

Abstract
Thanks to the advent of rapid DNA sequencing technology and its prevalence, many disease-associated genetic variants are rapidly identified in many genes from patient samples. However, the subsequent effort to experimentally validate and define their pathological roles is extremely slow. Consequently, the pathogenicity of most disease-associated genetic variants is solely speculated in silico, which is no longer deemed compelling. We developed an experimental approach to efficiently quantify the pathogenic effects of disease-associated genetic variants with a focus on SLC26A4, which is essential for normal inner ear function. Alterations of this gene are associated with both syndromic and nonsyndromic hereditary hearing loss with various severity. We established HEK293T-based stable cell lines that express pendrin missense variants in a doxycycline-dependent manner, and systematically determined their anion transport activities with high accuracy in a 96-well plate format using a high throughput plate reader. Our doxycycline dosage-dependent transport assay objectively distinguishes missense variants that indeed impair the function of pendrin from those that do not (functional variants). We also found that some of these putative missense variants disrupt normal mRNA splicing. Our comprehensive experimental approach helps determine the pathogenicity of each pendrin variant, which should guide future efforts to benefit patients.
Keywords: SLC26A4, pendrin, Pendred syndrome, DFNB4, hereditary hearing loss
INTRODUCTION
SLC26A4 encodes an anion transporter, pendrin, essential for normal inner ear function (Choi et al., 2011; Everett et al., 2001; Everett et al., 1997; Kim & Wangemann, 2010; Li et al., 2013b). It is the well-known causative gene for Pendred syndrome (PDS; MIM# 274600), which is the most common type of syndromic hereditary sensorineural hearing loss associated with enlarged vestibular aqueduct (EVA), a thyroid condition called goiter, and impaired iodide organification. SLC26A4 variants are also often associated with nonsyndromic hereditary sensorineural hearing loss with EVA (DFNB4; MIM# 600791). Hearing loss in PDS and DFNB4 patients is typically congenital, from moderate to profound in severity, and often fluctuating and progressive (Choi et al., 2009; Miyagawa et al., 2014; Pryor et al., 2005). This wide phenotypic spectrum suggests an underlying pathology that can be ascribed to partial to complete loss of pendrin’s anion transport activity. Therefore, it is crucial to quantify the detrimental effects of genetic alterations found in the pendrin gene in order to appreciate the pathology underlying various severity of PDS and DFNB4. This effort is also beneficial for excluding false-positively identified nonpathogenic functional variants from the disease-associated variant list. In this study, we established stable cell lines that express pendrin or its disease-associated exonic single nucleotide variants (putative missense variants) in a doxycycline-dependent manner so that we can reproducibly control the expressions of the pendrin constructs, and developed a plate reader-based anion transport assays that determines the transport activities of multiple pendrin constructs at once with high accuracy. We also examined the potential pathological effects of the putative missense variants on mRNA splicing by in vitro splicing assay.
In general, the rate of identifying novel disease-associated genetic variants far exceeds the subsequent time-consuming efforts to experimentally determine their pathogenicity. Consequently, the pathogenicity of many genetic variants has been solely assessed bioinformatically. Analyses of conservation and evolution of the primary sequences aided by machine learning are the basis of the computational pathogenicity prediction (Adzhubei et al., 2010; Bendl et al., 2014; Li et al., 2013a; Mottaz et al., 2010; Sasidharan Nair & Vihinen, 2013). However, due to the known limitation of these computational approaches (Miosge et al., 2015), such in silico predictions are not deemed compelling by the guidelines established by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) (Richards et al., 2015). The ACMG/AMP guidelines are adapted by the Hearing Loss Variant Curation Expert Panel, and a new ACMG/AMP-based classification rules recently set for genetic variants associated with hearing loss (Oza et al., 2018). These new guidelines are to improve consistency and accuracy of genetic diagnoses of patients, and underscore the importance of collecting experimental evidence supporting (or questioning) the pathogenicity of numerous “disease-associated” genetic variants. Our experimental approach described herein to systematically characterize a large number of pendrin variants thus fully meet the scope of these new guidelines.
MATERIALS AND METHODS
Subjects
Hearing and other clinical tests were conducted as described in a previous study (Okamoto et al., 2014). Genetic tests were approved by the Ethics Review Committee of the National Hospital Organization Tokyo Medical Center and conducted only after obtaining written informed consent from each subject.
Generation of stable cell lines
A cell line that constitutively expresses mVenus (Kremers et al., 2006) harboring p.His148Gln and p.Ile152Leu (Galietta et al., 2001) was established in HEK293T cells using the Sleeping Beauty transposon system (Kowarz et al., 2015). Briefly, a cDNA encoding mVenusp.H148Q/p.I152L was cloned in a pSBbi-Bla vector (60526, addgene), and introduced to HEK293T cells together with pCMV(CAT)T7-SB100 (34879, addgene) using Effectene transfection reagent (301425, Qiagen). Transfected cells were selected in a DMEM medium (11965, Thermo Fisher Scientific) supplemented with 10% FBS and 1 µg/mL blasticidin (A11139, Thermo Fisher Scientific). Cell lines that express recombinant human pendrin constructs (both wild-type and disease-associated missense pendrin variants) in a doxycycline dosage-dependent manner were established in a similar way using a pSBtet-Pur vector (60507, addgene). Transfected cells were selected in a DMEM medium supplemented with 10% FBS and 1 µg/mL puromycin (A11138, Thermo Fisher Scientific). For I−/Cl− antiport assay, the pendrin-expressing pSBtet-Pur vectors were transfected in the HEK293T cell line that constitutively express mVenusp.H148Q/p.I152L (see above).
HCO3−/Cl− antiport assay
Cells expressing pendrin (or its missense variants) were cultured in a 12-well plate, dissociated with Cell Dissociation Buffer (13150016, Thermo Fisher Scientific), and loaded with a pH indicator, SNARF-5F (S23923, Thermo Fisher Scientific), in a high Cl− buffer (140 mM NaCl, 4.5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 20 mM HEPES, pH 7.4, 320 mOsmol/L) for 30 min in the presence of 5% CO2 at room temperature. The cells were washed with the high Cl− buffer once, and resuspended in 100 μL of the high Cl− buffer. Portions of the cell suspensions (50 μL) were transferred to wells in a 96-well plate (~1.5×105/well). HCO3−/Cl− antiport assay was initiated by an automated injection of 200 μL of a low Cl− buffer (125 mM Na-gluconate, 5 mM K-gluconate, 1 mM MgCl2, 1 mM CaCl2, 20 mM HEPES, 25 mM NaHCO3, 5% CO2) in Synergy Neo2 plate reader equipped with dual top PMTs (BioTek). The fluorescence intensity of SNARF-5F was measured in a time dependent manner using the optical configuration shown in Supp. Fig. S1. The information of the excitation/emission filters and the dichroic mirrors used are provided in Supp. Fig. S1.
I−/Cl− antiport assay
Cells expressing both pendrin (or its missense variants) and iodide sensitive fluorescent protein, mVenusp.H148Q/p.I152L, were cultured in a 12-well plate, and dissociated with Cell Dissociation Buffer (13150016, Thermo Fisher Scientific) and resuspended in 200 μL of a high Cl− buffer (150 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 20 mM HEPES, pH 7.5, 320 mOsmol/L) for 30 min at room temperature. Portions of the cell suspensions (160 μL) were transferred to wells in a 96-well plate (~1.5×105/well). I−/Cl− antiport assay was initiated by an automated injection of 80 μL of a high I− buffer containing 150 mM NaI, 1 mM MgCl2, 1 mM CaCl2, 20 mM HEPES (pH 7.5) in Synergy Neo2. The fluorescence intensities of mVenusp.H148Q/p.I152L and mTq2 were simultaneously measured in a time dependent manner using the optical configuration shown in Supp. Fig. S1.
Confocal fluorescence imaging
Stable cell lines were grown on a cover glass in 24-well plates. After ~24 hrs post application of 1 µg/mL doxycycline hyclate (D9891, Sigma), the cells were fixed with 4% formaldehyde for 5 min at room temperature, washed in PBS, and mounted onto slides using Dako fluorescent mounting medium (DAKO). Images were captured using the Nikon A1R+ confocal laser microscope system with Plan Apo 60X oil objective (Nikon) at the Center for Advance Microscopy at Northwestern University. Figures were prepared using NIS-Elements (Nikon) and Photoshop (Adobe).
In vitro splicing assay
Human genomic DNA was extracted from hair bud using a Phire Tissue Direct PCR kit (F170-S, Thermo Fisher Scientific). Intronic regions flanking exons 2, 3, 4, 5, 6, 7/8, 9, 10, 11/12, 13, 14, 15, 16, 17, 18, and 19 (+150bp before and after) were cloned into a pET01 exon-trap vector (MoBiTec, Göttingen, Germany). The DNA sequences of the partial pendrin genes used for the splicing assay were confirmed to be identical to the NCBI reference sequence, NG_008489.1 (see Supporting Information). The day before transfection, HEK293T cells were seeded on a 6-well plate so that they were 60–70% confluent at the time of transfection. The pET01 constructs were introduced using Effectene (301425, Qiagen) following the manufacturer’s directions. After 40 – 48 hrs post transfection, total RNA was isolated from each well using a Quick-RNA MiniPrep Plus kit (R1057, Zymo Research) and cDNA synthesis was carried out with SuperScript III Reverse Transcriptase (18080044, Thermo Fisher Scientific). Retention or omission of an exon(s) were confirmed by PCR using Phusion High-Fidelity DNA polymerase (M0530, NEB) with DNA primer pairs, 5’- CCTGGCCTGCCCAGGCTTTTGTCAACA -3’ and 5’- CCACCTCCAGTGCCAAGGTCTGAAGGTCA -3’.
Statistical analyses
Statistical analyses were performed using Prism (GraphPad software). We compared the anion transport rates determined at five different doxycycline dosage conditions to that of noninduced control cells by ANOVA followed by uncorrected Fisher’s Least Significant Difference. Linear regressions were performed to assess the dependency of the transport rates on doxycycline dosage. F-tests were performed to find difference in the doxycycline-dependence between WT vs. pendrin missense variants. In all statistical tests, p < 0.05 was considered statistically significant.
RESULTS
Establishment of stable cell lines
Currently, 571 disease-associated genetic variants are found in SLC26A4, of which 344 are classified as missense variants (as of April 2019) (Stenson et al., 2017). In this study, we focused on 51 exonic single nucleotide variants found in the pendrin gene, which are presumed to cause missense changes (Table 1). Among these, the effects of 22 variants (p.Leu117Phe, p.Pro123Ser, p.Met147Val, p.Thr193Ile, p.Val239Asp, p.Asp266Asn, p.Phe354Ser, p.Lys369Glu, p.Ala372Val, p.Asn392Tyr, p.Ser408Phe, p.Arg409His, p.Thr410Met, p.Thr416Pro, p.Leu445Trp, p.Gly497Ser, p.Tyr556Cys, p.C565Tyr, p.Ser657Asn, p.Ser666Phe, p.Thr721Met, and p.His723Arg) on the anion transport function of pendrin were previously studied (Choi et al., 2009; Dai et al., 2009; de Moraes et al., 2016; Dossena et al., 2011a; Dossena et al., 2011b; Gillam et al., 2005; Ishihara et al., 2010; Jung et al., 2016; Kuwabara et al., 2018; Lee et al., 2014; Muskett et al., 2016; Scott et al., 2000; Taylor et al., 2002; Yoon et al., 2008), whereas the remaining 29 await experimental characterization. All these missense variants, except for p.Ser408Phe that was identified in mice in a mutagenesis screen (Dror et al., 2010), were found in human patients (Stenson et al., 2017), of which p.Tyr214Cys, p.Thr410Lys, p.Val483Glu, and p.Leu703Pro are novel pendrin missense variants described for the first time in this report (Supp. Tables S1 and S2).
Table 1.
Summaries of HCO3−/Cl− and l−/Cl− antiport assays and qualitative assessment of the fluorescence images of stable cell lines expressing the pendrin constructs generated in this study.
| ╲ | Transport activity [mean ± SD (n)] | Dox-dependence | Protein expression | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dox: 0.1 μg/mL | Dox: 0.3 μg/mL | Dox: 1 μg/mL | Dox: 3 μg/mL | Dox: 10 μg/mL | Transport activity/ log10 [Dox] (slope ± SE) | Comparison to WT (F-test) | At the cell membrane? | Intracellular puncta? | ||||
| WT | HCO3−/Cl− | 0.50 ± 0.21 (14) | 0.92 ± 0.30 (16) | 1.40 ± 0.28 (16) | 1.86 ± 0.41 (16) | 2.18 ± 0.41 (14) | 0.861 ± 0.055 | not applicable | Yes | Yes | ||
| [nM/sec] | p = 0.0080 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | ||||||
| l−/Cl− | 0.035 ± 0.010 (10) | 0.083 ± 0.017 (10) | 0.127 ± 0.018 (10) | 0.157 ± 0.038 (10) | 0.184 ± 0.038 (10) | 0.074 ± 0.005 | not applicable | |||||
| [mM/sec] | p = 0.18 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | ||||||
| N-terminal domain (NTD) | Exon 2 |
P.S28G (c.82A>G) |
HCO3−/Cl− | 0.50 ± 0.12 (3) | 0.67 ± 0.17 (3) | 0.98 ± 0.29 (3) | 0.90 ± 0.22 (3) | 1.02 ± 0.11 (3) | 0.251 ± 0.070 | Function impaired | Yes | Yes |
| [nM/sec] | p = 0.0016 | p = 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0034 | p < 0.0001 | |||||
| l−/Cl− | 0.052 ± 0.008 (3) | 0.104 ± 0.035 (3) | 0.097 ± 0.033 (3) | 0.116 ± 0.043 (3) | 0.131 ± 0.039 (3) | 0.034 ± 0.012 | Function impaired | |||||
| [mM/sec] | p = 0.26 | p = 0.0002 | p = 0.0005 | p < 0.0001 | p < 0.0001 | p = 0.0129 | p = 0.0011 | |||||
|
P.S49R (c.147C>G) |
HCO3−/Cl− | 0.90 ± 0.24 (3) | 1.40 ± 0.08 (3) | 2.09 ± 0.30 (3) | 2.23 ± 0.09 (3) | 2.73 ± 0.61 (3) | 0.896 ± 0.113 | WT-like | Yes | Yes | ||
| [nM/sec] | p = 0.0008 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.78 | |||||
| l−/Cl− | 0.078 ± 0.032 (3) | 0.106 ± 0.027 (3) | 0.148 ± 0.022 (3) | 0.215 ± 0.035 (3) | 0.178 ± 0.023 (3) | 0.062 ± 0.013 | WT-like | |||||
| [mM/sec] | p = 0.0003 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0003 | p = 0.29 | |||||
| Exon 3 |
p.P76S (c.226C>T) |
HCO3−/Cl− | 0.36 ± 0.08 (3) | 0.51 ± 0.16 (3) | 0.80 ± 0.10 (3) | 0.80 ± 0.22 (3) | 0.87 ± 0.08 (3) | 0.263 ± 0.052 | Function impaired | Yes | No | |
| [nM/sec] | p = 0.0022 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0002 | p < 0.0001 | |||||
| l−/Cl− | 0.038 ± 0.004 (3) | 0.059 ± 0.012 (3) | 0.070 ± 0.006 (3) | 0.074 ± 0.013 (3) | 0.068 ± 0.013 (3) | 0.015 ± 0.004 | Function impaired | |||||
| [mM/sec] | p = 0.0036 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0052 | p < 0.0001 | |||||
| Transmembrane domain (TMD) |
p.S90L (c.269C>T) |
HCO3−/Cl− | 0.042 ± 0.008 (3) | 0.030 ± 0.003 (3) | 0.057 ± 0.020 (3) | 0.057 ± 0.022 (3) | 0.031 ± 0.032 (3) | 0.001 ± 0.008 | Function impaired | No | No | |
| [nM/sec] | p = 0.77 | p = 0.65 | p = 0.22 | p = 0.23 | p = 0.68 | p = 0.91 | p < 0.0001 | |||||
| l−/Cl− | 0.032 ± 0.004 (3) | 0.027 ± 0.008 (3) | 0.020 ± 0.004 (3) | 0.020 ± 0.002 (3) | 0.019 ± 0.005 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.0048 | p = 0.057 | p = 0.88 | p = 0.72 | p = 0.56 | ns* | p < 0.0001 | |||||
|
p.T99R (c.296C>G) |
HCO3−/Cl− | 0.071 ± 0.023 (3) | 0.055 ± 0.031 (3) | 0.059 ± 0.030 (3) | 0.057 ± 0.005 (3) | 0.059 ± 0.044 (3) | slope < 0 | Function impaired | No | No | ||
| [nM/sec] | p = 0.12 | p = 0.40 | p = 0.30 | p = 0.36 | p = 0.31 | p = 0.65 | p < 0.0001 | |||||
| l−/Cl− | 0.023 ± 0.005 (3) | 0.017 ± 0.007 (3) | 0.015 ± 0.006 (3) | 0.011 ± 0.002 (3) | 0.013 ± 0.002 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.44 | p = 0.34 | p = 0.14 | < control | < control | ns* | p < 0.0001 | |||||
| Exon 4 |
p.L117F (c.349C>T) |
HCO3−/Cl− | 0.68 ± 0.34 (8) | 1.16 ± 0.33 (9) | 1.63 ± 0.53 (9) | 2.07 ± 0.54 (9) | 2.28 ± 0.34 (8) | 0.822 ± 0.094 | WT-like | Yes | Yes | |
| [nM/sec] | p = 0.0087 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.78 | |||||
| l−/Cl− | 0.048 ± 0.016 (5) | 0.078 ± 0.008 (5) | 0.121 ± 0.019 (5) | 0.165 ± 0.041 (5) | 0.164 ± 0.015 (5) | 0.064 ± 0.007 | WT-like | |||||
| [mM/sec] | p = 0.0123 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.24 | |||||
|
p.P123S (c.367C>T) |
HCO3−/Cl− | 0.045 ± 0.026 (3) | 0.048 ± 0.053 (3) | 0.062 ± 0.031 (3) | 0.087 ± 0.021 (3) | 0.088 ± 0.027 (3) | 0.025 ± 0.011 | Function impaired | No | No | ||
| [nM/sec] | p = 0.75 | p = 0.66 | p = 0.29 | p = 0.047 | p = 0.042 | p = 0.039 | p < 0.0001 | |||||
| l−/Cl− | 0.033 ± 0.012 (3) | 0.027 ± 0.009 (3) | 0.017 ± 0.003 (3) | 0.016 ± 0.005 (3) | 0.018 ± 0.002 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.0061 | p = 0.14 | p = 0.49 | p = 0.27 | p = 0.55 | ns* | p < 0.0001 | |||||
|
p.G131V (c.392G>T) |
HCO3−/Cl− | 0.012 ± 0.014 (3) | 0.065 ± 0.010 (3) | 0.093 ± 0.063 (3) | 0.025 ± 0.029 (3) | 0.039 ± 0.019 (3) | 0.003 ± 0.015 | Function impaired | No | No | ||
| [nM/sec] | p = 0.29 | p = 0.23 | p = 0.025 | p = 0.59 | p = 0.92 | p = 0.86 | p < 0.0001 | |||||
| l−/Cl− | 0.024 ± 0.012 (3) | 0.020 ± 0.009 (3) | 0.021 ± 0.008 (3) | 0.019 ± 0.005 (3) | 0.016 ± 0.008 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.48 | p = 0.90 | p = 0.86 | p = 0.74 | p = 0.32 | p = 0.24 | p < 0.0001 | |||||
|
p.S133T (c.397T>A) |
HCO3−/Cl− | 0.049 ± 0.005 (3) | 0.042 ± 0.019 (3) | 0.078 ± 0.064 (3) | 0.077 ± 0.074 (3) | 0.082 ± 0.059 (3) | 0.020 ± 0.017 | Function impaired | No | No | ||
| [nM/sec] | p = 0.73 | p = 0.90 | p = 0.25 | p = 0.25 | p = 0.20 | p = 0.25 | p < 0.0001 | |||||
| l−/Cl− | 0.026 ± 0.003 (3) | 0.021 ± 0.007 (3) | 0.019 ± 0.003 (3) | 0.017 ± 0.003 (3) | 0.015 ± 0.006 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.098 | p = 0.96 | p = 0.67 | p = 0.26 | p = 0.11 | ns* | p < 0.0001 | |||||
| Exon 5 |
p-G139A (c.416G>C) |
HCO3−/Cl− | 0.053 ± 0.034 (3) | 0.061 ± 0.037 (3) | 0.051 ± 0.019 (3) | 0.036 ± 0.003 (3) | 0.056 ± 0.018 (3) | slope < 0 | Function impaired | No | No | |
| [nM/sec] | p = 0.40 | p = 0.21 | p = 0.46 | p = 0.94 | p = 0.33 | p = 0.66 | p < 0.0001 | |||||
| l−/Cl− | 0.023 ± 0.006 (3) | 0.020 ± 0.006 (3) | 0.016 ± 0.009 (3) | 0.015 ± 0.004 (3) | 0.014 ± 0.006 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.52 | p = 0.85 | p = 0.25 | p = 0.17 | p = 0.10 | ns* | p < 0.0001 | |||||
|
p.M147T (c.440T>C) |
HCO3−/Cl− | 0.24 ± 0.10 (3) | 0.21 ± 0.02 (3) | 0.26 ± 0.08 (3) | 0.25 ± 0.07 (3) | 0.23 ± 0.08 (3) | 0.005 ± 0.024 | Function impaired | No | Yes | ||
| [nM/sec] | p = 0.0008 | p = 0.0021 | p = 0.0003 | p = 0.0005 | p = 0.010 | p = 0.84 | p < 0.0001 | |||||
| l−/Cl− | 0.025 ± 0.005 (3) | 0.027 ± 0.001 (3) | 0.031 ± 0.003 (3) | 0.032 ± 0.003 (3) | 0.028 ± 0.004 (3) | 0.002 ± 0.001 | Function impaired | |||||
| [mM/sec] | p = 0.11 | p = 0.051 | p = 0.002 | p = 0.0009 | p = 0.017 | p = 0.13 | p < 0.0001 | |||||
|
p.M147V (c.439A>G) |
HCO3−/Cl− | 0.38 ± 0.20 (3) | 0.41 ± 0.18 (4) | 0.51 ± 0.24 (4) | 0.51 ± 0.28 (4) | 0.46 ± 0.20 (3) | 0.055 ± 0.073 | Function impaired | No | Yes | ||
| [nM/sec] | p = 0.029 | p = 0.012 | p = 0.026 | p = 0.0025 | p = 0.0090 | p = 0.46 | p < 0.0001 | |||||
| l−/Cl− | 0.027 ± 0.010 (3) | 0.027 ± 0.003 (3) | 0.031 ± 0.004 (3) | 0.038 ± 0.011 (3) | 0.035 ± 0.004 (3) | 0.005 ± 0.002 | Function impaired | |||||
| [mM/sec] | p = 0.11 | p = 0.15 | p = 0.023 | p = 0.0005 | p = 0.0022 | p = 0.049 | p < 0.0001 | |||||
|
p.V163l (c.487G>A) |
HCO3−/Cl− | 0.61 ± 0.29 (4) | 1.03 ± 0.36 (4) | 1.52 ± 0.42 (4) | 1.69 ± 0.51 (4) | 2.54 ± 1.20 (4) | 0.905 ± 0.191 | WT-like | Yes | Yes | ||
| [nM/sec] | p = 0.15 | p = 0.019 | p = 0.0011 | p = 0.0004 | p < 0.0001 | p = 0.0002 | p = 0.76 | |||||
| l−/Cl− | 0.045 ± 0.017 (3) | 0.076 ± 0.020 (3) | 0.118 ± 0.016 (3) | 0.167 ± 0.022 (3) | 0.182 ± 0.064 (3) | 0.073 ± 0.011 | WT-like | |||||
| [mM/sec] | p = 0.13 | p = 0.0018 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.88 | |||||
|
p.V186F (c.556G>T) |
HCO3−/Cl− | 0.10 ± 0.02 (3) | 0.11 ± 0.01 (3) | 0.13 ± 0.06 (3) | 0.087 ± 0.028 (3) | 0.083 ± 0.033 (3) | slope < 0 | Function impaired | No | No | ||
| [nM/sec] | p = 0.015 | p = 0.0055 | p = 0.0009 | p = 0.049 | p = 0.065 | p = 0.35 | p < 0.0001 | |||||
| l−/Cl− | 0.016 ± 0.003 (3) | 0.016 ± 0.005 (3) | 0.017 ± 0.005 (3) | 0.017 ± 0.002 (3) | 0.017 ± 0.003 (3) | 0.000 ± 0.001 | Function impaired | |||||
| [mM/sec] | p = 0.20 | p = 0.16 | p = 0.28 | p = 0.27 | p = 0.23 | p = 0.82 | p < 0.0001 | |||||
|
p.T193l (c.578C>T) |
HCO3−/Cl− | 0.023 ± 0.030 (4) | 0.024 ± 0.016 (4) | 0.023 ± 0.024 (4) | 0.020 ± 0.025 (4) | 0.046 ± 0.068 (4) | 0.008 ± 0.011 | Function impaired | No | No | ||
| [nM/sec] | p = 0.57 | p = 0.57 | p = 0.55 | p = 0.47 | p = 0.71 | p = 0.46 | p < 0.0001 | |||||
| l−/Cl− | 0.015 ± 0.007 (3) | 0.015 ± 0.005 (3) | 0.014 ± 0.006 (3) | 0.011 ± 0.006 (3) | 0.022 ± 0.007 (3) | 0.002 ± 0.002 | Function impaired | |||||
| [mM/sec] | p = 0.15 | p = 0.15 | p = 0.12 | p = 0.032 | p = 0.65 | p = 0.32 | p < 0.0001 | |||||
| Exon 6 |
p.Y214C (c.641A>G) |
HCO3−/Cl− | 0.47 ± 0.15 (3) | 0.79 ± 0.03 (3) | 0.89 ± 0.21 (3) | 1.01 ± 0.09 (3) | 1.04 ± 0.05 (3) | 0.270 ± 0.049 | Function impaired | Yes | Yes | |
| [nM/sec] | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.055 ± 0.022 (3) | 0.071 ± 0.019 (3) | 0.118 ± 0.012 (3) | 0.099 ± 0.006 (3) | 0.109 ± 0.008 (3) | 0.027 ± 0.007 | Function impaired | |||||
| [mM/sec] | p = 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.023 | p < 0.0001 | |||||
|
p.V239D (c.716T>A) |
HCO3−/Cl− | 0.12 ± 0.03 (4) | 0.19 ± 0.04 (4) | 0.21 ± 0.05 (4) | 0.17 ± 0.07 (4) | 0.23 ± 0.10 (4) | 0.042 ± 0.020 | Function impaired | Yes? | No | ||
| [nM/sec] | p = 0.053 | p = 0.0011 | p = 0.0004 | p = 0.0032 | p < 0.0001 | p = 0.055 | p < 0.0001 | |||||
| l−/Cl− | 0.022 ± 0.006 (3) | 0.024 ± 0.007 (3) | 0.033 ± 0.007 (3) | 0.038 ± 0.013 (3) | 0.041 ± 0.015 (3) | 0.010 ± 0.003 | Function impaired | |||||
| [mM/sec] | p = 0.80 | p = 0.50 | p = 0.035 | p = 0.0044 | p = 0.0010 | p = 0.0079 | p < 0.0001 | |||||
| Exon 7 |
p.D266N (c.796G>A) |
HCO3−/Cl− | 0.82 ± 0.07 (3) | 1.11 ± 0.08 (3) | 2.15 ± 0.24 (3) | 2.19 ± 0.58 (3) | 2.57 ± 0.53 (3) | 0.919 ± 0.141 | WT-like | Yes | Yes | |
| [nM/sec] | p = 0.0043 | p = 0.0004 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.67 | |||||
| l−/Cl− | 0.042 ± 0.005 (3) | 0.073 ± 0.005 (3) | 0.129 ± 0.055 (3) | 0.167 ± 0.020 (3) | 0.176 ± 0.023 (3) | 0.072 ± 0.010 | WT-like | |||||
| [mM/sec] | p = 0.12 | p = 0.0008 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.86 | |||||
| Exon 8 |
p.T307A (c.919A>G) |
HCO3−/Cl− | 0.73 ± 0.20 (4) | 1.06 ± 0.32 (4) | 1.70 ± 0.48 (4) | 2.19 ± 0.45 (4) | 2.43 ± 0.44 (4) | 0.908 ± 0.117 | WT-like | Yes | Yes | |
| [nM/sec] | p = 0.0085 | p = 0.0003 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.69 | |||||
| l−/Cl− | 0.051 ± 0.009 (3) | 0.091 ± 0.006 (3) | 0.122 ± 0.002 (3) | 0.194 ± 0.013 (3) | 0.212 ± 0.066 (3) | 0.085 ± 0.011 | WT-like | |||||
| [mM/sec] | p = 0.045 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.37 | |||||
|
p.N324Y (c.970A>T) |
HCO3−/Cl− | 0.83 ± 0.08 (3) | 1.30 ± 0.21 (3) | 1.88 ± 0.11 (3) | 2.33 ± 0.47 (3) | 2.45 ± 0.91 (3) | 0.856 ± 0.159 | WT-like | Yes | Yes | ||
| [nM/sec] | p = 0.017 | p = 0.0007 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0001 | p = 0.98 | |||||
| l−/Cl− | 0.034 ± 0.004 (3) | 0.085 ± 0.029 (3) | 0.133 ± 0.009 (3) | 0.166 ± 0.012 (3) | 0.215 ± 0.040 (3) | 0.089 ± 0.008 | WT-like | |||||
| [mM/sec] | p = 0.22 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.19 | |||||
|
p.G334V (c.1001G>T) |
HCO3−/Cl− | 0.67 ± 0.15 (3) | 0.93 ± 0.18 (3) | 1.19 ± 0.39 (3) | 1.61 ± 0.28 (3) | 1.61 ± 0.17 (3) | 0.512 ± 0.087 | Function impaired | Yes | Yes | ||
| [nM/sec] | p = 0.0011 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0073 | |||||
| l−/Cl− | 0.045 ± 0.005 (3) | 0.075 ± 0.015 (3) | 0.116 ± 0.020 (3) | 0.151 ± 0.011 (3) | 0.143 ± 0.010 (3) | 0.054 ± 0.007 | WT-like | |||||
| [mM/sec] | p = 0.013 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.058 | |||||
| Exon 9 |
p.F354S (c.1061T>C) |
HCO3−/Cl− | 0.87 ± 0.19 (3) | 1.25 ± 0.09 (3) | 1.78 ± 0.07 (3) | 2.37 ± 0.62 (3) | 2.39 ± 0.62 (3) | 0.832 ± 0.140 | WT-like | Yes | Yes | |
| [nM/sec] | p = 0.0049 | p = 0.0002 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.83 | |||||
| l−/Cl− | 0.030 ± 0.002 (3) | 0.063 ± 0.013 (3) | 0.146 ± 0.045 (3) | 0.140 ± 0.011 (3) | 0.146 ± 0.022 (3) | 0.062 ± 0.011 | WT-like | |||||
| [mM/sec] | p = 0.42 | p = 0.011 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0001 | p = 0.30 | |||||
|
p.K369E (c.1105A>G) |
HCO3−/Cl− | 0.42 ± 0.01 (3) | 0.59 ± 0.10 (4) | 0.76 ± 0.05 (4) | 0.94 ± 0.11 (4) | 1.09 ± 0.13 (3) | 0.339 ± 0.029 | Function impaired | Yes | Yes | ||
| [nM/sec] | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.032 ± 0.006 (3) | 0.056 ± 0.011 (3) | 0.070 ± 0.028 (3) | 0.104 ± 0.004 (3) | 0.104 ± 0.041 (3) | 0.038 ± 0.008 | Function impaired | |||||
| [mM/sec] | p = 0.29 | p = 0.0035 | p = 0.0002 | p < 0.0001 | p < 0.0001 | p = 0.0003 | p = 0.0013 | |||||
|
p.A372V (c.11150T) |
HCO3−/Cl− | 0.072 ± 0.029 (4) | 0.064 ± 0.022 (4) | 0.081 ± 0.024 (4) | 0.076 ± 0.034 (4) | 0.069 ± 0.022 (4) | 0.001 ± 0.008 | Function impaired | Yes? | No? | ||
| [nM/sec] | p = 0.063 | p = 0.14 | p = 0.022 | p = 0.040 | p = 0.087 | p = 0.88 | p < 0.0001 | |||||
| l−/Cl− | 0.046 ± 0.014 (3) | 0.030 ± 0.007 (3) | 0.022 ± 0.004 (3) | 0.022 ± 0.002 (3) | 0.022 ± 0.002 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p < 0.0001 | p = 0.033 | p = 0.68 | p = 0.77 | p = 0.78 | ns* | p < 0.0001 | |||||
| Exon 10 |
p.N392Y (c.1174A>T) |
HCO3−/Cl− | 0.058 ± 0.019 (3) | 0.048 ± 0.015 (3) | 0.041 ± 0.015 (3) | 0.069 ± 0.024 (3) | 0.057 ± 0.007 (3) | 0.004 ± 0.006 | Function impaired | No | No | |
| [nM/sec] | p = 0.17 | p = 0.44 | p = 0.78 | p = 0.040 | p = 0.18 | p = 0.56 | p < 0.0001 | |||||
| l−/Cl− | 0.039 ± 0.014 (3) | 0.028 ± 0.006 (3) | 0.022 ± 0.006 (3) | 0.020 ± 0.007 (3) | 0.015 ± 0.005 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.0006 | p = 0.11 | p = 0.67 | p = 0.86 | p = 0.27 | ns* | p < 0.0001 | |||||
|
p.S399P (c.1195T>C) |
HCO3−/Cl− | 0.70 ± 0.12 (3) | 1.22 ± 0.21 (3) | 1.67 ± 0.36 (3) | 1.83 ± 0.40 (3) | 2.10 ± 0.12 (3) | 0.679 ± 0.096 | WT-like | Yes | Yes | ||
| [nM/sec] | p = 0.0013 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.16 | |||||
| l−/Cl− | 0.050 ± 0.007 (3) | 0.096 ± 0.010 (3) | 0.132 ± 0.010 (3) | 0.146 ± 0.018 (3) | 0.150 ± 0.031 (3) | 0.050 ± 0.008 | Function impaired | |||||
| [mM/sec] | p = 0.0026 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.027 | |||||
|
p.S408F (c.1223C>T) |
HCO3−/Cl− | 0.044 ± 0.011 (3) | 0.039 ± 0.040 (3) | 0.056 ± 0.051 (3) | 0.038 ± 0.027 (3) | 0.064 ± 0.014 (3) | 0.008 ± 0.011 | Function impaired | Yes | Yes | ||
| [nM/sec] | p = 0.76 | p = 0.95 | p = 0.41 | p = 0.97 | p = 0.24 | p = 0.47 | p < 0.0001 | |||||
| l−/Cl− | 0.013 ± 0.001 (3) | 0.021 ± 0.002 (3) | 0.019 ± 0.002 (3) | 0.022 ± 0.002 (3) | 0.023 ± 0.003 (3) | 0.004 ± 0.001 | Function impaired | |||||
| [mM/sec] | < control | p = 0.84 | p = 0.71 | p = 0.65 | p = 0.32 | p = 0.0011 | p < 0.0001 | |||||
|
p.R409H (c.1226G>A) |
HCO3−/Cl− | 0.018 ± 0.030 (3) | 0.051 ± 0.033 (3) | 0.081 ± 0.079 (3) | 0.091 ± 0.079 (3) | 0.058 ± 0.078 (3) | 0.024 ± 0.022 | Function impaired | Yes | Yes | ||
| [nM/sec] | p = 0.64 | p = 0.74 | p = 0.30 | p = 0.20 | p = 0.62 | p = 0.29 | p < 0.0001 | |||||
| l−/Cl− | 0.019 ± 0.006 (3) | 0.019 ± 0.008 (3) | 0.020 ± 0.003 (3) | 0.021 ± 0.010 (3) | 0.021 ± 0.006 (3) | 0.001 ± 0.002 | Function impaired | |||||
| [mM/sec] | p = 0.74 | p = 0.65 | p = 0.84 | p = 0.92 | p = 0.84 | p = 0.56 | p < 0.0001 | |||||
|
p.T410K (c.1229C>A) |
HCO3−/Cl− | 0.031 ± 0.030 (3) | 0.017 ± 0.022 (3) | 0.029 ± 0.010 (3) | 0.060 ± 0.021 (3) | 0.051 ± 0.029 (3) | 0.016 ± 0.009 | Function impaired | No | No | ||
| [nM/sec] | p = 0.75 | p = 0.26 | p = 0.65 | p = 0.21 | p = 0.44 | p = 0.078 | p < 0.0001 | |||||
| l−/Cl− | 0.014 ± 0.010 (3) | 0.019 ± 0.007 (3) | 0.013 ± 0.000 (3) | 0.013 ± 0.004 (3) | 0.012 ± 0.001 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.080 | p = 0.72 | p = 0.062 | p = 0.068 | < control | p = 0.34 | p < 0.0001 | |||||
|
p.T410M (c.1229C>T) |
HCO3−/Cl− | 0.062 ± 0.029 (3) | 0.088 ± 0.062 (3) | 0.095 ± 0.044 (3) | 0078 ± 0.049 (3) | 0.097 ± 0.035 (3) | 0.012 ± 0.015 | Function impaired | No | No | ||
| [nM/sec] | p = 0.42 | p = 0.10 | p = 0.073 | p = 0.19 | p = 0.065 | p = 0.44 | p < 0.0001 | |||||
| l−/Cl− | 0.025 ± 0.006 (3) | 0.021 ± 0.004 (3) | 0.017 ± 0.004 (3) | 0019 ± 0.007 (3) | 0.018 ± 0.007 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.19 | p = 0.90 | p = 0.37 | p = 0.72 | p = 0.59 | p = 0.14 | p < 0.0001 | |||||
|
p.T416P (c.1246A>C) |
HCO3−/Cl− | 0.060 ± 0.024 (3) | 0.083 ± 0.017 (3) | 0.101 ± 0.020 (3) | 0.089 ± 0.010 (3) | 0.105 ± 0.008 (3) | 0.019 ± 0.006 | Function impaired | No | Yes | ||
| [nM/sec] | p = 0.13 | p = 0.0056 | p = 0.0005 | p = 0.0025 | p = 0.0003 | p = 0.0098 | p < 0.0001 | |||||
| l−/Cl− | 0.015 ± 0.001 (3) | 0.015 ± 0.002 (3) | 0.019 ± 0.002 (3) | 0.019 ± 0.001 (3) | 0.019 ± 0.005 (3) | 0.002 ± 0.001 | Function impaired | |||||
| [mM/sec] | p = 0.057 | p = 0.090 | p = 0.67 | p = 0.63 | p = 0.60 | p = 0.029 | p < 0.0001 | |||||
|
p.Q421L (c.1262A>T) |
HCO3−/Cl− | 0.28 ± 0.04 (3) | 0.44 ± 0.12 (3) | 0.54 ± 0.13 (3) | 0.57 ± 0.16 (3) | 0.52 ± 0.18 (3) | 0.121 ± 0.050 | Function impaired | Yes? | Yes | ||
| [nM/sec] | p = 0.012 | p = 0.0003 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.030 | p < 0.0001 | |||||
| l−/Cl− | 0.031 ± 0.007 (3) | 0.040 ± 0.006 (3) | 0.045 ± 0.016 (3) | 0.050 ± 0.019 (3) | 0.058 ± 0.017 (3) | 0.013 ± 0.005 | Function impaired | |||||
| [mM/sec] | p = 0.16 | p = 0.012 | p = 0.002 | p = 0.0004 | p < 0.0001 | p = 0.013 | p < 0.0001 | |||||
| Exon 11 |
p.I426N (c.1277T>A) |
HCO3−/Cl− | 0.60 ± 0.21 (3) | 0.97 ± 0.24 (3) | 1.46 ± 0.16 (3) | 1.65 ± 0.21 (3) | 1.64 ± 0.29 (3) | 0.549 ± 0.090 | Function impaired | Yes? | Yes | |
| [nM/sec] | p = 0.0011 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.016 | |||||
| l−/Cl− | 0.036 ± 0.005 (3) | 0.062 ± 0.012 (3) | 0.089 ± 0.002 (3) | 0.104 ± 0.013 (3) | 0.111 ± 0.003 (3) | 0.038 ± 0.004 | Function impaired | |||||
| [mM/sec] | p = 0.0032 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0007 | |||||
|
p.L445W (c.1334T>G) |
HCO3−/Cl− | 0.077 ± 0.035 (3) | 0.102 ± 0.065 (3) | 0.127 ± 0.060 (3) | 0.070 ± 0.038 (3) | 0.079 ± 0.047 (3) | slope < 0 | Function impaired | No | No | ||
| [nM/sec] | p = 0.24 | p = 0.065 | p = 0.016 | p = 0.34 | p = 0.22 | p = 0.75 | p < 0.0001 | |||||
| l−/Cl− | 0.018 ± 0.005 (3) | 0.012 ± 0.002 (3) | 0.011 ± 0.002 (3) | 0.016 ± 0.003 (3) | 0.013 ± 0.001 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.43 | < control | < control | p = 0.14 | < control | p = 0.38 | p < 0.0001 | |||||
| Exon 12 |
p.N457K (c.1371C>A) |
HCO3−/Cl− | 0.29 ± 0.07 (3) | 0.52 ± 0.16 (3) | 0.74 ± 0.03 (3) | 0.88 ± 0.13 (3) | 0.89 ± 0.11 (3) | 0.313 ± 0.043 | Function impaired | Yes | Yes | |
| [nM/sec] | p = 0.0023 | p < 0. 001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.035 ± 0.008 (3) | 0.051 ± 0.003 (3) | 0.073 ± 0.014 (3) | 0.079 ± 0.002 (3) | 0.074 ± 0.013 (3) | 0.021 ± 0.004 | Function impaired | |||||
| [mM/sec] | p = 0.0099 | p < 0.0001 | p < 0.0001 | p < 0.001 | p < 0.0001 | p = 0.0002 | p < 0.0001 | |||||
|
p.R470H (c.1409G>A) |
HCO3−/Cl− | 0.65 ± 0.18 (5) | 1.01 ± 0.27 (5) | 1.70 ± 0.60 (5) | 2.16 ± 0.78 (5) | 2.64 ± 0.81 (5) | 1.030 ± 0.156 | WT-1ike | Yes | Yes | ||
| [nM/sec] | p = 0.086 | p = 0.0089 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.19 | |||||
| l−/Cl− | 0.065 ± 0.027 (3) | 0.098 ± 0.016 (3) | 0.135 ± 0.014 (3) | 0.197 ± 0.010 (3) | 0.224 ± 0.071 (3) | 0.083 ± 0.012 | WT-1ike | |||||
| [mM/sec] | p = 0.013 | p = 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.45 | |||||
| Exon 13 |
p.V483E (c.1448T>A) |
HCO3−/Cl− | 0.32 ± 0.13 (3) | 0.95 ± 0.19 (3) | 1.40 ± 0.25 (3) | 1.42 ± 0.22 (3) | 1.62 ± 0.15 (3) | 0.609 ± 0.092 | WT-1ike | Yes? | Yes | |
| [nM/sec] | p = 0.033 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0004 | p = 0.052 | |||||
| l−/Cl− | 0.049 ± 0.023 (3) | 0.089 ± 0.025 (3) | 0.131 ± 0.050 (3) | 0.148 ± 0.014 (3) | 0.148 ± 0.021 (3) | 0.051 ± 0.011 | Function impaired | |||||
| [mM/sec] | p = 0.052 | p = 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0004 | p = 0.046 | |||||
|
p.G497S (c.1489G>A) |
HCO3−/Cl− | 0.11 ± 0.04 (3) | 0.14 ± 0.02 (3) | 0.22 ± 0.05 (3) | 0.26 ± 0.07 (3) | 0.27 ± 0.13 (3) | 0.084 ± 0.024 | Function impaired | Yes? | Yes | ||
| [nM/sec] | p = 0.13 | p = 0.037 | p = 0.0017 | p = 0.0003 | p = 0.0002 | p = 0.0042 | p < 0.0001 | |||||
| l−/Cl− | 0.019 ± 0.004 (4) | 0.030 ± 0006 (4) | 0.037 ± 0.009 (4) | 0.048 ± 0.007 (4) | 0.045 ± 0.005 (4) | 0.014 ± 0.002 | Function impaired | |||||
| [mM/sec] | p = 0.65 | p = 0.011 | p = 0.9001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| C-terminal domain (CTD) | Exon 14 |
p.T527P (c.1579A<C) |
HCO3−/Cl− | 0.17 ± 003 (3) | 0.44 ± 0.09 (3) | 0.66 ± 0.19 (3) | 0.78 ± 0.14 (3) | 0.91 ± 0.24 (3) | 0.362 ± 0.054 | Function impaired | Yes | Yes |
| [nM/sec] | p = 0.19 | p = 0.0010 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0001 | |||||
| l−/Cl− | 0.023 ± 0.007 (4) | 0.056 ± 0.014 (4) | 0.088 ± 0.014 (4) | 0.095 ± 0.027 (4) | 0.100 ± 0.023 (4) | 0.039 ± 0.006 | Function impaired | |||||
| [mM/sec] | p = 0.76 | p = 0.0004 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0003 | |||||
|
p.l529S (c.1586T>G) |
HCO3−/Cl− | 0.18 ± 0.04 (5) | 0.28 ± 0.10 (5) | 0.36 ± 0.10 (5) | 0.40 ± 0.09 (5) | 0.46 ± 0.12 (5) | 0.139 ± 0.025 | Function impaired | Yes | Yes | ||
| [nM/sec] | p = 0.016 | p = 0.0002 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.028 ± 0.007 (3) | 0.042 ± 0.012 (3) | 0.060 ± 0.004 (3) | 0.065 ± 0.017 (3) | 0.064 ± 0.003 (3) | 0.019 ± 0.004 | Function impaired | |||||
| [mM/sec] | p = 0.18 | p = 0.0006 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0003 | p < 0.0001 | |||||
| Exon 15 |
p.Y556C (c.1667A>G) |
HCO3−/Cl− | 0.053 ± 0.041 (5) | 0.082 ± 0.017 (5) | 0.10 ± 0.03 (5) | 0.13 ± 0.05 (5) | 0.17 ± 0.05 (5) | 0.056 ± 0.011 | Function impaired | Yes? | Yes | |
| [nM/sec] | p = 0.53 | p = 0.081 | p = 0.016 | p = 0.0006 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.011 ± 0.004 (3) | 0.017 ± 0.002 (3) | 0.020 ± 0.006 (3) | 0.022 ± 0.003 (3) | 0.025 ± 0.007 (3) | 0.006 ± 0.002 | Function impaired | |||||
| [mM/sec] | < control | p = 0.33 | p = 0.86 | p = 0.76 | p = 0.22 | p = 0.0017 | p 0.0001 | |||||
|
p.C565Y (c.1694G>A) |
HCO3−/Cl− | 0.73 ± 0.07 (3) | 1.06 ± 0.16 (3) | 1.55 ± 0.05 (3) | 1.86 ± 0.30 (3) | 2.07 ± 0.37 (3) | 0.696 ± 0.077 | WT-11ke | Yes | Yes | ||
| [nM/sec] | p = 0.0091 | p = 0.0059 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.19 | |||||
| l−/Cl− | 0.043 ± 0.019 (3) | 0.044 ± 0.012 (3) | 0.064 ± 0.014 (3) | 0.108 ± 0.017 (3) | 0.131 ± 0.017 (3) | 0.048 ± 0.006 | Function impaired | |||||
| [mM/sec] | p = 0.0002 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0142 | |||||
| Exon 17 |
p.S657N (c.1970G>A) |
HCO3−/Cl− | 0.38 ± 007 (3) | 0.67 ± 0.14 (3) | 0.83 ± 0.12 (3) | 0.91 ± 0.11 (3) | 0.97 ± 0.18 (3) | 0.284 ± 0.049 | Function impaired | Yes | Yes | |
| [nM/sec] | p = 0.0007 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.020 ± 0.003 (3) | 0.042 ± 0.017 (3) | 0.060 ± 0.012 (3) | 0.086 ± 0.016 (3) | 0.095 ± 0.012 (3) | 0.039 ± 0.004 | Function impaired | |||||
| [mM/sec] | p = 0.97 | p = 0.0034 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0009 | |||||
|
p.V659L (c.1975G>C) |
HCO3−/Cl− | 0.59 ± 0.12 (5) | 0.82 ± 0.18 (5) | 0.90 ± 0.09 (5) | 1.17 ± 0.38 (5) | 1.23 ± 0.33 (5) | 0.326 ± 0.067 | Function impaired | Yes | Yes | ||
| [nM/sec] | p = 0.0008 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
| l−/Cl− | 0.048 ± 0.003 (3) | 0.086 ± 0.005 (3) | 0.081 ± 0.022 (3) | 0.111 ± 0.018 (3) | 0.096 ± 0.011 (3) | 0.024 ± 0.007 | Function impaired | |||||
| [mM/sec] | p = 0.0008 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0031 | p < 0.0001 | |||||
|
p.S666F (c.1997C>T) |
HCO3−/Cl− | 0.19 ± 0.05 (3) | 0.32 ± 0.07 (3) | 0.43 ± 0.15 (3) | 0.53 ± 0.19 (3) | 0.47 ± 0.12 (3) | 0.153 ± 0.045 | Function impaired | No? | Yes | ||
| [nM/sec] | p = 0.069 | p = 0.0032 | p = 0.0002 | p < 0.0001 | p < 0.0001 | p = 0.0050 | p < 0.0001 | |||||
| l−/Cl− | 0.030 ± 0.002 (3) | 0.039 ± 0.007 (3) | 0.053 ± 0.009 (3) | 0.065 ± 0.022 (3) | 0.073 ± 0.008 (3) | 0.022 ± 0.004 | Function impaired | |||||
| [mM/sec] | p = 0.12 | p = 0.0052 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | |||||
|
p.D669E (c.2007C>A) |
HCO3−/Cl− | 0.054 ± 0.020 (5) | 0.073 ± 0.043 (5) | 0.073 ± 0.033 (5) | 0.070 ± 0.030 (5) | 0.057 ± 0.046 (5) | 0.000 ± 0.010 | Function impaired | No? | Yes | ||
| [nM/sec] | p = 0.46 | p = 0.11 | p = 0.11 | p = 0.15 | p = 0.37 | p = 0.96 | p < 0.0001 | |||||
| l−/Cl− | 0.014 ± 0.006 (3) | 0.013 ± 0.006 (3) | 0.014 ± 0.007 (3) | 0.016 ± 0.004 (3) | 0.016 ± 0.004 (3) | 0.001 ± 0.002 | Function impaired | |||||
| [mM/sec] | p = 0.083 | p = 0.063 | p = 0.097 | p = 0.21 | p = 0.23 | p = 0.48 | p < 0.0001 | |||||
| Exon 18 |
p.F683S (c.2048T>C) |
HCO3−/Cl− | 0.012 ± 0.021 (3) | 0.051 ± 0.002 (3) | 0.022 ± 0.023 (3) | 0.052 ± 0.013 (3) | 0.071 ± 0.095 (3) | 0.024 ± 0.015 | Function impaired | No | Yes | |
| [nM/sec] | p = 0.41 | p = 0.64 | p = 0.61 | p = 0.63 | p = 0.27 | p = 0.15 | p < 0.0001 | |||||
| l−/Cl− | 0.013 ± 0.010 (3) | 0.010 ± 0.005 (3) | 0.008 ± 0.001 (3) | 0.008 ± 0.002 (3) | 0.012 ± 0.005 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | < control | < control | < control | < control | < control | p = 0.85 | p < 0.0001 | |||||
|
p.F692L (c.2074T>C) |
HCO3−/Cl− | 0.42 ± 0.18 (5) | 0.68 ± 0.23 (5) | 0.78 ± 0.26 (5) | 1.02 ± 0.47 (5) | 1.03 ± 0.26 (5) | 0.310 ± 0.080 | Function impaired | No | No | ||
| [nM/sec] | p = 0.034 | p = 0.0010 | p = 0.0002 | p < 0.0001 | p < 0.0001 | p = 0.0008 | p < 0.0001 | |||||
| l−/Cl− | 0052 ± 0011 (3) | 0.083 ± 0.015 (3) | 0.112 ± 0.020 (3) | 0.117 ± 0.038 (3) | 0.110 ± 0.026 (3) | 0.030 ± 0.009 | Function impaired | |||||
| [mM/sec] | p = 0.0108 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p < 0.0001 | p = 0.0059 | p = 0.0001 | |||||
| Exon 19 |
p.L703P (c.2108T>C) |
HCO3−/Cl− | 0.036 ± 0.029 (5) | 0.044 ± 0.027 (5) | 0.041 ± 0032 (5) | 0.056 ± 0.024 (5) | 0038 ± 0027 (5) | 0.003 ± 0.008 | Function impaired | No | Yes | |
| [nM/sec] | p = 0.93 | p = 0.68 | p = 0.81 | p = 0.28 | p = 0.97 | p = 0.69 | p < 0.0001 | |||||
| l−/Cl− | 0.022 ± 0.018 (4) | 0.024 ± 0.014 (4) | 0.032 ± 0.034 (4) | 0.020 ± 0.014 (4) | 0.023 ± 0.017 (4) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.87 | p = 0.74 | p = 0.23 | p = 0.99 | p = 0.83 | p = 0.94 | p < 0.0001 | |||||
|
p.T721M (c.2162C>T) |
HCO3−/Cl− | 0.10 ± 0.05 (4) | 0.14 ± 0.04 (4) | 0.12 ± 0.06 (4) | 0.13 ± 0.05 (4) | 0.12 ± 0.05 (4) | 0.005 ± 0.015 | Function impaired | No | Yes | ||
| [nM/sec] | p = 0.045 | p = 0:0043 | p = 0.016 | p = 0.0090 | p = 0.0139 | p = 0.75 | p < 0.0001 | |||||
| l−/Cl− | 0.017 ± 0.005 (3) | 0.022 ± 0.004 (3) | 0.020 ± 0.004 (3) | 0.019 ± 0.002 (3) | 0.025 ± 0.004 (3) | 0.003 ± 0.001 | Function impaired | |||||
| [mM/sec] | p = 0.34 | p = 0.64 | p = 0.87 | p = 0.74 | p = 0.16 | p = 0.11 | p < 0.0001 | |||||
|
p.H723R (c.2168A>G) |
HCO3−/Cl− | 0.29 ± 0.11 (4) | 0.35 ± 0.11 (4) | 0.43 ± 0.18 (4) | 0.45 ± 0.21 (4) | 0.53 ± 0.25 (4) | 0.115 ± 0.053 | Function impaired | No | Yes | ||
| [nM/sec] | p = 0.032 | p = 0.0095 | p = 0.0019 | p = 0.0013 | p = 0.0002 | p = 0.042 | p < 0.0001 | |||||
| l−/Cl− | 0.038 ± 0.013 (3) | 0.017 ± 0.005 (3) | 0.020 ± 0.012 (3) | 0.025 ± 0.005 (3) | 0.029 ± 0.008 (3) | slope < 0 | Function impaired | |||||
| [mM/sec] | p = 0.019 | p = 050 | p = 0.06 | p = 0.35 | p = 0.085 | p = 0.67 | p < 0.0001 | |||||
The conventional fluorescence microscopy-based anion transport assay (Keller et al., 2013; Soleimani et al., 2001) has an advantage in controlling the ionic composition of the extracellular solution easily by perfusion. Also, the assay is not affected by the presence of untransfected cells, as they can be visually identified and excluded before data analysis. However, such imaging-based assay is typically low throughput, and thus not ideal for characterizing a large number of samples. For this reason, we opted for a high throughput plate reader-based assay that allows measurement of the anion transport activities of multiple pendrin constructs at once. In order for our assay not to be complicated by indistinguishable contribution of untransfected cells, we established doxycycline-inducible stable cell lines for wild-type (WT) pendrin and all 51 pendrin missense variants. These cell lines allow reproducible control of the expressions of the pendrin constructs and measurement of their anion transport activities in a doxycycline dosage-dependent manner, which increases confidence in detecting transport activity (see below). Since these cell lines can also be used for other purposes in future studies (e.g., high throughput drug screen), efforts in establishing a cell line for each pendrin variant is worthwhile.
In this study, doxycycline-induced expression of the pendrin constructs was routinely determined by measuring the fluorescence of mTurquoise2 (mTq2) attached to the C-termini of the pendrin constructs using a plate reader. We also confirmed the expression and subcellular localization of the pendrin constructs by confocal fluorescence microscopy (Fig. 1). As anticipated, WT pendrin construct was found at the outermost regions of cells, suggesting successful targeting of WT-pendrin to the plasma membrane (Fig. 1a). We also found punctated mTq2 fluorescence inside the cells regardless of doxycycline dosage (Supp. Fig. S2). Similar intracellular puncta were also observed in HEK293T cells heterologously expressing ECFP-tagged prestin (SLC26A5) (Takahashi et al., 2016). A recent study also found that SLC26A9 is intracellularly retained when heterologously expressed (Walter et al., 2019). This study by Walter et al. demonstrated that two intrinsically disordered regions (IDRs) within the C-terminal domain of the SLC26 protein are responsible for the intracellular retention, and that elimination of these IDRs greatly increased membrane targeting (Walter et al., 2019). It is likely that the C-terminal domains of SLC26 proteins physically interact with intracellular components. These observations suggest that the intracellular retention of SLC26 proteins is not owing to overproduction and resulting saturation of the SLC26 proteins in the cell membrane. In fact, we found unsaturated doxycycline-dependent increase of the transport activity that correlates with overall protein expression (see below, Fig. 2e). In any case, since cells expressing only the mTq2 moiety did not show such subcellular localizations (Fig. 1b), we considered the manifestation of the subcellular localizations found in WT-pendrin-expressing cells as indications of normal production, maturation, and cell membrane targeting. We confirmed the expression of pendrin missense variants in all 51 of our stable cell lines using mTq2 fluorescence, and qualitatively evaluated whether the pendrin variants exhibited similar expression patterns to WT pendrin (summarized in Table 1). Although highly qualitative, these simple microscopic assessments provide some insights as to the pathogenicity of the missense variants. However, even small amounts of pendrin variants targeted to the cell membrane may exhibit significant transport activities, we performed functional assays on all 51 variants to quantify how overall transport activity is affected by their presumed amino acid changes regardless of the results of the fluorescence microscopic observation.
Figure 1.
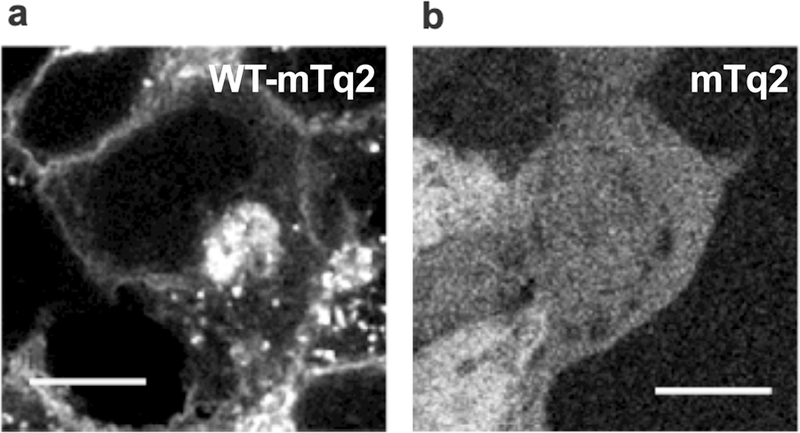
Stable cell lines heterologously expressing WT-pendrin-mTq2 (a) and mTq2 (b). Fluorescence of mTq2 was imaged using a confocal microscope. The scale bars indicate 10 µm. Fluorescence images of cells expressing pendrin missense variants are provided in Supp. Fig. S3. The subcellular localizations of all pendrin constructs were qualitatively assessed and summarized in Table 1.
Figure 2.

HCO3−/Cl− antiport assay. (a) Principle of the HCO3−/Cl− antiport assay. The chemical gradients of HCO3− and Cl− across the cell membrane drive exchange of these anions passively mediated by pendrin (green circle). Influx of HCO3− sucks up intracellular H+ and concomitantly increases intracellular pH, and vice versa. (b) The optical configuration of Synergy Neo2 plate reader used for the assay. Gray boxes indicate filters and dichroic mirrors (see Supp. Fig. S1 for details). (c) Response of a ratiometric fluorescence indicator, SNARF-5F, to pH. F1 and F2 are fluorescence intensities of SNARF-5F measured using two different fluorescence band-pass filters (Supp. Fig. S1). (d) The ratio of F1 and F2 (F2/F1) is plotted against pH. (e and f) Examples of HCO3−/Cl− antiport assay for WT- and p.Arg409His-pendrin-expressing cells, respectively. The broken lines indicate the initial rates (see the main text). The lower panels show summaries of the transport assay. The broken lines indicate the basal transport activity of non-induced cells (negative control). The expressions of the pendrin constructs were determined by measuring mTq2 fluorescence corrected for the density of cells (OD660nm) (blue triangles).
HCO3−/Cl− antiport assay
In the context of pendrin-associated hereditary hearing loss, the most pathophysiologically relevant anion substrates would be HCO3− and Cl−. Pendrin passively and reversibly exchanges HCO3− with Cl− depending on the chemical gradients of these anions across the cell membrane (Lee et al., 2014; Yoon et al., 2008). Since efflux or influx of HCO3− changes intracellular pH (Fig. 2a), pendrin-mediated HCO3−/Cl− antiport function can be monitored by measuring the intracellular pH. To this end, we used a ratiometric fluorescent pH indicator, SNARF-5F. The use of this emission ratiometric indicator allows simultaneous detection of its pH-sensitive fluorescence at F1 and F2 (Figs. 2b, 2c, and S1), which is the key to make our assay perfectly immune to photobleaching and a slight change of illumination area, if any, between measurements. This experimental configuration also allowed usage of suspended cells to greatly increase the number of cells in each well (~1.5 × 105/well) so that the signal-to-noise ratio is further increased. The assay was initiated by an automated injection of a low Cl−-containing solution. It should be noted that atmospheric CO2 affects HCO3− concentration, and that unequilibrated conversion of HCO3− to CO2 (and vice versa) would compromise the transport assay that relies on intracellular pH measurement because CO2 rapidly permeates the cell membrane and thus affects intracellular pH independently of pendrin activity. To eradicate these complications, we kept the atmospheric CO2 concentration at 5% before and during measurement. The F2/F1 fluorescence ratio of SNARF-5F was converted into H+ concentration using the calibration curve shown in Fig. 2d . The expressions of the pendrin constructs were controlled by the dosage of doxycycline added to the culture medium 1-day prior to the experiment and quantified by the fluorescence of C-terminally attached mTq2 corrected for the density of cells (OD660nm).
Examples of our HCO3−/Cl− antiport assays conducted for WT and p.Arg409His are shown in Figs. 2e and 2f. As expected, WT-pendrin-expressing cells showed doxycycline dosage-dependent transport activities (Fig. 2e). Since the chemical gradients of anions across the cell membrane (i.e., HCO3−, Cl−) that drives the transport of the very anions changes during measurement and greatly diminishes eventually, it is important to determine the initial transport rates in order to properly quantify loss of transport function; transport activities of fully or partially functional pendrin constructs can be easily underestimated otherwise. We determined the transport rate using the data points collected between 0 – 120 sec. We found that the time courses of [H+] in this time window can be sufficiently interpreted by a single exponential function, (0 ≤ t ≤ 120 sec, solid lines in Fig. 2e upper panel). This expedient exercise allowed objective determination of the initial slope, −kA, which is the time derivative of the single exponential function at t = 0 sec (shown by broken lines in Fig. 2e upper panel and filled circles in Fig. 2e lower panel). Unlike WT, the transport activities of pendrinp.R409H-expressing cells were very small and doxycycline dosage-independent (Figs. 2f), suggesting complete loss of HCO3−/Cl− antiport function. Summaries of our doxycycline dosage-dependent HCO3−/Cl− antiport assay are provided in Fig. 3. Representative examples of the assay for all 52 pendrin constructs (including WT) along with images of cells expressing these pendrin constructs are provided in Supp. Fig. S3. The numerical data used to generate Fig. 3 are provided in Table 1. Transport activities that were statistically indistinguishable from noninduced cells (p ≥ 0.05) are highlighted in gray in Table 1. Doxycycline-dependence of HCO3−/Cl− antiport activities, i.e., the slopes of linear regressions shown in Fig. 3 (transport activity vs. log10 of doxycycline dosage), and associated p-values are also provided in Table 1. Since the steepness of a slope reflects the magnitude of HCO3−/Cl− antiport activity, and since each slope is determined by multiple data points collected at five different doxycycline dosages, we use the slope values as the most reliable metrics of relative HCO3−/Cl− antiport activity. The p-values determined by the linear regressions are also useful to statistically determine the presence of HCO3−/Cl− antiport activity. We considered the absence of doxycycline dosage-dependence (p ≥ 0.05) as objective evidence for complete loss of HCO3−/Cl− antiport activity (highlighted in gray in Table 1). We also ran F-test to compare the doxycycline-dependence between WT vs. missense variants (Table 1). We considered p ≥ 0.05 as WT-like.
Figure 3.

A summary of HCO3−/Cl− antiport assay. The initial transport rates were determined as shown in Figs. 2e and 2f, and plotted against doxycycline concentrations. The error bars indicate the standard deviations. The solid lines indicate linear regressions (log10 of doxycycline dosage vs. transport rate). The broken lines indicate the transport activity of non-induced cells (negative control). The numerical data used to generate these graphs are provided in Table 1.
We also determined the HCO3−/Cl− antiport rates under a high extracellular Cl− condition after preincubating cells in a low Cl− condition (low-to-high Cl−, Supp. Fig. S4). As expected, this “reversed” procedure induced the opposite intracellular pH response (increase of [H+]) (Supp. Fig. S4). Nevertheless, the transport rates determined by the two opposite procedures correlate with each other to a high degree (r = 0.96, Supp. Fig. S4), suggesting that our effort to determine the relative anion transport rates of pendrin variants is not compromised by the large intracellular pH changes during the assay. We adopted the “high-to-low Cl−” procedure due to ~10 times lower basal transport activity of noninduced HEK293T cells observed with this procedure (0.037 ± 0.024 nM/sec (high-to-low Cl−) vs. 0.34 ± 0.05 nM/sec (low-to-high Cl−), Supp. Fig. S4).
I−/Cl− antiport assay
Pendrin also exchanges I− with Cl− (Scott et al., 1999). Pendrin-mediated iodide transport is thought to be important for iodide organification in thyroid cells because pendrin is expressed in the apical membrane of thyrocytes, and because patients with PDS often show a thyroid phenotype. Although this putative role of pendrin in thyroid cells has recently been questioned (Bizhanova & Kopp, 2011; Twyffels et al., 2011), we also determined I−/Cl− antiport activities for all 51 pendrin missense variants.
In order to monitor the transport of I−, we co-expressed an I−-sensitive mVenus p.H148Q/p.I152L (Fig. 4a). The p.H148Q/p.I152L double amino acid changes were reported to greatly increase the affinity of a yellow fluorescent protein (YFP) to I− (Galietta et al., 2001). The fluorescence intensities of both mTq2 (F1) and co-expressed mVenus p.H148Q/p.I152L (F2) were simultaneously measured (Figs. 4b and S1). The bleed-through fluorescence of mTq2, αF1, which is detected by the mVenus p.H148Q/p.I152L fluorescence channel and thus results in overestimation of F2, was subtracted offline, and the ratio of the fluorescence intensities of mTq2 and mVenus p.H148Q/p.I152L computed to be . The bleed-through correction constant, α, was determined to be 0.29 using cells expressing mTq2 but not mVenus p.H148Q/p.I152L. I−/Cl− antiport assay was initiated by an automated injection of a high I−/low Cl−-containing solution. Time-dependent reduction of the bleed-through-corrected fluorescence ratio of mTq2 and mVenus p.H148Q/p.I152L was normalized using the very ratio at time zero, , and converted into intracellular I− concentration as shown in Fig. 4c using the calibration shown in Fig. 4d. As in the HCO3−/Cl− antiport assay, we determined the initial rate of I−/Cl− antiport by the expedient single exponential procedure using the data points collected between 0 – 120 sec (Figs. 4e – 4g). A summary of the I−/Cl− antiport activities and representative examples of the assay for all the pendrin constructs are provided in Fig. 5 and Supp. Fig. S5, respectively. A numerical summary of the I−/Cl− antiport activities is provided in Table 1. We also determined the doxycycline-dependence of I−/Cl− antiport activities by linear regressions and associated p-values, and compared them with WT by F-test (Table 1).
Figure 4.

I−/Cl− antiport assay. (a) A fluorescence microscopic image of cells expressing WT-pendrin-mTq2 (doxycycline-induced) and mVenus p.H148Q/p.I152L (constitutively expressed). The fluorescence of mVenus p.H148Q/p.I152L is sensitive to I−, and thus can be used as an intracellular I− indicator. (b) The optical configuration of Synergy Neo2 plate reader used for the assay. See also Supp. Fig. S1 for detail. (c) Time-dependent change of mVenus p.H148Q/p.I152L fluorescence (after correction, black circles) and intracellular iodide (red triangle). See the main text for detail. (d) Response of mVenus p.H148Q/p.I152L fluorescence to I−. The affinity of mVenus p.H148Q/p.I152L to I− was determined to be 10.6 mM. This constant was used to convert the corrected mVenus p.H148Q/p.I152L fluorescence into intracellular I− concentration. (e and f) I−/Cl− antiport assays for WT- and p.Arg409His-pendrin-expressing cells, respectively. The broken lines indicate the initial rates. (g) Summaries of doxycycline dosage-dependent I−/Cl− antiport assay shown in panels e and f. The broken lines indicate the basal transport activity determined for non-induced cells (negative control).
Figure 5.

A summary of I−/Cl− antiport assay. The initial transport rates were determined as shown in Figs. 4e and 4f, and plotted against doxycycline concentrations. The error bars indicate the standard deviations. The solid lines indicate linear regressions (log10 of doxycycline dosage vs. transport rate). The broken lines indicate the transport activity of non-induced cells (negative control). The numerical data used to generate these graphs are provided in Table 1.
It should be noted that mVenus (and other YFP derivatives) are also sensitive to Cl− and pH (Seward et al., 2013). Accurate determination of intracellular I− concentrations using the calibration shown in Fig. 4d assumes that the contribution of Cl− to mVenus p.H148Q/p.I152L fluorescence is negligible and that there is little or no pH change during the assay. Although the affinity of mVenus p.H148Q/p.I152L to Cl− is very low (KCl = 240 mM) compared to I− (KI = 10.6 mM), it is likely that the fluorescence of mVenus p.H148Q/p.I152L was partly quenched before initiating I−/Cl− antiport assay because cells were cultured and subsequently preincubated in high Cl−-containing medium/solution. For example, influx of 1 mM I− would be underestimated to be 0.8 mM if the intracellular Cl− concentration were 50 mM. Furthermore, we found that intracellular pH slightly decreases during I−/Cl− antiport assay (Supp. Fig. S6). This small but non-negligible acidification is probably due to efflux of endogenous HCO3−. This endogenous HCO3− cannot be eliminated before the assay, because the concentration of HCO3−, which is in equilibrium with ~400 ppm atmospheric CO2, is estimated to be ~170 µM in our assay solutions containing 20 mM HEPES-NaOH (pH7.5) at room temperature (Supp. Fig. S6). Collectively, the transport rates determined by this assay using mVenus p.H148Q/p.I152L (and similar assay using YFP) should be regarded as relative activity values reflecting I−/Cl− antiport function that are to be compared with that of WT measured under the same condition.
Incidentally, the homogeneous cytosolic expression mVenus p.H148Q/p.I152L (e.g., Fig. 4a) aided us to locate the boundaries of cells, i.e., the cell membrane, and thus facilitated our effort to qualitatively assess membrane targeting of the pendrin constructs.
Effects of putative missense variants on the mRNA splicing
Exonic splicing enhancers (ESEs) and silencers (ESSs) are short nucleotide sequences that play important roles in constitutive and alternative splicing. As suggested by their names, these cis-regulatory elements enhance (ESEs) or silence (ESSs) splicing of precursor mRNAs (Lee & Rio, 2015). The pathogenicity assessment based on our functional study depends on the premise that putative missense variants indeed affect amino acid sequences. However, it may be possible that some of these exonic single nucleotide variants might primarily affect splicing. To explore this possibility, we performed in vitro splicing assay (Booth et al., 2018). Briefly, mini-gene constructs containing one or two human pendrin exons were created in a pET01 vector for WT and variants and transcribed in HEK293T cells (Supporting Information). The transcripts were extracted, reverse transcribed, and amplified by PCR to see if the exon(s) is retained or skipped (spliced out). Complete omission of an exon(s) results in 190-bp band, which is indistinguishable from the pET01vector control. Successful retention of an exon(s) produces a larger band depending on the size of the exon(s).
In order to assure the ability of this in vitro assay to discern pathophysiologically relevant mis-splicing for the pendrin gene, we generated a construct containing exons 7 and 8 with c.919–2A>G. This intronic pendrin variant, which was first described as 1143–2A>G (Coucke et al., 1999), is frequently found in Asian patients with PDS and experimentally demonstrated to cause skipping of exon 8 (Yang et al., 2005). The omission of exon 8 by c.919–2A>G was also recapitulated in a mouse model that shows pendrin null-like severe hearing phenotype (Lu et al., 2011). Consistently with these previous observations, we found that our mini-gene construct containing c.919–2A>G produced a shorter band that can be explained by the loss of exon 8 (83-bp). We also found a faint band at ~190-bp, suggesting an additional minor effect of c.919–2A>G to skip both exon 7 (153-bp) and 8 (Fig. 6). We also performed the mini-gene assay for two additional previously reported intronic pendrin variants, c.601–1G>A and c.1707+5G>A, which are predicted to affect the splice acceptor site of intron 5 (exon 6) and the splice donor site of intron 15 (exon 15), respectively (Park et al., 2005; Tsukamoto et al., 2003). The construct containing c.601–1G>A produced a slightly shorter band compared to the WT control, implying mis-splicing that results in a partial loss of exon 5 (Fig. 6). In fact, we confirmed that the PCR product lacked the first 21 bases of exon 6 that is presumed to encode a portion of a membrane-crossing α helix. The mini-gene construct harboring c.1707+5G>A produced a ~190-bp band (Fig. 6), suggesting a complete loss of exon 15 that encodes a significant portion (31-aa) of the highly conserved Sulphate Transporter and Anti-Sigma factor antagonist (STAS) domain present in the C-terminal domain of pendrin. These observations strongly suggest that both c.601–1G>A and c.1707+5G>A are indeed pathogenic, and reaffirm the ability of the mini-gene assay to successfully detect splicing defects for the pendrin gene.
Figure 6.

In vitro splicing assay. Mini-gene constructs containing one or two human pendrin exons with flanking introns (150-bp) were transcribed in HEK293T cells. The transcripts were reverse transcribed and amplified by PCR. Omission (skipping) of an exon(s) results in 190-bp band as pET01vector control does. Successful retention of an exon(s) produces a larger band depending on the size of the exon(s). Pendrin variants that produced clearly detectable misspliced band(s) are enclosed by gray squares. The DNA sequences of the mini-gene constructs are provided in Supporting Information.
We repeated this mini-gene assays for selected pendrin missense variants (Fig. 6). Of 33 (out of 51) missense variants tested, p.Gly131Val (c.392G>T) and p.Gly334Val (c.1001G>T) showed signs of mis-splicing. The clearly detectable ~190-bp band produced by c.392G>T suggests the omission of exon 4. Although a correctly spliced band was also detected, our transport assays demonstrated that pendrin p.G131V is barely functional (Table 1); that is, the high likelihood of p.Gly131Val (c.392G>T) to be pathogenic would not be overturned regardless of the knowledge whether c.392G>T compromises mRNA splicing or not. For this reason, most missense variants that almost completely abrogate the transport function of pendrin in our transport assays using pendrin constructs, which were expressed from cDNAs (Table 1), were dismissed from the assay. The codon that encodes Gly334 is separately located on exons 8 (3’ end) and 9 (5’ end). We found that c.1001G>T predominantly produced a band larger than WT, implying aberrant inclusion of a partial intronic sequence present between the two exons. Sequencing of this large PCR product confirmed inclusion of 40 bases of the intronic region flanking the 3’-end of exon 8. Since inclusion of this partial intronic sequence introduces an in-frame stop codon (TGA) right after exon 8, this mis-splicing should not produce a functional pendrin protein even if it is translated. Importantly, c.1001G>T also produces a correctly spliced band (confirmed by sequencing), suggesting that the pendrin gene harboring p.Gly334Val (c.1001G>T) can still produce a functional pendrin protein (Table 1) but its production may be significantly reduced compared to WT in vivo. The mini-gene construct containing exon 18 with p.Phe692Leu (c.2074T>C) also produced a PCR product larger than expected (245-bp). However, the WT control of exon 18 also showed the same large PCR product (Fig. 6). It is likely that intron 18 contains a cryptic exon, as there is a polypyrimidine tract followed by a potential splicing acceptor sequence (AGAT) (Supporting Information). It is conceivable that this cryptic exon was not spliced out due to elimination of an intronic regulatory element in our construct design that keeps only 150-bp intronic regions flanking the exon. In any case, since the result of the assay for c.2074T>C was WT-like, it is unlikely that c.2074T>C disrupts normal mRNA splicing.
The mis-splicing demonstrated for p.Gly131Val (c.392G>T) and p.Gly334Val (c.1001G>T) underscores the importance of experimentally determining the effects of exonic single nucleotide variants (putative missense variants) on mRNA splicing especially for those that only partly or barely affect the anion transport function of a recombinantly produced pendrin protein.
DISCUSSION
The large phenotypic spectrum of PDS and DFNB4 implies the presence of pathogenic variants that partially impair the function of pendrin to various degrees. Thus, quantification of the effects of genetic alterations of pendrin on its anion transport function would be crucial for appreciating the pathology of pendrin-associated hearing loss. By measuring HCO3−/Cl− and I−/Cl− antiport activities of pendrin missense variants in a doxycycline-dependent manner, we rigorously determined their relative transport abilities with respect to WT. The slope values provided in Table 1, which were determined from our doxycycline-dependent transport assays (Figs. 3 and 5), can be plotted in a two-dimensional (2D) plane (Fig. 7) so that the relationship between HCO3−/Cl− antiport activity vs. I−/Cl− antiport activity is visually inspected. The strong positive correlation found between the two measures (r = 0.97, Fig. 7) suggest that pendrin uses the same molecular mechanism for HCO3−/Cl− and I−/Cl− antiport. This 2D representation is also useful for visually appreciating the relative magnitudes of HCO3−/Cl− and I−/Cl− antiport activities. The p-values (calculated by F-test) provided in Table 1 statistically distinguish missense variants that significantly impair the anion transport function of pendrin (p < 0.05) from those that do not (WT-like, p ≥ 0.05). It is, of course, important to exclude and separately evaluate the pathogenicity of missense variants that barely or only partially impair the transport function when recombinantly expressed from cDNAs but impair mRNA splicing (e.g., p.Gly334Val).
Figure 7.
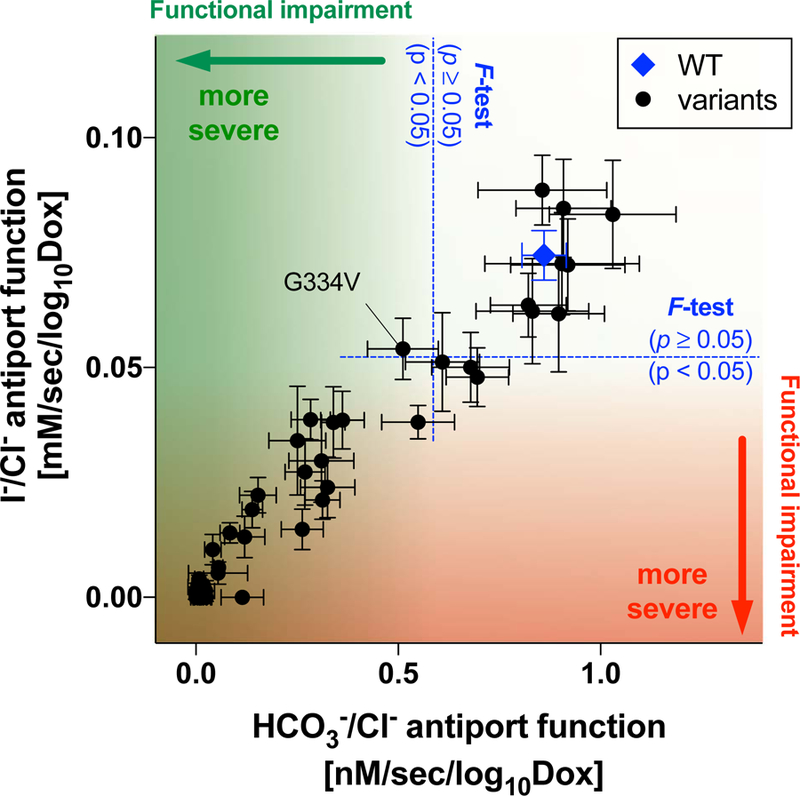
A two-dimensional representation of HCO3−/Cl− and I−/Cl− antiport activities (the slope values provided in Table 1) determined in this study for WT (indicated by blue diamond) and 51 pendrin missense variants. The error bars indicate the standard errors associated with linear regressions. F-tests were performed to objectively assess the pathological effects of the pendrin variants (see the main text for detail). Note that p.Gly334Val (c.1001G>T) was found to disrupt mRNA splicing (Fig. 6).
Pendrin and other vertebrate SLC26 proteins form tetramers (Hallworth et al., 2013). However, physiological importance of this multimerization is unknown. Each SLC26 protein subunit (single polypeptide) is presumed to have its own ion translocating pathway (Geertsma et al., 2015). It is likely that each subunit is independently functional because it is known that the function of WT-prestin (SLC26A5) does not change upon multimerization with virtually nonfunctional mutated prestin (Homma et al., 2013). The autosomal recessive inheritance pattern of all disease-associated genetic variants identified so far in the pendrin gene points to non-existence of dominant negative effect and thus further supports functional independence of a pendrin subunit. Therefore, we propose to use our transport activity data additively to estimate the overall pendrin activity for both monoallelic and biallelic pendrin variants. Given that one copy of WT-pendrin gene is sufficient for supporting normal inner ear function, it seems unlikely that pendrin variants with WT-like transport activities (e.g., p.Ser49Arg, p.Leu117Phe, p.Val163Ile, p.Asp266Asn, p.Thr307Ala, p.Asn324Tyr, p.Phe354Ser, p.Arg470His) are pathogenic. We speculate that these are nonpathogenic functional variants that co-segregate with a truly pathogenic genetic alteration such as those reported in previous studies (Ben Said et al., 2012; Chattaraj et al., 2017; Pique et al., 2014; Yang et al., 2009; Yang et al., 2007). This possibility also needs to be entertained for biallelic pendrin variants that only partly impair the overall pendrin-mediated transport activity, especially when the sums of the transport activities of the two pendrin variants are comparable to or exceeds that of WT. Exclusion of false-positively identified functional variants from the disease-associated variant list is very important in order to avoid unnecessary future clinical efforts to genetically treat patients with PDS/DFNB4.
Mouse models that do not express a pendrin protein (Everett et al., 2001; Lu et al., 2011) manifest severe inner ear phenotypes. Another pendrin mouse model expressing pendrin p.S408F (Dror et al., 2014; Dror et al., 2010) also shows severe inner ear phenotypes, which is consistent with our observation that p.Ser408Phe almost completely abrogates the anion transport activity of pendrin (Table 1). Studies using these mouse models unequivocally demonstrated the essentiality of pendrin-mediated anion transport function for normal inner ear function. However, in order to understand the pathology underlying the large phenotypic spectrum of PDS and DFNB4, it is also important to know how much pendrin-mediated transport activity is minimally required for normal inner ear function. Mouse models allow pathological examination of a monogenic pendrin variant in a clearly defined genetic background. However, it is conceivable that the tolerance of mice to a reduction of pendrin function may be greatly different from that of humans, because mice expressing pendrinp.H723R, whose HCO3−/Cl− antiport activity is only ~13% of WT (Table 1), do not show any inner ear phenotype even in a compound heterozygote having only one copy of pendrin p.H723R allele (pendrin p.H723R/-) (Lu et al., 2014). This surprisingly high tolerance of mice to a huge reduction of pendrin activity may be ascribed to the smaller inner ear volume compared to humans. In any case, mouse models may not be appropriate for assessing the severity of human pendrin missense variants. Thus, it is important to elaborate efforts in identifying human genetic alterations that are casually associated with PDS and DFNB4. Due to potentially polygenic nature of these disease phenotypes, it is also important to consider the possibility that a particular pendrin allele only partly contributes to manifestation of a certain disease phenotype. Therefore, rigorous quantification of the functional effects of genetic alterations of the pendrin gene demonstrated in this study is crucial to assess its relative pathological contribution when combined with another one.
It would not be so important to distinguish pendrin variants with very small transport activity from those with no transport activity for the purpose of assessing the pathogenicity of a variant. However, the ability to discern this subtle difference would be crucial for prioritizing future efforts to efficiently identify potentially rescuable pathogenic variants and finding pharmacological solutions against them. The p-values determined from linear regressions objectively determine the presence of the anion transport activity (p < 0.05) regardless of its magnitude, and thus useful for detecting small but statistically non-negligible antiport activity. For example, pendrin p.P123S exhibits statistically significant HCO3−/Cl− antiport activities only at two doxycycline conditions (3 and 10 µg/ml) (Table 1), but it shows a positive doxycycline-dependence (p = 0.039). These results suggest the p.Pro123Ser missense variant to be probably pathogenic, but the presence of the small HCO3−/Cl− antiport activity implies that this pendrin variant may be rescuable. In fact, a previous study demonstrated that pendrin p.P123S is pharmacologically rescuable at least partially (Ishihara et al., 2010).
If a missense variant is causally associated with a disease, it must somehow impair the function of a protein. In other words, a pathogenic variant would tell us which residue is critical for the function of a protein. Therefore, functional characterization of disease-associated missense changes also provides significant insights into the molecular mechanism of a protein. Although the tertiary structure of pendrin has not been defined, the recently solved structure of a mammalian SLC26A9 (PDB: 6RTF) (Walter et al., 2019) allows generation of a structural model for human pendrin (Figs. 8a – 8d). The transmembrane regions of SLC26 proteins are comprised of the gate and the core domains (Fig. 8d), and it is thought that an anion translocating pathway is located between these domains (shown by red broken circle in Fig. 8d) (Chang & Geertsma, 2017; Geertsma et al., 2015; Gorbunov et al., 2014; Walter et al., 2019). The locations of the amino acids focused in this study are also shown in the pendrin model (Figs. 8a – 8c). The missense alteration that result in > 80% reduction of HCO3−/Cl− antiport activity (compared to WT) are shown in red, while the others (≤ 80%) shown in blue. The presence of many red-colored sites within or in the vicinity of the putative anion translocating pathway implies the functional importance of this region, and thus in turn supports the pendrin model (Figs. 8). Given the probable structural and mechanistical similarities shared among the SLC26 family of proteins, a systematic characterization of pendrin missense variants has great potential to provide a common basis for understanding pathologies of disease-associated variants in pendrin and other SLC26 proteins.
Figure 8.

A structural model of pendrin viewed from the lateral (a), extracellular (b), and intracellular (c and d) sides. This structural model was generated using Phyre2 (Kelley et al., 2015) based on the recently solved structure of murine SLC26A9 (Walter et al., 2019). Regions corresponding to Met1-Cys54, Val577-Pro654, and Val733-Ser780 are not shown, because they were not modeled with high confidence. In panels a-c, the locations of missense changes that result in > 80% reduction of HCO3−/Cl− antiport activity are indicated as red, while the others (≤ 80%) as blue. In panel d, the core, gate, and C-terminal cytosolic domains are shown in blue, pink, and gray, respectively. The broken red circle indicates the putative anion translocating pathway.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Jing Zheng for providing laboratory resources for generating the pendrin constructs used in this study. This work is supported by NIH grants (DC014553 and DC017482 to K.H., DC011813 to J.Z.), JSPS Grant-in-Aid for Scientific Research (18K16869 to K.W.), and the Hugh Knowles Center. Imaging work was performed at the Northwestern University Center for Advanced Microscopy generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center.
Grant information
NIH: DC014553; DC017482; DC011813; NCI CCSG P30 CA060553
JSPS Grant-in-Aid for Scientific Research: 18K16869
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests. All DNA constructs and cell lines generated in this study are available to the scientific community upon request.
DATA AVAILABILITY STATEMENT
The SLC26A4 variant data have been deposited to and openly available from ClinVar (www.ncbi.nlm.nih.gov/clinvar, Accession IDs: SCV000994858 - SCV000994912).
REFERENCES
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Said M, Dhouib H, BenZina Z, Ghorbel A, Moreno F, Masmoudi S, … Hmani-Aifa M (2012). Segregation of a new mutation in SLC26A4 and p.E47X mutation in GJB2 within a consanguineous Tunisian family affected with Pendred syndrome. Int J Pediatr Otorhinolaryngol, 76(6), 832–836. 10.1016/j.ijporl.2012.02.053 [DOI] [PubMed] [Google Scholar]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, … Damborsky J (2014). PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol, 10(1), e1003440 10.1371/journal.pcbi.1003440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizhanova A, & Kopp P (2011). Controversies concerning the role of pendrin as an apical iodide transporter in thyroid follicular cells. Cell Physiol Biochem, 28(3), 485–490. 10.1159/000335103 [DOI] [PubMed] [Google Scholar]
- Booth KT, Azaiez H, Kahrizi K, Wang D, Zhang Y, Frees K, … Smith RJ (2018). Exonic mutations and exon skipping: Lessons learned from DFNA5. Hum Mutat, 39(3), 433–440. 10.1002/humu.23384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YN, & Geertsma ER (2017). The novel class of seven transmembrane segment inverted repeat carriers. Biol Chem, 398(2), 165–174. 10.1515/hsz-2016-0254 [DOI] [PubMed] [Google Scholar]
- Chattaraj P, Munjal T, Honda K, Rendtorff ND, Ratay JS, Muskett JA, … Griffith AJ (2017). A common SLC26A4-linked haplotype underlying non-syndromic hearing loss with enlargement of the vestibular aqueduct. J Med Genet, 54(10), 665–673. 10.1136/jmedgenet-2017-104721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BY, Kim HM, Ito T, Lee KY, Li X, Monahan K, … Griffith AJ (2011). Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J Clin Invest, 121(11), 4516–4525. 10.1172/JCI59353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BY, Stewart AK, Madeo AC, Pryor SP, Lenhard S, Kittles R, … Griffith AJ (2009). Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat, 30(4), 599–608. 10.1002/humu.20884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coucke PJ, Van Hauwe P, Everett LA, Demirhan O, Kabakkaya Y, Dietrich NL, … Van Camp G (1999). Identification of two different mutations in the PDS gene in an inbred family with Pendred syndrome. J Med Genet, 36(6), 475–477. [PMC free article] [PubMed] [Google Scholar]
- Dai P, Stewart AK, Chebib F, Hsu A, Rozenfeld J, Huang D, … Wu BL (2009). Distinct and novel SLC26A4/Pendrin mutations in Chinese and U.S. patients with nonsyndromic hearing loss. Physiol Genomics, 38(3), 281–290. 10.1152/physiolgenomics.00047.2009 [DOI] [PubMed] [Google Scholar]
- de Moraes VCS, Bernardinelli E, Zocal N, Fernandez JA, Nofziger C, Castilho AM, … Dossena S (2016). Reduction of cellular expression levels is a common feature of functionally affected pendrin (SLC26A4) protein variants. Mol Med, 22(1), 41–53. 10.2119/molmed.2015.00226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossena S, Bizhanova A, Nofziger C, Bernardinelli E, Ramsauer J, Kopp P, & Paulmichl M (2011a). Identification of allelic variants of pendrin (SLC26A4) with loss and gain of function. Cell Physiol Biochem, 28(3), 467–476. 10.1159/000335108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossena S, Nofziger C, Brownstein Z, Kanaan M, Avraham KB, & Paulmichl M (2011b). Functional characterization of pendrin mutations found in the Israeli and Palestinian populations. Cell Physiol Biochem, 28(3), 477–484. 10.1159/000335109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror AA, Lenz DR, Shivatzki S, Cohen K, Ashur-Fabian O, & Avraham KB (2014). Atrophic thyroid follicles and inner ear defects reminiscent of cochlear hypothyroidism in Slc26a4-related deafness. Mamm Genome, 25(7–8), 304–316. 10.1007/s00335-014-9515-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror AA, Politi Y, Shahin H, Lenz DR, Dossena S, Nofziger C, … Avraham KB (2010). Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J Biol Chem, 285(28), 21724–21735. 10.1074/jbc.M110.120188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, … Green ED (2001). Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet, 10(2), 153–161. [DOI] [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, … Green ED (1997). Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet, 17(4), 411–422. 10.1038/ng1297-411 [DOI] [PubMed] [Google Scholar]
- Galietta LJ, Haggie PM, & Verkman AS (2001). Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett, 499(3), 220–224. [DOI] [PubMed] [Google Scholar]
- Geertsma ER, Chang YN, Shaik FR, Neldner Y, Pardon E, Steyaert J, & Dutzler R (2015). Structure of a prokaryotic fumarate transporter reveals the architecture of the SLC26 family. Nat Struct Mol Biol, 22(10), 803–808. 10.1038/nsmb.3091 [DOI] [PubMed] [Google Scholar]
- Gillam MP, Bartolone L, Kopp P, & Benvenga S (2005). Molecular analysis of the PDS gene in a nonconsanguineous Sicilian family with Pendred’s syndrome. Thyroid, 15(7), 734–741. 10.1089/thy.2005.15.734 [DOI] [PubMed] [Google Scholar]
- Gorbunov D, Sturlese M, Nies F, Kluge M, Bellanda M, Battistutta R, & Oliver D (2014). Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nat commun, 5, 3622 10.1038/ncomms4622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallworth R, Stark K, Zholudeva L, Currall BB, & Nichols MG (2013). The conserved tetrameric subunit stoichiometry of slc26 proteins. Microsc Microanal, 19(4), 799–807. 10.1017/S1431927613000457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma K, Duan C, Zheng J, Cheatham MA, & Dallos P (2013). The V499G/Y501H mutation impairs fast motor kinetics of prestin and has significance for defining functional independence of individual prestin subunits. J Biol Chem, 288(4), 2452–2463. 10.1074/jbc.M112.411579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara K, Okuyama S, Kumano S, Iida K, Hamana H, Murakoshi M, … Wada H (2010). Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear Res, 270(1–2), 110–118. 10.1016/j.heares.2010.08.015 [DOI] [PubMed] [Google Scholar]
- Jung J, Kim J, Roh SH, Jun I, Sampson RD, Gee HY, … Lee MG (2016). The HSP70 co-chaperone DNAJC14 targets misfolded pendrin for unconventional protein secretion. Nat commun, 7, 11386 10.1038/ncomms11386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller J, Homma K, & Dallos P (2013). Pixels as ROIs (PAR): A Less-Biased and Statistically Powerful Approach for Gleaning Functional Information from Image Stacks. PLoS ONE, 8(7), e69047 10.1371/journal.pone.0069047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, & Sternberg MJ (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc, 10(6), 845–858. 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, & Wangemann P (2010). Failure of fluid absorption in the endolymphatic sac initiates cochlear enlargement that leads to deafness in mice lacking pendrin expression. PLoS ONE, 5(11), e14041 10.1371/journal.pone.0014041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowarz E, Loscher D, & Marschalek R (2015). Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J, 10(4), 647–653. 10.1002/biot.201400821 [DOI] [PubMed] [Google Scholar]
- Kremers GJ, Goedhart J, van Munster EB, & Gadella TW Jr. (2006). Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry, 45(21), 6570–6580. 10.1021/bi0516273 [DOI] [PubMed] [Google Scholar]
- Kuwabara MF, Wasano K, Takahashi S, Bodner J, Komori T, Uemura S, … Homma K (2018). The extracellular loop of pendrin and prestin modulates their voltage-sensing property. J Biol Chem, 293(26), 9970–9980. 10.1074/jbc.RA118.001831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Jung J, Shin JW, Song MH, Kim SH, Lee JH, … Park HJ (2014). Correlation between genotype and phenotype in patients with bi-allelic SLC26A4 mutations. Clin Genet, 86(3), 270–275. 10.1111/cge.12273 [DOI] [PubMed] [Google Scholar]
- Lee Y, & Rio DC (2015). Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem, 84, 291–323. 10.1146/annurev-biochem-060614-034316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Kwan JS, Bao SY, Yang W, Ho SL, Song YQ, & Sham PC (2013a). Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet, 9(1), e1003143 10.1371/journal.pgen.1003143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Sanneman JD, Harbidge DG, Zhou F, Ito T, Nelson R, … Wangemann P (2013b). SLC26A4 targeted to the endolymphatic sac rescues hearing and balance in Slc26a4 mutant mice. PLoS Genet, 9(7), e1003641 10.1371/journal.pgen.1003641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Wu CC, Shen WS, Yang TH, Yeh TH, Chen PJ, … Hsu CJ (2011). Establishment of a knock-in mouse model with the SLC26A4 c.919-2A>G mutation and characterization of its pathology. PLoS ONE, 6(7), e22150 10.1371/journal.pone.0022150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Wu CC, Yang TH, Lin YH, Yu IS, Lin SW, … Hsu CJ (2014). Differences in the pathogenicity of the p.H723R mutation of the common deafness-associated SLC26A4 gene in humans and mice. PLoS ONE, 8(6), e64906 10.1371/journal.pone.0064906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, … Andrews TD (2015). Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci U S A, 112(37), E5189–5198. 10.1073/pnas.1511585112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagawa M, Nishio SY, & Usami S (2014). Mutation spectrum and genotype-phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J Hum Genet, 59(5), 262–268. 10.1038/jhg.2014.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottaz A, David FP, Veuthey AL, & Yip YL (2010). Easy retrieval of single amino-acid polymorphisms and phenotype information using SwissVar. Bioinformatics, 26(6), 851–852. 10.1093/bioinformatics/btq028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskett JA, Chattaraj P, Heneghan JF, Reimold FR, Shmukler BE, Brewer CC, … Griffith AJ (2016). Atypical patterns of segregation of familial enlargement of the vestibular aqueduct. Laryngoscope, 126(7), E240–247. 10.1002/lary.25737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y, Mutai H, Nakano A, Arimoto Y, Sugiuchi T, Masuda S, … Matsunaga T (2014). Subgroups of enlarged vestibular aqueduct in relation to SLC26A4 mutations and hearing loss. Laryngoscope, 124(4), E134–140. 10.1002/lary.24368 [DOI] [PubMed] [Google Scholar]
- Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, … ClinGen Hearing Loss Clinical Domain Working, G. (2018). Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat, 39(11), 1593–1613. 10.1002/humu.23630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, Jang HS, … Koo SK (2005). Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet, 67(2), 160–165. 10.1111/j.1399-0004.2004.00386.x [DOI] [PubMed] [Google Scholar]
- Pique LM, Brennan ML, Davidson CJ, Schaefer F, Greinwald J Jr., & Schrijver I (2014). Mutation analysis of the SLC26A4, FOXI1 and KCNJ10 genes in individuals with congenital hearing loss. PeerJ, 2, e384 10.7717/peerj.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, … Griffith AJ (2005). SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet, 42(2), 159–165. 10.1136/jmg.2004.024208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan Nair P, & Vihinen M (2013). VariBench: a benchmark database for variations. Hum Mutat, 34(1), 42–49. 10.1002/humu.22204 [DOI] [PubMed] [Google Scholar]
- Scott DA, Wang R, Kreman TM, Andrews M, McDonald JM, Bishop JR, … Sheffield VC (2000). Functional differences of the PDS gene product are associated with phenotypic variation in patients with Pendred syndrome and non-syndromic hearing loss (DFNB4). Hum Mol Genet, 9(11), 1709–1715. [DOI] [PubMed] [Google Scholar]
- Scott DA, Wang R, Kreman TM, Sheffield VC, & Karniski LP (1999). The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet, 21(4), 440–443. 10.1038/7783 [DOI] [PubMed] [Google Scholar]
- Seward HE, Basran J, Denton R, Pfuhl M, Muskett FW, & Bagshaw CR (2013). Halide and proton binding kinetics of yellow fluorescent protein variants. Biochemistry, 52(14), 2482–2491. 10.1021/bi3016839 [DOI] [PubMed] [Google Scholar]
- Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, & Burnham CE (2001). Pendrin: an apical Cl−/OH−/HCO3- exchanger in the kidney cortex. Am J Physiol Renal Physiol, 280(2), F356–364. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, … Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet, 136(6), 665–677. 10.1007/s00439-017-1779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Cheatham MA, Zheng J, & Homma K (2016). The R130S mutation significantly affects the function of prestin, the outer hair cell motor protein. J Mol Med (Berl), 94(9), 1053–1062. 10.1007/s00109-016-1410-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Metcalfe RA, Watson PF, Weetman AP, & Trembath RC (2002). Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab, 87(4), 1778–1784. 10.1210/jcem.87.4.8435 [DOI] [PubMed] [Google Scholar]
- Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S, & Usami S (2003). Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet, 11(12), 916–922. 10.1038/sj.ejhg.5201073 [DOI] [PubMed] [Google Scholar]
- Twyffels L, Massart C, Golstein PE, Raspe E, Van Sande J, Dumont JE, … Kruys V (2011). Pendrin: the thyrocyte apical membrane iodide transporter? Cell Physiol Biochem, 28(3), 491–496. 10.1159/000335110 [DOI] [PubMed] [Google Scholar]
- Walter JD, Sawicka M, & Dutzler R (2019). Cryo-EM structures and functional characterization of murine Slc26a9 reveal mechanism of uncoupled chloride transport. Elife, 8 10.7554/eLife.46986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JJ, Tsai CC, Hsu HM, Shiao JY, Su CC, & Li SY (2005). Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutation in the PDS gene. Hear Res, 199(1–2), 22–30. 10.1016/j.heares.2004.08.007 [DOI] [PubMed] [Google Scholar]
- Yang T, Gurrola JG 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, & Smith RJ (2009). Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet, 84(5), 651–657. 10.1016/j.ajhg.2009.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Vidarsson H, Rodrigo-Blomqvist S, Rosengren SS, Enerback S, & Smith RJ (2007). Transcriptional control of SLC26A4 is involved in Pendred syndrome and nonsyndromic enlargement of vestibular aqueduct (DFNB4). Am J Hum Genet, 80(6), 1055–1063. 10.1086/518314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JS, Park HJ, Yoo SY, Namkung W, Jo MJ, Koo SK, … Lee MG (2008). Heterogeneity in the processing defect of SLC26A4 mutants. J Med Genet, 45(7), 411–419. 10.1136/jmg.2007.054635 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.