

Nucleoside diphosphate kinase (NDK), a ubiquitous enzyme, is known for its critical role in homeostasis of cellular nucleotide pools. However, NDK has now emerged as a molecule with pleiotropic effects in DNA repair, protein phosphorylation, gene expression, tumor metastasis, development, and pathogen virulence and persistence inside the host. In this study, we reveal an unexpected role of NDK in genome instability because of its activity in converting 8-O-dGDP to 8-O-dGTP. This observation has important consequences in escalating A-to-C mutations in Escherichia coli. The severity of NDK in enhancing these mutations may be higher in the organisms challenged with high oxidative stress, which promotes 8-O-dGDP/8-O-dGTP production.
KEYWORDS: dNTP, 8-oxo-dGTP (8-O-dGTP), MutT, NDK, mutation rate and mutation frequency
ABSTRACT
The chemical integrity of the nucleotide pool and its homeostasis are crucial for genome stability. Nucleoside diphosphate kinase (NDK) is a crucial enzyme that carries out reversible conversions from nucleoside diphosphate (NDP) to nucleoside triphosphate (NTP) and deoxynucleoside diphosphate (dNDP) to deoxynucleoside triphosphate (dNTP). Guanosine nucleotides (GDP, GTP, dGDP, and dGTP) are highly susceptible to oxidative damage to 8-oxo-GDP (8-O-GDP), 8-O-dGTP, 8-O-GTP, and 8-O-dGTP. MutT proteins in cells hydrolyze 8-O-GTP to 8-O-GMP or 8-O-dGTP to 8-O-dGMP to avoid its incorporation in nucleic acids. In Escherichia coli, 8-O-dGTP is also known to be hydrolyzed by RibA (GTP cyclohydrolase II). In this study, we show that E. coli NDK catalyzes the conversion of 8-O-dGDP to 8-O-dGTP or vice versa. However, the rate of NDK-mediated phosphorylation of 8-O-dGDP to 8-O-dGTP is about thrice as efficient as the rate of dephosphorylation of 8-O-dGTP to 8-O-dGDP, suggesting an additive role of NDK in net production of 8-O-dGTP in cells. Consistent with this observation, the depletion of NDK (Δndk) in E. coli ΔmutT or ΔmutT ΔribA strains results in a decrease of A-to-C mutations. These observations suggest that NDK contributes to the physiological load of MutT in E. coli.
IMPORTANCE Nucleoside diphosphate kinase (NDK), a ubiquitous enzyme, is known for its critical role in homeostasis of cellular nucleotide pools. However, NDK has now emerged as a molecule with pleiotropic effects in DNA repair, protein phosphorylation, gene expression, tumor metastasis, development, and pathogen virulence and persistence inside the host. In this study, we reveal an unexpected role of NDK in genome instability because of its activity in converting 8-O-dGDP to 8-O-dGTP. This observation has important consequences in escalating A-to-C mutations in Escherichia coli. The severity of NDK in enhancing these mutations may be higher in the organisms challenged with high oxidative stress, which promotes 8-O-dGDP/8-O-dGTP production.
INTRODUCTION
The fidelity of genome replication and repair is dependent on the cellular ability to maintain an optimal balance and integrity of the deoxynucleoside triphosphate (dNTP) pools. A number of enzymes contribute to the synthesis of dNTPs. Ribonucleotide reductase (RNR), an allosterically regulated enzyme, catalyzes the conversion of nucleoside diphosphates (NDPs) into deoxynucleoside diphosphates (dNDPs) (1–4). Nucleoside diphosphate kinase (NDK) then converts dNDPs into dNTPs, and it also maintains their optimal relative concentrations by interconversions between dNDPs and dNTPs (5–7). The chemical nature of the dNTPs is also at constant risk of incurring damages by reactive oxygen species (ROS) and reactive nitrogen species (RNI) generated by the exogenous and endogenous sources. Among the most common chemical modification of dNTPs is the oxidation of dGTP to 8-oxo-dGTP (8-O-dGTP) (8, 9). Also, deamination of dCTP to dUTP occurs by dCTP deaminase (10, 11). MutT and Dut hydrolyze 8-O-dGTP and dUTP, respectively, into their monophosphate forms and prevent their incorporation in DNA. Although the incorporation of dUTP in DNA (opposite A) is not directly mutagenic, any deficiencies in MutT activity may lead to incorporation of 8-O-dGTP in DNA opposite A or C and result in T-to-G (or A-to-C) or to a lesser extent C-to-A (or G-to-T) transversion mutations (12, 13). An excessive incorporation of 8-O-dGTP in the genome could also be lethal because of the increased double-stranded DNA breaks (14). RNA polymerases are also known to bypass 8-O-G in the DNA, giving rise to aberrant transcripts (15). Moreover, RNA polymerase can utilize 8-O-GTP as a substrate for incorporation in RNA, leading to erroneous translation. MutT proteins also act on the 8-O-GTP to avoid its incorporation in RNA (16, 17). Living systems have evolved a variety of mechanisms to reduce the adverse effects of 8-O-G. MutM, a DNA glycosylase, excises 8-O-G from DNA and initiates base excision repair (18). Another DNA glycosylase, MutY, excises the misincorporated A (opposite 8-O-G) from DNA (19, 20). Together, the activities of MutM and MutY avoid C-to-A transversions in the genome (21). In Escherichia coli, 8-O-dGTP can also be hydrolyzed by GTP cyclohydrolase II (ribA product), and its overexpression is known to rescue the ΔmutT strain of E. coli. Interestingly, the Nudix box motif, which is essential for the catalytic activity of MutT, is absent from GTP cyclohydrolase II (22). The presence of multiple MutT proteins in humans (23–25), and in G+C rich organisms like mycobacteria (26–29), further highlights the importance of 8-O-dGTP detoxification mechanisms in the maintenance of genome stability.
NDK, a housekeeping enzyme with broad substrate specificity, catalyzes reversible phosphorylation to generate dNTPs from the corresponding dNDPs. Deficiency of NDK decreases dATP and increases dCTP and dGTP levels, leading to altered dNTP pool composition. However, the levels of nucleoside triphosphates (NTPs) remain unaffected (6, 7). The loss of nucleotide pool homeostasis has been reasoned to be the basis for the contribution of NDK to the mutator phenotype (30). However, such a role for NDK remains unclear. For example, the human NDK homolog complements the mutator phenotype of the E. coli Δndk strain but without restoring its dNTP pools (31). On the other hand, a synergistic increase in mutation frequency of dut1 Δndk strain and occurrence of high-level mutations at A·T base pairs in the genome argues for the role of NDK in reducing dUTP levels (31). While the primary role of NDK is in generating optimum levels of dNTPs and NTPs, it has also been shown to convert 8-O-dGDP into 8-O-dGTP (32, 33). Such an activity of NDK on 8-O-dGDP could be counterproductive for genome integrity during oxidative stress and compromised nucleotide pool sanitization. In this study, we addressed the role of NDK in the metabolism of 8-O-dGDP/8-O-dGTP and its impact on genome stability. We show that NDK favors the formation of 8-O-dGTP. Mutational analysis for the specific transversions arising from the incorporation of 8-O-dGTP shows that NDK contributes to the mutator phenotype of the MutT-deficient strains.
RESULTS
Purification of EcoNDK.
To examine the role of NDK in metabolism of 8-O-dGTP and 8-O-dGDP, which are otherwise well-known substrates of MutT (NudA), we overexpressed and purified EcoNDK from an E. coli MG1655 ΔmutT strain to eliminate any contamination from EcoMutT in the preparation. C-terminally His-tagged EcoNDK was expressed in the soluble fraction and purified by affinity chromatography (Ni-nitrilotriacetic acid [Ni-NTA]). The analysis of purified protein on a 15% SDS-PAGE gel revealed a single band (Fig. 1A) close to its expected molecular weight (with a His tag) of 16.5 kDa.
FIG 1.

Purification of EcoNDK from the E. coli mutT null strain and activity analysis on 8-O-dGTP. (A) Representative 15% SDS-PAGE gel showing the purity and migration of the purified EcoNDK. M stands for the protein size markers. (B) Chromatograms indicate NDK-catalyzed reaction on 8-O-dGTP. Reaction analytes were separated on a DNAPac column through high-performance liquid chromatography (HPLC), using a gradient of 1 M LiCl (0% to 40%). The retention times of different analytes are shown on the x axis and the corresponding peak intensities (given in arbitrary units) are shown on the y axis. (i) Control. Shown is the migration profile of ADP and 8-O-dGTP. (ii) Reaction in the presence of NDK. Shown is the emergence of ATP and 8-O-dGDP peaks due to NDK-mediated transfer of terminal phosphate from 8-O-dGTP to ADP.
Enzymatic activities on mutagenic substrates and determination of initial rates.
The function of NDK has been established in maintaining pools of dNTPs and NTPs by catalyzing reversible phosphorylation reactions between nucleoside diphosphates and triphosphates. While catalyzing the reaction, NDK hydrolyzes the terminal pyrophosphate bond of the donor nucleoside triphosphate for transfer of the terminal phosphate to the acceptor nucleoside diphosphate to convert it into nucleoside triphosphate. We tested the activity of EcoNDK in hydrolyzing the mutagenic nucleotide 8-O-dGTP in the presence of ADP (an acceptor). We maintained a five times higher concentration of normal nucleoside derivatives in comparison to that of the modified nucleoside derivatives (in the cellular context, the abundance of the modified nucleoside derivatives will be much less compared to that of the normal nucleoside derivatives). On incubation of EcoNDK with 8-O-dGTP and ADP, we detected the presence of two new peaks corresponding to 8-O-dGDP and ATP (Fig. 1B), indicating that EcoNDK utilizes the modified nucleotide as a phosphate donor. As NDK-mediated reactions are reversible, we next analyzed the rates for the forward (generation of 8-O-dGTP by transfer of phosphate from ATP to 8-O-dGDP) and reverse (hydrolysis of 8-O-dGTP to transfer the phosphate from 8-O-dGTP to ADP) reactions because a difference in the rate of the two reactions would elucidate the role of EcoNDK in the homeostasis of 8-O-dGTP nucleotides. The initial rate analyses showed that EcoNDK-catalyzed formation of 8-O-dGTP is 2 to 3 times more efficient than its hydrolysis (compare Table 1, rows A and B). As a control, under the same experimental conditions, we also calculated the rates for the generation and hydrolysis of a normal nucleotide (GTP), and we observed that EcoNDK-mediated forward and reverse reaction rates for a normal nucleotide are not much different (compare Table 1, rows C and D). We conclude that EcoNDK favors generation of 8-O-dGTP rather than its hydrolysis (Fig. 2).
TABLE 1.
Rates of NDK-catalyzed reactions from two independent (I and II) experiments
| Reaction label | NDK-catalyzed reactions (forward/reverse) | Rate (μmol/min/mg) for expt: |
|
|---|---|---|---|
| I | II | ||
| A | 8-O-dGDP→8-O-dGTP | 5.81 ± 0.10 | 6.78 ± 0.23 |
| B | 8-O-dGTP→8-O-dGDP | 2.69 ± 0.14 | 2.24 ± 0.06 |
| C | GDP→GTP | 11.36 ± 0.35 | 16.57 ± 0.78 |
| D | GTP→GDP | 9.67 ± 0.45 | 14.35 ± 0.60 |
FIG 2.
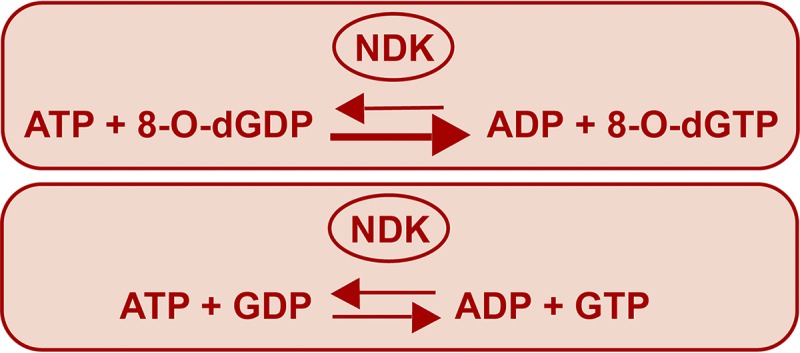
Summary of the reaction rate analysis.
Mutation frequency analysis upon overexpression of EcoNDK.
To investigate the physiological consequences of our biochemical findings (Table 1), we introduced plasmids overexpressing either EcoMutT (control) or EcoNDK (test) in an MG1655 ΔmutT::kan strain to examine their impact on the mutation frequencies of the strain (Rifr phenotype). We observed a mutation frequency of 2.78 × 10−5 in the MG1655 ΔmutT::kan strain. As expected, complementation of the strain by EcoMutT rescued the mutation frequency to 9.21 × 10−7 (30-fold reduction). However, overexpression of EcoNDK resulted in a 4.6-fold increase in mutation frequency (12.85 × 10−5) of the MG1655 ΔmutT::kan strain (Fig. 3A). As seen from the Coomassie blue-stained SDS-PAGE gel (Fig. 3B), EcoMutT and EcoNDK were expressed sufficiently upon induction by 0.02% arabinose.
FIG 3.

Mutation frequency analyses. (A) Mutation frequency analysis of MG1655 ΔmutT::kan strain harboring a plasmid (pBAD or pBAD_Eco_mutT or pBAD_Eco_ndk [refer to Table S2]). Mutation frequencies (using 4 replicates for each strain) were determined by dividing the number of colonies that appeared on the LB plus Amp plate containing rifampin by the corresponding viable counts obtained on the LB plus Amp plate. (B) The expression of plasmid-borne EcoMutT or EcoNDK in the MG1655 ΔmutT::kan strain was verified under the experimental conditions.
Assessment of A-to-C mutations on overexpression of EcoNDK.
In the experiment shown in Fig. 3, we evaluated the overall mutation frequency of the strain, inclusive of non-A-to-C mutations. To score specifically for A-to-C (or T-to-G) mutations, we tested the function of EcoNDK in the CC101 ΔmutT::kan strain. This strain produces inactive β-galactosidase due to an amber mutation (G to T) in the lacZ gene. The absence of mutT enables the strain to undergo T-to-G reversions due to increased misincorporations of 8-O-dGTP in the genome, leading to the gain of an Lac-positive (Lac+) phenotype. As shown in Fig. 4A, the CC101 ΔmutT::kan strain had a reversion frequency of 5.21 × 10−6. The introduction of EcoMutT decreased it to an undetectable level. However, the introduction of EcoNDK did not significantly alter the frequency of A-to-C mutations (5.7 × 10−6). Such an observation implies that either the biochemical observation of EcoNDK-mediated production of 8-O-dGTP is not physiologically important or that, for the in vivo conversion of 8-O-dGDP to 8-O-dGTP, the cellular levels of EcoNDK are already adequate as far as the A-to-C mutations are concerned. Moreover, in these experiments, the possible lack of a discernible impact of EcoNDK overexpression in the CC101 ΔmutT::kan strain (for A-to-C mutations) due to the variable levels of NDK expression in the two different strain backgrounds (compare Fig. 3B with Fig. 4B) could also not be ruled out.
FIG 4.

Mutation frequency analyses. (A) Analysis of A-to-C (or T-to-G) mutation frequency of the CC101 ΔmutT::kan strain harboring the pBAD vector or its derivatives expressing EcoMutT or EcoNDK (refer to Table S3). Reversion frequencies (using 9 or 10 replicates for each strain) were calculated by dividing the number of colonies that appeared on the minimal medium Amp plate containing lactose by the corresponding CFU on the minimal medium Amp plate containing glucose. (B) The expression of plasmid-borne EcoMutT or EcoNDK in the CC101 ΔmutT::kan strain was confirmed during the assay conditions.
Deletion of ndk and the study of A-to-C transversions by mutation frequency and mutation rate analyses.
To further investigate the biological significance of the in vitro activity of EcoNDK in contributing to the cellular levels of 8-O-dGTP, we wished to analyze the A-to-C (or T-to-G) mutations in CC101 ΔmutT and CC101 ΔmutT Δndk strains. To generate the CC101 ΔmutT Δndk strain, we first created an unmarked deletion of mutT in the CC101 ΔmutT::kan strain, followed by transduction of the Δndk::kan allele (see the supplemental material), and validated the strains by amplifying the two loci using flanking primers (Fig. S1). In two independent experiments, we noted that CC101 ΔmutT Δndk strain has 3.4-fold (from 3.41 × 10−6 to 1.00 × 10−6)- and 2.5-fold (6.19 × 10−6 to 2.45 × 10−6)-reduced reversion frequencies in comparison to those of the CC101 ΔmutT strain (Fig. 5). These observations clearly support the biochemical role of EcoNDK in contributing to the cellular pools of 8-O-dGTP.
FIG 5.

Comparisons of Lac+ reversion frequencies between CC101 ΔmutT and CC101 ΔmutT Δndk strains. Panels A and B show two independent data sets. Reversion frequencies were calculated by dividing the number of spontaneous revertants by the corresponding viable counts. The plotted median and confidence intervals are calculated with ≥95% of the confidence of the coefficient, using 10 replicates for each strain (refer to Table S4).
Importantly, RibA has also been reported to hydrolyze 8-O-dGTP. In an E. coli mutT deletion background, deletion of ribA was shown to enhance mutation frequencies by 2- to 4-fold, whereas its overexpression decreased the mutation frequency by 200-fold (22). Thus, to further validate the biochemical activity of EcoNDK in building the cellular levels of 8-O-dGTP (from 8-O-dGDP), we compared the reversion frequencies of the CC101 ΔmutT ΔribA strain in the presence and absence of native ndk. The CC101 ΔmutT ΔribA strain was created by disrupting the ribA gene in the ΔmutT strain with a chloramphenicol marker. We then transduced the Δndk::kan allele into the CC101 ΔmutT ΔribA strain to obtain the CC101 ΔmutT ΔribA Δndk strain (see the supplemental material) and confirmed the strains by PCR amplification of the respective loci (Fig. S2).
In two independent reversion frequency comparisons, we observed decreases of 5.4-fold (from 1.85 × 10−5 to 3.39 × 10−6) and 3.5-fold (from 1.33 × 10−5 to 3.73 × 10−6) upon deletion of native ndk in the ΔmutT ΔribA background (Fig. 6A and B). As an additional validation of this observation, we assessed A-to-C transversion rates, which showed a 3.0-fold decrease in appearance of Lac+ colonies (4.32 × 10−7 to 1.41 × 10−7) upon deletion of ndk in the CC101 ΔmutT ΔribA strain (Fig. 6C), suggesting a three-times-higher spontaneous transversion rate due to the presence of ndk. These in vivo assays confirm the role of EcoNDK in promoting A-to-C mutations and suggest its role in generating 8-O-dGTP in the physiological conditions.
FIG 6.

Comparisons between CC101 ΔmutT ΔribA and CC101 ΔmutT ΔribA Δndk strains. Panels A and B represent two independent comparisons of Lac+ reversion frequencies. Reversion frequencies were obtained by dividing the number of Lac+ revertants by the respective viable counts. (C) Lac+ reversion rates were estimated as described in Materials and Methods. Each comparative plot represents median and ≥95% confidence intervals estimated using 10 or 11 replicates of each strain (refer to Tables S5 and S6).
DISCUSSION
Owing to its low redox potential, guanines (G) in DNA or the guanosine nucleotides are particularly susceptible to oxidative damage, which results in the generation of 8-O-G, a mutagenic base (34). Interestingly, the overexpression of MutT has also been shown to rescue the effect of oxidative damages to G (14, 35). This observation emphasizes the significance of nucleotide pool sanitization in mutation prevention. In this study, we investigated the role of EcoNDK in the metabolism of 8-O-dGTP. The ndk is an antimutator gene whose product (EcoNDK) regulates the levels of NTPs/dNTPs in response to dynamic cellular needs and its absence results in a complex mutation spectrum, not fully understood from its in vitro activities. Nonetheless, the imbalances in the nucleotide pool (6, 7, 30) and increased insertion of uracil in the genome (31) have been proposed as possible explanations. A marked decrease in mutation frequencies under anaerobic conditions in comparison to that in aerobic conditions was also noticed for the E. coli Δndk strain (6). A similar phenotype has been reported previously for the E. coli ΔmutT strain and is ascribed to the extent of 8-O-dGTP incorporation in DNA (36). However, it remained undetermined whether the same molecular phenomenon is responsible for the decrease in mutation frequency of the ndk-deficient strain. Besides, an increase in the GC/AT ratio is also observed in the genome of the ΔmutT strain upon repeated subculturing (37). Thus, we were interested in understanding the function of NDK in the possible homeostasis of 8-O-dGTP, in order to gain better understanding for its mutator phenotype. Earlier studies on the metabolism of 8-O-dGTP using mammalian and E. coli systems showed that 8-O-dGTP is hydrolyzed into 8-O-dGMP by MutT. The return of the 8-O-dGMP into the dNTP pool is prevented by the absence of guanylate kinase activity, which is specific for conversion of cellular dGMP into dGDP. It was emphasized that dGDP present in the cellular pools can be oxidized to 8-O-dGDP. Interestingly, in mycobacteria, some of the MutT proteins (e.g., MutT1) also convert 8-O-dGTP to 8-O-dGDP (27). Broad-specificity enzymes like NDK and adenylate kinase (ADK) can facilitate reconversion of 8-O-dGDP to 8-O-dGTP. Ribonucleotide reductase, the enzyme which catalyzes the conversion of NDP into dNDP, is inactive on 8-O-GDP (17, 32, 33). However, the physiological consequences of the role of NDK in returning cellular pools of 8-O-dGDP to 8-O-dGTP had not been investigated. In this study, we have shown that NDK promotes the generation of 8-O-dGTP from 8-O-dGDP (however, as a control, the rates for reversible conversions between GTP and GDP were approximately the same). Furthermore, using an in vivo E. coli model compromised in 8-O-dGTP sanitation mechanisms, we have validated the results obtained from the biochemical assays and provided evidence that native NDK increases the cellular pools of 8-O-dGTP, which are mutagenic in the absence of the antimutator functions. NudB (orf17) has been reported to hydrolyze 8-O-dGTP and serves as backup for the MutT function in E. coli (38, 39). It should be noted that orf17 was unperturbed in our strain background. Similarly to DNA polymerase III-mediated incorporation of 8-O-dGTP in DNA opposite A (40), we have demonstrated a mechanism wherein the ubiquitously present NDK increases A-to-C mutations in the genome. Our findings have implications for organisms such as Mycobacterium tuberculosis that not only possess a G+C rich genome but also sustain an ROI-rich niche of the host macrophages that promotes oxidative damage of dGTP to 8-O-dGTP. Interestingly, in these organisms a true homolog of EcoMutT is yet to be identified. The MutT proteins identified in mycobacteria so far either are catalytically inefficient as MutT or convert 8-O-dGTP to 8-O-dGDP (26–29).
MATERIALS AND METHODS
Bacterial strains, plasmids, and DNA oligomers.
Strains and plasmids used in this study are listed in Table 2. Sequences of DNA oligomers are provided in Table S1 in the supplemental material. Enzymes used for DNA manipulations were procured from Thermo Fisher and New England Biolabs.
TABLE 2.
Description of strains and plasmids used in the study
| Strain or plasmid | Relevant details | Reference or source |
|---|---|---|
| Strains | ||
| E. coli MG1655 | K strain, F− λ− rph-1 | 44 |
| E. coli MG1655 ΔmutT::kan | MG1655 strain, where the mutT gene is disrupted with a kan cassette | 28 |
| E. coli BW25113 | F− Δ(araD araB)567 ΔlacZ4787(::rrnB-3) λ− rph-1 Δ(rhaD rhaB)568 hsdR514 | 45 |
| E. coli JW2502-1 Δndk::kan | BW25113 strain with Δndk-764::kan | 45 |
| E. coli CC101 | F′ ara-600 Δ(gpt lac)5 λ− relA1 spoT1 thiE1 F128− (used to screen for A-to-C or T-to-G mutation) | 42 |
| E. coli CC101 ΔmutT::kan | CC101 strain with ΔmutT::kan allele | 28 |
| E. coli CC101 ΔmutT | A derivative of CC101 ΔmutT::kan strain in which the kanamycin marker cassette has been removed from the ΔmutT::kan locus to create an unmarked deletion of mutT | This study |
| E. coli CC101 ΔmutT Δndk | CC101 ΔmutT cured strain, in which the ndk allele has been disrupted by insertion of kan cassette | This study |
| Plasmids | ||
| pQE60 | An E. coli expression vector harboring ColE1 origin of replication, ampicillin resistance marker, and T5 promoter (containing the lac operator) and provides a C-terminal His tag | Invitrogen |
| pQE60_Eco_ndk | pQE60 plasmid harboring Eco_ndk cloned (NcoI, BglII) under T5 promoter | This study |
| pBADHisB (pBAD) | An E. coli expression vector harboring ColE1 origin of replication, ampicillin resistance marker, and an arabinose-inducible promoter | Invitrogen |
| pBAD_Eco_mutT | Derivative of pBAD plasmid harboring Eco_mutT cloned (NcoI, NheI) under arabinose promoter | 28 |
| pBAD_Eco_ndk | A derivative of pBAD plasmid harboring Eco_ndk cloned (NcoI, BglII) under arabinose promoter | This study |
Strains, media, and growth conditions.
E. coli strains were cultured in Luria-Bertani (LB) medium. For growth on the solid surface, 1.6% agar was added to liquid medium. Minimal medium plates used for reversion assays contained 1× M9 salts (1.28% Na2HPO4, 0.3% KH2PO4, 0.05% NaCl, and 0.1% NH4Cl), 2 mM MgSO4, 0.1 mM CaCl2, 5 μg/ml thiamine, 0.2% glucose (or lactose), and 1.6% agar. Media were supplemented with ampicillin (Amp; 100 μg/ml), kanamycin (Kan; 25 μg/ml), hygromycin (Hyg; 150 μg/ml), or gentamycim (Gm; 20 μg/ml) as required. Medium components were obtained from BD Difco (Franklin Lakes, NJ).
Cloning of Eco_ndk.
The gene sequence of Eco_ndk was PCR amplified from E. coli MG1655 genomic DNA using the primers Eco_ndk_NcoI_Fp and Eco_ndk_BglII_Rp. The PCR mixture (20 μl) contained 250 ng of E. coli genomic DNA, 0.5 units of Pfu DNA polymerase, 1× Pfu buffer, 250 μM dNTPs, and 10 pmol each of the forward and reverse primers. Initial denaturation was done at 94°C for 4 min, followed by 30 cycles of 94°C for 1 min, 52°C for 35 s, 70°C for 1 min, and a final extension of 10 min at 70°C. The PCR amplicon (450 bp) was digested with NcoI/BglII restriction enzymes and ligated with the similarly digested pQE60 vector to obtain the pQE60_Eco_ndk construct. For generating the pBAD_Eco_ndk construct, the open reading frame (ORF) of ndk was amplified using the primers Eco_ndk_NcoI_Fp and Eco_ndk_BglII_Rp_Stop (amplifies the ORF with its stop codon) and cloned into the pBAD vector using NcoI/BglII restriction sites. All constructs were verified by DNA sequence analysis.
Expression and purification of EcoNDK.
EcoNDK was expressed and purified using pQE60_Eco_ndk construct in MG1655 ΔmutT strain. To prepare primary culture, a single transformant was inoculated in 50 ml of LB plus Kan and Amp and grown to the stationary phase at 37°C, under shaking at 180 rpm for 12 h. Primary culture (1%) was inoculated in 3 liters of LB plus Amp and incubated at 37°C under shaking at 180 rpm until the optical density at 600 nm (OD600) reached 0.6, followed by induction with 0.05 mM isopropyl-β-d-galactopyranoside (IPTG) for 2 h. Cells were harvested and resuspended into 30 ml of buffer A (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 10% glycerol, and 2 mM β-mercaptoethanol). The successive steps were performed at 4°C. The cells were lysed using 4 or 5 cycles of sonication at 36% amplitude. Each 1-min sonication cycle comprised 2-s pulses with on/off and a 5-min gap between 2 consecutive cycles. The clarified cell lysate was obtained by centrifugation at 24,000 rpm at 4°C for 2 h, loaded on a Ni-NTA column preequilibrated with buffer A, and washed with 10 column volumes of buffer B (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 10% glycerol, 2 mM β-mercaptoethanol, and 20 mM imidazole). The proteins were eluted with a gradient (0 to 1 M) of imidazole in buffer C (20 mM Tris-HCl pH 7.5, 500 mM NaCl, 10% glycerol, 2 mM β-mercaptoethanol) at a flow rate of 1 ml/min in 30 min. The fractions containing protein purified to near homogeneity (as judged by a single band in SDS-PAGE) were pooled, dialyzed against buffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl, 50% glycerol, and 2 mM β-mercaptoethanol), stored at −20°C, and quantified using bovine serum albumin (BSA) as standard.
Enzyme activity assays.
Assays were performed in 10-μl volumes containing 1× reaction buffer (20 mM Tris-HCl [pH 8.0], 8 mM MgCl2, 5 mM dithiothreitol [DTT], 40 mM NaCl, and 2% glycerol), and 100 ng of protein and substrates specific to the forward and the reverse reactions. For the forward reaction, 1 mM donor nucleoside triphosphate (ATP) and 0.2 mM acceptor nucleoside diphosphate (8-O-dGDP or GDP) were added, whereas 0.2 mM donor nucleoside triphosphate (8-O-dGTP or GTP) and 1 mM acceptor nucleoside diphosphate (ADP) were included for the reverse reaction. The reaction mixture was incubated at 37°C for 30 min, followed by termination of the reaction by adding 10 μl of 0.1% SDS. Samples were separated on a DNAPac column (PA200 analytical, 4 by 250 mm) by high-performance liquid chromatography (HPLC) using 25 mM Tris-HCl (pH 8.0) as a binding buffer with a gradient of 1 M LiCl (0% to 40%) for 25 min at a flow rate of 0.5 ml/min. While determining the rates for the forward and the reverse reactions, 0.2 to 2 ng of EcoNDK was used and the time points between 0 to 6 min were taken. Reaction components were detected by UV absorbance at 252 nm (ATP, ADP, GTP, GDP 8-O-dGTP, and 8-O-dGDP) and quantified by determining the area under the peak using Chromeleon software (Dionex). Modified nucleotides (8-O-dGTP and 8-O-dGDP) and normal nucleotides (ATP, ADP, GTP, and GDP) were purchased from Jena Bioscience and Sigma, respectively.
Mutation frequency determination using rifampin resistance as reporter.
Strains were generated by introducing respective plasmids in the MG1655 ΔmutT::kan strain. Four replicates of each strain were inoculated in 2 ml LB medium containing 0.02% arabinose and Amp and allowed to grow at 37°C and 180 rpm for 12 h. For individual cultures, serial dilutions were made and 100-μl aliquots from 10−5 and 10−6 dilutions were plated on LB plates containing Amp to estimate the viable counts. To evaluate the number of spontaneously arising mutants, cells from undiluted cultures (1 ml for MG1655 ΔmutT::kan/pBAD_Eco_mutT and 100 μl for rest of the strains) were plated on LB plate containing rifampin (50 μg/ml) and Amp. After 24 h of incubation at 37°C, the colonies were counted, and mutation frequencies of the strains were calculated by dividing the number of colonies obtained on rifampin plate by the corresponding total viable counts (obtained from the Amp plate). Using replicate values, the median and confidence of intervals were calculated (41), and fold differences were expressed by comparing the medians. Tables comprising raw values and statistical significance parameters are provided as supplemental data (Tables S2 to S6).
Analysis of A-to-C mutations using Lac− to Lac+ reversion assay.
Reversion assay was carried out in E. coli CC101 strain background, which has a point mutation in the lacZ gene that results in a growth-deficient phenotype on lactose as the sole carbon source. The mutT deletion allows the strain to undergo enhanced reversions, leading to a Lac+ phenotype and growth on lactose (42). To execute the assay, strains were obtained by either fresh transformation of specific plasmids in CC101 ΔmutT::kan strain or streaking from glycerol stocks of recently generated mutant strains and grown on an LB plate containing 50 μg/ml X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; specific for the CC101 strain) and respective antibiotics. Riboflavin at a concentration of 0.25 μg/ml was maintained while culturing ΔribA strains. For each strain, 10 replicates were inoculated in 2 ml LB medium containing 0.02% arabinose (only where ectopic expression of genes from an arabinose-inducible promoter is required) and an appropriate antibiotic and grown at 37°C and 180 rpm for 12 h (CC101 ΔmutT and CC101 ΔmutT Δndk) to 16 h (CC101 ΔmutT ΔribA and CC101 ΔmutT ΔribA Δndk). Serial dilutions were made for each culture, and 100-μl fractions of 10−5 and 10−6 dilutions were spread on a minimal medium plate containing 0.2% glucose and respective antibiotic for determining the viable counts. For estimating Lac+ revertants, cells from 100 μl culture were spread on a minimal medium plate containing 0.2% lactose and a respective antibiotic. The number of colonies were enumerated after an incubation period of 24 h (CC101 ΔmutT and CC101 ΔmutT Δndk) to 48 h (CC101 ΔmutT ΔribA and CC101 ΔmutT ΔribA Δndk) at 37°C. The reversion frequency was assessed by dividing the number of colonies obtained on the lactose plate by the corresponding number of colonies on the glucose plate.
Analysis of A-to-C reversion rates.
Reversion rates were determined as described before (43). Briefly, 11 replicates for each strain were inoculated in LB supplemented with riboflavin and chloramphenicol and incubated for 16 h to achieve saturation. Aliquots of 50 μl from a 10−5 dilution were used to determine viable counts (N0; plated on minimal medium containing 0.2% glucose, riboflavin, and chloramphenicol), spontaneous mutants (number of mutants in the seeding culture [M0]; plated on minimal medium containing 0.2% lactose, riboflavin, and chloramphenicol), and also seeded in 2 ml LB supplemented with riboflavin and chloramphenicol. After 24 h of incubation, cultures were used to estimate the number of cells after 24 h (Nt) and the number of mutants after 24 h (Mt). Reversion rates were calculated according to the equation a = 2 ln2[(Mt/Nt) − (M0/N0)]/n, where a refers to mutation rate and n stands for the number of generations. N0 corresponds to the number of cells in the seeding culture. The value of n was calculated using (log Nt/log N0)/0.301.
Supplementary Material
ACKNOWLEDGMENTS
We thank our laboratory colleagues for their suggestions on the manuscript. We also thank Arpan Amlan Jyoti Das for the technical help and Krishna Kurthkoti for the critical discussions on the experimental designs.
This work was supported by funds received from the Department of Biotechnology (DBT), Ministry of Science and Technology; the Department of Science and Technology (DST), Ministry of Science and Technology; and the DST J.C. Bose Fellowship (to U.V.). We acknowledge the support of the DBT-IISc partnership program, Centre of Advanced Studies of the University Grants Commission, New Delhi, and the DST-FIST level II infrastructure to carry out this work.
We declare that there are no conflicts of interest.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Stubbe J. 1990. Ribonucleotide reductases. Adv Enzymol Relat Areas Mol Biol 63:349–419. [DOI] [PubMed] [Google Scholar]
- 2.Stubbe J. 1990. Ribonucleotide reductases: amazing and confusing. J Biol Chem 265:5329–5332. [PubMed] [Google Scholar]
- 3.Hofer A, Crona M, Logan DT, Sjöberg BM. 2012. DNA building blocks: keeping control of manufacture. Crit Rev Biochem Mol Biol 47:50–63. doi: 10.3109/10409238.2011.630372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ahmad MF, Dealwis CG. 2013. The structural basis for the allosteric regulation of ribonucleotide reductase. Prog Mol Biol Transl Sci 117:389–410. doi: 10.1016/B978-0-12-386931-9.00014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lascu I, Gonin P. 2000. The catalytic mechanism of nucleoside diphosphate kinases. J Bioenerg Biomembr 32:237–246. doi: 10.1023/A:1005532912212. [DOI] [PubMed] [Google Scholar]
- 6.Lu Q, Zhang X, Almaula N, Mathews CK, Inouye M. 1995. The gene for nucleoside diphosphate kinase functions as a mutator gene in Escherichia coli. J Mol Biol 254:337–341. doi: 10.1006/jmbi.1995.0620. [DOI] [PubMed] [Google Scholar]
- 7.Shen R, Wheeler LJ, Mathews CK. 2006. Molecular interactions involving Escherichia coli nucleoside diphosphate kinase. J Bioenerg Biomembr 38:255–259. doi: 10.1007/s10863-006-9041-2. [DOI] [PubMed] [Google Scholar]
- 8.Dizdaroglu M. 1992. Oxidative damage to DNA in mammalian chromatin. Mutat Res 275:331–342. doi: 10.1016/0921-8734(92)90036-O. [DOI] [PubMed] [Google Scholar]
- 9.Jaruga P, Dizdaroglu M. 1996. Repair of products of oxidative DNA base damage in human cells. Nucleic Acids Res 24:1389–1394. doi: 10.1093/nar/24.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomita F, Takahashi I. 1969. A novel enzyme, dCTP deaminase, found in Bacillus subtilis infected with phage PBS I. Biochim Biophys Acta 179:18–27. doi: 10.1016/0005-2787(69)90117-8. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Weiss B. 1992. dcd (dCTP deaminase) gene of Escherichia coli: mapping, cloning, sequencing, and identification as a locus of suppressors of lethal dut (dUTPase) mutations. J Bacteriol 174:5647–5653. doi: 10.1128/jb.174.17.5647-5653.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maki H, Sekiguchi M. 1992. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355:273–275. doi: 10.1038/355273a0. [DOI] [PubMed] [Google Scholar]
- 13.Yanofsky C, Cox EC, Horn V. 1966. The unusual mutagenic specificity of an E. coli mutator gene. Proc Natl Acad Sci U S A 55:274–281. doi: 10.1073/pnas.55.2.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. 2012. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science 336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charlet-Berguerand N, Feuerhahn S, Kong SE, Ziserman H, Conaway JW, Conaway R, Egly JM. 2006. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J 25:5481–5491. doi: 10.1038/sj.emboj.7601403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taddei F, Hayakawa H, Bouton M, Cirinesi A, Matic I, Sekiguchi M, Radman M. 1997. Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 278:128–130. doi: 10.1126/science.278.5335.128. [DOI] [PubMed] [Google Scholar]
- 17.Hayakawa H, Hofer A, Thelander L, Kitajima S, Cai Y, Oshiro S, Yakushiji H, Nakabeppu Y, Kuwano M, Sekiguchi M. 1999. Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry 38:3610–3614. doi: 10.1021/bi982361l. [DOI] [PubMed] [Google Scholar]
- 18.Tchou J, Kasai H, Shibutani S, Chung MH, Laval J, Grollman AP, Nishimura S. 1991. 8-Oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc Natl Acad Sci U S A 88:4690–4694. doi: 10.1073/pnas.88.11.4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Au KG, Clark S, Miller JH, Modrich P. 1989. Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc Natl Acad Sci U S A 86:8877–8881. doi: 10.1073/pnas.86.22.8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nghiem Y, Cabrera M, Cupples CG, Miller JH. 1988. The mutY gene: a mutator locus in Escherichia coli that generates G·C→T·A transversions. Proc Natl Acad Sci U S A 85:2709–2713. doi: 10.1073/pnas.85.8.2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaels ML, Cruz C, Grollman AP, Miller JH. 1992. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci U S A 89:7022–7025. doi: 10.1073/pnas.89.15.7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kobayashi M, Ohara-Nemoto Y, Kaneko M, Hayakawa H, Sekiguchi M, Yamamoto K. 1998. Potential of Escherichia coli GTP cyclohydrolase II for hydrolyzing 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem 273:26394–26399. doi: 10.1074/jbc.273.41.26394. [DOI] [PubMed] [Google Scholar]
- 23.Fujikawa K, Kamiya H, Yakushiji H, Fujii Y, Nakabeppu Y, Kasai H. 1999. The oxidized forms of dATP are substrates for the human MutT homologue, the hMTH1 protein. J Biol Chem 274:18201–18205. doi: 10.1074/jbc.274.26.18201. [DOI] [PubMed] [Google Scholar]
- 24.Kamiya H, Hori M, Arimori T, Sekiguchi M, Yamagata Y, Harashima H. 2009. NUDT5 hydrolyzes oxidized deoxyribonucleoside diphosphates with broad substrate specificity. DNA Repair (Amst) 8:1250–1254. doi: 10.1016/j.dnarep.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 25.Takagi Y, Setoyama D, Ito R, Kamiya H, Yamagata Y, Sekiguchi M. 2012. Human MTH3 (NUDT18) protein hydrolyzes oxidized forms of guanosine and deoxyguanosine diphosphates: comparison with MTH1 and MTH2. J Biol Chem 287:21541–21549. doi: 10.1074/jbc.M112.363010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dos Vultos T, Blazquez J, Rauzier J, Matic I, Gicquel B. 2006. Identification of Nudix hydrolase family members with an antimutator role in Mycobacterium tuberculosis and Mycobacterium smegmatis. J Bacteriol 188:3159–3161. doi: 10.1128/JB.188.8.3159-3161.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patil AG, Sang PB, Govindan A, Varshney U. 2013. Mycobacterium tuberculosis MutT1 (Rv2985) and ADPRase (Rv1700) proteins constitute a two-stage mechanism of 8-oxo-dGTP and 8-oxo-GTP detoxification and adenosine to cytidine mutation avoidance. J Biol Chem 288:11252–11262. doi: 10.1074/jbc.M112.442566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sang PB, Varshney U. 2013. Biochemical properties of MutT2 proteins from Mycobacterium tuberculosis and M. smegmatis and their contrasting antimutator roles in Escherichia coli. J Bacteriol 195:1552–1560. doi: 10.1128/JB.02102-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arif SM, Patil AG, Varshney U, Vijayan M. 2017. Biochemical and structural studies of Mycobacterium smegmatis MutT1, a sanitization enzyme with unusual modes of association. Acta Crystallogr D Struct Biol 73:349–364. doi: 10.1107/S2059798317002534. [DOI] [PubMed] [Google Scholar]
- 30.Schaaper RM, Mathews CK. 2013. Mutational consequences of dNTP pool imbalances in E. coli. DNA Repair (Amst) 12:73–79. doi: 10.1016/j.dnarep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nordman J, Wright A. 2008. The relationship between dNTP pool levels and mutagenesis in an Escherichia coli NDP kinase mutant. Proc Natl Acad Sci U S A 105:10197–10202. doi: 10.1073/pnas.0802816105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayakawa H, Taketomi A, Sakumi K, Kuwano M, Sekiguchi M. 1995. Generation and elimination of 8-oxo-7, 8-dihydro-2′-deoxyguanosine 5′-triphosphate, a mutagenic substrate for DNA synthesis, in human cells. Biochemistry 34:89–95. doi: 10.1021/bi00001a011. [DOI] [PubMed] [Google Scholar]
- 33.Sekiguchi T, Ito R, Hayakawa H, Sekiguchi M. 2013. Elimination and utilization of oxidized guanine nucleotides in the synthesis of RNA and its precursors. J Biol Chem 288:8128–8135. doi: 10.1074/jbc.M112.418723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steenken S, Jovanovic SV. 1997. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J Am Chem Soc 119:617–618. doi: 10.1021/ja962255b. [DOI] [Google Scholar]
- 35.Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 36.Fowler RG, Erickson JA, Isbell RJ. 1994. Activity of the Escherichia coli mutT mutator allele in an anaerobic environment. J Bacteriol 176:7727–7729. doi: 10.1128/jb.176.24.7727-7729.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cox EC, Yanofsky C. 1967. Altered base ratios in the DNA of an Escherichia coli mutator strain. Proc Natl Acad Sci U S A 58:1895–1902. doi: 10.1073/pnas.58.5.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hori M, Fujikawa K, Kasai H, Harashima H, Kamiya H. 2005. Dual hydrolysis of diphosphate and triphosphate derivatives of oxidized deoxyadenosine by Orf17 (NtpA), a MutT-type enzyme. DNA Repair (Amst) 4:33–39. doi: 10.1016/j.dnarep.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Hori M, Asanuma T, Inanami O, Kuwabara M, Harashima H, Kamiya H. 2006. Effects of overexpression and antisense RNA expression of Orf17, a MutT-type enzyme. Biol Pharm Bull 29:1087–1091. doi: 10.1248/bpb.29.1087. [DOI] [PubMed] [Google Scholar]
- 40.Yamada M, Shimizu M, Katafuchi A, Grúz P, Fujii S, Usui Y, Fuchs RP, Nohmi T. 2012. Escherichia coli DNA polymerase III is responsible for the high level of spontaneous mutations in mutT strains. Mol Microbiol 86:1364–1375. doi: 10.1111/mmi.12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nair KR. 1940. Table of confidence interval for the median in samples from any continuous population. Indian J Stat 4:551–558. [Google Scholar]
- 42.Cupples CG, Miller JH. 1989. A set of lacZ mutations in Escherichia coli that allow rapid detection of each of the six base substitutions. Proc Natl Acad Sci U S A 86:5345–5349. doi: 10.1073/pnas.86.14.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.David HL. 1970. Probability distribution of drug-resistant mutants in unselected populations of Mycobacterium tuberculosis. Appl Microbiol 20:810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blattner FR, Plunkett G III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. 1997. The complete genome sequence of Escherichia coli K-12. Science 277:1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 45.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.