

ABSTRACT
To identify the mechanism and functions of IRX1 in heart failure (HF) and provide evidence for new therapies. Bioinformatic analysis was performed to select target genes in HF cells compared to normal groups. Experimental rats were treated in a controllable manner to explore how IRX1 methylation accounted for this disease in vivo. Cardiac ultrasonic and morphologic examinations were conducted to test the mouse heart and evaluate the degree of cardiac impairment at in the level of organization. GSEA analysis revealed the relative enrichment of functions. Immunofluorescent assays, western blotting and qRT-PCR were used to determine the DNA methylation and expression levels. IRX1 was hypermethylated in heart failure and identified as a target gene by bioinformatic analysis. Transverse aortic constriction (TAC) induced heart failure in rats, while 5-aza-2ʹ-deoxycytidine (5-Aza) alleviated heart failure in rats according to medical cardiac indexes. Western blotting and qRT-PCR revealed that a conspicuous difference in the expression of IRX1 and CXCL14 between HF and normal cardiac cells. As a result of gene methylation, left ventricular hypertrophy and cardiac fibrosis is usually accompanied by heart failure. Moreover, is the results implied that the demethylation of IRX1 improves heart failure in vivo and in vitro. IRX1 methylation induced damaged cardiac function and even heart failure, which has important implications for HF treatment and diagnosis.
KEYWORDS: DNA methylation, heart failure, 5-aza-2’-deoxycytidine
Introduction
Cardiovascular diseases (CVD) represent considerable morbidity and mortality, of which heart failure is often observed as the end stage [1]. Heart failure, as a growing health concern, affects over 20% of persons and contributes to 11% of deaths worldwide [2]. Clinical medicine diagnoses heart failure as a complex syndrome accompanied by reduced cardiac output, insufficient blood supply and increased risk of cardiac arrest [3]. As fetal genes, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) have been confirmed to correlate well with the clinical severity and prognosis of this disease [4]. Despite tremendous scientific efforts during the past several decades, heart failure remains high-risk and fatal, with an incidence that is expected to increase.
Specialists classify the current treatments for heart failure into five categories: drug treatment [5], auxiliary device, heart transplantation, cell transplantation and gene therapy, among which medicine is the most widely used [6]. Most drugs primarily target neuroendocrine mechanisms, including angiotensin-converting enzyme (ACE) inhibitors, angiotension receptor type 1 (AT1) antagonists, b-adrenoceptor antagonists (BAA), aldosterone receptor antagonists, diuretics and digoxin [7]. Given that evolving therapeutic strategies fail to generate a satisfactory declining prevalence of HF, new molecular targets are urgently needed.
The development and progression of heart failure cannot be separated from epigenetic changes [8]. Unlike genetic alterations, the potential reversibility and heritability of chromosomal component modifications make epigenetic mechanisms attractive and promising targets for therapeutic intervention [9]. Epigenetic mechanisms, such as microRNA and DNA methylation, have been implicated in the aberrant gene expression observed heart failure in previous studies [10]. Moreover, several studies have shown that DNA methylation can silence or activate expression patterns of genes that drive the CVD process, including left ventricular hypertrophy (LVH) and myocardial fibrosis (MFS) [11]. However, the detailed mechanism and potential clinical application of DNA methylation in HF needs further analysis.
Iroquois homeobox 1 (IRX1), a transcription factor, plays a crucial role in embryonic development. Recently, IRX1 has emerged as a potential suppressor gene in rheumatoid arthritis (RA), congenital heart disease (CHD) and tumors, such as gastric cancer (GC) and osteosarcoma [12–14]. Changlong Guo et al. linked an aberrant non synonymous mutation in IRX1 to irregular cardiac function and IRX1 hypermethylation has been frequently detected in HF cell lines [13]. These findings suggest that IRX1 likely plays a role in the precise molecular mechanisms of heart failure. In addition, the association of IRX1 methylation with myocardial function in HF patients is of interest.
Chemokines are a superfamily of small chemotactic cytokines, Chemokine (C-X-C motif) ligand 14 (CXCL14) is a member of the BRAK superfamily [15] Previous studies have reported the CXC chemokine superfamily plays an important role in inflammatory, autoimmune and tumorigenic functions [16,17]. Normal human epithelial cells constitutively express CXCL14, its expression is frequently reduced in cervical, prostate, and oral cancers [18,19]. Consistently, many studies have reported that CXCL14 was highly expressed in normal tissues, especially in normal kidney tissues, but absent in the tumor cell lines and primary tumors [18,20]. CXCL14 is generally considered a tumor suppressor [21,22], but several recent studies have reported the opposite results. For example, the upregulation of CXCL14 in pancreatic cancer promotes the invasion of tumor cells [23].
Furthermore, IRX1 and CXCL14 may have interactive effects on cardiac function and hence provide a new approach to heart failure.
In our study, IRX1 was epigenetically activated in HF samples and its expression was negatively associated with promoter hypermethylation. In addition, the downregulation of IRX1 resulted in decreased CXCL14 expression levels. More importantly, this study is a significant step forward in ascertaining a potential target for epigenetic-based heart failure therapy.
Methods
Bioinformatic analysis
Here, the ChiP Analysis Methylation Pipeline (ChAMP) package, differential analysis of limma package, and gene set enrichment analysis (GSEA) were applied to show significant differences between two biological states based on the NCBI GEO DataSets database (GSE54681, https://www.ncbi.nlm.nih.gov/geo/). We identified the differential gene expression between HF and normal tissues by comparing the reads of each sample to the genome according to screening conditions (|fold change| > 2 and P value < 0.05). Upregulated signal pathways were predicted and analyzed by GSEA, and then related to differentially expressed genes [24].
Animal preparation
Male six-month-old Sprague–Dawley rats (225 to 250 g) were purchased from the Chinese Academy of Sciences of Shanghai. Animal experiments were approved by the Animal Care and Use Committee and were performed according to the official recommendations of the National Institutes of Health guidelines and the Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine.
TAC surgery
Six-week-old rats underwent the TAC operation. All animals were anesthetized with 2.0% inhalational isoflurane, and the aortic arch was tied with a 4/0 silk suture between the brachiocephalic and left common artery with an overlaying 20-gauge needle. After ligation, the needle was quickly removed, and the skin was closed. The sham operation was identical, except that the thread was not ligated [25,26]. We randomly divided twenty rats into 4 groups: (i) Sham control, (ii) TAC, (iii) Sham+5-Aza, (iv) TAC+5-Aza. Rat heart failure was induced by TAC for 4 weeks. 5-Aza was administered intraperitoneally during the last 2 weeks.
Echocardiography
Echocardiography was performed using the VIVO 2100 system (Visual Sonics, Canada) to evaluate cardiac function. During the procedure, the animals were under inhaled anesthesia (isoflurane 2%), and body temperature was maintained using a heat mat. The cine loops were analyzed for left ventricular developed pressure (LVDP), ejection fraction (EF) and maximal rate of the increase in left ventricular pressure (dp/dt max).
Histological analysis
Whole hearts were fixed with 10% formalin, embedded in paraffin, and sectioned at 7-μm intervals. Each slide had 10 sections, which started at the apex and ended at the suture ligation site (approximately 50 slides). Every 4th slide was stained with picrosirius red staining to identify areas of fibrosis [27]. Hematoxylin and eosin staining was performed by Rapid Chrome hematoxylin and eosin staining kit (Thermo Scientific, USA) according to the manufacturer’s instructions [28].
Measures of apoptosis
A TUNEL Apoptosis Detection Kit (FITC) (YEASEN, China) was used to label apoptotic bodies according to the manufacturer’s guidelines. The TUNEL positive cells in the stained images were counted to indicate the apoptotic rate [27].
Isolation of cardiac myocytes
The rats were adequately anaesthetized with 75 mg/kg ketamine and 5 mg/kg xylazine injected intramuscularly, and the respiration rate of the rats was closely monitored to determine the degree of anesthesia. Then, the hearts were removed and maintained in a suitable environment. Cardiomyocytes were obtained by Langendorff perfusion with 0.25 mg/mL Liberase enzyme solution (DH Research Grade, Roche, and Basel, Switzerland). Then cardiac myocytes were plated (1× penicillin, streptomycin, glutamine, 10% horse serum, 5% newborn calf serum, 1.68% M199 salts in DMEM) for 24 h and transferred to DMEM containing 0.1% insulin/transferrin/sodium selenite supplement [29].
Global DNA methylation measurement
Genomic DNA was isolated from left ventricles and incubated with a mixture of phosphodiesterse I, benzonase nuclease, and shrimp alkaline phosphatase. HPLC coupled with tandem mass spectrometry (HPLC–MS, SHIMADZU) was detected to quantify methyl-cytosine (Cm) and cytosine (C) in multiple reaction monitoring mode. The results are represented as the Cm/C ratio [11].
Western blotting
We resolved proteins on 10% SDS-PAGE gels followed by transfer to PVDF membranes (Millipore). Subsequently, the proteins bands were detected according to standard lab protocols. The membranes were incubated with Anti-IRX1 antibody (ab98343, Abcam), Anti-CXCL14 antibody (ab36622, Abcam), Anti-GAPDH antibody (ab181602, Abcam) [30].
qRT-PCR
Transcript levels were determined by RT-PCR using a QuantiTect SYBR Green real-time PCR Kit (11,746,500, Invitrogen). Total RNA was extracted from samples with Trizol reagent (A33252, Invitrogen) following the manufacturer’s instructions. Reverse transcription was performed at 50°C for 20 min, and the cDNA was amplified in 20 μl reaction volumes using 10 pmol of primers for 37 cycles at 94°C for 10 s, 57°C for 15 s, and 72°C for 5 s. Notably, GAPDH acted as an internal control to calculate the relative abundance of the mRNAs [31]. The primer sequences are listed in Table 1.
Table 1.
Primers used in this study.
| Gene |
|
Sequence (5’-3’) |
| IRX1 | Forward | GAAATCCTGGGCACAGTCCA |
| Reverse | CACAGTGGTTTTGCTCGGTG | |
| CXCL14 | Forward | GCAGGGCAATTGGGAACAAG |
| Reverse | GGGTGCAGGGTCATCCTAAC | |
| ANP | Forward | CCGGTACCGAAGATAACAGC |
| Reverse | CCGGTACCGAAGATAACAGC | |
| BNP | Forward | CTGGGAAGTCCTAGCCAGTCTCCA |
| Reverse | GCGACTGACTGCGCCGATCCGGTC | |
| α-MHC | Forward | ACAGAGTGCTTCGTGCCTGAT |
| Reverse | CGAATTTCGGAGGGTTCTGC | |
| β-MHC | Forward | GCTACCCAACCCTAAGGATGC |
| Reverse | TCTGCCTAAGGTGCTGTTTCA | |
| GAPDH | Forward | AACGACCCCTTCATTGACCT |
| Reverse | CCCCATTTGATGTTAGCGGG | |
| MSP(M) | Forward | TTTTTTTAAGAGTAAGGGGGTTAGC |
| Reverse | TAAAATATTCGAATAAATCAACGCT | |
| MSP(UM) | Forward | TTTTTAAGAGTAAGGGGGTTAGTGA |
| Reverse | TAAAATATTCAAATAAATCAACACT | |
| shIRX1-A | Forward | CACCGCCTGCTTATTATCCCTATGG CGAACCATAGGGATAATAAGCAGGC |
| Reverse | AAAAGCCTGCTTATTATCCCTATGG TTCGCCATAGGGATAATAAGCAGGC | |
| shIRX1-B | Forward | CACCGGAGAGCATTGACATCGATCA CGAATGATCGATGTCAATGCTCTCC |
| Reverse | AAAAGGAGAGCATTGACATCGATCA TTCGTGATCGATGTCAATGCTCTCC |
M: methylation primer; UM: unmethylation primer.
Quantitative methylation-specific PCR
DNA collected from samples was treated with sodium bisulfite at 55 °C for 16 hours. Bisulfite-treated DNA was used as a template for real-time fluorogenic methylation-specific PCR (MSP) using primers created to amplify promoter binding sites containing possible methylation sites based on MethPrimer [32] and listed in Table 1.
Gene ontology (GO) annotation analysis
For the GO functional annotation analysis of the genes targeted by IRX1 and differentially expressed genes, we applied the R package Metascape (http://metascape.org/gp/index.html#/main/step1). The R package Go plot was used for analysis visualization. The protein-protein interaction (PPI) network and the cluster of differentially expressed genes were drawn using Cytoscape software (version: 3.3.0).
Microarray
All sample labeling and microarray chip processing were performed at the Veterans Medical Research Foundation Microarray & NGS Core in San Diego. RNA integrity was also validated by the ratio of 28S/18S ribosomal RNA using an Agilent 2100 Bioanalyzer. cDNA was synthesized from 1 µg of total RNA (pooled 500 ng from each rat) using an Affymetrix GeneChip WT PLUS Reagent Kit. Whole transcriptome microarray analysis was performed using the GeneChip Rat Gene 2.0 ST array (Affymetrix). Colocalization was measured using in-house software to measure distances between clusters [33].
Immunohistochemical staining
Frozen sections of rat cardiac tissues were stained with anti-IRX1 antibody (ab180860, Abcam), DAPI (Beyotime) and WGA (Invitrogen). Immunofluorescence images showed the membranes (green) [12].
Statistical analysis
Graph Prism 6.0 software (GraphPad Prism, San Diego, CA) was used for data analysis and diagram generation. Values are shown as the mean ± standard deviation (SD). Depending on the situation, the differences among groups were evaluated by one-way analysis of variance (ANOVA) or two-tailed Student’s t-test. All data were derived from at least three repetitions for each type of experiment. The data group with a P-value<0.05 was regarded as a significantly different group [34].
Results
The CpG sites methylated abnormally in HF compared to normal groups
Multi-dimensional scaling (MDS) identified 1000 variable positions, revealing significant differences between HF and normal tissues (Figure S1(a)). Different beta value distributions were displayed by Type-I and Type-II assays and normalization procedures were used to adjust differences between probe types, in which 0 represented unmethylated sites while 1 represented fully methylated sites (Figure S1(b)). Figure S1C shows the comparison of the beta value density plots from each sample and identified the poor performing arrays basing on a large deviation from the rest of the samples. The dendrogram for 481,173 probes intuitively displayed a clear distinction between normal and HF tissues (Figure S1(d)). For a heatmap of 1000 variable CpGs revealed hypermethylation in HF tissues and hypomethylation in normal groups (Figure S1(e)).
IRX1 and its relevant pathway were identified
As the heatmap of the top 20 differently methylated genes exhibited, several genes (CHL1, PTRF, SOX1 and GREB1L) were less methylated in normal tissues than in HF tissues (Figure 1(a)) and IRX1 was selected as the target gene. Upregulated pathways in normal and HF tissues were explored (Figure 1(b)) and the position in the ranked list of genes according to GSEA analysis (Figure 1(c)) indicated a significant correlation between IRX1 expression and C-X-C motif chemokine ligand 14/nuclear factor-kappa B.
Figure 1.
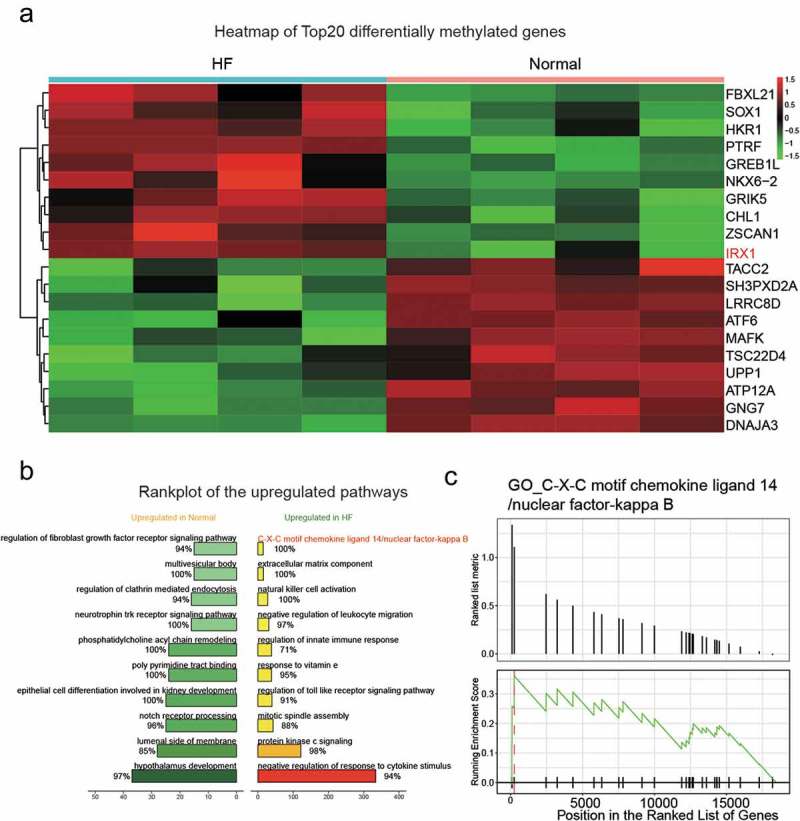
Heatmap of the methylation level and target pathway in HF and normal pairs.
(a) The heatmap of the top 20 DEGs. IRX1 showed a higher level of methylation in HF than normal. HF: heart failure; DEGs: differentially expressed genes. (b) Rank plot of the upregulated pathway in normal and HF. (c) C-X-C motif chemokine 14(CXCL14)/nuclear factor-kappa B (NF-kB) was identified through GO.
5-Aza-2ʹ-deoxycytidine suppressed IRX1 hypermethylation and restored gene expression in heart failure rats
Echocardiography by direct LV catheterization assessed the cardiac function in experimental rats (Figure 2(a–f)). Pathological hypertrophy was triggered over a 4-week period through an ascending TAC-induced pressure overload, producing a growth in the left ventricle to body weight ratio, while dramatically decreases in left ventricular development pressure (LVDP), dP/dtmax (Figure 2(a–c)) and ejection fraction (Figure 2(e-f)). Strikingly, the rats received a complete reversal of TAC-induced heart injuries due to treatment with 5-Aza (Figure 2(a–c,e,f)). Additionally, hematoxylin and eosin staining showed that TAC had disordered arrangement of cardiomyocytes, which was alleviated by 5-Aza treatment (Figure 2(d)). These predictable differences showed representative symptoms of heart failure.
Figure 2.
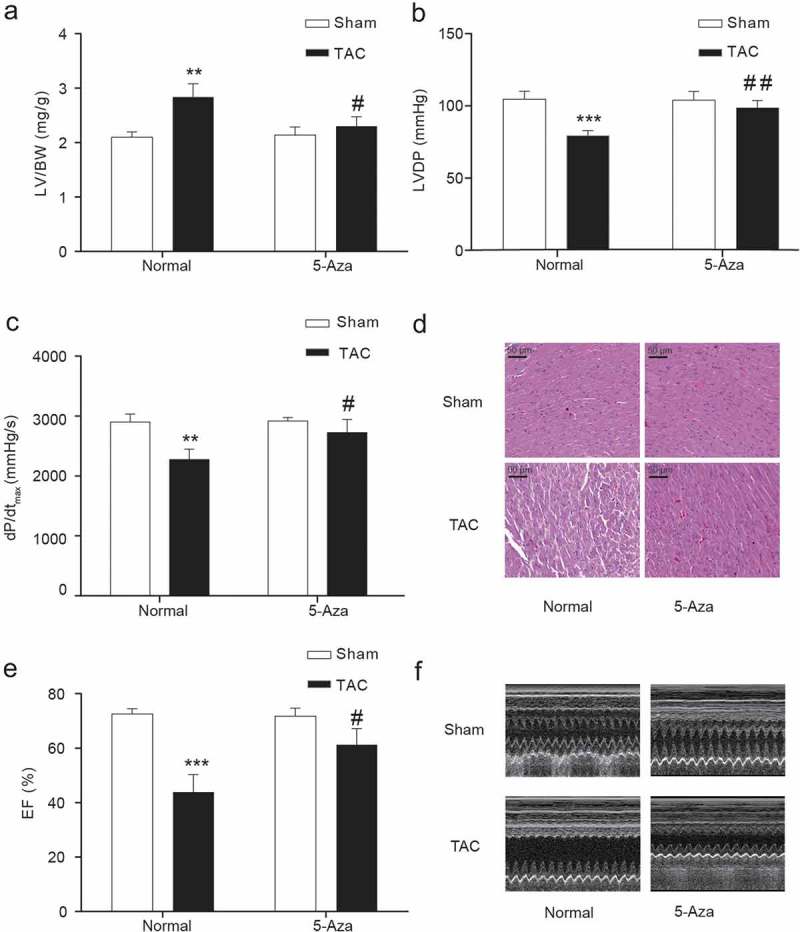
Cardiac functions of rats in each group.
(a) LV/BW (b) LVDP (c) The maximal rate of the increase in left ventricular pressure (dp/dt max) (d) HE staining of cardiac tissue. (E-F) Ejection fraction (%) measured by echocardiography. LV, left ventricle; BW, body weight, EF, ejection fraction. Data are presented as the mean ± SD of five animals; **P < 0.01, ***P < 0.001 vs. Sham-Normal group, # P < 0.05, ## P < 0.01 vs. TAC-Normal group.
TAC-induced heart failure enhanced global genomic DNA methylation (Figure 3(a)) and a gene-specific IRX1methylation (Figure 3(b)). Significant increases in the mRNA expression of genes the ANP and BNP genes in the left ventricle symbolize the occurrence of heart failure (Figure 3(c,d)), Additionally, 5-aza-2ʹ-deoxycytidine treatment reversed the expression of α-MHC and β-MHC, two markers of pathological cardiac hypertrophy (Figure 3(e,f)). Moreover, the abundant IRX1 mRNA (Figure 4(a)) and protein (Figure 4(b)) inhibition were observed with heart failure. However, the treatment of rats with 5-aza-2ʹ-deoxycytidine reversed the low-expression of IRX1 induced by TAC and normalized to the Sham group (Figure 4(a–c)). That is, epigenetic changes such as DNA methylation are always reversible in diseases.
Figure 3.
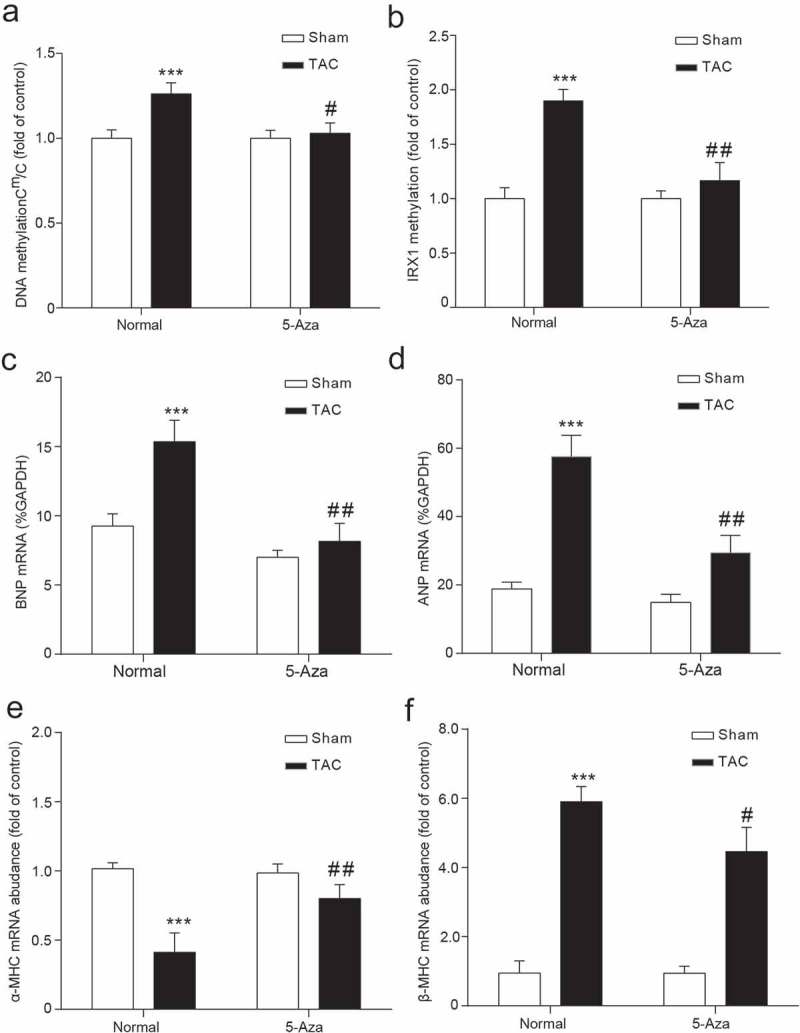
Differential gene expression and methylation in HF rats.
(a-b) ANP and BNP are significant symbols of HF. (c) Cm/C in LV. (d) IRX1 methylation was determined by MSP. (e) α-MHC mRNA expression. (f) β-MHC mRNA expression. Data are presented as the mean ± SD of five animals; ***P < 0.001 vs. Sham-Normal group, # P < 0.05, ## P < 0.01 vs. TAC-Normal group.
Figure 4.
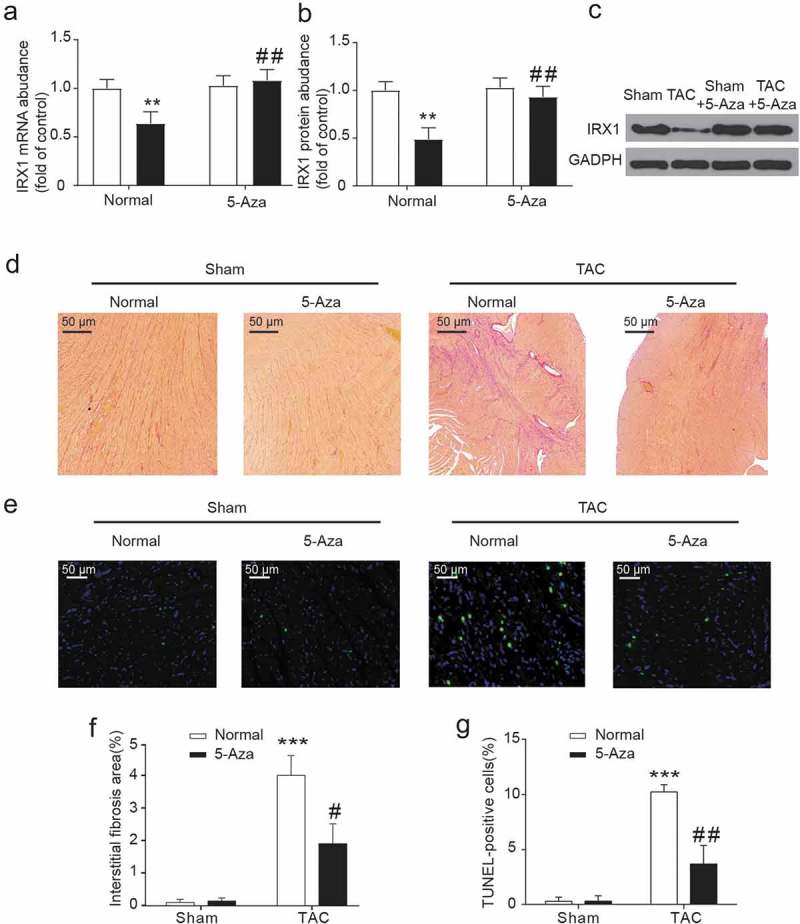
5-Aza-2ʹ-deoxycytidine reversed cardiac hypertrophy and IRX1 downregulation in TAC-induced heart failure.
(a) qRT-PCR determined IRX1 mRNA expression in the left ventricles. (b-c) Western blot analysis determined IRX1 protein expression in the left ventricles. (d, f) Sirius red staining and morphometric analysis identified interstitial cardiac fibrosis. (e) TdT-mediated dUTP-biotin nick end labeling (TUNEL) staining to perceive detect apoptotic cells. (g) TUNEL positive cells ratio. Data are presented as the mean ± SD of five animals; **P < 0.01, ***P < 0.001, vs. Sham-Normal group, # P < 0.05, ## P < 0.01 vs. TAC-Normal group.
5-aza-2ʹ-deoxycytidine protected tac-induced myocardial fibrosis and apoptosis by upregulating CXCL14 expression
To investigate whether 5-aza-2ʹ-deoxycytidine protected cardiac function by reducing myocardial fibrosis and myocardial apoptosis, we detected the fibrotic area and myocardial apoptosis after 2 weeks of 5-Aza treatment. IRX1 hypomethylation facilitated maladaptive cardiac remodeling because 5-Aza-2ʹ-deoxycytidine significantly abrogated fibrotic (Figure 4(d,f)) and apoptotic (Figure 4(e,g)) responses to TAC-induced heart failure.
To probe the relationship between IRX1 and CXCL14, qRT-PCR and western blotting were used to determine the mRNA and protein expression of CXCL14 (Figure 5(a,b)). To further reveal the relationship between IRX1 and CXCL14, we generated 2 lentiviral constructs encoding IRX1-targeting shRNAs, and western blot assay confirmed that the ShIRX1 lentiviral construct could inhibit the protein expression level of IRX1, and the over expression lentiviral could increase the protein expression level of IRX1 (Figure 5(c-d)). The lentivirus was transfected into cardiac myocytes, and CXCL14 mRNA and protein expression were detected. As expected, we found that IRX1 knockdown reduced the expression of both CXCL14 mRNA and protein (Figure 5(e,g,h)). We further confirmed the relationship between IRX1 and CXCL14 through gain-of-function studies, and found that CXCL14 mRNA and protein levels were upregulated by IRX1 overexpression (Figure 5(f–h)). According to the experimental results obtained, there is a positive regulatory relationship between IRX1 and CXCL14. Therefore, IRX1 may activate CXCL14 expression to protect TAC-induced myocardial fibrosis and apoptosis.
Figure 5.
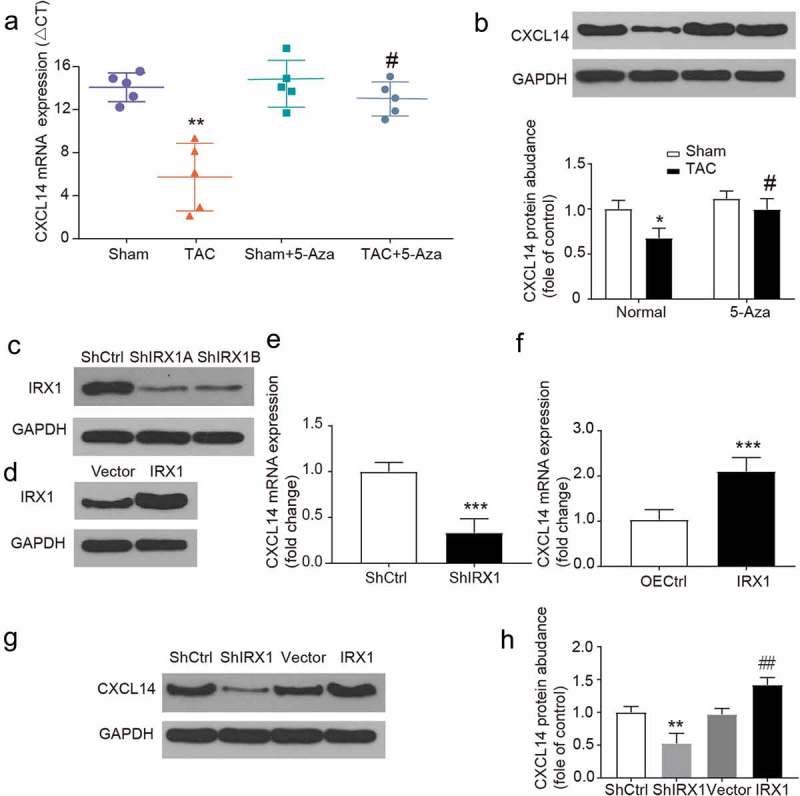
Repression of CXCL14 gene expression is relevant to IRX1.
(A-B) CXCL14 mRNA and protein expression was downregulated in TAC rats. Data are presented as the mean ± SD of five animals; **P < 0.01, vs. Sham-Normal group, # P < 0.05, vs. TAC-Normal group. (C) The effect of IRX1-targeting shRNAs was confirmed by western blot analysis. (D) The efficiency of IRX1 overexpression was confirmed by Western blot analysis. (E) CXCL14 mRNA levels were downregulated by IRX1 shRNA transfection. Data are presented as the mean ± SD, ***P < 0.001 (F) CXCL14 mRNA levels were upregulated by IRX1 overexpression. Data are presented as the mean ± SD, ***P < 0.001 (G-H) CXCL14 protein levels were downregulated by IRX1 shRNA transfection and upregulated by IRX1 over expression. Data are presented as the mean ± SD, ***P < 0.001, vs. ShCtrl group, ## P < 0.01 vs. Vector group.
Discussion
In our study, IRX1 was identified as a hypermethylation-activated gene and the suppression of IRX1 decreased CXCL14 expression levels in rat models of heart failure. Our data show that the 5-Aza-induced hypomethylation of IRX1 positively regulates adaptation during heart failure. Therefore, we concluded that the therapeutic targeting of the IRX1 pathway will be a predictable strategy for suppressing heart failure. A Large amount of evidence indicates that DNA methylation plays a vital role in the progression of disease initiation and progression due to epigenetic adjustment [35]. To understand its role in regulating gene expression and cell phenotype, high-throughput sequencing or array platforms focused on the methylation of the promoter CpG islands of genes have enabled the profiling of methylation marks at a genome-wide scale [36]. Recent studies have shown that DNA methylation is altered during cardiomyocyte proliferation or apoptosis and ventricular remodeling [37]. On the other hand, 5-Aza, as a cytidine nucleoside analog, is the most popular demethylation agent that blocks DNA methylation by forming a covalent bond in which DNMTs are removed from the active nuclear pool [38]. Here, the results of bioinformatic analysis showed that the DNA methylation level of genes in HF tissues was significantly different from that in normal tissues. In further animal experiments, 5-Aza enhanced IRX1 expression and inhibited the development of HF, which implied that IRX1 might become a molecular target of HF.
The Iroquois homeobox family plays multiple roles in drug discovery, disease investigation and cell-based therapies[39]. For example, IRX3 downregulation in breast cancer was linked to poor clinical outcomes, and interaction between IRX4 and IRX5 controls cardiac potassium channel Kv4.2 gene transcription [40,41]. Importantly, variants of the IRX1 gene were reported to contribute to the increased susceptibility of CHD [13]. Therefore, treatment strategies that target IRX1 signaling must consider its various roles to avoid increasing the risk of other diseases [42]. Our study showed that IRX1 was expressed at a low level in the cardiomyocytes of HF rats and this protein functions as a controlling gene, suggesting that IRX1 might specifically function in heart development.
Accurately replicating heart failure in animal models is necessary for the exploration of promising disease targets [43]. Decades of experience remind us that clinical advances are usually aroused from relatively simple experimental design of animal models [44]. A recent study documented that echocardiography data could easily be interpreted by human echocardiographers, suggesting that the molecular imaging of animal hearts are compatible with human technology despite technological limits in some areas [45,46]. In this study, rat models of heart failure were established by TAC induced pressure overload over 4 weeks, with echocardiography to detect cardiac function.
Cardiomyocyte fibrosis is as an important cause of various cardiovascular diseases that develop into heart failure [47]. Notably, understanding the molecular mechanisms of cardiac fibrosis can lead to new approaches for HF prevention or management [48]. Fibroblast growth factors, secreted into the extracellular and interstitial space by cardiac cells, promote and sustain, cardiac fibrosis [49]. Immunochemistry staining in the present study showed that myocardial fibrosis was higher in the heart failure group than in the normal group. Our data also indicated that CXCL14 exerts a synergistic effect with IRX1 to attenuate myocardial fibrosis and cardiomyocyte apoptosis. Therefore, we proposed that IRX1 inhibited fibrosis formation, thereby improving cardiac function. This study showed that the reversible regulation of IRX1 was essential during the development of heart failure. IRX1 hypermethylation decreased the expression of CXCL14 and increased fibrosis formation, which dramatically impaired cardiac function. However, the exact regulatory mechanisms for IRX1 silencing and how IRX1 and CXCL14cooperate in heart failure remain elusive. Nevertheless, these observations will have potential implications for recovery from heart failure in the future.
Supplementary Material
Funding Statement
This work was supported by the Project of Hangzhou Science and Technology (Grant Number: 20180533B97).
Disclosure statement
No potential conflict of interest was reported by the authors.
Ethics approval and consent to participate
The study protocol conforms to the official recommendations of the National Institutes of Health guidelines and the Affiliated Hangzhou First People’s Hospital, Zhejiang University School of Medicine.
Authors’ contributions
Contributing to the conception and design: LH Z, NY G and JY C; Analyzing and interpreting data: LH Z, NY G, GY J and YK Z; Drafting the article: LH Z, JY C and YK Z; Collection of funding: YK Z; Revising it critically for important intellectual content: YK Z; Approving the final version to be published: All authors.
References
- [1].Hwee DT, Kennedy AR, Hartman JJ, et al. The small-molecule fast skeletal troponin activator, CK-2127107, improves exercise tolerance in a rat model of heart failure. J Pharmacol Exp Ther. 2015;353:159–168. [DOI] [PubMed] [Google Scholar]
- [2].Genet M, Lee LC, Baillargeon B, et al. Modeling pathologies of diastolic and systolic heart failure. Ann Biomed Eng. 2016;44:112–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chaudhry SP, Stewart GC.. New pharmacological and technological management strategies in heart failure. Vasc Health Risk Manag. 2017;13:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hohl M, Wagner M, Reil JC, et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123:1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rossignol P, Hernandez AF, Solomon SD, et al. Heart failure drug treatment. Lancet. 2019;393:1034–1044. [DOI] [PubMed] [Google Scholar]
- [6].Deng B, Wang JX, Hu XX, et al. Nkx2.5 enhances the efficacy of mesenchymal stem cells transplantation in treatment heart failure in rats. Life Sci. 2017;182:65–72. [DOI] [PubMed] [Google Scholar]
- [7].Thorup L, Simonsen U, Grimm D, et al. Ivabradine: current and future treatment of heart failure. Basic Clin Pharmacol Toxicol. 2017;121:89–97. [DOI] [PubMed] [Google Scholar]
- [8].Mano H. Epigenetic abnormalities in cardiac hypertrophy and heart failure. Environ Health Prev Med. 2008;13:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lu J, Song G, Tang Q, et al. IRX1 hypomethylation promotes osteosarcoma metastasis via induction of CXCL14/NF-kappaB signaling. J Clin Invest. 2015;125:1839–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Movassagh M, Choy MK, Goddard M, et al. Differential DNA methylation correlates with differential expression of angiogenic factors in human heart failure. PLoS One. 2010;5:e8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xiao D, Dasgupta C, Chen M, et al. Inhibition of DNA methylation reverses norepinephrine-induced cardiac hypertrophy in rats. Cardiovasc Res. 2014;101:373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu X, Zhang J, Liu L, et al. Protein arginine methyltransferase 5-mediated epigenetic silencing of IRX1 contributes to tumorigenicity and metastasis of gastric cancer. Biochimica Et Biophysica Acta Molecular Basis of Disease. 2018;1864:2835–2844. [DOI] [PubMed] [Google Scholar]
- [13].Guo C, Wang Q, Wang Y, et al. Exome sequencing reveals novel IRXI mutation in congenital heart disease. Mol Med Rep. 2017;15:3193–3197. [DOI] [PubMed] [Google Scholar]
- [14].Park SH, Kim SK, Choe JY, et al. Hypermethylation of EBF3 and IRX1 genes in synovial fibroblasts of patients with rheumatoid arthritis. Mol Cells. 2013;35:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hromas R, Broxmeyer HE, Kim C, et al. Cloning of BRAK, a novel divergent CXC chemokine preferentially expressed in normal versus malignant cells. Biochem Biophys Res Commun. 1999;255:703–706. [DOI] [PubMed] [Google Scholar]
- [16].Moser B, Loetscher P. Lymphocyte traffic control by chemokines. Nat Immunol. 2001;2:123–128. [DOI] [PubMed] [Google Scholar]
- [17].Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002;118:915–922. [DOI] [PubMed] [Google Scholar]
- [18].Frederick MJ, Henderson Y, Xu X, et al. In vivo expression of the novel CXC chemokine BRAK in normal and cancerous human tissue. Am J Pathol. 2000;156:1937–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shurin GV, Ferris RL, Tourkova IL, et al. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174:5490–5498. [DOI] [PubMed] [Google Scholar]
- [20].Shellenberger TD, Wang M, Gujrati M, et al. BRAK/CXCL14 is a potent inhibitor of angiogenesis and a chemotactic factor for immature dendritic cells. Cancer Res. 2004;64:8262–8270. [DOI] [PubMed] [Google Scholar]
- [21].Schwarze SR, Luo J, Isaacs WB, et al. Modulation of CXCL14 (BRAK) expression in prostate cancer. Prostate. 2005;64:67–74. [DOI] [PubMed] [Google Scholar]
- [22].Ozawa S, Kato Y, Komori R, et al. BRAK/CXCL14 expression suppresses tumor growth in vivo in human oral carcinoma cells. Biochem Biophys Res Commun. 2006;348:406–412. [DOI] [PubMed] [Google Scholar]
- [23].Wente MN, Mayer C, Gaida MM, et al. CXCL14 expression and potential function in pancreatic cancer. Cancer Lett. 2008;259:209–217. [DOI] [PubMed] [Google Scholar]
- [24].Tian Y, Morris TJ, Webster AP, et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics. 2017;33:3982–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou Y, Shiok TC, Richards AM, et al. MicroRNA-101a suppresses fibrotic programming in isolated cardiac fibroblasts and in vivo fibrosis following trans-aortic constriction. J Mol Cell Cardiol. 2018;121:266–276. [DOI] [PubMed] [Google Scholar]
- [26].Kaimoto S, Hoshino A, Ariyoshi M, et al. Activation of PPAR-alpha in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H305–H13. [DOI] [PubMed] [Google Scholar]
- [27].Lee PJ, Rudenko D, Kuliszewski MA, et al. Survivin gene therapy attenuates left ventricular systolic dysfunction in doxorubicin cardiomyopathy by reducing apoptosis and fibrosis. Cardiovasc Res. 2014;101:423–433. [DOI] [PubMed] [Google Scholar]
- [28].Huang CY, Kuo WW, Lo JF, et al. Doxorubicin attenuates CHIP-guarded HSF1 nuclear translocation and protein stability to trigger IGF-IIR-dependent cardiomyocyte death. Cell Death Dis. 2016;7:e2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mayer SC, Gilsbach R, Preissl S, et al. Adrenergic repression of the epigenetic reader MeCP2 facilitates cardiac adaptation in chronic heart failure. Circ Res. 2015;117:622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jeong D, Lee MA, Li Y , et al. Matricellular protein CCN5 reverses established cardiac fibrosis. J Am Coll Cardiol. 2016;67:1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leach JP, Heallen T, Zhang M, et al. Hippo pathway deficiency reverses systolic heart failure after infarction Nature. 2017;550:260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- [33].Wassenaar JW, Gaetani R, Garcia JJ, et al. Evidence for mechanisms underlying the functional benefits of a myocardial matrix hydrogel for post-MI treatment. J Am Coll Cardiol. 2016;67:1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sun X, Sui Q, Zhang C, et al. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol Cancer Ther. 2013;12:2885–2896. [DOI] [PubMed] [Google Scholar]
- [35].Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. [DOI] [PubMed] [Google Scholar]
- [36].Chatterjee A, Rodger EJ, Morison IM, et al. Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. Methods Mol Biol. 2017;1537:249–277. [DOI] [PubMed] [Google Scholar]
- [37].Reuter JA, Spacek DV, Snyder MP. High-throughput sequencing technologies. Mol Cell. 2015;58:586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ribas L, Vanezis K, Imues MA, et al. Treatment with a DNA methyltransferase inhibitor feminizes zebrafish and induces long-term expression changes in the gonads. Epigenetics Chromatin. 2017;10:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liu LC, Damman K, Lipsic E, et al. Heart failure highlights in 2012-2013. Eur J Heart Fail. 2014;16:122–132. [DOI] [PubMed] [Google Scholar]
- [40].He W, Jia Y, Takimoto K. Interaction between transcription factors Iroquois proteins 4 and 5 controls cardiac potassium channel Kv4.2 gene transcription. Cardiovasc Res. 2009;81:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Asaka S, Fujimoto T, Akaishi J, et al. Genetic prognostic index influences patient outcome for node-positive breast cancer. Surg Today. 2006;36:793–801. [DOI] [PubMed] [Google Scholar]
- [42].Houweling AC, Dildrop R, Peters T, et al. Gene and cluster-specific expression of the Iroquois family members during mouse development. Mech Dev. 2001;107:169–174. [DOI] [PubMed] [Google Scholar]
- [43].Blenck CL, Harvey PA, Reckelhoff JF, et al. The importance of biological sex and estrogen in rodent models of cardiovascular health and disease. Circ Res. 2016;118:1294–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kianmeher M, Ghorani V, Boskabady MH. Animal model of asthma, various methods and measured parameters: A methodological review. Iran J Allergy Asthma Immunol. 2016;15:445–465. [PubMed] [Google Scholar]
- [45].Schaefer A, Schneeberger Y, Stenzig J, et al. A new animal model for investigation of mechanical unloading in hypertrophic and failing hearts: combination of transverse aortic constriction and heterotopic heart transplantation. PLoS One. 2016;11:e0148259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Breckenridge R. Heart failure and mouse models. Dis Model Mech. 2010;3:138–143. [DOI] [PubMed] [Google Scholar]
- [47].Piek A, de Boer RA, Sillje HH. The fibrosis-cell death axis in heart failure. Heart Fail Rev. 2016;21:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Santiago JJ, McNaughton LJ, Koleini N, et al. High molecular weight fibroblast growth factor-2 in the human heart is a potential target for prevention of cardiac remodeling. PLoS One. 2014;9:e97281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hu Y, Li L, Shen L, et al. FGF-16 protects against adverse cardiac remodeling in the infarct diabetic heart. Am J Transl Res. 2017;9:1630–1640. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.