

ABSTRACT
Complement-dependent cytotoxicity (CDC) is a potent effector mechanism, engaging both innate and adaptive immunity. Although strategies to improve the CDC activity of antibody therapeutics have primarily focused on enhancing the interaction between the antibody crystallizable fragment (Fc) and the first subcomponent of the C1 complement complex (C1q), the relative importance of intrinsic affinity and binding valency of an antibody to the target antigen is poorly understood. Here we show that antibody binding affinity to a cell surface target antigen evidently affects the extent and efficacy of antibody-mediated complement activation. We further report the fundamental role of antibody binding valency in the capacity to recruit C1q and regulate CDC. More specifically, an array of affinity-modulated variants and functionally monovalent bispecific derivatives of high-affinity anti-epidermal growth factor receptor (EGFR) and anti-human epidermal growth factor receptor 2 (HER2) therapeutic immunoglobulin Gs (IgGs), previously reported to be deficient in mediating complement activation, were tested for their ability to bind C1q by biolayer interferometry using antigen-loaded biosensors and to exert CDC against a panel of EGFR and HER2 tumor cells of various histological origins. Significantly, affinity-reduced variants or monovalent derivatives, but not their high-affinity bivalent IgG counterparts, induced near-complete cell cytotoxicity in tumor cell lines that had formerly been shown to be resistant to complement-mediated attack. Our findings suggest that monovalent target engagement may contribute to an optimal geometrical positioning of the antibody Fc to engage C1q and deploy the complement pathway.
Keywords: CDC, intrinsic affinity, binding valency, IgG, DuetMab, C1q, Fc
Introduction
Monoclonal antibodies (mAbs), which represent the fastest-growing class of biological therapeutics, have led to a paradigm shift in the treatment of several hematologic and solid malignances.1–4 Unconjugated therapeutic mAbs, upon binding to a target antigen, manifest their biological activity either through antigen-binding fragment (Fab)-dependent agonism or antagonism of a broad network of signaling pathways or by their ability to recruit and activate several Fc-dependent immune effector mechanisms, such as antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cell-mediated phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC).5,6 The latter is considered to be a potent defense cascade of innate immunity,7,8 however, the role of complement activation in the anti-tumor efficacy of many therapeutic mAbs remains elusive.9,10 To date, the vital role of CDC in the control and eradication of malignant cells has been best illustrated by rituximab, an anti-CD20 mAb approved for various indications.11–14
Activation of the classical complement pathway is triggered by the binding of C1q, the first subcomponent of the C1 complement complex, to the Fc region of a cell-bound antibody.15,16 Subsequent recruitment of a series of complement proteins that are abundantly present in serum results in the formation of a membrane attack complex (MAC) that mediates the lysis of target cells.15,16
Strategies for enhancing the affinity between antibody Fc and activating FcγRs expressed on effector cells are widely employed, and Fc-engineered antibodies with improved ADCC routinely enter clinical development.17–19 Nevertheless, the enhancement of antibody-mediated complement activation has proven to be challenging, primarily due to the low affinity of Fc to C1q and the structural intricacy of the IgG1-C1q complex.9,20–22
Attempts to improve the inherent interaction of antibody Fc to C1q, leading to enhanced activation of the complement pathway, have predominantly focused on Fc mutagenesis or glycoengineering.19,23–30 Alternative approaches that have aimed to augment the CDC activity of therapeutic mAbs have included use of combinations of IgG antibodies targeting distinct, nonoverlapping epitopes of the target antigen31–33 or the neutralization of complement regulatory proteins that are overexpressed in tumor cells, using blocking antibodies or small interfering RNAs (siRNAs).34,35 Previously, Diebolder et al. reported a novel approach to promote antibody-dependent complement activation.36 In that monumental study, the authors demonstrated that, upon binding to a cell surface target antigen, IgG antibodies form hexameric structures through noncovalent Fc interactions that can better engage C1q. It was subsequently demonstrated that specific point mutations in the Fc region can stimulate IgG hexamerization and induce potentiated complement activation.37 More recently, Strasser et al. demonstrated that the cell surface antigen density and membrane mobility may dramatically affect the pathways and kinetics of IgG oligomerization and complement activation.38 Despite these efforts, the interplay of IgG’s intrinsic affinity and binding valency to the target antigen in relation to the capacity to recruit C1q and mediate CDC is poorly understood.
We recently reported that the intrinsic affinity of antibody to the target antigen directly influenced the extent and efficiency of ADCC.39 More specifically, using an array of affinity-modulated anti-CD4, epidermal growth factor receptor (EGFR), and human epidermal growth factor receptor 2 (HER2) model mAbs, we have shown that at saturating antibody concentrations, IgG variants with moderate intrinsic affinities (dissociation constant [KD] ~10–150 nM), similar to those generated by the primary humoral immune response, exhibited ADCC that was superior to that of their affinity-improved counterparts (KD ≤1 nM). We further demonstrated that reformatting these high-affinity bivalent IgGs into monovalent formats resulted in substantial augmentation of ADCC.39 We speculated that IgG antibodies with faster off-rates are likely to dissociate each binding arm more rapidly, resulting in a higher propensity of monovalent engagement with the target cell. This binding mode promotes enhanced opsonization of the target cell, leading to improved recruitment of effector cells. In the same study, we also unexpectedly found that affinity-reduced anti-CD4 IgG variants induced near-complete CDC but the high-affinity parental IgG failed to elicit CDC, suggesting that factors other than improved target cell opsonization may have contributed to the boost in CDC activity.39 However, the underlying mechanisms were not thoroughly interrogated.
As reported here, we sought to understand how the intrinsic affinity and binding valency of antibody to a cell surface target antigen affect its ability to interact with C1q and promote CDC. To that end, we selected a series of high-affinity anti-EGFR and anti-HER2 therapeutic mAbs that have been previously shown to be deficient in triggering CDC.32,40,41 We then evaluated the capacity of affinity-modulated variants and monovalent derivatives of the high-affinity bivalent IgGs to engage C1q and induce CDC against a panel of EGFR and HER2 tumor cell lines that were formerly shown to be resistant to complement-mediated attack.35,42,43 We show that antibody intrinsic affinity to the target antigen clearly influenced the extent and efficiency of antibody-mediated complement activation. We further report the pivotal role of antibody binding valency in the ability to recruit C1q and regulate CDC. Finally, we discuss the implications of our findings in the development of clinically optimized antibody therapeutics.
Results
Reducing IgG intrinsic affinity enhances CDC activity
To determine the effect of antibody binding affinity to target antigen on the capacity to promote CDC, we selected an in-house high-affinity anti-EGFR antibody, QD6, and two pre-affinity-matured variants thereof, Tdev4 and GLH4. These IgG variants differ by only point mutations in the variable heavy and light chain domains and target an identical epitope on the extracellular domain of EGFR (data not shown). The intrinsic binding kinetics of the three IgG antibodies to EGFR were determined by Octet analysis. We found that variants Tdev4 and GLH4 exhibited approximately 20- and 100-fold reduced affinity, respectively, compared with the high-affinity QD6 IgG (Table 1). For assessment of cellular binding and CDC, we selected three tumor cell lines of different histological origins, expressing high levels of EGFR as determined by receptor density analysis (Supplementary Table S1). Notably, MDA-MB-468 and A431 cell lines were previously shown to be insensitive to CDC after treatment with various high-affinity anti-EGFR IgGs, even though both cells express high levels of EGFR antigen.32,40,41 Before the three anti-EGFR variants were tested for CDC, their cellular binding properties were determined by flow cytometry. As we previously demonstrated for other affinity-modulated anti-EGFR antibodies,39 the low-affinity variants, Tdev4 and GLH4, exhibited lower mean fluorescence intensity (MFI) values than the high-affinity QD6 IgG (Figure 1(a–c)).
Table 1.
Binding kinetics of IgG and DuetMab antibodies to target antigensa.
| Intrinsic affinity |
|||||
|---|---|---|---|---|---|
| Antibody | Antigen | kon (M–1 sec–1) |
koff (sec–1) |
KD (nM) |
Epitope groupb |
| QD6 IgG | EGFR | (1.8 ± 0.04) × 105 | (2.9 ± 0.33) × 10–4 | 1.6 ± 0.19 | 1 |
| Tdev4 IgG | EGFR | (1.2 ± 0.03) × 105 | (3.1 ± 0.05) × 10–3 | 27.0 ± 0.81 | |
| GLH4 IgG | EGFR | (1.1 ± 0.05) × 105 | (8.2 ± 0.19) × 10–3 | 76.2 ± 4.1 | |
| QD6/NMGC DuetMab | EGFR | (1.7 ± 0.08) × 105 | (2.9 ± 0.88) × 10–4 | 1.7 ± 0.52 | |
| GA201 IgG | EGFR | (2.5 ± 0.14) × 105 | (1.6 ± 0.53) × 10–4 | 0.6 ± 0.21 | 1 |
| GA201/NMGC DuetMab | EGFR | (5.1 ± 0.25) × 105 | (4.0 ± 0.67) × 10–4 | 0.8 ± 0.14 | |
| Cetuximab IgG | EGFR | (4.4 ± 0.09) × 105 | (3.2 ± 0.05) × 10–3 | 7.2 ± 0.19 | 2 |
| Cetuximab/NMGC DuetMab | EGFR | (5.5 ± 0.18) × 105 | (3.3 ± 0.14) × 10–3 | 5.9 ± 0.32 | |
| Trastuzumab IgG | HER2 | (3.4 ± 0.11) × 105 | (4.1 ± 2.2) × 10–4 | 1.2 ± 0.65 | 3 |
| Trastuzumab/NMGC DuetMab | HER2 | (3.4 ± 0.08) × 105 | (3.3 ± 0.48) × 10–4 | 1.0 ± 0.14 | |
| Pertuzumab IgG | HER2 | (9.8 ± 0.27) × 104 | (1.5 ± 0.05) × 10–3 | 15.5 ± 0.66 | 4 |
| Pertuzumab/NMGC DuetMab | HER2 | (9.2 ± 0.5) × 104 | (2.1 ± 0.1) × 10–3 | 22.3 ± 1.61 | |
| B1D2 IgG | HER2 | (2.3 ± 0.04) × 105 | (2.3 ± 0.59) × 10–4 | 1.0 ± 0.26 | 4 |
| B1D2/NMGC DuetMab | HER2 | (3.6 ± 0.07) × 105 | (2.6 ± 0.14) × 10–4 | 0.7 ± 0.04 | |
aKinetic measurements to soluble monomeric forms of EGFR and HER2 were performed with an Octet384 instrument. Dissociation constants (KDs), were calculated as the ratio of koff/kon from a nonlinear fit of the data.
bAntibodies were categorized by their epitopes, determined with an Octet competitive binding assay. QD6 and GA201 antibodies recognize a similar epitope, whereas cetuximab binds a distinct epitope. Pertuzumab and B1D2 recognize a similar epitope, whereas trastuzumab binds a distinct epitope.
Figure 1.
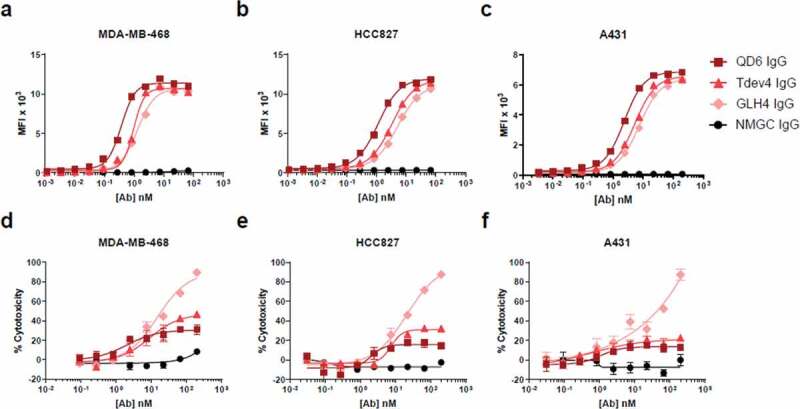
Cell binding and CDC activity of anti-EGFR IgG variants. (a–c) Cell binding of anti-EGFR IgG variants to EGFR-expressing cell lines MDA-MB-468 (a), HCC827 (b), and A431 (c) cells. (d–f) CDC activity of anti-EGFR IgG variants against MDA-MB-468 (d), HCC827 (e), and A431 (f) cells, using rabbit complement at a final concentration of 10% (vol/vol). In (d-f), % of cell lysis was determined 4 hrs after addition of cells, antibodies and serum. Antibody-specific complement dependent cytotoxicity was calculated using the formula: % cytotoxicity = 100 x (1 – E/S), where E is the luminescence with experimental antibody, and S is the luminescence without antibody, but with the same concentration of serum. NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars. See Table 2 for statistical analysis and P values.
For CDC analysis, antibodies were incubated with target cells in the presence of complement at a final concentration of 10% (vol/vol), and cytotoxicity was determined by a luminescence viability assay. In alignment with our previous findings for affinity-modulated anti-CD4 IgGs,39 at higher antibody concentrations the low-affinity anti-EGFR variants mediated a greater degree of cell cytotoxicity relative to the high-affinity QD6 IgG (Figure 1(d–f)). Particularly, the level of CDC activity was inversely correlated with the reduced intrinsic affinity to EGFR. At maximal antibody concentration, the lowest-affinity variant, GLH4, exhibited statistically significant superior cytotoxicity (P < .0001) against all three EGFR-expressing cells in comparison with QD6, whereas the variant Tdev4 mediated statistically significant improved CDC against MDA-MB-468 (P < .01) and HCC827 (P < .0002) cell lines (Table 2). In agreement with data previously reported for other high-affinity anti-EGFR mAbs,32,40,41 the high-affinity QD6 IgG induced very modest CDC against all three cell lines. Consistent with our previous findings with ADCC,39 there was an inverse correlation between the cell binding signals observed at maximal antibody concentration and CDC potency. This disparity may stem from the different nature of the assays. Cell binding assays are subjected to rigorous cycles of washing after incubation of the antibodies with target cells, and thus antibodies with faster dissociation rates are more susceptible to being washed out from the cell surface than are high-affinity mAbs. In contrast, the CDC assay described in this study is a non-wash assay, allowing low-affinity binders to be retained on the cell surface.
Table 2.
CDC activity of anti-EGFR antibodies against EGFR tumor cell lines.
| MDA-MB-468 |
HCC827 |
A431 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Antibody | EC50 (nM) | % Cytotoxicity at maximal antibody concentration | Pa | EC50 (nM) | % Cytotoxicity at maximal antibody concentration | P | EC50 (nM) | % Cytotoxicity at maximal antibody concentration | P |
| QD6 IgG | NA | 30.8 | NA | 14.5 | NA | 12.8 | |||
| Tdev4 IgG | 9.0 | 46.7 | 0.0174 | 7.5 | 32.1 | 0.0002 | 0.6 | 22.7 | NS |
| GLH4 IgG | 15.2 | 89.5 | <0.0001 | 21.2 | 87.6 | <0.0001 | NA | 87.4 | <0.0001 |
| QD6/NMGC DuetMab | 4.9 | 81.6 | <0.0001 | 7.5 | 78.9 | <0.0001 | 3.1 | 89.7 | <0.0001 |
| GA201 IgG | NA | 17.4 | NA | 15.5 | NA | 8.0 | |||
| GA201/NMGC DuetMab | 10.7 | 86.5 | <0.0001 | 11.1 | 74.9 | <0.0001 | 7.0 | 48.4 | 0.0008 |
| Cetuximab IgG | NA | 6.2 | NA | 39.0 | NA | 15.6 | |||
| Cetuximab/NMGC DuetMab | 2.7 | 54.0 | 0.0005 | 7.1 | 69.7 | 0.0043 | 3.4 | 59.1 | 0.0006 |
aP: One-way analysis of variance and unpaired t test were used to determine statistically significant CDC at maximal antibody concentration tested. Statistical significance was accepted for P < 0.05 at 95% confidence interval.
NA = not available; NS = not significant.
To confirm that the improved CDC observed with the low-affinity IgG variants was not affected by complement concentration, we compared the CDC activity induced by the anti-EGFR variants in the presence of a lower (5%) and a higher (15%) serum concentration, which again revealed significantly enhanced CDC activity for Tdev4 and GLH4 (data not shown). Furthermore, no CDC activity was detected when the antibodies were incubated in the presence of heat-inactivated (HI) serum (data not shown). Taken together, our findings demonstrate that the intrinsic affinity of QD6 and variants thereof for their target antigen EGFR clearly regulate the extent and efficiency of CDC on tumor cell lines.
Monovalent binding to target antigen augments CDC activity
Inspired by our previous demonstration that reformatting of monospecific bivalent IgG antibodies into monovalent formats resulted in substantial augmentation of ADCC,39 we tested whether conversion of target-specific IgG antibodies into monovalent derivatives would lead to improved CDC. Using our previously described monovalent bispecific DuetMab platform,44 we generated monovalent formats of three high-affinity anti-EGFR mAbs, QD6, GA201, and cetuximab,45,46 and three high-affinity anti-HER2 IgGs, trastuzumab, pertuzumab, and B1D2.47–49 These monovalent DuetMab molecules carried the Fab domain of their IgG counterparts paired with a Fab of a non-binding isotype control IgG (NMGC).39,44,50,51 Thus, these bispecific DuetMab derivatives are functionally monovalent to their target antigens. The corresponding DuetMab antibodies were produced from mammalian cells, and their oligomeric state and purity were determined as previously described.44,50 The intrinsic binding kinetics of the IgG and DuetMab pairs were determined by Octet analysis. It was found that the monovalent DuetMabs retained the intrinsic binding kinetics of the bivalent IgGs from which they were derived (Table 1).
We selected HER2 as a second target antigen because previous attempts to demonstrate CDC in solid tumor cells with high-affinity anti-HER2 IgGs failed to produce potent activity, suggesting that the classical complement pathway is not likely to be a significant mechanism of action by which HER2 antibodies elicit their biological activity.35,42,43 The HER2-positive tumor cell lines SK-BR-3, BT474, and Calu-3, which were previously shown to be resistant to antibody-mediated CDC using trastuzumab and pertuzumab as monotherapies,35 were selected for this study. The levels of HER2 antigen on these target cells were determined by receptor density analysis (Supplementary Table S1). The cellular binding properties of the IgGs and their respective DuetMabs to EGFR and HER2 target cells were determined by flow cytometry. At saturating antibody concentrations, the monovalent DuetMabs displayed approximately two-fold higher binding intensities than their bivalent IgG counterparts (Supplementary Figure S1). The enhanced MFI recorded for the monovalent formats stems from an increased number of antibody Fc domains on the cell surface, owing to a 1:1 ratio of Fab to antigen, as opposed to a 2:1 ratio as in the case of the bivalent IgGs. We then compared the levels of CDC activity mediated by the high-affinity IgGs and their corresponding monovalent DuetMab molecules. Consistent with results previously reported for high-affinity EGFR32,40,41 and HER235,42,43 mAbs, the high-affinity anti-EGFR (Figure 2) and anti-HER2 (Figure 3) as bivalent IgGs exhibited no or minimal CDC activity. In contrast, the corresponding monovalent anti-EGFR (Figure 2, Table 2) and anti-HER2 (Figure 3, Table 3) DuetMabs exhibited statistically significant improved CDC at maximal antibody concentration compared with their bivalent IgG counterparts. Intriguingly, the enhanced CDC activity observed after reformatting of the bivalent IgGs into monovalent formats or by the low-affinity EGFR variant GLH4 was more significant than expected, suggesting that factors other than improved target cell opsonization may account for the boost in CDC activity. More specifically, the monovalent binding nature of the DuetMab molecules should yield two-fold excess of antibody Fc available for C1q engagement, which in theory should enhance CDC by only about two-fold. However, all DuetMabs exhibited substantially more than two-fold improvement in CDC compared with their respective IgGs (Figure 2, 3). Particularly at saturating antibody concentrations, the QD6/NMGC DuetMab induced near-complete cell cytotoxicity against all three EGFR-expressing cells, whereas the bivalent QD6 IgG mediated only marginal cell cytotoxicity. Comparable results were observed for the anti-EGFR GA201 IgG and GA201/NMGC DuetMab pair against MDA-MB-468 and HCC827 cells and for the anti-HER2 trastuzumab IgG and trastuzumab/NMGC DuetMab pair against SK-BR-3 and BT474 cells. Similarly, the low-affinity variant GLH4 also demonstrated near-complete cytotoxicity against all three EGFR-expressing cells. Our findings are in agreement with those reported by Diebolder et al., in which the conversion of a high-affinity anti-EGFR IgG into a monovalent format strongly enhanced its CDC activity.36
Figure 2.
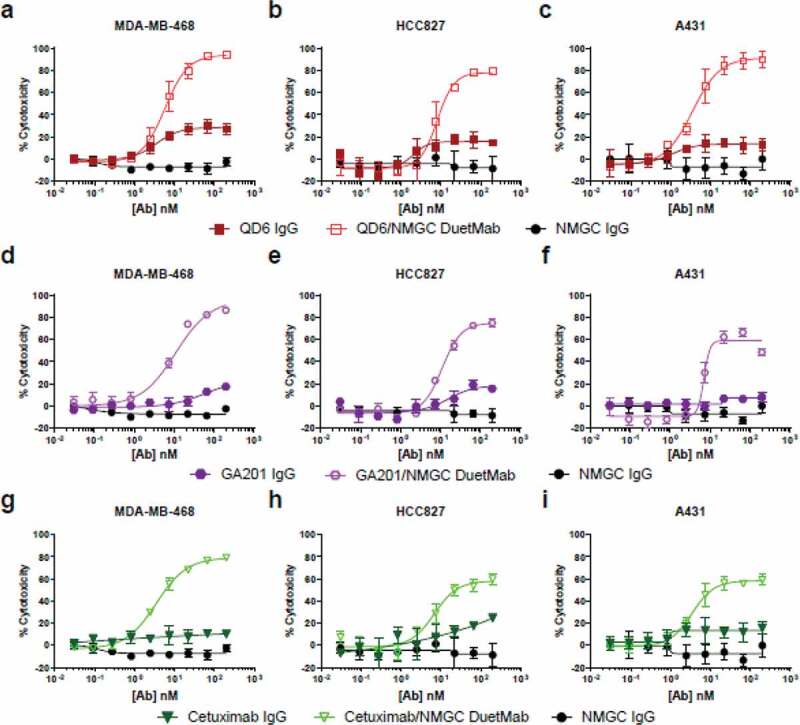
CDC activity of bivalent anti-EGFR IgGs and their respective monovalent bispecific DuetMabs. (a–c) CDC activity of QD6 IgG and QD6/NMGC DuetMab against MDA-MB-468 (a), HCC827 (b), and A431 (c) cells at 10%, 10%, and 5% serum concentrations. (d–f) CDC activity of GA201 IgG, GA201/NMGC DuetMab against MDA-MB-468 (d), HCC827 (e), and A431 (f) cells at the corresponding serum concentrations as for the QD6 antibody set. (g–i) CDC activity of cetuximab IgG, cetuximab/NMGC DuetMab against MDA-MB-468 (g), HCC827 (h), and A431 (i) cells at the corresponding serum concentrations as for the QD6 antibody set. NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars. See Table 2 for statistical analysis and P values.
Figure 3.
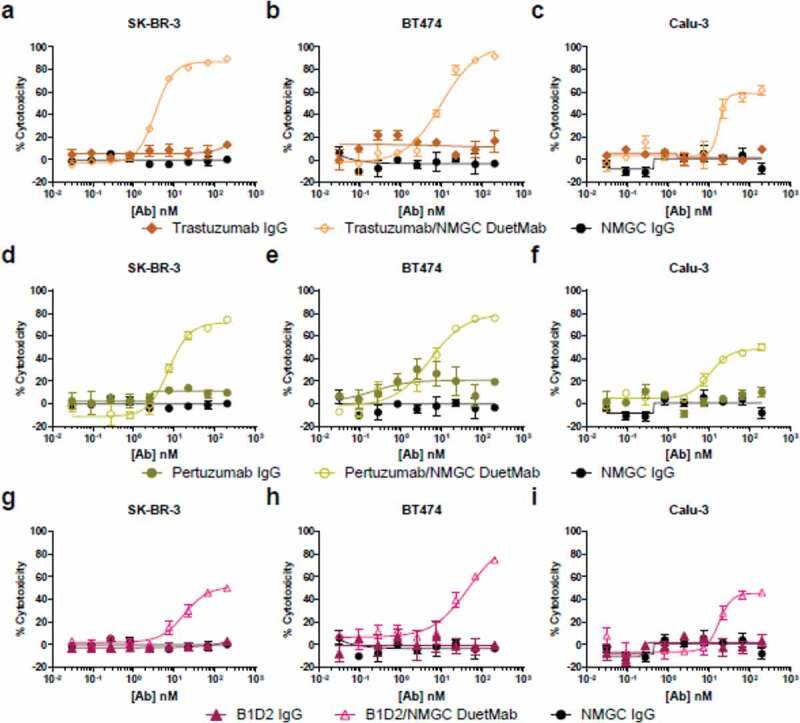
CDC activity of bivalent anti-HER2 IgGs and their respective monovalent bispecific DuetMabs. (a–c) CDC activity of trastuzumab IgG and trastuzumab/NMGC DuetMab against SK-BR-3 (a), BT474 (b), and Calu-3 (c) cells at 10%, 5%, and 10% serum concentrations. (d–f) CDC activity of pertuzumab IgG and pertuzumab/NMGC DuetMab against SK-BR-3 (d), BT474 (e), and Calu-3 (f) cells at the corresponding serum concentrations as for the trastuzumab antibody set. (g–i) CDC activity of B1D2 IgG and B1D2/NMGC DuetMab against SK-BR-3 (g), BT474 (h), and Calu-3 (i) cells at the corresponding serum concentrations as for the trastuzumab antibody set. NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars. See Table 3 for statistical analysis and P values.
Table 3.
CDC activity of anti-HER2 antibodies against HER2 tumor cell lines.
| SK-BR-3 |
BT474 |
Calu-3 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Antibody | EC50 (nM) | % Cytotoxicity at maximal antibody concentration | Pa | EC50 (nM) | % Cytotoxicity at maximal antibody concentration | P | EC50 (nM) | % Cytotoxicity at maximal antibody concentration | P |
| Trastuzumab IgG | NA | 15.9 | NA | 16.8 | NA | 9.3 | |||
| Trastuzumab/NMGC DuetMab | 4.1 | 84.2 | 0.0001 | 9.7 | 91.4 | 0.0015 | 17.8 | 61.4 | 0.0003 |
| Pertuzumab IgG | NA | 13.4 | NA | 19.4 | NA | 10.4 | |||
| Pertuzumab/NMGC DuetMab | 7.5 | 53.2 | 0.0002 | 5.2 | 75.9 | <0.0001 | 10.5 | 49.9 | 0.0021 |
| B1D2 IgG | NA | 1.5 | NA | –0.3 | NA | 3.2 | |||
| B1D2/NMGC DuetMab | 18.0 | 39.3 | 0.0002 | 43.8 | 75.0 | 0.0004 | 17.6 | 46.2 | 0.0034 |
aP: Unpaired t test was used to determine statistically significant CDC at maximal antibody concentration tested. Statistical significance was accepted for P < 0.05 at 95% confidence interval.
NA = not available.
To further elucidate the role of monovalent versus bivalent binding in the capacity of a mAb to induce potentiated CDC, we compared the levels of cytotoxicity mediated by the DuetMabs and their respective high-affinity IgGs when tested alone or in combination. A 1:1 mixture of the monovalent DuetMab with its paired bivalent IgG resulted in a significant reduction in cell cytotoxicity compared with the activity of the DuetMab alone (Figure 4). These results suggest that a high-affinity IgG, via strong avidity mediated by bivalent binding to a target antigen, can readily out-compete and impair the activity mediated by its monovalent bispecific counterpart.
Figure 4.
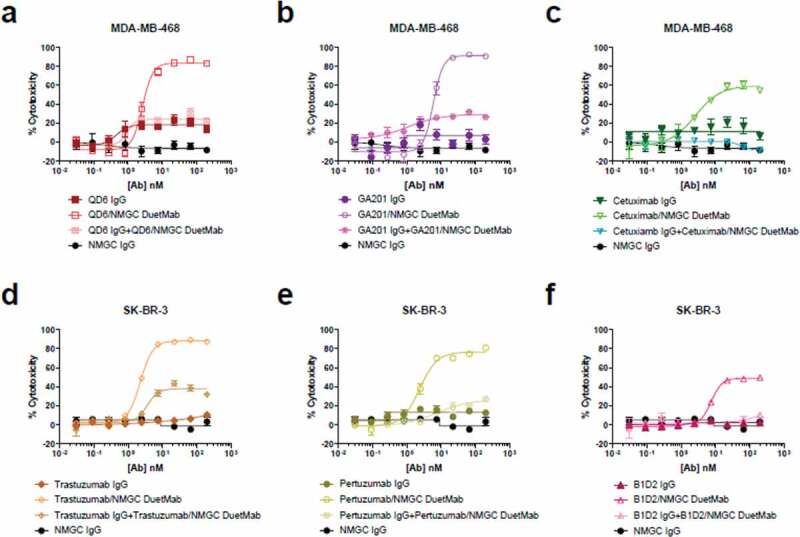
Competition CDC studies. (a–c) CDC activity of anti-EGFR IgGs and their respective monovalent DuetMabs when tested alone or in combination against MDA-MB-468 cells at a final serum concentration of 10% (vol/vol). (d–f) CDC activity of anti-HER2 IgGs and their respective monovalent DuetMabs when tested alone or in combination against SK-BR-3 cells at a final serum concentration of 10% (vol/vol). NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars.
Collectively, our results suggest that mechanisms other than enhanced target cell opsonization contribute to the enhanced CDC activity of monovalent DuetMabs and affinity-reduced IgG variants. We further demonstrated that reformatting high-affinity bivalent IgGs, which were previously reported to be deficient in triggering CDC, into a monovalent format facilitated efficient eradication of tumor cells that were formerly considered to be insensitive to complement-mediated killing.32,35,41
Combinations of IgGs targeting distinct epitopes exhibit similar potentiated CDC of monovalent DuetMabs
Previous studies have demonstrated that combinations of anti-EGFR31,32 or anti-folate receptor33 IgGs, targeting distinct epitopes could substantially enhance their CDC activity, whereas combinations of cross-competing mAbs did not induce significant CDC. In this experiment, we sought to understand how format valency affects the capacity of antibody combinations to regulate CDC.
We first categorized the two groups of anti-EGFR and anti-HER2 IgGs based on their binding epitopes. Antibody epitope binning studies were performed on an Octet by sequential binding to antigen-loaded biosensors (Supplementary Figure S2). We identified two epitope groups for each target antigen (Table 1). The anti-EGFR antibodies QD6 and GA201 target a similar epitope, as the two IgGs compete with each other’s binding, whereas cetuximab binds a distinct epitope. For the anti-HER2 antibodies, pertuzumab and B1D2 recognize a similar epitope, whereas trastuzumab binds a unique epitope.
We then examined the levels of cytotoxicity mediated by a 1:1 mixture of IgGs or DuetMabs targeting distinct or similar epitopes. Combinations of IgGs that targeted noncompeting epitopes (for anti-EGFR, QD6 + cetuximab and GA201 + cetuximab; for anti-HER2, trastuzumab + pertuzumab and trastuzumab + B1D2) displayed strong synergism, resulting in significantly enhanced CDC compared with the activity mediated by each IgG alone (Figure 5). In contrast, combinations of IgGs targeting overlapping epitopes (for anti-EGFR, QD6 + GA201; for anti-HER2, pertuzumab + B1D2) did not induce significant CDC. Our findings are in agreement with data previously reported for other anti-EGFR and anti-folate receptor IgGs.31–33
Figure 5.
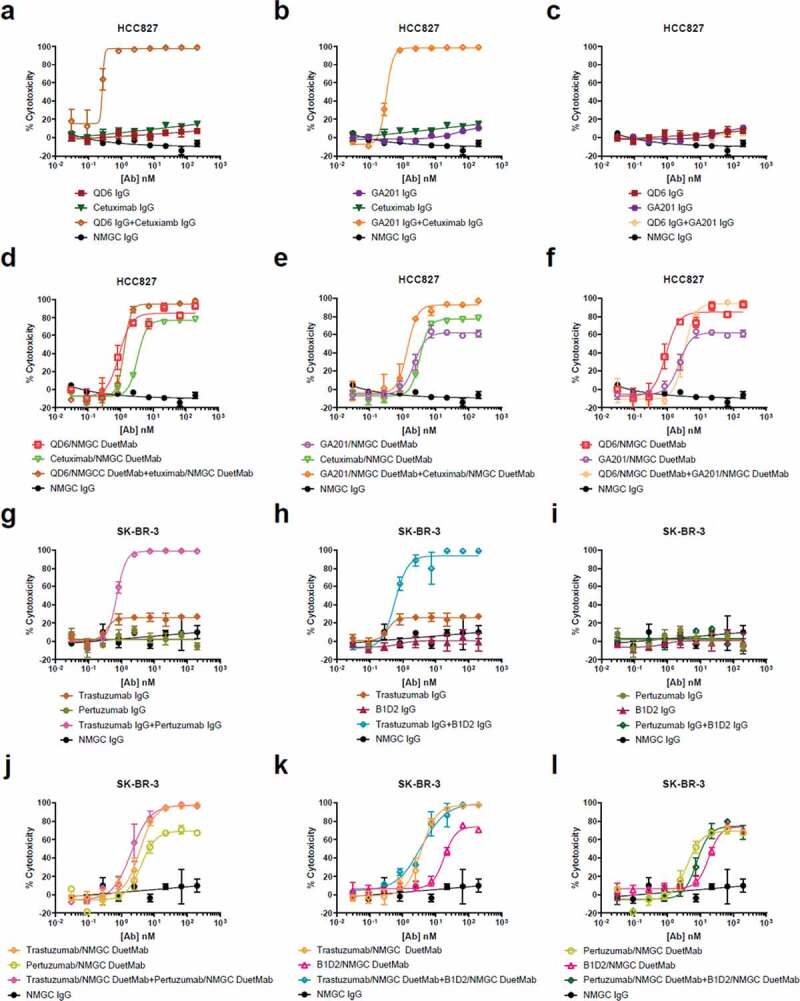
CDC activity of IgG and DuetMab combinations targeting distinct or overlapping epitopes. (a–c) CDC activity of QD6 IgG and cetuximab IgG alone or in combination (a), GA201 IgG and cetuximab IgG alone or in combination (b), and QD6 IgG and GA201 alone or in combination (c) against HCC827 cells at a final serum concentration of 10% (vol/vol). (d–f) CDC activity of QD6/NMGC DuetMab and cetuximab/NMGC DuetMab alone or in combination (d), GA201/NMGC DuetMab and cetuximab/NMGC DuetMab alone or in combination (e), and QD6/NMGC DuetMab and GA201/NMGC DuetMab alone or in combination (f) against HCC827 cells at a final serum concentration of 10% (vol/vol). (g–i) CDC activity of trastuzumab IgG and pertuzumab IgG alone or in combination (g), trastuzumab IgG and B1D2 IgG alone or in combination (h), and pertuzumab IgG and B1D2 IgG alone or in combination (i) against BT474 cells at a final serum concentration of 5% (vol/vol). (j–l) CDC activity of trastuzumab/NMGC DuetMab and pertuzumab/NMGC DuetMab alone or in combination (j), trastuzumab/NMGC DuetMab and B1D2/NMGC DuetMab alone or in combination (k), and pertuzumab/NMGC DuetMab and B1D2/NMGC DuetMab alone or in combination (l) against BT474 cells at a final serum concentration of 5% (vol/vol). NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars.
Conversely, combinations of DuetMabs targeting distinct or similar epitopes did not further augment the potent CDC activity induced by each DuetMab alone. These results suggest that when combined, IgG antibodies that target distinct epitopes may adopt structural rearrangements that are similar to those of their monovalent DuetMab counterparts. We speculate that at saturating antibody concentrations, the crowded cell surface environment that is formed after opsonization of two noncompeting IgGs generates substantial steric hindrance, which may force the antibodies into a dominant monovalent binding mode. This imposed binding orientation may create an optimal geometrical positioning of the Fc to better interact with C1q. In contrast, two IgG antibodies with overlapping epitopes are likely to compete for binding, resulting in the higher-affinity antibody interacting bivalently with the target antigen because of a stronger binding avidity, and thus no enhanced CDC was observed. In comparison, the monovalent binding mode of the DuetMab format does not change, regardless of whether the two molecules are targeting distinct or overlapping epitopes, and hence no further augmentation in cytotoxicity was observed.
Complement regulatory proteins impede CDC activity of bivalent IgGs but not monovalent DuetMabs
To further comprehend the role of format valency in the capacity of mAbs to regulate CDC, we examined the effect of silencing membrane-bound complement regulatory proteins (mCRPs) in tumor cells on the ability of monovalent and bivalent antibody formats to elicit CDC. Complement regulatory proteins, including membrane cofactor protein (CD46), decay accelerating factor (CD55), and protectin (CD59), are a group of cell surface proteins that can significantly inhibit CDC.10,52 These membrane regulators are indispensable for keeping CDC function under check and for preventing complement-mediated attack of normal cells.10,52 Previous studies have shown that many tumor cells overexpress mCRPs to attenuate CDC.42,52–54 Neutralization of mCRPs by either blocking antibodies or down-regulation with siRNAs was recently reported to enhance the CDC activity of anti-tumor antibodies.34,35
In this experiment, we compared the levels of cytotoxicity mediated by the bivalent IgGs and their corresponding monovalent DuetMabs after siRNA silencing of CD46, CD55, and CD59 mCRPs in the EGFR and HER2 tumor cells used in this study. We first confirmed basal expression of mCRPs on the EGFR and HER2 tumor cell lines by flow cytometry (Supplementary Figure S3). Next, the tumor cells were transfected with a mixture of siRNAs targeting CD46, CD55, and CD59. Three days after transfection, all six cell lines exhibited significant reduction of mCRP expression as determined by flow cytometry (Supplementary Figure S3). We then compared the levels of CDC activity induced by the anti-EGFR and anti-HER2 IgGs and their respective DuetMabs against siRNA-treated and untreated cells. Remarkably, the effect of mCRP silencing on the capacity of anti-EGFR and anti-HER2 IgGs to promote CDC was quite variable and seemed to be both antibody and cell line dependent, whereas the impact of mCRP knockdown on the enhanced potency of all DuetMabs was insignificant (Figure 6, 7). Specifically, for the anti-EGFR IgGs, QD6 exhibited improved cytotoxicity against siRNA-treated HCC827 cells, but had no significant effect on other tumor cells (Figure 6(a)). GA201 demonstrated significantly enhanced cytotoxicity against siRNA-treated MDA-MB-468 cells and marginally improved activity against siRNA-treated A431 cells (Figure 6(b)), whereas cetuximab displayed enhanced cytotoxicity against both siRNA-treated MDA-MB-468 and HCC827 cells (Figure 6(c)). For the anti-HER2 IgGs, trastuzumab and pertuzumab exhibited significantly enhanced cytotoxicity only against siRNA-treated Calu-3 cells, whereas B1D2 had no significant effect on any of the siRNA-treated cells (Figure 7(a–c)–c)). Notably, there was no correlation between the relative expression of mCRPs on the cancer cell lines or the binding epitope recognized by the antibody and the ability of the IgG format to mediate CDC upon mCRP silencing. In contrast, downregulation of the mCRPs did not further significantly improve the potent activity mediated by all EGFR and HER2 DuetMabs (Figure 6(d–f), 7(d–f)). Taken together, our findings indicate that monovalent binding to the target antigen seems to play a fundamental role in the capacity of the antibody Fc to interact and recruit C1q and can override the inhibitory effect of mCRPs expressed by target cells.
Figure 6.
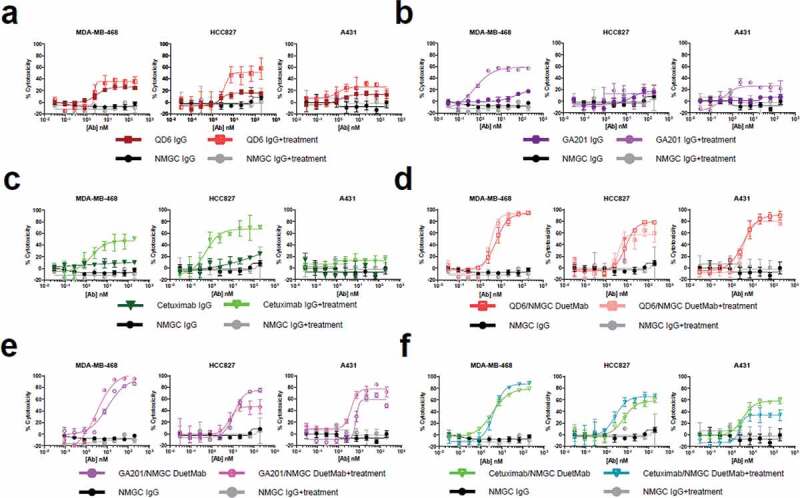
CDC activity of anti-EGFR IgGs and their respective DuetMabs against EGFR tumor cells upon mCRP silencing. (a–c) CDC activity of QD6 (a), GA201 (b), and cetuximab (c) IgGs against siRNA-treated and untreated MDA-MB-468 at 10% (left), HCC827 at 10% (middle), and A431 cells at 5% (right) serum concentrations. (d–f) CDC activity of QD6/NMGC (d), GA201/NMGC (e), and cetuximab/NMGC (f) DuetMabs against siRNA-treated and untreated MDA-MB-468 at 10% (left), HCC827 at 10% (middle), and A431 cells at 5% (right) serum concentrations. NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars.
Figure 7.
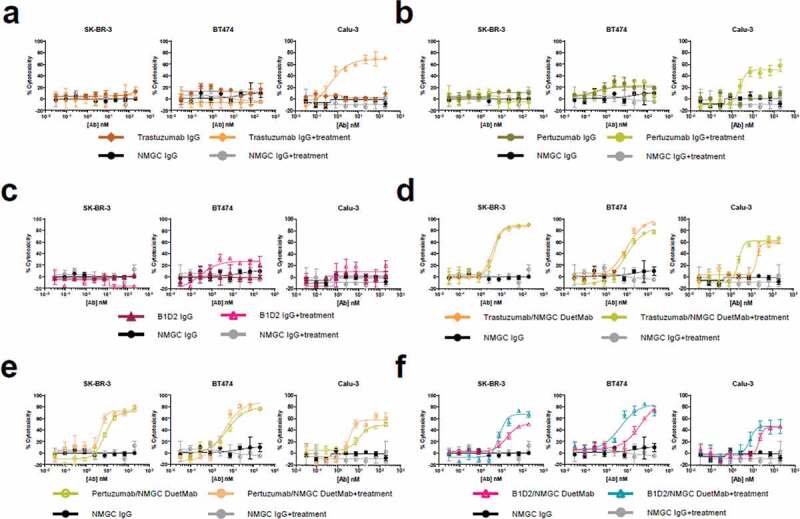
CDC activity of anti-HER2 IgGs and their respective DuetMabs against HER2 tumor cells upon mCRPs silencing. (a–c) CDC activity of trastuzumab (a), pertuzumab (b), and B1D2 (c) IgGs against siRNA-treated and untreated SK-BR-3 at 10% (left), BT474 at 5% (middle), and Calu-3 cells at 10% (right) serum concentrations. (d–f) CDC activity of trastuzumab/NMGC (d), pertuzumab/NMGC (e), and B1D2/NMGC (f) DuetMabs against siRNA-treated and untreated SK-BR-3 at 10% (left), BT474 at 5% (middle), and Calu-3 cells at 10% (right) serum concentrations. NMGC was used as an isotype control. Each point represents the mean values of triplicate wells, and the standard error of the mean is represented by error bars.
Monovalent DuetMabs exhibit improved binding association to C1q
To interrogate the underlying mechanisms that contribute to the significant enhancement in CDC activity after the reformatting of high-affinity bivalent IgGs into monovalent formats, we sought to analyze the intrinsic kinetics of monovalent and bivalent antibody formats to C1q. Previous studies have shown that, upon engagement with a target antigen, IgG antibodies assemble at the cell surface into hexameric clusters that can better engage with C1q.20,36,38,55 It was further demonstrated that several Fc point mutations can stimulate IgG hexamerization and thus enhance CDC.36,37 We reasoned that enhanced target cell opsonization due to monovalent binding is probably not the sole contributor to improved CDC, and speculated that the geometrical positioning of the Fc upon monovalent target engagement may be more favorable for C1q binding. Such favorable Fc configuration should enhance the association of C1q with Fc bound to the cell surface. Because accurate measurement of Fc-C1q kinetics on a cell surface is quite challenging to achieve, we attempted to recapitulate cell surface Fc-C1q interactions by using antigen-loaded biosensors on an Octet. To that end, biotinylated antigens were first loaded onto streptavidin sensors. Next, the monovalent and bivalent formats of the anti-EGFR QD6 and anti-HER2 trastuzumab antibodies were allowed to interact with their respective target antigens before association and dissociation measurements were conducted with C1q. The monovalent DuetMabs exhibited enhanced association rates (kon) compared with their bivalent IgG counterparts (Table 4, Supplementary Figure S4). Specifically, the monovalent QD6 antibody exhibited an approximately three-fold increase in kon relative to the bivalent IgG, whereas trastuzumab as monovalent DuetMab exhibited an approximately four- to five-fold increase in kon when loaded to either a higher density (200 nM) or a lower density (55 nM), respectively, compared with the bivalent IgG counterpart. The latter concentration confirmed that the improved kon values recorded for the monovalent format were not due to DuetMab molecules on the sensors having higher densities than IgGs. For the low-affinity IgGs, we could not obtain stable baselines for accurate determination of Fc-C1q binding kinetics due to their rapid dissociation rates (koff) (data not shown).
Table 4.
Binding kinetics of C1q to Fc domain of IgG and DuetMab antibodiesa.
| Antibody | Antigen concentration (nM) | Apparent affinity |
||
|---|---|---|---|---|
| kon (M–1 sec–1) | koff (sec–1) | KD (µM) | ||
| QD6 IgG | 200 | (2.7 ± 0.63) × 105 | 2.9 ± 0.61 | 10.8 ± 0.45 |
| NMGC-QD6 DuetMab | 200 | (9.6 ± 0.43) × 105 | 2.4 ± 0.35 | 2.5 ± 0.62 |
| Trastuzumab IgG | 200 | (2.1 ± 0.23) × 104 | (1.9 ± 0.32) × 10–1 | 9.4 ± 0.27 |
| Trastuzumab/NMGC DuetMab | 200 | (8.9 ± 0.28) × 104 | (2.3 ± 0.33) × 10–1 | 2.6 ± 0.87 |
| Trastuzumab/NMGC DuetMab | 55 | (1.0 ± 0.23) × 105 | (1.8 ± 0.22) × 10–1 | 1.7 ± 0.44 |
aKinetic measurements to soluble C1q were performed with an Octet 384 instrument. Dissociation constants (KD) were calculated as the ratio of koff/kon from a nonlinear fit of the data.
Taken together, these results suggest that monovalent binding to a cell surface target antigen may constitute an optimal geometrical positioning of the antibody Fc to interact with C1q and deploy the complement cascade.
Discussion
Fc-mediated immune effector functions, including ADCC, ADCP, and CDC, are important mechanisms of action and form an essential link between innate and adaptive immunity.5,6 Novel Fc-engineered antibodies with tailored effector mechanisms that aim to further boost efficacy now frequently enter clinical development.17–19 Although extensive efforts have been made to improve the interaction of antibody Fc to C1q, enhancement of antibody-dependent complement activation has proven to be challenging, and, to the best of our knowledge, no CDC-enhanced antibodies have entered clinical development. In this study we systemically interrogated the collective role of antibody intrinsic affinity and binding valency to the target antigen in relation to the ability to recruit C1q and regulate CDC. Using an array of high-affinity anti-EGFR and anti-HER2 mAbs, including affinity-modulated variants and monovalent bispecific derivatives, we demonstrated that antibody intrinsic affinity to the target antigen clearly influenced the extent and efficiency of CDC. We further demonstrated that reformatting high-affinity bivalent IgGs previously shown to be deficient in triggering CDC into monovalent bispecific DuetMabs resulted in near-complete eradication of tumor cell lines that were formerly considered to be resistant to complement-mediated attack.
To accentuate the fundamental role of antibody binding valency in the capacity to regulate CDC, we demonstrated that combinations of IgGs targeting distinct epitopes, but not overlapping epitopes, exhibited potentiated CDC activity similar to that of the monovalent DuetMab format, suggesting that, when combined, IgG antibodies that target distinct epitopes may be forced into monovalent binding conformation owing to a significant steric hindrance formed at the crowded cell surface environment. We speculate that this imposed binding orientation may create an optimal geometrical positioning of the antibody Fc to interact with C1q and exert complement activation. Remarkably, the favorable Fc configuration constituted upon monovalent target engagement could override the inhibitory effect of mCRPs because the CDC potency of all anti-EGFR and anti-HER2 DuetMabs was not significantly improved after siRNA silencing of CD46, CD55, and CD59 mCRPs. In contrast, the impact of mCRP knockdown on the ability of the bivalent IgGs to induce CDC was rather variable and appeared to be mutually antibody- and cell line-dependent. To elucidate the underlining mechanisms contributing to the significant improvement in CDC upon reformatting of high-affinity bivalent IgGs into monovalent DuetMabs, we compared the binding kinetics of monovalent and bivalent antibody formats to C1q. We showed here for the first time that the association rates (kon) of C1q to the Fc of antigen-bound DuetMabs were higher than those recorded for the corresponding antigen-bound IgGs. These results further exemplified the critical role monovalent target engagement plays in founding an optimal docking conformation for the antibody Fc to interact with C1q. Our observations also argue that conditional, target binding-dependent approaches to improve Fc-C1q interaction, may be advantageous over strategies focused on Fc mutagenesis or glycoengineering that enhance the affinity of antibody Fc to C1q independent of target binding, and thus may result in suboptimal complement activation due to binding of the therapeutic IgG in solution to C1q proteins that are abundantly present in the serum.
Asymmetric monovalent bispecific antibodies that resemble natural IgG are currently the dominant bispecific format in clinical development.56,57 The monovalent binding nature of this class of molecules is believed to promote improved target selectivity via cross-arm avidity targeting of antigen double-positive cells over single-positive normal tissue.58–61 This feature is of substantial importance because it provides attractive opportunities of enhanced efficacy coupled with reduced systemic toxicity that can potentially lead to better drug safety and improved therapeutic index. However, with respect to the ability of monovalent bispecific antibodies to mediate Fc-dependent immune effector functions as a mode of action, the findings we presented here, combined with data we recently reported in relation to the effect of antibody binding valency on the capacity to regulate ADCC, may suggest that monovalent binding of a bispecific antibody to a cell surface antigen on single-positive normal tissue may correspond with substantial augmentation of immune effector functions and potential damaging of non-target cells. In contrast, concurrent bivalent binding of the bispecific antibody to double-positive target cells will resemble the binding orientation of a natural IgG, and thus may not trigger enhanced Fc-dependent effector mechanisms. Such a scenario may lead to elevated systemic toxicity and overall reduced therapeutic index. A possible solution to decouple Fc-mediated effector functions from the intended mechanisms of action of the bispecific antibody can be achieved by engineering the Fc with a set of mutations that abrogate immune effector mechanisms.62–64
In conclusion, the results of our study provide a deeper understanding of factors that regulate CDC and underline the pivotal roles of antibody’s intrinsic affinity and binding valency to the target antigen in the capacity to recruit C1q and mediate complement activation. We suggest a careful examination of these key design parameters when developing clinically relevant antibody and bispecific antibody therapeutics. Experiments are in progress to determine the in vivo implications of these findings.
Materials and methods
Cells and media
EGFR-expressing cell lines MDA-MB-468, HCC827, and A431, and HER2-expressing cell lines SK-BR-3, BT474, and Calu-3 were obtained from the American Type Culture Collection. MDA-MB-468 cells were cultured in Leibovitz L-15 medium (Gibco) supplemented with 10% HI fetal bovine serum (FBS) (Gibco). HCC827 and BT474 cells were cultured in RPMI 1640 medium (Gibco) supplemented with 10% HI FBS. A431 cells were cultured in Dulbecco modified Eagle medium (Gibco) supplemented with 10% HI FBS. SK-BR-3 cells were cultured in McCoy’s 5a medium (Gibco) supplemented with 10% HI FBS. Calu-3 cells were cultured in Eagle’s minimal essential medium (Gibco) supplemented with 10% HI FBS. All cell lines were maintained at 37°C with 5% CO2 except for MDA-MB-468 cells, which were maintained at 37°C without CO2.
Antibodies and sera
The panel of anti-EGFR and anti-HER2 antibodies, whose amino acid sequences were previously reported,45–49,65 were transiently expressed in CHO-K1 cells. Ten days after transfection, culture supernatants were harvested, filtered, and loaded on a 5-mL MabSelect SuRe column (GE Healthcare). After extensive washing, bound antibodies were eluted with IgG elution buffer (Life Technologies) and immediately neutralized with 1 M Tris HCl, pH 8.0 (Gibco). After buffer exchange in phosphate-buffered saline (PBS), pH 7.2 (Life Technologies), antibodies were loaded on a Superdex 200 16/600 column (GE Healthcare) and the monomer fractions were collected. The purity and oligomeric state of the purified antibodies were determined by analytical size exclusion chromatography and Bioanalyzer A (Agilent). The stringent multistep purification process ensured that only highly pure antibodies were obtained and used in the study, and that interference by impurities, especially aggregates, on CDC was excluded, as antibody effector functions are extremely sensitive to aggregates.66 As a final quality control, before every CDC assay, analytical size exclusion chromatography was used to ensure that every antibody contained more than 99% monomer. For CDC assays, baby rabbit complement (Cedarlane) was used.
Cell binding by antibodies
For all anti-EGFR and anti-HER2 antibodies, flow cytometry was used to determine cell binding profiles. Cells were harvested and resuspended in fluorescence-activated cell sorting buffer (Dulbecco’s PBS, pH 7.2 [Life Technologies]; 2% HI FBS; 0.1% NaN3; and 2 mM ethylenediaminetetraacetic acid). A total of 2 × 105 cells were transferred into each well of a 96-well U-bottomed plate (Corning) and incubated with three-fold serially diluted antibodies starting at 180 nM at 4°C for 1 hour. After washing, cells were incubated with 1:200 diluted fluorescein isothiocyanate (FITC)–conjugated goat anti-human Fc polyclonal antibody (Jackson ImmunoResearch, Catalog No.: 109-096-098) at 4°C for 45 minutes. After another wash, the plate was run on a BD LSRII flow cytometer (Becton Dickinson). FlowJo software (FlowJo) was used to analyze the binding profiles.
Receptor density quantification
Flow cytometry was used to quantify EGFR or HER2 receptor density on all cell lines. Anti-EGFR GA201 and anti-HER2 trastuzumab IgGs were first labeled with an Alexa Fluor 647 labeling kit (Invitrogen), and their concentrations and fluorochrome-to-protein ratios were measured. Cells were then collected and resuspended in fluorescence-activated cell sorting buffer, after which 2 × 106 cells were incubated with AF647-conjugated antibodies at saturating concentrations (≥20 μg/mL) at 4°C for 1 hour. After washing, cells were fixed in 1.8% paraformaldehyde and bound antibodies were detected with MACSQuant VYB (Miltenyl Biotec). At the same time, Quantum AF647 MESF (molecules of equivalent soluble fluorochrome) beads (Bangs Laboratories) were analyzed to establish a standard curve. Using the QuickCal program, the calculated MESF was then divided by the antibody fluorochrome-to-protein ratio to give a corrected antibody binding capacity.
CDC
CellTiter-Glo Luminescent Cell Viability Assay (Promega) was used to assess the CDC activity of anti-EGFR and anti-HER2 antibodies. Cells were first harvested and resuspended in assay medium composed of RPMI 1640 medium supplemented with 10% HI FBS. To minimize antibody-independent nonspecific cytotoxicity, every 1 mL of baby rabbit complement was pre-absorbed on 1 × 106 target cells at 4°C for 20 minutes. A total of 5 × 103 fresh target cells in 50 μL of assay medium; 25 μL of threefold serially diluted antibodies starting at 30 μg/mL; and 5 (5% serum concentration), 10 (10% serum concentration), or 15 μL (15% serum concentration) of cleared complement after pre-absorption were added sequentially to each well of a 96-well flat-bottomed white plate, and the volume was increased to 100 μL with assay medium. After 4 hours of incubation at 37°C, the plates were taken out, the supernatant was discarded, and 50 μL of fresh assay medium was added to each well. After the addition of 50 μL of CellTiter-Glo reagents, the plates were incubated at room temperature for 20 minutes and read for luminescence on a Perkin Elmer plate reader. All antibodies were tested in triplicate.
For CDC with HI serum, baby rabbit complement was heat inactivated at 56°C for 30 minutes before use.
Antibody-specific complement dependent cytotoxicity was calculated using the formula: % cytotoxicity = 100 x (1 – E/S), where E is the luminescence with experimental antibody, and S is the luminescence without antibody but with the same concentration of serum.
mCRP knockdown
siRNAs against CD46, CD55, and CD59 were obtained from Dharmacon as ON-TARGETplus SMARTpool. A total of 106 target cells were seeded into each well of a six-well tissue culture plate (Corning) and transfected with all three siRNA pools, using Lipofectamine RNAiMAX (Life Technologies). At 3 days after transfection, cells were harvested, verified for knockdown of mCRPs, and then used in the CDC assay.
Flow cytometry was used to compare expression of CD46, CD55, and CD59 before and after siRNA treatment. siRNA-treated and untreated cells were incubated with mouse anti-human CD46, CD55, and CD59 antibodies (BD Biosciences, Catalog No.: 555948, 555691, and 555761, respectively) and then stained with FITC-conjugated goat anti-mouse IgG polyclonal antibody (Jackson ImmunoResearch, Catalog No.: 115-095-146). The plate was run on a BD LSRII flow cytometer, and FlowJo software was used to analyze the data.
Binding kinetics measurements
Kinetic measurements to soluble monomeric forms of EGFR (R&D Systems) and HER2 (eBioscience) ligands were measured by biolayer interferometry on an Octet384 instrument (ForteBio). 10 μg/mL of antibodies in Octet buffer composed of PBS, pH 7.2, 3 mg/ml bovine serum albumin, and 0.05% (vol/vol) Tween 20 were loaded on anti-human Fc sensors (ForteBio). After washing, fourfold serially diluted EGFR or HER2 proteins starting at 100 nM were loaded to determine association and dissociation rates.
For assessment of C1q-Fc interaction, 2 μg/ml of biotinylated EGFR or HER2 proteins were loaded on streptavidin sensors (ForteBio), after which anti-EGFR antibodies QD6 IgG and QD6/NMGC DuetMab or anti-HER2 antibodies trastuzumab IgG and trastuzumab/NMGC DuetMab were loaded on the antigen-loaded sensors. After washing, two- or threefold serially diluted C1q (Quidel, Catalog No.: A400) starting at 1 µM was loaded on the sensors to characterize association and dissociation between C1q and Fc.
For epitope mapping of anti-EGFR and anti-HER2 antibodies, 2 μg/mL of biotinylated EGFR or HER2 proteins were loaded on streptavidin sensors, and then the first antibody was loaded on the antigen-loaded sensors. After the sensors were saturated with the first antibody, the mixture of the two antibodies that needed to be mapped were loaded to detect whether there was further binding.
Statistical analysis
Statistical analysis was performed with one-way analysis of variance followed by Holm-Sidak multiple comparisons test for comparison of three or more parametric variables or unpaired t test for comparison of two parametric variables, in GraphPad version 8 (Prism). Statistical significance was accepted for P < .05 at 95% confidence interval.
Acknowledgments
We thank Zachary Britton, Keith Rickert, and Arnita Barnes for their help with antibody purification.
Disclosure of potential conflicts of interest
All authors are employees of AstraZeneca with stock interests in the company.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Scott AM, Wolchok JD, Old LJ.. Antibody therapy of cancer. Nat. Rev. Cancer. 2012;12:278–15. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 2.Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341:1192–98. doi: 10.1126/science.1241145. [DOI] [PubMed] [Google Scholar]
- 3.Chester C, Marabelle A, Houot R, Kohrt HE. Dual antibody therapy to harness the innate anti-tumor immune response to enhance antibody targeting of tumors. Curr. Opin. Immunol. 2015;33:1–8. doi: 10.1016/j.coi.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 4.Shuptrine CW, Surana R, Weiner LM. Monoclonal antibodies for the treatment of cancer. Semin. Cancer Biol. 2012;22:3–13. doi: 10.1016/j.semcancer.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter PJ. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006;6:343–57. doi: 10.1038/nri1837. [DOI] [PubMed] [Google Scholar]
- 6.Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010;10:317–27. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walport MJ. Complement. First of two parts. N. Engl. J. Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 8.Ehrnthaller C, Ignatius A, Gebhard F, Huber-Lang M. New insights of an old defense system: structure, function, and clinical relevance of the complement system. Mol. Med. 2011;17:317–29. doi: 10.2119/molmed.2010.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kellner C, Otte A, Cappuzzello E, Klausz K, Peipp M. Modulating cytotoxic effector functions by Fc engineering to improve cancer therapy. Transfus Med. Hemother. 2017;44:327–36. doi: 10.1159/000479980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melis JP, Strumane K, Ruuls SR, Beurskens FJ, Schuurman J, Parren PW. Complement in therapy and disease: regulating the complement system with antibody-based therapeutics. Mol. Immunol. 2015;67:117–30. doi: 10.1016/j.molimm.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 11.Weiner GJ. Rituximab: mechanism of action. Semin. Hematol. 2010;47:115–23. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou X, Hu W, Qin X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: implications for therapy. Oncologist. 2008;13:954–66. doi: 10.1634/theoncologist.2008-0089. [DOI] [PubMed] [Google Scholar]
- 13.Manches O, Lui G, Chaperot L, Gressin R, Molens JP, Jacob MC, Sotto -J-J, Leroux D, Bensa J-C, Plumas J, et al. In vitro mechanisms of action of rituximab on primary non-Hodgkin lymphomas. Blood. 2003;101:949–54. doi: 10.1182/blood-2002-02-0469. [DOI] [PubMed] [Google Scholar]
- 14.van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clin. Cancer Res. 2006;12:4027–35. doi: 10.1158/1078-0432.CCR-06-0066. [DOI] [PubMed] [Google Scholar]
- 15.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gros P, Milder FJ, Janssen BJ. Complement driven by conformational changes. Nat. Rev. Immunol. 2008;8:48–58. doi: 10.1038/nri2231. [DOI] [PubMed] [Google Scholar]
- 17.Bang YJ, Giaccone G, Im SA, Oh DY, Bauer TM, Nordstrom JL, Li H, Chichili GR, Moore PA, Hong S, et al. First-in-human phase 1 study of margetuximab (MGAH22), an Fc-modified chimeric monoclonal antibody, in patients with HER2-positive advanced solid tumors. Ann. Oncol. 2017;28:855–61. doi: 10.1093/annonc/mdx002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desjarlais JR, Lazar GA. Modulation of antibody effector function. Exp. Cell Res. 2011;317:1278–85. doi: 10.1016/j.yexcr.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 19.Kellner C, Derer S, Valerius T, Peipp M. Boosting ADCC and CDC activity by Fc engineering and evaluation of antibody effector functions. Methods. 2014;65:105–13. doi: 10.1016/j.ymeth.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 20.Wang G, de Jong RN, van den Bremer ET, Beurskens FJ, Labrijn AF, Ugurlar D, Gros P, Schuurman J, Parren PHI, Heck AR, et al. Molecular basis of assembly and activation of complement component C1 in complex with immunoglobulin G1 and antigen. Mol. Cell. 2016;63:135–45. doi: 10.1016/j.molcel.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 21.Hughes-Jones NC, Gardner B. Reaction between the isolated globular sub-units of the complement component C1q and IgG-complexes. Mol. Immunol. 1979;16:697–701. doi: 10.1016/0161-5890(79)90010-5. [DOI] [PubMed] [Google Scholar]
- 22.Feinstein A, Richardson N, Taussig MI. Immunoglobulin flexibility in complement activation. Immunol. Today. 1986;7:169–74. [DOI] [PubMed] [Google Scholar]
- 23.Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, Mulkerrin MG.. Engineered antibodies with increased activity to recruit complement. J. Immunol. 2001;166:2571–75. doi: 10.4049/jimmunol.166.4.2571. [DOI] [PubMed] [Google Scholar]
- 24.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. 2010;2:181–89. doi: 10.4161/mabs.2.2.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, Münz C, Nimmerjahn F, Dalakas MC, Lünemann JD, et al. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J. Clin. Invest. 2015;125:4160–70. doi: 10.1172/JCI82695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Natsume A, Niwa R, Satoh M. Improving effector functions of antibodies for cancer treatment: enhancing ADCC and CDC. Drug Des. Devel. Ther. 2009;3:7–16. [PMC free article] [PubMed] [Google Scholar]
- 27.Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr. Opin. Biotechnol. 2009;20:685–91. doi: 10.1016/j.copbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 28.Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, Wakitani M, Ohta S, Satoh M, Shitara K, et al. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res. 2008;68:3863–72. doi: 10.1158/0008-5472.CAN-07-6297. [DOI] [PubMed] [Google Scholar]
- 29.Raju TS. Terminal sugars of Fc glycans influence antibody effector functions of IgGs. Curr. Opin. Immunol. 2008;20:471–78. doi: 10.1016/j.coi.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Ferrara C, Brunker P, Suter T, Moser S, Puntener U, Umana P. Modulation of therapeutic antibody effector functions by glycosylation engineering: influence of Golgi enzyme localization domain and co-expression of heterologous beta1, 4-N-acetylglucosaminyltransferase III and Golgi alpha-mannosidase II. Biotechnol. Bioeng. 2006;93:851–61. doi: 10.1002/bit.20777. [DOI] [PubMed] [Google Scholar]
- 31.Klausz K, Berger S, Lammerts van Bueren JJ, Derer S, Lohse S, Dechant M, van de Winkel JGJ, Peipp M, Parren PWHI, Valerius T, et al. Complement-mediated tumor-specific cell lysis by antibody combinations targeting epidermal growth factor receptor (EGFR) and its variant III (EGFRvIII). Cancer Sci. 2011;102:1761–68. doi: 10.1111/j.1349-7006.2011.02019.x. [DOI] [PubMed] [Google Scholar]
- 32.Dechant M, Weisner W, Berger S, Peipp M, Beyer T, Schneider-Merck T, Lammerts van Bueren JJ, Bleeker WK, Parren PWHI, van de Winkel JGJ, et al. Complement-dependent tumor cell lysis triggered by combinations of epidermal growth factor receptor antibodies. Cancer Res. 2008;68:4998–5003. doi: 10.1158/0008-5472.CAN-07-6226. [DOI] [PubMed] [Google Scholar]
- 33.Macor P, Mezzanzanica D, Cossetti C, Alberti P, Figini M, Canevari S, Tedesco F. Complement activated by chimeric anti-folate receptor antibodies is an efficient effector system to control ovarian carcinoma. Cancer Res. 2006;66:3876–83. doi: 10.1158/0008-5472.CAN-05-3434. [DOI] [PubMed] [Google Scholar]
- 34.Macor P, Tripodo C, Zorzet S, Piovan E, Bossi F, Marzari R, Amadori A, Tedesco F. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer Res. 2007;67:10556–63. doi: 10.1158/0008-5472.CAN-07-1811. [DOI] [PubMed] [Google Scholar]
- 35.Mamidi S, Cinci M, Hasmann M, Fehring V, Kirschfink M. Lipoplex mediated silencing of membrane regulators (CD46, CD55 and CD59) enhances complement-dependent anti-tumor activity of trastuzumab and pertuzumab. Mol. Oncol. 2013;7:580–94. doi: 10.1016/j.molonc.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJR, et al. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260–63. doi: 10.1126/science.1248943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, Horstman W, Engelberts PJ, Oostindie SC, Wang G, et al. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLoS Biol. 2016;14:e1002344. doi: 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strasser J, de Jong RN, Beurskens FJ, Wang G, Heck AJR, Schuurman J, Parren PWHI, Hinterdorfer P, Preiner J.Unraveling the macromolecular pathways of IgG oligomerization and complement activation on antigenic surfaces. Nano. Lett. 2019;19:4787–96. doi: 10.1021/acs.nanolett.9b02220. [DOI] [PubMed] [Google Scholar]
- 39.Mazor Y, Yang C, Borrok MJ, Ayriss J, Aherne K, Wu H, Dall’Acqua WF. Enhancement of immune effector functions by modulating IgG’s intrinsic affinity for target antigen. PLoS One. 2016;11:e0157788. doi: 10.1371/journal.pone.0157788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bleeker WK, Lammerts van Bueren JJ, van Ojik HH, Gerritsen AF, Pluyter M, Houtkamp M, Halk E, Goldstein J, Schuurman J, van Dijk MA, et al. Dual mode of action of a human anti-epidermal growth factor receptor monoclonal antibody for cancer therapy. J. Immunol. 2004;173:4699–707. doi: 10.4049/jimmunol.173.7.4699. [DOI] [PubMed] [Google Scholar]
- 41.Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007;98:1275–80. doi: 10.1111/j.1349-7006.2007.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gelderman KA, Tomlinson S, Ross GD, Gorter A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004;25:158–64. doi: 10.1016/j.it.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 43.Spiridon CI, Ghetie MA, Uhr J, Marches R, Li JL, Shen GL, Vitetta ES. Targeting multiple Her-2 epitopes with monoclonal antibodies results in improved antigrowth activity of a human breast cancer cell line in vitro and in vivo. Clin. Cancer Res. 2002;8:1720–30. [PubMed] [Google Scholar]
- 44.Mazor Y, Oganesyan V, Yang C, Hansen A, Wang J, Liu H, Sachsenmeier K, Carlson M, Gadre DV, Borrok MJ, et al. Improving target cell specificity using a novel monovalent bispecific IgG design. MAbs. 2015;7:377–89. doi: 10.1080/19420862.2015.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wong SF. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin. Ther. 2005;27:684–94. doi: 10.1016/j.clinthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Gerdes CA, Nicolini VG, Herter S, van Puijenbroek E, Lang S, Roemmele M, Moessner E, Freytag O, Friess T, Ries CH, et al. GA201 (RG7160): a novel, humanized, glycoengineered anti-EGFR antibody with enhanced ADCC and superior in vivo efficacy compared with cetuximab. Clin. Cancer Res. 2013;19:1126–38. doi: 10.1158/1078-0432.CCR-12-0989. [DOI] [PubMed] [Google Scholar]
- 47.Boekhout AH, Beijnen JH, Schellens JH. Trastuzumab. Oncologist. 2011;16:800–10. doi: 10.1634/theoncologist.2010-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Capelan M, Pugliano L, De Azambuja E, Bozovic I, Saini KS, Sotiriou C, Loi S, Piccart-Gebhart MJ. Pertuzumab: new hope for patients with HER2-positive breast cancer. Ann. Oncol. 2013;24:273–82. doi: 10.1093/annonc/mds328. [DOI] [PubMed] [Google Scholar]
- 49.Tang Y, Lou J, Alpaugh RK, Robinson MK, Marks JD, Weiner LM. Regulation of antibody-dependent cellular cytotoxicity by IgG intrinsic and apparent affinity for target antigen. J. Immunol. 2007;179:2815–23. doi: 10.4049/jimmunol.179.5.2815. [DOI] [PubMed] [Google Scholar]
- 50.Mazor Y, Sachsenmeier KF, Yang C, Hansen A, Filderman J, Mulgrew K, Wu H, Dall’Acqua WF. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 2017;7:40098. doi: 10.1038/srep40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mazor Y, Hansen A, Yang C, Chowdhury PS, Wang J, Stephens G, Wu H, Dall’Acqua WF. Insights into the molecular basis of a bispecific antibody’s target selectivity. MAbs. 2015;7:461–69. doi: 10.1080/19420862.2015.1022695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogers LM, Veeramani S, Weiner GJ. Complement in monoclonal antibody therapy of cancer. Immunol. Res. 2014;59:203–10. doi: 10.1007/s12026-014-8542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan J, Allendorf DJ, Li B, Yan R, Hansen R, Donev R. The role of membrane complement regulatory proteins in cancer immunotherapy. Adv. Exp. Med. Biol. 2008;632:159–74. [PubMed] [Google Scholar]
- 54.Fishelson Z, Donin N, Zell S, Schultz S, Kirschfink M. Obstacles to cancer immunotherapy: expression of membrane complement regulatory proteins (mCRPs) in tumors. Mol. Immunol. 2003;40:109–23. doi: 10.1016/S0161-5890(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 55.Ugurlar D, Howes SC, de Kreuk BJ, Koning RI, de Jong RN, Beurskens FJ, Schuurman J, Koster AJ, Sharp TH, Parren PWHI, et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science. 2018;359:794–97. doi: 10.1126/science.aao4988. [DOI] [PubMed] [Google Scholar]
- 56.Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug. Discov. 2019. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 57.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9:182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng S, Moores S, Jarantow S, Pardinas J, Chiu M, Zhou H, Wang W. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. MAbs. 2016;8:551–61. doi: 10.1080/19420862.2015.1136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schubert I, Saul D, Nowecki S, Mackensen A, Fey GH, Oduncu FS. A dual-targeting triplebody mediates preferential redirected lysis of antigen double-positive over single-positive leukemic cells. MAbs. 2014;6:286–96. doi: 10.4161/mabs.26768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Supernat A, Lapinska-Szumczyk S, Majewska H, Gulczynski J, Biernat W, Wydra D, Żaczek AJ. Tumor heterogeneity at protein level as an independent prognostic factor in endometrial cancer. Transl. Oncol. 2014;7:613–19. doi: 10.1016/j.tranon.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dimasi N, Gao C, Fleming R, Woods RM, Yao XT, Shirinian L, Kiener PA, Wu H. The design and characterization of oligospecific antibodies for simultaneous targeting of multiple disease mediators. J. Mol. Biol. 2009;393:672–92. doi: 10.1016/j.jmb.2009.08.032. [DOI] [PubMed] [Google Scholar]
- 62.Oganesyan V, Gao C, Shirinian L, Wu H, Dall’Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr. D Biol. Crystallogr. 2008;64:700–04. doi: 10.1107/S0907444908007877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Couch JA, Yu YJ, Zhang Y, Tarrant JM, Fuji RN, Meilandt WJ, Solanoy H, Tong RK, Hoyte K, Luk W, et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 2013;5(183ra57):1–12. doi: 10.1126/scitranslmed.3005338. [DOI] [PubMed] [Google Scholar]
- 64.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJY, et al. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. J. Biol. Chem. 2017;292:3900–08. doi: 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018. 46:D1074–D82. doi: 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J. Immunol. 2013;190:4315–23. doi: 10.4049/jimmunol.1200501. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.