] [Google Scholar]
- 188. Alomari E., Bruno S., Ronda L., Paredi G., Bettati S., and Mozzarelli A. (2018) Protein carbonylation detection methods: a comparison. Data Brief 19, 2215–2220 10.1016/j.dib.2018.06.088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Mirzaei H., and Regnier F. (2005) Affinity chromatographic selection of carbonylated proteins followed by identification of oxidation sites using tandem mass spectrometry. Anal. Chem. 77, 2386–2392 10.1021/ac0484373 [DOI] [PubMed] [Google Scholar]
- 190. Mirzaei H., and Regnier F. (2007) Identification of yeast oxidized proteins: chromatographic top-down approach for identification of carbonylated, fragmented and cross-linked proteins in yeast. J. Chromatogr. A 1141, 22–31 10.1016/j.chroma.2006.11.009 [DOI] [PubMed] [Google Scholar]
- 191. Bollineni R. C., Fedorova M., Blüher M., and Hoffmann R. (2014) Carbonylated plasma proteins as potential biomarkers of obesity induced type 2 diabetes mellitus. J. Proteome Res. 13, 5081–5093 10.1021/pr500324y [DOI] [PubMed] [Google Scholar]
- 192. Bollineni R. C., Hoffmann R., and Fedorova M. (2014) Proteome-wide profiling of carbonylated proteins and carbonylation sites in HeLa cells under mild oxidative stress conditions. Free Radic. Biol. Med. 68, 186–195 10.1016/j.freeradbiomed.2013.11.030 [DOI] [PubMed] [Google Scholar]
- 193. Choi J., Rees H. D., Weintraub S. T., Levey A. I., Chin L. S., and Li L. (2005) Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 280, 11648–11655 10.1074/jbc.M414327200 [DOI] [PubMed] [Google Scholar]
- 194. Tiwari M. K., Hägglund P. M., Møller I. M., Davies M. J., and Bjerrum M. J. (2019) Copper ion/H2O2 oxidation of Cu/Zn-superoxide dismutase: implications for enzymatic activity and antioxidant action. Redox Biol. 26, 101262 10.1016/j.redox.2019.101262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Masson N., Keeley T. P., Giuntoli B., White M. D., Puerta M. L., Perata P., Hopkinson R. J., Flashman E., Licausi F., and Ratcliffe P. J. (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 365, 65–69 10.1126/science.aaw0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Sobotta M. C., Liou W., Stöcker S., Talwar D., Oehler M., Ruppert T., Scharf A. N., and Dick T. P. (2015) Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 11, 64–70 10.1038/nchembio.1695 [DOI] [PubMed] [Google Scholar]
- 197. Stöcker S., Maurer M., Ruppert T., and Dick T. P. (2018) A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 14, 148–155 10.1038/nchembio.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Stöcker S., Van Laer K., Mijuskovic A., and Dick T. P. (2018) The conundrum of hydrogen peroxide signaling and the emerging role of peroxiredoxins as redox relay hubs. Antioxid. Redox Signal. 28, 558–573 10.1089/ars.2017.7162 [DOI] [PubMed] [Google Scholar]
- 199. Rahuel-Clermont S., and Toledano M. B. (2018) Parsing protein sulfinic acid switches. Nat. Chem. Biol. 14, 991–993 10.1038/s41589-018-0151-z [DOI] [PubMed] [Google Scholar]
- 200. Giridharan S. S., and Caplan S. (2014) MICAL-family proteins: complex regulators of the actin cytoskeleton. Antioxid. Redox Signal. 20, 2059–2073 10.1089/ars.2013.5487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Frémont S., Romet-Lemonne G., Houdusse A., and Echard A. (2017) Emerging roles of MICAL family proteins–from actin oxidation to membrane trafficking during cytokinesis. J. Cell Sci. 130, 1509–1517 10.1242/jcs.202028 [DOI] [PubMed] [Google Scholar]
- 202. Pattison D. I., and Davies M. J. (2006) Reactions of myeloperoxidase-derived oxidants with biological substrates: gaining insight into human inflammatory diseases. Curr. Med. Chem. 13, 3271–3290 10.2174/092986706778773095 [DOI] [PubMed] [Google Scholar]
- 203. Liu H., Ponniah G., Neill A., Patel R., and Andrien B. (2013) Accurate determination of protein methionine oxidation by stable isotope labeling and LC-MS analysis. Anal. Chem. 85, 11705–11709 10.1021/ac403072w [DOI] [PubMed] [Google Scholar]
- 204. Hollemeyer K., Heinzle E., and Tholey A. (2002) Identification of oxidized methionine residues in peptides containing two methionine residues by derivatization and matrix-assisted laser desorption/ionization mass spectrometry. Proteomics 2, 1524–1531 10.1002/1615-9861(200211)2:11<1524::AID-PROT1524>3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 205. Shipman J. T., Go E. P., and Desaire H. (2018) Method for quantifying oxidized methionines and application to HIV-1 Env. J. Am. Soc. Mass Spectrom. 29, 2041–2047 10.1007/s13361-018-2010-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Vogt W. (1995) Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic. Biol. Med. 18, 93–105 10.1016/0891-5849(94)00158-G [DOI] [PubMed] [Google Scholar]
- 207. Tarrago L., Oheix E., Péterfi Z., and Gladyshev V. N. (2018) Monitoring of methionine sulfoxide content and methionine sulfoxide reductase activity. Methods Mol. Biol. 1661, 285–299 10.1007/978-1-4939-7258-6_20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Onorato J. M., Thorpe S. R., and Baynes J. W. (1998) Immunohistochemical and ELISA assays for biomarkers of oxidative stress in aging and disease. Ann. N.Y. Acad. Sci. 854, 277–290 10.1111/j.1749-6632.1998.tb09909.x [DOI] [PubMed] [Google Scholar]
- 209. Malle E., Hazell L., Stocker R., Sattler W., Esterbauer H., and Waeg G. (1995) Immunologic detection and measurement of hypochlorite-modified LDL with specific monoclonal antibodies. Arterioscl. Thromb. Vasc. Biol. 15, 982–989 10.1161/01.ATV.15.7.982 [DOI] [PubMed] [Google Scholar]
- 210. Chapman A. L., Senthilmohan R., Winterbourn C. C., and Kettle A. J. (2000) Comparison of mono- and dichlorinated tyrosines with carbonyls for detection of hypochlorous acid modified proteins. Arch. Biochem. Biophys. 377, 95–100 10.1006/abbi.2000.1744 [DOI] [PubMed] [Google Scholar]
- 211. Cai H., Chuang C. Y., Vanichkitrungruang S., Hawkins C. L., and Davies M. J. (2019) Hypochlorous acid-modified extracellular matrix contributes to the behavioural switching of human coronary artery smooth muscle cells. Free Radic. Biol. Med. 134, 516–526 10.1016/j.freeradbiomed.2019.01.044 [DOI] [PubMed] [Google Scholar]
- 212. Hazell L. J., Arnold L., Flowers D., Waeg G., Malle E., and Stocker R. (1996) Presence of hypochlorite-modified proteins in human atherosclerotic lesions. J. Clin. Invest. 97, 1535–1544 10.1172/JCI118576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Hazell L. J., Baernthaler G., and Stocker R. (2001) Correlation between intima-to-media ratio, apolipoprotein B-100, myeloperoxidase, and hypochlorite-oxidized proteins in human atherosclerosis. Free Radic. Biol. Med. 31, 1254–1262 10.1016/S0891-5849(01)00717-1 [DOI] [PubMed] [Google Scholar]
- 214. Buss H., Chan T. P., Sluis K. B., Domigan N. M., and Winterbourn C. C. (1997) Protein carbonyl measurement by a sensitive ELISA method. Free Radic. Biol. Med. 23, 361–366 10.1016/S0891-5849(97)00104-4 [DOI] [PubMed] [Google Scholar]
- 215. Aksenov M. Y., Aksenova M. V., Butterfield D. A., Geddes J. W., and Markesbery W. R. (2001) Protein oxidation in the brain in Alzheimer's disease. Neuroscience 103, 373–383 10.1016/S0306-4522(00)00580-7 [DOI] [PubMed] [Google Scholar]
- 216. Nakamura A., and Goto S. (1996) Analysis of protein carbonyls with 2,4-dinitrophenyl hydrazine and its antibodies by immunoblot in two-dimensional gel electrophoresis. Biochemistry 119, 768–774 10.1093/oxfordjournals.jbchem.a021306 [DOI] [PubMed] [Google Scholar]
- 217. Conrad C. C., Choi J., Malakowsky C. A., Talent J. M., Dai R., Marshall P., and Gracy R. W. (2001) Identification of protein carbonyls after two-dimensional electrophoresis. Proteomics 1, 829–834 10.1002/1615-9861(200107)1:7<829::AID-PROT829>3.0.CO;2-R [DOI] [PubMed] [Google Scholar]
- 218. Augustyniak E., Adam A., Wojdyla K., Rogowska-Wrzesinska A., Willetts R., Korkmaz A., Atalay M., Weber D., Grune T., Borsa C., Gradinaru D., and Chand Bollineni R., Fedorova M., and Griffiths H. R. (2015) Validation of protein carbonyl measurement: a multi-centre study. Redox Biol. 4, 149–157 10.1016/j.redox.2014.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Weber D., Davies M. J., and Grune T. (2015) Determination of protein carbonyls in plasma, cell extracts, tissue homogenates, isolated proteins: focus on sample preparation and derivatization conditions. Redox Biol. 5, 367–380 10.1016/j.redox.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. van Dalen C. J., Aldridge R. E., Chan T., Senthilmohan R., Hancox R. J., Cowan J. O., Taylor D. R., Town G. I., and Kettle A. J. (2009) Bromotyrosines in sputum proteins and treatment effects of terbutaline and budesonide in asthma. Ann. Allergy Asthma Immunol. 103, 348–353 10.1016/S1081-1206(10)60536-4 [DOI] [PubMed] [Google Scholar]
- 221. Wu W., Chen Y., d'Avignon A., and Hazen S. L. (1999) 3-Bromotyrosine and 3,5-dibromotyrosine are major products of protein oxidation by eosinophil peroxidase: potential markers for eosinophil-dependent tissue injury in vivo. Biochemistry 38, 3538–3548 10.1021/bi982401l [DOI] [PubMed] [Google Scholar]
- 222. Beckmann J. S., Ye Y. Z., Anderson G., Chen J., Accavitti M. A., Tarpey M. M., and White C. R. (1994) Extensive nitration of protein tyrosines in human atherosclerosis detected by immunohistochemistry. Biol. Chem. Hoppe-Seyler 375, 81–88 10.1515/bchm3.1994.375.2.81 [DOI] [PubMed] [Google Scholar]
- 223. Gieseg S. P., Simpson J. A., Charlton T. S., Duncan M. W., and Dean R. T. (1993) Protein-bound 3,4-dihydroxyphenylalanine is a major reductant formed during hydroxyl radical damage to proteins. Biochemistry 32, 4780–4786 10.1021/bi00069a012 [DOI] [PubMed] [Google Scholar]
- 224. Gaut J. P., Byun J., Tran H. D., and Heinecke J. W. (2002) Artifact-free quantification of free 3-chlorotyrosine, 3-bromotyrosine, and 3-nitrotyrosine in human plasma by electron capture-negative chemical ionization gas chromatography mass spectrometry and liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Biochem. 300, 252–259 10.1006/abio.2001.5469 [DOI] [PubMed] [Google Scholar]
- 225. Paz M. A., Flückiger R., Boak A., Kagan H. M., and Gallop P. M. (1991) Specific detection of quinoproteins by redox-cycling staining. J. Biol. Chem. 266, 689–692 [PubMed] [Google Scholar]
- 226. Hecht E. S., Oberg A. L., and Muddiman D. C. (2016) Optimizing mass spectrometry analyses: a tailored review on the utility of design of experiments. J. Am. Soc. Mass Spectrom. 27, 767–785 10.1007/s13361-016-1344-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Leinisch F., Mariotti M., Hägglund P., and Davies M. J. (2018) Structural and functional changes in RNase A originating from tyrosine and histidine cross-linking and oxidation. Free Radic. Biol. Med. 126, 73–86 10.1016/j.freeradbiomed.2018.07.008 [DOI] [PubMed] [Google Scholar]
- 228. Bachi A., Dalle-Donne I., and Scaloni A. (2013) Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chem. Rev. 113, 596–698 10.1021/cr300073p [DOI] [PubMed] [Google Scholar]
- 229. Rathore D., Faustino A., Schiel J., Pang E., Boyne M., and Rogstad S. (2018) The role of mass spectrometry in the characterization of biologic protein products. Expert Rev. Proteomics 15, 431–449 10.1080/14789450.2018.1469982 [DOI] [PubMed] [Google Scholar]
- 230. Baez N. O., Reisz J. A., and Furdui C. M. (2015) Mass spectrometry in studies of protein thiol chemistry and signaling: opportunities and caveats. Free Radic. Biol. Med. 80, 191–211 10.1016/j.freeradbiomed.2014.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Cravatt B. F., Simon G. M., and Yates J. R. 3rd. (2007) The biological impact of mass-spectrometry-based proteomics. Nature 450, 991–1000 10.1038/nature06525 [DOI] [PubMed] [Google Scholar]
- 232. Aebersold R., and Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 10.1038/nature01511 [DOI] [PubMed] [Google Scholar]
- 233. Roepstorff P., and Fohlman J. (1984) Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 11, 601 10.1002/bms.1200111109 [DOI] [PubMed] [Google Scholar]
- 234. Müller T., and Winter D. (2017) Systematic evaluation of protein reduction and alkylation reveals massive unspecific side effects by iodine-containing reagents. Mol. Cell. Proteomics 16, 1173–1187 10.1074/mcp.M116.064048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Nybo T., Cai H., Chuang C. Y., Gamon L. F., Rogowska-Wrzesinska A., and Davies M. J. (2018) Chlorination and oxidation of human plasma fibronectin by myeloperoxidase-derived oxidants, and its consequences for smooth muscle cell function. Redox Biol. 19, 388–400 10.1016/j.redox.2018.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Nybo T., Davies M. J., and Rogowska-Wrzesinska A. (2019) Analysis of protein chlorination by mass spectrometry. Redox Biol. 26, 101236 10.1016/j.redox.2019.101236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Hains P. G., and Robinson P. J. (2017) The impact of commonly used alkylating agents on artifactual peptide modification. J. Proteome Res. 16, 3443–3447 10.1021/acs.jproteome.7b00022 [DOI] [PubMed] [Google Scholar]
- 238. Chen H. J., Yang Y. F., Lai P. Y., and Chen P. F. (2016) Analysis of chlorination, nitration, and nitrosylation of tyrosine and oxidation of methionine and cysteine in hemoglobin from type 2 diabetes mellitus patients by nanoflow liquid chromatography tandem mass spectrometry. Anal. Chem. 88, 9276–9284 10.1021/acs.analchem.6b02663 [DOI] [PubMed] [Google Scholar]
- 239. Kagan H. M. (1994) Lysyl oxidase: mechanism, regulation and relationship to liver fibrosis. Pathol. Res. Pract. 190, 910–919 10.1016/S0344-0338(11)80995-7 [DOI] [PubMed] [Google Scholar]
- 240. Bhave G., Cummings C. F., Vanacore R. M., Kumagai-Cresse C., Ero-Tolliver I. A., Rafi M., Kang J.-S., Pedchenko V., Fessler L. I., Fessler J. H., and Hudson B. G. (2012) Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 8, 784–790 10.1038/nchembio.1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. McCall A. S., Cummings C. F., Bhave G., Vanacore R., Page-McCaw A., and Hudson B. G. (2014) Bromine is an essential trace element for assembly of collagen IV scaffolds in tissue development and architecture. Cell 157, 1380–1392 10.1016/j.cell.2014.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Burzio L. A., and Waite J. H. (2000) Cross-linking in adhesive proteins: studies with model decapeptides. Biochemistry 39, 11147–11153 10.1021/bi0002434 [DOI] [PubMed] [Google Scholar]
- 243. Waite J. H. (1990) The phylogeny and chemical diversity of quinone-tanned glues and varnishes. Comp. Biochem. Physiol. B 97, 19–29 10.1016/0305-0491(90)90172-P [DOI] [PubMed] [Google Scholar]
- 244. Kato Y., Dozaki N., Nakamura T., Kitamoto N., Yoshida A., Naito M., Kitamura M., and Osawa T. (2009) Quantification of modified tyrosines in healthy and diabetic human urine using liquid chromatography/tandem mass spectrometry. J. Clin. Biochem. Nutr. 44, 67–78 10.3164/jcbn.08-185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Abdelrahim M., Morris E., Carver J., Facchina S., White A., and Verma A. (1997) Liquid chromatographic assay of dityrosine in human cerebrospinal fluid. J. Chromatogr. B 696, 175–182 10.1016/S0378-4347(97)00248-X [DOI] [PubMed] [Google Scholar]
- 246. DiMarco T., and Giulivi C. (2007) Current analytical methods for the detection of dityrosine, a biomarker of oxidative stress, in biological samples. Mass Spectrom. Rev. 26, 108–120 10.1002/mas.20109 [DOI] [PubMed] [Google Scholar]
- 247. Marvin L. F., Delatour T., Tavazzi I., Fay L. B., Cupp C., and Guy P. A. (2003) Quantification of o,o′-dityrosine, o-nitrotyrosine, and o-tyrosine in cat urine samples by LC/electrospray ionization-MS/MS using isotope dilution. Anal. Chem. 75, 261–267 10.1021/ac020309w [DOI] [PubMed] [Google Scholar]
- 248. Wells-Knecht M. C., Huggins T. G., Dyer D. G., Thorpe S. R., and Baynes J. W. (1993) Oxidized amino acids in lens protein with age. Measurement of o-tyrosine and dityrosine in the aging human lens. J. Biol. Chem. 268, 12348–12352 [PubMed] [Google Scholar]
- 249. Leeuwenburgh C., Rasmussen J. E., Hsu F. F., Mueller D. M., Pennathur S., and Heinecke J. W. (1997) Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipoprotein isolated from human atherosclerotic plaques. J. Biol. Chem. 272, 3520–3526 10.1074/jbc.272.6.3520 [DOI] [PubMed] [Google Scholar]
- 250. Carroll L., Pattison D. I., Davies J. B., Anderson R. F., Lopez-Alarcon C., and Davies M. J. (2017) Formation and detection of oxidant-generated tryptophan dimers in peptides and proteins. Free Radic. Biol. Med. 113, 132–142 10.1016/j.freeradbiomed.2017.09.020 [DOI] [PubMed] [Google Scholar]
- 251. Carroll L., Pattison D. I., Davies J. B., Anderson R. F., Lopez-Alarcon C., Davies M. J. (2018) Superoxide radicals react with peptide-derived tryptophan radicals with very high rate constants to give hydroperoxides as major products. Free Radic Biol Med. 118, 126–136 10.1016/j.freeradbiomed.2018.02.033 [DOI] [PubMed] [Google Scholar]
- 252. Fuentes-Lemus E., Silva E., Barrias P., Aspee A., Escobar E., Lorentzen L. G., Carroll L., Leinisch F., Davies M. J., and López-Alarcon C. (2018) Aggregation of α- and β-caseins induced by peroxyl radicals involves secondary reactions of carbonyl compounds as well as di-tyrosine and di-tryptophan formation. Free Radic. Biol. Med. 124, 176–188 10.1016/j.freeradbiomed.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 253. Mukherjee S., Kapp E. A., Lothian A., Roberts A. M., Vasil'ev Y. V., Boughton B. A., Barnham K. J., Kok W. M., Hutton C. A., Masters C. L., Bush A. I., Beckman J. S., Dey S. G., and Roberts B. R. (2017) Characterization and identification of dityrosine cross-linked peptides using tandem mass spectrometry. Anal. Chem. 89, 6136–6145 10.1021/acs.analchem.7b00941 [DOI] [PubMed] [Google Scholar]
- 254. Xu C. F., Chen Y., Yi L., Brantley T., Stanley B., Sosic Z., and Zang L. (2017) Discovery and characterization of histidine oxidation initiated cross-links in an IgG1 monoclonal antibody. Anal. Chem. 89, 7915–7923 10.1021/acs.analchem.7b00860 [DOI] [PubMed] [Google Scholar]
- 255. Fritzsche R., Ihling C. H., Götze M., and Sinz A. (2012) Optimizing the enrichment of cross-linked products for mass spectrometric protein analysis. Rapid Commun. Mass Spectrom. 26, 653–658 10.1002/rcm.6150 [DOI] [PubMed] [Google Scholar]
- 256. Tinnefeld V., Venne A. S., Sickmann A., and Zahedi R. P. (2017) Enrichment of cross-linked peptides using charge-based fractional diagonal chromatography (ChaFRADIC). J. Proteome Res. 16, 459–469 10.1021/acs.jproteome.6b00587 [DOI] [PubMed] [Google Scholar]
- 257. Schmidt R., and Sinz A. (2017) Improved single-step enrichment methods of cross-linked products for protein structure analysis and protein interaction mapping. Anal. Bioanal. Chem. 409, 2393–2400 10.1007/s00216-017-0185-1 [DOI] [PubMed] [Google Scholar]
- 258. Hajkova D., Rao K. C., and Miyagi M. (2006) pH dependency of the carboxyl oxygen exchange reaction catalyzed by lysyl endopeptidase and trypsin. J. Proteome Res. 5, 1667–1673 10.1021/pr060033z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Reynolds K. J., Yao X., and Fenselau C. (2002) Proteolytic 18O labeling for comparative proteomics: evaluation of endoprotease Glu-C as the catalytic agent. J. Proteome Res. 1, 27–33 10.1021/pr0100016 [DOI] [PubMed] [Google Scholar]
- 260. Wallis T. P., Pitt J. J., and Gorman J. J. (2001) Identification of disulfide-linked peptides by isotope profiles produced by peptic digestion of proteins in 50% 18O water. Protein Sci. 10, 2251–2271 10.1110/ps.15401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Rose K., Savoy L. A., Simona M. G., Offord R. E., and Wingfield P. (1988) C-terminal peptide identification by fast atom bombardment mass spectrometry. Biochem. J. 250, 253–259 10.1042/bj2500253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Mariotti M., Leinisch F., Leeming D. J., Svensson B., Davies M. J., and Hägglund P. (2018) Mass-spectrometry-based identification of cross-links in proteins exposed to photo-oxidation and peroxyl radicals using 18O labeling and optimized tandem mass spectrometry fragmentation. J. Proteome Res. 17, 2017–2027 10.1021/acs.jproteome.7b00881 [DOI] [PubMed] [Google Scholar]
- 263. Liu M., Zhang Z., Cheetham J., Ren D., and Zhou Z. S. (2014) Discovery and characterization of a photo-oxidative histidine-histidine cross-link in IgG1 antibody utilizing 18O-labeling and mass spectrometry. Anal. Chem. 86, 4940–4948 10.1021/ac500334k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Medinas D. B., Gozzo F. C., Santos L. F., Iglesias A. H., and Augusto O. (2010) A ditryptophan cross-link is responsible for the covalent dimerization of human superoxide dismutase 1 during its bicarbonate-dependent peroxidase activity. Free Radic. Biol. Med. 49, 1046–1053 10.1016/j.freeradbiomed.2010.06.018 [DOI] [PubMed] [Google Scholar]