

Abstract
Twenty-one natural and unnatural phenolic compounds containing a carbohydrate moiety were synthesized and their structure–activity relationship (SAR) was evaluated for α-glucosidase inhibition and antioxidative activity. Varying the position of the galloyl unit on the 1,5-anhydro-d-glucitol (1,5-AG) core resulted in changes in the α-glucosidase inhibitory activity and notably, particularly strong activity was demonstrated when the galloyl unit was present at the C-2 position. Furthermore, increasing the number of the galloyl units significantly affected the α-glucosidase inhibition, and 2,3,4,6-tetra-galloyl-1,5-AG (54) and 2,3,4,6-tetra-galloyl-d-glucopyranose (61) exhibited excellent activities, which were more than 13-fold higher than the α-glucosidase inhibitory activity of acertannin (37). Moreover, a comparative structure-activity study suggested that a hemiacetal hydroxyl functionality in the carbohydrate core and a biaryl bond of the 4,6-O-hexahydroxydiphenoyl (HHDP) group, which are components of ellagitannins including tellimagrandin I, are not necessary for the α-glucosidase inhibitory activity. Lastly, the antioxidant activity increased proportionally with the number of galloyl units.
Keywords: 1,5-AG; tellimagrandin I; acertannin; maplexin; ginnalin; polyphenol; α-glucosidase; antioxidant
1. Introduction
Impaired glucose tolerance increases the risk of vascular events such as atherosclerotic coronary artery disease [1,2]. Particularly, postprandial hyperglycemia is a serious risk factor for cardiovascular diseases and is believed to be the cause of oxidative stress that leads to vascular events [3,4,5,6,7]. Thus, controlling postprandial hyperglycemia is an important target to prevent diabetes as well as diabetic complications. In clinical medicine, α-glucosidase inhibitors such as acarbose, miglitol, and voglibose, belong to the class of antidiabetic drugs used for improving postprandial hyperglycemia [8,9,10,11]. Currently, natural products and their derivatives constitute more than half of the drugs in the clinic [12,13,14,15]. Therefore, finding inspiration in nature to develop more efficient and effective medicines has attracted significant interest.
Trees belonging to the Acer species have been used as traditional medicinal plants for many years and are widely known for their sap, which can be concentrated to produce maple syrup [16]. It has been demonstrated that Acer extracts display various bioactivities such as anti-cancer [17,18], antioxidant [19,20,21,22], and antihyperglycemic effects [23,24]. A. Honma et al. identified a compound from Acer saccharum extracts able to suppress hyperglycemia, namely acertannin, and revealed that its effects are a consequence of potent inhibitory activity toward α-glucosidase [25]. The structural components of acertannin include the characteristic 1,5-anhydro-d-glucitol (1,5-AG) sugar moiety, which lacks the hemiacetal hydroxyl group present in d-glucose, as the carbohydrate core, and two gallic acid functionalities as the phenolic units (Figure 1) [26]. However, only a few plants belonging to the Acer genus produce the 1,5-AG core containing polyphenols [27,28]. To date, maplexin A–J and ginnalin A–C have been isolated and characterized. The molecules possess varying numbers and positions of the phenol units esterified with the 1,5-AG core [29,30,31,32]. These polyphenols were shown to exhibit different bioactivities such as α-glucosidase inhibition and antioxidant activity. It is noteworthy that different numbers, positions, and types of the phenol units on the 1,5-AG core display non-identical bioactivities [32,33,34,35,36].
Figure 1.
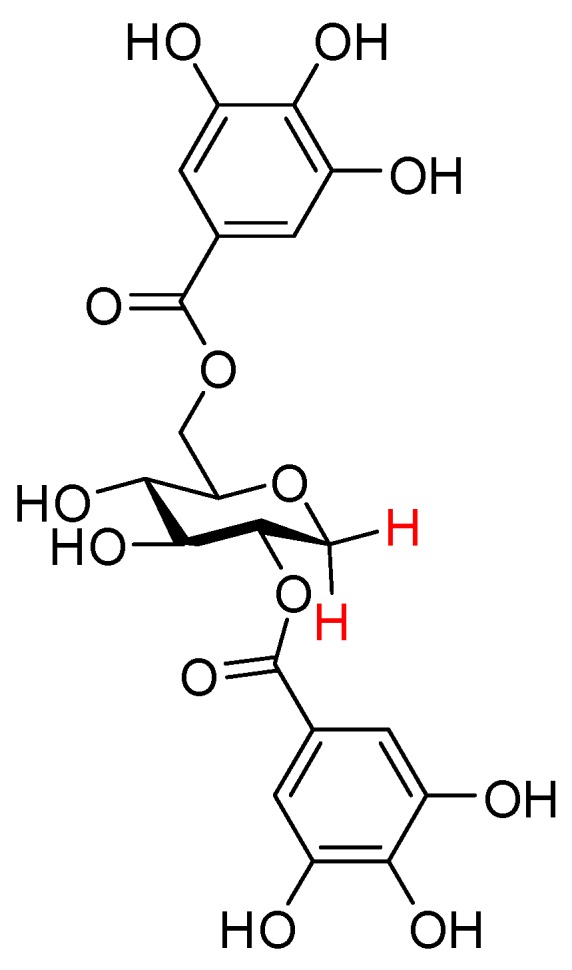
Structure of acertannin.
Tellimagrandin I, which belongs to ellagitannins, has also been demonstrated to be an α-glucosidase inhibitor and to show antioxidant activity [37,38]. The molecule is characterized by the presence of a hexahydroxydiphenoyl (HHDP) group, two galloyl units and the d-glucose core possesses a hemiacetal hydroxyl functionality (Figure 2) [39]. The HHDP group provides structural diversity in polyphenols, and the macro-lactone structure is considered to be the element responsible for the pharmacological activity [40]. Nonetheless, to our knowledge, no reports on the evaluation of the synthesis and/or bioactivity of compounds comprising the HHDP functionality on the 1,5-AG core have been reported so far. Furthermore, the hemiacetal hydroxyl group is a fundamental moiety in the carbohydrate chemistry; however, its effects on the bioactivity remain largely unexplored.
Figure 2.

The structural components of tellimagrandin I. HHDP, hexahydroxydiphenoyl.
In the present study, we report the synthesis of a series of 21 carbohydrate-based phenolic compounds to investigate the structure–activity relationship (SAR). α-glucosidase inhibition and antioxidant activity were examined by studying the effects of (1) the position and number of galloyl units, (2) the type of phenol units, (3) the existence the 4,6-O-HHDP group, and (4) the presence of the hemiacetal hydroxyl group.
2. Results
2.1. Syntheses of 1,5-AG-Based Polyphenols
2.1.1. Syntheses of Galloylated 1,5-AGs
Recently, A. Kamori et al. reported the synthesis of various natural and unnatural acertannin derivatives and evaluated their SAR against ceramidase and ceramide synthase enzymes [35]. In addition, we have also previously reported a facile method for the preparation of 1,5-anhydroalditol via treatment of per-O-TMS-glycopyranosyl iodide with LiBH4 [41]. In total, 1,5-AG, which can be easily synthesized from d-glucose on multi-gram scale in three days, possesses four hydroxyl groups; hence, 15 different combinations are possible for mono-, di-, tri-, and tetra-galloylation of 1,5-AG. In the current study, we attempted the synthesis of all of these galloylated compounds starting from 1,5-AG.
Firstly, 4,6-O-benzylidene-1,5-AG (1) [42] was protected with benzyl (Bn) group using BnBr and NaH to afford di-benzylated compound 2 [42]. However, reacting 1 in a 2-phase dichloromethane (DCM)/5% NaOH system with BnBr, NaI, and tetra-n-butylammonium hydrogen sulfate provided the 3-OH analog 3 [35] and the 2-OH analog 4 [35] as a mixture of products, which could be separated by column chromatography (C.C.) (Scheme 1) [43].
Scheme 1.

Selective protection of 1,5-AG.
Subsequently, the 4,6-O-benzylidene groups on the 1,5-AG derivatives 1–4 were converted to the corresponding hydroxyl moieties using an acid or reducing reagent (Scheme 2). Compounds 2–4 were then deprotected with 80% acetic acid/ solution to give the corresponding diol analogs 5–7 [35,42]. Reduction of 1, 3, and 4 by BH3/THF and trimethylsilyl trifluoromethanesulfonate (TMSOTf) [44], and reduction of 2 by diisobutylaluminium hydride (DIBAL-H) [45] resulted in the formation of the 6-OH derivatives 8–11 [35,46,47]. Moreover, compounds 1–4 were treated with triethylsilane and trifluoroacetic acid (TFA) [48] to afford the 4-OH derivatives 12–15 [35,49]. Esterification of these -OH derivatives with Bn-protected gallic acid (16) [50] using N,N-dimethyl-4-aminopyridine (DMAP) and triethylamine (TEA), as well as 2-chloro-1-methylpyridinium iodide as the condensing reagent, provided 1,5-AG-based galloylated derivatives 17–30. Finally, hydrogenolysis using Pd(OH)2 as the catalyst in a MeOH/THF solvent mixture gave 14 types of the 1,5-AG core containing polyphenol analogs 31–44 (Scheme 3).
Scheme 2.

Deprotection of 1–4.
Scheme 3.

Synthesis of galloylated 1,5-AGs.
2.1.2. Synthesis of Maplexin J Analogs
Maplexin J (54) [32,50], which is per-galloylated 1,5-AG, could be easily synthesized from 1,5-AG. Therefore, we attempted to synthesize of maplexin J analogs, which in addition to the galloyl moiety, contained different numbers of phenolic groups at various positions intending to elucidate the effect of this functionality on the bioactivity. Phenol derivatives 45–48 [51,52,53] were prepared in 2-steps from commercially available methyl esters (Scheme 4). Subsequently, 1,5-AG was condensed with 16 [50], and 45–48 using DMAP, TEA, and 2-chloro-methylpyridinium iodide to obtain the corresponding Bn-protected 1,5-AG analogs 49–53. Deprotection under hydrogenolysis conditions provided compounds 54–58 (Scheme 5). However, esterification of benzyl glucoside 59 [54] with 16 using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and DMAP resulted in the formation of intermediate 60. Following hydrogenolysis, tetra-galloylated d-glucopyranose 61 containing a hemiacetal moiety was formed. Notably, the structure of 61 [55] is analogous to tellimagrandin I; however, the biaryl bond of the HHDP group is missing (Scheme 6).
Scheme 4.

Preparation of the Bn-protected phenol units 45–48.
Scheme 5.

Synthesis of maplexin J analogs 54–58.
Scheme 6.

Synthesis of tellimagrandin I analog 61.
2.1.3. Synthesis of a 1,5-AG-Based Tellimagrandin I Analog
To consider the effect of the hemiacetal group on the activity of tellimagrandin I, we also focused on the synthesis of the 1-deoxy analog 67. Deprotection of benzylidene acetal in 21 in a methanol/DCM solvent system using iodine, according to the method reported by Feldman et al. [56], gave diol 62 in a high yield of 93% (Scheme 7). Intermediate 62 was then condensed with the gallic acid derivative 63 [57] to afford the galloylated compound 64. The subsequent removal of the methoxymethyl (MOM) protecting groups provided 65 in 93% yield.
Scheme 7.

Synthesis of the 1,5-AG-based tellimagrandin I analog 67.
The construction of the HHDP group was performed in accordance with the approach previously reported by Yamada et al. [57,58,59,60]. Compound 65 was treated with and to provide the biaryl derivative 66. Finally, hydrogenation of 66 in MeOH/THF with Pd(OH)2 as the catalyst gave the desired 1,5-AG-based tellimagrandin I analog 67.
2.2. Evaluation of α-Glucosidase Inhibition and Antioxidant Activity
2.2.1. The α-Glucosidase Inhibitory Activity
The α-glucosidase inhibitory activity of all samples was assayed utilizing a commercially available FUJIFILM α-glucosidase inhibitory activity assay kit. In total, 25 μL of each sample (25–2000 μg/mL in ) or acarbose (0.5–8 μg/mL in ), 50 μL of 18.5 mM maltose diluted in maleic anhydride buffer (100 mM, pH = 6.0), and 25 μL of the rat α-glucosidase solution were incubated in a micro-tube at 37 °C for 30 min. Subsequently, 400 μL of purified water was added to the solution and the reaction mixture was boiled for 3 min to deactivate α-glucosidase. The generated glucose was measured by LabAssay™ glucose (mutarotase-GOD method). In total, 100 μL of the reaction solution and 150 μL of the coloring solution were incubated in 96-well plates at 37 °C for 10 min, and the absorbance was recorded at 505 nm using a microplate reader (Bio-Rad, Model 680). The inhibition percentage was calculated using the following Equation:
where As is the absorbance of the analyzed sample, Ab is the absorbance of the blank (immediate deactivation), and Ac is the absorbance of the control (without α-glucosidase). Acarbose was used as the positive control.
2.2.2. Evaluation of Antioxidant Activity
The antioxidant activity was assessed by employing the 2,2-diphenyl-1-picrylhydrazyl (DPPH) radical scavenging assay, according to the previously published method [61]. In total, 100 μL of each sample (1000–10 μg/mL in 50%EtOH/) or trolox (160–0 μM in 50% EtOH/), and 50 μL of 2-(N-morpholino)ethanesulfonic acid (MES)-NaOH buffer (200 mM, pH = 6.0), and 50 μL of an 800 μM DPPH diluted in 99.5% EtOH were mixed in 96-well plates. Following shaking the microplate for 15 min in the dark, the absorbance was recorded at 520 nm using a microplate reader (Bio-Rad, Model 680). Effective percentages (%) were calculated as follows:
where As is the absorbance of the sample and Ab is the absorbance of the blank (without samples, 50% EtOH/ solution was used instead).
In addition, correlation of the absorbance on the y-axis and the concentrations on the x-axis resulted in the formation of an approximately straight-line plot. The trolox-equivalent (sample-mol/trolox-mol) was calculated as follows:
where Ss is the slope of the sample and St is the slope of trolox.
3. Discussion
The results of the biological evaluation of the 21 synthesized compounds considering the α-glucosidase inhibitory and antioxidant activities are summarized in Table 1.
Table 1.
α-Glucosidase inhibitory and antioxidant activities.
| Compounds | α-Glucosidase | Antioxidant | |
|---|---|---|---|
| IC50 | EC50 | ||
| Mono- | |||
| 2-galloyl (Ginnalin C) [31] 31 | 95.1 ± 0.22 | 19.40 ± 1.02 | 2.04 ± 0.08 |
| 3-galloyl (Maplexin A) [29] 32 | 137.9 ± 1.50 | 20.50 ± 2.05 | 2.38 ± 0.24 |
| 4-galloyl (Maplexin B) [29] 33 | 143.9 ± 0.52 | 20.80 ± 1.54 | 2.09 ± 0.19 |
| 6-galloyl (Ginnalin B) [31] 34 | 127.1 ± 0.88 | 22.90 ± 1.61 | 2.13 ± 0.12 |
| Di- | |||
| 2,3-galloyl (Maplexin C) [29] 35 | 20.50 ± 0.50 | 11.30 ± 0.98 | 4.29 ± 0.23 |
| 2,4-galloyl (Maplexin D) [29] 36 | 48.30 ± 1.25 | 14.30 ± 0.29 | 3.36 ± 0.13 |
| 2,6-galloyl (Acertannin) [25] 37 | 35.60 ± 2.58 | 15.20 ± 0.53 | 3.32 ± 0.37 |
| 3,4-galloyl 38 | 91.40 ± 6.56 | 14.80 ± 0.56 | 3.55 ± 0.42 |
| 3,6-galloyl 39 | 55.00 ± 8.84 | 11.90 ± 0.71 | 3.67 ± 0.19 |
| 4,6-galloyl 40 | 26.60 ± 2.37 | 13.00 ± 1.12 | 3.78 ± 0.33 |
| Tri- | |||
| 2,3,4-galloyl 41 | 6.72 ± 0.21 | 7.94 ± 0.39 | 5.59 ± 0.26 |
| 2,3,6-galloyl (Maplexin F) [30] 42 | 5.34 ± 0.55 | 8.09 ± 0.41 | 5.75 ± 0.18 |
| 2,4,6-galloyl (Maplexin E) [29] 43 | 9.34 ± 0.99 | 10.30 ± 0.76 | 4.31 ± 0.35 |
| 3,4,6-galloyl 44 | 12.60 ± 0.61 | 8.48 ± 0.21 | 5.36 ± 0.20 |
| Tetra- | |||
| 2,3,4,6-galloyl (Maplexin J) [32] 54 | 2.56 ± 0.10 | 6.61 ± 0.68 | 6.77 ± 0.63 |
| 3′,4′-dihydroxybenzoyl 55 | 3.28 ± 0.16 | 6.99 ± 0.11 | 6.54 ± 0.15 |
| 3′,5′-dihydroxybenzoyl 56 | 9.34 ± 0.02 | 70< | n.d |
| 3′-hydroxybenzoyl 57 | 78< | n.d | |
| 4′-hydroxybenzoyl 58 | 78< | n.d | |
| 1-OH-2,3,4,6-galloyl 61 | 1.68 ± 0.21 | 5.60 ± 0.28 | 8.33 ± 0.49 |
| 2,3-galloyl-4,6-HHDP 67 | 3.22 ± 0.51 | 6.79 ± 0.21 | 6.44 ± 0.24 |
| Tellimagrandin I | 3.37 ± 0.04 | 6.31 ± 0.27 | 7.04 ± 0.37 |
| Methyl gallate | 90.50 ± 6.97 | 19.60 ± 0.11 | 2.42 ± 0.27 |
| Methyl 3,4-dihydroxybenzoate | 300<< | 21.10 ± 1.08 | 2.20 ± 0.05 |
| Methyl 3,5-dihydroxybenzoate | 300<< | n.d | n.d |
| Methyl 3-hydroxybenzoate | 660<< | n.d | n.d |
| Methyl 4-hydroxybenzoate | 660<< | n.d | n.d |
| Acarbose | 0.11 ± 0.02 | - | - |
| Trolox | - | 49.40 ± 3.87 | - |
IC50 data represents mean ± S.D. of n = 2. EC50 and trolox-eq data represents mean ± S.D. of n = 3. Novel compound. Did not dissolve in water. n.d.: not detected.
The comparison of the 1,5-AG-based polyphenol analogs 31–44 and 54 revealed that the α-glucosidase inhibitory activity significantly increased with the number of the galloyl units in the compounds and the highest inhibition was observed for tetra-O-galloyl-1,5-AG (maplexin J) 54 (IC50 = 2.56 μM). This result is in accordance with the previous reports [32]. In addition, different the position of the galloyl unit on the 1,5-AG core appeared to influence the α-glucosidase inhibitory activity, even for the compounds with the same number of these moieties. Analogous outcomes were noted for 2-galloyl-1,5-AG (31) and methyl gallate (IC50 = 95.1, 90.5 μM, respectively). Conversely, differing α-glucosidase inhibitory activities were obtained for mono-galloyl analogs 32–34, which are weaker inhibitors than methyl gallate (IC50 = 127.1–143.9 μM). Furthermore, compound 33 (galloyl unit at the C-4 position) exhibited lowest activity (IC50 = 143.9 μM). Higher inhibitory activity was detected for di-galloylated analogs 35–37 (IC50 = 20.5–48.3 μM), which possessed the galloyl unit at the C-2 position than for analogs 38–40 (IC50 = 26.6–91.4 μM). In particular, compound 35 that contains esterified galloyl units at the C-2 and C-3 positions exhibited three-times higher inhibitory activity than gallic acid. Moreover, analog 38, galloylated at the C-3 and C-4 positions, displayed significantly lower activity (IC50 = 91.4). Among the tri-galloyllated analogs, compound 42 without a galloyl unit at the C-4 position showed stronger inhibitory activity (IC50 = 5.34 μM) than analogs 41, 43, and 44 (IC50 = 6.72, 9.34, and 12.6 μM, respectively). In addition, analog 44 without a galloyl moiety at the C-2 position displayed weak activity. Thus, our results suggested that a galloyl unit at the C-2 position considerably increases the α-glucosidase inhibitory activity, while the presence of this group at the C-4 position causes a decrease in the activity.
Subsequently, we compared maplexin J (54) and its analogs (55 and 56) to elucidate the effect of the phenolic hydroxyl group. The 3′,4′-di-hydroxybenzoyl analog 55 exhibited good inhibitory activity, whereas the 3′,5′-di-hydroxybenzoyl analog 56 was a weaker inhibitor of the α-glucosidase enzyme (IC50 = 3.28, 9.34 μM, respectively). Consequently, these results implied that the presence of two adjacent phenolic hydroxyl groups is essential for the desired activity. We then focused on the evaluation of the influence of the hemiacetal hydroxyl and the HHDP functionalities against the α-glucosidase inhibitory activity. The effect of the hemiacetal hydroxyl moiety can be observed by the comparison of the activity of maplexin J (54) and its analog 61 (IC50 = 1.68, 2.56 μM, respectively). Furthermore, tellimagrandin I and 67 showed analogous inhibitory activity (IC50 = 3.37, 3.22 μM, respectively). Therefore, the obtained outcomes suggested that the presence of a hemiacetal hydroxyl group did not have a significant effect on the α-glucosidase inhibitory activity. Lastly, to examine the influence of the HHDP group, the results for maplexin J (54) and the 4,6-O-HHDP analog 67 were compared and it transpired that maplexin J (54) displayed marginally higher activity than its analog 67 (IC50 = 2.56, 3.22 μM, respectively). Likewise, the assessment of the tellimagrandin I activity and the activity of analog 61 without the HHDP group revealed that 61 was a stronger inhibitor than tellimagrandin I (IC50 = 3.37, 1.68 μM, respectively). Intriguingly, our results suggested that the 4,6-O-HHDP group has a weakening effect on the α-glucosidase inhibitory activity [38,62].
Meanwhile, we also investigated the antioxidant activity of these polyphenols and their analogs. Firstly, the mono-galloylated analogs 31–34 showed nearly equivalent antioxidative activity with methyl gallate (EC50 = 19.4–22.9 μM, TE = 2.04–2.42). Moreover, the activity improved as the number of galloyl units increased, with di-galloylated analogs (EC50 = 11.9–15.2 μM, TE = 3.32–4.29) exhibiting lower activity than the tri-galloylated analogs (EC50 = 7.94–10.3 μM, TE = 4.31–5.75) and the tetra-galloylated analogs 54, 61 displaying the highest activity (EC50 = 5.60, 6.61 μM, TE = 8.33, 6.77, respectively). Unlike the significant increase observed for the α-glucosidase inhibitory activity, the antioxidant activity increased proportionally to the number of galloyl units. Moreover, the antioxidant activity was not affected by the position of the galloyl units on the carbohydrate core. In addition, the comparison of compounds 54, 61, and 67 with tellimagrandin I revealed similar activity (EC50 = 6.61, 5.60, 6.79, and 6.31 μM, TE = 6.77, 8.33, 6.44, and 7.04, respectively). This implied that the antioxidative activity was not influenced by the presence of a hemiacetal hydroxyl and the 4,6-O-HHDP groups. The presence of the phenolic hydroxyl groups in 3,4-dihydroxybenzoyl analogs 55 and methyl 3,4-dihydroxybenzoate appeared to result in improved antioxidative activity (EC50 = 6.99 μM, TE = 6.54); however, no antioxidant activity was observed for 3,5-dihydroxybenzoyl analog 56 and the monohydroxybenzoyl analogs (57 and 58). Likewise, the activity of methyl 3,4-dihydroxybenzoate was comparable with methyl gallate (EC50 = 21.1 μM, TE = 2.20), whereas methyl 3,5-dihydroxybenzoate, methyl 3- and 4-hydroxybenzoate did not exhibit any notable activity. This data therefore suggested that the presence of two adjacent phenolic hydroxyl groups is necessary for the antioxidant activity, as was the case with α-glucosidase inhibitory activity.
4. Conclusions
We synthesized 21 carbohydrate-based phenolic analogs including a series of compounds containing all possible combinations of galloylation on 1,5-AG. The α-glucosidase inhibition and antioxidant activities of these compounds were further studied to evaluate the SAR. Our results suggested that the α-glucosidase inhibitory activity; 1) is significantly enhanced with the increasing number of galloyl units, and changing the position of the galloyl moiety substitution on the 1,5-AG unit tends to affect the activity; particularly, the presence of this functionality at the C-2 position improves the α-glucosidase inhibition, whereas substitution at the C-4 position reduces it, 2) requires two adjacent phenolic hydroxyl groups, 3) is not affected by the presence of the biaryl bond on the 4,6-O-HHDP group, 4) is not influenced by the hemiacetal hydroxyl functionality on the carbohydrate unit. Moreover, the following trends were determined for the antioxidant activity; 1) the activity is dependent on the number of galloyl units; however, it is not affected by their position, 2) the presence of two adjacent phenolic hydroxyl groups is significant, 3) the activity is not affected by the HHDP group or the hemiacetal hydroxyl group. The α-glucosidase inhibitory activity is undoubtedly affected by the position of the galloyl group on 1,5-AG. This outcome indicates that the carbohydrate core is not only a store unit for the galloyl moiety but can also act as a carrier to the biological targets. Thus, derivatives modified at the C-2 or C-4 positions on the carbohydrate unit, which is not d-glucose, have the potential to exhibit stronger antidiabetic activity. The synthesis and profiling of further analogs will be reported in due course.
5. Materials and Methods
5.1. General Information
Proton nuclear magnetic resonance ( NMR) spectra were recorded on JEOL JNM-ECX600 spectrometers. Chemical shifts are reported relative to internal standard (tetramethylsilane; 0.00, ; 7.26). Data are presented as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant and integration. Carbon nuclear magnetic resonance NMR) spectra were recorded on JEOL JNM-ECX600 (150 MHz) spectrometers. The following internal reference was used: (tetramethylsilane: δ 0.00, δ 77.0, ; δ 29.8, ; δ 49.0). Optical rotations were measured on a JASCO P-1030 digital polarimeter at the sodium D line (589 nm). Electron impact (EI) mass analyses and fast atom bombardment (FAB) mass analyses were carried out with a JEOL JMS-GCMATE. C.C. was carried out on Kanto Silica gel 60N spherical (63–210 mesh). Analytical thin-layer chromatography (TLC) was carried out using Merck Kieselgel 60 F254 plates with visualization by ultraviolet light or stained by 8% /EtOH solution on hot-plate. Tellimagrandin I was purchased from Nagara Science (Gifu, Japan). Methyl gallate, methyl 3,4-dihydroxybenzoate, methyl 3,5-dihydroxybenzoate, methyl 3-hydroxybenzoate, methyl 4-hydroxybenzoate, Trolox and MES were purchased from TOKYO CHEMICAL INDUSTRY CO., LTD (Tokyo, Japan). DPPH was purchased from Sigma-Aldrich Japan (Tokyo, Japan).
5.2. Chemical Synthesis
2-O-Benzyl-4,6-O-benzylidene-1,5-anhydro-d-glucitol (3) [35] and 3-O-Benzyl-4,6-O-benzylidene-1,5-anhydro-d-glucitol (4) [35]; Compound 1 (2.5 g, 10 mmol) and (0.68 g, 2.0 mmol) in 160 mL of DCM and 14 mL of 5% NaOH was stirred at rt. Then, BnBr (2.1 mL, 17 mmol) was slowly added, and the mixture was refluxed for 30 h. After the addition 50 mL of water, the mixture was extracted with DCM (3 × 100). The combined organic layers were washed with brine, dried over , filtered and concentrated under reduced pressure. The crude product was separated by C.C (Hex/EtOAc = 4/1) to obtain 2-O-Bn compound 3 (2.0 g, 59%) as colorless needles and 3-O-Bn compound 4 (1.0 g, 30% yield) as a white solid. Compound 3: m.p. 163 °C; = −3.16 (c 1.0, ); NMR (, 600 MHz) δ 7.49–7.47 (m, 2H), 7.38–7.29 (m, 8H), 5.50 (s, 1H), 4.76, 4.67 (ABq, J = 11.7 Hz, 2H), 4.30 (dd, J = 10.5, 5.0 Hz, 1H), 4.01 (dd, J = 11.3, 5.5 Hz, 1H), 3.84 (m, 1H), 3.65 (t, J = 10.3 Hz, 1H), 3.59–3.55 (m, 1H), 3.45 (t, J = 9.3 Hz, 1H), 3.35 (m, 1H), 3.30 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 138.02, 137.05, 129.23, 128.56, 128.33, 128.03, 127.89, 126.29, 101.88, 81.05, 77.74, 74.81, 73.43, 70.91, 68.79, 68.45. Compound 4: m.p. 137 °C; = +5.3 (c 1.0, ); NMR ( 600 MHz) δ 7.50–7.48 (m, 2H), 7.41–7.29 (m, 8H), 5.58 (s, 1H), 5.03, 4.72 (ABq, J = 11.3 Hz, 2H), 4.34 (dd, J = 10.5, 5.0 Hz, 1H), 4.06 (dd, J = 11.2, 5.7 Hz, 1H), 3.80–3.76 (m, 1H), 3.72 (t, J = 10.3 Hz, 1H), 3.66 (t, J = 9.1 Hz, 1H), 3.58 (t, J = 8.8 Hz, 1H), 3.44–3.40 (m, 1H), 3.34 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 138.30, 137.32, 129.00, 128.61, 128.30, 128.13, 128.01, 125.98, 101.21, 82.66, 82.13, 74.71, 71.53, 69.91, 69.79, 68.88.
2,3-Di-O-benzyl-1,5-anhydro-d-glucitol (5) [42]; Compound 2 (0.86g, 2.0 mmol) in 10 mL of 80% AcOH/ solution was stirred at 80 °C for 5 h. After the addition 5 mL of saturated aq. , the reaction solution was extracted by EtOAc (3 × 30). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by recrystallization (EtOAc/Hex) to obtain 5 (0.65 g, 95%) as colorless needles. m.p. 129 °C; = −10.4 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.29 (m, 10H), 5.03, 4.70 (ABq, J = 11.7 Hz, 2H), 4.64, 4.03 (ABq, J = 11.3, 2H), 3.86–3.82 (m, 1H), 3.72–3.68 (m, 1H), 3.63–3.59 (m, 1H), 3.50–3.44 (m, 2H), 3.28–3.23 (m, 2H); NMR (, 150 MHz) δ 138.54, 137.97, 128.67, 128.53, 127.98, 127.93, 127.83, 85.30, 79.53, 78.29, 75.10, 73.07, 70.45, 67.94, 62.89.
1,5-Anhydro-2-O-benzyl-d-glucitol (6) [35]; Compound 3 (860 mg, 2.5 mmol) in 10 mL of 80% AcOH/ solution was stirred at 80 °C for 5 h. By the same procedure previously described for the preparation of compound 5, compound 6 (503 mg, 76%) was obtained as colorless needles. m.p. 131 °C; = +8.4 (c 1.00, MeOH); NMR (, 600 MHz); δ 7.40–7.36 (m, 2H), 7.33–7.31 (m, 2H), 7.28–7.25 (m, 1H), 4.75, 4.64 (ABq, J = 11.7 Hz, 2H), 3.96 (dd, J = 11.0, 5.2 Hz, 1H), 3.81 (dd, J = 11.9, 2.2 Hz, 1H), 3.59 (dd, J = 11.9, 6.0 Hz, 1H), 3.43 (t, J = 8.9 Hz, 1H), 3.39–3.35 (m, 1H), 3.26–3.22 (m, 1H), 3.15–3.11 (m, 2H); NMR (, 150 MHz); δ 140.09, 129.37, 129.05, 128.76, 82.42, 79.31, 79.27, 74.14, 71.92, 68.95, 63.07.
1,5-Anhydro-3-O-benzyl-d-glucitol (7); Compound 4 (680 mg, 2.0 mmol) in 10 mL of 80% AcOH/ solution was stirred at 80 °C for 5 h. By the same procedure previously described for the preparation of compound 5, compound 7 (410 mg, 80%) was obtained as colorless needles. m.p. 153 °C; = +28.6 (c 1.00, MeOH); NMR (, 600 MHz); δ 7.44 (m, 2H), 7.32–7.30 (m, 2H), 7.26–7.23 (m, 1H), 4.90 (overlap, 2H), 3.89 (dd, J = 11.3, 5.5 Hz, 1H), 3.83 (dd, J = 11.9, 2.2 Hz, 1H), 3.62–3.57 (m, 2H), 3.37 (dd, J = 18.0, 8.71 Hz, 1H), 3.31–3.28 (overlap, 1H), 3.22–3.16 (m, 2H); NMR (, 150 MHz); δ 140.53, 129.20, 129.09, 128.49, 88.20, 82.68, 76.10, 71.67, 71.54, 71.09, 63.05. HRMS (ESI, m/z): , calcd for : 277.1052; found 277.1052.
1,5-Anhydro-4-O-benzyl-d-glucitol (8) [35]; Compound 1 (0.76 g, 3.0 mmol) in 15 mL of DCM was stirred at 0 °C. Then, 15 mL of borane-THF (ca. 1M THF solution) and TMSOTf (0.11 mL, 0.60 mmol) were successively added to the mixture. The mixture was allowed to stir for 4 h and added MeOH carefully. After the addition 1 mL of saturated aq. , the reaction solution was extracted with DCM (5 × 40 mL). The combined organic layer was dried over , filtered and concentrated under reduced pressure. The crude product was purified by C.C. /MeOH = 8/1) to obtain 8 (0.64 g, 74%) as a white solid. = +27.7 (c 0.45, ); NMR (, 600 MHz) δ 7.38–7.25 (m, 5H), 4.94, 4.64 (ABq, J = 11.0 Hz, 2H), 3.89 (dd, J = 13.3, 5.4 Hz, 1H), 3.78 (dd, J = 12.0, 2.1 Hz, 1H), 3.60 (dd, 1H, J = 11.4, 5.73 Hz,), 3.49–3.46 (m, 2H), 3.33–3.30 (overlap, 1H), 3.20 (ddd, J = 9.7, 5.2, 2.1 Hz, 1H), 3.14 (t, J = 10.7, 1H); NMR (, 150 MHz); δ 140.09, 129.33, 129.14, 128.70, 81.75, 80.43, 79.50, 75.90, 71.77, 70.93, 62.72.
2,4-Di-O-benzyl-1,5-anhydro-d-glucitol (9) [46]; Compound 3 (1.0 g, 3.0 mmol) in 15 mL of DCM was successively added 15 mL of borane-THF (ca. 1M THF solution) and TMSOTf (0.11 mL, 0.6 mmol) at 0 °C. The mixture was allowed to stir at rt for 10 h and added 1 mL of MeOH carefully. After the addition 1 mL of saturated aq. , the mixture was extracted with DCM (3 × 40 mL). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by recrystallization (Hex/EtOAc) to obtain 9 (820 mg, 82%) as colorless needles. m.p. 112 °C; = +34.6 (c 1.00, ); NMR (, 600 MHz) δ 7.37–7.28 (m, 10H), 4.86, 4.70 (ABq, J = 11.3 Hz, 2H), 4.65 (s, 2H), 3.99 (dd, J = 11.3, 5.2 Hz, 1H), 3.84 (ddd, J = 11.8, 5.8, 2.7 Hz, 1H), 3.74 (td, J = 8.9, 2.1 Hz, 1H), 3.68–3.64 (m, 1H), 3.45–3.41 (m, 1H), 3.41 (t, J = 9.2 Hz, 1H), 3.26 (ddd, J = 9.6, 4.5, 2.7 Hz, 1H), 3.19 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 138.18, 138.00, 128.60, 128.56, 128.09, 127.98, 127.85, 79.34, 78.18, 77.97, 77.39, 74.74, 73.05, 67.44, 62.27.
3,4-Di-O-benzyl-1,5-anhydro-d-glucitol (10) [47]; Compound 4 (850 g, 2.5 mmol) in 15 mL of DCM was successively added 12.5 mL of borane-THF (ca. 1M THF solution) and TMSOTf (90 μL, 0.5 mmol) at 0 °C. The mixture was allowed to stir at rt for 7 h and added 5 mL of MeOH carefully. After the addition 1 mL of saturated aq. , the mixture was extracted with DCM (3 × 40 mL). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by recrystallization (Hex/EtOAc) to obtain 10 (523 mg, 61%) as colorless needles. m.p. 99 °C; = +48.3 (c 1.40, ); NMR (, 600 MHz) δ 7.38–7.30 (m, 10H), 4.97, 4.77 (ABq, J = 11.3 Hz, 2H), 4.86, 4.68 (ABq, J = 11.0 Hz, 2H), 3.98 (dd, J = 11.2, 5.3 Hz, 1H), 3.84 (ddd, J = 11.9, 5.5, 2.6 Hz, 1H), 3.71–3.65 (m, 2H), 3.52 (t, J = 9.1 Hz, 1H), 3.47 (t, J = 8.8 Hz, 1H), 3.32–3.29 (m, 1H), 3.23 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 138.45, 137.84, 128.71, 128.56, 128.02, 127.86, 86.73, 79.92, 77.79, 75.25, 74.95, 70.16, 69.30, 61.99.
2,3,4-Tri-O-benzyl-1,5-anhydro-d-glucitol (11) [46]; Compound 2 (860 mg, 2.0 mmol) in 10 mL of toluene was stirred at rt. The reaction solution was added DIBAL-H (ca. 1M toluene solution, 6 mL) and stirred at rt for 20 h. The reaction solution was slowly added 4.2 mL of MeOH and 7.2 mL of 30% Rochelle salt solution and stirred for 1 h. After the addition 20 mL of EtOAc, the mixture was extracted with 30% Rochelle salt solution (3 × 15 mL). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C. (Hex/EtOAc = 4/1–2/1) to obtain 11 (760 mg, 90% yield) as colorless needles. m.p. 83 °C; = +8.9 (c 0.90, ); NMR (, 600 MHz) δ 7.37–7.28 (m, 15H), 4.98–4.64 (m, 6H), 3.99 (dd, J = 11.3, 5.2 Hz, 1H), 3.84–3.81 (m, 1H), 3.67–3.58 (m, 3H), 3.48 (t, J = 9.3 Hz, 1H), 3.29–3.25 (m, 1H), 3.23 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 138.61, 138.09, 138.01, 128.48, 128.41, 128.05, 127.89, 127.83, 127.64, 86.17, 79.69, 78.55, 77.57, 75.55, 75.13, 73.34, 67.96, 62.25.
1,5-Anhydro-6-O-benzyl-d-glucitol (12) [35]; Compound 1 (760 mg, 3.0 mmol) in 15 mL of DCM was added triethylsilane (2.4 mL, 15 mmol) and trifluoracetic acid (1.2 mL, 15 mmol) at 0 °C. The reaction solution was stirred at rt for 8 h. After the addition 10 mL of saturated aq. , the mixture was extracted with DCM (5 × 30 mL). The combined organic layer was dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (/MeOH = 100/1–10/1) to obtain 12 (471 mg, 62%) as a colorless oil. = +8.9 (c 0.30, ); NMR (, 600 MHz) δ 7.35–7.25 (m, 5H), 4.55 (d, J = 2.1 Hz, 2H), 3.88 (dd, J = 11.0, 5.5 Hz, 1H), 3.77 (dd, J = 10.8, 1.9 Hz, 1H), 3.60 (dd, J = 10.8, 5.7 Hz, 1H), 3.48–3.42 (m, 1H), 3.32–3.24 (overlap, 3H), 3.15 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 139.57, 129.34, 128.90, 128.67, 81.38, 79.95, 74.48, 71.85, 71.31, 71.19, 70.95.
2,3,6-Tri-O-benzyl-1,5-anhydro-d-glucitol (13) [49]; Compound 2 (860 mg, 2.0 mmol) in 10 mL of DCM was added triethylsilane (1.6 mL, 10 mmol) and tifluoroacetic acid (0.8 mL, 10 mmol) at 0 °C. The reaction solution was stirred at rt for 15 h. After the addition 5 mL of saturated aq. , the mixture was extracted with DCM (3 × 30 mL). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude product was purified by C.C. (Hex/EtOAc = 4/1) to obtain 13 (505 mg, 59%) as a colorless oil. = −7.0 (c 1.00, ); NMR (, 600 MHz) δ 7.37–7.25 (m, 15H), 5.00, 4.76 (ABq, J = 11.5 Hz, 2H), 4.69, 4.63 (ABq, J = 11.7 Hz, 2H), 4.58, 4.54 (ABq, J = 12.2 Hz, 2H), 4.04 (dd, J = 11.3, 5.2 Hz, 1H), 3.70 (dd, J = 10.5, 3.3 Hz, 1H), 3.64–3.60 (m, 2H), 3.54 (td, J = 9.2, 2.1 Hz, 1H), 3.44 (t, J = 8.9 Hz, 1H), 3.37–3.34 (m, 1H), 3.23 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 138.64, 138.05, 137.84, 128.58, 128.48, 128.39, 127.95, 127.90, 127.87, 127.83, 127.77, 127.70, 85.40, 78.75, 78.04, 75.10, 73.65, 73.05, 70.87, 69.98, 68.06.
2,6-Di-O-benzyl-1,5-anhydro-d-glucitol (14); Compound 3 (860 mg, 2.5 mmol) in 15 mL of DCM was added triethylsilane (2.0 mL, 12.5 mmol) and tifluoroacetic acid (1.0 mL, 12.5 mmol) at 0 °C. The reaction solution was stirred at rt for 8 h. After the addition 10 mL of saturated aq. , the mixture was extracted with DCM (3 × 40 mL). The organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude product was purified by C.C. /MeOH = 8/1) to obtain 14 (500 mg, 58%) as a white solid. = +17.4 (c 1.00, ); NMR (, 600 MHz) δ 7.36–7.27 (m, 10H), 4.64 (s, 2H), 4.59, 4.54 (ABq, J = 12.0 Hz, 2H), 4.02 (dd, J = 11.2, 5.0 Hz, 1H), 3.69 (dd, J = 10.5, 3.6 Hz, 1H), 3.65 (dd, J = 10.5, 5.0 Hz, 1H), 3.55 (td, J = 8.8, 2.1 Hz, 1H), 3.50 (td, J = 9.0, 2.6 Hz, 1H), 3.47–3.43 (m, 1H), 3.37–3.34 (m, 1H), 3.19 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 137.96, 137.71, 128.59, 128.46, 128.07, 127.90, 127.81, 78.31, 77.74, 77.41, 73.68, 72.95, 71.34, 70.01, 67.70. HRMS (ESI, m/z): , calcd for : 367.1521; found 367.1521.
1,5-Anhydro-3,6-di-O-benzyl-d-glucitol (15); Compound 4 (680 mg, 2.0 mmol) in 15 mL of DCM was added triethylsilane (1.6 mL, 10 mmol) and trifluoroacetic acid (0.80 mL, 10 mmol) at 0 °C. The reaction solution was stirred at rt for 4 h. After the addition 10 mL of saturated aq. , the mixture was extracted with DCM (3 × 40 mL). The combined organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude product was purified by C.C. /MeOH = 8/1) to obtain 15 (420 mg, 62%) as a white solid. = +22.7 (c 1.00, ); NMR (, 600 MHz) δ 7.39–7.27 (m, 10H), 4.91, 4.83 (ABq, J = 11.7 Hz, 2H), 4.59, 4.54 (ABq, J = 12.2 Hz, 2H), 3.96 (dd, J = 11.2, 5.3 Hz, 1H), 3.72–3.66 (m, 3H), 3.61 (dt, J = 9.1, 2.6 Hz, 1H), 3.39–3.36 (m, 1H), 3.31 (t, J = 8.8 Hz, 1H), 3.21 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 138.60, 137.61, 128.70, 128.47, 128.02, 127.93, 127.86, 127.82, 86.47, 78.47, 74.87, 73.74, 72.11, 70.38, 69.79, 69.57. HRMS (ESI, m/z): , calcd for : 367.1521; found 367.1522.
1,5-Anhydro-2-O-(3′,4′,5′-tribenzyloxybenzoyl)-3-O-benzyl-4,6-O-benzylidene-d-glucitol (17); Compound 3 (340 mg, 1.0 mmol), compound 16 [39] (650 mg, 1.5 mmol), 2-chloro-1-methylpyridinium iodide (380 mg, 1.5 mmol), DMAP (37 mg, 0.30 mmol), TEA (416 μL, 3.0 mmol) in 15 mL of DCM was stirred at rt for 20 h. After the addition 100 mL of saturated aq. , the reaction solution was extracted with DCM (3 × 60 mL). The combined organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C. (DCM/MeOH = 500/1 − 200/1) to obtain 17 (350 mg, 41%) as a colorless amorphous oil. = +8.4 (c 1.04, ); NMR (, 600 MHz) δ 7.53–7.11 (m, 29H), 5.61 (s, 1H), 5.27–5.20 (m, 1H), 5.15 (s, 2H), 5.09 (s, 4H), 4.86, 4.71 (ABq, J = 12.0 Hz, 2H), 4.36 (dd, J = 10.3, 4.8 Hz, 1H), 4.19 (dd, J = 11.0, 5.8 Hz, 1H), 3.87 (t, J = 9.1 Hz, 1H), 3.79–3.74 (m, 2H), 3.45 (td, J = 9.7, 4.9 Hz, 1H), 3.36 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.06, 152.55, 142.74, 138.10, 137.31, 137.28, 136.56, 129.03, 128.57, 128.30, 128.25, 128.22, 128.07, 128.02, 127.89, 127.58, 127.47, 126.02, 124.50, 109.41, 101.32, 81.96, 79.12, 75.14, 74.33, 71.50, 71.30, 68.76, 67.68; HRMS (ESI, m/z): , calcd for : 787.2883; found 787.2881.
1,5-Anhydro-2-O-benzyl-3-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-O-benzylidene-d-glucitol (18); Compound 2 (0.51 g, 1.5 mmol), compound 16 (1.0 g, 2.3 mmol), 2-chloro-1-methylpyridinium iodide (0.59 g, 2.3 mmol), DMAP (28 mg, 0.23 mmol), TEA (0.62 mL, 4.5 mmol) in 20 mL of DCM was stirred at rt for 20 h. By the same procedure previously described for the preparation of compound 17, compound 18 (0.87 g, 68%) was obtained as a colorless amorphous oil. = −45.6 (c 0.95, ); NMR (, 600 MHz) δ 7.24–7.44 (m, 24H), 7.20–7.14 (m, 5H), 5.53 (t, J = 9.3 Hz, 1H), 5.46 (s, 1H), 5.15–5.10 (m, 6H), 4.55, 4.46 (ABq, J = 12.4 Hz, 2H), 4.35 (dd, J = 10.5, 5.0 Hz, 1H), 4.13 (dd, J = 11.3, 5.5 Hz, 1H), 3.76–3.72 (m, 1H), 3.70 (t, J = 10.3 Hz, 1H), 3.65 (t, J = 9.5 Hz, 1H), 3.51 (td, J = 9.7, 4.8 Hz, 1H), 3.48 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.16, 152.47, 142.58, 137.53, 137.39, 136.69, 128.95, 128.54, 128.38, 128.21, 128.16, 128.02, 127.98, 127.87, 127.53, 126.14, 125.05, 109.55, 101.35, 79.18, 75.62, 75.15, 74.94, 72.97, 71.48, 71.33, 68.80, 68.74; HRMS (ESI, m/z): , calcd for : 787.2883; found 787.2882.
1,5-Anhydro-2,3,6-tris-O-benzyl-4-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (19); Compound 13 (340 mg, 0.78 mmol), compound 16 (530 mg, 1.2 mmol), 2-chloro-1-methylpyridinium iodide (307 mg, 1.2 mmol), DMAP (95 g, 0.78 mmol), TEA (315 μL, 2.3 mmol) in 15 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 19 (607 mg, 91%) was obtained as a colorless amorphous oil. = −32.0 (c 0.95, ); NMR (, 600 MHz) δ 7.44–7.26 (m, 20H), 7.23–7.15 (m, 7H), 7.11–7.03 (m, 5H), 5.18–5.15 (m, 3H), 5.13–5.07 (m, 4H), 4.76, 4.54 (ABq, J = 11.3 Hz, 2H), 4.74, 4.65 (ABq, J = 11.7 Hz, 1H), 4.45 (s, 2H), 4.08 (dd, J = 11.3, 5.2 Hz, 1H), 3.75–3.71 (m, 1H), 3.65 (t, J = 9.1 Hz, 1H), 3.59–3.56 (m, 1H), 3.50 (dd, J = 10.8, 2.6 Hz, 1H), 3.45 (dd, J = 10.7, 5.8 Hz, 1H), 3.29 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 164.83, 152.41, 142.58, 138.03, 137.58, 137.37, 136.62, 128.57, 128.50, 128.23, 128.14, 128.06, 128.01, 127.94, 127.86, 127.83, 127.58, 127.49, 127.45, 124.67, 109.34, 83.08, 78.18, 78.08, 75.13, 74.94, 73.67, 73.40, 71.24, 69.47, 68.23; HRMS (ESI, m/z): , calcd for : 879.3509; found 879.3511.
1,5-Anhydro-2,3,4-tris-O-benzyl-6-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (20); Compound 11 (0.43 g, 1.0 mmol), compound 16 (0.66 g, 1.5 mmol), 2-chloro-1-methylpyridinium iodide (0.38 g, 1.5 mmol), DMAP (0.18 g, 1.5 mmol), TEA (0.42 mL, 3.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 20 (0.50 g, 58%) was obtained as a colorless amorphous oil. = +27.9 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.18 (m, 32H), 5.14–5.10 (m, 6H), 5.03, 4.88 (ABq, J = 10.7 Hz, 2H), 4,85, 4.53 (ABq, J = 11.8 Hz, 2H), 4.74, 4.67 (ABq, J = 11.7 Hz, 2H), 4.52 (dd, J = 12.0, 2.1 Hz, 1H), 4.37 (dd, J = 12.0, 4.5 Hz, 1H), 4.03 (dd, J = 11.3, 5.2 Hz, 1H), 3.69 (t, J = 8.8 Hz, 1H), 3.66–3.62 (m, 1H), 3.52–3.49 (m, 1H), 3.46 (t, J = 8.9 Hz, 1H), 3.23 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.82, 152.41, 142.46, 138.55, 138.04, 137.69, 137.37, 136.65, 128.59, 128.53, 128.48, 128.46, 128.20, 128.08, 128.01, 127.96, 127.93, 127.85, 127.76, 127.45, 124.91, 109.30, 86.33, 78.48, 77.83, 77.57, 75.73, 75.23, 75.09, 73.29, 71.17, 68.12, 63.86; HRMS (ESI, m/z): , calcd for : 879.3509; found 879.3509.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-O-benzylidene-d-glucitol (21); Compound 1 (380 mg, 1.5 mmol), compound 16 (2.0 g, 4.5 mmol), 2-chloro-1-methylpyridinium iodide (1.2 g, 4.5 mmol), DMAP (0.55 g, 4.5 mmol), TEA (1.3 mL, 9.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 21 (1.6 g, 88%) was obtained as a colorless amorphous oil. = +46.4 (c 1.00, ); NMR ( , 600 MHz) δ 7.44–7.42 (m, 6H), 7.34–7.31 (m, 26H), 7.25–7.22 (m, 4H), 5.80 (t, J = 9.5 Hz, 1H), 5.56 (s, 1H), 5.27–5.23 (m, 1H), 5.11–4.93 (m, 12H), 4.45–4.41 (m, 2H), 3.87 (t, J = 16.0 Hz, 1H), 3.82 (t, J = 17.0 Hz, 1H), 3.65–3.62 (m, 1H), 3.55 (t, J = 10.5 Hz, 1H); NMR (, 150 MHz) δ 165.6, 165.4, 152.6, 142.9, 142.8, 137.4, 136.8, 136.5, 129.1, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.5, 126.2, 109.3, 109.1, 101.6, 78.8, 75.1, 73.0, 71.9, 71.2, 71.2, 71.1, 68.7, 67.7; HRMS (ESI, m/z): , calcd for : 1119.3932; found 1119.3901.
1,5-Anhydro-2,4-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-3,6-bis-O-benzyl-d-glucitol (22); Compound 15 (0.30 mg, 0.87 mmol), compound 16 (1.3 g, 3.0 mmol), 2-chloro-1-methylpyridinium iodide (0.77 g, 3.0 mmol), DMAP (0.37 g, 3.0 mmol), TEA (0.83 mL, 6.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 22 (0.93 g, 77%) was obtained as a colorless amorphous oil. = +7.87 (c 1.00, ); NMR NMR (, 600 MHz) δ 7.58–7.14 (m, 45H), 7.08–6.95 (m, 5H), 5.32–5.25 (m, 2H), 5.19–5.08 (m, 13H), 4.61–4.45 (m, 4H), 4.29 (dd, J = 11.3, 5.5 Hz, 1H), 3.94 (t, J = 9.1 Hz, 1H), 3.72–3.68 (m, 1H), 3.58–3.53 (m, 2H), 3.39 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 164.87, 164.71, 152.61, 152.51, 142.88, 142.79, 137.54, 137.52, 137.32, 137.27, 136.56, 136.49, 128.59, 128.57, 128.55, 128.28, 128.24, 128.22, 128.16, 128.10, 128.05, 128.02, 127.85, 127.76, 127.65, 127.53, 127.49, 124.51, 109.43, 80.72, 78.29, 75.16, 74.16, 73.72, 72.12, 71.35, 71.32, 69.44, 67.03; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4554.
1,5-Anhydro-2,6-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-3,4-bis-O-benzyl-d-glucitol (23); Compound 10 (0.45 g, 1.3 mmol), compound 16 (1.8 g, 4.0 mmol), 2-chloro-1-methylpyridinium iodide (1.0 g, 4.0 mmol), DMAP (0.49 g, 4.0 mmol), TEA (1.1 mL, 8.0 mmol) in 30 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 23 (1.4 g, 92%) was obtained as a colorless amorphous oil. = +51.2 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.18 (m, 44H), 5.27–5.21 (m, 1H), 5.17–5.07 (m, 12H), 4.82, 4.52 (ABq, J = 11.0 Hz, 2H), 4.77, 4.70 (ABq, J = 11.3 Hz, 2H), 4.56 (m, 1H), 4.42–4.39 (m, 1H), 4.19 (dd, J = 11.2, 5.7 Hz, 1H), 3.85–3.82 (m, 1H), 3.60–3.56 (m, 2H), 3.31 (t, J = 10.7 Hz, 1H); NMR (, 150 MHz) δ 165.80, 165.05, 152.56, 152.43, 142.76, 142.47, 137.88, 137.49, 137.37, 137.28, 136.66, 136.53, 128.62, 128.60, 128.57, 128.51, 128.39, 128.21, 128.12, 128.06, 128.01, 127.88, 127.80, 127.40, 127.37, 124.80, 124.49, 109.32, 109.25, 84.32, 77.91, 77.42, 75.45, 75.26, 75.13, 75.09, 72.22, 71.26, 71.15, 67.11, 63.52; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4550.
1,5-Anhydro-2,6-bis-O-benzyl-3,4-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (24); Compound 14 (380 mg, 1.1 mmol), compound 16 (1.5 g, 3.3 mmol), 2-chloro-1-methylpyridinium iodide (0.84 g, 3.3 mmol), DMAP (0.91 g, 3.3 mmol), TEA (0.91 mL, 6.6 mmol) in 30 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 24 (1.2 g, 92%) was obtained as a colorless amorphous oil. = −80.8 (c 1.00, ); NMR (, 600 MHz) δ 7.41–7.14 (m, 44H), 5.58 (t, J = 9.5 Hz, 1H), 5.31 (t, J = 9.8 Hz, 1H), 5.09–5.00 (m, 12H), 4.54, 4.48 (ABq, J = 12.2 Hz, 2H), 4.52, 4.47 (ABq, J = 12.0 Hz, 2H), 4.19 (dd, J = 11.5, 5.3 Hz, 1H), 3.79 (td, J = 9.9, 5.2 Hz, 1H), 3.73 (m, 1H), 3.59 (dd, J = 10.7, 2.4 Hz, 1H), 3.51 (dd, J = 10.8, 5.3 Hz, 1H), 3.46 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.62, 165.18, 152.43, 142.67, 142.53, 137.56, 137.48, 137.43, 137.39, 136.57, 136.56, 128.49, 128.41, 128.38, 128.36, 128.27, 128.16, 127.98, 127.89, 127.87, 127.84, 127.65, 127.55, 124.67, 124.20, 109.15, 77.99, 76.35, 75.22, 75.10, 75.08, 73.70, 72.96, 71.12, 70.07, 69.06, 68.30; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4558.
1,5-Anhydro-2,4-bis-O-benzyl-3,6-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (25); Compound 9 (0.52 mg, 1.5 mmol), compound 16 (2.0 g, 4.5 mmol), 2-chloro-1-methylpyridinium iodide (1.2 g, 4.5 mmol), DMAP (0.55 g, 4.5 mmol), TEA (1.2 mL, 9.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 25 (1.6 g, 91%) was obtained as a colorless amorphous oil. = +16.1 (c 1.00, ); NMR (, 600 MHz) δ 7.43–7.24 (m, 34H), 7.18–7.05 (m, 10H), 5.50 (t, J = 9.3 Hz, 1H), 5.19–5.08 (m, 12H), 4.56, 4.43 (ABq, J = 12.4 Hz, 2H), 4.55 (dd, J = 12.0 Hz, 2.0 Hz, 1H), 4.45 (dd, J = 12.0, 5.5 Hz, 1H) 4.41 (s, 2H), 4.11 (dd, J = 11.3, 5.2 Hz, 1H), 3.67–3.62 (m, 2H), 3.54 (t, J = 9.5 Hz, 1H), 3.36 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.84, 165.22, 152.49, 142.66, 137.65, 137.31, 136.99, 136.59, 128.56, 128.36, 128.22, 128.16, 128.05, 127.94, 127.83, 127.52, 127.47, 124.96, 124.88, 109.54, 109.51, 78.19, 77.87, 76.42, 75.26, 75.13, 74.68, 72.63, 71.32, 67.97, 63.91; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4557.
1,5-Anhydro-2,3-bis-O-benzyl-4,6-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (26); Compound 5 (0.34 g, 1.0 mmol), compound 16 (1.3 g, 3.0 mmol), 2-chloro-1-methylpyridinium iodide (0.77 g, 3.0 mmol), DMAP (0.37 g, 3.0 mmol), TEA (0.83 mL, 6.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 26 (1.0 g, 87%) was obtained as a colorless amorphous oil. = +13.7 (c 1.00, ); NMR (, 600 MHz) δ 7.45–7.21 (m, 39H), 7.12–7.03 (m, 5H), 5.32 (t, J = 9.5 Hz, 1H), 5.15–5.08 (m, 8H), 5.03–5.01 (m, 4H), 4.79, 4.58 (ABq, J = 11.5, 2H), 4.76, 4.66 (ABq, J = 11.5 Hz, 2H), 4.63 (dd, J = 12.0, 2.8 Hz, 1H), 4.12 (dd, J = 12.2, 5.3 Hz, 1H), 4.07 (dd, J = 11.5, 5.0 Hz, 1H), 3.70–3.68 (m, 2H), 3.70 (t, J = 8.9 Hz, 1H), 3.30 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.80, 164.76, 152.48, 152.44, 142.77, 142.42, 137.98, 137.91, 137.47, 137.37, 136.73, 136.56, 128.53, 128.50, 128.47, 128.20, 128.17, 128.07, 128.03, 128.01, 127.94, 127.84, 127.60, 127.53, 124.75, 124.54, 109.40, 109.14, 82.95, 78.22, 76.56, 75.15, 75.08, 73.47, 71.24, 71.06, 70.96, 68.32, 63.44; HRMS (ESI, m/z): , calcd for : 1211.4558; found 1211.4553.
1,5-Anhydro-2,3,4-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-6-O-benzyl-d-glucitol (27); Compound 12 (180 mg, 0.70 mmol), compound 16 (1.4 g, 3.2 mmol), 2-chloro-1-methylpyridinium iodide (0.82 g, 3.2 mmol), DMAP (0.39 g, 3.2 mmol), TEA (0.89 mL, 6.4 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 27 (0.94 g, 91%) was obtained as a colorless amorphous oil. = −4.9 (c 0.65, ); NMR (, 600 MHz) δ 7.43–7.16 (m, 56H), 5.82 (t, J = 9.6 Hz, 1H), 5.55 (t, J = 10.0 Hz, 1H), 5.29 (td, J = 10.0, 5.5 Hz, 1H), 5.13–4.96 (m, 14H), 4.90 (s, 4H), 4.58, 4.53 (ABq, J = 12.0, 2H), (dd, J = 10.2, 5.6 Hz, 1H), 3.88–3.84 (m, 1H), 3.67 (dd, J = 11.0, 2.4 Hz, 1H), 3.61 (dd, J = 10.8, 5.3 Hz, 1H), 3.57 (t, J = 10.8 Hz, 1H) NMR (, 150 MHz) δ 165.92, 165.17, 165.02, 152.54, 152.50, 142.88, 142.82, 142.71, 137.44, 137.34, 136.49, 136.45, 136.36, 128.55, 128.51, 128.39, 128.32, 128.27, 128.17, 128.15, 128.10, 128.06, 128.02, 127.95, 127.92, 127.89, 127.81, 127.72, 127.56, 127.52, 124.08, 124.03, 109.20, 109.11, 109.02, 78.36, 75.12, 75.09, 75.07, 74.65, 73.77, 71.18, 71.10, 71.02, 70.75, 69.71, 69.00, 67.26; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5601.
1,5-Anhydro-2,3,6-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-4-O-benzyl-d-glucitol (28); Compound 8 (0.38 mg, 1.5 mmol), compound 16 (3.0 g, mmol), 2-chloro-1-methylpyridinium iodide (1.8 g, 7.0 mmol), DMAP (0.12 g, 1.0 mmol), TEA (1.9 mL, 14 mmol) in 30 mL of DCM was stirred at rt for 1 d. By the same procedure previously described for the preparation of compound 17, compound 28 (1.0 g, 45% yield) was obtained as a colorless amorphous oil. = +47.2 (c 0.40, ); NMR (, 600 MHz) δ 7.47–7.19 (m, 51H), 7.11–7.05 (m, 5H), 5.75 (t, J = 9.1 Hz, 1H), 5.19–5.03 (m, 15H), 5.00, 4.92 (ABq, J = 11.7 Hz, 4H), 4.60–4.53 (m, 2H), 4.45 (s, 2H), 4.41 (dd, J = 11.3, 5.5 Hz, 1H), 3.75–3.70 (m, 2H), 3.46 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.84, 165.51, 165.41, 152.59, 152.54, 142.94, 142.75, 142.65, 137.35, 137.32, 137.27, 136.86, 136.62, 136.51, 136.39, 128.60, 128.54, 128.46, 128.44, 128.38, 128.22, 128.18, 128.14, 128.08, 128.06, 128.01, 127.97, 127.90, 127.61, 127.50, 127.43, 124.81, 124.40, 124.05, 109.54, 109.29, 109.03, 78.11, 76.54, 76.04, 75.14, 75.11, 75.09, 74.96, 71.34, 71.20, 71.06, 70.81, 67.07, 63.58; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5604.
1,5-Anhydro-2,4,6-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-3-O-benzyl-d-glucitol (29); Compound 7 (0.33 mg, 1.3 mmol), compound 16 (2.6 g, 6.0 mmol), 2-chloro-1-methylpyridinium iodide (1.5 g, 6.0 mmol), DMAP (0.73 g, 6.0 mmol), TEA (1.7 mL, 12 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, compound 29 (1.3 g, 65% yield) was obtained as a colorless amorphous oil. = +26.7 (c 0.95, ); NMR (, 600 MHz) δ 7.46–7.21 (m, 49H), 7.07–6.96 (m, 5H), 5.48 (t, J = 9.5 Hz, 1H), 5.34–5.29 (m, 1H), 5.17–5.07 (m, 14H), 5.04 (s, 4H), 4.68 (dd, J = 12.0, 3.1 Hz, 1H), 4.62, 4.54 (ABq, J = 11.7 Hz, 2H), 4.30 (dd, J = 11.2, 5.7 Hz, 1H), 4.22 (dd, J = 12.2, 5.0 Hz, 1H), 3.99 (t, J = 9.1 Hz, 1H), 3.90–3.87 (m, 1H), 3.42 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.81, 164.83, 164.61, 152.63, 152.55, 152.46, 142.94, 142.49, 137.46, 137.41, 137.33, 137.24, 136.67, 136.50, 136.46, 128.57, 128.53, 128.51, 128.46, 128.21, 128.19, 128.14, 128.10, 128.06, 128.02, 127.95, 127.85, 127.80, 127.62, 127.56, 127.47, 124.64, 124.41, 124.38, 109.47, 109.44, 109.13, 80.62, 76.53, 75.16, 75.08, 74.34, 72.02, 71.35, 71.30, 71.05, 70.98, 67.05, 63.33; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5599.
1,5-Anhydro-2-O-benzyl-3,4,6-tris-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (30); Compound 6 (0.38 g, 1.5 mmol), compound 16 (3.0 g, 6.8 mmol), 2-chloro-1-methylpyridinium iodide (1.7 g, 6.8 mmol), DMAP (0.83 g, 6.8 mmol), TEA (2.0 mL, 14 mmol) in 30 mL of DCM was stirred at rt for 2 days. By the same procedure previously described for the preparation of compound 17, compound 30 (2.0 g, 89% yield) was obtained as a colorless amorphous oil. = −36.2 (c 1.00, ); NMR (, 600 MHz) δ 7.42–7.14 (m, 54H), 5.67 (t, J = 9.6 Hz, 1H), 5.41 (t, J = 9.8 Hz, 1H), 5.13–4.94 (m, 18H), 4.69 (dd, J = 12.2, 2.9 Hz, 1H), 4.59, 4.51 (ABq, J = 12.4 Hz, 2H), 4.23 (dd, J = 12.4, 5.5 Hz, 1H), 4.19 (dd, J = 11.5, 5.3 Hz, 1H) 3.92 (m, 1H), 3.83 (td, J = 9.8, 5.3 Hz, 1H), 3.48 (t, J = 11.0 Hz, 1H); NMR (, 150 MHz) δ 165.68, 165.65, 165.29, 152.51, 152.48, 152.40, 142.87, 142.66, 142.53, 137.54, 137.43, 137.38, 136.63, 136.46, 128.51, 128.46, 128.43, 128.38, 128.29, 128.15, 128.11, 127.98, 127.92, 127.89, 127.87, 127.80, 127.78, 127.57, 127.52, 124.65, 124.53, 123.97, 109.18, 76.69, 76.16, 75.17, 75.08, 72.95, 71.13, 71.03, 70.20, 68.38, 63.54; HRMS (ESI, m/z): , calcd for : 1543.5606; found 1543.5607.
1,5-Anhydro-2-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (31) [31]; on C (20 wt.%, 20 mg) was added to a solution of 17 (270 mg, 0.35 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. After the replaced argon atmosphere to hydrogen gas, the suspension was stirred at rt for 6 h. The reaction mixture was filtered and concentrated under reduced pressure to obtain purple amorphous oil. The purple amorphous oil was dissolved by 2 mL acetone and filtered through a whatman™ puradisc 0.1 μM TF and concentrated under reduced pressure. In addition, the purple amorphous oil dissolved with 2 mL of MeOH and added acidic resin until becoming a clear solution. After the filtered through the whatman™ puradisc 0.1 μm TF, the solution was concentrated under reduced pressure to give 31 (106 mg, 96%) as pale yellow amorphous oil. = +58.5 (c 0.70, MeOH); NMR (, 600 MHz) δ 7.08 (d, J = 5.5 Hz, 2H), 4.87–4.83 (overlap, 1H), 4.08 (dd, J = 10.5, 5.5 Hz, 1H), 3.86 (dd, J = 11.9, 2.2 Hz, 1H), 3.66–3.63 (m, 2H), 3.36 (t, J = 9.5 Hz, 1H), 3.29 (t, J = 10.7 Hz, 1H), 3.24 (ddd, J = 9.7, 5.9, 2.3 Hz, 1H). NMR (, 150 MHz) δ 167.80, 146.42, 139.92, 121.14, 110.25, 82.56, 77.06, 73.28, 71.97, 67.84, 62.93; HRMS (, m/z): , calcd for : 315.0716; found 315.0718. Please find NMR, NMR spectra of compounds 31–44, 54–58, 61 and 67 in the Supplementary Materials.
1,5-Anhydro-3-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (32) [29]; on C (20 wt.%, 20 mg) was added to a solution of compound 18 (270 mg, 0.35 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 32 (110 mg, quant.) was obtained as a colorless amorphous oil. = +24.8 (c 1.0 MeOH); NMR , 600 MHz) δ 7.13 (s, 2H), 5.04 (t, J = 9.3 Hz, 1H), 3.97 (dd, J = 11.3, 5.5 Hz, 1H), 3.85 (dd, J = 11.9, 2.2 Hz, 1H), 3.73–3.69 (m, 1H), 3.66 (dd, J = 12.0, 5.8 Hz, 1H), 3.51 (t, J = 9.5 Hz, 1H), 3.31–3.27 (overlap, 2H). NMR (, 150 MHz) δ 168.50, 146.39, 139.64, 121.88, 110.34, 82.51, 81.20, 70.95, 69.98, 69.89, 62.75; HRMS (, m/z): , calcd for : 315.0716; found 315.0721.
1,5-Anhydro-4-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (33) [29]; on C (20 wt.%, 20 mg) was added to a solution of compound 19 (280 mg, 0.33 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 33 (100 mg, 96%) was obtained as a colorless amorphous oil. = −5.7 (c 0.90, MeOH); NMR (600 MHz) δ 7.14 (s, 2H), 4.88 (t, J = 9.3 Hz, 1H), 3.94 (dd, J = 11.2, 5.3 Hz, 1H), 3.68 (t, J = 8.9 Hz, 1H), 3.64–3.60 (m, 1H), 3.57 (dd, J = 12.0, 2.1 Hz, 1H), 3.51–3.44 (m, 2H), 3.25 (t, J = 10.8 Hz, 1H); δ 166.59, 145.90, 138.86, 121.46, 110.07, 80.56, 77.44, 72.70, 71.38, 70.45, 62.54; HRMS (, m/z): , calcd for : 315.0716; found 315.0718.
1,5-Anhydro-6-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (34) [31]; on C (20 wt.%, 20 mg) was added to a solution of compound 20 (210 mg, 0.25 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 55 (74 mg, 96%) was obtained as a colorless amorphous oil. = +30.3 (c 1.00, MeOH); NMR (, 600 MHz) δ 7.09 (s, 2H), 4.54 (dd, J = 11.9, 1.9 Hz, 1H), 4.35 (dd, J = 12.0, 5.5 Hz, 1H), 3.93 (dd, J = 11.2, 5.3 Hz, 1H), 3.51–3.55 (m, 1H), 3.48–3.45 (m, 1H), 3.40 (t, J = 8.6 Hz, 1H), 3.37 (t, J = 8.6 Hz, 1H), 3.23 (t, J = 10.8 Hz, 1H). NMR (, 150 MHz) δ 168.32, 146.36, 139.71, 121.26, 110.09, 79.91, 79.63, 71.59, 71.20, 70.86, 65.02; HRMS (, m/z): , calcd for : 315.0716; found 315.0718.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (35) [29]; on C (20 wt.%, 50 mg) was added to a solution of compound 21 (210 mg, 0.25 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 35 (313 mg, quant.) was obtained as a colorless amorphous oil. = +139.7 (c 1.00, MeOH); NMR (, 600 MHz) δ 7.05 (s, 2H), 6.96 (s, 2H), 5.41 (t, J = 9.5 Hz, 1H), 5.08 (td, J = 10.1, 5.3 Hz, 1H), 4.20 (dd, J = 11.0, 5.5 Hz, 1H), 3.90 (dd, J = 11.9, 2.2 Hz, 1H), 3.74–3.69 (m, 2H), 3.45 (t, J = 10.8 Hz, 1H), 3.40 (ddd, J = 9.6, 5.5, 2.1 Hz, 1H); NMR (, 150 MHz) δ 168.18, 167.38, 146.38, 146.34, 140.08, 139.85, 121.29, 120.55, 110.30, 110.25, 82.65, 77.91, 71.39, 69.83, 67.81, 62.57; HRMS (, m/z): , calcd for : 467.0826; found 467.0831.
1,5-Anhydro-2,4-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (36) [29]; on C (20 wt.%, 50 mg) was added to a solution of compound 22 (680 mg, 0.57 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 36 (267 mg, quant.) was obtained as a colorless amorphous oil. = +11.3 (c 0.70, MeOH); NMR (600 MHz) δ 7.17 (s, 2H), 7.14 (dd, J = 9.8, 4.0 Hz, 2H), 5.04 (t, J = 9.5 Hz, 1H), 5.01–4.97 (m, 1H), 4.15–4.11 (m, 2H), 3.63–3.54 (m, 3H), 3.41 (t, J = 10.8 Hz, 1H); δ 166.39, 166.21, 146.00, 145.98, 138.99, 138.94, 121.43, 121.38, 110.15, 110.09, 80.72, 74.30, 73.19, 72.73, 67.38, 62.49; HRMS (, m/z): , calcd for : 467.0826; found 467.0827.
1,5-Anhydro-2,6-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (37) [26]; on C (20 wt.%, 50 mg) was added to a solution of compound 23 (440 mg, 0.37 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 37 (170 mg, quant.) was obtained as a yellow amorphous oil. = +19.4 (c 1.00 in MeOH); NMR (600 MHz) δ 7.16 (s, 2H), 7.14 (s, 2H), 4.92–4.88 (m, 1H), 4.57 (d, J = 10.7 Hz, 1H), 4.41–4.35 (m, 1H), 4.07 (dd, J = 10.8, 5.3 Hz, 1H), 3.83–3.78 (m, 1H), 3.61–3.57 (m, 2H), 3.37 (t, J = 10.7 Hz, 1H); δ 166.63, 166.30, 146.02, 145.96, 138.89, 138.77, 121.74, 121.46, 110.07, 109.85, 79.47, 76.57, 72.94, 71.60, 67.45, 64.54; HRMS (, m/z): , calcd for : 467.0826; found 467.0831.
1,5-Anhydro-3,4-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (38); on C (20 wt.%, 50 mg) was added to a solution of compound 24 (710 mg, 0.60 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 38 (280 mg, quant.) was obtained as a yellow amorphous oil. = −78.2 (c 1.00, MeOH); NMR (600 MHz) δ 7.06 (s, 2H), 7.04 (s, 2H), 5.40 (t, J = 9.5 Hz, 1H), 5.16 (t, J = 9.6 Hz, 1H), 4.06 (dd, J = 11.3, 5.5 Hz, 1H), 3.98–3.93 (m, 1H), 3.71–3.64 (m, 2H), 3.58 (dd, J = 12.5, 5.7 Hz, 1H), 3.45 (t, J = 10.8 Hz, 1H); δ 166.41, 166.12, 145.83, 145.72, 139.04, 138.66, 121.52, 120.78, 110.05, 110.01, 80.29, 78.04, 70.50, 70.17, 69.50, 62.25; HRMS (, m/z): , calcd for : 467.0826; found 467.0829.
1,5-Anhydro-3,6-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (39) [35]; on C (20 wt.%, 50 mg) was added to a solution of compound 25 (800 mg, 0.67 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 39 (310 mg, quant.) was obtained as a yellow amorphous oil. = +28.7 (c 0.60, MeOH); NMR (600 MHz) δ 7.17 (s, 2H), 7.16 (s, 2H), 5.11 (t, J = 9.1 Hz, 1H), 4.57 (dd, J = 11.9, 1.9 Hz, 1H), 4.40 (dd, J = 11.9, 5.3 Hz, 1H), 3.99 (dd, J = 11.0, 5.5 Hz, 1H), 3.84–3.79 (m, 1H), 3.72 (t, J = 9.5 Hz, 1H), 3.65–3.63 (m, 1H), 3.38 (t, J = 10.8 Hz, 1H); δ 166.90, 166.63, 145.97, 145.82, 138.71, 138.52, 122.16, 121.76, 110.10, 109.85, 81.10, 79.61, 70.73, 69.66, 69.46, 64.54; HRMS (, m/z): , calcd for : 467.0826; found 467.0828.
1,5-Anhydro-4,6-bis-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (40) [35]; on C (20 wt.%, 50 mg) was added to a solution of compound 26 (430 mg, 0.36 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 40 (160 mg, 95%) was obtained as a yellow amorphous oil. = +41.7 (c 0.60, MeOH); NMR (600 MHz) δ 7.15 (s, 2H), 7.14 (s, 2H), 5.09 (t, J = 9.5 Hz, 1H), 4.41 (dd, J = 12.0, 2.1 Hz, 1H), 4.13 (dd, J = 12.0, 5.8 Hz, 1H), 3.97 (dd, J = 11.2, 5.3 Hz, 1H), 3.82–3.79 (m, 1H), 3.75 (t, J = 9.1 Hz, 1H), 3.71–3.66 (m, 1H), 3.33 (t, J = 10.8 Hz, 1H); δ 166.47, 166.13, 145.92, 138.90, 138.77, 121.53, 121.38, 110.12, 109.91, 77.56, 77.42, 72.24, 71.28, 70.51, 69.79, 63.98, 55.32; HRMS (, m/z): , calcd for : 467.0826; found 467.0829.
1,5-Anhydro-2,3,4-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (41) [35]; on C (20 wt.%, 50 mg) was added to a solution of compound 27 (520 mg, 0.34 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 41 (210 mg, quant.) was obtained as a yellow amorphous oil. = −4.8 (c 1.00, MeOH); NMR (600 MHz) δ 7.07 (d, J = 3.8 Hz, 4H), 7.01 (d, J = 3.8 Hz, 2H), 5.77 (t, J = 9.6 Hz, 1H), 5.36 (t, J = 9.8 Hz, 1H), 5.23 (td, J = 10.1, 5.4 Hz, 1H), 4.28 (dd, J = 11.2, 5.7 Hz, 1H), 3.83–3.80 (m, 1H), 3.72 (dd, J = 12.4, 2.1 Hz, 1H), 3.62–3.65 (m, 2H); δ 166.12, 165.91, 165.89, 145.80, 145.65, 139.00, 138.76, 120.93, 120.70, 120.62, 110.03, 109.96, 109.91, 80.37, 74.46, 70.83, 69.96, 67.27, 61.99; HRMS (, m/z): , calcd for : 619.0935; found 619.0934.
1,5-Anhydro-2,3,6-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (42) [30]; on C (20 wt.%, 50 mg) was added to a solution of compound 28 (410 mg, 0.27 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 42 (170 mg, quant.) was obtained as a yellow amorphous oil. = +80.9 (c 0.70, MeOH); NMR (600 MHz) δ 7.19 (s, 2H), 7.10 (s, 2H), 7.04 (s, 2H), 5.50 (t, J = 9.5 Hz, 1H), 5.13–5.09 (m, 1H), 4.60 (dd, J = 11.9, 1.9 Hz, 1H), 4.47 (dd, J = 12.0, 4.8 Hz, 1H), 4.20 (dd, J = 11.0, 5.5 Hz, 1H), 3.93 (t, J = 9.5 Hz, 1H), 3.79–3.77 (m, 1H), 3.56 (t, J = 10.8 Hz, 1H) δ 166.60, 166.39, 166.03, 146.05, 145.96, 145.87, 139.15, 138.86, 138.81, 121.64, 121.56, 120.75, 110.06, 110.00, 109.89, 79.67, 77.07, 70.84, 69.60, 67.44, 64.23; HRMS (, m/z): , calcd for : 619.0935; found 619.0931.
1,5-Anhydro-2,4,6-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (43) [29]; on C (20 wt.%, 50 mg) was added to a solution of compound 29 (480 mg, 0.32 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. B By the same procedure previously described for the preparation of compound 31, desired compound 37 (200 mg, quant.) was obtained as a yellow amorphous oil. = +36.3 (c 1.00, MeOH); NMR (600 MHz) δ 7.17 (s, 2H), 7.16 (s, 2H), 7.15 (s, 2H), 5.25 (dd, J = 10.0, 9.3 Hz, 1H), 5.07–5.03 (m, 1H), 4.46 (dd, J = 12.2, 1.9 Hz, 1H), 4.20–4.16 (m, 3H), 3.95–3.92 (m, 1H), 3.50 (t, J = 10.8 Hz, 1H); δ 166.44, 166.19, 165.95, 145.98, 138.98, 138.83, 121.56, 121.35, 121.31, 110.20, 110.11, 109.96, 77.63, 74.27, 72.96, 72.22, 67.45, 63.76; HRMS (, m/z): , calcd for : 619.0935; found 619.0934.
1,5-Anhydro-3,4,6-tris-O-(3′,4′,5′-trihydroxybenzoyl)-d-glucitol (44) [35]; on C (20 wt.%, 50 mg) was added to a solution of compound 30 (765 mg, 0.50 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 37 (310 mg, quant.) was obtained as a colorless amorphous oil. = −6.12 (c 0.80, MeOH); NMR (600 MHz) δ 7.20 (s, 2H), 7.07 (s, 2H), 7.06 (s, 2H), 5.46 (t, J = 9.5 Hz, 1H), 5.34 (t, J = 9.8 Hz, 1H), 4.47 (dd, J = 12.4, 2.1 Hz, 1H), 4.27 (dd, J = 12.2, 5.3 Hz, 1H), 4.12 (dd, J = 11.3, 5.5 Hz, 1H), 4.04–4.00 (m, 2H), 3.55 (t, J = 10.8 Hz, 1H) δ 166.39, 166.35, 165.72, 145.83, 145.73, 145.66, 138.98, 138.74, 138.64, 121.44, 121.38, 120.67, 110.05, 109.97, 109.94, 77.86, 77.41, 70.54, 69.73, 69.41, 63.52; HRMS (, m/z): , calcd for : 619.0935; found 619.0941.
3,4-bis(Benzyloxy)benzoic acid (45) [51]; Methyl 3,4-dihydroxybenzoate (2.5 g, 15 mmol) and (8.2 g, 60 mmol) and KI (2.0 g, 12 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (4.2 mL, 36 mmol) and refluxed for 7 h. TLC indicated full conversion of the start material, added MeOH and stirred 1 h. The reaction mixture was filtered by celite and filtrate was evaporated under reduced pressure. The residue was purified by recrystallization with hexane, and the mother liquid was purified by C.C (Hex/EtOAc = 100/1–4/1) to afford methyl ester of 45 (total 5.0 g, 97%) as a white solid. Further on, methyl ester of 45 (3.0 g, 9.0 mmol), KOH (5.0 g, 90 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. TLC indicated full conversion of the start material, the reaction mixture was cooled to 0 °C and slowly added 6 M HCl until pH = 1. The precipitating muddy suspension was filtrated, and the white residue was washed by water and MeOH until pH = 7. The white solid was dried in vacuo, purified by recrystallizing with MeOH to obtain desired compound 45 (2.4 g, 79%) as colorless needles. m.p. 211 °C; NMR (DMSO-d6, 600 MHz) δ 7.56–7.15 (m, 13H), 5.22 (s, 2H), 5.18 (s, 2H); NMR (, 150 MHz) δ 166.88, 151.94, 147.50, 136.93, 136.62, 128.36, 128.30, 127.82, 127.71, 127.46, 127.34, 123.36, 123.22, 114.43, 112.97, 69.87, 69.73.
3,5-bis(Benzyloxy)benzoic acid (46) [53]; Methyl 3,5-dihydroxybenzoate (2.5 g, 15 mmol) and (8.2 g, 60 mmol) and KI (2.0 g, 12 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (4.2 mL, 36 mmol) and refluxed for 9 h. By the same procedure previously described for the preparation of compound 45, methyl ester of 46 (5.2 g, quant.) was obtained as a white solid. Further on, methyl ester of 46 (3.0 g, 9.0 mmol), KOH (5.0 g, 90 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. By the same procedure previously described for the preparation of compound 45, desire compound 46 (2.5 g, 81%) was obtained as colorless needles. m.p. 185 °C; NMR (, 600 MHz) δ 7.46–7.30 (m, 11H), 7.19–7.16 (m, 2H), 6.93–6.93 (m, 1H), 5.15 (s, 4H); NMR (, 150 MHz) δ 166.80, 159.30, 136.64, 132.80, 128.36, 127.79, 127.58, 107.87, 106.43, 69.38.
3-(Benzyloxy)benzoic acid (47) [52]; Methyl 3-hydroxybenzoate (2.3 g, 15 mmol) and (4.2 g, 30 mmol) and KI (1.0 g, 6.0 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (2.1 mL, 18 mmol) and refluxed for 10 h. By the same procedure previously described for the preparation of compound 45, methyl ester of 47 (3.7 g, 95%) was obtained as a white solid. Further on, methyl ester of 47 (1.9 g, 7.4 mmol), KOH (4.2 g, 74 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. By the same procedure previously described for the preparation of compound procedure described for previously 45 preparation, desire compound 47 (1.4 g, 85%) was obtained as colorless needles. m.p. 136 °C; NMR (600 MHz, DMSO-d6) δ 7.57–7.53 (m, 2H), 7.48–7.39 (m, 5H), 7.35–7.32 (m, 1H), 7.28–7.26 (m, 1H), 5.17 (s, 2H); -NMR (150 MHz, DMSO-d6) δ 167.12, 158.34, 136.84, 132.23, 129.79, 128.50, 127.92, 127.70, 121.82, 119.77, 114.89, 69.35.
4-(Benzyloxy)benzoic acid (48) [51]; Methyl 4-hydroxybenzoate (2.3 g, 15 mmol) and (4.2 g, 30 mmol) and KI (1.0 g, 6.0 mmol) in 100 mL of acetone was stirred at rt. The reaction suspension was slowly added BnCl (2.1 mL, 18 mmol) and refluxed for 9 h. By the same procedure previously described for the preparation of compound 45, methyl ester of 48 (3.6 g, quant.) was obtained as a white solid. Further on, methyl ester of 48 (2.3 g, 9.0 mmol), KOH (5.0 g, 90 mmol) in 90 mL of 1,4-dioxane and 30 mL of MeOH was stirred at 85 °C for 2 h. By the same procedure previously described for the preparation of compound procedure described for previously 45 preparation, desired compound 48 (1.4 g, 85%) was obtained as colorless needles. m.p. 192 °C; NMR (600 MHz, DMSO-d6) δ 7.95–7.90 (m, 2H), 7.48–7.46 (m, 2H), 7.42–7.40 (m, 2H), 7.36–7.34 (m, 1H), 7.12–7.09 (m, 2H), 5.19 (s, 2H); -NMR (150 MHz, DMSO-d6) δ 166.90, 161.83, 136.43, 131.27, 128.40, 127.92, 127.73, 123.09, 114.50, 69.35.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (49) [50]; 1,5-AG (82 mg, 0.50 mmol), compound 16 (1.1 g, 2.4 mmol), 2-chloro-1-methylpyridinium iodide (0.61 g, 2.4 mmol), DMAP (0.29 g, 2.4 mmol), TEA (0.67 mL, 4.8 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desire compound 49 (0.86 g, 92%) was obtained as a colorless amorphous oil. = +7.0 (c 1.00, ); NMR (600 MHz, CHLOROFORM-D) δ 7.56–7.07 (m, 68H), 5.89 (t, J = 9.8 Hz, 1H), 5.63 (t, J = 9.8 Hz, 1H), 5.35–5.31 (m, 1H), 5.18–4.94 (m, 22H), 4.88–4.84 (m, 4H), 4.76 (dd, J = 12.2, 2.9 Hz, 1H), 4.52 (dd, J = 11.3, 5.5 Hz, 1H), 4.30 (dd, J = 12.4, 5.2 Hz, 1H), 4.06–4.03 (m, 1H), 3.60 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.91, 165.71, 165.14, 165.10, 152.54, 152.43, 143.06, 142.93, 142.79, 142.62, 137.41, 137.31, 136.59, 136.44, 136.35, 136.25, 128.54, 128.45, 128.44, 128.37, 128.27, 128.23, 128.14, 128.11, 128.08, 128.02, 128.00, 127.95, 127.90, 127.87, 127.81, 127.55, 127.52, 124.56, 123.95, 123.79, 109.22, 109.14, 109.03, 75.10, 75.06, 74.53, 71.18, 71.11, 71.05, 70.51, 69.78, 67.31, 63.36 HRMS (ESI, m/z): , calcd for : 1875.6644; found 1875.6653.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,4′-dibenzyloxybenzoyl)-d-glucitol (50); 1,5-AG (0.12 g, 0.7 mmol), compound 45 (1.4 g, 4.2 mmol), 2-chloro-1-methylpyridinium iodide (1.1 g, 4.2 mmol), DMAP (0.52 g, 4.2 mmol), TEA (1.1 mL, 8.0 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desired compound 50 (0.61 g, 72%) was obtained as a colorless amorphous oil. = +18.8 (c 0.90, ); NMR (, 600 MHz) δ 7.65–7.23 (m, 48H), 6.89–6.86 (m, 2H), 6.82–6.80 (m, 1H), 6.77–6.75 (m, 1H), 5.81 (t, J = 9.8 Hz, 1H), 5.56 (t, J = 9.6 Hz, 1H), 5.29 (td, J = 10.1, 5.5 Hz, 1H), 5.22–5.03 (m, 14H), 4.99 (s, 2H), 4.63 (dd, J = 12.4, 2.7 Hz, 1H), 4.43 (dd, J = 11.3, 5.5 Hz, 1H), 4.29 (dd, J = 12.4, 5.2 Hz, 1H), 3.97–3.94 (m, 1H), 3.53 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.83, 165.69, 165.17, 164.91, 153.26, 153.14, 152.96, 148.27, 148.17, 136.87, 136.68, 136.51, 136.38, 128.57, 128.53, 128.46, 128.40, 127.94, 127.86, 127.46, 127.41, 127.09, 127.01, 126.98, 124.36, 124.25, 121.79, 115.37, 115.20, 113.01, 76.91, 73.90, 71.07, 71.04, 70.95, 70.70, 70.64, 70.16, 69.38, 67.29, 63.16; HRMS (ESI, m/z): , calcd for : 1451.4980; found 1451.4977.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,5′-dibenzyloxybenzoyl)-d-glucitol (51); 1,5-AG (0.15 g, 0.9 mmol), compound 46 (1.8 g, 5.4 mmol), 2-chloro-1-methylpyridinium iodide (1.3 g, 5.4 mmol), DMAP (0.66 g, 5.4 mmol), TEA (1.5 mL, 10.8 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desired compound 51 (0.69 g, 63%) was obtained as a colorless amorphous oil. = +8.3 (c 0.90, ); NMR (, 600 MHz) δ 7.45–7.18 (m, 48H), 6.79–6.76 (m, 2H), 6.72–6.69 (m, 2H), 5.89 (t, J = 9.6 Hz, 1H), 5.62 (t, J = 9.8 Hz, 1H), 5.37 (td, J = 10.1, 5.6 Hz, 1H), 5.06–4.98 (m, 8H), 4.95–4.84 (m, 8H), 4.66 (dd, J = 12.2, 2.9 Hz, 1H), 4.49 (dd, J = 11.3, 5.5 Hz, 1H), 4.40 (dd, J = 12.2, 5.3 Hz, 1H), 4.04–4.01 (m, 1H), 3.58 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.84, 165.80, 165.20, 165.06, 159.76, 159.75, 159.70, 136.44, 136.31, 136.26, 136.22, 131.45, 130.91, 130.87, 130.71, 128.60, 128.57, 128.51, 128.09, 128.06, 127.59, 127.57, 108.53, 108.49, 108.41, 108.11, 107.74, 107.70, 107.51, 74.28, 70.41, 70.23, 70.19, 69.73, 67.21, 63.51; HRMS (ESI, m/z): , calcd for : 1451.4980; found 1451.4980.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′-benzyloxybenzoyl)-d-glucitol (52); 1,5-AG (0.10 g, 0.6 mmol), compound 47 (0.82 g, 3.6 mmol), 2-chloro-1-methylpyridinium iodide (0.81 g, 3.6 mmol), DMAP (0.44 g, 3.6 mmol), TEA (1.0 mL, 7.2 mmol) in 20 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desire compound 52 (0.56 g, 92%) was obtained as a colorless amorphous oil. = +22.7 (c 1.00, ); NMR (, 600 MHz) δ 7.68–7.05 (m, 36H), 5.90 (t, J = 9.6 Hz, 1H), 5.65 (t, J = 9.8 Hz, 1H), 5.40 (td, J = 10.1, 5.6 Hz, 1H), 5.02–5.13 (m, 4H), 5.00 (s, 2H), 4.96 (s, 2H), 4.65 (dd, J = 12.0, 2.7 Hz, 1H), 4.48 (dd, J = 11.2, 5.7 Hz, 1H), 4.42 (dd, J = 12.2, 5.3 Hz, 1H), 4.02–4.05 (m, 1H), 3.59 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 166.03, 165.85, 165.34, 165.11, 158.66, 136.57, 136.37, 130.91, 130.30, 130.12, 129.56, 129.49, 128.61, 128.56, 128.13, 127.62, 127.58, 122.52, 121.12, 120.85, 120.71, 120.53, 115.16, 115.12, 115.00, 114.84, 76.89, 74.07, 70.27, 70.12, 69.50, 67.21, 63.31; HRMS (ESI, m/z): , calcd for : 1027.3306; found 1027.3301.
1,5-Anhydro-2,3,4,6-tetrakis-O-(4′-benzyloxybenzoyl)-d-glucitol (53); 1,5-AG (0.16 g, 1.0 mmol), compound 48 (1.4 g, 6.1 mmol), 2-chloro-1-methylpyridinium iodide (1.4 g, 6.0 mmol), DMAP (0.73 g, 6.0 mmol), TEA (1.7 mL, 12 mmol) in 30 mL of DCM was stirred at rt for 2 d. By the same procedure previously described for the preparation of compound 17, desire compound 53 (0.99 g, 98%) was obtained as a colorless amorphous oil. = +38.2 (c 0.90, ); NMR (, 600 MHz) δ 8.06–7.96 (m, 2H), 7.92–7.86 (m, 6H), 7.44–7.17 (m, 20H), 6.93–6.78 (m, 8H), 5.89 (t, J = 9.5 Hz, 1H), 5.63 (t, J = 9.8 Hz, 1H), 5.40 (td, J = 10.0, 5.7 Hz, 1H), 5.02–4.85 (m, 8H), 4.61–4.59 (m, 1H), 4.41 (td, J = 12.5, 5.4 Hz, 2H), 3.99–3.96 (m, 1H), 3.54 (t, J = 10.8 Hz, 1H); NMR (, 150 MHz) δ 165.83, 165.55, 165.15, 164.86, 162.78, 162.62, 162.57, 136.22, 136.07, 131.93, 131.86, 128.63, 128.61, 128.17, 127.46, 127.41, 127.19, 122.30, 121.65, 121.59, 121.46, 114.52, 114.47, 114.43, 114.38, 73.69, 69.96, 69.90, 69.24, 67.23, 63.05; HRMS (ESI, m/z): , calcd for : 1027.3306; found 1027.3304.
2,3,4,6-tetrakis-O-(3′,4′,5′-Trihydroxybenzoyl)-d-glucitol (54) [50]; on C (20 wt.%, 50 mg) was added to a solution of compound 50 (860 mg, 0.46 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 54 (321 mg, 90%) was obtained as a yellow amorphous oil. = +58.0 (c 1.04, MeOH); NMR (600 MHz) δ 7.19 (s, 2H), 7.06 (s, 2H) × 2, 6.99 (s, 2H), 5.81 (t, J = 9.6 Hz, 1H), 5.50 (t, J = 9.8 Hz, 1H), 5.28–5.24 (m, 1H), 4.49 (dd, J = 12.4, 2.1 Hz, 1H), 4.33–4.29 (m, 2H), 4.17–4.15 (m, 1H), 3.72 (t, J = 10.8 Hz, 1H); δ 166.36, 166.05, 165.90, 165.63, 146.00, 145.97, 145.92, 145.80, 139.22, 138.97, 138.90, 121.50, 120.90, 120.63, 110.17, 110.08, 110.01, 109.98, 77.57, 74.31, 70.74, 69.61, 67.44, 63.32; HRMS-ESI (m/z): , calcd for : 795.1021; found 795.1019.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3′,4′-dihydroxybenzoyl)-d-glucitol (55); on C (20 wt.%, 50 mg) was added to a solution of compound 50 (365 mg, 0.30 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 55 (196 mg, 92%) was obtained as a colorless amorphous oil. = +48.3 (c 0.65, MeOH); NMR (600 MHz); δ 7.56 (d, J = 2.1 Hz, 1H), 7.49 (dd, J = 8.4, 1.9 Hz, 1H), 7.43–7.33 (m, 6H), 6.90 (d, J = 8.2 Hz, 1H), 6.84 (dd, J = 11.3, 8.2 Hz, 2H), 6.76 (d, J = 8.2 Hz, 1H), 5.85 (t, J = 9.6 Hz, 1H), 5.55 (t, J = 9.8 Hz, 1H), 5.30 (td, J = 10.1, 5.4 Hz, 1H), 4.52 (dd, J = 12.0, 2.4 Hz, 1H), 4.37–4.32 (m, 2H), 4.20–4.17 (m, 1H), 3.75 (t, J = 10.8 Hz, 1H); δ 166.22, 166.00, 165.76, 165.56, 151.21, 151.01, 145.55, 145.39, 123.78, 123.71, 123.62, 122.53, 121.96, 121.74, 117.41, 117.29, 117.19, 117.09, 115.74, 115.68, 77.53, 74.40, 70.78, 69.87, 67.48, 63.52; HRMS (, m/z): , calcd for : 707.1248; found 707.1255.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3,5-dihydroxybenzoyl)-d-glucitol (56); on C (20 wt.%, 50 mg) was added to a solution of compound 51 (365 mg, 0.30 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 56 (210 mg, quant.) was obtained as a colorless amorphous oil. = +39.1 (c 0.50, MeOH); NMR (600 MHz); δ 7.09 (s, 2H), 6.95 (d, J = 1.4 Hz, 4H), 6.89 (d, J = 1.7 Hz, 2H), 6.63–6.49 (m, 4H), 5.90 (t, J = 9.6 Hz, 1H), 5.59 (t, J = 9.6 Hz, 1H), 5.37 (td, J = 10.0, 5.6 Hz, 1H), 4.55 (d, J = 12.4 Hz, 1H), 4.43 (dd, J = 12.2, 4.6 Hz, 1H), 4.37 (dd, J = 11.3, 5.5 Hz, 1H), 4.25 (dd, J = 9.8, 4.3 Hz, 1H), 3.80 (t, J = 10.8 Hz, 1H) δ 166.33, 166.13, 165.82, 165.67, 159.36, 159.31, 159.24, 132.73, 132.10, 131.91, 129.67, 128.96, 108.81, 108.74, 108.65, 108.39, 108.24, 108.08, 77.25, 74.65, 70.85, 69.79, 67.25, 63.45; HRMS (, m/z): , calcd for : 707.1248; found 707.1255.
1,5-Anhydro-2,3,4,6-tetrakis-O-(3-hydroxybenzoyl)-d-glucitol (57); on C (20 wt.%, 50 mg) was added to a solution of compound 52 (300 mg, 0.30 mmol) in 20 mL of MeOH and 20 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 57 (181 mg, 94%) was obtained as a colorless amorphous oil. = +36.4 (c 0.70, MeOH); NMR 600 MHz); δ 7.55–6.97 (m, 20H), 5.94 (t, J = 9.5 Hz, 1H), 5.65 (t, J = 9.6 Hz, 1H), 5.41 (td, J = 10.1, 5.4 Hz, 1H), 4.57 (dd, J = 12.2, 2.6 Hz, 1H), 4.47 (dd, J = 12.4, 4.8 Hz, 1H), 4.39 (dd, J = 11.2, 5.7 Hz, 1H), 4.29–4.26 (m, 1H), 3.83 (t, J = 10.8 Hz, 1H); δ 166.38, 166.23, 165.90, 165.82, 158.46, 158.25, 132.14, 131.57, 131.43, 130.49, 130.44, 130.40, 129.72, 129.00, 126.08, 121.43, 121.39, 121.21, 121.08, 117.04, 116.93, 116.88, 116.73, 77.23, 74.83, 70.96, 70.23, 67.33, 63.83; HRMS (, m/z): , calcd for : 643.1452; found 643.1459.
1,5-Anhydro-2,3,4,6-tetrakis-O-(4-hydroxybenzoyl)-d-glucitol (58); on C (20 wt.%, 50 mg) was added to a solution of compound 54 (288 mg, 0.29 mmol) in 15 mL of MeOH and 15 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desired compound 58 (190 mg, quant.) was obtained as a colorless amorphous oil. = +45.3 (c 0.50, MeOH); NMR (600 MHz); δ 7.93–7.76 (m, 8H), 6.92–6.77 (m, 8H), 5.89 (t, J = 9.6 Hz, 1H), 5.61 (t, J = 9.8 Hz, 1H), 5.34 (td, J = 10.0, 5.3 Hz, 1H), 4.55 (dd, J = 12.4, 2.7 Hz, 1H), 4.41–4.35 (m, 2H), 4.23–4.20 (m, 1H), 3.77 (t, J = 10.8 Hz, 1H); δ 166.17, 165.99, 165.69, 165.54, 163.17, 163.13, 162.88, 162.82, 132.75, 132.68, 132.61, 132.58, 121.99, 121.44, 121.21, 121.19, 116.09, 116.05, 116.00, 115.94, 77.45, 74.47, 70.76, 70.05, 67.50, 63.65; HRMS (, m/z): , calcd for : 643.1452; found 643.1459.
Benzyl 2,3,4,6-tetrakis-O-(3′,4′,5′-tribenzyloxybenzoyl)-α,β-d-glucopyranoside (60); Compound 59 [43] (211 mg, 0.78 mmol), EDC·HCl (1.2 g, 6.3 mmol), compound 16 (2.06 g, 4.7 mmol), DMAP (47.7 mg, 0.39 mmol) in 30 mL of DCM was stirred at rt for overnight. After the addition 20 mL of water, the reaction mixture was extracted with DCM (20 mL). The combined organic layer was washed with water (30 mL × 4) and brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (/MeOH = 200/1) to obtain desired compound 60 (791 mg, 52%) as a colorless amorphous oil. = +18.1 (c 1.00, ); NMR (, 600 MHz) (assigned for the major anomer; α) δ 7.42–7.16 (m, 80H), 6.21 (t, J = 10.0, 1H), 5.69 (t, J = 10.0 Hz, 1H), 5.45 (d, J = 3.8 Hz, 1H), 5.12–4.95 (m, 16H), 4.73 (dd, J = 3.1, 12.4 Hz, 1H), 4.54–4.51 (m, 1H), 4.73 (dd, J = 3.1, 12.4 Hz, 1H); NMR (, 150 MHz) (assigned for the major anomer; α) δ 165.68, 165.16, 152.55, 152.52, 152.49, 143.00, 142.86, 142.69, 142.64, 137.41, 137.36, 137.32, 136.56, 136.53, 136.48, 136.40, 136.29, 128.54, 128.46, 128.38, 128.29, 128.24, 128.16, 128.12, 128.09, 128.05, 127.98, 127.94, 127.92, 127.88, 127.81, 127.58, 127.54, 127.52, 124.64, 124.15, 124.02, 123.85, 109.23, 109.16, 109.01, 95.27, 75.11, 75.09, 75.06, 72.17, 71.18, 71.15, 71.05, 70.16, 69.96, 67.96, 63.17. HRMS (ESI, m/z): , calcd for : 1981.7073; found 1981.7067.
2,3,4,6-tetrakis-O-(3′,4′,5′-Trihydroxybenzoyl)-α,β-d-glucopyranose (61) [55]; on C (20 wt.%, 20 mg) was added to a solution of compound 54 (100 mg, 0.51 mmol) in 10 mL of MeOH and 10 mL of THF under the argon. By the same procedure previously described for the preparation of compound 31, desire compound 58 (34 mg, 85%) was obtained as a colorless amorphous oil. = +54.4 (c 0.30, MeOH); NMR (, 600 MHz) δ [α-form] 7.20 (s, 2H), 7.09 (s, 2H), 7.07 (s, 2H), 7.00 (s, 2H), 6.11 (t, J = 9.6 Hz, 1H), 5.62 (d, J = 3.4 Hz, 1H), 5.56 (t, J = 10.2 Hz, 1H), 5.16 (dd, J = 10.3, 3.5 Hz, 1H), 4.47 (dd, J = 2.4, 11.4 Hz, 1H), 4.35 (dd, J = 12.6, 4.8 Hz, 1H), 4.32–4.29 (m, 1H); [β-form] 7.19 (s, 2H), 7.08 (s, 2H), 7.05 (s, 2H), 6.95 (s, 2H), 5.80 (t, J = 9.6 Hz, 1H), 5.51 (t, J = 9.6 Hz, 1H), 5.29 (t, J = 9.0 Hz, 1H), 5.24 (d, J = 9.0 Hz, 1H), 4.50 (d, J = 1.7, 11.4 Hz, 1H), 4.31 (m, 2H); (assigned for the major anomer; α) δ 166.48, 166.22, 165.99, 165.73, 146.04, 145.99, 145.84, 139.27, 139.24, 139.00, 138.93, 121.61, 121.02, 120.78, 120.67, 110.21, 110.17, 110.06, 110.01, 91.04, 73.02, 70.70, 69.88, 68.55, 63.27; HRMS (ESI, m/z): , calcd for : 811.0970; found 811.0970.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-d-glucitol (62); Compound 21 (3.5 g, 3.2 mmol), iodine (0.71 g, 5.6 mmol) in 100 mL of DCM and 50 mL of MeOH was stirred at 70 °C for 7 days. The reaction mixture was washed with sodium thiosulfate solution and brine. The crude product was purified by C.C (DCM/MeOH = 4/1) to obtain desired compound 62 (3.0 g, 93%) as a white solid. = +80.3 (c 1.06, ); NMR (, 600 MHz) δ 7.38–7.21 (m, 32H), 5.34 (t, J = 9.0 Hz, 1H), 5.28–5.26 (m, 1H), 5.07–4.97 (m, 12H), 4.35–4.32 (m, 1H), 4.00–3.99 (m, 1H), 3.91–3.88 (m, 2H), 3.51–3.48 (m, 2H), 3.07 (d, J = 4.0 Hz, 1H), 2.01 (t, J = 6.0 Hz, 1H); NMR (, 150 MHz) δ 167.2, 165.3, 152.6, 143.1, 142.9, 137.3, 136.5, 136.4, 128.5, 128.4, 128.2, 128.1, 128.0, 127.7, 127.5, 109.3, 109.1, 80.4, 78.5, 75.1, 71.1, 67.0, 69.8, 66.9, 62.4; HRMS-ESI (m/z): , calcd for : 1031.3612; found 1031.3619.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-bis-O-(3′,5′-dimetoxymetoxy-4′-benzyloxybenzoyl)-d-glucitol (64); Compound 62 (2.5 g, 2.5 mmol), 63 [46] (2.5 g, 6.2 mmol), EDC·HCl (1.4 g, 7.2 mmol), DMAP (0.15 g, 1.2 mmol) in 15 mL of DCM was stirred at rt for 18 h. After the addition 30 mL of water, the reaction mixture was extracted with EtOAc (3 × 50 mL). The combined organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (/acetone = 100/1) to obtain 64 (2.3 g, 55% yield) as a colorless amorphous oil. = +24.7 (c 2.15 in ); NMR (, 600 MHz) δ 7.58 (s, 2H), 7.46–7.25 (m, 50H), 5.85 (t, J = 10Hz, 1H), 5.61 (t, J = 10.0Hz, 1H), 5.28–5.26 (m, 1H), 5.22–4.93 (m, 24H), 4.73–4.71 (m, 1H), 4.50–4.47 (m, 1H), 4.35–4.32 (m, 1H), 4.06–4.03 (m, 1H), 3.56 (t, J = 11.0 Hz, 1H), 3.48 (s, 6H), 3.42(s, 6H); NMR (, 150 MHz) δ 165.7, 165.5, 165.1, 164.8, 152.5, 150.9, 150.8, 143.7, 143.4, 142.8, 142.7, 137.4, 137.3, 137.2, 136.5, 136.4, 132.4, 130.8, 128.7, 128.5, 128.4, 128.3, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 125.1, 124.2, 124.1, 124.0, 112.4, 112.3, 109.1, 109.0, 95.4, 75.2, 75.1, 75.0, 74.4, 71.1, 71.0, 70.6, 69.3, 68.1, 67.1, 63.3, 56.4; HRMS-ESI (m/z): , calcd for : 1691.5824; found 1691.5825.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-tribenzyloxybenzoyl)-4,6-bis-O-(3′,5′-dihydroxy-4′-benzyloxybenzoyl)-d-glucitol (65); Compound 64 (2.0 g, 1.2 mmol) in 8 mL of THF solution was added 8.8 mL of 2-propanol and 0.2 mL of conc. HCl solution, and the mixture was stirred at 60 °C for 6 h. After the addition 3 mL of , the mixture was extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with brine, dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (/MeOH = 1000/1–500/1) to obtain 65 (1.7 g, 93% yield) as a white amorphous oil. = +26.8 (c 2.36, ); NMR (, 600 MHz) δ 7.43–7.14 (m, 48H), 6.00 (s, 4H), 5.82 (t, J = 9.5 Hz, 1H), 5.59 (t, J = 9.5 Hz, 1H), 5.37–5.30 (m, 1H), 5.10–4.82 (m, 16H), 4.70–4.68 (m, 1H), 4.54–4.52 (m, 1H), 4.45–4.42 (m, 1H), 3.97–3.95 (m, 1H), 3.57 (t, J = 10.5 Hz, 1H); NMR (, 150 MHz) δ 166.2, 165.8, 165.5, 165.2, 152.5, 149.0, 148.9, 142.8, 142.7, 138.2, 137.8, 137.3, 137.2, 136.6, 136.4, 136.2,128.8, 128.7, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.7, 127.5, 124.5, 123.9, 123.8, 109.9, 109.8, 109.1, 108.9, 77.6, 75.2, 75.1, 75.0, 74.3, 71.1, 71.0, 70.7, 69.7, 68.6, 67.3, 62.6; HRMS-ESI (m/z): , calcd for : 1515.4775; found 1515.4777.
Benzyl protected 1,5-AG-tellimagrandin analog (66); To a solution of (135 mg, 1.0 mmol) in 10 mL of MeOH was added n-butylamine (400 μL, 4.0 mmol). After the stirred at rt for 1.5 h, the mixture was added to solution of compound 65 (500 mg, 0.34 mmol) in 20 mL of 1,2-dichloroethane (DCE) and stirred at rt for 30 min. The reaction mixture was diluted with 50 mL of diethyl ether, and 50 mL of 5M aq. HCl and 50 mL of diethyl ether were added. The separating organic layer washed with water, and brine. The organic layer was dried over , filtered and concentrated under reduced pressure. The crude was purified by C.C (DCM/MeOH = 250/1) and HPLC (column, FNED01048, 250 × 20 mm, SG80–5 μm, eluant DCM/MeOH = 200/1) to afford 66 (237 mg, 48%) as a peal yellow amorphous oil. = +69.5 (c 1.43, ); NMR (, 600 MHz) δ 7.43–7.23 (m, 50H), 6.75 (s, 1H), 6.67 (s, 1H), 5.67 (t, J = 10.0 Hz, 1H), 5.37–5.34 (m, 1H), 5.29 (t, J = 10.0 Hz, 1H), 5.25–5.24 (m, 1H), 5.13–4.85 (m, 16H), 4.47–4.45 (m, 1H), 3.98 (d, J = 12.5 Hz, 1H), 3.96–3.94 (m. 1H), 3.46 (t, J = 11.0 Hz, 1H); δ 167.1, 166.6, 165.8, 165.2, 152.5, 149.0, 147.0, 142.8, 137.4, 137.3, 136.5, 136.4, 136.3, 136.2, 135.7, 135.5, 130.3, 129.6, 129.0, 128.9, 128.6, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 127.8, 127.6, 127.5, 123.9, 113.7, 113.2, 109.2, 108.4, 107.9, 75.6, 75.1, 75.0, 74.4, 71.1, 71.0, 70.8, 70.1, 67.7, 63.6; HRMS-ESI (m/z): , calcd for ; found 1513.4620.
1,5-Anhydro-2,3-bis-O-(3′,4′,5′-trihydroxybenzoyl)-4,6-O-hexahydroxydiphenyl-d-glucitol (67); on C (20 wt.%, 20 mg) was added to a solution of compound 66 (60 mg, 0.40 mmol) in 2 mL of MeOH and 2 mL of THF under the argon. By the same procedure previously described for the preparation of compound procedure described for previously 31 preparation, desired compound 67 (32 mg, quant.) was obtained as a peal yellow amorphous oil. = +113.3 (c 0.29, ); NMR (600 MHz) δ 7.00 (s, 2H), 6.95 (s, 2H), 6.60 (s, 1H), 6.40 (s, 1H), 5.61 (t, J = 9.5 Hz, 1H), 5.27–5.19 (m, 2H), 5.03 (t, J = 10.0 Hz, 1H), 4.25–4.22 (m, 1H), 4.08–4.07 (m, 1H), 3.80 (t, J = 13.0 Hz, 1H), 3.54 (t, J = 11.0 Hz, 1H); NMR ( 150 MHz); 171.1, 170.7, 168.8, 168.3, 150.6, 147.6, 147.1, 144.6, 143.5, 141.8, 139.7, 126.8, 126.7, 126.3, 120.7, 120.3, 120.1, 119.4, 111.1, 110.8, 108.3, 107.7, 78.7, 78.5, 76.2, 72.1, 72.0, 71.8, 71.4, 71.3, 69.4, 68.7, 64.7; HRMS (ESI, m/z): , calcd for : 793.0864; found 793.0865.
Acknowledgments
We thank Koichi Metori (Chemical Analysis Center, School of Pharmacy, Nihon University) for performing mass measurements.
Supplementary Materials
The following are available online NMR, NMR spectra of compounds 31–44, 54–58, 61 and 67.
Author Contributions
Experiment: S.M. (Shota Machida), S.M. (Saki Mukai), R.K. and M.F.; Writing—Original Draft preparation: S.M. (Shota Machida); Writing—Review and Editing: S.M. (Shota Machida), H.S. and T.U.; project administration: T.U.
Funding
This study was partly supported by Japan Society for the Promotion of Science (JSPS), 18K06725 (T.U.) and Nihon University Mutidisciplinary Research Grant for 2019 (19-016).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Oizumi T., Daimon M., Jimbu Y., Wada K., Kameda W., Susa S., Yamaguchi H., Ohnuma H., Tominaga M., Kato T. Impaired Glucose Tolerance is a Risk Factor for Stroke in a Japanese Sample-the Funagata Study. Metabolism. 2008;57:333–338. doi: 10.1016/j.metabol.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Bornfeldt K.E., Tabas I. Insulin Resistance, Hyperglycemia, and Atherosclerosis. Cell Metab. 2011;14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ceriello A., Bortolotti N., Motz E., Pieri C., Marra M., Tonutti L., Lizzio S., Feletto F., Catone B., Taboga C. Meal-Induced Oxidative Stress and Low-Density Lipoprotein Oxidation in Diabetes: The Possible Role of Hyperglycemia. Metabolism. 1999;48:1503–1508. doi: 10.1016/S0026-0495(99)90237-8. [DOI] [PubMed] [Google Scholar]
- 4.Ten Bruggencate S.J., Frederiksen P.D., Pedersen S.M., Floris-Vollenbroek E.G., Lucas-van de Bos E., van Hoffen E., Wejses P.L. Dietary Milk-Fat-Globule Membrane Affects Resistance to Diarrheagenic Escherichia Coli in Healthy Adults in a Randomized, Placebo-Controlled, Double-Blind Study. J. Nutr. 2016;146:249–255. doi: 10.3945/jn.115.214098. [DOI] [PubMed] [Google Scholar]
- 5.Ceriello A., Genovese S. Atherogenicity of Postprandial Hyperglycemia and Lipotoxicity. Rev. Endocr. Metab. Dis. 2016;17:111–116. doi: 10.1007/s11154-016-9341-8. [DOI] [PubMed] [Google Scholar]
- 6.Mah E., Bruno R.S. Postprandial Hyperglycemia on Vascular Endothelial Function: Mechanisms and Consequences. Nutr. Res. 2012;32:727–740. doi: 10.1016/j.nutres.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Kitasato L., Tojo T., Hatakeyama Y., Kameda R., Hashikata T., Yamaoka-Tojo M. Postprandial Hyperglycemia and Endothelial Function in Type 2 Diabetes: Focus on Mitiglinide. Cardiovasc. Diabetol. 2012;11:79. doi: 10.1186/1475-2840-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derosa G., Maffioli P. α-Glucosidase Inhibitors and their Use in Clinical Practice. Arch. Med. Sci. 2012;8:899–906. doi: 10.5114/aoms.2012.31621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuccori M., Convertino I., Galiulo Maria T., Marino A., Capogrosso-Sansone A., Blandizzi C. Diabetes Drugs and the Incidence of Solid Cancers: A Survey of the Current Evidence. Expert Opin. Drug Saf. 2017;16:1133–1148. doi: 10.1080/14740338.2017.1361401. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z., Ma S. Recent Advances in Synthetic α-Glucosidase Inhibitors. Chem. Med. Chem. 2017;12:819–829. doi: 10.1002/cmdc.201700216. [DOI] [PubMed] [Google Scholar]
- 11.Singla R.K., Singh R., Dubey A.K. Important Aspects of Post-Prandial Antidiabetic Drug, Acarbose. Curr. Top Med. Chem. 2016;16:2625–2633. doi: 10.2174/1568026616666160414123500. [DOI] [PubMed] [Google Scholar]
- 12.Newman D.J., Cragg G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 13.Che C.-T., Zhang H. Plant natural products for human health. Int. J. Mol. Sci. 2019;20:830. doi: 10.3390/ijms20040830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wright G.D. Opportunities for Natural Products in 21(st) Century Antibiotic Discovery. Nat. Prod. Rep. 2017;34:694–701. doi: 10.1039/C7NP00019G. [DOI] [PubMed] [Google Scholar]
- 15.Zhao C., Yang C.F., Wai S.T.C., Zhang Y.B., Portillo M.P., Paoli P., Wu Y., Cheang W.S., Liu B., Carpéné C., et al. Regulation of Glucose Metabolism by Bioactive Phytochemicals for the Management of Type 2 Diabetes Mellitus. Crit. Rev. Food Sci. 2019;59:830–847. doi: 10.1080/10408398.2018.1501658. [DOI] [PubMed] [Google Scholar]
- 16.Bi W., Gao Y., Shen J., He C., Liu H., Peng Y., Zhang C., Xiao P. Traditional Uses, Phytochemistry, and Pharmacology of the Genus Acer (Maple): A review. J. Ethnopharmacol. 2016;189:31–60. doi: 10.1016/j.jep.2016.04.021. [DOI] [PubMed] [Google Scholar]
- 17.González-Sarrías A., Li L., Seeram N.P. Effects of Maple (Acer) Plant Part Extracts on Proliferation, Apoptosis and Cell Cycle Arrest of Human Tumorigenic and Non-Tumorigenic Colon Cells. Phyther. Res. 2012;26:995–1002. doi: 10.1002/ptr.3677. [DOI] [PubMed] [Google Scholar]
- 18.Bi W., Liu H., Shen J., Zhang L.H., Li P., Peng B., Cao L., Zhang P., He C., Xiao P. Chemopreventive Effects of Ku-Jin Tea Against AOM-Induced Precancerous Colorectal Lesions in Rats and Metabolomic Analysis. Sci. Rep. 2017;7:15893. doi: 10.1038/s41598-017-16237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Han S.S., Lo S.C., Choi Y.W., Kim J.H., Baek S.H. Antioxidant Activity of Crude Extract and Pure Compounds of Acer Ginnala Max. Bull. Korean Chem. Soc. 2004;25:389–391. [Google Scholar]
- 20.Royer M., Diouf P.N., Stevanovic T. Polyphenol Contents and Radical Scavenging Capacities of Red Maple (Acer Rubrum L.) Extracts. Food Chem. Toxicol. 2011;49:2180–2188. doi: 10.1016/j.fct.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe M., Prasad D.H. Antioxidant Phenolic Constituents from the Leaves of Acer Ginnala var Aidzuense. J. Nat. Remedies. 2017;17:9–12. doi: 10.18311/jnr/2017/15632. [DOI] [Google Scholar]
- 22.Bi W., Shen J., Gao Y., He C., Peng Y., Xiao P. Ku-Jin Tea (Acer Tataricum Subsp. Ginnala or A. Tataricum Subsp. Theiferum), an Underestimated Functional Beverage Rich in Antioxidant Phenolics. J. Funct. Foods. 2016;24:75–84. doi: 10.1016/j.jff.2016.04.002. [DOI] [Google Scholar]
- 23.Apostolidis E., Li L., Lee C., Seeram N.P. In Vitro Evaluation of Phenolic-Enriched Maple Syrup Extracts for Inhibition of Carbohydrate Hydrolyzing Enzymes Relevant to Type 2 Diabetes Management. J. Funct. Foods. 2011;3:100–106. doi: 10.1016/j.jff.2011.03.003. [DOI] [Google Scholar]
- 24.Honma A., Koyama T., Yazawa K. Antihyperglycemic effects of Japanese maple Acer amoenum leaf extract and its constituent corilagin. J. Wood Sci. 2010;56:507–512. doi: 10.1007/s10086-010-1130-5. [DOI] [Google Scholar]
- 25.Honma A., Koyama T., Yazawa K. Anti-Hyperglycemic Effects of Sugar Maple Acer Saccharum and its Constituent Acertannin. Food Chem. 2010;123:390–394. doi: 10.1016/j.foodchem.2010.04.052. [DOI] [Google Scholar]
- 26.Bock K., LaCour N.F., Jensen S.R., Nielsen B.J. The structure of acertannin. Phytochemistry. 1980;19:2033. doi: 10.1016/0031-9422(80)83034-2. [DOI] [Google Scholar]
- 27.Bate-Smith E.C. Astringent Tannins of Acer Species. Phytochemistry. 1977;16:1421–1426. doi: 10.1016/S0031-9422(00)88795-6. [DOI] [Google Scholar]
- 28.Hatano T., Hattori S., Ikeda Y., Shingu T., Okuda T. Tannins of Aceraceous Plants. Part II. Gallotannins Having a 1,5-Anhydro-D-Glucitol Core and Some Ellagitannins from Acer Species. Chem. Pharm. Bull. 1990;38:1902–1905. doi: 10.1248/cpb.38.1902. [DOI] [Google Scholar]
- 29.Wan C., Yuan T., Li L., Kandhi V., Cech N.B., Xie M., Seeram N.P. Maplexins, New α-Glucosidase Inhibitors from Red Maple (Acer Rubrum) Stems. Bioorganic Med. Chem. Lett. 2012;22:597–600. doi: 10.1016/j.bmcl.2011.10.073. [DOI] [PubMed] [Google Scholar]
- 30.Yuan T., Wan C., Liu K., Seeram N.P. New Maplexins F-I and Phenolic Glycosides from Red Maple (Acer Rubrum) Bark. Tetrahedron. 2012;68:959–964. doi: 10.1016/j.tet.2011.11.062. [DOI] [Google Scholar]
- 31.Honma A., Koyama T., Yazawa K. Anti-Hyperglycaemic Effects of the Japanese Red Maple Acer Pycnanthum and its Constituents the Ginnalins B and C. J. Enzyme Inhib. Med. Chem. 2011;26:176–180. doi: 10.3109/14756366.2010.486795. [DOI] [PubMed] [Google Scholar]
- 32.Ma H., Wang L., Niesen D.B., Cai A., Cho B.P., Tan W., Gu Q., Xu J., Seeram N.P. Structure Activity Related, Mechanistic, and Modeling Studies of Gallotannins Containing a Glucitol-Core and α-Glucosidase. RSC Adv. 2015;5:107904–107915. doi: 10.1039/C5RA19014B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.González-Sarrías A., Yuan T., Seeram N.P. Cytotoxicity and Structure Activity Relationship Studies of Maplexins A-I, Gallotannins from Red Maple (Acer Rubrum) Food Chem. Toxicol. 2012;50:1369–1376. doi: 10.1016/j.fct.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 34.González-Sarrías A., Ma H., Edmonds M.E., Seeram N.P. Maple Polyphenols, Ginnalins A-C, Induce S- and G2/M-Cell Cycle Arrest in Colon and Breast Cancer Cells Mediated by Decreasing Cyclins A and D1 Levels. Food Chem. 2013;136:636–642. doi: 10.1016/j.foodchem.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 35.Kamori A., Kato A., Miyawaki S., Koyama J., Nash R.J., Fleet G.W.J., Miura D., Ishikawa F., Adachi I. Dual Action of Acertannins as Potential Regulators of Intracellular Ceramide Levels. Tetrahedron: Asymmetry. 2016;27:1177–1185. doi: 10.1016/j.tetasy.2016.09.006. [DOI] [Google Scholar]
- 36.Kato A., Koyama J., Shinzawa K., Imaeda S., Adachi I., Nash R.J., Fleet G.W.J., Shintani M., Takeuchi C., Ishikawa F. Ginnalin B Induces Differentiation Markers and Modulates the Proliferation/Differentiation Balance via the Upregulation of NOTCH1 in Human Epidermal Keratinocytes. Bioorganic Med. Chem. 2019;27:2172–2180. doi: 10.1016/j.bmc.2019.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Yokozawa T., Chen C.P., Dong E., Tanaka T., Nonaka G., Nishioka I. Study on the Inhibitory Effect of Tannins and Flavonoids Against the 1,1-Diphenyl-2-Picrylhydrazyl Radical. Biochem. Pharmacol. 1998;56:213–222. doi: 10.1016/S0006-2952(98)00128-2. [DOI] [PubMed] [Google Scholar]
- 38.Lee D.Y., Kim H.W., Yang H., Sung S.H. Hydrolyzable Tannins from the Fruits of Terminalia Chebula Retz and their α-Glucosidase Inhibitory Activities. Phytochemistry. 2017;137:109–116. doi: 10.1016/j.phytochem.2017.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Wilkins C.K., Bohm B.A. Ellagitannins from Tellima Grandiflora. Phytochemistry. 1976;15:211–214. doi: 10.1016/S0031-9422(00)89087-1. [DOI] [PubMed] [Google Scholar]
- 40.Zheng S., Laraia L., O’Connor C.J., Sorrell D., Tan Y.S., Xu Z., Venkitaraman A.R., Wu W., Spring D.R. Synthesis and Biological Profiling of Tellimagrandin I and Analogues Reveals that the Medium Ring Can Significantly Modulate Biological Activity. Org. Biomol. Chem. 2012;10:2590–2593. doi: 10.1039/c2ob25065a. [DOI] [PubMed] [Google Scholar]
- 41.Uchiyama T., Shishikura K., Ogawa K., Ohshima Y., Miyairi S. An Efficient Method for the Preparation of 1,5-Anhydroalditol from Unprotected Carbohydrates via Glycopyranosyl Iodide. Tetrahedron Lett. 2016;57:5294–5296. doi: 10.1016/j.tetlet.2016.10.063. [DOI] [Google Scholar]
- 42.Nie X., Wang G. Synthesis of a Ring-Oxygenated Variant of the 2-Carboxy-6-Hydroxyoctahydroindole Core of Aeruginosin 298-A from Glucose. J. Org. Chem. 2005;70:8687–8692. doi: 10.1021/jo0507901. [DOI] [PubMed] [Google Scholar]
- 43.Lonnecker A.T., Lim Y.H., Felder S.E., Besset C.J., Wooley K.L. Four Different Regioisomeric Polycarbonates Derived from One Natural Product, D-Glucose. Macromolecules. 2016;49:7857–7867. doi: 10.1021/acs.macromol.6b00591. [DOI] [Google Scholar]
- 44.Tanaka N., Ogawa I., Yoshigase S., Nokami J. Regioselective Ring Opening of Benzylidene Acetal Protecting Group(s) of Hexopyranoside Derivatives by DIBAL-H. Carbohydr. Res. 2008;343:2675–2679. doi: 10.1016/j.carres.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 45.Daragics K., Fügedi P. Regio- and Chemoselective Reductive Cleavage of 4,6-O-Benzylidene-Type Acetals of Hexopyranosides Using BH3·THF-TMSOTf. Tetrahedron Lett. 2009;50:2914–2916. doi: 10.1016/j.tetlet.2009.03.194. [DOI] [Google Scholar]
- 46.Haslam E., Radford T. Synthesis of 1,3-Anhydro-D-Glucitol and Some Derivatives of 1,5-Anhydro-D-Glucitol. Carbohydr. Res. 1966;2:301–314. doi: 10.1016/S0008-6215(00)82565-3. [DOI] [Google Scholar]
- 47.Qian P., Yao W., Huang L., Meng X., Li Z. A Mild and Efficient Method for the Selective Cleavage of Primary p-Methoxybenzyl Protecting Group of Saccharides by Co2(CO)8–Me2PhSiH–CO System. Tetrahedron Lett. 2015;56:5238–5241. doi: 10.1016/j.tetlet.2015.07.051. [DOI] [Google Scholar]
- 48.DeNinno M.P., Etienne J.B., Duplantier K.C. A Method for the Selective Reduction of Carbohydrate 4,6-O-Benzylidene Acetals. Tetrahedron Lett. 1995;36:669–672. doi: 10.1016/0040-4039(94)02348-F. [DOI] [Google Scholar]
- 49.Cocinero E.J., Gamblin D.P., Davis B.G., Simons J.P. The Building Blocks of Cellulose: The Intrinsic Conformational Structures of Cellobiose, its Epimer, Lactose, and their Singly Hydrated Complexes. J. Am. Chem. Soc. 2009;131:11117–11123. doi: 10.1021/ja903322w. [DOI] [PubMed] [Google Scholar]
- 50.Ren Y., Himmeldirk K., Chen X. Synthesis and Structure–Activity Relationship Study of Antidiabetic Penta-O-galloyl-d-glucopyranose and its Analogues. J. Med. Chem. 2006;49:2829–2837. doi: 10.1021/jm060087k. [DOI] [PubMed] [Google Scholar]
- 51.Tranchimand S., Tron T., Gaudin C., Iacazio G. First Chemical Synthesis of Three Natural Depsides Involved in Flavonol Catabolism and Related to Quercetinase Catalysis. Synth. Commun. 2006;36:587–597. doi: 10.1080/00397910500406534. [DOI] [Google Scholar]
- 52.Fergusson K.M., Hird M. The Dramatic Influence of the Location of Bend and of Lateral Fluoro Substitution on the Mesomorphic Properties of Angular Chiral Esters Based on a 1,3-Disubstituted Benzene Ring. J. Mater. Chem. 2010;20:3069–3078. doi: 10.1039/b923267b. [DOI] [Google Scholar]
- 53.Hawker C.J., Lee R., Frechet J.M.J. One-Step Synthesis of Hyperbranched Dendritic Polyesters. J. Am. Chem. Soc. 1991;113:4583–4588. doi: 10.1021/ja00012a030. [DOI] [Google Scholar]
- 54.Furukawa M., Kamo S., Makino M., Kurita M., Tabata K., Matsuzaki K., Suzuki T., Uchiyama T. Triterpenoid Glycosides from Ladenbergia Hexandra Klotzsch. Phytochemistry. 2017;136:147–155. doi: 10.1016/j.phytochem.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 55.Gramshaw J.W., Haslam E., Haworth R.D., Searle T. 571. Gallotannins. Part, V. The Structure of Penta- and Tetra-O-Galloylglucoses, and Some Observations on the Molecular Weights of the Gallotannins. J. Chem. Soc. 1962:2944. doi: 10.1039/jr9620002944. [DOI] [Google Scholar]
- 56.Feldman K.S., Ensel S.M., Minard R.D. Ellagitannin Chemistry. The First Total Chemical Synthesis of an Ellagitannin Natural Product, Tellimagrandin I. J. Am. Chem. Soc. 1994;116:1742–1745. doi: 10.1021/ja00084a015. [DOI] [Google Scholar]
- 57.Michihata N., Kaneko Y., Kasai Y., Tanigawa K., Hirokane T., Higasa S., Yamada H. High-Yield Total Synthesis of (-)-Strictinin through Intramolecular Coupling of Gallates. J. Org. Chem. 2013;78:4319–4328. doi: 10.1021/jo4003135. [DOI] [PubMed] [Google Scholar]
- 58.Yamada H., Nagao K., Dokei K., Kasai Y., Michihata N. Total Synthesis of (-)-Corilagin. J. Am. Chem. Soc. 2008;130:7566–7567. doi: 10.1021/ja803111z. [DOI] [PubMed] [Google Scholar]
- 59.Kasai Y., Michihata N., Nishimura H., Hirokane T., Yamada H. Total Synthesis of (+)-Davidiin. Angew. Chemie. Int. Ed. 2012;51:8026–8029. doi: 10.1002/anie.201203305. [DOI] [PubMed] [Google Scholar]
- 60.Asakura N., Fujimoto S., Michihata N., Nishii K., Imagawa H., Yamada H. Synthesis of Chiral and Modifiable Hexahydroxydiphenoyl Compounds. J. Org. Chem. 2011;76:9711–9719. doi: 10.1021/jo201750d. [DOI] [PubMed] [Google Scholar]
- 61.Oki T., Masuda M., Furuta S., Nishiba Y., Suda I. Radical Scavenging Activity of Fried Chips Made from Purple-Fleshed Sweet Potato. J. Jpn. Soc. Food Sci. 2011;48:926–932. doi: 10.3136/nskkk.48.926. [DOI] [Google Scholar]
- 62.Toda M., Kawabata J., Kasai T. α-Glucosidase Inhibitors from Clove (Syzgium aromaticum) Biosci. Biotechnol. Biochem. 2000;64:294–298. doi: 10.1271/bbb.64.294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.