

Abstract
The Rosetta software suite for macromolecular modeling is a powerful computational toolbox for protein design, structure prediction, and protein structure analysis. The development of novel Rosetta‐based scientific tools requires two orthogonal skill sets: deep domain‐specific expertise in protein biochemistry and technical expertise in development, deployment, and analysis of molecular simulations. Furthermore, the computational demands of molecular simulation necessitate large scale cluster‐based or distributed solutions for nearly all scientifically relevant tasks. To reduce the technical barriers to entry for new development, we integrated Rosetta with modern, widely adopted computational infrastructure. This allows simplified deployment in large‐scale cluster and cloud computing environments, and effective reuse of common libraries for simulation execution and data analysis. To achieve this, we integrated Rosetta with the Conda package manager; this simplifies installation into existing computational environments and packaging as docker images for cloud deployment. Then, we developed programming interfaces to integrate Rosetta with the PyData stack for analysis and distributed computing, including the popular tools Jupyter, Pandas, and Dask. We demonstrate the utility of these components by generating a library of a thousand de novo disulfide‐rich miniproteins in a hybrid simulation that included cluster‐based design and interactive notebook‐based analyses. Our new tools enable users, who would otherwise not have access to the necessary computational infrastructure, to perform state‐of‐the‐art molecular simulation and design with Rosetta.
Keywords: conda, containerization, dask, de novo protein design, disulfide‐rich miniprotein, elastic cloud services, high performance computing, jupyter, python, Rosetta
Abbreviations
- API
application programming interface
- dict
dictionary
- DSL
domain specific language
- HPC
high performance computing
- I/O
input/output
- JSON
JavaScript object notation
- MPI
message passing interface
- XML
extensible markup language
- XSD
XML schema definition
1. INTRODUCTION
The Rosetta software suite for macromolecular modeling is a powerful computational toolbox for protein design, structure prediction, and protein structure analysis. Effectively leveraging Rosetta requires both extensive computational resources and deep knowledge of the inner workings of the software. To make the software accessible to a broader range of scientists, the suite exposes multiple, high‐level interfaces including RosettaScripts,1 PyRosetta,2 and protocol‐specific command‐line executables.3 Current academic efforts to make Rosetta more accessible, such as Robetta4 or ROSIE,5 focus on exposing specific, pre‐existing functionality for a general use audience, while commercial efforts focus on the development of integrated, end‐to‐end biomolecular modeling environments.6
The Rosetta suite implements a shared molecular energy function and fundamental “Pose” data structure, a complex object representing a full molecular system optimized for modeling operations. The high‐level protocols layer of the suite consists of composable submodules that either modify (“Movers”) or evaluate scalar‐valued scores (“Filters”) on the Pose.7 The rapid adoption of this model is a testament to its utility, with a majority of Rosetta‐based tools making broad use of these shared internal abstractions accessed via the RosettaScripts programming interface (Figure 1a).
Figure 1.
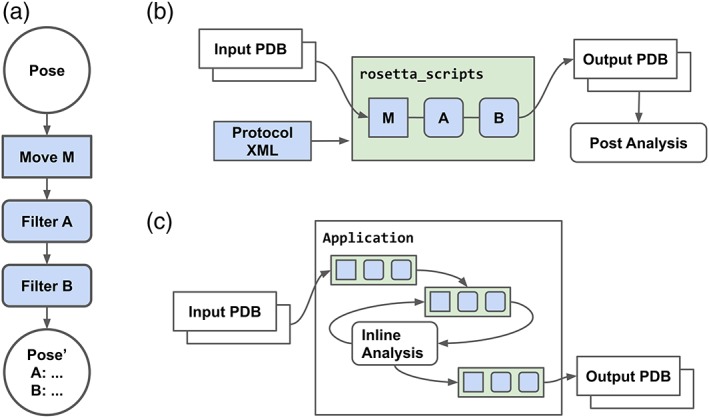
Rosetta protocols execution model and RosettaScripts workflows. Components managed by conda‐based redistributable packages are highlighted in green, and RosettaScripts‐defined protocol components are highlighted in blue. (a) The Rosetta protocols‐level pipelined execution model. An input Pose (P) is modified by Mover (M) and Filter objects A and B, resulting in modified Pose P′ and score values A, B. (b) In a standard rosetta_scripts based workflow, a protocol pipeline is applied, and output is post‐processed via external scripts. (c) In the integrated workflow described in this work, multiple RosettaScripts‐encoded protocols and additional components are combined in an arbitrary dataflow within a single application
Despite this shared high‐level architecture, Rosetta‐based scientific protocols often rely on multi‐step simulation workflows, iterating between execution of modeling protocols via the rosetta_scripts interpreter and data post‐processing and analysis via statistical tools such as Pandas, R, and Scikit‐learn (Figure 1b). This workflow results in nontrivial development overhead for scientific end users and a profusion of fragile ad hoc data munging, job management, and tool interface solutions. To address these deficiencies in computational workflows, the broader scientific community has embraced interactive computing “notebooks” (e.g., Jupyter notebooks). Notebooks allow users to interactively execute code blocks and display rich output (such as inline plots, formatted tables, or other visualizations) intermingled with typeset prose explaining the work and results, capturing the simulation and analysis for a scientific workflow in a single document. A limitation of typical notebook‐based workflows is the assumption that calculations are performed on a single machine running the notebook. However, for nearly all Rosetta‐based workflows, an individual simulation may require thousands of core‐hours of compute and must be split across many networked computers (distributed computing) to finish in a reasonable length of time. Thus, traditional notebook‐based workflows are unusable.
Molecular modeling workflows are typically run in high performance computing (HPC) environments using a shared file system to organize the input and output of simulations run by many worker machines using specialized code for inter‐worker communication (e.g., via message passing interface [MPI]). Together, distributed execution and data management create significant barriers for scientists performing molecular modeling tasks, as they must manage deployment of software on the HPC system, simulation execution, and subsequent tracking of a simulation and its associated analyses.
In addition to the rise of notebooks, the decade following Rosetta3's initial development has seen dramatic growth of distributed data processing tools running on elastically scaled cloud infrastructure. Distributed data processing tools (e.g., MapReduce, Spark, Dask, etc.) express workflows as many isolated task “nodes” with explicit data “edges” connecting task outputs and inputs into a computational graph (commonly referred to as task‐based parallelism). A scheduling engine then assigns tasks to distributed workers to compute the full workflow result. Unlike traditional MPI applications, in which the entire distributed application fails if a single worker fails, task‐based parallelism can accommodate failed workers by retrying the worker's isolated tasks. This allows large‐scale distributed execution on “unreliable” workers, such as inexpensive preemptible or spot instances provided in cloud environments. A substantial, freely available scientific software ecosystem has been developed around this computing paradigm. However, despite advantages in cost and throughput, there was no way to cleanly integrate these distributed computing tools with the community standard RosettaScripts interface.
Here, we describe deployment of the Rosetta suite via the conda package manager and the implementation of primary python programming interfaces using RosettaScripts components. We developed infrastructure to standardize the build process of Rosetta so that it can be easily and reproducibly deployed on a wide variety of compute environments. We then extended Rosetta's RosettaScripts interface to support task‐based parallelism and simplified integration with existing scientific computing libraries. Together, these allow (a) flexible deployment to multiple computing providers, including elastic cloud infrastructure; (b) reproducible specification of complete Rosetta based protocols and analysis; (c) extensive integration with existing components of the scientific software ecosystem; and (d) notebook‐amenable workflows that can drive simulations on a distributed computing system. We demonstrate the utility of this approach through two real‐world examples: (a) de novo design and analysis of disulfide‐rich miniproteins and (b) large‐scale protein relaxation simulations.
2. RESULTS AND DISCUSSION
Below, we report two proof‐of‐concept workflows: an end‐to‐end pipeline of a disulfide stabilized miniprotein design protocol8 combining classic HPC and notebook‐based analysis, and a benchmark of distribution engines on a community‐standard parallel model relaxation task.9 These results were generated using our novel conda packages and programming interfaces, implemented via a hybrid batch‐interactive workflow and distributed task‐based parallelism.
2.1. Miniprotein batch design and interactive analysis
De novo protein design with Rosetta currently requires significant compute resources, as backbone generation necessitates large amounts of conformational sampling. Generally, the process begins by designing thousands of structures in silico. Then, a portion of these designs are selected for production and characterization in vitro; design selection is commonly performed using quantitative measurements of protein structure (e.g., amount of nonpolar surface area sequestered in the protein interior) combined with heuristics and human intuition.8, 10, 11, 12 Thus, interactive data analysis greatly facilitates the de novo protein design process. To demonstrate the utility of the computational infrastructure we describe below, we generated a library of 1,000 disulfide‐rich mini‐proteins by running an automated RosettaScripts XML protocol with batch execution on a cluster, followed by analysis and selection via a Jupyter notebook (Figure 2a).
Figure 2.
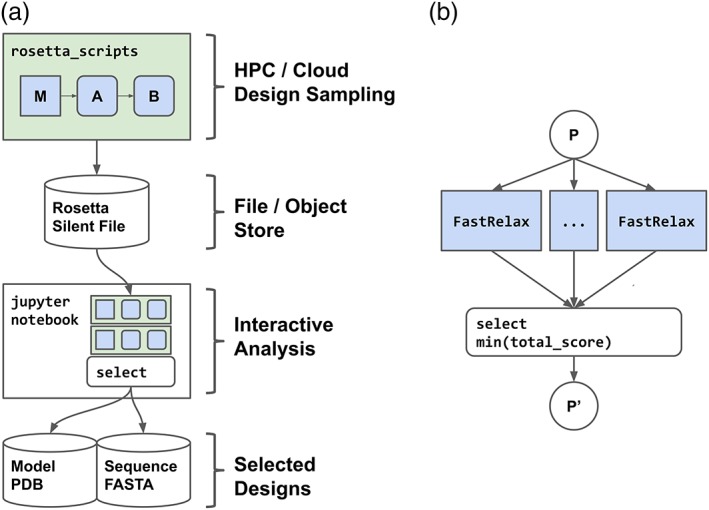
Example applications implemented in this work. (a) A multi‐application batch‐to‐interactive disulfide‐rich mini‐protein de novo design protocol. (b) Scatter–gather job implemented via dask.distributed for a parallel‐relax protocol
Independent design trajectories begin by generating a protein main chain through fragment‐based monte‐carlo sampling, and a blueprint file is used to specify the desired secondary and tertiary structure. Protein “backbones” (i.e., a poly‐valine model of protein structure) that pass a set of quality filters are then used for disulfide discovery. All possible disulfide positions are identified, and the combinations of disulfides are rank‐ordered by disulfide entropy, a metric of how well distributed the disulfide connections are within the protein backbone.13 Disulfide‐bonded backbones are then subjected to iterative rounds of structure minimization and sidechain‐based rotamer optimization followed by filtering based on the full‐atom model before output; the goal is to identify a low energy primary structure that will direct the protein to adopt the designed tertiary structure. (File S1).
Following batch simulation, results are collected from an intermediate store for interactive analysis. The collection of models, and associated filter values, are aggregated and used to develop application‐specific selection criteria, including features such as sequence or structural diversity. Within the analysis notebook, the dask.distributed.LocalCluster interface is used to distribute analysis tasks across multiple worker cores. The final selected designs are visually inspected, stored as PDB models for later analysis, and the primary amino acid sequences are output for reverse translation and gene synthesis. (File S1).
2.2. Relax benchmarking
Scalability describes a system's ability to effectively utilize additional computational resources. A distributed system is described as “horizontally scalable” if adding worker resources results in a commensurate increase in throughput, for example, total tasks per second throughout the system. All distributed task engines introduce a per‐task overhead. This overhead is constant for small system sizes, and it increases nonlinearly with system size. Thus, at the engine's scaling limit, additional worker resources are consumed by overhead and no longer provide increased throughput.
To demonstrate the multi‐worker scalability of our new infrastructure, we compared the performance of traditional multithreaded job execution versus the distributed task model. As an example, we chose to perform a common modeling task: Relax‐based refinement of a crystallographic structure. This step is required for all modeling or design work with Rosetta that makes use of experimentally determined protein models. Because the Relax operation is stochastic, it requires repeating sampling followed by model selection. This exposes trajectory level parallelism, and a distributed task model is ideal for this type of computational workflow. Here, we utilized the dask.distributed library to express individual trajectories as independent tasks, followed by aggregation and model selection by the resulting score (Figure 2b; File S1).
When executed via a single, multithreaded worker process, performance scaled effectively to eight concurrent tasks. At 12–24 concurrent tasks, scaling decreased due to multithread overhead in underlying PyRosetta components. On the other hand, multi‐node distributed execution scaled effectively to 96 concurrent tasks run via 16 processes on 4 AWS m5.24xlarge worker instances with no performance loss (Figure 3; File S1)
Figure 3.
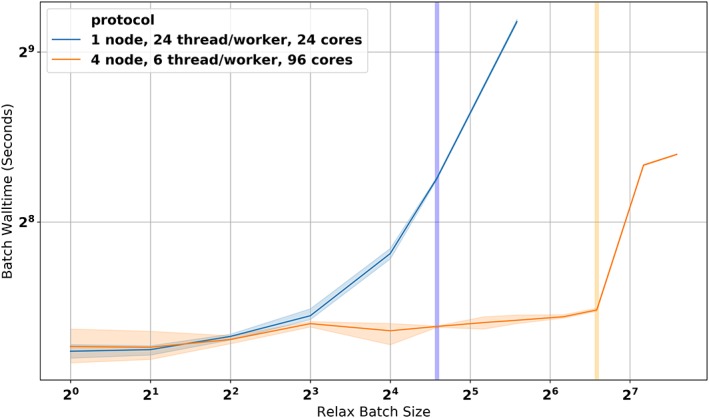
Scaling via distributed execution. (a) Parallel FastRelax walltime in multithreaded and distributed Dask clusters via pyrosetta.distributed. Vertical bars indicate batch sizes equivalent to one task‐per‐core. Single‐process multithreaded execution (blue) shows decreased performance beyond eight threads per process before saturating the 24 available cores. Cross‐node distributed execution (orange) limited to six threads‐per‐process and four process‐per‐node scales to saturate all distributed cores without additional overhead. All benchmarks were run on AWS m5.12xlarge instances with 24 physical cores per node, default network settings, and hyperthreading disabled
As runtime of an individual protocol step executes on the order of seconds to minutes, the millisecond‐scale per‐task constant overhead does not appreciably impact the total throughput. Thus, scaling to significantly larger cluster sizes is limited only by the application‐specific task architecture and the underlying dask.distributed computing library, which has been demonstrated on thousands of concurrent cores.14, 15
3. CONCLUSION
Currently, incorporating Rosetta into scientific workflows requires effort to manage the source code, compilation modes, system libraries, and other dependencies. This consumes time and energy, and it substantially increases the difficulty of reproducing computational scientific results. Post‐deployment, the need to learn a highly specialized set of APIs presents additional barriers to innovation. To address these critical problems in the field, we provided conda packages for Rosetta executables and PyRosetta, and we integrated the RosettaSuite with the ubiquitous python programming interface.
By using Conda packages, scientists can describe a complete computing environment in an easily shareable file, allowing trivial reconstruction and deployment of this environment on multiple platforms. The portability and reliability of these environments creates a pathway for renewed emphasis on reproducibility, and it enables scientists to spend their time focusing on research questions instead of managing software dependencies.
Rather than create a new standard, we integrated the RosettaSuite with common programming interfaces in order to make Rosetta compatible with established third party tools used in scientific computing. Relying on a broader standard facilitates the development of workflows wherein a Rosetta simulation may be one step in a larger computational pipeline. These larger pipelines can be executed interactively through a Jupyter notebook that can, in turn, scale simulations via an HPC or cloud‐based scheduler and collect results.
The developments reported here position Rosetta to be easily deployed across multiple computing providers, adopt state‐of‐art reproducibility of results, and integrate with existing components of the scientific software ecosystem when available. Together, this enables users, who would otherwise not have access to the necessary computational infrastructure, to perform state‐of‐the‐art molecular simulation and design with Rosetta, and it enables robust development of complex simulation workflows.
4. COMPONENTS
4.1. Conda—Adaptable packaging
Originally developed for managing the Python scientific software stack, conda has evolved into a de facto general purpose standard for scientific software management, with notable growth in the life sciences via the Bioconda project.16 Conda deploys applications into isolated software environments decoupled from the underlying operating system. This approach enables per‐project dependency management, and precise definition and reproduction of a software environment without system‐wide administrator privileges. Relocatable binary packages, built from a well‐defined recipe in a strictly isolated environment, can be deployed from shared distribution channels into multiple environments.
To support both existing and novel workflows, we developed Conda recipes packing both Rosetta suite command‐line executables and a PyRosetta build variant supporting the pyrosetta.distributed namespace (described below). For Rosetta users, integration via conda packages provides support for simplified multi‐platform deployment, reproducible capture of existing protocols, and simplified use of external tools in novel protocols.
Reproducible and broadly redeployable packages can be built via a containerized build process derived from the conda‐forge build infrastructure (see Installation and Support).17 For users who wish to incorporate Rosetta into their workflows, these packages eliminate the time‐consuming resolution of source code, compilation modes, system libraries and other dependencies. For protocol developers, these packages can be used to capture development variants for scientific testing.
4.2. Pyrosetta.distributed—RosettaScripts/python interface integration
The PyRosetta distribution has two primary requirements: an exhaustive presentation of the internal components of the Rosetta suite, and a zero‐dependency deployment on multiple platforms; this has successfully filled important roles for both pedagogical and development prototyping applications. Simultaneously, the ease‐of‐access, rapid prototyping support, reusability, and the speed of execution of RosettaScripts has allowed it to become the lingua franca of Rosetta‐based protein design applications.
A lack of cross‐compatibility between these tools has led to common, but fragmented, workflows in which rosetta_scripts executable outputs are reprocessed via ad hoc data analysis scripts using Pandas, Scikit‐Learn, and other components of the PyData software stack. Many recent advances in Rosetta‐based protein modeling have been implemented as Mover and Filter components or as full RosettaScripts protocols. This has led to an increased gap in the fraction of the suite's functionality exposed via PyRosetta, as well as increased development friction in integrating RosettaScript components into python‐based secondary analysis tools.
We sought to bridge this gap for two primary scenarios: notebook‐based interactive development and large‐scale scientific computing. Exposing the RosettaScripts DSL via a high‐level programming interface allows users to utilize the nearly ubiquitous Jupyter interface for prototyping and data analysis, capturing multi‐step workflows in a single document. We then tailored these APIs to support the core python interfaces that are required for distributed execution; this allows transition from notebook prototypes to large‐scale simulation applications that integrate Rosetta with multiple external libraries. We hypothesized that, in a manner similar to our conda‐based deployment integration, targeting a widely adopted platform would enable broad utilization of existing community supported tools and infrastructure.
4.3. Integration components
The python software ecosystem relies on a small set of shared core interfaces utilizing primitive language‐native data structures, pure function invocation, and object serialization to provide loosely coupled interoperability between independent software components. Our component, the “pyrosetta.distributed” namespace, utilizes established elements of the Rosetta internal architecture: the Pose model and score representation, RosettaScript protocols, and Pose serialization.
The adoption of a small set of core interfaces supports integration with an array of scientific computing tools, including support for interactive development environments, common record‐oriented data formats, statistical analysis, and machine learning packages, and multiple distributed computing packages. The pyrosetta.distributed package provides example integrations with several preferred packages for data analysis (Pandas), distributed computing (Dask), and interactive development (Jupyter Notebook), but is loosely coupled to allow later integration with additional libraries. Updated code examples for the components below are available under the Supporting Information—http://distributed_overview.ipynb.
4.4. Data structures (pyrosetta.distributed.packed_pose)
“Primitive” datatypes form a primary interface between many python libraries and, though not strictly defined, typically include the built‐in scalar types (string, int, bool, float, …), key‐value dicts, and lists. Libraries operating on more complex user‐defined classes often expose routines interconverting to and from primitive datatypes, and primitive datatypes can be efficiently serialized in multiple formats.
For interaction between Rosetta protocol components and external libraries, we developed the pyrosetta.distributed.packed_pose namespace. This implements an isomorphism between the Pose object and dict‐like records of the molecular model and scores. The Pose class represents a mutable, full‐featured molecular model with nontrivial memory footprint. A Pose may be inexpensively interconverted to a compact binary encoding via cereal‐based serialization, originally developed for the suite's internal job distribution engines. This serialized format is used to implement the PackedPose class, an immutable record containing model scores and the encoded model, which is isomorphic to a dict‐based record. Adaptor functions within the packed_pose namespace freely adapt between collections of Pose (packed_pose.to_pose), PackedPose (packed_pose.to_packed), dict‐records (packed_pose.to_dict), and pandas.DataFrame objects (Figure 4a).
Figure 4.
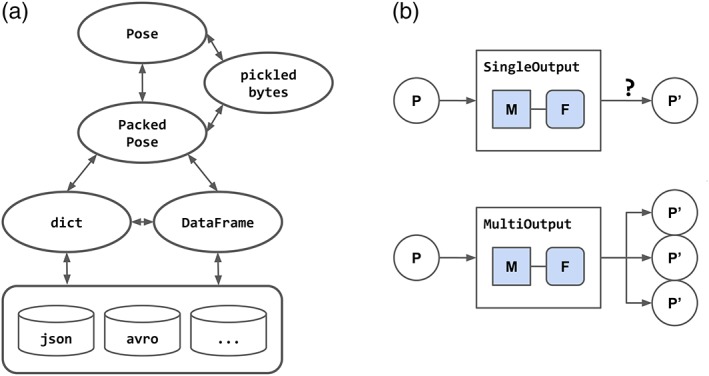
The pyrosetta.distributed data types and task types. (a) pyrosetta.distributed.packed_pose datatypes freely interconvert between mutable Pose and immutable PackedPose, dict‐record, and DataFrame representations, allowing integration with external libraries. (b) pyrosetta.distributed.tasks supports task‐based distributed computing via serialization of immutable task and Pose objects
A dict‐record and DataFrame interface provides zero‐friction integration with a wide variety of data analysis tools and storage formats. For example, the record‐oriented format can be passed through statsmodels or scikit‐learn based filtering and analysis and written to any json‐encoded text file, avro record‐oriented storage, or parquet column‐oriented storage. The http://pyrosetta.distributed.io namespace implements functions that mirror the pyrosetta.io namespace, providing conversion between PackedPose and the PDB, mmCIF, and Rosetta binary silent‐file formats.
Critically, the PackedPose record format can also be transparently serialized, stored with a minimal memory footprint, and transmitted between processes in a distributed computing context. This allows a distributed system to process PackedPose records as plain data, storing and transmitting numerous model decoys while only unpacking a small working set into heavyweight Pose objects.
4.5. Protocol components (pyrosetta.distributed.tasks)
RosettaScripts uses an XML‐based DSL to tersely encode molecular modeling protocols with a pipeline‐like dataflow. The rosetta_scripts interpreter functions by parsing, XSD‐validating and initializing a single RosettaScripts protocol. It then applies this protocol to input structures repeatedly to produce simulation output. Recent work has expanded support for more complex dataflow, including multi‐stage operations and additional logic; however, RosettaScripts is not intended to be a general purpose programming language.
The pyrosetta.distributed.tasks namespace encapsulates the RosettaScripts interface, allowing the DSL to be utilized within python processes. Protocol components are represented as “task” objects containing an XML encoded script. Task objects are serializable via the standard pickle interface, and they use a simple caching strategy to perform on‐demand initialization of the underlying protocol object as needed for task application.
Task components accept any valid pose‐equivalent data structure and return immutable PackedPose data structures by (a) deserializing the input into a short‐lived Pose object, (b) applying the parsed protocol to the Pose and (c) serializing the resulting model as a PackedPose. Two task classes, SingleOutputRosettaScriptsTask and MultipleOutputRosettaScriptsTask define either a one‐to‐one function returning a single output, or a one‐to‐many protocol component returning a lazy iterator of outputs. All tasks operate as “pure functions,” returning a modified copy rather than directly manipulating input data structures (Figure 4b).
4.6. Multithreaded and distributed execution
Rosetta‐based simulations frequently involve execution of numerous independent monte‐carlo sampling trajectories that all begin from a single starting structure; in other words, they are “embarrassingly” or “trivially” parallel. The Rosetta suite implements a job distribution framework (“JD2” and “JD3”) to manage I/O and task scheduling for parallelizable workloads of this type; this allows the rosetta_scripts interpreter to operate as a single process or within MPI, BOINC, and other distributed computing frameworks. Semantics of the RosettaScripts language have also evolved to incorporate nontrivial forms of parallelism, including support for multi‐stage scatter/gather protocols (“Multistage RosettaScripts”). Though fully functional, this framework is optimized for operation as a standalone application and does not provide straightforward integration with third party tools or generalized program logic.
The combination of immutable data structures and pure function interfaces implemented in the pyrosetta.distributed namespace provides an alternative approach to job parallelization by integrating RosettaScripts as a submodule that is compatible with dask.distributed and other task‐based distributed computing frameworks. By virtue of reliance on standard python primitives, the pyrosetta.distributed namespace is not tightly coupled to a single execution engine. Single‐node scheduling may be managed via the standard multiprocessing or concurrent. Futures interfaces, providing a zero‐dependency solution for small‐scale sampling or analysis tasks. Execution via MPI‐based HPC deployments may be managed via the mpi4py interface.
To support effective distributed execution, the pyrosetta.distributed namespace is intended to be installed via a build configuration of PyRosetta, provided by conda packages described above, supporting multithreaded execution. This variant utilizes existing work establishing thread‐safety in the suite, and it releases the CPython global interpreter lock when calling compiled Rosetta interfaces. This enables multi‐core concurrent execution of independent modeling trajectories via python‐managed threads, as well as python‐level operations such as network I/O and process heartbeats to occur concurrently with long‐running Rosetta API calls.
4.7. Interactive analysis and notebook‐based computing
Notebook‐based interactive analysis, typified by the Jupyter project,18 has become a dominant tool in modern data science software development. In this model, data, code, output, and visualization are combined in a single document, which is viewed and edited through a browser‐based interface to a remote execution environment.
To facilitate interactive analysis, we extended the PyRosetta Pose interface to expose total, residue one‐body, and residue‐pair two‐body terms of the Rosetta score function as NumPy structured arrays. Combined with the pandas.DataFrame representation offered in pyrosetta.distributed.packed_pose, this provides an expressive interface for interactive model analysis and selection. We also integrated existing documentation into the pyrosetta.distributed.docs namespace to allow introspection‐based exploration of Mover and Filter RosettaScripts components. Existing tools for web‐based biomolecular visualization, such as py3dmol19 and NGLview,20 extend this interface to a fully‐featured biomolecular simulation, analysis, and visualization environment.
Remote notebook execution has the distinct advantage of allowing a user to access computational resources far beyond the capabilities of a single workstation. By using tools such as Dask via the integrations described above, a remote notebook interface can be used to manage a distributed simulation spanning hundreds of cores for rapid model analysis, and it offers a viable alternative to traditional batch‐based computing for some classes of simulation.
4.8. Limitations and extensions
The pyrosetta.distributed namespace repurposes features of the existing RosettaScripts implementation to provide a task‐based computing model. However, as the suite was not originally designed and implemented around this model, notable limitations exist. Most obviously, the suite's global, static command‐line options system is not effectively exposed or managed in the pyrosetta.distributed namespace. Simulations requiring options‐based configuration can be supported via application‐wide initialization from a single set of flags. However, components that require options‐based specification of individual modeling steps cannot currently be represented as tasks. An implementation that delegates to a task‐specific subprocess, which may initialize the suite with arbitrary command‐line options, could overcome this limitation. Additionally, some protocol components of the suite may be tightly coupled to the job distributor, utilize global state, maintain internal caches, or violate thread safety assumptions in ways that are incompatible this framework. Integration and unit testing should be used before large‐scale simulation to ensure a protocol is functioning as anticipated.
The Rosetta suite's existing Pose serialization format directly encodes the structure of many internal classes without a guarantee of forward or reverse compatibility. As a result, the packed data formats described in this work are not suitable for use across differing versions of the Rosetta suite, and cross‐version compatibility should be considered fortuitous at best. Compatibility across application versions, as would be required for persistent storage or use in an incrementally deployed distributed system, requires persistence through alternative formats such as PDB, mmCIF, or Rosetta silent files. Development of a robust, cross version serialization format through a tool such as Protocol Buffers, Thrift, or a defined MessagePack or JSON schema could be used to address this limitation in a performant manner.
5. INSTALLATION AND SUPPORT
5.1. Package access
The Rosetta suite is freely available for academic and non‐profit users, and it is available for commercial licensing. The suite can be licensed via https://els.comotion.uw.edu/express_license_technologies/rosetta and downloaded in source form at https://www.rosettacommons.org/. Binary conda packages described in this work can be built from source, with documentation located under main/source/conda/README.md.
Binary packages for weekly source releases are also available at https://conda.graylab.jhu.edu, licensed at https://els.comotion.uw.edu/express_license_technologies/pyrosetta and downloaded via instructions at http://www.pyrosetta.org/dow.
As of this writing, build configurations are provided for Linux and MacOS platforms. Execution on Windows is supported via Conda and the Windows Subsystem for Linux.
5.2. Relax benchmark
The parallel relax performance benchmark was performed via http://aws-parallelcluster. The benchmark generation script is available in the supporting information.
5.3. Miniprotein design protocol
Disulfide‐rich miniprotein de novo design was performed on the O2 cluster at Harvard Medical School and GCP. The XML protocol used for design, protein blueprint file, job submission scripts, and library of designed protein models generated in this study are available in the supporting information.
Supporting information
File S1. supporting information.
ACKNOWLEDGMENTS
Portions of this research were conducted on the O2 High Performance Compute Cluster, supported by the Research Computing Group, at Harvard Medical School. See http://rc.hms.harvard.edu for more information.
The authors would like to thank and acknowledge Sergey Lyskov for development and maintenance of the PyRosetta project, Jason Klima, Stacey Gerben and members of the Institute for Protein Design for gracious testing and revisions, David Baker for mentorship, and the larger Rosetta Commons community.
Ford AS, Weitzner BD, Bahl CD. Integration of the Rosetta suite with the python software stack via reproducible packaging and core programming interfaces for distributed simulation. Protein Science. 2020;29:43–51. 10.1002/pro.3721
Statement of importance: Development and application of novel biomolecular modeling protocols using the Rosetta suite involves challenges in both software development and distributed execution on cluster and cloud computing platforms. Integration with existing python programming interfaces with the community standard RosettaScripts language allows accelerated development and scalable execution via reuse of an existing scientific software ecosystem, while conda‐based package management enables reproducible deployment of modeling protocols.
Funding information Harvard Medical School, Grant/Award Number: O2
Contributor Information
Alexander S. Ford, Email: alex.ford@proteininnovation.org.
Christopher D. Bahl, Email: chris.bahl@proteininnovation.org.
REFERENCES
- 1. Fleishman SJ, Leaver‐Fay A, Corn JE, et al. RosettaScripts: A scripting language interface to the Rosetta macromolecular modeling suite. PLoS One. 2011;6:e20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chaudhury S, Lyskov S, Gray JJ. PyRosetta: A script‐based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bender BJ, Cisneros A III, Duran AM, et al. Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry. 2016;55:4748–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004;32:W526–W531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moretti R, Lyskov S, Das R, Meiler J, Gray JJ. Web‐accessible molecular modeling with rosetta: The rosetta online server that includes everyone (ROSIE). Protein Sci. 2018;27:259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cyrus Biotech|Molecular Modeling and Design. Available from: https://cyrusbio.com/
- 7. Leaver‐Fay A, Tyka M, Lewis SM, et al. ROSETTA3: An object‐oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhardwaj G, Mulligan VK, Bahl CD, et al. Accurate de novo design of hyperstable constrained peptides. Nature. 2016;538:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014;23:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buchko GW, Pulavarti SVSRK, Ovchinnikov V, et al. Cytosolic expression, solution structures, and molecular dynamics simulation of genetically encodable disulfide‐rich de novo designed peptides. Protein Sci. 2018;27:1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rocklin GJ, Chidyausiku TM, Goreshnik I, et al. Global analysis of protein folding using massively parallel design, synthesis, and testing. Science. 2017;357:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nivón LG, Bjelic S, King C, Baker D. Automating human intuition for protein design. Proteins. 2014;82:858–866. [DOI] [PubMed] [Google Scholar]
- 13. Harrison PM, Sternberg MJ. Analysis and classification of disulphide connectivity in proteins. The entropic effect of cross‐linkage. J Mol Biol. 1994;244:448–463. [DOI] [PubMed] [Google Scholar]
- 14.Rocklin M Dask Scaling Limits. Available from: http://matthewrocklin.com/blog/work/2018/06/26/dask-scaling-limits
- 15. Lunacek M, Braden J, Hauser T The scaling of many‐task computing approaches in python on cluster supercomputers. In: 2013 IEEE International Conference on Cluster Computing (CLUSTER). ; 2013. pp. 1–8. Available from: 10.1109/CLUSTER.2013.6702678 [DOI]
- 16. Grüning B, Dale R, Sjödin A, et al. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat Methods. 2018;15:475–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conda‐forge: community driven packaging for conda. Available from: https://conda-forge.org/
- 18. Perkel JM. Why Jupyter is data scientists' computational notebook of choice. Nature. 2018;563:145–146. [DOI] [PubMed] [Google Scholar]
- 19. Rego N, Koes D. 3Dmol.js: Molecular visualization with WebGL. Bioinformatics. 2015;31:1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nguyen H, Case DA, Rose AS. NGLview‐interactive molecular graphics for Jupyter notebooks. Bioinformatics. 2018;34:1241–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
File S1. supporting information.