

Abstract
Our genomic DNA is under constant assault from endogenous and exogenous sources, which needs to be resolved to maintain cellular homeostasis. The eukaryotic DNA repair enzyme Tyrosyl-DNA phosphodiesterase I (Tdp1) catalyzes the hydrolysis of phosphodiester bonds that covalently link adducts to DNA-ends. Tdp1 utilizes two catalytic histidines to resolve a growing list of DNA-adducts. These DNA-adducts can be divided into two groups: small adducts, including oxidized nucleotides, RNA, and non-canonical nucleoside analogs, and large adducts, such as (drug-stabilized) topoisomerase- DNA covalent complexes or failed Schiff base reactions as occur between PARP1 and DNA. Many Tdp1 substrates are generated by chemotherapeutics linking Tdp1 to cancer drug resistance, making a compelling argument to develop small molecules that target Tdp1 as potential novel therapeutic agents. Tdp1’s unique catalytic cycle, which is centered on the formation of Tdp1-DNA covalent reaction intermediate, allows for two principally different targeting strategies: (1) catalytic inhibition of Tdp1 catalysis to prevent Tdp1-mediated repair of DNA-adducts that enhances the effectivity of chemotherapeutics; and (2) poisoning of Tdp1 by stabilization of the Tdp1- DNA covalent reaction intermediate, which would increase the half-life of a potentially toxic DNA-adduct by preventing its resolution, analogous to topoisomerase targeted poisons such as topotecan or etoposide. The catalytic Tdp1 mutant that forms the molecular basis of the autosomal recessive neurodegenerative disease spinocerebellar ataxia with axonal neuropathy best illustrates this concept; however, no small molecules have been reported for this strategy. Herein, we concisely discuss the development of Tdp1 catalytic inhibitors and their results.
Keywords: Tdp1, small molecules, DNA topoisomerases, Camptothecins, oxidative DNA damage, DNA adducts, Etoposide, chain terminating nucleotides/nucleoside analogs, DNA metabolism, drug development
Introduction
Tyrosyl-DNA phosphodiesterase I (Tdp1) is a eukaryotic DNA repair enzyme which is a member of the phospholipase D superfamily and hydrolyzes the phosphodiester bond that links an adduct to the end of a nicked DNA strand[1-3]. Tdp1 was discovered as an enzyme activity able to hydrolyze a 3’ phospho-tyrosyl linkage, which is the chemical bond between the active site tyrosine of Tyrosine-recombinases and eukaryotic DNA Topoisomerase I (Topo1), and the 3’ phosphoryl-end of a DNA strand[3]. Over the last two decades, a broad spectrum of phosphodiester linked 3’ and 5’ DNA-adducts were identified as Tdp1 substrates[4]. Tdp1 natural substrates can be divided into two groups: small adducts consisting of damaged nucleotides, DNA inserted ribonucleotides, and non-canonical nucleotide/nucleoside analogs, and large adducts including covalent protein-DNA adducts that are generated as a transient reaction intermediate by, for example, DNA topoisomerases and Tdp1, or protein fragments (peptides) as a result of failed Schiff base linked proteins such as proteolytically processed poly (ADP-ribose) polymerase 1 (PARP1)-DNA adducts [Table 1]. These transient protein-DNA adducts can be stabilized by chemotherapeutics including camptothecins (CPT), epipodophyllotoxins (e.g., etoposide), and local DNA perturbations introduced by, for example, irradiation and endogenously generated reactive oxygen species [Table 1]. Moreover, Tdp1 is localized in both the nuclear and mitochondrial compartments to catalyze the hydrolysis of phosphodiester linked DNA adducts[4-7]. This reveals the general role of Tdp1 in maintaining nuclear and mitochondrial genome stability and chemotherapeutic-resistance.
Table 1.
Therapeutic and endogenous agents generating Tdp1 substrates
| Agent | Substrate(s) | Ref. |
|---|---|---|
| Reactive oxygen species | 3’ abasic site | [8-14] |
| Chain terminating nucleoside analogs1 | 3’ nucleoside adducts | [10] |
| Irradiation/Bleomycin | 3’ phospho-glycolate, 3’ abasic sites | [15-19] |
| MMS, TMZ2 | Methylated bases | [9,20] |
| PARP1 inhibitors3 | PARP1-DNA Schiff base | [21-23] |
| Camptothecins4 | 3’ phospho-tyrosine | [2,3] |
| Etoposide, doxorubicin5 | 5’ phospho-tyrosine | [20,24,25] |
| Tdp1 mutants | 3’ phospho-histidine | [1,16,26-29] |
1Arcyclovir (ACV), cytarabine (Ara-C), zidovudine (AZT), zalcitabine (ddC), and sapacitabine; 2methyl methanesulfonate (MMS) and temozolomide (TMZ); 3BMN673, Olaparib, and Rucaparip; 4U.S. Food and Drug Administration (FDA) approved analogs: topotecan and irinotecan; 5epipodophyllotoxins such as etoposide and anthracyclins, with Doxorubicin as an example, are Topo2-DNA stabilizing agents with different mechanisms of action[30]. Tdp1: Tyrosyl-DNA phosphodiesterase I
Tdp1 utilizes two highly conserved histidine-lysine-aspartate (HxKx4D; x being any amino acid) motifs in two coordinated SN2 nucleophilic attacks to hydrolyze the phosphodiester linkage. First, His263 nucleophilically attacks the phosphodiester bond to release the adduct/protein by forming a transient Tdp1-DNA adduct, which is broken by the general acid/base His493 mediated hydrolysis via activation of water releasing Tdp1 from the DNA-end [Figure 1][2,3,11,16,26,28,31,32]. The intriguing fact is that cells risk the formation of second potentially toxic enzyme-DNA reaction intermediate to resolve the primary toxic insult. This potential danger to the cell is highlighted by the catalytic human Tdp1 (H493R) mutant, which is the molecular basis for the rare autosomal recessive neurodegenerative disease spinocerebellar ataxia with axonal neuropathy (SCAN1)[29,33]. Tdp1 is expressed in all human tissues at a low level, probably due to its potential danger. However, elevated levels of Tdp1 are detected in a heterologous distribution in virtually all tumors[7,34]. Elevated Tdp1 levels stimulate chromosome instability[35] and increase cell sensitivity to DNA damaging agents[24,26,35-37]. Thought-provoking is that deletion of Tdp1 in yeast, DT40 chicken cells, HEK293 cells, and mice also results in enhanced cell sensitivity to DNA damaging agents[2,5,16,20,27,31,38-40]. Overall, this supports the two different therapeutic strategies hypothesized: (1) no Tdp1 activity or catalytic inhibition of Tdp1 that prevents repair of DNA-adducts leads to cytotoxicity; and (2) accumulation of Tdp1-DNA adducts via poisoning/stabilization of the protein-DNA complex, or increase of Tdp1 levels, results in cytotoxicity [Figure 1]. This review focuses on the current development of Tdp1 as a therapeutic target to improve treatment response with FDA approved chemotherapeutics - a topoisomerase targeting drug[30].
Figure 1.
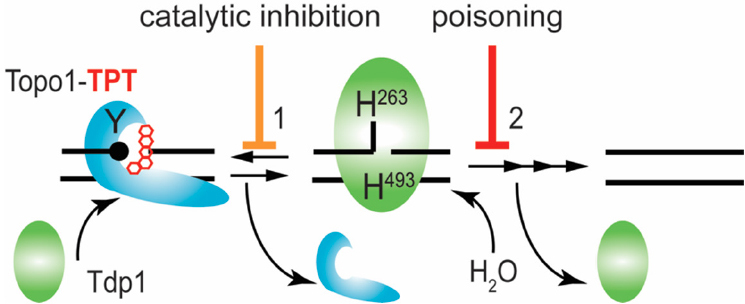
Tdp1 catalytic cycle. Tdp1 utilizes two catalytic histidines to hydrolyze the phosphodiester bond that link adducts to the DNA. Here, we show the example of removal of a Topo1-DNA adduct-3’ phospho-tyrosyl linkage - stabilized by Topotecan. Tdp1 interacts with the Topo1-DNA adduct to initiate Step 1: the nucleophilic attack by His263 - that is, the nucleophilic histidine (in yeast Tdp1 His182) - on the 3’ phospho-tyrosyl linkage forms a 3’ phospho-hystidyl linkage or Tdp1-DNA adduct that releases the tyrosine and by extension Topo1. For Step 2, the general acid/base histidine His493 (His432 in yeast Tdp1) will activate a water molecule to hydrolyze the 3’ phospho-histidyl linkage dissociating Tdp1 from the DNA. However, a single strand nick is left behind by Tdp1 with 5’ hydroxyl and 3’ phosphoryl chemical groups that are processed (reversed) by polynucleotide kinase/phosphatase to facilitate DNA ligase III to regulate the DNA strands. Topo1: topoisomerase I; TPT: topotecan; Tdp1: tyrosyl-DNA phosphodiesterase I
Tdp1 as therapeutic target
The potential of Tdp1 as a therapeutic target for catalytic inhibitors was directly proposed by Nash and co-workers in its discovery paper[3]. Over the last five years, the search for Tdp1 inhibitors has been rapidly growing with the therapeutic focus to combine catalytic Tdp1 inhibiting agents with the current FDA-approved Topo1 targeting chemotherapeutics. Besides catalytic inhibition, we champion a second strategy to poison Tdp1 or to pharmacologically stabilize the enzyme-DNA adduct turning Tdp1 into a cellular toxin, similar to topoisomerase inhibitors[4,30]. The principle of this strategy is supported by the SCAN1 HisgabArg-mutant and other Tdp1 catalytic mutants tested[1,16,26,28,29,36,37,41]. Currently, no small molecules for this strategy have been reported and, herein, we focus only on catalytic Tdp1 inhibitors.
Catalytic inhibitors
To date, many catalytic Tdp1 inhibitors have been identified, yet only a few were tested in cell or cancer models. Interestingly, many of these Tdp1 catalytic inhibitors are based on natural products isolated from fungi, plants, nucleoside analogs, and bile acids showing inhibition in the low micromolar to the high nanomolar range. The first reported catalytic Tdp1 inhibitors were the non-specific transition metals vanadate (VO43-) and tungstate at millimolar concentrations and were used to resolve the crystal structure of the transition state complex of hTdp1 catalytic core domain with VO4-DNA-hTopo1 peptide fragments[42-44]. Vanadate and tungstate are known general inhibitors for protein phosphotyrosyl phosphatases[45]. Yves Pommier and collaborators advanced the search for catalytic inhibitors by exploring existing compounds and developing in vitro/alpha-screen based high-throughput screens (HTSs) using oligonucleotides with a 3’ phospho-linked fluorescent adduct to mimic the 3’ phospho-tyrosyl bond. They exploited antibiotic ribosomal inhibitors and identified as the first pharmacological active Tdp1 inhibitors Neomycin, an aminoglycoside analog, and non-aminoglycosides such as puromycin and thiostrepton with IC50 ranging 2-30 mmol/L[46]. They further identified rolitetracycline and potentially other tetracycline analogs to inhibit Tdp1 catalysis at micromolar levels[47]. Huang et al.[48] successfully translated this observation by repurposing minocycline in combination with irinotecan to treat high grade ovarian cancer cell lines and an orthotopic xenograft mouse model of human ovarian carcinomatosis. Intriguingly, minocycline decreased Tdp1 expression levels that enhanced irinotecan toxicity in platinum resistant cell lines, while in vivo this combination reduced micro-metastases to improve overall survival. Why minocycline specifically reduces Tdp1 protein levels and/or inhibits Tdp1 activity as suggested for another tetracycline analog - rolitetracycline - is currently unknown[47,48]. How tetracyclines inhibit eukaryotic protein synthesis is still questionable - the most popular explanation is ribosome inhibition[49], but the reduction of one protein (Tdp1) by minocycline remains an enigma.
The diamidine analogue furamidine [2,5-bis (4-amidinophenyl) furan] was the first inhibitor identified in a HTS at National Institutes of Health (NIH)[50]. Furamidine is used to treat Trypanosomiasis and leishmaniasis, which express Tdp1, but it is unclear if Tdp1 inhibition is part of the antiparasitic activity[51]. An interesting twist is that furamidine combined with irinotecan suppresses murine lupus nephritis[52]. Additional HTSs at NIH identified additional micromolar to high nanomolar Tdp1 inhibitors: progesterone derivative NSC88915 (3,20-dioxopregn-4-en-21-yl 4-bromobenzenesulfonate) and phospho-tyrosyl mimetics such as suramin and dephostatin derivative methyl-3,4-dephostatin (3,4-Dihydroxy-N-methyl-N-nitrosoaniline)[53,54]. The Pommier and Wang collaboration identified arylidene thioazolidinone derivatives as high nanomolar inhibitors of Tdp1 catalysis[55].
The identification of non-hydrolysable phospho-tyrosyl mimetics that perfectly docked in the catalytic pocket led to the first in silico-docking screen to classify 46 additional potential Tdp1 inhibitors that dock the catalytic cavities [Figure 2], as such competing with Tdp1 substrates[56]. However, these compounds were not verified for their potential biochemical and biological activity. Subsequent in silico-docking screens by other groups did verify their compounds; Gushchina et al.[57] found thioether sulfo-heterocyclic linked compounds that docket into the catalytic pocket and inhibited catalysis in the high micromolar range. Waugh’s group recently reported an impressive crystallographic verification of 11 phthalic acids and quinolone-based fragment ligands identified in their in silico-docking screen[58]. They demonstrated in silico docking within Tdp1 catalytic pocket, inhibition of Tdp1 catalysis in the mid micromolar to high millimolar range, and resolved 11 independent crystal structures of the Tdp1 catalytic domain in complex with these compounds to confirm their in silico-docking screen results, concluding they identified competitive Tdp1 inhibitors[58]. This approach provides a great basis for structure-based drug design to further develop these inhibitors and test them in cell-based/mice models.
Figure 2.
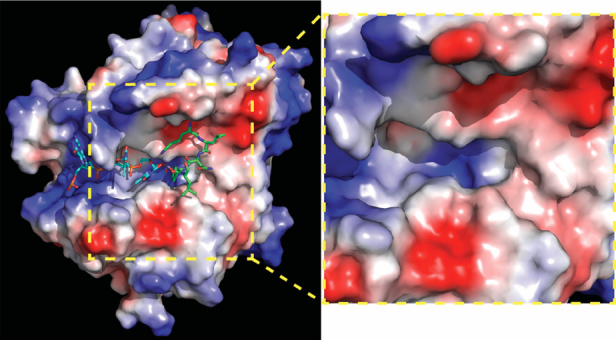
Human Tdp1 electrostatic surface distribution. Electrostatic surface potential of human Tdp1 is shown in a gradient from negative (red) through neutral (white) to positive (blue) and was degenerated by PyMol. Shown in cyan is the DNA and in green the Topo1-peptide fragment that is bonded to the DNA via phosphate. In the used structure, the phosphate was replaced by a vanadate to capture the Tdp1-Topo1-DNA complex (PDB file: 1NOP Davies 2003). A positively charged “DNA-gorge/cleft” fits single strand DNA with the adducted end located in the catalytic pocket from which a “funnel cone” shape pocket emerges that facilitates docking of the protein/peptide adduct. The yellow zoom box highlights the electrostatic charge distribution of the DNA-gorge, catalytic pocket, and funnel cone in which a potential inhibitor will need to bind to prevent Tdp1 interaction with and hydrolyzes of a DNA-adduct. The figure was generated using MacPyMol (DeLano Scientific, San Carlos CA). Topo1: topoisomerase I; Tdp1: tyrosyl-DNA phosphodiesterase I
However, except for furamidine and minocycline, none of these compounds were verified in cell-based or animal models of cancer. The first report using cell-based screens for Tdp1 inhibitors exploited Tdp1-deficient DT40 chicken cells complemented with and without human TDP1 to identify compounds that demonstrate a synergistic effect with CPT[22]. This approach, however, identified PARP1 inhibitors and not Tdp1 inhibitors. The authors identified five compounds after a primary (DT40tdp1-/- + hTDP1) and secondary (DT40tdp1-/- vs. DT40tdp1-/- + hTDP1) screen that did not show Tdp1 inhibition but inhibited PARP1-activity analyzed by ELISA and immunoblotting for PARylation. Moreover, this revealed how tricky cell-based screens and the tight cellular interplay of DNA repair-DNA damage response proteins are. This tight interplay can also foster alternative treatment strategies. For example, Pfeifer and collaborators reasoned that Tdp1 inhibitors will be synergistic with CPT but also with PARP1 inhibitors[34]. This is mechanistically supported by the reported observation that Tdp1 and PARP1 are epistatic for the repair of Topo1-DNA adducts[9,59]. They identified an alkylidene barbiturate derivative [CD00509; 5-(2-Furylmethylidene)-2-thioxohexahydropyrimidine-4,6-dione] in a biochemical-screen and verified this compound in TDP1 and tdp1-/- MEF-cells, showing that TDP1-MEFs with CD00509 showed a similar CPT sensitivity as tdp1-/- MEFs without CD00509. Moreover, CD00509 combined with CPT or Rucaparib in MCF7 breast cancer cells resulted in more toxicity for both combinations compared to the agent alone[34]. Thus, Tdp1 inhibition together with PARP1 inhibition is a successful treatment strategy taking advantage of additional impeding mutations that cancer cells maintain. Current knowledge adds an additional explanation: PARP1 inhibitors not only inhibit PARP1 catalysis but also stabilize the PARP1-DNA intermediate[60], the reaction intermediate of a Schiff base reaction, which itself induces cytotoxicity. Moreover, the phosphodiester bond linking the damaged nucleotide to the PARP1 peptide is a substrate for Tdp1 hydrolyses[23]. Pommier teamed up with the Malhorta group and broadened cell-based screening by testing 15 newly synthesized piperidinyl sulfamide derivatives in the NCI60 cell line panel[61]. The advantage of this NCI60 screen is that, in addition to screening for compound induced cytotoxicity, it may reveal potential cancer specificity/cell type and may reveal potential response pathways, since the NCI60 panel has been molecular characterized over the years. Moreover, the inclusion of R- and S-stereoisomers will reveal differences in biological activity, which was observed for the only compound that induced a significant cell toxicity and inhibited Tdp1 catalysis, namely piperidyl sulfamide-18 [NSC750706; (R)-Methyl 2-(N-(1-(4-fluorobenzyl)piperidin-4-yl)-N-(3-fluorophenyl) sulfamoylamino)-3-methyl butanoate], while its S-stereoisomer, NSC764209, induced no phenotype[61].
Another forerunner in the hunt for Tdp1 catalytic inhibitors is Olga Lavrik and her collaborators. These researchers synthesized their derivatives and evaluated the compounds in: (1) Tdp1 catalytic assay; (2) in silico docking; and (3) cell toxicity/growth inhibition studies. They identified a wide variety of chemical scaffolds that include the Benzopentathiepines derivative 2-(Dibutylamino)-N-(8-(trifluoromethyl)benzo[f]-[1,2,3,4,5]pentathiepin-6-yl)acetamide, which inhibits catalysis at high nanomolar concentrations and induces MCF7 cytotoxicity (IC50 ~ 28 μmol/L) via DNA fragmentation and apoptosis[62]. They combined a 7-hydroxycoumarin with monoterpenoid moieties resulting in 7-(((1S,5R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methoxy)-2,3-dihydrocyclopenta[c]chromen-4(1H)-one that exhibits a high nanomolar catalytic inhibition (IC50 = 675 ± 7 nmol/L), a MCF7 CC50 of 180 nmol/L (in combination with CPT) and increased MCF7 CPT sensitivity, but had no effect on RPMI-8226 human multiple melanoma cells that maintain lower Tdp1 levels than MCF7 cells[63]. They concluded that the induced toxicity is Tdp1-dependent; however, this would have been better supported by knockdown of Tdp1 levels, since many other different factors, including PARP1 activity, can contribute to a lack of effect/phenotype. Using the Structure-Activity Relationship of octahydro-2H-chromen-4-ol scaffold, the Lavrik group developed a series of 3(4S)- and 3(4R)-diastereomers derivative with different bulky side-groups that inhibit Tdp1 catalysis in the low micromolar range while in silico-docking showed that each of these six ligands binds Tdp1 in more than one location within the Tdp1 catalytic cavity [Figure 2][64]. Recently, Lavrik and colleagues reported 15 monoterpenoid and adamantane fragments, which are able to inhibit Tdp1 catalysis (0.86-4.08 μmol/L). Of these 15 fragments, 3,7-Dimethyloctyl adamantane-1-carboxylate in combination with topotecan induced synergistic toxicity in A549 human lung carcinoma cells[65].
In addition to arrays of synthesized compounds, Lavrik and co-workers also utilized natural product scaffolds in their search for potential Tdp1 catalytic inhibitors. They synthesized 29 aryliden- and hetarylidenfuranone derivatives of usnic acid (a metabolite found in various lichens) that inhibit Tdp1 in the low nanomolar range. These compounds also induced A549 cytotoxicity with IC50 between 5 and 20 μmol/L and potentiated topotecan toxicity[66]. Their subsequent synthesized hydrazinothiazole usnic acid derivative (R,E)-2-acetyl-6-(2-(2-(4-bromobenzyliden)hydrazinyl)thiazol-4-yl)-3,7,9-trihydroxy-8, 9b-dimethyldibenzo[b,d]furan-1(9bH)-one is an effective Tdp1 catalytic inhibitor that increased topotecan toxicity in a Lewis lung carcinoma cell model and was the first potential Tdp1 inhibitor to show, in combination with topotecan, an anti-tumor and anti-metastatic effect in a mouse model of Lewis Lung Carcinoma[67]. This compound is now entering the preclinical trial phase. This group also used semi-synthetic derivatives of bile acids and disaccharide nucleosides as a scaffold for the development of Tdp1 catalytic inhibitors[68,69]. The bile acid derivatives were tested by in silico-docking and in a catalytic assay showing inhibition in the 300 to 500 nmol/L ranges with N-(2’’-(3’,5’-Di-tret-buthyl-4’-hydroxyphenyl)-ethyl)-3a,12a-diacetoxy-5b-cholan-24-amide as the most promising compound[69]. Disaccharide nucleosides were explored as Tdp1 inhibitors following reports showing that pyrimidine disaccharide derivatives including nicotinamide adenine dinucleotide (NAD+)-mimetics catalytically inhibited PARP1[70] and that PARP1 synthesized free PAR-monomers and -polymers that inhibit, for example, XPC-RAD23B[71]. Disaccharide nucleoside analogs inhibited wild type Tdp1 catalysis (low micromolar to high nanomolar range) but interestingly not the Tdp1H493R-SCAN1-mutant[68]. Why these compounds do not show inhibition of the SCAN1-mutant enzyme is unknown and cannot be explained from the reported experimental results. However, some of these compounds potentiated topotecan induced toxicity in A549 cells and non-cancerous WI-38 (fibroblasts derived from lung tissue of a three months gestation female fetus) cells, suggesting to induce “normal” cell toxicity. These active derivatives can be divided into three classes: (1) (1’-2’)-glycosidic bond (2’-O-pentafuranosyl nucleosides); (2) b(1’-3’)-glycosidic bond (3’-O-b-D-ribofuranosyl nucleosides); and (3) b(1’-5’)-glycosidic bond (5’-O-β-D-ribofuranosyl nucleosides). They induce catalytic inhibition in the low micromolar to high nanomolar range, but need further development[68]. Quinn and co-workers exploited 3,4-dimethoxyphenol-1-β-D-(6’-O-galloyl) glucopyranoside and 3-(4-hydroxy-3-methoxyphenyl)propane-1,2-diol2-β-D-(6-O-galloyl) glucopyranoside from Macropteranthes leichhardtii, and achyrodimer F from the teleomorphic fungus family Cortinariaceae. Both compounds inhibit Tdp1 in the low micromolar range[72,73]. Takagi et al.[74] isolated JBIR21 from an unidentified anamorphic fungus RF-13305 culture that showed catalytic inhibition with IC50 of 18 μmol/L and induced growth inhibition of cervical carcinoma HeLa cells, malignant mesothelioma NCI-H2052 cells, colon adenocarcinoma HT-29 cells, and lymphoblastoid namalva cells with an IC50 range of 3.5-3 μmol/L. JBIR21 also showed an antitumor effect in a HT-29 xenograft model (treatment-to-control ratio of 0.51) without noticeable toxicity or other adverse effects, suggesting that JBIR21 forms a highly potential scaffold for further development of a clinically applicable compound. Figure 3 shows examples of structures of potential Tdp1 inhibitors discussed above.
Figure 3.
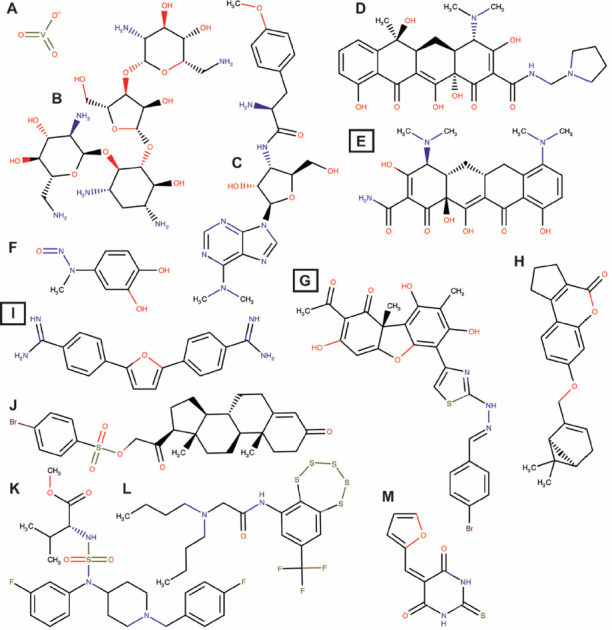
Structures of potential Tdp1 inhibitors. A: Vanadate; B: Neomycin; C: Puromycin; D: Rolitetracycline; E: Minocycline; F: methyl-3,4-dephostatin; G: (R,E)-2-acetyl-6-(2-(2-(4-bromobenzyliden)hydrazinyl)thiazole-4-yl)-3,7,9-trihydroxy-8,9b-dimethyl dibenzo[b,d]furan-1(9bH)-one; H: 7-(((1S,5R)-6,6-Dimethylbicyclo[3.1.1]hept-2-en-2-yl)methoxy)-2,3-dihydrocyclopenta[c]chromen-4(1H)-one; J: 3,20-Dioxopregn-4-en-21-yl 4-bromobenzenesulfonate; K: 2-(Dibutylamino)-N-(8-(trifluoromethyl)benzo[f]-[1,2,3,4,5] pentathiepin-6-yl)acetamide; L: (R)-Methyl 2-(N-(1-(4-fluorobenzyl)piperidin-4-yl)-N-(3-fluorophenyl) sulfamoyl amino)-3-methylbutanoate; M: 5-(2-Furyl Methylidene)-2-thioxo hexahydro pyrimidine-4,6-dione. Box letters are compounds tested in vivo. Chemical structures were drawn using MarvinSketch (17.3.13.0) ChemAxon (http://www.chemaxon.com)
Conclusion
Over the last two decades, the development of Tdp1 catalytic inhibitors has produced active compounds that showed a high potential to be tested in (pre-)clinical trials. Although these compounds were originally selected for their ability to inhibit Tdp1 catalysis and modeled-docking of the compounds into the Tdp1 catalytic pocket, the more current and promising compounds were tested in combination with DNA Topo1 inhibitors (topotecan or irinotecan) in cell and xenograft mouse models of cancer. However, Tdp1 specificity is still unclear and has not been addressed yet for all these compounds. Moreover, the potential development of Tdp1-poisons, compounds that selectively increase the lifetime of Tdp1-DNA adducts similar to Tdp1 catalytic mutants such as the SCAN1 mutant, would be a welcome addition for combinational treatment options for anti-cancer therapy. These molecules form a promising base for further development to join the fight against cancer. The development of catalytic Tdp1 inhibitors might also help patients with other diseases such as SCAN1, which appears to be a common founder mutation in the Arab population and even Lupus nephritis[29,33,52]. Hence, the “SCAN1” Tdp1H493R mutant enzyme performs the first step [Figure 1] with similar kinetics as the wild type Tdp1[26]. However, the rate of the second step is dramatically reduced, resulting in a prolonged life-time of the toxic enzyme-DNA covalent reaction intermediate. Thus, catalytic inhibition of this SCAN1 Tdp1 mutant would prevent the formation of the toxic Tdp1SCAN1-DNA intermediate and the subsequent induction of cerebellar atrophy, which could stabilize disease progression and symptoms.
Declarations
Acknowledgments
We want to thank the present and past van Waardenburg lab-members and our collaborators for contributing to increase our knowledge of Tdp1 and Topoisomerases and Ms. Lisa Park for critical reading and editing of this manuscript. Our apologies to colleagues whose work we did not mention, due to space limitations. RCAMvW greatly appreciates the current and past financial support from the UAB Department of Pharmacology and Toxicology, UAB O’Neil Comprehensive Cancer center, UAB ACS-IRG Junior Faculty Development, DOD-OCRP pilot award, UAB School of Medicine, the Norma Livingston Ovarian Cancer Foundation and the Alabama Drug Discovery Alliance.
Authors’ contributions
Collected data, wrote the review and edited the revision: Brettrager EJ, van Waardenburg RCAM
Availability of data and materials
Not applicable.
Financial support and sponsorship
RCAMvW was in part funded by American Cancer Society UAB ACS-IRG Junior Faculty Development Grant (ACS-IRG-60-001-53); Department of Defense OCRP pilot award W81XWH-15-1-0198; and the National Institutes of Health Cancer Center Core Support Grant (P30CA013148).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
References
- 1.Interthal H, Pouliot JJ, Champoux JJ. The tyrosyl-DNA phosphodiesterase Tdp1 is a member of the phospholipase D superfamily. Proc Natl Acad Sci U S A. 2001;98:12009–14. doi: 10.1073/pnas.211429198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pouliot JJ, Yao KC, Robertson CA, Nash HA. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science. 1999;286:552–5. doi: 10.1126/science.286.5439.552. [DOI] [PubMed] [Google Scholar]
- 3.Yang SW, Burgin AB Jr, Huizenga BN, Robertson CA, Yao KC, et al. A eukaryotic enzyme that can disjoin dead-end covalent complexes between DNA and type I topoisomerases. Proc Natl Acad Sci U S A. 1996;93:11534–9. doi: 10.1073/pnas.93.21.11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comeaux EQ, van Waardenburg RC. Tyrosyl-DNA phosphodiesterase I resolves both naturally and chemically induced DNA adducts and its potential as a therapeutic target. Drug Metab Rev. 2014;46:494–507. doi: 10.3109/03602532.2014.971957. [DOI] [PubMed] [Google Scholar]
- 5.Ben Hassine S, Arcangioli B. Tdp1 protects against oxidative DNA damage in non-dividing fission yeast. EMBO J. 2009;28:632–40. doi: 10.1038/emboj.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc Natl Acad Sci U S A. 2010;107:19790–5. doi: 10.1073/pnas.1009814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fam HK, Chowdhury MK, Walton C, Choi K, Boerkoel CF, et al. Expression profile and mitochondrial colocalization of Tdp1 in peripheral human tissues. J Mol Histol. 2013;44:481–94. doi: 10.1007/s10735-013-9496-5. [DOI] [PubMed] [Google Scholar]
- 8.Al Abo M, Sasanuma H, Liu X, Rajapakse VN, Huang SY, et al. TDP1 is critical for the repair of DNA breaks induced by sapacitabine, a nucleoside also targeting ATM- and BRCA-deficient tumors. Mol Cancer Ther. 2017;16:2543–51. doi: 10.1158/1535-7163.MCT-17-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alagoz M, Wells OS, El-Khamisy SF. TDP1 deficiency sensitizes human cells to base damage via distinct topoisomerase I and PARP mechanisms with potential applications for cancer therapy. Nucleic Acids Res. 2014;42:3089–103. doi: 10.1093/nar/gkt1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang SY, Murai J, Dalla Rosa I, Dexheimer TS, Naumova A, et al. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013;41:7793–803. doi: 10.1093/nar/gkt483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Interthal H, Chen HJ, Champoux JJ. Human Tdp1 cleaves a broad spectrum of substrates, including phosphoamide linkages. J Biol Chem. 2005;280:36518–28. doi: 10.1074/jbc.M508898200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lebedeva NA, Rechkunova NI, El-Khamisy SF, Lavrik OI. Tyrosyl-DNA phosphodiesterase 1 initiates repair of apurinic/apyrimidinic sites. Biochimie. 2012;94:1749–53. doi: 10.1016/j.biochi.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lebedeva NA, Rechkunova NI, Ishchenko AA, Saparbaev M, Lavrik OI. The mechanism of human tyrosyl-DNA phosphodiesterase 1 in the cleavage of AP site and its synthetic analogs. DNA Repair (Amst) 2013;12:1037–42. doi: 10.1016/j.dnarep.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Lebedeva NA, Rechkunova NI, Lavrik OI. AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. FEBS Lett. 2011;585:683–6. doi: 10.1016/j.febslet.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 15.El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007;6:1485–95. doi: 10.1016/j.dnarep.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 16.He X, van Waardenburg RC, Babaoglu K, Price AC, Nitiss KC, et al. Mutation of a conserved active site residue converts tyrosyl-DNA phosphodiesterase I into a DNA topoisomerase I-dependent poison. J Mol Biol. 2007;372:1070–81. doi: 10.1016/j.jmb.2007.07.055. [DOI] [PubMed] [Google Scholar]
- 17.Inamdar KV, Pouliot JJ, Zhou T, Lees-Miller SP, Rasouli-Nia A, et al. Conversion of phosphoglycolate to phosphate termini on 3’ overhangs of DNA double strand breaks by the human tyrosyl-DNA phosphodiesterase hTdp1. J Biol Chem. 2002;277:27162–8. doi: 10.1074/jbc.M204688200. [DOI] [PubMed] [Google Scholar]
- 18.Zhou T, Akopiants K, Mohapatra S, Lin PS, Valerie K, et al. Tyrosyl-DNA phosphodiesterase and the repair of 3’-phosphoglycolate-terminated DNA double-strand breaks. DNA Repair (Amst) 2009;8:901–11. doi: 10.1016/j.dnarep.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou T, Lee JW, Tatavarthi H, Lupski JR, Valerie K, et al. Deficiency in 3’-phosphoglycolate processing in human cells with a hereditary mutation in tyrosyl-DNA phosphodiesterase (TDP1). Nucleic Acids Res. 2005;33:289–97. doi: 10.1093/nar/gki170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murai J, Huang SY, Das BB, Dexheimer TS, Takeda S, et al. Tyrosyl-DNA phosphodiesterase 1 (TDP1) repairs DNA damage induced by topoisomerases I and II and base alkylation in vertebrate cells. J Biol Chem. 2012;287:12848–57. doi: 10.1074/jbc.M111.333963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–99. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murai J, Marchand C, Shahane SA, Sun H, Huang R, et al. Identification of novel PARP inhibitors using a cell-based TDP1 inhibitory assay in a quantitative high-throughput screening platform. DNA Repair (Amst) 2014;21:177–82. doi: 10.1016/j.dnarep.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prasad R, Horton JK, Dai DP, Wilson SH. Repair pathway for PARP-1 DNA-protein crosslinks. DNA Repair (Amst) 2019;73:71–7. doi: 10.1016/j.dnarep.2018.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, et al. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem. 2004;279:55618–25. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- 25.Nitiss KC, Malik M, He X, White SW, Nitiss JL. Tyrosyl-DNA phosphodiesterase (Tdp1) participates in the repair of Top2-mediated DNA damage. Proc Natl Acad Sci U S A. 2006;103:8953–8. doi: 10.1073/pnas.0603455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gajewski S, Comeaux EQ, Jafari N, Bharatham N, Bashford D, et al. Analysis of the active-site mechanism of tyrosyl-DNA phosphodiesterase I: a member of the phospholipase D superfamily. J Mol Biol. 2012;415:741–58. doi: 10.1016/j.jmb.2011.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hawkins AJ, Subler MA, Akopiants K, Wiley JL, Taylor SM, et al. In vitro complementation of Tdp1 deficiency indicates a stabilized enzyme-DNA adduct from tyrosyl but not glycolate lesions as a consequence of the SCAN1 mutation. DNA Repair (Amst) 2009;8:654–63. doi: 10.1016/j.dnarep.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, et al. SCAN1 mutant Tdp1 accumulates the enzyme - DNA intermediate and causes camptothecin hypersensitivity. EMBO J. 2005;24:2224–33. doi: 10.1038/sj.emboj.7600694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32:267–72. doi: 10.1038/ng987. [DOI] [PubMed] [Google Scholar]
- 30.Cuya SM, Bjornsti MA, van Waardenburg R. DNA topoisomerase-targeting chemotherapeutics: what’s new? Cancer Chemother Pharmacol. 2017;80:1–14. doi: 10.1007/s00280-017-3334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pouliot JJ, Robertson CA, Nash HA. Pathways for repair of topoisomerase I covalent complexes in Saccharomyces cerevisiae. Genes Cells. 2001;6:677–87. doi: 10.1046/j.1365-2443.2001.00452.x. [DOI] [PubMed] [Google Scholar]
- 32.Raymond AC, Rideout MC, Staker B, Hjerrild K, Burgin AB Jr. Analysis of human tyrosyl-DNA phosphodiesterase I catalytic residues. J Mol Biol. 2004;338:895–906. doi: 10.1016/j.jmb.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Scott P, Al Kindi A, Al Fahdi A, Al Yarubi N, Bruwer Z, et al. Spinocerebellar ataxia with axonal neuropathy type 1 revisited. J Clin Neurosci. 2019;67:139–44. doi: 10.1016/j.jocn.2019.05.060. [DOI] [PubMed] [Google Scholar]
- 34.Dean RA, Fam HK, An J, Choi K, Shimizu Y, et al. Identification of a putative Tdp1 inhibitor (CD00509) by in vitro and cell-based assays. J Biomol Screen. 2014;19:1372–82. doi: 10.1177/1087057114546551. [DOI] [PubMed] [Google Scholar]
- 35.Duffy S, Fam HK, Wang YK, Styles EB, Kim JH, et al. Overexpression screens identify conserved dosage chromosome instability genes in yeast and human cancer. Proc Natl Acad Sci U S A. 2016;113:9967–76. doi: 10.1073/pnas.1611839113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Comeaux EQ, Cuya SM, Kojima K, Jafari N, Wanzeck KC, et al. Tyrosyl-DNA phosphodiesterase I catalytic mutants reveal an alternative nucleophile that can catalyze substrate cleavage. J Biol Chem. 2015;290:6203–14. doi: 10.1074/jbc.M114.635284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuya SM, Comeaux EQ, Wanzeck K, Yoon KJ, van Waardenburg RC. Dysregulated human Tyrosyl-DNA phosphodiesterase I acts as cellular toxin. Oncotarget. 2016;7:86660–74. doi: 10.18632/oncotarget.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirano R, Interthal H, Huang C, Nakamura T, Deguchi K, et al. Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007;26:4732–43. doi: 10.1038/sj.emboj.7601885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, et al. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007;26:4720–31. doi: 10.1038/sj.emboj.7601869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Summerlin M, Nitiss KC, Nitiss JL, Hanakahi LA. TDP1 is required for efficient non-homologous end joining in human cells. DNA Repair (Amst) 2017;60:40–9. doi: 10.1016/j.dnarep.2017.10.003. [DOI] [PubMed] [Google Scholar]
- 41.El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature. 2005;434:108–13. doi: 10.1038/nature03314. [DOI] [PubMed] [Google Scholar]
- 42.Davies DR, Interthal H, Champoux JJ, Hol WG. Insights into substrate binding and catalytic mechanism of human tyrosyl-DNA phosphodiesterase (Tdp1) from vanadate and tungstate-inhibited structures. J Mol Biol. 2002;324:917–32. doi: 10.1016/s0022-2836(02)01154-3. [DOI] [PubMed] [Google Scholar]
- 43.Davies DR, Interthal H, Champoux JJ, Hol WG. Crystal structure of a transition state mimic for Tdp1 assembled from vanadate, DNA, and a topoisomerase I-derived peptide. Chem Biol. 2003;10:139–47. doi: 10.1016/s1074-5521(03)00021-8. [DOI] [PubMed] [Google Scholar]
- 44.Davies DR, Interthal H, Champoux JJ, Hol WG. Explorations of peptide and oligonucleotide binding sites of tyrosyl-DNA phosphodiesterase using vanadate complexes. J Med Chem. 2004;47:829–37. doi: 10.1021/jm030487x. [DOI] [PubMed] [Google Scholar]
- 45.Swarup G, Cohen S, Garbers DL. Inhibition of membrane phosphotyrosyl-protein phosphatase activity by vanadate. Biochem Biophys Res Commun. 1982;107:1104–9. doi: 10.1016/0006-291x(82)90635-0. [DOI] [PubMed] [Google Scholar]
- 46.Liao Z, Thibaut L, Jobson A, Pommier Y. Inhibition of human tyrosyl-DNA phosphodiesterase by aminoglycoside antibiotics and ribosome inhibitors. Mol Pharmacol. 2006;70:366–72. doi: 10.1124/mol.105.021865. [DOI] [PubMed] [Google Scholar]
- 47.Huang SN, Pommier Y, Marchand C. Tyrosyl-DNA phosphodiesterase 1 (Tdp1) inhibitors. Expert Opin Ther Pat. 2011;21:1285–92. doi: 10.1517/13543776.2011.604314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang HC, Liu J, Baglo Y, Rizvi I, Anbil S, et al. Mechanism-informed repurposing of minocycline overcomes resistance to topoisomerase inhibition for peritoneal carcinomatosis. Mol Cancer Ther. 2018;17:508–20. doi: 10.1158/1535-7163.MCT-17-0568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chukwudi CU. rRNA binding sites and the molecular mechanism of action of the tetracyclines. Antimicrob Agents Chemother. 2016;60:4433–41. doi: 10.1128/AAC.00594-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antony S, Marchand C, Stephen AG, Thibaut L, Agama KK, et al. Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 2007;35:4474–84. doi: 10.1093/nar/gkm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banerjee B, Roy A, Sen N, Majumder HK. A tyrosyl DNA phosphodiesterase 1 from kinetoplastid parasite Leishmania donovani (LdTdp1) capable of removing topo I-DNA covalent complexes. Mol Microbiol. 2010;78:119–37. doi: 10.1111/j.1365-2958.2010.07318.x. [DOI] [PubMed] [Google Scholar]
- 52.Keil A, Frese-Schaper M, Steiner SK, Korner M, Schmid RA, et al. The Topoisomerase I inhibitor irinotecan and the Tyrosyl-DNA phosphodiesterase 1 inhibitor furamidine synergistically suppress murine lupus nephritis. Arthritis Rheumatol. 2015;67:1858–67. doi: 10.1002/art.39119. [DOI] [PubMed] [Google Scholar]
- 53.Dexheimer TS, Gediya LK, Stephen AG, Weidlich I, Antony S, et al. 4-Pregnen-21-ol-3,20-dione-21-(4-bromobenzenesulfonate) (NSC 88915) and related novel steroid derivatives as tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors. J Med Chem. 2009;52:7122–31. doi: 10.1021/jm901061s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marchand C, Lea WA, Jadhav A, Dexheimer TS, Austin CP, et al. Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Mol Cancer Ther. 2009;8:240–8. doi: 10.1158/1535-7163.MCT-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sirivolu VR, Vernekar SK, Marchand C, Naumova A, Chergui A, et al. 5-Arylidenethioxothiazolidinones as inhibitors of tyrosyl-DNA phosphodiesterase I. J Med Chem. 2012;55:8671–84. doi: 10.1021/jm3008773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weidlich IE, Dexheimer T, Marchand C, Antony S, Pommier Y, et al. Inhibitors of human tyrosyl-DNA phospodiesterase (hTdp1) developed by virtual screening using ligand-based pharmacophores. Bioorg Med Chem. 2010;18:182–9. doi: 10.1016/j.bmc.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gushchina IV, Nilov DK, Zakharenko AL, Lavrik OI, Svedas VK. Structure modeling of human Tyrosyl-DNA phosphodiesterase 1 and screening for its inhibitors. Acta Naturae. 2017;9:59–66. [PMC free article] [PubMed] [Google Scholar]
- 58.Lountos GT, Zhao XZ, Kiselev E, Tropea JE, Needle D, et al. Identification of a ligand binding hot spot and structural motifs replicating aspects of tyrosyl-DNA phosphodiesterase I (TDP1) phosphoryl recognition by crystallographic fragment cocktail screening. Nucleic Acids Res. 2019;47:10134–50. doi: 10.1093/nar/gkz515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das BB, Huang SY, Murai J, Rehman I, Ame JC, et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014;42:4435–49. doi: 10.1093/nar/gku088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433–43. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jun JH, Kumar V, Dexheimer TS, Wedlich I, Nicklaus MC, et al. Synthesis, anti-cancer screening and tyrosyl-DNA phosphodiesterase 1 (Tdp1) inhibition activity of novel piperidinyl sulfamides. Eur J Pharm Sci. 2018;111:337–48. doi: 10.1016/j.ejps.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zakharenko A, Khomenko T, Zhukova S, Koval O, Zakharova O, et al. Synthesis and biological evaluation of novel tyrosyl-DNA phosphodiesterase 1 inhibitors with a benzopentathiepine moiety. Bioorg Med Chem. 2015;23:2044–52. doi: 10.1016/j.bmc.2015.03.020. [DOI] [PubMed] [Google Scholar]
- 63.Khomenko T, Zakharenko A, Odarchenko T, Arabshahi HJ, Sannikova V, et al. New inhibitors of tyrosyl-DNA phosphodiesterase I (Tdp 1) combining 7-hydroxycoumarin and monoterpenoid moieties. Bioorg Med Chem. 2016;24:5573–81. doi: 10.1016/j.bmc.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 64.Li-Zhulanov NS, Zakharenko AL, Chepanova AA, Patel J, Zafar A, et al. A novel class of Tyrosyl-DNA phosphodiesterase 1 inhibitors that contains the octahydro-2H-chromen-4-ol scaffold. Molecules. 2018;23:E2468. doi: 10.3390/molecules23102468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mozhaitsev ES, Zakharenko AL, Suslov EV, Korchagina DV, Zakharova OD, et al. Novel inhibitors of DNA repair enzyme TDP1 combining monoterpenoid and adamantane fragments. Anticancer Agents Med Chem. 2019;19:463–72. doi: 10.2174/1871520619666181207094243. [DOI] [PubMed] [Google Scholar]
- 66.Zakharova O, Luzina O, Zakharenko A, Sokolov D, Filimonov A, et al. Synthesis and evaluation of aryliden- and hetarylidenfuranone derivatives of usnic acid as highly potent Tdp1 inhibitors. Bioorg Med Chem. 2018;26:4470–80. doi: 10.1016/j.bmc.2018.07.039. [DOI] [PubMed] [Google Scholar]
- 67.Zakharenko AL, Luzina OA, Sokolov DN, Kaledin VI, Nikolin VP, et al. Novel tyrosyl-DNA phosphodiesterase 1 inhibitors enhance the therapeutic impact of topotesmall es, Cyrillican on in vivo tumor models. Eur J Med Chem. 2019;161:581–93. doi: 10.1016/j.ejmech.2018.10.055. [DOI] [PubMed] [Google Scholar]
- 68.Komarova AO, Drenichev MS, Dyrkheeva NS, Kulikova IV, Oslovsky VE, et al. Novel group of tyrosyl-DNA-phosphodiesterase 1 inhibitors based on disaccharide nucleosides as drug prototypes for anti-cancer therapy. J Enzyme Inhib Med Chem. 2018;33:1415–29. doi: 10.1080/14756366.2018.1509210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Salomatina OV, Popadyuk II, Zakharenko AL, Zakharova OD, Fadeev DS, et al. Novel semisynthetic derivatives of bile acids as effective Tyrosyl-DNA phosphodiesterase 1 inhibitors. Molecules. 2018;23:E679. doi: 10.3390/molecules23030679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Efremova AS, Zakharenko AL, Shram SI, Kulikova IV, Drenichev MS, et al. Disaccharide pyrimidine nucleosides and their derivatives: a novel group of cell-penetrating inhibitors of poly(ADP-ribose) polymerase 1. Nucleosides Nucleotides Nucleic Acids. 2013;32:510–28. doi: 10.1080/15257770.2013.827793. [DOI] [PubMed] [Google Scholar]
- 71.Maltseva EA, Rechkunova NI, Sukhanova MV, Lavrik OI. Poly(ADP-ribose) polymerase 1 modulates interaction of the nucleotide excision repair factor XPC-RAD23B with DNA via Poly(ADP-ribosyl)ation. J Biol Chem. 2015;290:21811–20. doi: 10.1074/jbc.M115.646638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tian LW, Feng Y, Tran TD, Shimizu Y, Pfeifer T, et al. Tyrosyl-DNA phosphodiesterase I inhibitors from the Australian plant macropteranthes leichhardtii. J Nat Prod. 2015;78:1756–60. doi: 10.1021/acs.jnatprod.5b00211. [DOI] [PubMed] [Google Scholar]
- 73.Tian LW, Feng Y, Tran TD, Shimizu Y, Pfeifer T, et al. Achyrodimer F, a tyrosyl-DNA phosphodiesterase I inhibitor from an Australian fungus of the family Cortinariaceae. Bioorg Med Chem Lett. 2017;27:4007–10. doi: 10.1016/j.bmcl.2017.07.062. [DOI] [PubMed] [Google Scholar]
- 74.Takagi M, Ueda JY, Hwang JH, Hashimoto J, Izumikawa M, et al. Tyrosyl-DNA phosphodiesterase 1 inhibitor from an anamorphic fungus. J Nat Prod. 2012;75:764–7. doi: 10.1021/np2007389. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.