

Abstract
We report the case of a woman with syncope and persistently prolonged QTc interval. Screening of congenital long QT syndrome (LQTS) genes revealed that she was a heterozygous carrier of a novel KCNH2 mutation, c.G238C. Electrophysiological and biochemical characterizations unveiled the pathogenicity of this new mutation, displaying a 2‐fold reduction in protein expression and current density due to a maturation/trafficking‐deficient mechanism. The patient's phenotype can be fully explained by this observation. This study illustrates the importance of performing genetic analyses and mutation characterization when there is a suspicion of congenital LQTS. Identifying mutations in the PAS domain or other domains of the hERG1 channel and understanding their effect may provide more focused and mutation‐specific risk assessment in this population.
Ann Noninvasive Electrocardiol 2011;16(2):213–218
Keywords: electrophysiology, long QT syndrome, clinical, ion channels and membrane transporters, basic, molecular biology/genetics
INTRODUCTION
Congenital long QT syndrome (LQTS) is a genetic disorder primarily resulting from mutation‐induced loss‐of‐function of cardiac ion channels or mutations in channel‐associated proteins, and characterized by the electrocardiographic finding of QT interval prolongation and T wave abnormalities. 1 This syndrome affects an estimated 1 in 2500 people 2 and typically presents with syncope, seizures or sudden death. Congenital LQTS types are named depending on the gene affected. There are currently 13 genes reported and among them 6 encode potassium channel subunits. 3 , 4 , 5 KCNQ1 and KCNH2 genes encode voltage‐gated potassium channels essential for the repolarizing currents (IKs and IKr) of the cardiac action potential and thus its duration. They are the most common genes mutated in congenital LQTS, type1 (LQT1) and 2 (LQT2), respectively. Nearly 600 LQT2‐mutations have been reported in KCNH2, the gene encoding the hERG1 protein (http://www.HGMD.cf.ac.uk). Recently, several mutations in the amino‐terminus PAS‐domain were reported, all showing defective maturation and/or trafficking of hERG1 channels to the cell membrane as well as reduced channel currents. 6 , 7 , 8 , 9 , 10
In the present study, we report the case of a young woman with syncope and a persistently prolonged QTc interval. Screening of congenital‐LQTS genes revealed that she was a heterozygous carrier of a yet‐undescribed KCNH2 mutation in the PAS domain. Electrophysiological and biochemical characterizations unveiled the pathogenicity of this mutation and highlighted the importance of trafficking‐deficient mechanisms affecting PAS‐domain mutations of hERG1.
CASE PRESENTATION
A 20‐year‐old female of Caucasian origin was referred to our hospital after having 2 episodes of syncope. The first syncopal event occurred 10 months prior with clonical‐type movements, but without tongue bite or urinary loss. No cardiac assessment was performed at that time. The second episode occurred a few days prior to presentation. According to the patient's report a short malaise preceded a 5‐minute long fainting period, but no one witnessed the event. Both episodes were postprandial, while the patient was at rest and without adrenergic stimulation. Outpatient cardiac evaluation showed a prolonged QT interval, QTc values of 536 ms to 575 ms, and variations in T wave morphology. Echocardiography revealed very mild mitral valve regurgitation with no evidence of valvular prolapse. Cardiac assessment upon admission to our hospital confirmed the prolonged QT interval (Fig. 1, QT = 560 ms) and showed bifid T waves in precordial leads. During exercise testing no further QT prolongation was measured (Fig. 2), suggesting a limited risk during exercise activities in accordance with the clinical history of the patient. Because patient's ECG recorded at admission was consistent with congenital LQTS, and more specifically LQT2 because of the characteristic bifid T wave morphology, 11 , 12 the patient was started on beta‐blocker treatment (metoprolol, 25 mg daily), with no further episodes reported since. The clinical diagnosis of congenital LQTS was followed by genetic screening of the main genes known to be involved in LQTS, leading to the identification of a new heterozygous missense mutation in KCNH2 (c.G238C, p.A80P).
Figure 1.
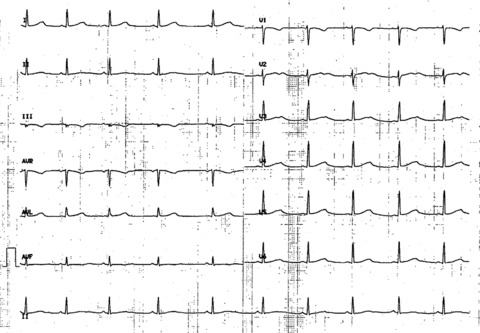
12‐lead ECG recorded at admission. Sinus rhythm 58 bpm. PR: 140 ms. QRS: 80 ms, normal axis. Prolongation of QT interval with an obvious bifid T wave in precordial lead V2‐V3, and a subtle bifid T wave in V4‐V6. QTc: 560 ms.
Figure 2.
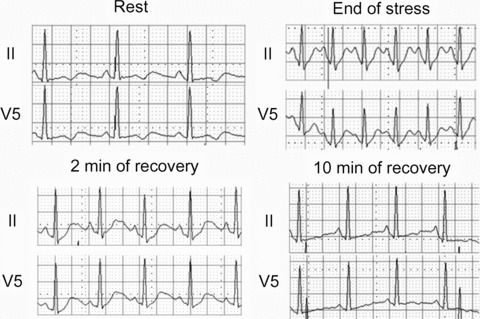
12‐lead ECG recorded before and during exercise test. QTUc measurement in leads II and V5: at rest (570 ms), at the end of stress (520 ms), after 2 min (550 ms) and 10 min of recovery (550 ms).
The first degree family history revealed that the patient's parents and two siblings were in good health. The enlarged family history showed only one case (a maternal great‐uncle) of a relatively premature and undetermined death reported to be due to myocardial infarction. A family letter was given to the patient to inform her relatives regarding LQTS condition and mode of inheritance. Information was given regarding incomplete penetrance, diagnostic and therapeutic options. 13 The proband's father and sister underwent cardiologic examination and their ECG showed no abnormalities. Both declined genetic testing. Other relatives did not undertake cardiologic or genetic testing until now.
METHODS
A complete version of the methods is presented in Supporting Information file 1.
MOLECULAR AND FUNCTIONAL ANALYSES
Genetic screening of the proband for the three main LQTS genes revealed the presence of a heterozygous point mutation in KCNH2, with no abnormal variants in KCNQ1 or SCN5A. The KCNH2 c.G238C mutation translates into the missense p.A80P mutation at the protein level, which affects a residue of the amino‐terminal PAS domain of the hERG1 channel.
Generation of the plasmid encoding the hERG1‐A80P mutant allowed for its electrophysiological and biochemical characterization. Patch‐clamp technique in whole cell configuration was used to measure the current density in CHO cells transiently expressing wild‐type (WT) and mutant hERG1 channels (Fig. 3A). When compared with hERG1‐WT condition (100%± 10% in Fig. 3A), the heterozygous‐mimicking condition (hERG1‐WT/A80P) elicited only half of the tail current at −20mV (52%± 11%), whereas complete hERG1‐A80P elicited almost no current (4%± 2%). These results are consistent with a complete loss of function of the hERG1‐A80P mutant in a haploinsufficient phenotype. Protein expression was assessed using Western blot analysis (Fig. 3B), where hERG1‐WT protein appears with a lower band at 135kDa (nonmature form) and a higher band at 155kDa (fully glycosylated and mature form). Functional channels at the cell surface are mainly constituted by fully glycosylated subunits. There was a clear decrease of the fully glycosylated band in the heterozygous‐mimicking condition (46%± 5% of hERG1‐WT 155kDa band), and no signal was observed for the A80P‐mutant homozygous condition (0.6%± 2.8% of hERG1‐WT 155kDa band). These results are in line with the functional assay of current measurement. Interestingly, the nonmature band was still present for the homozygous‐mutant condition. The signal intensity for the 135kDa band of hERG1‐A80P was 22%± 6% of hERG1‐WT, and heterozygous condition presented intermediate intensity values of 66%± 7% (Fig. 3B).
Figure 3.
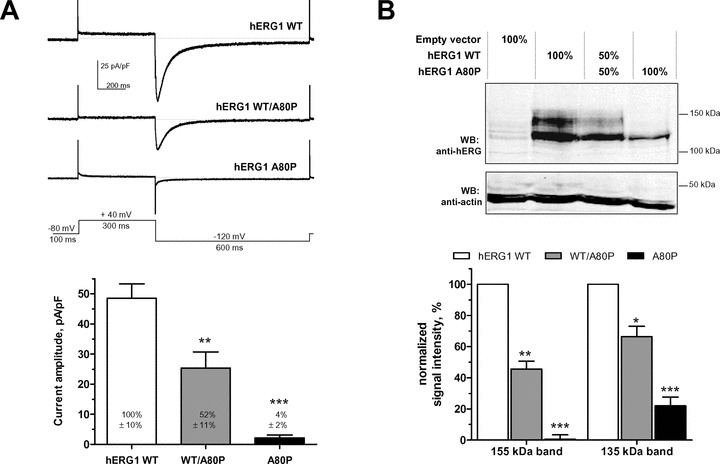
Electrophysiological and biochemical characterization of hERG1‐A80P mutant (A) Upper panel: patch‐clamp current recordings of hERG1‐WT, heterozygous (hERG1‐WT/A80P) and homozygous hERG1‐A80P mutant obtained in whole cell configuration according to the depicted protocol. Lower panel: Quantification of ‐120‐mV peak tail currents of hERG1‐WT, heterozygous (hERG1‐WT/A80P) and homozygous hERG1‐A80P mutant for n = 12–18 cells per condition and at least 3 independent experiments. (B) Upper panel: Protein expression of hERG1‐WT, heterozygous (hERG1‐WT/A80P) and homozygous hERG1‐A80P mutant with Western blot analyses. Lower panel: Fully glycosylated (mature, 155kDa band) and core‐glycosylated (nonmature, 135kDa band) protein quantification of hERG1‐WT, heterozygous (hERG1‐WT/A80P) and homozygous hERG1‐A80P mutant upon 3 independent experiments.
DISCUSSION
The PAS domain is a highly structured domain in the amino‐terminus of the hERG1 channel. It interacts with the core of the channel and influences the rate of deactivation. 9 Due to its important packing of β‐sheets and α‐helices, the amino‐terminus of hERG1 could be crystallized and is available in the Protein Data Bank (pdb entry: 1byw), which enabled us to precisely locate the residue affected in the largest α‐helix of the PAS domain (αC). Recently, Shimizu et al. 14 investigated the effect of location, coding type, and topology of hERG1 mutations on clinical phenotype in a cohort of 858 LQT2 patients. Patients with missense mutations in the transmembrane pore region and amino‐terminus had a significantly greater risk for first cardiac events than the ones with carboxy‐terminal mutations (HR 2.87 and 1.86, respectively). 14 With regard to the topology of mutation, the QTc interval was longer and cardiac events were more frequent among patients with mutations located in α‐helical domains than among patients with mutations in either β‐sheets or other uncategorized locations. 14 Regarding the missense mutation in the αC helix of the PAS domain described in this study, note that a proline residue is different from other amino acids because of its distinctive cyclic structure that locks the dihedral angle and provides important conformational rigidity. Such a mutation in an α‐helix can disrupt the proper folding of the amino‐terminus, and the misfolded protein may be recognized and degraded by the cellular quality‐control machinery. Functional analysis and expression patterns of the hERG1‐A80P protein allows us to conclude that the mutant channel is biosynthesized and expressed in the endoplasmic reticulum (ER), although to a lesser extent than the wild type, as seen by the weaker 135kDa band. In addition maturation in the Golgi complex, where the final glycosylation takes place, is not possible since the protein may be recognized and destroyed by the ER‐associated degradation pathway. Therefore, in the heterozygous state, the ability of mutant hERG1‐A80P proteins to form heterotetramers with wild‐type subunits is lost. Only the complete WT hERG1 channels are able to reach the cell membrane and elicit current, leading to haploinsufficiency. Chen and et al. 15 described a similar mutation, p.A78P, which showed accelerated deactivation but no decrease in current. This PAS mutant, however, was electrophysiologically characterized in Xenopus oocytes. It has been shown that mutant proteins can be rescued and trafficked toward the plasma membrane after incubation at temperatures lower that 37°C, which is the case when using Xenopus oocytes. 16 Thus, it remains possible the p.A78P mutation exhibits defective trafficking in mammalian cells as seen with p.A80P. Interestingly, Anderson et al. 17 reported that missense mutations located in a highly ordered structure as α‐helices or β‐sheet correlated with a trafficking‐deficient phenotype. Pharmacological chaperones, usually channel blockers, also have the ability to rescue some hERG1 mutants. Pharmacological rescue is not only domain‐dependent (e.g. cNBD mutations are not rescued), but also mutation‐dependent. The PAS‐domain mutant p.T65P 6 was rescued with E4031 and cisapride incubations, but a 7‐amino‐acid duplication in the PAS domain 10 was not rescued upon astemizole treatment. Importantly, abnormal protein trafficking appears as the most common mechanism for decreased ionic current caused by KCNH2 mutations. 17 Improved knowledge on chemical chaperone binding sites and the rescue mechanisms of protein trafficking might lead to the development of specific pharmacological chaperones for the treatment of LQT2. Recently, Balijepalli et al. 18 pointed out the importance of assessing in vitro, and in adequate cellular models, an identified LQT2 mutation. Indeed, in case of trafficking‐deficient mutants leading to haploinsufficiency, hERG1 channel activators may have greater benefits than pharmacological chaperones in future treatments. 18
In conclusion, we identified a 20‐year‐old woman with LQTS carrying a previously undescribed hERG1 PAS‐domain heterozygous mutation. This mutation proved to be pathogenic, as coexpression of WT and mutant alleles in mammalian cells led to a ∼50% decrease of the hERG1 protein at the cell surface as well a decrease in its current. The patient's LQTS phenotype may be fully explained by these observations. This additional mutation in the PAS domain, leading to impaired channel maturation and trafficking, underlines this structure as a nonpore mutational “hot spot.”
ABBREVIATIONS
WT: wild‐type; LQTS: long QT syndrome; LQT1: long QT syndrome type 1; LQT2: long QT syndrome type 2; ECG: electrocardiogram; cNBD: cyclic nucleotide binding domain
CONSENT
Written informed consent was obtained from the patient for publication of this case report and accompanying images. The genetic investigation conformed to the principles outlined in the Declaration of Helsinki, and the research protocol was approved by the ethics committee of the Faculty of Biology and Medicine of the University of Lausanne.
Supporting information
Method
Supporting info item
Acknowledgments
Acknowledgments: Authors thank Sophie Roy for her help with the biochemistry assays and Dr. Francine Thonney and Séverine Arcioni for their molecular analysis of the LQTS genes.
This work was supported by grants of the CardioMet Centre of the Faculty of Biology and Medicine (CardioGene grant 2007–2009), University of Lausanne, and the Swiss National Science Foundation (310000–120707 to HA).
REFERENCES
- 1. Shimizu W. The long QT syndrome: Therapeutic implications of a genetic diagnosis. Cardiovasc Res 2005;67:347–356. [DOI] [PubMed] [Google Scholar]
- 2. Ackerman MJ. Cardiac channelopathies: It's in the genes. Nat Med 2004;10:463–464. [DOI] [PubMed] [Google Scholar]
- 3. Markiewicz‐Loskot G, Moric‐Janiszewska E, Mazurek U. The risk of cardiac events and genotype‐based management of LQTS patients. Ann Noninvas Electro 2009;14:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ueda K, Valdivia C, Medeiros‐Domingo A, et al Syntrophin mutation associated with long QT syndrome through activation of the nNOS‐SCN5A macromolecular complex. Proc Natl Acad Sci USA 2008;105:9355–9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang Y, Yang Y, Liang B, et al Identification of a Kir3.4 mutation in congenital long QT syndrome. Am J Hum Genet 2010;86:872–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Paulussen A, Raes A, Matthijs G, et al A novel mutation (T65P) in the PAS domain of the human potassium channel HERG results in the long QT syndrome by trafficking deficiency. J Biol Chem 2002;277:48610–48616. [DOI] [PubMed] [Google Scholar]
- 7. Shushi L, Kerem B, Goldrait M, et al Clinical, genetic, and electrophysiologic characteristics of a new pas‐domain HERG mutation (M124R) causing long QT syndrome. Ann Noninvas Electro 2005;10:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rossenbacker T, Mubagwa K, Jongbloed RJ, et al Novel Mutation in the Per‐Arnt‐Sim Domain of KCNH2 Causes a Malignant Form of Long‐QT Syndrome. Circulation 2005;111:961–968. [DOI] [PubMed] [Google Scholar]
- 9. Morais Cabral JH, Lee A, Cohen SL, et al Crystal structure and functional analysis of the HERG potassium channel N terminus: A eukaryotic PAS domain. Cell 1998;95:649–655. [DOI] [PubMed] [Google Scholar]
- 10. Sintra Grilo L, Pruvot E, Grobaty M, et al Takotsubo cardiomyopathy and congenital long QT syndrome in a patient with a novel duplication in the Per‐Arnt‐Sim (PAS) domain of hERG1. Heart Rhythm 2010;7:260–265. [DOI] [PubMed] [Google Scholar]
- 11. Moss AJ, Zareba W, Benhorin J, et al ECG T‐wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995;92:2929–2934. [DOI] [PubMed] [Google Scholar]
- 12. Zareba W. Genotype‐specific ECG patterns in long QT syndrome. J Electrocardiol 2006;39(4 Suppl):S101–S106. [DOI] [PubMed] [Google Scholar]
- 13. Sauer AJ, Moss AJ, McNitt S, et al Long QT syndrome in adults. J Am Coll Cardiol 2007;49:329–337. [DOI] [PubMed] [Google Scholar]
- 14. Shimizu W, Moss AJ, Wilde AA, et al Genotype‐phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol 2009;54:2052–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen J, Zou A, Splawski I, et al Long QT syndrome‐associated mutations in the Per‐Arnt‐Sim (PAS) Domain of HERG potassium channels accelerate channel deactivation. J Biol Chem 1999;274:10113–10118. [DOI] [PubMed] [Google Scholar]
- 16. Ficker E, Thomas D, Viswanathan PC, et al Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am J Physio Heart Circ Physiol 2000;279:H1748–H1756. [DOI] [PubMed] [Google Scholar]
- 17. Anderson CL, Delisle BP, Anson BD, et al Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking‐deficient) mechanism. Circulation 2006;113:365–373. [DOI] [PubMed] [Google Scholar]
- 18. Balijepalli SY, Anderson CL, Lin EC, et al Rescue of mutated cardiac ion channels in inherited arrhythmia syndromes. J Cardiovasc Pharmacol 2010;56:113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Method
Supporting info item