

Abstract
Adaptive metabolic response to injury includes the utilization of alternative energy substrates – such as ketone bodies (KB) – to protect the brain against further damage. Here, we examined cerebral ketone metabolism in patients with traumatic brain injury (TBI; n = 34 subjects) monitored with cerebral microdialysis to measure total brain interstitial tissue KB levels (acetoacetate and β-hydroxybutyrate). Nutrition – from fasting vs. stable nutrition state – was associated with a significant decrease of brain KB (34.7 [10th–90th percentiles 10.7–189] µmol/L vs. 13.1 [6.5–64.3] µmol/L, p < 0.001) and blood KB (668 [168.4–3824.9] vs. 129.4 [82.6–1033.8] µmol/L, p < 0.01). Blood KB correlated with brain KB (Spearman’s rho 0.56, p = 0.0013). Continuous feeding with medium-chain triglycerides-enriched enteral nutrition did not increase blood KB, and provided a modest increase in blood and brain free medium chain fatty acids. Higher brain KB at the acute TBI phase correlated with age and brain lactate, pyruvate and glutamate, but not brain glucose. These novel findings suggest that nutritional ketosis was the main determinant of cerebral KB metabolism following TBI. Age and cerebral metabolic distress contributed to brain KB supporting the hypothesis that ketones might act as alternative energy substrates to glucose. Further studies testing KB supplementation after TBI are warranted.
Keywords: Traumatic brain injury, ketones, cerebral metabolism, glucose, nutrition
Introduction
Adaptive metabolic response to injury includes the utilization of alternative energy substrates by the brain, such as lactate1 and ketones.2 Production of ketone bodies (KB, including acetoacetate [AcAc] and β-hydroxybutyrate [BHB]) upon mobilization of fatty acids during fasting or adrenergic stress (endogenous ketosis or ketogenesis) provides alternative energy substrate in conditions of increased demand,3 while simultaneously preserving limited glucose reserves.4–9 This rescue mechanism may be an advantage in conditions of impaired glycolysis and potentially limited cerebral glucose availability, such as after traumatic brain injury (TBI).10,11 Besides their key role in regulating cerebral energy metabolism, KB have other important neuroprotective properties, including attenuation of oxidative stress,12,13 apoptotic cell death14 and microglial activation.15,16 Increasing KB metabolism through fasting or diet-induced ketosis promotes brain resistance to stress and injury2 and attenuates acute cerebral damage.16–18
Ketone body supplementation, e.g. with the use of medium-chain triglycerides (MCTs) ketogenic diets, has therefore emerged as a potential non-pharmacological neuroprotective therapy.19 Apart from KB supplementation, MCT-enriched diets result in elevated circulating levels of free medium chain fatty acids (MCFAs, including octanoic acid [C8] and decanoic acid [C10]), which may have additional protective effects against brain damage, mainly via in situ production of brain KB by astrocytes15 or via the inhibition of glutamate receptors and the promotion of mitochondrial biogenesis.20 Whether this holds true in humans was not demonstrated, therefore analyzing free C8 and C10 is an important component of MCT-driven KB supplementation and provides further insights into their importance in modified enteral formulations. Finally, KB supplementation can be achieved intravenously via the administration of sodium BHB infusion.21
Growing evidence on the collective therapeutic potential of KB supplementation has accrued over the last decade, through clinical investigation showing that ketosis improves cognitive performance3,19 and reduces seizure activity.19,22,23 Experimental injury models, particularly after TBI, showing that KB supplementation might attenuate brain damage,13,14,23–26 provide the rationale for further clinical investigation of cerebral ketone metabolism in this setting.
To the best of our knowledge, no clinical study investigated cerebral ketone metabolism in patients with acute brain injury in general, and specifically in those with TBI. We therefore designed a study to examine the modulation of cerebral ketone metabolism in TBI subjects who underwent cerebral microdialysis (CMD) monitoring for the measurement of brain interstitial tissue biochemistry and dynamic changes of cerebral metabolic state.27,28 The main study endpoint was to analyze TBI changes of brain interstitial tissue KB levels according to the patient nutritional state (fasted vs. fed state), and the contribution of endogenous plasma ketosis and systemic glucose to brain KB. Additionally, free MCFA C8 and C10 levels were measured in plasma and CMD to investigate MCT pharmacokinetics. We further explored the potential relationship of fasted brain KB release at the acute phase following TBI with patient age and brain energy metabolism, using CMD markers of increased glycolysis (lactate and pyruvate) and excitatory amino acid release (glutamate).
Material and methods
Patients
A total of 34 TBI subjects from two clinical studies were included in the present analysis. Subjects were admitted to the Department of Intensive Care Medicine, Centre Hospitalier Universitaire Vaudois (CHUV) – Lausanne University Hospital, Switzerland, following TBI and had a post-resuscitation Glasgow Coma Scale (GCS) <9 with an abnormal CT scan (defined by the presence of intracranial lesions [contusions, hematoma]). All patients underwent multimodal brain monitoring with CMD in combination with intracranial pressure (ICP; Codman®, Raynham, MA, USA) and brain tissue oxygen tension (PbtO2; Licox®, Integra Neurosciences, Plainsboro, NJ, USA) probes, as part of standard patient care.
Study 1 consisted of 24 TBI subjects who were part of an observational cohort study to investigate acute cerebral ketone metabolism (study nr 314/14). Study 2 consisted of 10 TBI subjects with the same inclusion criteria as in Study 1, who participated to a study designed to test an MCT-enriched enteral formula and to measure prospectively brain interstitial tissue and circulating plasma concentrations of KB and free MCFA (clinical trials.gov study nr NCT02716532). Approval for the studies was obtained by the Ethical Committee of the University of Lausanne, Switzerland, with an authorization for a waiver of consent for Study 1 and a signed informed consent obtained from each patient next-of-kin for Study 2. The study was in compliance with STROBE guidelines for reporting observational studies.
CMD
CMD consisted of an intra-parenchymal (sub-cortical white matter, visually normal brain) catheter (20 kDa cut-off CMA 70®, CMA Microdialysis AB, Solna, Sweden) that was inserted in the operating room. The catheter was constantly perfused (rate: 0.3 µL/min) with a sterile solution mimicking cerebrospinal fluid (CSF) content through a pump (CMA 106®, CMA Microdialysis AB), as described in our previous studies,29,30 and in line with recent consensus guidelines.27 CMD samples were collected hourly and analyzed immediately at the bedside using a kinetic enzymatic analyzer (ISCUS Flex®, CMA Microdialysis AB). Data from the first 2 h of monitoring were discarded due to stabilization time of the ISCUS analyzer. Routine cerebral metabolites included lactate, pyruvate, glucose and glutamate.
Analysis of KB
CMD and heparinized plasma samples were immediately frozen at −20℃ and then stored at −80℃ for subsequent analysis of concentrations of total KB – including AcAc and BHB, C8 and C10. Analytes concentrations were measured by liquid chromatography coupled to mass spectrometry (LC-MS). Prior to analysis, plasma samples (50 µL) were de-proteinized by addition of isopropanol (1:9). After centrifugation, the supernatant was dried under vacuum, reconstituted in a solution of internal standards in eluent A, and immediately analyzed. Pooled CMD samples (approximately 40 µL) were diluted 2-fold with a solution of internal standards in eluent A and immediately analyzed. Chromatographic separation was performed on a Waters ACQUITY UPLC BEH C8 Column (1.7 µm, 100 × 2.1 mm) using a linear gradient of water + 0.1% acetic acid (eluent A) and acetonitrile/isopropanol (1:1) + 0.1% acetic acid (eluent B). The gradient was as follows: 0.0–1.0 min at 0% B, 1.0–6.5 min from 0% to 100% B, 6.5–8.5 min 100% B, followed by 2 min of equilibration at initial conditions. Flow rate was set to 450 µL/min, column oven temperature to 55℃ and the injection volume to 1 µL. The UPLC system was coupled to an Orbitrap Q Exactive Plus mass spectrometer (ThermoFisher Scientific, Bremen, Germany) operating in negative full scan mode with a resolving power of 35,000 (at m/z = 200). AcAc, BHB and free MCFA C8 and C10 chromatograms were extracted using a 5 ppm mass tolerance and quantified against an external calibration curve in Xcalibur software 2.2 SP1 (ThermoFisher Scientific, Bremen, Germany). For CMD samples, analytes quantification ranges were the following: 1.0–200.0 µmol/L for AcAc, 0.2–200.0 µmol/L for BHB, 0.4–200 µmol/L for C8, and 0.4–100.0 µmol/L for C10.
Nutrition and metabolic control
Nutrition was administered according to our standard of care, aiming at starting progressive feeding, ideally within the first 48 h from ICU admission, targeting an energy delivery of 20–25 Kcal/kg/d energy and a protein intake of 1.2 g/kg/d. The amount of calories was defined based on enteral nutritional formulas only, without accounting for non-nutritional products (mainly, propofol). No patients received intravenous glucose solutions. Enteral nutrition for Study 1 (n = 24 patients) consisted of standard Promote Fibres Plus® (Abbott Nutrition, Switzerland; 130 Kcal/100 mL; 8.1 g protein [25%], 14 g carbohydrates [43%], 4.3 g lipids [30%], MCT 6.5 g/1000 Kcal). For Study 2 (n = 10 patients), MCT-enriched Peptamen AF® (Nestlé Health Science, Switzerland; 150 Kcal/100 mL; 9.4 g protein [25%], 13.5 g carbohydrates [36%], 6.5 g lipids [39%], MCT 23 g/1000 Kcal) was used. Sedation consisted of 2–3 mg/kg/h of 2% propofol (Disoprivan PFS®; 20 mg/mL propofol, containing 0.1 g lipid/mL).
Glycaemic targets were set to keep arterial blood glucose at 6–8 mmol/L (110-145 mg/dL), using continuous intravenous insulin infusion as needed.
Avoidance of fever (defined as a body temperature >38℃) was managed by using external surface cooling.
Data collection and processing
Study design is summarized in Figure 1. The main endpoint was to examine the dynamic changes of total brain KB according to patient nutritional state. For this purpose, pooled 4-hourly CMD samples (≈ 40 µL) were collected from each patient (N = 34 subjects) and subsequently analyzed for the measurement of total brain KB at three pre-defined time-points: (1) fasting (0 Kcal), (2) intermediate nutrition (≈7.5 Kcal/kg), and (3) stable nutrition (≈15 Kcal/kg). In a subset of the total patient cohort (Study 2, n = 10 subjects, who underwent enteral nutrition with Peptamen AF® as part of a prospective study) plasma and brain concentrations of total KB (AcAc and BHB) and free MCFA (C8 and C10) were measured simultaneously at each pre-defined time-point.
Figure 1.

Study design. Pooled 4-hourly brain interstitial tissue samples (≈ 40 µL) were collected using cerebral microdialysis (CMD) from a total of 34 subjects and analyzed for the measurement of total ketone bodies (KB, including acetoacetate [AcAc] and β-hydroxybutyrate [BHB]) at three separate time-points following traumatic brain injury (TBI): (1) fasting (0 Kcal; median time 37 [interquartile range 24–47] h from hospital admission), (2) intermediate nutrition (≈7.5 Kcal/kg; median 55 [46–64] h), (3) stable nutrition (≈15 Kcal/kg; median 85 [73–118] h). In a subset of patients (n = 10), blood (plasma) samples were collected simultaneously to CMD for the measurement of blood total KB concentrations and medium chain fatty acids (MCFA; C8 and C10).
Patient CMD samples from the total TBI cohort were also collected during the acute phase (time 0 to immediately before nutrition start; see Figure 1) to explore the correlations of total brain KB with blood arterial and brain glucose, and with brain interstitial tissue markers of increased glycolysis (CMD lactate and pyruvate) and excitatory amino acid release (CMD glutamate).
Statistical analysis
Data are presented as median [10th–90th percentiles], except when otherwise stated. Normality of the distributions was evaluated with the Shapiro–Wilk test. Univariate comparisons were performed using ANOVA for repeated measures and Wilcoxon-ranked paired test. Linear correlations were assessed with the non-parametric Spearman’s rho coefficient test. Statistical analyses were conducted using JMP 13 software® (JMP, Cary, NC, USA).
Results
Patient characteristics and outcome
A total of pooled 1063 CMD samples from 34 TBI patients were studied for KB analysis. Patient baseline characteristics and six-month outcome are summarized in Table 1. GCS on site was median 6 [3–14]. Two patients had type 2 diabetes. Maximum body temperature during the study period was 37.9 ± 0.6℃; six patients had refractory fever (>38.3℃). During the whole study period, median concentration of total brain KB was 17.0 µmol/L [6.1–62.6 µmol/L]; median concentrations of other brain interstitial tissue metabolites (including lactate, pyruvate, glucose, glutamate) are also shown in Table 2.
Table 1.
Patient baseline demographics and outcome.
| Variables | Value |
|---|---|
| Patient number | 34 |
| Age, years | 51 [24–76] |
| Female gender, number (%) | 8 (24) |
| Glasgow coma scale on site, n | 6 [3–14] |
| Marshall admission CT score, n | 3 [2–5] |
| 1 | 1 |
| 2 | 14 |
| 3 | 7 |
| 4 | 4 |
| 5 | 5 |
| 6 | 3 |
| Glasgow outcome score (GOS)a, n | |
| 1 | 6 |
| 2 | 4 |
| 3 | 5 |
| 4 | 10 |
| 5 | 5 |
| 6-month favorable outcome (GOS 4 and 5), n (%) | 15 (50) |
| 6-month mortality, n (%) | 6 (18) |
Note: Data are presented as median (10th–90th percentiles) or as number (percentage).
Four patients were lost to six-months follow-up.
Table 2.
Overall median values of total brain interstitial tissue ketone bodies (including acetoacetate and β-hydroxybutyrate) and other brain metabolic variables during the whole study period, measured with cerebral microdiaylsis.
| Variables | Value |
|---|---|
| Total brain ketone bodies, µmol/L | 17.0 (6.1–62.6) |
| Brain glutamate, µmol/L | 3.0 (0.9–24.2) |
| Brain glucose, mmol/L | 1.1 (0.5–2.7) |
| Brain pyruvate, µmol/L | 104.0 (65.5–166.8) |
| Brain lactate, mmol/L | 2.9 (1.8–5.4) |
| Brain lactate/pyruvate ratio | 29 (20–46) |
Note: Median values obtained from 1063 cerebral microdialysis samples among 34 patients with traumatic brain injury. Data are expressed as median (10th–90th percentiles).
Modulation of cerebral ketone metabolism by nutritional state
Nutrition started a median of 31 (interquartile range 24–47) h following hospital admission (Figure 1). As shown in Figure 2, feeding was associated with a significant decrease in total brain interstitial tissue KBs, from fasting 34.7 [10.7–189] µmol/L vs. 17.3 [7.1–96] µmol/L (intermediate nutrition; p = 0.01) vs. 13.1 [6.5–64.3] µmol/L (stable nutrition; p = 0.001). A median 2.8 fold decrease of total KB was observed from fasting to stable nutrition. Delayed time from admission to nutrition start did not correlate with total KB level measured at fasting time (p = 0.83).
Figure 2.
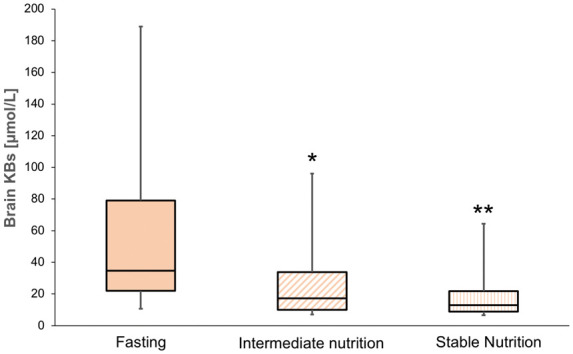
Changes of total brain ketone bodies during nutrition. Brain interstitial tissue concentrations of total ketone bodies (KB, including acetoacetate [AcAc] and (β-hydroxybutyrate [BHB]) were measured with cerebral microdialysis (CMD) in patients with TBI (n = 34) at baseline fasting vs. intermediate nutrition ((7.5 Kcal/kg) and stable nutrition (15 Kcal/kg)). Box-plots illustrate median (midline), 25–75th percentiles (lower and upper part of the boxes), 10–90th percentiles (lower and upper part of the whiskers). **p = 0.001; *p = 0.01 for comparison of fasting state vs. nutrition states.
Brain KB decrease from fasting vs. stable nutrition did not differ significantly in patients receiving standard nutrition vs. those who received MCT-enriched nutrition (p = 0.69).
Lactate/pyruvate ratio (27 [17–44] vs. 27 [16–48] vs. 28 [18–45]), brain tissue PO2 (29 [19–39] vs. 29.5 [19–36] vs. 29 [21–35] mm Hg) and ICP (10 [3–20] vs. 11 [5–17] vs. 12 [3–18] mm Hg) did not differ significantly at each time-point tested (all p > 0.2).
The impact of endogenous circulating ketosis
In Study 2, a concomitant decrease of plasma KB was observed, from fasting 668 [168.4–3824.9] µmol/L vs. 459.3 [195.6–1098.8] µmol/L (p = 0.08) vs. 129.4 [82.6–1033.8] µmol/L (p = 0.01) (Figure 3(a)), representing a median 4.7-fold decrease of blood KB from fasting to stable nutrition state. Median individual plasma/brain KB ratio was 11.
Figure 3.

Changes of total blood ketone bodies and free MCFA during nutrition. Concentrations of total plasma ketone bodies (KB, including acetoacetate [AcAc] and (β-hydroxybutyrate [BHB]); panel A) were measured in patients with TBI (n = 10; who received MCT enriched continuous enteral feeding) at baseline fasting vs. intermediate nutrition ((7.5 Kcal/kg) and stable nutrition (15 Kcal/kg)). Panel B illustrating the positive linear correlation between brain and blood KB (N = 30 samples from 10 TBI patients); Spearman’s rho linear correlation coefficient. Changes in the concentrations of plasma-free medium chain fatty acids (MCFA; C8 and C10) from fasting to nutrition are shown in panel C. Box-plots illustrate median (midline), 25–75th percentiles (lower and upper part of the boxes), 10–90th percentiles (lower and upper part of the whiskers). **p = 0.001; *p = 0.01; #p = 0.08 for comparison of fasting state vs. nutrition states.
A positive linear correlation was found between blood and brain KB (r = 0.56, p < 0.0013) (Figure 3(b)).
The effect of MCT-enriched enteral nutrition on circulating free MCFA
MCT-enriched continuous enteral feeding was associated with a significant increase of plasma-free C8 (from fasting 1.25 [0.81–1.8] µmol/L vs. intermediate nutrition 18.2 [6.1–51.4] µmol/L vs. stable nutrition 16.3 [9.1–51.5] µmol/L) and plasma-free C10 (7.9 [2.5–11.8] µmol/L vs. 18.7 [7.2–34.6] µmol/L vs. 15.2 [10.4–41] µmol/L; all p < 0.001) (Figure 3(c)). Brain C8 and C10 ranged between 1 and 2 µmol/L and also increased significantly (both p < 0.05) during nutrition.
Correlation of age, energy metabolism and blood glucose with brain KB at the acute phase of TBI
CMD samples (n = 196; 34 patients) collected at the acute phase of TBI, between hospital admission and nutrition initiation, were further examined for correlations of patient age, blood glucose and brain energy metabolites with brain KB. As shown in Table 3, acute brain interstitial tissue KB levels were positively correlated with increased age (r = 0.48, p = 0.004), with a non-significant trend found between age and time to feeding initiation (p = 0.06). Lower blood arterial glucose correlated with higher brain KB (r = –0.39, p < 0.0001), while no correlation was found between brain KB and brain glucose (r = 0.11, p = 0.16). Acute phase brain KB levels were positively correlated with brain glutamate, lactate and pyruvate.
Table 3.
Correlations of brain ketone bodies with age, arterial blood, brain glucose, glutamate, lactate and pyruvate at the acute fasted phase of TBI.
| Variable | Spearman’s ρ | p | |
|---|---|---|---|
| Total brain ketone bodies vs: | Age | 0.48 | 0.004 |
| Blood glucose | −0.39 | <0.0001 | |
| Brain glucose | 0.11 | 0.16 | |
| Brain glutamate | 0.29 | <0.0001 | |
| Brain lactate | 0.14 | 0.05 | |
| Brain pyruvate | 0.23 | 0.002 |
Note: Brain interstitial tissue (measured with cerebral microdialysis) and arterial blood data were collected during the acute TBI phase (time from hospital admission to immediately before nutrition start; see also Figure 1).
TBI: traumatic brain injury.
Discussion
To the best of our knowledge, this is the first study exploring cerebral ketone metabolism following TBI in humans. We first show that brain KB levels can be detected using CMD in TBI patients, with concentrations that appear comparable to brain KB levels reported by previous studies using other methods and brain conditions.31–35 Feeding – from fasting to stable nutrition state – was associated with a significant progressive decrease in brain and blood KB. Blood KB levels were also driven by changes in nutritional state (from fasting to feeding), and correlated with brain KB levels. Provision of high level of MCT by continuous feeding did not increase blood KB, and yielded a statistically significant modest increase in blood-free MCFA. Furthermore, levels of brain interstitial tissue KB at the acute fasted phase of TBI were associated with increased age, suggesting that nutrition status might not be the only determinant contributing to brain KB. Positive correlations of brain KB with glutamate, lactate, pyruvate – but not brain glucose – support the hypothesis that increased cerebral metabolic distress might trigger KB release, which may then potentially act as alternative substrates to glucose at the early phase of TBI.
Ketone metabolism and the injured brain
Fasting and nutritional modulation (ketogenic diet) increase KB availability.36 In this setting, KB act as alternative energy substrates to support the brain, independently from glycolysis, and allow glucose and protein sparing.37 Acute brain injury is associated with impaired glycolytic metabolism and reduced cerebral glucose availability,11 therefore the utilization of alternative substrates such as KB might impart a significant advantage.25 In addition to their role as brain energy fuel, accumulating evidences demonstrate that KB have several neuroprotective effects, including anti-seizure activity, improvement of cognitive function and motor performance, protection against oxidative stress and decrease in traumatic and ischemic cerebral damage.14,38
Cerebral ketone levels in humans with TBI
There are very limited data about brain KB levels in humans, and no study examined KB levels with the use of CMD. Overall, our data indicate that most of the CMD KB concentrations were within a range of 6 to 60 µmol/L. One study in mice fed with ketogenic diet34 reported CMD KB levels ranging from 40 to 50 µmol/L. Owen et al.31 reported KB levels in the CSF of about 40 µmol/L in healthy volunteers,31 while more recently White et al.35 – in a mixed population of brain-injured patients (n = 6) – found CSF KB concentrations in the average of 90 µmol/L. Cerebral KB levels were previously measured in healthy human subjects using high field 1H magnetic resonance spectroscopy, showing levels ranging from 50 to 100 µmol/L.32,33,36 Considering the dialysate recovery is estimated to be ≈ 70% of actual brain concentrations at a standard 0.3 µL/min perfusate flow rate,28 CMD KB concentrations appear comparable to brain KB levels reported by previous studies.
Modulation of brain KB by nutritional ketosis
We found a close relationship between nutritional state and KB levels. Feeding was associated with a significant and progressive decrease of KB levels in the brain and the blood, compared to baseline fasting, irrespective of the composition of the enteral formula used (standard vs. MCT enriched).
In the subset of 10 patients in whom plasma samples were collected, we found strikingly elevated plasma KB levels, up to 2–3 mmol/L. Previous data demonstrated that fasting results in increased circulating free fatty acids due to increased insulin inhibition of the adipose tissue hormone sensitive lipase. Free fatty acids serve as a substrate for ketosis in the hepatocytes, where the ultimate products KB are secreted.37 Cerebral KB transfer is regulated by the monocarboxylic acid transporters 1 (MCT1).36 Our data in TBI subjects are in line with previous findings in healthy volunteers,32 demonstrating a very close link between brain and blood KB and indicate that circulating plasma ketones are the main source of cerebral KB supplementation following TBI.
Additional factors that might trigger brain KB at the acute phase of TBI
First, we found a significant inverse correlation between acute fasted brain KB and blood glucose – but not brain glucose – supporting the fact that limited glucose availability is an important systemic driver of ketone production at the acute phase of TBI.39 We also identified a significant positive correlation between acute fasted cerebral KB levels and patient age. Our data are in line with previous observations in healthy subjects, showing increased cerebral ketone metabolism secondary to reduced glucose metabolism with advancing age,40 whereby increased brain KB compensate for glucose shortage by the aging brain.4,41
Furthermore, the positive correlations of brain KB increase with elevated CMD lactate, pyruvate and glutamate (but not CMD glucose) suggest that increased cerebral metabolic distress might trigger KB release, which may then potentially function as alternative energy substrates to glucose at the acute phase of TBI. Global cerebral ketone metabolic rate was not measured, therefore we cannot speculate about potential KB utilization. However, our findings are hypothesis generating and support the notion that cerebral KB may act as energy substrates to sustain the injured brain in conditions of limited carbohydrate availability from the circulation and/or when cerebral metabolic demand is increased due to injury.
Potential clinical implications of this study
Based on a number of large multicenter studies, the practice of early feeding in acutely ill patients has been challenged and replaced by a new paradigm of permissive underfeeding,42 where caloric restriction may be tolerated.43 Our findings favor this view that KB may support the injured brain with additional energy fuel suggesting that hypocaloric feeding may not be detrimental after TBI, because of increased KB compensation. Whether energy metabolic switch from glucose to KB by fasting periods (i.e. by using intermittent rather than continuous feeding) may be beneficial after TBI needs further study.
Apart from the amount and timing of nutrition, optimal feeding formulation in patients with acute brain injury patients also remains unclear. In the subset of patients who received enteral nutrition with high level MCT enteral formula, we found a modest – but statistically significant – increase in plasma C8 and C10, although this level of MCFA did not result in an increase of plasma KB level or a greater brain KB level at stable nutrition state. When brain KB levels were compared to those observed while using a standard formula with three-fold lower MCT level, no difference was observed either. This indicates that administration of MCT by continuous enteral feeding is ineffective at raising KB to therapeutically relevant levels.
Therapeutic KB supplementation in patients with TBI
An alternative to MCT ketogenic diets and more effective way to increase blood KB is via the direct enteral administration of drinks, containing exogenous dietary ketones, such as ketone esters and ketone salts, that can rapidly achieve blood KB concentrations of up to 3 mmol/L.44 Higher blood KB levels (4–5 mmol/L) may also be reached exogenously via intravenous infusion of sodium BHB solutions.7,21,45 Using such approaches, supplemental KB therapy has proven efficacious in reducing brain injury in animal models of TBI and ischemia14 and thus appears a promising avenue for future clinical investigation.45
Study limitations
The main limitation of this study was the single-center design and the fact that nutritional time points (7.5 and 15 Kcal/kg) were defined somewhat arbitrarily. These definitions, however, were convenient for the specific purpose of this study and provided novel insights on the modulation of cerebral ketone metabolism at the acute phase of TBI, which also represents the time window of opportunity for potential neuroprotective therapies. Another advantage of restricting our analysis to the early phase, where patients did not receive intravenous insulin yet, was to avoid a potential interaction between KB and insulin infusion.
Plasma samples were available only in a subset of 10 subjects who participated in a prospective study, but on the other hand were sufficient to reveal very robust correlations between blood and brain KB and therefore to conclude that endogenous ketosis plays an important role in the modulation of cerebral ketone metabolism. The decrease of brain and blood KB over time with the use of substrates provided by nutrition was evident. However, because the natural course of KB was not examined in control unfed patients, a causal link is not entirely proven by our data. CMD glucose or KB levels do not provide a dynamic value of cerebral metabolic rate as other measurements such as positron emission tomography do,46 and might provide only a partial information on brain energy metabolism.47 Finally, due to the sample size, this study did not explore possible relationships between ketone metabolism and different nutritional products with patient prognosis, which will require further investigation.
Conclusion
This was the first study in humans with TBI exploring cerebral ketone metabolism at the acute injury phase, and showing that endogenous ketosis upon fasting and low blood glucose is the main determinant of KB transfer from the circulation to the injured brain. Acute fasted brain KB levels were positively correlated with patient age suggesting that other factors beyond nutritional ketosis are implicated in the regulation of cerebral KB metabolism. The concomitant increase in brain KB, lactate, pyruvate and glutamate, but not brain glucose, suggests that higher cerebral metabolic demand might contribute to KB release. In concert, these novel findings support the view of an adaptive response, whereby ketones may be mobilized by systemic and local cerebral factors to potentially act as supplemental energy fuel to sustain the injured human brain. Our findings also establish a physiologic rationale and provide important data for future clinical interventional studies aiming to test the effect of exogenous KB therapy in patients with TBI and other acute cerebral conditions.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by research grants from the Swiss National Science Foundation (grants nr 32003B_155957 and 31NE30_173675) and Nestlé Health Science Research, Switzerland.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
AB was involved in study conduct, data analysis, statistical analysis, data interpretation and writing of the report; DS, JPM, PM, LC were involved in data acquisition and coordination, data analysis, and revising the report; SAM was involved in study conduct and coordination, and revising the report; MM, NC, MB, FH, PE were involved in data analysis, data interpretation and revising the report; MH was involved in statistical analysis, data interpretation and revising the report; BC was involved in the study design and concept, study conduct, data coordination and analysis, data interpretation and revising the report critically; MO was the lead investigator and conceived the study design and concept, was involved in study conduct, data coordination, data analysis and interpretation, and writing the report. All authors approved the final version of this manuscript.
References
- 1.Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci 2018; 19: 235–249. [DOI] [PubMed] [Google Scholar]
- 2.Mattson MP, Moehl K, Ghena N, et al. Intermittent metabolic switching, neuroplasticity and brain health. Nat Rev Neurosci 2018; 19: 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox PJ, Kirk T, Ashmore T, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab 2016; 24: 256–268. [DOI] [PubMed] [Google Scholar]
- 4.Courchesne-Loyer A, Croteau E, Castellano CA, et al. Inverse relationship between brain glucose and ketone metabolism in adults during short-term moderate dietary ketosis: a dual tracer quantitative positron emission tomography study. J Cereb Blood Flow Metab 2017; 37: 2485–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang L, Mason GF, Rothman DL, et al. Cortical substrate oxidation during hyperketonemia in the fasted anesthetized rat in vivo. J Cereb Blood Flow Metab 2011; 31: 2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LaManna JC, Salem N, Puchowicz M, et al. Ketones suppress brain glucose consumption. Adv Exp Med Biol 2009; 645: 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Svart M, Gormsen LC, Hansen J, et al. Regional cerebral effects of ketone body infusion with 3-hydroxybutyrate in humans: reduced glucose uptake, unchanged oxygen consumption and increased blood flow by positron emission tomography. A randomized, controlled trial. PLoS One 2018; 13: e0190556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Valdebenito R, Ruminot I, Garrido-Gerter P, et al. Targeting of astrocytic glucose metabolism by beta-hydroxybutyrate. J Cereb Blood Flow Metab 2016; 36: 1813–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Kuang Y, Xu K, et al. Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab 2013; 33: 1307–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glenn TC, Kelly DF, Boscardin WJ, et al. Energy dysfunction as a predictor of outcome after moderate or severe head injury: indices of oxygen, glucose, and lactate metabolism. J Cereb Blood Flow Metab 2003; 23: 1239–1250. [DOI] [PubMed] [Google Scholar]
- 11.Jalloh I, Carpenter KL, Grice P, et al. Glycolysis and the pentose phosphate pathway after human traumatic brain injury: microdialysis studies using 1,2-(13)C2 glucose. J Cereb Blood Flow Metab 2015; 35: 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maalouf M, Sullivan PG, Davis L, et al. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007; 145: 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prins ML. Cerebral ketone metabolism during development and injury. Epilepsy Res 2012; 100: 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prins ML, Matsumoto JH. The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J Lipid Res 2014; 55: 2450–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh S, Castillo E, Frias ES, et al. Bioenergetic regulation of microglia. Glia 2018; 66: 1200–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu K, Ye L, Sharma K, et al. Diet-induced ketosis protects against focal cerebral ischemia in mouse. Adv Exp Med Biol 2017; 977: 205–213. [DOI] [PubMed] [Google Scholar]
- 17.Brownlow ML, Jung SH, Moore RJ, et al. Nutritional ketosis affects metabolism and behavior in Sprague-Dawley rats in both control and chronic stress environments. Front Mol Neurosci 2017; 10: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis LM, Pauly JR, Readnower RD, et al. Fasting is neuroprotective following traumatic brain injury. J Neurosci Res 2008; 86: 1812–1822. [DOI] [PubMed] [Google Scholar]
- 19.Augustin K, Khabbush A, Williams S, et al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol 2018; 17: 84–93. [DOI] [PubMed] [Google Scholar]
- 20.Chang P, Augustin K, Boddum K, et al. Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 2016; 139: 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.White H, Venkatesh B. Clinical review: ketones and brain injury. Crit Care 2011; 15: 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neal EG, Chaffe H, Schwartz RH, et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol 2008; 7: 500–506. [DOI] [PubMed] [Google Scholar]
- 23.Prins M. Diet, ketones, and neurotrauma. Epilepsia 2008; 49(Suppl 8): 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appelberg KS, Hovda DA, Prins ML. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J Neurotrauma 2009; 26: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prins ML. Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab 2008; 28: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prins ML, Hovda DA. The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J Neurotrauma 2009; 26: 1083–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hutchinson PJ, Jalloh I, Helmy A, et al. Consensus statement from the 2014 International Microdialysis Forum. Intensive Care Med 2015; 41: 1517–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oddo M, Hutchinson PJ. Understanding and monitoring brain injury: the role of cerebral microdialysis. Intensive Care Med. Epub ahead of print 23 December 2017. DOI: 10.1007/s00134-017-5031-6. [DOI] [PubMed] [Google Scholar]
- 29.Patet C, Quintard H, Suys T, et al. Neuroenergetic response to prolonged cerebral glucose depletion after severe brain injury and the role of lactate. J Neurotrauma 2015; 32: 1560–1566. [DOI] [PubMed] [Google Scholar]
- 30.Sala N, Suys T, Zerlauth JB, et al. Cerebral extracellular lactate increase is predominantly nonischemic in patients with severe traumatic brain injury. J Cereb Blood Flow Metab 2013; 33: 1815–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owen OE, Reichard GA, Jr, Boden G, et al. Comparative measurements of glucose, beta-hydroxybutyrate, acetoacetate, and insulin in blood and cerebrospinal fluid during starvation. Metabolism 1974; 23: 7–14. [DOI] [PubMed] [Google Scholar]
- 32.Pan JW, Rothman TL, Behar KL, et al. Human brain beta-hydroxybutyrate and lactate increase in fasting-induced ketosis. J Cereb Blood Flow Metab 2000; 20: 1502–1507. [DOI] [PubMed] [Google Scholar]
- 33.Pan JW, Telang FW, Lee JH, et al. Measurement of beta-hydroxybutyrate in acute hyperketonemia in human brain. J Neurochem 2001; 79: 539–544. [DOI] [PubMed] [Google Scholar]
- 34.Samala R, Klein J, Borges K. The ketogenic diet changes metabolite levels in hippocampal extracellular fluid. Neurochem Int 2011; 58: 5–8. [DOI] [PubMed] [Google Scholar]
- 35.White H, Venkatesh B, Jones M, et al. Serial changes in plasma ketone concentrations in patients with acute brain injury. Neurol Res 2017; 39: 1–6. [DOI] [PubMed] [Google Scholar]
- 36.Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis 2005; 28: 109–121. [DOI] [PubMed] [Google Scholar]
- 37.Bouteldja N, Andersen LT, Moller N, et al. Using positron emission tomography to study human ketone body metabolism: a review. Metabolism 2014; 63: 1375–1384. [DOI] [PubMed] [Google Scholar]
- 38.Puchalska P, Crawford PA. Multi-dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab 2017; 25: 262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolahan SM, Prins ML, McArthur DL, et al. Influence of glycemic control on endogenous circulating ketone concentrations in adults following traumatic brain injury. Neurocrit Care 2017; 26: 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.London ED, Margolin RA, Duara R, et al. Effects of fasting on ketone body concentrations in healthy men of different ages. J Gerontol 1986; 41: 599–604. [DOI] [PubMed] [Google Scholar]
- 41.Croteau E, Castellano CA, Fortier M, et al. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer's disease. Exp Gerontol 2018; 107: 18–26. [DOI] [PubMed] [Google Scholar]
- 42.Casaer MP, Ziegler TR. Nutritional support in critical illness and recovery. Lancet Diabetes Endocrinol 2015; 3: 734–745. [DOI] [PubMed] [Google Scholar]
- 43.Al-Dorzi HM, Albarrak A, Ferwana M, et al. Lower versus higher dose of enteral caloric intake in adult critically ill patients: a systematic review and meta-analysis. Crit Care 2016; 20: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stubbs BJ, Cox PJ, Evans RD, et al. On the metabolism of exogenous ketones in humans. Front Physiol 2017; 8: 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White H, Venkatesh B, Jones M, et al. Effect of a hypertonic balanced ketone solution on plasma, CSF and brain beta-hydroxybutyrate levels and acid-base status. Intensive Care Med 2013; 39: 727–733. [DOI] [PubMed] [Google Scholar]
- 46.Cunnane SC, Courchesne-Loyer A, Vandenberghe C, et al. Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of Alzheimer's disease. Front Mol Neurosci 2016; 9: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dienel GA, Rothman DL, Nordstrom CH. Microdialysate concentration changes do not provide sufficient information to evaluate metabolic effects of lactate supplementation in brain-injured patients. J Cereb Blood Flow Metab 2016; 36: 1844–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]