

Abstract
Uveal melanoma (UVM) is the most common primary intraocular malignancy in adults. With over 50% of patients developing metastatic disease, there is an unmet need for improved diagnostic and therapeutic options. Efforts to understand the molecular biology of the disease have revealed several markers that correlate with patient prognosis, including the copy number of chromosome 3, genetic alterations in BAP1, EIF1AX and SF3B1 genes, and other transcriptional features. Here, we expand upon previous reports by comprehensively characterizing the short RNA-ome in 80 primary UVM tumor samples. In particular, we describe a previously-unseen complex network involving numerous regulatory molecules that comprise microRNA (miRNAs), novel UVM specific miRNA loci, miRNA isoforms (isomiRs), and tRNA-derived fragments (tRFs). Importantly, we show that the abundance profiles of isomiRs and tRFs associate with various molecular phenotypes, metastatic disease, and patient survival. Our findings suggest deep involvement of isomiRs and tRFs in the disease etiology of UVM. We posit that further study and characterization of these novel molecules will improve understanding of the mechanisms underlying UVM, and lead to the development of new diagnostic and therapeutic approaches.
Keywords: microRNAs, isomiRs, transfer RNA, tRFs, melanoma, uveal melanoma, MicroRNAs, RNA, tRNA fragments, transcriptome, noncoding RNA, isomiR
INTRODUCTION
Uveal melanoma (UVM) arises from melanocytes of the uveal tract and is the most common primary intraocular malignancy in adults. With approximately 2,500 new U.S. cases annually (Kaliki and Shields, 2017), UVM accounts for ~5% of diagnosed melanomas. UVMs occur anywhere within the uveal tract, but the choroid and ciliary bodies are the most frequent locations (~90%) (Kaliki and Shields, 2017). Approximately 40% patients develop metastases within 10 years, irrespective of treatment type, but this strongly correlates to tumor size and stage of tumor at diagnosis (Shields et al., 2009; Shields et al., 2013; Shields et al., 2015). Currently, the primary plaque brachytherapy or enucleation. Despite treatment options, uveal melanoma shows a high propensity for liver metastasis (Spagnolo et al., 2016), with less than 12 month median survival.
Molecular disease features strongly correlate with clinical outcomes. For example, chromosomal abnormalities monosomy of chromosome 3 (M3) are commonly detected (Robertson et al., 2018; Shields et al., 2017a; Shields et al., 2017b) and associated with increased metastasis and poorer overall prognosis. Loss of function of the 3p21 BRCA Associated Protein 1 (BAP1) gene, either through mutations or decreased expression, correlates with M3 phenotype. Conversely SRSF2/SF3B1 and EIF1AX mutant tumors (Robertson et al., 2018) have distinct copy number alterations and DNA methylation profiles that associate with the better overall prognosis of the disomy 3 (D3) phenotype.
While the genome’s contributions to the disease have been well-studied, it is clear that additional unknown disease mechanisms remain. One such mechanism may relate to short non-coding RNAs (ncRNAs). Two categories of short ncRNAs with demonstrated regulatory roles and emerging strong relevance for UVM are the isoforms of microRNAs (miRNAs), and transfer RNA (tRNA) derived fragments.
MiRNAs are the best-studied ncRNAs to date. These ~22 nucleotide (nts) RNAs modulate the abundance of messenger RNAs (mRNAs) and long non-coding RNAs (lncRNAs) in a sequence-dependent manner (Bartel, 2009). As we showed previously, one miRNA can target hundreds to thousands of genes (Clark et al., 2014), in the coding sequence (Tay et al., 2008), 5´ untranslated region (UTR), (Zhou and Rigoutsos, 2014), or 3´-UTR of an mRNA. MiRNAs operate through several mechanisms, including translational inhibition (Djuranovic et al., 2012), disruption of cap–tail interactions (Cui et al., 2006), or exonuclease-mediated mRNA degradation (Ramachandran and Chen, 2008).
Early deep-sequencing studies reported expression of multiple isoforms of miRNAs (isomiRs) that were initially dismissed as aberrant. We showed that isomiRs are produced constitutively and exhibit tissue-specific abundance profiles (Loher et al., 2014; Telonis and Rigoutsos, 2017). We also found that some miRNA arms produce more than 30 distinct isomiRs (Telonis and Rigoutsos, 2017). Importantly, we demonstrated that isomiRs from the same miRNA target different mRNAs (Telonis et al., 2015b). Thus, isomiR expression greatly increases the number of target mRNAs regulated by miRNAs. Notably, we found that the most abundant isomiR from a given miRNA locus generally varies amongst different tissues (Telonis et al., 2016a). Presumably, this tissue-dependence corresponds to specific regulatory needs.
This last observation becomes even more relevant when considering that the tissue-specific patterns of isomiR expression are modulated further by individual attributes such as a person’s sex, race/ethnicity, and population origin, as well as disease type and subtype, as we also showed (Loher et al., 2014; Telonis et al., 2016a; Telonis et al., 2015b). We recently described several thousand novel human-specific and tissue-specific miRNA loci (Londin et al., 2015). These novel miRNA loci produce isomiRs that drive tissue-specific regulatory events that are currently-uncharacterized and cannot be captured by mouse models.
It is now known that tRNAs produce short ncRNAs, named tRNA-derived fragments (tRFs) (Cole et al., 2009; Lee et al., 2009). Studies by others and our team have shown that tRFs are not products of random degradation (Goodarzi et al., 2015; Honda et al., 2015; Kumar et al., 2014; Kumar et al., 2015; Magee et al., 2018; Pliatsika et al., 2018; Shigematsu and Kirino, 2017; Telonis et al., 2015a; Telonis and Rigoutsos, 2017; Telonis and Rigoutsos, 2018). In fact, some tRFs target mRNAs in a miRNA-like manner (Kuscu et al., 2018). In our work with human tissues, we showed that tRFs are produced constitutively and their profiles depend on a person’s sex, race/ethnicity, and population origin, as well as on tissue, tissue state, and disease (Telonis et al., 2016a; Telonis et al., 2015a), in complete analogy to our isomiR findings. tRFs have also been shown to decoy RNA-binding proteins (Goodarzi et al., 2015), and to displace mRNAs from the active ribosome (Gebetsberger et al., 2016).
While earlier studies looked at miRNAs from the standpoint of locus expression (Robertson et al., 2018; Triozzi et al., 2016), we are not aware of any efforts to investigate the roles of isomiRs and tRFs in UVM. In what follows, we present the findings from our analysis of these two categories of mRNA regulators across the 80 samples of the TCGA UVM cohort. Specifically, we examined whether isomiR/tRF profiles correlate with clinical outcomes. We also sought genomic loci that harbor previously unreported miRNA precursors with UVM-specific transcription. IsomiRs, tRFs, and previously-unreported miRNA loci represent novel and uncharacterized regulators of UVM biology.
METHODS
Datasets:
Short RNA-seq data and associated clinical information for 80 primary UVM samples were downloaded from the Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/).
Sequence read mapping:
For miRNA characterization, sequence reads were quality trimmed using the cutadapt tool (Martin, 2011), and reads were mapped unambiguously using SHRIMP2 (David et al., 2011). We mapped the reads to the human genome assembly hg19/GRCh37 in order to enable comparisons with the TCGA consortium’s report on UVM (Robertson et al., 2018). No insertions or deletions, and at most one replacement was permitted during mapping. For tRF mining, we used the MINTmap algorithm (Loher et al., 2017; Telonis et al., 2016b).
Identification of novel miRNAs:
To identify novel miRNAs, we processed each UVM dataset independently with miRDeep2 (Friedlander et al., 2012), using default parameter settings and a score threshold of ≥ 1. For all novel miRNA precursors, we identified the most abundant isomiR and labeled it the ‘archetype’ sequence for the corresponding locus, as we did previously (Londin et al., 2015).
Identification of isomiRs:
IsomiRs were identified as in our prior work (Loher et al., 2014; Telonis et al., 2016a; Telonis et al., 2015b; Telonis and Rigoutsos, 2017). We use the isomiR nomenclature scheme that we introduced previously (Loher et al., 2014). For example, the isomiR whose 5´ terminus begins one position to the right (+1) of the archetype’s 5´ terminus and whose 3´ terminus ends two positions to the left (−2) of the archetype’s 3´ terminus is labeled “+1|−2”.
Quantification of miRNAs and isomiRs:
We quantified isomiR abundance in reads per million (RPM). Only reads that passed quality trimming and filtering and could aligned exactly to miRNA arms were used in the denominator of this calculation. The abundance of a miRNA arm is calculated as the sum of normalized abundances of all isomiRs from the arm. We identified statistically significant miRNAs/isomiRs with Threshold-seq algorithm (Magee et al., 2017), which adapts to sequencing-depth (“adaptive thresholding”). This thresholding was carried out separately for each dataset. Additionally, we enforced strict criteria and required that a miRNA or isomiR exceed Threshold-seq’s threshold in at least 20 (25%) of the 80 TCGA UVM samples before it entered our analysis.
Quantification of tRFs:
RPM values for tRFs were determined as with miRNAs/isomiRs. For parity, all MINTmap-identified tRFs were subjected to the same Threshold-seq threshold that we established for the isomiRs of the corresponding dataset. As with the miRNAs/isomiRs, we considered only tRFs that were present in at least 20 of the 80 TCGA UVM samples.
Identification of differentially abundant miRNAs, isomiRs, and tRFs:
This analysis was performed using 5000 permutations of the Significance Analyses of Microarrays (SAM) (Tusher et al., 2001) algorithm and using a stringent False Discovery Rate (FDR) of 1% and a log2 fold change threshold of absolute value ≥ 1 (i.e., ≥ 2x fold change).
Overall Survival Analysis:
To determine overall survival associations with specific miRNAs, isomiRs and tRFs, we performed Kaplan-Meier survival analyses. Briefly, we split patient samples into an “above” and “below” group, according to whether a molecule was expressed above or below the total mean in each sample. We then stratified the samples into these two groups and computed the time to development of metastasis in each group. Log-rank tests describe the significance of any emergent differences.
RESULTS
The miRNAs have characteristic profiles in UVM.
Using stringent criteria (see Methods), we characterized isomiRs present in the TCGA UVM cohort. Four hundred and seven unique miRNA arms, producing one or more isomiRs, are expressed in at least 25% of the 80 samples. Interestingly, 32 appear UVM-specific: they have not been reported in any other context. Collectively, 407 arms produce 1,677 statistically-significant isomiRs (Table 1; Supplemental File 1 lists full miRNA characteristics). Of 407 miRNA arms, 112 produce only one isomiR. Thirty-seven arms produce ≥ 10 isomiRs. Fig. 1A shows the top 20 isomiR-producing miRNA arms. Notably, some of the best-studied miRNA loci (e.g. miR-21–5p, miR-183–5p, miR-143–3p) produce many isomiRs. Similar to our previous findings (Loher et al., 2014; Magee et al., 2018), we observe increased 3´ isomiR variability in UVM, compared to 5´ endpoints. Notably, the top-producer of UVM isomiRs is MD2.ID00112–5p, a novel miRNA that we previously reported (Londin et al., 2015); MD2.ID00112 currently remains uncharacterized.
Table 1:
Summary of the UVM miRNAome
| Category | # |
|---|---|
| miRNA arms previously characterized in UVM | 375 |
| miRNA arms not previously characterized in UVM | 32 |
| total isomiRs expressed in UVM | 1,677 |
| isomiRs previously linked in UVM (i.e., miRbase’s 0|0) | 327 |
| isomiRs not previously linked with UVM | 1,350 |
| isomiRs differing from 0|0 only in their 3´ end | 831 |
| isomiRs differing from 0|0 only in their 5´ end | 187 |
| isomiRs differing from 0|0 in both their 5´ and 3´ ends | 332 |
| miRNA arms with a single isomiR | 112 |
| miRNA arms with ≥ 2 isomiRs | 263 |
| miRNA arms not expressing miRbase (0|0) isoform | 79 |
| miRNA arms where miRbase isoform not most abundant | 174 |
Figure 1: Profiling of UVM isomiRs.
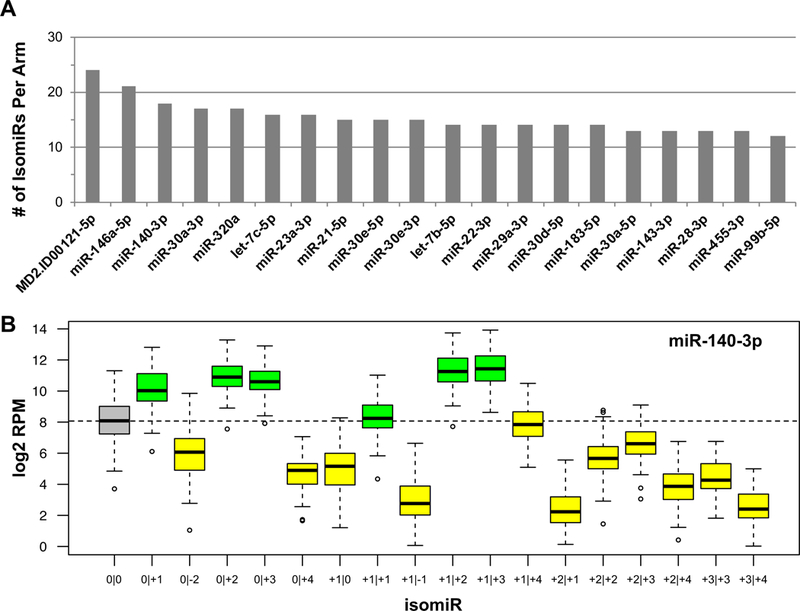
(A) Each bar is a miRNA arm and the y-axis is the number of isomiRs produced at that locus. Shown are the top 20 producing loci. (B) An example of the expression of isomiRs from the miR-140–3p locus. The y-axis is a log2 RPM value for each individual isomiR. The grey box is the ‘0|0’ isomiR, green boxes indicate those isomiRs in which the median expression level is greater than the median expression level of the ‘0|0’ isomiR, while the yellow bars represents those isomiRs in which the median expression level is less than the ‘0|0’ isomiR. The dotted line shows the median expression of the ‘0|0’ isomiR.
We stress that for 165 (44%) of the 375 isomiR-producing miRNA arms listed in miRBase, the most abundant isomiR in UVM is not the ‘archetype’. miR-140–3p presents an illustrative example in UVM (Fig. 1B). Four of the five isomiRs that are more abundant than the archetype have “seeds” that differ from the archetype’s. We stress that because of the different seeds, these four isomiRs are expected to target genes that differ from the genes targeted by miR-140–3p’s “0|0” isomiR, thereby contributing to UVM biology in currently uncharacterized ways. Moreover, for another 80 (21.3%) of the 375 miRNA arms in miRBase, the archetype is not present at all in UVM (Table 1). These observations mirror our earlier findings in other tissues and cell types (Loher et al., 2014; Magee et al., 2018), and lead to an important corollary: focusing on “0|0” isomiRs (miRBase entries) will ignore important regulatory molecules in UVM. Clearly, many miRNA regulators exist in UVM with currently unknown roles.
Newly-discovered miRNA loci are UVM-specific.
We discovered 32 new miRNA loci in these 80 samples (Supp. File 1). When compared to the other 31 TCGA cancer types, many of these loci produce isomiRs that are either exclusively present in UVM, or prominently abundant in UVM. These novel sequences are ideal candidates to serve as candidate UVM biomarkers.
The tRFs have characteristic profiles in UVM.
Across the 80 UVM samples, we find 3,780 unique tRFs. Of these tRFs, 2,286 (60%) are exclusive to the tRNA space, and can only have arisen from tRNA genes. 2,847 (75%) of the tRFs originate in nuclearly-encoded tRNAs, whereas the remaining 933 (25%) derive from mitochondrially-encoded tRNAs. Although the absolute number of tRFs from nuclear tRNAs seems to outweigh that from MT tRNAs, the situation is actually reversed: isodecoder for isodecoder, the 22 MT-encoded tRNAs contribute nine times as many tRFs as the 610 nuclearly-encoded tRNAs.
The tRFs that overlap the mature tRNA sequence belong to one of five structural categories (A Sobala, 2011): 5´-tRFs, i-tRFs, 3´-tRFs, 5´-tRNA halves (5´-tRHs), and 3´-tRNA halves (3´-tRHs). In the UVM samples, the majority of MT-derived tRFs are either 3´-tRFs or i-tRFs (Fig. 2A). On the other hand, nuclearly-encoded tRNAs give rise predominantly to 5´-tRFs (Fig. 2A). The 5´- and 3´-tRHs are under-represented in the UVM samples: the TCGA sequencing protocol specifically enriched for miRNAs (20–24 nts) and ran for only 30 cycles (Robertson et al., 2018). Given that tRHs are among the most abundant molecules in cells, it follows that many additional, as yet uncharacterized, tRF species exist within the UVM transcriptome.
Figure 2: Profiling of UVM tRFs.
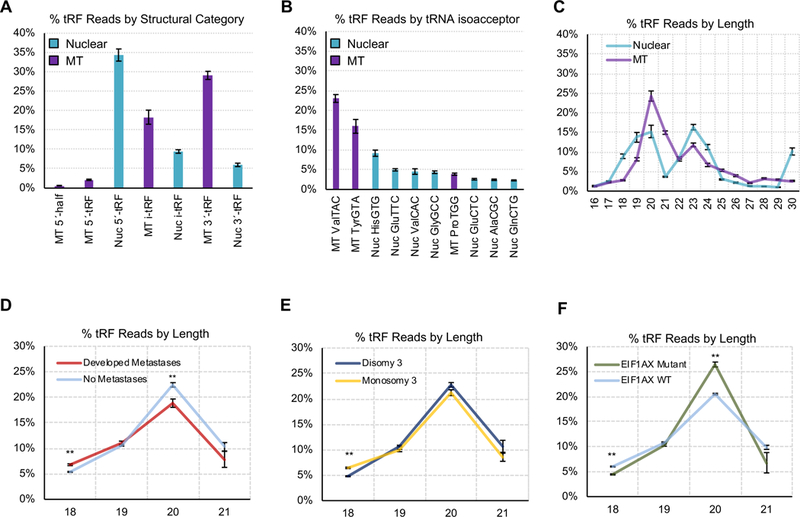
(A) Percentage of tRFs based upon the genomic origin - mitochondrial (MT) or nuclear (Nuc) - and structural type. Here, we show the sum of all reads of each structural type and genomic origin, as a percentage of the sum of all reads. (B) Percentage of tRF reads aligning to the different isoacceptors. Here, we show the sum of all reads from each isoacceptor, as a percentage of the sum of all reads. (C) Distribution of tRFs based upon read length for both nuclear and MT tRFs. Here, we compute the sum of tRFs of each length from 16–30nt, in each individual sample. We then convert these sums to percentages of the sum of reads in that sample, and plot the mean and standard deviations of each percentage for lengths 16–30nt, across all 80 UVM samples. We next analyzed tRF abundance as it relates to clinical characteristics. We show the % of tRFs at varying lengths (18–21 nts), by expression. In each plot, the x-axis shows tRF length (nt), while the y-axis is the percentage sum of sequence reads mapping to tRFs of each specific length. (D) Differentiates patients who did or did not develop metastasis, (E) differentiates M3 or D3 patients, and (F) differentiates patients with or without a mutation in either the EIF1AX or SF3B1 genes. P-values were determined by t-tests.
Remarkably, 73.15% of the sequenced tRFs are produced by only 10 of the 61 nuclear and 22 MT isoacceptors: MT tRNAValTAC, MT tRNATyrGTA, Nuc tRNAHisGTG, Nuc tRNAGluTTC, Nuc tRNAValCAC, Nuc tRNAGlyGCC, MT tRNAProTGG, Nuc tRNAGluCTC, Nuc tRNAAlaCGC, and Nuc tRNAGlnCTG (Fig. 2B). This suggests outsized roles for the respective tRNAs. tRFs from MT tRNAs are generally shorter (20–21 nts) whereas tRFs from nuclear tRNAs show peaks at 20 and 23 nts (Fig. 2C). Notably, our previous work on the TCGA prostate cancer datasets showed a prominent peak at 18 nts, whereas the triple negative breast cancer TCGA datasets showed the prominent peak at 19 nts. These observations indicate tissue-specific differences in the production of tRFs.
IsomiRs can serve as candidate markers of disease progression and patient survival.
We stratified the UVM datasets by M3 status and somatic BAP1 mutation status (Supplemental Table 1) and searched for miRNA loci and isomiRs that are differentially abundant (DA) between the two groups. While we find several miRNA arms to be DA between these two groups (data not shown), many more molecules are found to be DA when we look at individual isomiRs (Fig. 3). Specifically, the expression of the miR-508/514 miRNA cluster from the X chromosome decreases considerably in M3 or BAP1 mutant patients. The miR-508/514 cluster is not located on chromosome 3. IsomiRs from multiple other miRNA loci (e.g. miR-199a/b whose paralogues are located on chromosomes 1, 9 and 19) increase in abundance in M3 and BAP1 mutant patients (Fig. 3). isomiRs from several de novo miRNA loci or from novel miRNA loci from our prior work are also DA (Fig. 3). This further supports the notion that these previously-unreported regulatory molecules are linked mechanistically to UVM.
Figure 3: Differential abundance of isomiRs.
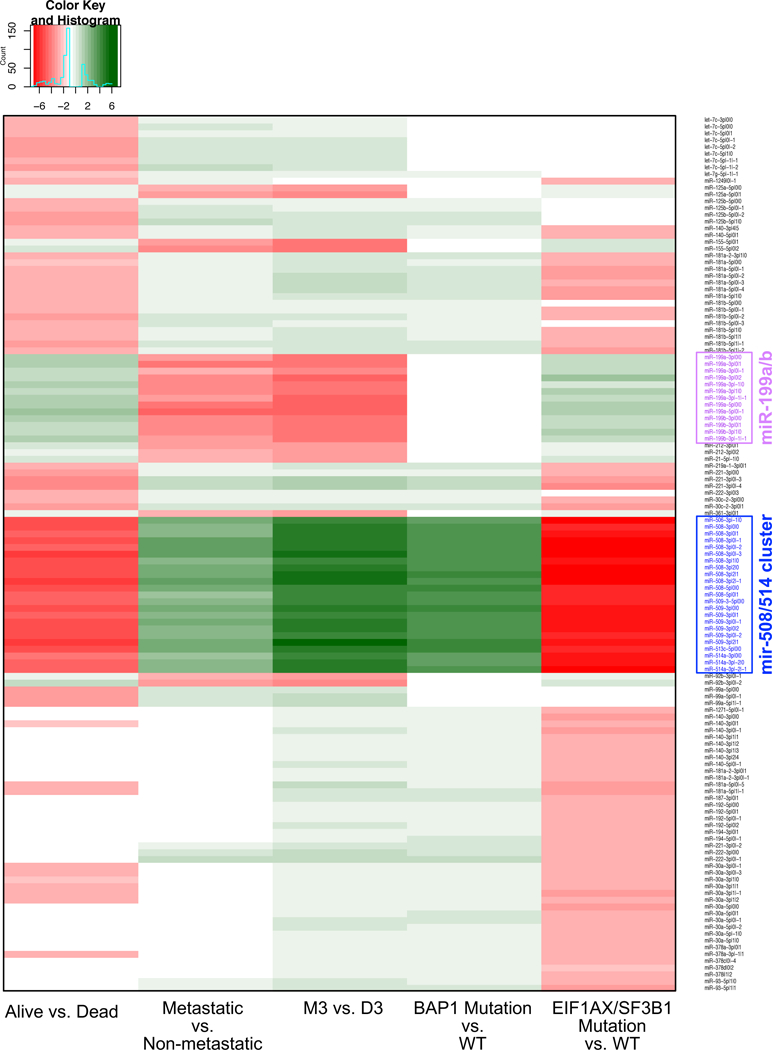
Heatmap showing the most differentially abundant isomiRs for the classification of UVM based upon different clinical attributes (columns): patients with or without a BAP1 mutation, M3 or D3 patients, patients who developed metastatic disease versus those who do not, patients with or without a mutation in either the EIF1AX or SF3B1 genes, and patients who have died versus those who were still alive at last clinical visit. Shown is the log2 of the fold change for each comparison. Each row represents a different isomiR. The isomiR labels highlighted in blue are those of the miR-508/514 cluster, and those highlighted in magenta are isomiRs of the miR-199a/b locus. White cells indicate that the isomiR was not DA for that particular comparison.
Red bars indicate those isomiRs with increased expression in the relevant category, while those isomiRs with decreased expression are represented by green bars. In each graph the x-axis represents a log2 of the fold change.
Stratifying the UVM datasets based on protective SRSF2/SF3B1 or EIF1AX mutations reveals the opposite trend (Fig. 3). isomiRs from the miR-508/514 miRNA cluster of chromosome X show increased abundance, whereas isomiRs from miR-199a/b and miR-142 show decreased abundance with mutation status (Fig. 3). This is the exact opposite of the pattern observed in M3 or BAP1 mutant patients. The miR-187–3p locus offers another such example: its abundance is increased in patients with an EIF1AX or SRSF2 mutation, and decreased in patients with M3 or BAP1 mutations (Fig. 3).
Notably, isomiRs from the same locus sometimes exhibit differing behaviors and differing associations with outcome. Consequently, it follows that the ‘functionally important’ molecules from the various loci discussed here will be different in different patients and will depend upon the context. Supplemental Figure 1 shows a summary of the number of isomiRs that are found to be DA in each of the comparisons. Expanding the miRNA loci, yields a more complete picture of the molecules that associate to the disease phenotypes than collapsing the loci to a single miRNA arm (Supplemental Figure 2). Specifically, in many cases, isomiRs were discovered to be DA, yet their differences in abundances would therefore be missed when considering the mature locus as a whole. For example, many members of the let7 family of miRNAs have isomiRs that are DA (Fig. 3), while the locus as a whole does not display DA (Supplemental Figure 2).
For completeness, we also explored the possibility that isomiRs are DA with regard to other clinical and demographic attributes including patient sex, tumor stage according to the AJCC criteria, tumor cell type as identified by histopathology, and somatic copy number analysis (SCNA) lncRNA, and PARADIGM cluster, as identified in the TCGA UVM work (Robertson et al., 2018). While we did identify DA isomiRs to be DA (data not shown), they tended to be 3´ isomiRs (i.e., they shared miRNA seeds, and, thus, would predicted to have common mRNA targets) from loci that were described by the UVM TCGA consortium.
Biases in tRF lengths are correlated with clinical attributes.
Previous studies revealed that, for example, 18 nt tRFs can inhibit reverse transcriptase whereas 22 nt tRFs prevent their translation by blocking tRNA primer binding sites (Schorn et al., 2017). With this in mind, we sought to determine whether analogous differences exist in UVM. We found that patients who developed metastases showed a significantly higher proportion of 18-nt long tRFs (p-val=0.008) and a lower proportion of 20-nt-long tRFs (p-val=0.002) (Fig. 2D). Similarly, M3 patients showed increased proportions of 18-nt-long tRFs (p-val=0.002) (Fig. 2E). Conversely, EIF1AX mutant patients showed comparatively fewer 18-nt-long tRFs (p-val=0.021) and more 20-nt-long tRFs (p-val=0.003) (Fig. 2F). These observations suggest that tRF fragment lengths correlate with clinical features associated with patient prognosis.
Differential abundance of tRFs correlates with clinical attributes.
We found multiple tRFs that are associated with clinical stage, histology, development of metastatic disease, and the miRNA and lncRNA clusters reported in the TCGA UVM analyses (Table 2). Briefly, EIF1AX mutation status, sex, and development of metastasis are all binary classifications. SCNA, PARADIGM, miRNA, and lncRNA cluster designations derive from the original TCGA UVM analysis. Histology refers to the majority cell type comprising the primary tumor: either spindle, or epithelioid. Finally, AJCC staging represents primary tumor staging according to the AJCC tumor classification guidelines, with the T4 classification representing the worst prognosis (Kaliki and Shields, 2017). Supplemental file 1 lists all DA tRFs.
Table 2: Summary of DA tRFs by clinical comparison and tRF type.
We used Significance Analysis of Microarrays (SAM) to identify tRFs that were differentially abundant (DA) between several clinic contexts, including: EIF1AX mutation status, sex, and development of metastasis are all binary classifications. SCNA, PARADIGM, miRNA, and lncRNA clusters derive from the original TCGA UVM analysis (Robertson et al., 2018). Histology refers to the majority cell type comprising the primary tumor: either spindle or epithelioid. AJCC staging represents primary tumor staging according to the AJCC tumor classification guidelines, where T4 classification represents the worst prognosis2. In this table, we list the number of tRFs that were DA in each comparison, broken down by the type of tRF as determined by the genome of origin – nuclear or mitochondrial – combined by the structural type: 5´-half, 5´-tRF, 3´-half, 3´-tRF, or i-tRF.
| MT 5´-tRF |
MT i-tRF |
MT 3´-tRF |
MT 5´-tRH |
Nuc 5´-tRF |
Nuc i-tRF |
Nuc 3´-tRF |
Total | |
|---|---|---|---|---|---|---|---|---|
| AJCC Clinical Stage | 0 | 0 | 0 | 0 | 1 | 3 | 0 | 4 |
| EIF1AX | 0 | 0 | 0 | 0 | 40 | 4 | 2 | 46 |
| Sex (Male vs Female) | 0 | 0 | 0 | 0 | 9 | 0 | 0 | 9 |
| Histology | 0 | 0 | 0 | 0 | 30 | 3 | 24 | 57 |
| lncRNA_Cluster | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 2 |
| Metastatic Disease | 1 | 23 | 1 | 1 | 1 | 1 | 0 | 28 |
| miRNA Cluster | 1 | 33 | 2 | 0 | 3 | 0 | 0 | 39 |
| PARADIGM Cluster | 11 | 133 | 25 | 3 | 6 | 15 | 2 | 195 |
| SCNA Cluster | 7 | 56 | 7 | 0 | 52 | 74 | 3 | 199 |
PARADIGM and SCNA cluster stratification yields the highest number of DA tRFs. Of note, the majority of these DA tRFs are i-tRFs and arise from MT tRNAs. On the other hand, in SRSF2/SF3B1 or EIF1AX mutation carriers, the majority of DA tRFs that are statistically significant are nuclear 5´-tRFs. Several DA tRFs are DA across multiple clinical and phenotypical categories (Table 3). For example, SCNA and PARADIGM clusters share several dozen DA tRFs. Interestingly, 21 tRFs are DA between “metastatic disease” and “SCNA,” or, between “metastatic disease” and “PARADIGM.” While the functional impact of these two groups of tRFs is not understood, it is reasonable to hypothesize that they play important regulatory roles in UVM.
Table 3: Summary of the overlap of DA tRFs.
We used Significance Analysis of Microarrays (SAM) to identify tRFs that were differentially abundant (DA) between several clinic contexts, including: EIF1AX mutation status, sex, and development of metastasis are all binary classifications. SCNA, PARADIGM, miRNA, and lncRNA clusters derive from the original TCGA UVM analysis (Robertson et al., 2018). Histology refers to the majority cell type comprising the primary tumor: either spindle or epithelioid. Finally, AJCC staging represents primary tumor staging according to the AJCC tumor classification guidelines, where T4 classification represents the worst prognosis. In this table, we show the overlap of DA tRFs between each category. The diagonal shows the number of DA tRFs in each individual category.
| EIF1AX | Sex | SCNA | lncRNA | Meta static |
Para digm |
miRNA | Histo logy |
AJCC | |
|---|---|---|---|---|---|---|---|---|---|
| EIF1AX | 46 | 1 | 27 | 0 | 0 | 2 | 0 | 2 | 0 |
| Sex | 1 | 9 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| SCNA | 27 | 1 | 199 | 2 | 5 | 54 | 1 | 2 | 0 |
| lncRNA | 0 | 0 | 2 | 2 | 0 | 2 | 0 | 0 | 0 |
| Metastatic | 0 | 0 | 5 | 0 | 28 | 16 | 0 | 0 | 0 |
| Paradigm | 2 | 0 | 54 | 0 | 16 | 195 | 0 | 0 | 0 |
| miRNA | 0 | 0 | 1 | 0 | 0 | 1 | 39 | 0 | 0 |
| Histology | 2 | 0 | 2 | 0 | 0 | 0 | 0 | 47 | 0 |
| AJCC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 |
IsomiRs and tRFs associate with the development of metastases.
We next examined the link between individual isomiRs or tRFs and the development of metastases. We performed Kaplan-Meier analyses for time to first metastasis, grouping patients based on the mean RPM of the ncRNA in question (Fig. 4A–D). IsomiRs such as miR-21–5p|−1|0 and miR-29a-3p|−1|1 appear increased in abundance in patients with metastases (red curves in Fig. 4A–B). In contrast, isomiRs such as miR-99a-3p|1|1 and let-7c-5p|−1|1 are decreased in abundance in patients with metastases (Fig. 4C–D).
Figure 4: Survival analyses of isomiRs and tRFs.
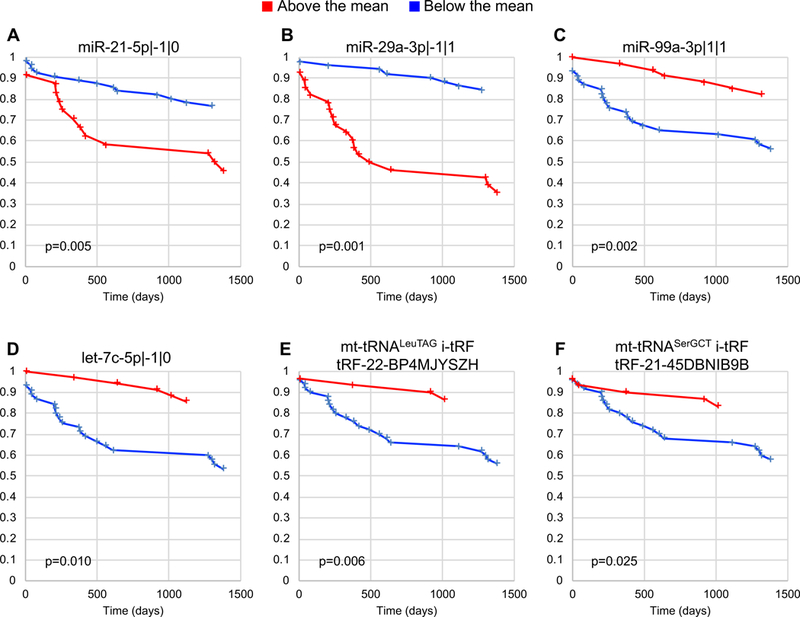
Kaplan-Meir curves for isomiRs and tRFs. In all cases, the samples were grouped based upon whether the expression of the isomiR in question was above the total mean (red lines) or below the total mean (blue lines) in that sample. The x-axis is the time to development of metastasis (days), while the y-axis shows the survival function for each sample group. Panels A-D show representative isomiRs whereas panels E and F shows the two exclusive tRFs that displayed significant DA across multiple clinical and phenotypical categories are shown.
Lastly, we examined tRFs that may associate with metastasis. Two MT i-tRFs, tRF-22-BP4MJYSZH from MT tRNA LeuTAG and tRF-21–45DBNIB9B from MT tRNA SerGCT, showed significant differences in time to first metastasis when we stratified patients based on mean RPM (Fig. 4E–F). Naturally, these tRFs and isomiRs are interesting targets for future experimental investigations.
DISCUSSION
The recent TCGA analysis of UVM identified four main clinical subcategories associated with patient outcomes (Robertson et al., 2018). The two categories with poorer prognosis were characterized by M3 status and BAP1 mutations. Patients with these characteristics show increased overall risk of progressing to metastatic disease. Patients with D3 and EIF1AX/SF3B1 mutations have decreased metastatic risk and better disease prognosis. To gain additional insights into the molecular architecture of this disease, we re-analyzed TCGA datasets, specifically focusing on characterizing the profiles and expression of isomiRs and tRFs. These two categories of short ncRNAs have received increased attention recently, because of their increasingly evident involvement in key regulatory processes.
Our study of the isomiR and tRF profiles reveals a complex regulatory network active in UVM. In several instances, we found that their expression correlates with distinct clinical outcomes. The work represents the first exploration of isomiRs and tRFs in this disease and has identified many novel features that can be further explored as biomarkers for diagnostic and therapeutic purposes.
Previous miRNA profiling endeavors in UVM characterized the expression of numerous highly abundant miRNA loci (Londin et al., 2015; Robertson et al., 2018). While our work corroborates these earlier observations, it goes significantly further than previous efforts in identifying novel and important features of the UVM miRNA-ome. For example, we showed that 72.5% of the 407 miRNA loci that are active in UVM express multiple isomiRs (Supp. File 1). In fact, for nearly half of these loci, the most abundant isoform is not the archetype isoform that is listed in the miRBase. Moreover, for 21.3% of these loci the archetype isoform listed in miRBase was not even present in the UVM samples. As a result, many of the isoforms that are important for UVM biology have not been studied before, while many of the isoforms that have been studied to date were either not the most relevant ones or absent altogether. These two observations strongly suggest that a very large part of the regulatory layer of UVM (isomiRs and their mRNA targets) has not been studied.
Adding to this last point, the 5´ termini of ~30% of all isomiRs that emerged from our analysis of UVM differ from the 5´ termini of the archetypal miRNAs that have been studied to date. Because a shifted 5´ terminus creates a change in the miRNAs’ seed sequence, the discovery of these isomiRs is of functional consequence. These isomiRs have mRNA targetomes whose contribution to the UVM biology is uncharacterized and differs from what has been studied to date. Indeed, as we showed previously in the context of breast cancer, different isomiRs from the same miRNA arm have largely non-overlapping mRNA targetomes (Telonis et al., 2015b).
The 3p arm of miR-140 (Fig. 1B) offers a characteristic example of the relevance of these studies. Of the 18 isomiRs from that this locus that are active in UVM, four are at least as abundant as the archetype and their 5´ terminus differs from that of the archetype. Previous work showed that a 5´-isomiR from miR-140–3p is increased in breast tumors compared to normal breast tissue, wherein it functions to suppress tumor growth and progression (Salem et al., 2016). The four highly abundant 5´-isomiRs we identified here share the same seed as the isomiR that was examined in the breast cancer study. Interestingly, these four isomiRs are increased in abundance in patients with EIFA1X or SF3B1 mutations, and decreased in M3 and BAP1 mutation carriers. This suggests that these isomiRs may play tumor-suppressive roles in UVM, just as in breast cancer, an observation that remains to be validated.
We previously reported thousands of novel miRNA loci that are both tissue- and primate-specific (Londin et al., 2015). We found that many of these previously-reported miRNA loci produce abundant isomiRs in UVM and that their expression correlates with clinical attributes (Fig. 3). De novo analyses of the UVM samples uncovered 32 novel miRNA loci (Supp. File 1). Several of these loci appear to be expressed in a tissue-specific manner. While the functional importance of these newly discovered miRNAs remains to be determined, they nonetheless have potential application as disease-specific biomarkers.
As shown in Fig. 3, multiple loci and isomiRs are DA across clinical attributes. The miR-508/514 locus stands out. Several of the 15 miRNAs in this cluster have been associated with functions upstream of several pathways involved in: tumor development, promotion of melanocyte transformation, and melanoma growth (Streicher et al., 2012). Furthermore, over-expression of some of these miRNAs in skin melanoma (SKCM) results in decreased cell proliferation and colony formation (Streicher et al., 2012). Individual miRNAs of this cluster have been associated with cellular phenotypes in other cancers. For example, miR-509–3p has been shown to inhibit cellular migration in ovarian cancer cells (Li et al., 2016) . Decreased expression of this miRNA, as seen in UVM, may increase metastatic potential. Similarly, low abundances of miR-508–5p in glioma have been associated with shorter overall survival. Inhibition of miR-508–5p results in increased cell proliferation and cellular migration. Interestingly, this cluster shows the opposite effect in patients with protective SIF1AX and SF3B1 mutations. Therefore, decreased expression here may be indicative of poor prognosis, perhaps arising from promotion of tumor growth. It should be noted that ~5% of all of the DA isomiRs reside on chromosome 3. This is notable as M3 status correlates with poorer overall survival and therefore we reasonable hypothesize that many of the miRNA loci residing on this chromosome would therefore be DA. In contrast, the limited number of DA isomiRs derived from chromosome 3 suggests that M3 status and the DA of isomiRs are independent events. Finally, we find that multiple isomiRs associate with patient survival (Fig. 4A–D). Combined, these collections of isomiRs have a potential application as disease biomarkers that may help determine patient outcomes and/or to decide when to begin new therapeutic interventions.
It is also worth mentioning here an interesting interplay between the abundances of individual isomiRs from a miRNA arm and the abundance of the arm as a whole. Specifically, we found multiple instances of isomiRs that are DA between two groups of patients, but whose corresponding miRNA arm as a whole did not show any change in abundance (data not shown). This important finding highlights the importance of studying miRNA regulation at the isomiR level and its ability to uncover new consequential players, in UVM biology and elsewhere. Doing so is likely to reveal novel disease dependencies that would have otherwise remained hidden from view.
In addition to exploring the UVM miRNA-ome, we also profiled and analyzed the endogenous tRFs, in line with our previous reporting of tRF dysregulation in other disease type (Telonis et al., 2015a; Telonis and Rigoutsos, 2017). Our analyses show that numerous tRFs are expressed at high levels in UVM. tRFs of nuclear origin are mostly 5′-tRFs, whereas MT tRFs are predominantly 3′-tRFs (Fig. 2A). Additionally, nearly three quarters of the UVM tRFs are produced by ten tRNA isoacceptors: MT ValTAC, MT TyrGTA, Nuc HisGTG, Nuc GluTTC, Nuc ValCAC, Nuc GlyGCC, MT ProTGG, Nuc GluCTC, Nuc AlaCGC, and Nuc GlnCTG (Fig. 2B). Considering our tRF studies in other contexts (Telonis et al., 2015a; Telonis and Rigoutsos, 2017), this particular tRNA isoacceptor bias is unique to UVM. It is conceivable that the corresponding tRFs are particularly important for key regulatory pathways in UVM and further work is required to determine the full functional consequences of this observation.
Our analysis also revealed a length bias in the production of tRFs (Fig. 2). MT-derived tRFs exhibit a unimodal distribution with a peak at 20 nts (Fig. 2C). Nuclear tRFs on the other hand exhibit a bimodal distribution with peaks at 20 and 23 nts (Fig. 2C). This length bias suggests that distinct processes produce tRFs from the MT and nuclear tRNAs. Recall here that the sequencing protocol employed by the TCGA (Robertson et al., 2018) likely prevents us from seeing tRFs longer than 30nt.
Patients who developed metastases have a significantly lower percentage of tRFs of length 18 nts and a higher percentage of tRFs of length 20 nts (Fig. 2C,D). Similarly, M3 patients show an increased percentage of 18-nt-long tRFs. Conversely, patients with protective EIF1AX mutations show a decrease in 18-nt-long tRFs and an increase in 20-nt-long tRFs. The mechanisms responsible for these cleavage patterns are not known. Previous work described a mechanism whereby 18-nt-long fragments block reverse transcription whereas longer tRFs (22 nts) prime RNAi mediated degradation (Schorn et al., 2017). Thus, the strong association between tRF length and clinical characteristics warrants determination of the functional roles of these tRFs in UVM.
Lastly, we also identified a large number of DA tRFs across clinical categories (Table 2). tRF profiles differ across the formerly-established somatic copy number analysis (SCNA) and mRNA paradigm cluster (PARADIGM) analysis described by the TCGA Consortium. Additionally, we found tRFs to be DA in the context of EIF1AX mutations, metastatic disease, and when considering the majority cell type of the underlying tumor. The vast majority of tRFs are DA in one category, but a number are shared across categories. tRF abundance also associates with overall patient survival (Fig. 4E–F). This evidence points toward differential activity of tRFs in each of these contexts. Unique tRFs may play a role in regulation of pro-metastatic pathways, whereas others may be active in the control and differentiation of uveal cells.
Taken together, our findings delineate complex relationships among the isomiRs and tRFs that are present in primary UVM. Our work identified many novel features that can be potentially leveraged to build novel diagnostic, prognostic, or therapeutic applications. While much additional work is needed to elucidate the roles of these molecules, the generated insights represent a first step towards making new inroads into improving patient outcomes in UVM.
Supplementary Material
Shown are the number of isomiRs that are differentially abundant for each comparison performed in Fig. 3. In both cases, the blue bars represent the number of molecules that decreased in abundance while the red bars indicate the number of molecules that increased in abundance.
Heatmap showing the most differentially abundant miRNA arms for the classification of UVM based upon different clinical attributes (columns): patients with or without a BAP1 mutation, M3 or D3 patients, patients who developed metastatic disease versus those who do not, patients with or without a mutation in either the EIF1AX or SF3B1 genes, and patients who have died versus those who were still alive at last clinical visit. Shown is the log2 of the fold change for each comparison. Each row represents a different miRNA arm. White cells indicate that the isomiR was not DA for that particular comparison.
Summary of clinicopathological phenotypes examined in this study.
SIGNIFICANCE.
Uveal melanoma (UVM) is the most common intraocular malignancy in adults. Much work has focused on understand the roles of genetic in the diseases, while less work has focused on the role of short RNAs in the disease etiology. Using the TCGA UVM dataset we describe a complex network involving numerous regulatory molecules comprised of miRNA isoforms (isomiRs), novel UVM specific miRNA loci, and tRNA-derived fragments (tRFs) that associate with various molecular phenotypes, metastatic disease, and patient survival. This work provides the first characterization isomiRs and tRFs in UVM and furthers our understanding of the molecular basis of this disease.
ACKNOWLEDGEMTS
This study was funded by NIH/NCI grant (grant number R21CA195204)
Footnotes
CONFLICTS OF INTEREST
None declared
REFERENCES
- A Sobala GH (2011). Transfer RNA-Derived Fragments: Origins, Processing, and Functions. Wiley Interdisciplinary Reviews RNA 2, 853–862. [DOI] [PubMed] [Google Scholar]
- Bartel DP (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PM, Loher P, Quann K, Brody J, Londin ER, and Rigoutsos I (2014). Argonaute CLIP-Seq reveals miRNA targetome diversity across tissue types. Sci Rep 4, 5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C, Sobala A, Lu C, Thatcher SR, Bowman A, Brown JW, Green PJ, Barton GJ, and Hutvagner G (2009). Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 15, 2147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Yu Z, Purisima EO, and Wang E (2006). Principles of microRNA regulation of a human cellular signaling network. Molecular Systems Biology 2, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David M, Dzamba M, Lister D, Ilie L, and Brudno M (2011). SHRiMP2: sensitive yet practical SHort Read Mapping. Bioinformatics 27, 1011–2. [DOI] [PubMed] [Google Scholar]
- Djuranovic S, Nahvi A, and Green R (2012). miRNA-mediated gene silencing by translational repression followed by mRNA deadenylation and decay. Science 336, 237–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, and Rajewsky N (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebetsberger J, Wyss L, Mleczko AM, Reuther J, and Polacek N (2016). A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi H, Liu X, Nguyen HC, Zhang S, Fish L, and Tavazoie SF (2015). Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 161, 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda S, Loher P, Shigematsu M, Palazzo JP, Suzuki R, Imoto I, Rigoutsos I, and Kirino Y (2015). Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc Natl Acad Sci U S A 112, E3816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliki S, and Shields CL (2017). Uveal melanoma: relatively rare but deadly cancer. Eye (Lond) 31, 241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Anaya J, Mudunuri SB, and Dutta A (2014). Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Mudunuri SB, Anaya J, and Dutta A (2015). tRFdb: a database for transfer RNA fragments. Nucleic Acids Res 43, D141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C, Kumar P, Kiran M, Su Z, Malik A, and Dutta A (2018). tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer independent manner. RNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Shibata Y, Malhotra A, and Dutta A (2009). A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev 23, 2639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wu H, Li W, Yin L, Guo S, Xu X, Ouyang Y, Zhao Z, Liu S, Tian Y, et al. (2016). Downregulated miR-506 expression facilitates pancreatic cancer progression and chemoresistance via SPHK1/Akt/NF-kappaB signaling. Oncogene 35, 5501–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loher P, Londin ER, and Rigoutsos I (2014). IsomiR expression profiles in human lymphoblastoid cell lines exhibit population and gender dependencies. Oncotarget 5, 8790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loher P, Telonis AG, and Rigoutsos I (2017). MINTmap: fast and exhaustive profiling of nuclear and mitochondrial tRNA fragments from short RNA-seq data. Sci Rep 7, 41184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Londin E, Loher P, Telonis AG, Quann K, Clark P, Jing Y, Hatzimichael E, Kirino Y, Honda S, Lally M, et al. (2015). Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc Natl Acad Sci U S A 112, E1106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee R, Loher P, Londin E, and Rigoutsos I (2017). Threshold-seq: a tool for determining the threshold in short RNA-seq datasets. Bioinformatics 33, 2034–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee RG, Telonis AG, Loher P, Londin E, and Rigoutsos I (2018). Profiles of miRNA Isoforms and tRNA Fragments in Prostate Cancer. Sci Rep 8, 5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17. [Google Scholar]
- Pliatsika V, Loher P, Magee R, Telonis AG, Londin E, Shigematsu M, Kirino Y, and Rigoutsos I (2018). MINTbase v2.0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all The Cancer Genome Atlas projects. Nucleic Acids Res 46, D152–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V, and Chen X (2008). Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science 321, 1490–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, Hess JM, Uzunangelov V, Walter V, Danilova L, et al. (2018). Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 33, 151. [DOI] [PubMed] [Google Scholar]
- Salem O, Erdem N, Jung J, Münstermann E, Wörner A, Wilhelm H, Wiemann S, and Körner C (2016). The highly expressed 5’isomiR of hsa-miR-140–3p contributes to the tumor-suppressive effects of miR-140 by reducing breast cancer proliferation and migration. BMC Genomics 17, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorn AJ, Gutbrod MJ, Leblanc C, and Martienssen R (2017). LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 170, 61–71 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields CL, Furuta M, Thangappan A, Nagori S, Mashayekhi A, Lally DR, Kelly CC, Rudich DS, Nagori AV, Wakade OA, et al. (2009). Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol 127, 989–98. [DOI] [PubMed] [Google Scholar]
- Shields CL, Kaliki S, Furuta M, Fulco E, Alarcon C, and Shields JA (2013). American Joint Committee on Cancer classification of posterior uveal melanoma (tumor size category) predicts prognosis in 7731 patients. Ophthalmology 120, 2066–71. [DOI] [PubMed] [Google Scholar]
- Shields CL, Kaliki S, Furuta M, Fulco E, Alarcon C, and Shields JA (2015). American Joint Committee on Cancer Classification of Uveal Melanoma (Anatomic Stage) Predicts Prognosis in 7,731 Patients: The 2013 Zimmerman Lecture. Ophthalmology 122, 1180–6. [DOI] [PubMed] [Google Scholar]
- Shields CL, Say E. a. T., Hasanreisoglu M, Saktanasate J, Lawson BM, Landy JE, Badami AU, Sivalingam MD, Hauschild AJ, House RJ, et al. (2017a). Personalized Prognosis of Uveal Melanoma Based on Cytogenetic Profile in 1059 Patients over an 8-Year Period: The 2017 Harry S. Gradle Lecture. Ophthalmology 124, 1523–1531. [DOI] [PubMed] [Google Scholar]
- Shields CL, Say E. a. T., Hasanreisoglu M, Saktanasate J, Lawson BM, Landy JE, Badami AU, Sivalingam MD, Mashayekhi A, Shields JA, et al. (2017b). Cytogenetic Abnormalities in Uveal Melanoma Based on Tumor Features and Size in 1059 Patients: The 2016 W. Richard Green Lecture. Ophthalmology 124, 609–618. [DOI] [PubMed] [Google Scholar]
- Shigematsu M, and Kirino Y (2017). 5’-Terminal nucleotide variations in human cytoplasmic tRNAHisGUG and its 5’-halves. RNA 23, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo F, Picasso V, Spano L, Tanda E, Venzano C, and Queirolo P (2016). Update on Metastatic Uveal Melanoma: Progress and Challenges. BioDrugs 30, 161–72. [DOI] [PubMed] [Google Scholar]
- Streicher KL, Zhu W, Lehmann KP, Georgantas RW, Morehouse CA, Brohawn P, Carrasco RA, Xiao Z, Tice DA, Higgs BW, et al. (2012). A novel oncogenic role for the miRNA-506–514 cluster in initiating melanocyte transformation and promoting melanoma growth. Oncogene 31, 1558–70. [DOI] [PubMed] [Google Scholar]
- Tay Y, Zhang J, Thomson AM, Lim B, and Rigoutsos I (2008). MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455, 1124–8. [DOI] [PubMed] [Google Scholar]
- Telonis A, Magee R, Loher P, Chervoneva I, Londin E, and Rigoutsos I (2016a). The presence or absence alone of miRNA isoforms (isomiRs) successfully discriminate amongst the 32 TCGA cancer types. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis AG, Loher P, Honda S, Jing Y, Palazzo J, Kirino Y, and Rigoutsos I (2015a). Dissecting tRNA-derived fragment complexities using personalized transcriptomes reveals novel fragment classes and unexpected dependencies. Oncotarget 6, 24797–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis AG, Loher P, Jing Y, Londin E, and Rigoutsos I (2015b). Beyond the one-locus-one-miRNA paradigm: microRNA isoforms enable deeper insights into breast cancer heterogeneity. Nucleic Acids Res 43, 9158–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis AG, Loher P, Kirino Y, and Rigoutsos I (2016b). Consequential considerations when mapping tRNA fragments. BMC Bioinformatics 17, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis AG, and Rigoutsos I (2017). Race disparities in the contribution of miRNA isoforms and tRNA-derived fragments to triple-negative breast cancer. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis AG, and Rigoutsos I (2018). Race Disparities in the Contribution of miRNA Isoforms and tRNA-Derived Fragments to Triple-Negative Breast Cancer. Cancer Res 78, 1140–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triozzi PL, Achberger S, Aldrich W, Crabb JW, Saunthararajah Y, and Singh AD (2016). Association of tumor and plasma microRNA expression with tumor monosomy-3 in patients with uveal melanoma. Clin Epigenetics 8, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, and Chu G (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98, 5116–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, and Rigoutsos I (2014). MiR-103a-3p targets the 5’ UTR of GPRC5A in pancreatic cells. RNA 20, 1431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Shown are the number of isomiRs that are differentially abundant for each comparison performed in Fig. 3. In both cases, the blue bars represent the number of molecules that decreased in abundance while the red bars indicate the number of molecules that increased in abundance.
Heatmap showing the most differentially abundant miRNA arms for the classification of UVM based upon different clinical attributes (columns): patients with or without a BAP1 mutation, M3 or D3 patients, patients who developed metastatic disease versus those who do not, patients with or without a mutation in either the EIF1AX or SF3B1 genes, and patients who have died versus those who were still alive at last clinical visit. Shown is the log2 of the fold change for each comparison. Each row represents a different miRNA arm. White cells indicate that the isomiR was not DA for that particular comparison.
Summary of clinicopathological phenotypes examined in this study.