

Abstract
Paclitaxel is a commonly used drug in the treatment of multiple solid tumors, including cancers of the breast, lung, and ovaries. Despite the established exposure–pharmacodynamic relationships for paclitaxel, treatment is associated with wide interindividual pharmacokinetic variability that leads to unpredictability of the agent's clinical activity and toxicity. We hypothesized that physiologically‐based modeling approaches could be employed to predict the human pharmacokinetics of paclitaxel following administration of the approved Cremophor‐based formulation (Taxol). The model was developed from tissue distribution studies performed in mice and applied to plasma concentration‐time data obtained in adult cancer patients receiving Taxol at the approved dose and schedule (175 mg/m2 by a 3‐hour intravenous infusion), taking into account interspecies differences in physiological parameters. The final model adequately captured the observed concentrations in patients and allowed prediction of paclitaxel distribution profiles in multiple target organs and can be applied to further refine the chemotherapeutic treatment with a clinically important agent.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
☑ Paclitaxel is commonly used in the treatment of multiple solid tumors. Although exposure–clinical activity and toxicity relationships for paclitaxel were established, the agent distribution profiles in multiple target organs have not been characterized in cancer patients.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ We leveraged physiologically‐based pharmacokinetic modeling to characterize multiple target organ distribution in cancer patients and used this to evaluate the suggested dosing regimens for clinical activity and toxicity in this population.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The model was developed from tissue distribution studies performed in mice and applied to plasma concentration‐time data obtained from adult cancer patients receiving paclitaxel, with interspecies differences in physiological parameters taken into account.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study exemplifies that the final model adequately captured the observed concentrations in patients and allowed prediction of paclitaxel distribution profiles in multiple target organs and can be applied to further refine the chemotherapeutic treatment with a clinically important agent.
Paclitaxel is an antineoplastic agent that exhibits antitumor activities toward a wide array of solid tumors,1, 2 including breast cancer and non‐small cell lung cancer,3, 4 by inhibiting cellular growth through promoting and stabilizing microtubule assembly by a noncovalent interaction with tubulin.5 The disposition properties of paclitaxel are highly variable and unpredictable and constitute an important cause of unwarranted, potentially life‐threatening toxicities that remain a significant pharmaceutical challenge in medical oncology. Despite the established relationships between measures of systemic exposure and pharmacodynamic outcome of treatment, the mechanisms underlying the agent's unpredictable pharmacokinetics remain largely unexplained. It has been suggested that unfavorable pharmaceutical and physicochemical properties, including high lipophilicity and low solubility6 as well as variability in paclitaxel inactivation through hepatic metabolism by Cytochrome P450 3A4 (CYP3A4) and Cytochrome P450 (CYP2C8) could contribute to differential tolerability observed with paclitaxel‐based treatment.
Previous studies involving drug concentration monitoring in plasma have demonstrated a large volume of distribution for paclitaxel, suggesting extensive tissue distribution, uptake, and retention.7 The pharmacokinetic profile of paclitaxel in mice is characterized by a distinct nonlinearity in plasma levels, whereas concentrations in tissues are strictly dose proportional within a clinically relevant range of doses.8 The nonlinear profile has been ascribed to a time‐dependent and dose‐dependent interaction of paclitaxel with its formulation excipient Cremophor EL within the systemic circulation that causes temporarily altered blood cell distribution, which in turn impacts total plasma concentration, but not the unbound concentration.9, 10, 11 As the pharmacodynamic actions exerted by paclitaxel are generally more closely related to unbound than the total concentration of drug,12 proper integration of knowledge on tissue distribution features of paclitaxel is critical to further understanding of the drug's toxicity and efficacy profiles.
The purpose of the present study was to develop a whole‐body physiologically‐based pharmacokinetic (PBPK) model for evaluating and predicting the distribution of paclitaxel into various peripheral tissues. The model was developed based on detailed plasma and tissue distribution data obtained in mice and was then extrapolated to humans to describe the plasma concentration‐time profile of paclitaxel in cancer patients and predict its distribution to human tissues.
Materials and Methods
Drugs and chemicals
Paclitaxel and paclitaxel formulated as a stock solution of 6 mg/mL (Taxol) in 50% Cremophor EL in dehydrated ethanol with U.S. Pharmacopeia (USP) standard were supplied by the Bristol Myers Squibb (Princeton, NJ). Docetaxel, used as an internal standard, was also provided by Bristol Myers Squibb. All chemicals were of analytical grade or better (methanol and acetonitrile were high performance liquid chromatography grade solvents) and were obtained from Merck (Darmstadt, Germany). Water was purified by the Milli‐Q Plus system (Waters, Milford, MA).
Preclinical pharmacokinetic study
Female FVB mice, 10–14 weeks of age with a body weight of 23–29 g, were used throughout all experiments and were given food and water ad libitum. The animals were housed according to institutional guidelines, and the experimental procedures were approved by the Laboratory Animal Resources and Animal Care and Use Committee. A commercially available preparation containing paclitaxel at a concentration of 6 mg/mL formulated in Cremophor EL and ethanol (1:1, v/v) was diluted with isotonic sodium chloride to a final paclitaxel concentration of 3 mg/mL. The administered volume of Cremophor EL was 1.67 mL/kg, and the maximum volume of the diluted drug solution injected was always less than 200 μL. Paclitaxel was administered at a dose of 20 mg/kg body weight by a single intravenous (i.v.) bolus injection in the tail vein.
Blood and tissue specimens were obtained from four animals per time point at 0.5, 1, 4, 8, and 24 hours after drug administration. Because of the fact that animals were euthanized at early times to allow for the collection of tissues, we did not expressly assess neutropenia in our mouse studies. Blood samples were obtained from the retro‐orbital venous plexus, collected in 1.5‐mL polypropylene microtubes (Eppendorf, Hamburg, Germany) containing 7 USP units lithium heparin, and centrifuged at 2100 g for 10 minutes at 4°C. The plasma supernatant was separated and stored at −20°C until analysis within 2 weeks. Brain, dorsal fat, colon, cecum, small intestine, stomach, liver, kidneys, lungs, spleen, and heart were dissected, homogenized at 4°C in 5 or 10 volumes of 4% (w/v) bovine serum albumin in water, and stored at −20°C. Preliminary data from these murine studies were reported previously.8
Clinical pharmacokinetic study
Pharmacokinetic data for paclitaxel were obtained from patients receiving single‐agent paclitaxel at a dose of 175 mg/m2 as a 3‐hour continuous i.v. infusion, and details have been reported previously.13 The clinical protocol was approved by the Rotterdam Cancer Institute Review Board, and all patients signed informed consent before entering the study. Briefly, all patients had a confirmed solid tumor, were eligible for paclitaxel‐based chemotherapy, and were all > 18 years of age. Blood samples of 5 mL each were obtained from all patients before paclitaxel administration, at 1 and 2 hours after the start of infusion, immediately at the end of infusion, and at 5, 15, 30, and 45 minutes and 1, 2, 4, 6, 8, 12, and 21 hours after the end of infusion. Plasma was separated by centrifugation (4,000g for 5 minutes) at 4°C and stored at −80°C until analysis.
Pharmacokinetic analysis
Paclitaxel concentrations in plasma and tissues were determined by reversed‐phase high performance liquid chromatography with ultraviolet detection, as reported in detail elsewhere.8, 13 The pharmacokinetics of paclitaxel were initially evaluated by a noncompartmental (model‐independent) pharmacokinetic analysis (NCA) using Phoenix WinNonlin version 8.3 (Certara, Princeton, NJ). The elimination rate constant was estimated from the slope of the terminal phase of the log plasma concentration‐time curve fitted by the method of least squares, and the terminal half‐life was calculated by 0.693/elimination rate constant. The peak plasma concentration and the time to reach peak plasma concentration were obtained by visual inspection of the data from the concentration‐time curves. The area under the plasma concentration–time curve (AUC) was calculated according to the linear trapezoidal rule.
PBPK model development: Overview
A whole‐body PBPK model was developed with the observed paclitaxel data obtained in the pharmacokinetic studies in mice and included consideration of plasma (p) as well as heart (h), brain (br), gut (g), lung (lu), liver (l), spleen (s), kidney (k), fat (f), and muscle (m) (Figure 1). Paclitaxel concentrations were not determined in some organs, such as the skin, eye, and bone, and these were therefore included in the combined remainder compartment (r). As the impact of residual blood on tissue could be relevant, especially for low‐distribution organs such as the brain, we estimated the contribution of residual blood‐to‐brain distribution based on a 0.4% plasma contamination14 and found that the brain concentrations are overestimated by about 10%. The actual contamination effect is likely less given the fact that we did not perform ligation and that leakage occurred postresection. Biotransformation via CYP3A4 and CYP2C8 in the liver was assumed the only relevant route of elimination, in agreement with the results of prior studies.15 The PBPK modeling was performed using Phoenix WinNonlin version 8.3 software. The average total body weight of the mice 25 g, and data on cardiac output (Q) were derived from a previous report.16 The distribution of paclitaxel into the eight tissues examined—brain, fat, muscle, kidney, lung, spleen, heart, and gut—were described by a perfusion‐limited model, and the drug concentration‐time profile in the remainder tissues was described with a permeability‐limited model, where the limiting process of drug permeation is the cell membrane,17 according to Eqs. (1), (2), (3), (4), (5), (6).
Figure 1.
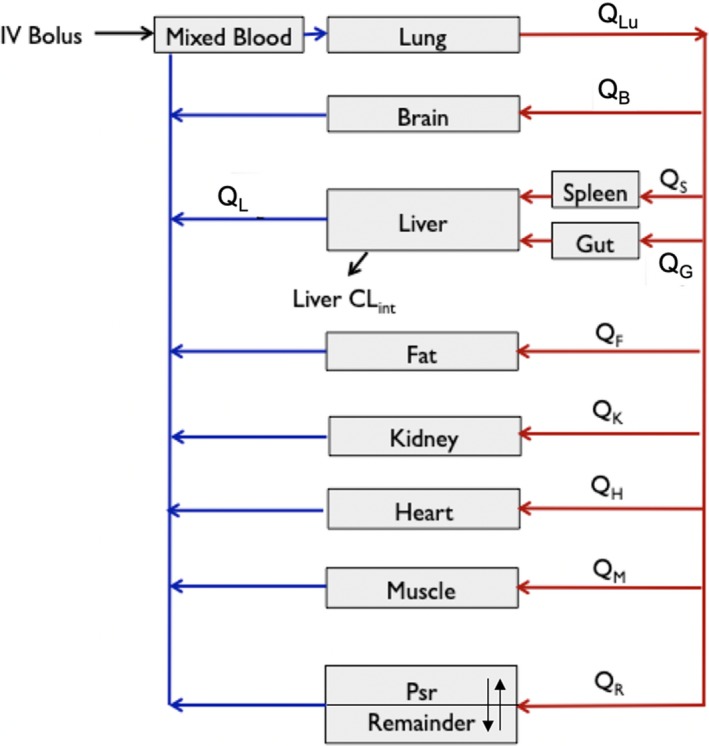
Schematic representation of a physiologically‐based pharmacokinetics model for paclitaxel in the mice. CLint, intrinsic clearance; IV, intravenous; Psr, permeability surface area conduct; QB, brain blood flow rate; QF, fat blood flow rate; QG, gut blood flow rate; QH, heart blood flow rate; QK, kidney blood flow rate; QL, liver blood flow rate; QLu, lung blood flow rate; QM, muscle blood flow rate; QS, spleen blood flow rate.
The applied equation for the perfusion rate‐limited model was:
| (1) |
The equation for liver as the eliminating organ was:
| (2) |
The equations for the permeability rate‐limited model were:
| (3) |
| (4) |
The equations for lung (Eq. 5) and arterial (Eq. 6) and venous (Eq. 7) plasma were:
| (5) |
| (6) |
| (7) |
In these equations, C t refers to the concentration of paclitaxel in tissue; C p to the plasma concentration in arteries; C b to the plasma concentration in venes; C lu to the concentration in lungs; Q t and V t to tissue blood flow rate and tissue volume, respectively; PSr to permeability surface area product; K pr to the partition coefficient of tissue; fu to the fraction unbound paclitaxel in plasma; C t,ISF and C t,c to drug concentration in the interstitial fluid and cells, respectively; and V t,c and V r ISF to the intracellular and extracellular volumes, respectively.
Model parameterization and scale‐up from mice to humans
Physiological parameters of each organ in mice and humans used in the model are listed in Table S1 ,16 and the fu of paclitaxel was fixed to a value of 0.05.18 All parameters were estimated by the full‐body PBPK model, and the K pt was calculated according to Eq. 8, where the values for AUCplasma, 0−last were calculated by NCA.
| (8) |
The initial estimates for CLint,H were calculated from the well‐stirred liver distribution model (Eq. 8).
| (9) |
In this equation, CLH and Q H represent the liver clearance and blood flow rate in mice, respectively; and fu is the fraction unbound paclitaxel. The CLint used in the human simulation was set to a value of 1,410 L/hour. Values for PSt in humans were derived by using an allometric growth scale equation PSt = A(M) B, where M represents organ weight, A is estimated by the PBPK model, and B was fixed to a value of 0.75 as the power function.
Model evaluation and sensitivity analysis
The full‐body PBPK models were evaluated by the Akaike information criterion and goodness‐of‐fit plots. Analysis using the Akaike information criterion was performed using Eqs. 10 and 11.
| (10) |
In the equation, Re are residuals, ∑ the sum, W a weighting factor, C i the observed concentration, and C aj the adjusted concentration.
| (11) |
In the equation, N represents the number of data points, Re are residual values, and P the number of pharmacokinetic parameters.
A normalized sensitivity analysis was conducted, as described,19 to evaluate the effect of each model parameter on the simulated paclitaxel plasma AUC for both mouse and human models. Normalized sensitivity coefficients (SC) were calculated as the ratio of percentage change in AUC as a result of 1% increase in a given model parameter: Each parameter was analyzed individually, keeping all other parameters fixed at their original levels. A small absolute value of SC indicates that the model output is insensitive to the parameter, whereas absolute values of SC > 1 suggest that there may amplify parameter errors.20 The model prediction check was performed using the Predictive Check of Run Options in Phoenix in WinNonlin. Simulations were performed on a total of 500 individuals, and the variability parameters included Klu, KCll, Kr, Kg, Km, Ks, Kl, Kk, Kh, Kf, Kbr, PSr, and Krt.
Results
The preclinical pharmacokinetics of paclitaxel were investigated in mice after i.v. administration of a dose of 20 mg/kg. Concentrations of paclitaxel in the plasma, spleen, and heart declined to undetectable levels 4–8 hours after drug administration, although in most organs paclitaxel was measurable even at 24 hours (Figure 2). The observed tissue‐to‐plasma partition coefficients of paclitaxel were in increasing order brain (K pt = 0.03) < muscle (K pt = 0.38) < heart (K pt = 0.49) < fat (K pt = 0.61) < spleen (K pt = 0.73) < lung (K pt = 0.78) < kidney (K pt = 1.03) < gut (K pt = 1.32) < liver (K pt = 2.74) (Table 1). A remainder compartment was included as a permeability rate‐limited model (Figure 3), which included the bone, skin, eye, and so on.
Figure 2.
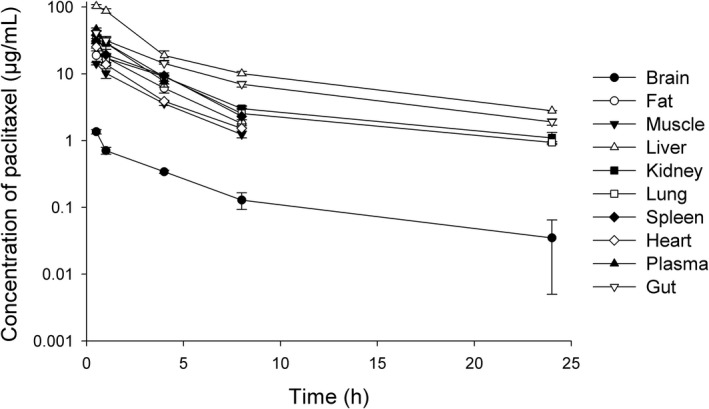
Paclitaxel concentrations in mice plasma and in nine examined tissues (brain, fat, gut, spleen, heart, kidney, liver, lung, muscle) following intravenous bolus administration of 20 mg/kg paclitaxel in mice (n = 4).
Table 1.
Paclitaxel pharmacokinetic parameter estimation and precision (CV%) in mouse plasma and tissue using the proposed PBPK model
| Organ | Parameter | NCA estimated | PBPK estimated | CV% | 2.5% CI | 97.5% CI | Difference, % |
|---|---|---|---|---|---|---|---|
| Spleen | K s | 0.73 | 0.83 | 14.06 | 0.59 | 1.06 | 10.97 |
| Kidney | K k | 1.03 | 1 | 13.41 | 0.73 | 1.28 | 2.34 |
| Heart | K h | 0.49 | 0.54 | 13.95 | 0.38 | 0.69 | 7.78 |
| Lung | K lu | 0.78 | 0.77 | 13.67 | 0.56 | 0.99 | 1.26 |
| Liver | K l | 2.74 | 2.8 | 13.66 | 2.02 | 3.58 | 1.99 |
| Clint,H (mL/hour) | 89.26 | 79.72 | 11.7 | 60.69 | 98.74 | 11.97 | |
| Gut | K g | 1.32 | 1.61 | 14.23 | 1.14 | 2.08 | 18.09 |
| Muscle | K m | 0.38 | 0.37 | 13.87 | 0.27 | 0.48 | 1.27 |
| Fat | K f | 0.61 | 0.58 | 13.95 | 0.42 | 0.75 | 5.6 |
| Brain | K br | 0.03 | 0.03 | 13.65 | 0.03 | 0.04 | 8.36 |
| Remainder | K r | 0.52 | 35.05 | 0.15 | 0.9 | ||
| PSr (mL/hour) | 43 | 14.85 | 29.98 | 56.02 | |||
| K ISF | 2.63 | 29.47 | 1.05 | 4.21 |
CI, confidence interval; CV%, coefficient of variation; NCA, noncompartmental pharmacokinetic analysis; PBPK, physiologically‐based pharmacokinetic; Ks, spleen partition coefficient; Kk, kidney partition coefficient; Kh, heart partition coefficient; Klu, lung partition coefficient; Kl, liver partition coefficient; Kg, gut partition coefficient; Km, muscle partition coefficient; Kf, fat partition coefficient; Hbr, brain partition coefficient; Kr, reminder partition coefficient; Clint,H, intrinsic clearance; PSr, permeability surface area conduct; KISF, interstitial fluid partition coefficient.
Figure 3.
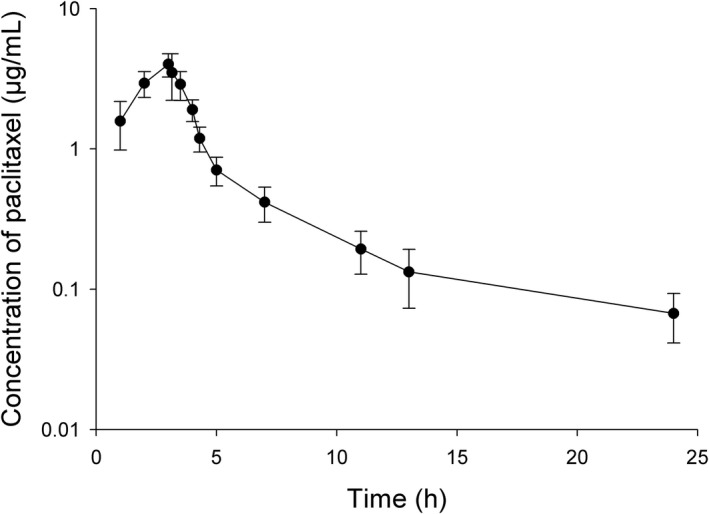
Paclitaxel concentrations in plasma concentration of paclitaxel following intravenous drug administration of a dose of 175 mg/m2 in patients with solid tumors (n = 14).
The applied preclinical PBPK model adequately described the concentration‐time profiles of paclitaxel in all tested tissues as well as in plasma as judged on the basis of goodness‐of‐fit plots (Figure 4), confirming that the experimental data were very similar to the PBPK‐based model predictions. As a measure of precision, the values for the coefficient of variation were always less than 15% in all of the tissues evaluated, except for the remainder compartment, where values were < 36%. When comparing values for AUC0−last from NCA to those estimated from the model, the differences were < 18% (Table 2).
Figure 4.
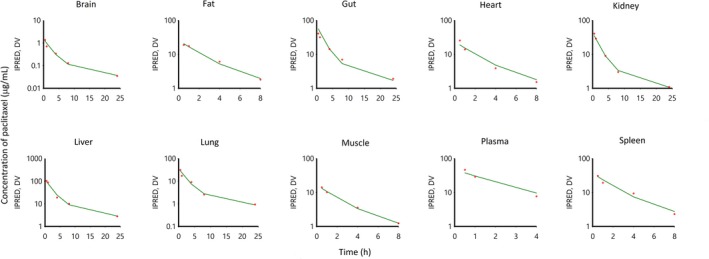
Observed mean paclitaxel data (dots) and predicted (lines) paclitaxel concentrations in plasma and various tissues in mice with a dose of 20 mg/kg paclitaxel by intravenous administration.
Table 2.
Paclitaxel pharmacokinetic parameters for mice in plasma and various tissues calculated based on the experimental data and PBPK predictions using NCA
| Parameter | Unit | NCA estimated | PBPK estimated | Difference, % |
|---|---|---|---|---|
| AUCspleen 0−last | Hour × µg/mL | 115.13 | 130.85 | 12.02 |
| AUCliver 0−last | Hour × µg/mL | 379.04 | 365.79 | 3.62 |
| AUCkidney 0−last | Hour × µg/mL | 138.83 | 137.84 | 0.72 |
| AUCheart 0−last | Hour × µg/mL | 52.45 | 56.09 | 6.48 |
| AUCplasma 0−last | Hour × µg/mL | 106.08 | 104.55 | 1.46 |
| AUClung 0−last | Hour × µg/mL | 82.75 | 87.19 | 5.09 |
| AUCgut 0−last | Hour × µg/mL | 139.98 | 169.42 | 17.38 |
| AUCmuscle 0−last | Hour × µg/mL | 40 | 39.35 | 1.64 |
| AUCfat 0−last | Hour × µg/mL | 65.03 | 61.22 | 6.22 |
| AUCbrain 0−last | Hour × µg/mL | 3.38 | 3.63 | 7.04 |
NCA, noncompartmental pharmacokinetic analysis; PBPK, physiologically‐based pharmacokinetic; AUCspleen, area under the curve for spleen; AUCliver, area under the curve for liver; AUCkidney, area under the curve for kidney; AUCheart, area under the curve for heart; AUCplasma, area under the curve for plasma; AUClung, area under the curve for lung; AUCgut, area under the curve for gut; AUCmuscle, area under the curve for muscle; AUCfat, area under the curve for fat; AUCbrain, area under the curve for brain.
The human PBPK model was created by incorporating known human physiological data, including blood flow rate and variability in organ volumes from a 70‐kg individual, and by assuming that the principal elimination pathway of paclitaxel is based on CYP3A4‐mediated and CYP2C8‐mediated hepatic metabolism. A comparison of the observed and PBPK‐model simulated plasma concentration‐time profiles of paclitaxel in 14 cancer patients receiving a standard dose of 175 mg/m2 (3‐hour i.v. infusion) demonstrated adequate predictions (Figure 5). The sensitivity coefficients for the mouse (20 mg/kg dose) and human (175 mg/m2 dose) PBPK model with regard to plasma AUC were < 0.01, indicating that the model output is insensitive to variation in the given parameter. The only parameter that stood out among others was the intrinsic clearance in the human PBPK model, but its absolute value for the sensitivity coefficient was still considered to be relatively small (0.5). We also tested the effect of parameter changes to plasma AUC and found minimum influences on AUC (<1%), even when parameters such as Ks, Kl, Kk, Kh, Klu, Kf, Kbr were changed by twofold. This further suggests that our model provides a reliable prediction of the paclitaxel plasma and tissue profiles irrespective of variability of the source.
Figure 5.
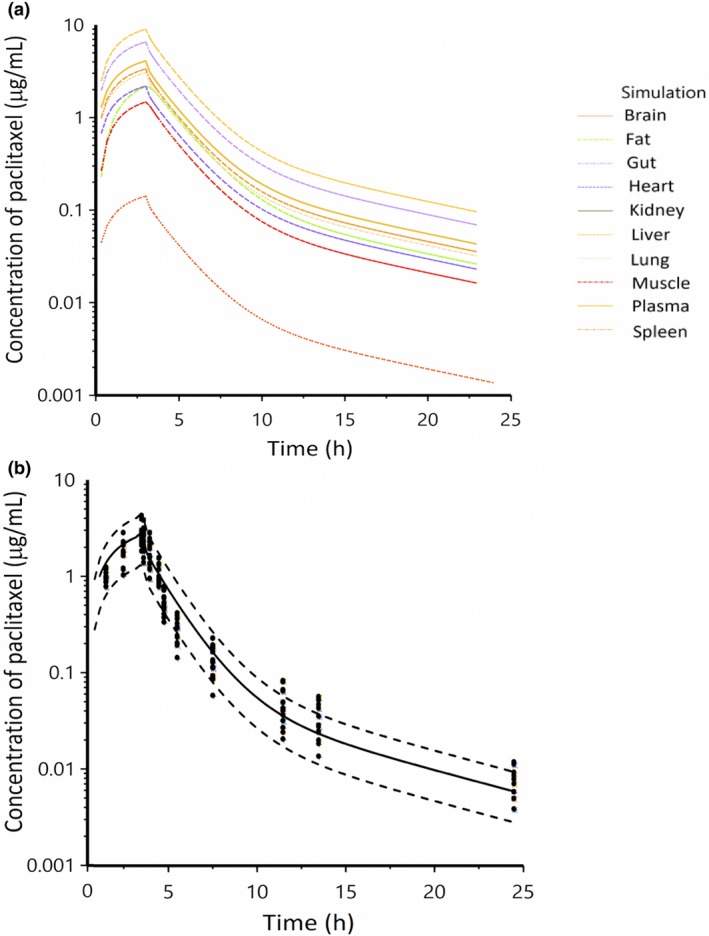
Observed and physiologically‐based pharmacokinetic (PBPK) model‐simulated concentrations of paclitaxel in tissues (a) and plasma (b) following a dose of 175 mg/m2 in patients with solid tumors. Black dots represent observed data, and solid lines and dash lines represent the mean and 90% confidence intervals of the simulations from the proposed PBPK model.
Discussion
PBPK modeling represents a useful and increasingly applied tool to evaluate the disposition of drugs in various tissues and organs. It can be employed to predict drug distribution profiles in as yet untested or untestable situations as well as to increase understanding of possible relationships between tissue exposure and drug effects. In the current study, we applied a PBPK modeling strategy to gain insights into the tissue distribution properties in mice and humans of the widely used anticancer agent paclitaxel. Using a clinically relevant drug dose of 20 mg/kg, we found that the concentration‐time profiles of paclitaxel in mice could be best fit with a perfusion‐limited kinetic in all organs. The distribution of paclitaxel was particularly extensive in highly perfused tissues, such as the gut and the liver, with partition coefficients ranging from 1.32 to 2.74. The extensive distribution of paclitaxel into the small intestine and liver are consistent with our previously reported transport mechanisms in which paclitaxel was demonstrated to undergo extensive intestinal ATP binding cassette subfamily B member 1 (ABCB1)‐mediated intestinal secretion after i.v. administration21 as well as organic anion‐transporting polypeptide 1B2 (OATP1B2)‐dependent hepatocellular uptake.22 In contrast, the partition coefficients of paclitaxel in the mouse brain was only 0.03, and these low concentrations in the brain are consistent with previous observations that paclitaxel exhibits poor penetration across the blood–brain barrier in both rodents23, 24 and cancer patients25 as a result of ATP binding cassette subfamily (ABC) transporter‐mediated efflux from brain capillaries.26 The distribution of paclitaxel across cell membranes in the remainder tissues was predicted to be permeability rate limited, although consideration of binding in some deep peripheral tissues did not further improve the fit and stability of the model.
The developed PBPK model was next extrapolated from mice to humans to fit the observed plasma concentration‐time profiles and to predict the distribution of paclitaxel into various tissues. We found that the model adequately predicted the plasma concentration‐time profiles of paclitaxel in the patients, with almost all observations falling within the 90% confidence interval of the simulated concentrations. Compared with more traditional NCA methods, the present analysis represents a more robust modeling that is less influenced by sampling, analytical, and/or dosing errors. In addition, in comparison with conventional allometric scaling to estimate human tissue drug distribution properties,27, 28 the predictions obtained here with our full‐body PBPK model is likely to be more precise and accurate because the latter is able to employ prior knowledge of physiological relevance, including specific parameters related to tissue volume and blood flow rate, and include experimental data on intrinsic clearance obtained in nonhuman animals. In addition, further refinements of the developed PBPK model in the future can also consider the activity, specificity, and potential species differences of drug‐metabolizing enzymes and transport mechanisms of relevance to drug distribution and elimination.
The ability to predict the time profile of paclitaxel concentrations in specific tissues in cancer patients provides a potential clinically useful tool to monitor exposure to paclitaxel as a function of treatment duration. This is particularly important in view of the notion that tissue‐specific distribution features associated with paclitaxel that are relevant to treatment‐related side effect profiles cannot necessarily be accurately predicted on the basis of plasma concentrations alone.29 In this context, it is worth pointing out that, in most common cancer types for which treatment with paclitaxel is indicated, the efficacy remains rather limited but is combined with a significant degree of toxicity. The major reasons for this have long presumed to have been the lack of detailed knowledge in tumor cell biology and inappropriate, poorly predictable preclinical models for the identification and testing of treatment regimens.30 Although the standard strategy still in use for the dose selection of paclitaxel is to apply the therapeutic dose originally determined in phase II trials and subsequently, at best, modify it for individual differences in body‐surface area,31 there is a wealth of experimental data indicating that both the efficacy and safety of paclitaxel treatment might be optimized if dosing strategies would take into consideration individual patient characteristics as they relate to the agent's unique pharmacokinetic profile.32 It is difficult to make specific recommendations for dosing changes of paclitaxel‐containing chemotherapeutic regimens on the basis of the current findings. Although monitoring of (unbound) paclitaxel plasma concentrations and dosage adjustment may be necessary to optimize treatment efficacy in cancer patients,33 therapeutic drug monitoring of paclitaxel is still not available for routine implementation. It is noted that our described model is without tumor analysis because of the fact that data were obtained from healthy mice. Recently, a novel statistical approach for a preclinical‐to‐clinical translation of first‐order net growth rate constant parameter became available that could potentially solve the issue of mice‐to‐human differences in cancer growth.34 It is our intention to apply this novel approach to further refine the model in the future by integrating data from our currently ongoing murine xenograft studies. Despite this limitation, the described PBPK model continues to increase our knowledge on this clinically important drug and provides the basis for designing future prospective investigations aimed at refining the model. Further validation of the developed model with additional, prospectively generated clinical pharmacokinetic data are needed, and it is a current ongoing effort in our lab.
In conclusion, the current PBPK analysis confirms a number of findings previously described by conventional pharmacological analyses as well as another recently reported PBPK model.35 This latter model was derived from published literature data on paclitaxel distribution in rodent tissues reported by other investigators and was captured by Plot Digitizer (GetData Graph Digitizer, Version 2.26). In contrast, we used our own tissue distribution data and had access to the actual raw data and pertinent information on experimental design that were absent in some of the previously reported findings. The absence of this information in the published PBPK model may have contributed to the exceptional extent of variability between the various preclinical studies. It should also be noted that, when compared with our current model, the preclinical studies used in the previous PBPK model did not include distribution profiles for paclitaxel to several important organs we had access to for model prediction, including muscle, fat, and brain. Regardless of these differences in the approach and final outcome, both of the presently available PBPK models have further enhanced our understanding of the complex interactions of biological and physiologic parameters that affect the pharmacokinetics of paclitaxel and provide a foundation for the reevaluation of the mechanisms involved in drug interactions with paclitaxel36 as well as for the assessment of alternative and potentially improved dosing regimens for this clinically important drug.
Funding
The project was supported in part by National Institutes of Health Grants R01CA215802 (A.S.) and R01CA238946 (S.H. and M.B.L) and by The Ohio State University Comprehensive Cancer Center using Pelotonia funds. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
Q.F., M.B.L., A.S., and S.H. wrote the manuscript. Q.F., A.S., and S.H. designed the research. Q.F. and X.S. performed the research. Q.F., X.S., A.S., and S.H. analyzed the data.
Supporting information
Table S1. Physiological and kinetic parameters for paclitaxel PBPK simulations in mice and humans.
Code S1.
Acknowledgments
We would like to acknowledge the excellent technical assistance of Alice Gibson and thank Sharyn Baker for a critical review of the manuscript.
References
- 1. McGuire, W.P. et al Taxol: a unique antineoplastic agent with significant activity in advanced ovarian epithelial neoplasms. Ann. Intern. Med. 111, 273–279 (1989). [DOI] [PubMed] [Google Scholar]
- 2. Wani, M.C. , Taylor, H.L. , Wall, M.E. , Coggon, P. & McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 93, 2325–2327 (1971). [DOI] [PubMed] [Google Scholar]
- 3. Chang, A.Y. et al Phase II study of taxol, merbarone, and piroxantrone in stage IV non‐small‐cell lung cancer: the Eastern Cooperative Oncology Group results. J. Natl. Cancer. Inst. 85, 388–394 (1993). [DOI] [PubMed] [Google Scholar]
- 4. Holmes, F.A. et al Phase II trial of Taxol, an active drug in the treatment of metastatic breast cancer. J. Natl. Cancer. Inst. 83, 1797–1805 (1991). [DOI] [PubMed] [Google Scholar]
- 5. Kumar, N. Taxol‐induced polymerization of purified tubulin. Mechanism of action. J. Biol. Chem. 256, 10435–10441 (1981). [PubMed] [Google Scholar]
- 6. ten Tije, A.J. , Verweij, J. , Loos, W.J. & Sparreboom, A. Pharmacological effects of formulation vehicles: implications for cancer chemotherapy. Clin. Pharmacokinet. 42, 665–685 (2003). [DOI] [PubMed] [Google Scholar]
- 7. Spencer, C.M. & Paclitaxel, F.D. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs 48, 794–847 (1994). [DOI] [PubMed] [Google Scholar]
- 8. Sparreboom, A. , van Tellingen, O. , Nooijen, W.J. & Beijnen, J.H. Tissue distribution, metabolism and excretion of paclitaxel in mice. Anticancer. Drugs. 7, 78–86 (1996). [DOI] [PubMed] [Google Scholar]
- 9. Gianni, L. et al Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J. Clin. Oncol. 13, 180–190 (1995). [DOI] [PubMed] [Google Scholar]
- 10. Sparreboom, A. , van Tellingen, O. , Nooijen, W.J. & Beijnen, J.H. Nonlinear pharmacokinetics of paclitaxel in mice results from the pharmaceutical vehicle Cremophor EL. Cancer. Res. 56, 2112–2115 (1996). [PubMed] [Google Scholar]
- 11. Sparreboom, A. et al Cremophor EL‐mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications. Cancer. Res. 59, 1454–1457 (1999). [PubMed] [Google Scholar]
- 12. Henningsson, A. et al Mechanism‐based pharmacokinetic model for paclitaxel. J. Clin. Oncol. 19, 4065–4073 (2001). [DOI] [PubMed] [Google Scholar]
- 13. van Zuylen, L. et al Pharmacokinetic modeling of paclitaxel encapsulation in Cremophor EL micelles. Cancer. Chemother. Pharmacol. 47, 309–318 (2001). [DOI] [PubMed] [Google Scholar]
- 14. Kaliss, N. & Pressman, D. Plasma and blood volumes of mouse organs, as determined with radioactive iodoproteins. Proc. Soc. Exp. Biol. Med. 75, 16–20 (1950). [DOI] [PubMed] [Google Scholar]
- 15. Rowinsky, E.K. , Cazenave, L.A. & Donehower, R.C. Taxol: a novel investigational antimicrotubule agent. J. Natl. Cancer. Inst. 82, 1247–1259 (1990). [DOI] [PubMed] [Google Scholar]
- 16. Davies, B. & Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 10, 1093–1095 (1993). [DOI] [PubMed] [Google Scholar]
- 17. Jones, H. & Rowland‐Yeo, K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics. Syst. Pharmacol. 2, e63 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brouwer, E. et al Measurement of fraction unbound paclitaxel in human plasma. Drug. Metab. Dispos. 28, 1141–1145 (2000). [PubMed] [Google Scholar]
- 19. Loccisano, A.E. , Campbell, J.L. Jr , Butenhoff, J.L. , Andersen, M.E. & Clewell, H.J. 3rd Comparison and evaluation of pharmacokinetics of PFOA and PFOS in the adult rat using a physiologically based pharmacokinetic model. Reprod. Toxicol. 33, 452–467 (2012). [DOI] [PubMed] [Google Scholar]
- 20. Hudachek, S.F. & Gustafson, D.L. Physiologically based pharmacokinetic model of lapatinib developed in mice and scaled to humans. J. Pharmacokinet. Pharmacodyn. 40, 157–176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sparreboom, A. et al Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P‐glycoprotein in the intestine. Proc. Natl. Acad. Sci. USA 94, 2031–2035 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nieuweboer, A.J. et al Influence of drug formulation on OATP1B‐mediated transport of paclitaxel. Cancer. Res. 74, 3137–3145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fellner, S. et al Transport of paclitaxel (Taxol) across the blood‐brain barrier in vitro and in vivo. J. Clin. Invest. 110, 1309–1318 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamasaki, Y. et al Characterization of P‐glycoprotein humanized mice generated by chromosome engineering technology: its utility for prediction of drug distribution to the brain in humans. Drug. Metab. Dispos. 46, 1756–1766 (2018). [DOI] [PubMed] [Google Scholar]
- 25. Glantz, M.J. et al Paclitaxel disposition in plasma and central nervous systems of humans and rats with brain tumors. J. Natl. Cancer. Inst. 87, 1077–1081 (1995). [DOI] [PubMed] [Google Scholar]
- 26. Kemper, E.M. et al Increased penetration of paclitaxel into the brain by inhibition of P‐Glycoprotein. Clin. Cancer. Res. 9, 2849–2855 (2003). [PubMed] [Google Scholar]
- 27. Rowland, M. , Peck, C. & Tucker, G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 51, 45–73 (2011). [DOI] [PubMed] [Google Scholar]
- 28. Tang, H. & Mayersohn, M. A global examination of allometric scaling for predicting human drug clearance and the prediction of large vertical allometry. J. Pharm. Sci. 95, 1783–1799 (2006). [DOI] [PubMed] [Google Scholar]
- 29. Leblanc, A.F. et al OATP1B2 deficiency protects against paclitaxel‐induced neurotoxicity. J. Clin. Invest. 128, 816–825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sparreboom, A. & Figg, W.D. Identifying sources of interindividual pharmacokinetic variability with population modeling. Clin. Cancer. Res. 12, 1951–1953 (2006). [DOI] [PubMed] [Google Scholar]
- 31. Smorenburg, C.H. et al Randomized cross‐over evaluation of body‐surface area‐based dosing versus flat‐fixed dosing of paclitaxel. J. Clin. Oncol. 21, 197–202 (2003). [DOI] [PubMed] [Google Scholar]
- 32. Joerger, M. , Huitema, A.D. , van den Bongard, D.H. , Schellens, J.H. & Beijnen, J.H. Quantitative effect of gender, age, liver function, and body size on the population pharmacokinetics of Paclitaxel in patients with solid tumors. Clin. Cancer. Res. 12, 2150–2157 (2006). [DOI] [PubMed] [Google Scholar]
- 33. Joerger, M. et al Validation of a commercial assay and decision support tool for routine paclitaxel therapeutic drug monitoring (TDM). Ther. Drug. Monit. 39, 617–624 (2017). [DOI] [PubMed] [Google Scholar]
- 34. Kay, K. , Dolcy, K. , Bies, R. & Shah, D.K. Estimation of solid tumor doubling times from progression‐free survival plots using a novel statistical approach. AAPS J. 21, 27 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zang, X. & Kagan, L. Physiologically‐based modeling and interspecies prediction of paclitaxel pharmacokinetics. J. Pharmacokinet. Pharmacodyn. 45, 577–592 (2018). [DOI] [PubMed] [Google Scholar]
- 36. Qiang, F. et al Effect of maceligan on the systemic exposure of paclitaxel: in vitro and in vivo evaluation. Eur. J. Pharm. Sci. 41, 226–231 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Physiological and kinetic parameters for paclitaxel PBPK simulations in mice and humans.
Code S1.