

Abstract
Isotropic chemical shifts measured by solution nuclear magnetic resonance (NMR) spectroscopy offer extensive insights into protein structure and dynamics. Temperature dependences add a valuable dimension; notably, the temperature dependences of amide proton chemical shifts are valuable probes of hydrogen bonding, temperature‐dependent loss of structure, and exchange between distinct protein conformations. Accordingly, their uses include structural analysis of both folded and disordered proteins, and determination of the effects of mutations, binding, or solution conditions on protein energetics. Fundamentally, these temperature dependences result from changes in the local magnetic environments of nuclei, but correlations with global thermodynamic parameters measured via calorimetric methods have been observed. Although the temperature dependences of amide proton and nitrogen chemical shifts are often well approximated by a linear model, deviations from linearity are also observed and may be interpreted as evidence of fast exchange between distinct conformational states. Here, we describe computational methods, accessible via the Shift‐T web server, including an automated tracking algorithm that propagates initial (single temperature) 1H—15N cross peak assignments to spectra collected over a range of temperatures. Amide proton and nitrogen temperature coefficients (slopes determined by fitting chemical shift vs. temperature data to a linear model) are subsequently calculated. Also included are methods for the detection of systematic, statistically significant deviation from linearity (curvature) in the temperature dependences of amide proton chemical shifts. The use and utility of these methods are illustrated by example, and the Shift‐T web server is freely available at http://meieringlab.uwaterloo.ca/shiftt.
Keywords: chemical shifts, curvature, NMR, temperature, temperature coefficients
Abbreviations
- DSS
4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid
- fALS
familial amyotrophic lateral sclerosis
- HSQC
heteronuclear single quantum coherence spectroscopy
- NMR
nuclear magnetic resonance
- SOD1
superoxide dismutase 1
- TCEP
tris(2‐carboxyethyl)phosphine
1. INTRODUCTION
Nuclear magnetic resonance (NMR) chemical shifts are sensitive probes of protein structure and dynamics. Logically, the determinants of chemical shifts must also be responsible for temperature dependences thereof; however, these determinants may not all vary with temperature to the same extent. Variable‐temperature NMR (VT‐NMR) therefore offers unique insights into the phenomena underlying chemical shift dispersion in general. VT‐NMR may also facilitate the resolution of peaks that are significantly overlapped, that is, they may separate due to differing temperature dependences. Here, we focus on the temperature dependences of amide proton and nitrogen chemical shifts, which may be easily and inexpensively measured via a series of 1H—15N heteronuclear single quantum coherence (HSQC) spectra collected at different temperatures.
Empirically, the temperature dependences of both amide proton and amide nitrogen chemical shifts are frequently linear.1 The linear temperature coefficients (i.e., slopes determined by fitting chemical shift vs. temperature data to a linear model) of amide protons have been used to probe the hydrogen bond status of individual amides,2, 3, 4 interpreted as a measure of temperature‐dependent loss of structure,1, 5, 6, 7, 8 and grouped sequentially to distinguish between ordered and disordered protein regions.7, 9, 10, 11 To our knowledge, no similarly straightforward interpretations of amide nitrogen temperature coefficients have been proposed,1, 12 although the determinants of amide nitrogen chemical shifts have been studied in some detail.13, 14, 15, 16 Systematic deviations from linearity (curvature) are also of great interest, as they may be interpreted in terms of fast exchange between distinct conformations,8, 17, 18, 19, 20, 21 but such curvature is often subtle and may be difficult to detect and validate.
In the following, we describe algorithms, accessible via the Shift‐T web server (Figure 1), for automated tracking of 1H—15N cross peaks (e.g., from 1H—15N HSQC spectra) over temperature, decomposition of this motion in the 1H—15N plane into amide proton and nitrogen chemical shift temperature dependences, and subsequent analysis in terms of linear temperature coefficients and curvature. Although not demonstrated here, it may also be feasible to use these methods to analyze other types of 2D NMR spectra, for example, 1H—13C HSQC or 1H—1H TOCSY. We illustrate the utility of these algorithms by applying them to disulfide‐reduced, unmetallated superoxide dismutase 1 (apoSOD12SH) VT‐NMR data. ApoSOD12SH is an immature form of human Cu, Zn SOD1, various mutants, and maturation states of which have been implicated in a progressive neurodegenerative disease called familial amyotrophic lateral sclerosis (fALS).22 Our analysis of this VT‐NMR data reveals conformational heterogeneity in the native ensemble and rationalizes differences in the biophysical characteristics of closely related variants.
Figure 1.
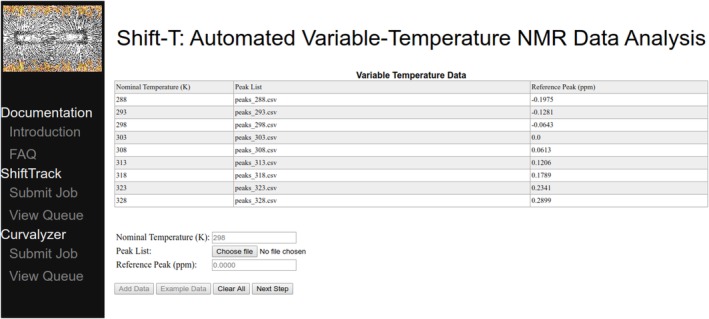
ShiftTrack job submission via the Shift‐T web server: peak lists (one per temperature) and temperature‐invariant reference peaks (e.g., DSS). The Shift‐T server can be found at http://meieringlab.uwaterloo.ca/shiftt
2. RESULTS AND DISCUSSION
2.1. Cross peak tracking and temperature coefficients
In order to combat slow drift in the strength of the field generated by the electromagnet, modern NMR spectrometers use a feedback control system (commonly referred to as the “lock”) that dynamically adjusts the strength of the magnetic field to maintain the resonant frequency of a particular nucleus at a fixed offset to the base frequency. In aqueous solutions such as those suitable for protein NMR, the resonance of deuterium nuclei from D2O/HDO is monitored for this purpose. The chemical shift of water (including H2O, D2O, and HDO) is intrinsically temperature dependent,23, 24 but the feedback control system compensates. Thus, the chemical shift of water appears to be temperature invariant, while all other peaks shift (because of change in the strength of the magnetic field) from their “true” positions by an amount equal to the temperature dependence of water. To recover 1H and 15N temperature‐dependent chemical shifts unbiased by this “deuterium lock artefact,” spectra must be referenced to a standard with negligible temperature dependence, for example, 4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid (DSS)24, 25 (Figure 2c). In spectra that have not been referenced to such a standard, the temperature dependence of water is often the largest contributor to the observed changes in chemical shifts (Figure 2a). Because the set point of the spectrometer temperature controller is not a sufficiently accurate measure of the actual temperature, the chemical shift of DSS (or apparent movement thereof induced by the deuterium lock artefact) may also be used in temperature calculations (see the Methods section).
Figure 2.
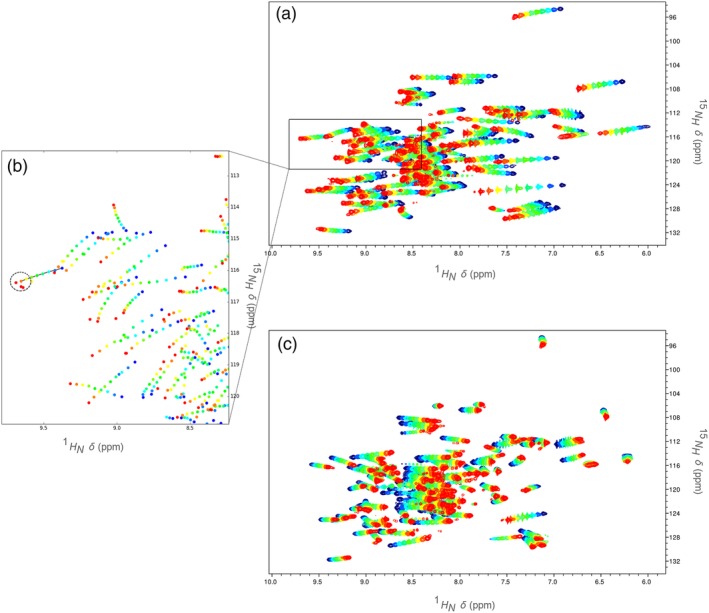
An example of overlaid 1H—15N data from HSQC spectra collected over a range of temperatures (blue: low; red: high). (a) unreferenced HSQC spectra; the deuterium lock artefact is evident. (b) peaks picked from the indicated region of the 1H—15N plane—line segments join peaks in a ShiftTrack candidate line, and the search radius for the final point is indicated by the dashed circle (peaks are tracked in unreferenced spectra in order to take advantage of deuterium lock artefact‐induced regularity). (c) HSQC spectra referenced to DSS, showing the true temperature dependences of amide chemical shifts
Empirically, movement of cross peaks in the 1H—15N plane is often approximately linear with respect to temperature, even after referencing all chemical shifts to DSS. However, prior to such referencing a much greater degree of regularity is apparent (Figure 2a); as temperature increases, cross peaks tend to move downfield in both dimensions in a highly linear fashion. From 1H—15N cross peak coordinates for a given amide, specified at a single temperature, our ShiftTrack algorithm finds the set of cross peaks (one per temperature, selected from peaks picked automatically from unassigned spectra, for example, by TopSpin; see the Methods section) with the smallest deviation from linearity, that is, the set for which a simple linear regression gives the lowest residual sum of squares, subject to a weak constraint on the spacings between points. We prefer to apply this algorithm before referencing the spectra to DSS, turning artefact‐induced regularity to our advantage.
ShiftTrack maintains a list of candidate “lines” (sets of points). As the unassigned peak list from each temperature is processed, new candidate lines are constructed through extension of existing candidates by a single point (Figure 2b). In order to avoid a combinatorial explosion, new candidates are constructed using only points found within a user‐defined radius of the cross peak from the previous temperature and filtered to remove candidates with pronounced nonlinearity from consideration. After peak lists from all temperatures have been processed, the list of candidate lines may contain entries with varying numbers of peaks, for example, if no suitable points with which to extend a given candidate were found at one or more temperatures. Preference is shown for full‐length (i.e., one peak per temperature) solutions. If no full‐length solutions below the residual sum of squares linearity threshold are found, shorter solutions are considered. If these are also rejected on the basis of the residual sum of squares linearity criterion, no solution will be reported for the assignment in question. The output of ShiftTrack includes plots of chemical shifts versus temperature, a line determined by simple linear regression, and regression residuals; these plots, particularly the residuals, facilitate verification of ShiftTrack solutions (Figure 3). At the discretion of the user, residuals more than two SDs from the mean (which is typically zero) may be flagged for manual review.
Figure 3.
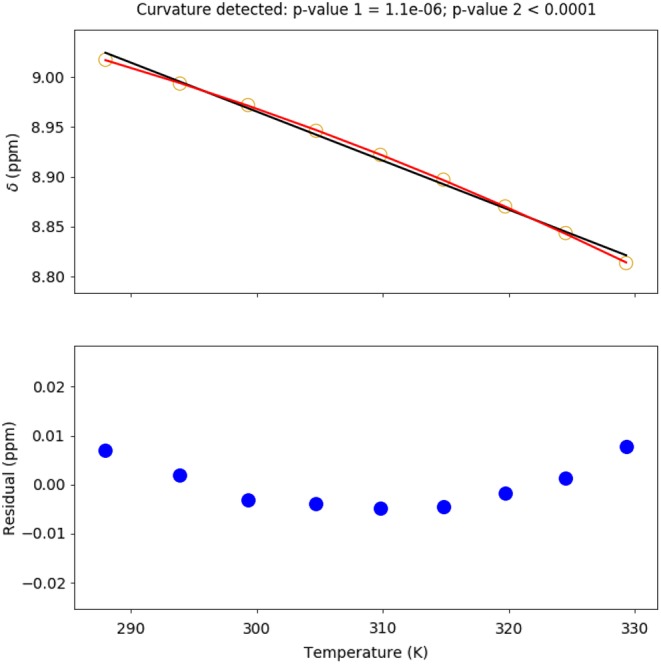
Chemical shifts (referenced to DSS; open circles, top) and residuals (filled circles, bottom) illustrating both temperature coefficient calculation (slope of black line) and curvature detection (red quadratic). The probability that the linear model is correct (from the extra sum of squares F test) is reported as p value 1. The probability that the curvature observed is the result of random errors (from numerical simulation of residual errors) is reported as p value 2
Amide proton temperature coefficients determined by the ShiftTrack algorithm for pseudo wild‐type (pWT) and H46R apoSOD12SH are illustrated in Figure 4. Both maturation state and mutations (such as H46R) of SOD1 have been associated with the onset and progression of ALS.22 Fully mature pWT SOD1 (holoSOD1) is an exceptionally thermostable (T m = 92.0°C)29 homodimeric metalloenzyme, each 153 amino acid subunit of which binds one Cu2+ and one Zn2+ with very high affinities. Also, the interface between the immunoglobulin‐like β‐barrel subunits is stabilized by a disulfide bond, and the “electrostatic loop” (Loop VII; Figure 4b) contributes to metal binding and function.30 In contrast, pWT apoSOD12SH adopts a dynamic, predominantly monomeric structure with much lower thermostability (T m = 47.6°C).31, 32
Figure 4.
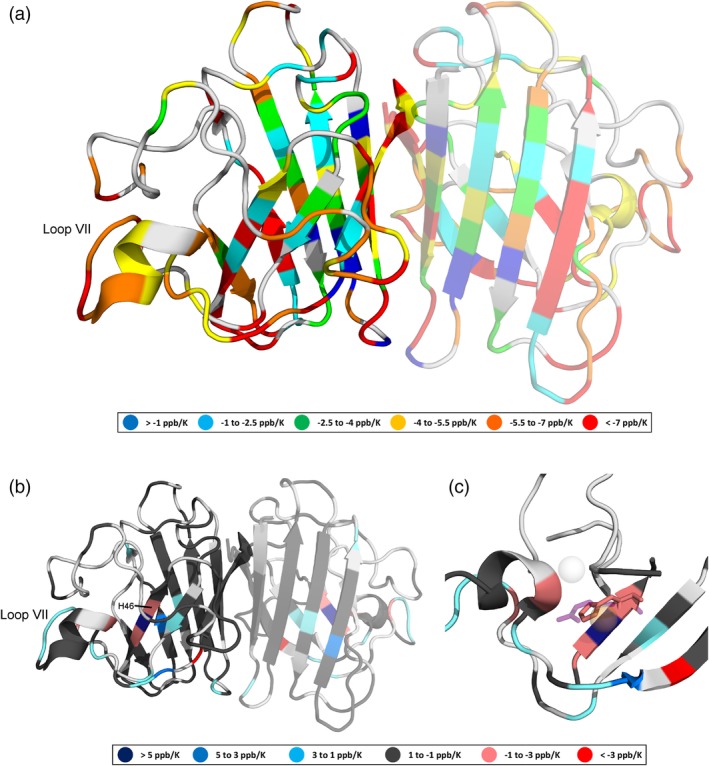
(a) Amide proton temperature coefficients (determined using chemical shifts referenced to DSS) for pWT apoSOD12SH, (b) temperature coefficient differences (relative to pWT) resulting from the H46R mutation, and (c) the metal‐binding region of apoSOD12SH showing overlaid H46 (pink) and R46 (purple, semitransparent) side chains. Light gray indicates that data are not available. The monomeric, rather than dimeric form of apoSOD12SH predominates, but in (a) and (b), we project the data onto a dimeric SOD1 structure (PDB ID: 2AF2)26 with a semitransparent second subunit in order to illustrate the residues that form the dimer interface and present two views (one rotated ∼ 180° relative to the other). In holoSOD1 (PDB ID: 1HL5),27 the H46 residue coordinates the Cu2+ ion (orange, semitransparent), while in H46R (PDB ID: 1OZT) the positively charged guanidinium group of the R46 side chain sits between the vacant Cu2+ and Zn2+ (light gray, semitransparent) binding sites. Figure prepared using PyMOL28
Consistent with this dynamism, we find that the magnitudes of pWT apoSOD12SH temperature coefficients (Figure 4a) are frequently greater than those of corresponding pWT holoSOD1 residues,8 indicating that the magnetic environments of the amide protons change more with temperature. Relative to pWT apoSOD12SH, some H46R apoSOD12SH temperature coefficients increase, while others decrease, with the greatest differences concentrated near the site of mutation and in the electrostatic loop (Loop VII; Figure 4b). Notably, H46R is an fALS‐associated mutation,22 and despite being more stable than pWT apoSOD12SH, H46R apoSOD12SH exhibits increased propensity to misfold and aggregate.31, 33 There is also evidence that the H46R apoSOD12SH electrostatic loop can participate in aberrant, non‐native intermolecular interactions33; interestingly, we show that the magnetic environments of amide protons in the H46R apoSOD12SH electrostatic loop actually change less with temperature than the corresponding pWT apoSOD12SH protons (Figure 4b), which may further clarify this possible mechanism of H46R‐induced disease.
We previously found that the average of the temperature coefficients can report on protein global stability changes.8 Here we compare the temperature coefficient averages of 103 amides for pWT and H46R apoSOD12SH, which are −5.30 ppb/K and − 5.10 ppb/K, respectively. Consistent with our previous results, the magnitude of the average temperature coefficient of pWT apoSOD12SH (T m = 47.6°C) is greater than that of the more stable H46R apoSOD12SH (T m = 52.6°C).31 Insight into the molecular basis for global stabilization of H46R apoSOD12SH relative to pWT apoSOD12SH is obtained from examination of individual temperature coefficients; the positively charged guanidinium group of R46 is situated between the vacant Zn2+ and Cu2+ binding sites in H46R apoSOD12SH 33 where it may electrostatically stabilize the protein.
2.2. Detection and statistical validation of curvature
Although the temperature dependences of amide proton chemical shifts are predominantly linear, curvature is not uncommon, and may be attributed to temperature‐dependent shifts in the population of distinct conformational states.8, 17, 18, 19, 20, 21 Here, we focus first on strategies for the detection of curvature, which may be subtle, then on statistical validation that the curvature detected is likely to result from changes in the population of conformational states rather than a confluence of random errors. Collectively, these tests (which we refer to as Curvalyzer) are designed to be quite stringent, as detailed later. Temperature‐dependent conformational changes that do not result in curvature are possible; absence of curvature is not particularly informative, therefore false negatives are unlikely to lead to incorrect conclusions. In contrast, false positives (inference of curvature where there is none) may result in serious errors.
Curvalyzer treats curvature detection as a “model selection” problem. Two nested models are considered for each set of chemical shift versus temperature data: linear and quadratic. There is no theoretical reason to believe that experimentally observed curvature should fit a quadratic model. However, as expected given our hypothesis that temperature‐dependent shifts in the relative population of conformational states are causative, curvature generally manifests as a gentle curve with a single minimum or maximum (Figure 3); thus, the quality of fit to a quadratic model is an adequate test of nonlinearity. Unless the temperature dependence is exactly linear, the quadratic model will have a lower sum of squared errors; however, if the improvement (relative to the linear model) is small, it may not be statistically significant. Curvalyzer uses an extra‐sum‐of‐squares F test to quantify the statistical significance of the improvement.34 The p value resulting from this F test is reported (using a significance threshold of 0.01), where the null hypothesis is that the linear model is correct. To increase the stringency of this test, we ensure that nonlinearity introduced by a single outlier (and therefore unlikely to be attributable to the sampling of distinct conformational states) does not introduce false positives by requiring that for each set of points generated by leave‐one‐out resampling without replacement (i.e., from a set of amide proton chemical shifts measured at N different temperatures, N subsets of N−1 points are possible), the quadratic fit must be significantly better (p value less than .01, per the extra sum of squares F test) than that of the linear fit in order for the temperature dependence to be considered curved.
Once the existence of curvature has been established, we must consider its source. The F test referenced above implicitly considers the possibility that deviations from linearity are caused by errors in the data, but each set (one per amide proton) of chemical shifts is evaluated separately. For the set of all chemical shifts (at all temperatures) of amide protons for which curvature was not detected, we can calculate residuals and use them to better estimate the distribution of errors. We find that distributions of residuals are bell shaped with means near zero, but may have heavy tails that indicate deviation from normality. We therefore fit residuals to a “t distribution,” which includes the normal distribution as a special case, but allows for the possibility of heavier tails (Figure S1).34 Plausible sets of residuals can be simulated by drawing random values from this fitted distribution; by doing so many (100000) times, we calculate the probability of observing curvature (due to random errors) of a given magnitude. In our analysis of experimentally observed curvature, these probabilities are equivalent to p values (using a significance threshold of .01) where the null hypothesis is that the curvature is due to measurement errors rather than shifts in the population of distinct conformational states. This statistical test complements the first by screening out cases where, although curvature was detected, there is a significant chance that it is a product of experimental or peak picking error (e.g., as a result of overlapping peaks).
Curvature detected in the temperature dependences of pWT apoSOD12SH amide proton chemical shifts is illustrated in Figure 5. Consistent with the aforementioned dynamism of apoSOD12SH, curvature is apparent throughout the protein, including within loops and regions of secondary structure. Because the VT‐NMR data were collected at temperatures at temperatures >10° C below the start of the global unfolding endotherm (measured by differential scanning calorimetry),31 we hypothesize that the distinct conformations contributing to curvature may be substates within the apoSOD12SH native ensemble; interestingly, curvature is observed in regions such as the dimer interface and electrostatic loop that were previously found to have high mobility by chemical exchange saturation transfer and Carr–Purcell–Meiboom–Gill relaxation dispersion experiments.32
Figure 5.
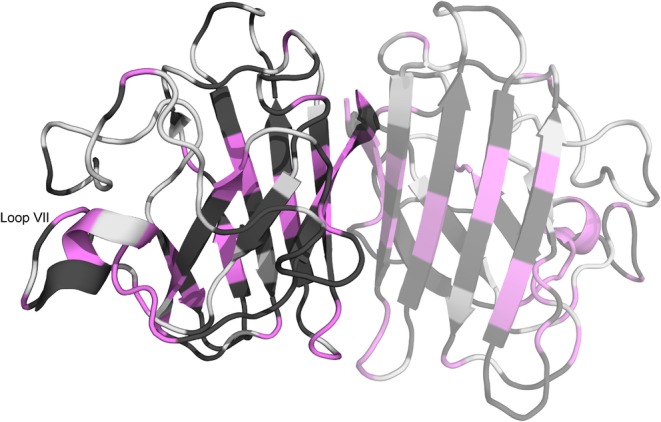
pWT apoSOD12SH curvature, as determined by Curvalyzer (using chemical shifts referenced to DSS). Curvature may be interpreted as evidence of exchange between distinct conformations, but the absence of curvature does not rule exchange out. The monomeric, rather than dimeric form of apoSOD12SH predominates, but here we project the data onto a dimeric SOD1 structure (PDB ID: 2AF2)26 with a semitransparent second subunit in order to illustrate which residues form the dimer interface and present two views (one rotated ∼ 180° relative to the other). Figure prepared using PyMOL28
3. CONCLUSIONS
Manually tracking 1H—15N cross peak movement with temperature is a relatively straightforward, yet labor‐intensive task. Here, we implement an algorithm that automates the bulk of this task, freeing the user to focus on ambiguous cases. We also automate the detection and statistical validation of curvature, which requires stringent tests in order to prevent false (curvature) positives. Overall, these methods form the foundation upon which our analyses of protein VT‐NMR data are built. In general, temperature coefficients and curvature may report on major loss of structure (e.g., unfolding), but here we analyze data collected at temperatures well below the onset of global unfolding. The results of analyses such as these may be widely useful for the characterization of hydrogen bonding in structured2, 3, 4, 32 or disordered7, 9, 10, 11 proteins, and identification of local changes in energetics associated with protein mutation, maturation, or function.1, 8
4. METHODS
4.1. Expression and purification
BL21 Escherichia coli cells were transformed with pHSOD1ASlacIq plasmids35 into which genes encoding pWT SOD1 (wild‐type protein with cysteines 6 and 111 replaced by alanine and serine, respectively22, 31, 35) or its H46R variant had been cloned. 15N‐labeled protein was prepared by growing these cells in M9 minimal media with 15NH4Cl as the sole nitrogen source. Cu,Zn‐SOD1 was purified using a modification of the procedure described by Getzoff et al.35 in which a POROS HP2 hydrophobic interaction column replaced the diethylaminoethyl column. Subsequently, disulfide‐reduced apo (metal free) SOD1 was prepared as previously described.31, 36
4.2. NMR sample preparation
Purified 15N‐labeled reduced apo SOD1 in buffer‐containing 20 mM HEPES pH 7.4, 1 mM tris(2‐carboxyethyl)phosphine (TCEP), 1 mM DSS, and 90% H2O/10% D2O was concentrated using an Amicon Ultra‐4 centrifugal filter (EMD Millipore) to ∼ 15 mg/ml.
4.3. Variable‐temperature NMR
Backbone resonance assignments were determined as previously described.32 Variable‐temperature 1H—15N HSQC (Bruker pulse program “hsqcetfpf3gpsi”37, 38, 39, 40) spectra were acquired using a Bruker AVANCE 600 MHz spectrometer equipped with a 5‐mm triple resonance TXI probe. Amide proton chemical shifts used for temperature coefficient calculations and curvature analysis were directly referenced to DSS (i.e., shifted by the same correction required to move the DSS peak in a 1D 1H spectrum, acquired with appropriate solvent suppression, to 0 ppm), while amide nitrogen chemical shifts are indirectly referenced to DSS41 using a 15N/1H Ξ ratio of 0.101329118. Nominal temperatures (Tnom) ranging from 279 K to 305 K (for pWT apoSOD12SH) or 283 K to 311 K (for H46R apoSOD12SH) in 2 K increments were programmed into the temperature controller via the Bruker TopSpin 1.3 spectrometer control software. Actual temperatures can be determined from the separation between “thermometer” resonances, for example, between H2O and DSS proton chemical shifts (the method used here) or the chemical shifts of methyl and hydroxyl deuterons in methanol‐d4,24, 42 and provided to the Shift‐T server. Alternatively, if spectra are collected over multiple temperatures without varying the center frequency, the Shift‐T server can be instructed to calculate temperature differentials from movement of the DSS peak (due to the deuterium lock artefact described above) and the known temperature dependence of water (−11.9 ppb/K)23 (Equation (1), where T ref is the reference temperature):
| (1) |
Data processing, including automatic peak picking (with parabolic interpolation) from unassigned 1H—15N spectra, was performed using the Bruker TopSpin 4.0.6 software. Peak lists were written to comma‐separated variable (CSV) text files. These data were analyzed by the ShiftTrack and Curvalyzer algorithms via the “Shift‐T: Automated Variable‐Temperature Data Analysis” web server (http://meieringlab.uwaterloo.ca/shiftt).
The ShiftTrack and Curvalyzer algorithms are implemented in Python, and the source files are freely available (https://github.com/meiering-lab/Shift-T). The primary output file format is comma‐separated variable (CSV), which can be edited using a spreadsheet program such as Microsoft Excel (an example output file is included in Appendix S1). The first five lines consist of a header specifying the project name, temperature data, and reference shifts (e.g., DSS). The remainder of the file is in a columnar format, with user‐provided residue names and peak coordinates, amide proton, and nitrogen temperature coefficients calculated by linear regression (along with the residual sum of square errors for each), and both raw (subject to the deuterium lock artefact) and re‐referenced (e.g., to DSS) chemical shifts at each temperature.
4.4. Practical Aspects
Here, we include general recommendations for acquiring data that are likely to yield easily analyzed results. Two important considerations are the number of temperatures at which data are acquired and the temperature range. In principle, temperature coefficients (linear slopes) can be calculated using data from as few as two temperatures. However, the ShiftTrack algorithm will work more reliably with data collected at five or more evenly spaced temperatures, and more points will yield better linear approximations in the presence of noise. The server will not currently accept submissions with data from fewer than four different temperatures.
Curvature detection may be substantially improved by increasing both the number of temperatures (we recommend a minimum of eight) and the temperature range. We advocate collecting data over the widest temperature range possible, given limitations imposed by your equipment (such as probe, temperature controller, spinner material) and the biophysical characteristics of your protein. Unless you wish to probe thermally unfolded states, it is advisable to select a temperature range that avoids heat‐ or cold‐induced global unfolding transitions; such temperature limits can be established using differential scanning calorimetry.
DSS or an equivalently temperature‐invariant standard for chemical shift referencing can often be added directly to the sample; however, DSS has been observed to participate in hydrophobic interactions with proteins.43 If your protein unfolds (fully or partially) during the course of your experiments or has exposed hydrophobic patches, you may wish to use a coaxial insert containing DSS instead.
Supporting information
Figure S1 A histogram (blue) showing the distribution of amide proton chemical shift residuals in the example data (from all residues for which curvature is not detected, at all temperatures). A t distribution (black line) fit to these residuals is superimposed.
Appendix S1: Supporting information
ACKNOWLEDGMENTS
This research was funded by a grant awarded to E.M.M and scholarships awarded to K.T., J.A.P., and D.W.S.M. by the National Sciences and Engineering Research Council of Canada (NSERC) and supported by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET; https://www.sharcnet.ca) and Compute Canada (https://www.computecanada.ca).
Trainor K, Palumbo JA, MacKenzie DWS, Meiering EM. Temperature dependence of NMR chemical shifts: Tracking and statistical analysis. Protein Science. 2020;29:306–314. 10.1002/pro.3785
Funding information Natural Sciences and Engineering Research Council of Canada
REFERENCES
- 1. Tomlinson JH, Williamson MP. Amide temperature coefficients in the protein G B1 domain. J Biomol NMR. 2012;52:57–64. [DOI] [PubMed] [Google Scholar]
- 2. Baxter NJ, Williamson MP. Temperature dependence of 1H chemical shifts in proteins. J Biomol NMR. 1997;9:359–369. [DOI] [PubMed] [Google Scholar]
- 3. Cierpicki T, Otlewski J. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J Biomol NMR. 2001;21:249–261. [DOI] [PubMed] [Google Scholar]
- 4. Cierpicki T, Zhukov I, Byrd RA, Otlewski J. Hydrogen bonds in human ubiquitin reflected in temperature coefficients of amide protons. J Magn Reson. 2002;157:178–180. [DOI] [PubMed] [Google Scholar]
- 5. Andersen NH, Chen C, Marschner TM, Krystek SR Jr, Bassolino DA. Conformational isomerism of endothelin in acidic aqueous media: A quantitative NOESY analysis. Biochemistry. 1992;31:1280–1295. [DOI] [PubMed] [Google Scholar]
- 6. Andersen NH, Neidigh JW, Harris SM, Lee GM, Liu Z, Tong H. Extracting information from the temperature gradients of polypeptide NH chemical shifts. 1. the importance of conformational averaging. J Am Chem Soc. 1997;119:8547–8561. [Google Scholar]
- 7. Bouvignies G, Vallurupalli P, Cordes MH, Hansen DF, Kay LE. Measuring 1HN temperature coefficients in invisible protein states by relaxation dispersion NMR spectroscopy. J Biomol NMR. 2011;50:13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doyle CM, Rumfeldt JA, Broom HR, Sekhar A, Kay LE, Meiering EM. Concurrent increases and decreases in local stability and conformational heterogeneity in Cu, Zn superoxide dismutase variants revealed by temperature‐dependence of amide chemical shifts. Biochemistry. 2016;55:1346–1361. [DOI] [PubMed] [Google Scholar]
- 9. Bah A, Vernon RM, Siddiqui Z, et al. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature. 2015;519:106–109. [DOI] [PubMed] [Google Scholar]
- 10. Okazaki H, Matsuo N, Tenno T, et al. Using 1HN amide temperature coefficients to define intrinsically disordered regions: An alternative NMR method. Protein Sci. 2018;27:1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suárez IP, Gauto DF, Hails G, et al. Conformational sampling of the intrinsically disordered dsRBD‐1 domain from Arabidopsis thaliana DCL1. Phys Chem Chem Phys. 2018;20:11237–11246. [DOI] [PubMed] [Google Scholar]
- 12. Miletti T, Di Trani J Jr, Levros LC, Mittermaier A. Conformational plasticity surrounding the active site of NADH oxidase from Thermus thermophilus. Protein Sci. 2015;24:1114–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Dios AC, Pearson JG, Oldfield E. Secondary and tertiary structural effects on protein NMR chemical shifts: An ab initio approach. Science. 1993;260:1491–1496. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Jardetzky O. Investigation of the neighboring residue effects on protein chemical shifts. J Am Chem Soc. 2002;124:14075–14084. [DOI] [PubMed] [Google Scholar]
- 15. Wang Y, Jardetzky O. Predicting 15N chemical shifts in proteins using the preceding residue‐specific individual shielding surfaces from φ, ψ i−1, and χ 1 torsion angles. J Biomol NMR. 2004;28:327–340. [DOI] [PubMed] [Google Scholar]
- 16. Wang L, Markley JL. Empirical correlation between protein backbone 15N and 13C secondary chemical shifts and its application to nitrogen chemical shift re‐referencing. J Biomol NMR. 2009;44:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baxter NJ, Hosszu LL, Waltho JP, Williamson MP. Characterisation of low free‐energy excited states of folded proteins. J Mol Biol. 1998;284:1625–1639. [DOI] [PubMed] [Google Scholar]
- 18. Williamson MP. Many residues in cytochrome c populate alternative states under equilibrium conditions. Proteins. 2003;53:731–739. [DOI] [PubMed] [Google Scholar]
- 19. Krishna Mohan P, Mukherjee S, Chary KV. Differential native state ruggedness of the two Ca2+‐binding domains in a Ca2+ sensor protein. Proteins. 2008;70:1147–1153. [DOI] [PubMed] [Google Scholar]
- 20. Chandra K, Sharma Y, Chary K. Characterization of low‐energy excited states in the native state ensemble of non‐myristoylated and myristoylated neuronal calcium sensor‐1. Biochim Biophys Acta Proteins Proteomics. 2011;1814:334–344. [DOI] [PubMed] [Google Scholar]
- 21. Bari KJ, Sharma S, Chary KV. On identifying low energy conformational excited states with differential ruggedness in human γs‐crystallin promoting severe infantile cataracts. Biochem Biophys Res Commun. 2019;514:946–952. [DOI] [PubMed] [Google Scholar]
- 22. Broom HR, Rumfeldt JA, Meiering EM. Many roads lead to Rome? multiple modes of Cu, Zn superoxide dismutase destabilization, misfolding and aggregation in amyotrophic lateral sclerosis. Essays Biochem. 2014;56:149–165. [DOI] [PubMed] [Google Scholar]
- 23. Wishart DS, Bigam CG, Yao J, et al. 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. [DOI] [PubMed] [Google Scholar]
- 24. Hoffman RE. Standardization of chemical shifts of TMS and solvent signals in NMR solvents. Magn Reson Chem. 2006;44:606–616. [DOI] [PubMed] [Google Scholar]
- 25. Cross BP, Schleich T. Temperature dependence of the chemical shifts of commonly employed proton NMR reference compounds. Org Magn Reson. 1977;10:82–85. [Google Scholar]
- 26. Banci L, Bertini I, Cantini F, D'Amelio N, Gaggelli E. Human SOD1 before harboring the catalytic metal solution structure of copper‐depleted, disulfide‐reduced form. J Biol Chem. 2006;281:2333–2337. [DOI] [PubMed] [Google Scholar]
- 27. Strange RW, Antonyuk S, Hough MA, et al. The structure of holo and metal‐deficient wild‐type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–891. [DOI] [PubMed] [Google Scholar]
- 28. Schrödinger, LLC (2015) The PyMOL molecular graphics system, version 1.8.
- 29. Stathopulos PB, Rumfeldt JA, Karbassi F, Siddall CA, Lepock JR, Meiering EM. Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis‐associated Gly‐93 mutants of superoxide dismutase. J Biol Chem. 2006;281:6184–6193. [DOI] [PubMed] [Google Scholar]
- 30. Valentine JS, Doucette PA, Zittin Potter S. Copper‐zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. [DOI] [PubMed] [Google Scholar]
- 31. Vassall KA, Stubbs HR, Primmer HA, et al. Decreased stability and increased formation of soluble aggregates by immature superoxide dismutase do not account for disease severity in ALS. Proc Natl Acad Sci U S A. 2011;108:2210–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sekhar A, Rumfeldt JA, Broom HR, et al. Thermal fluctuations of immature SOD1 lead to separate folding and misfolding pathways. Elife. 2015;4:e07296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elam JS, Taylor AB, Strange R, et al. Amyloid‐like filaments and water‐filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Mol Biol. 2003;10:461–467. [DOI] [PubMed] [Google Scholar]
- 34. DeGroot MH, Schervish MJ. Probability and statistics. Boston, MA: Pearson Education, 2012. [Google Scholar]
- 35. Getzoff ED, Cabelli DE, Fisher CL, et al. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358:347–351. [DOI] [PubMed] [Google Scholar]
- 36. Vassall KA, Stathopulos PB, Rumfeldt JA, Lepock JR, Meiering EM. Equilibrium thermodynamic analysis of amyotrophic lateral sclerosis‐associated mutant apo Cu,Zn superoxide dismutases. Biochemistry. 2006;45:7366–7379. [DOI] [PubMed] [Google Scholar]
- 37. Palmer AG III, Cavanagh J, Wright PE, Rance M. Sensitivity improvement in proton‐detected two‐dimensional heteronuclear correlation NMR spectroscopy. J Magn Reson. 1991;93:151–170. [Google Scholar]
- 38. Kay L, Keifer P, Saarinen T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc. 1992;114:10663–10665. [Google Scholar]
- 39. Grzesiek S, Bax A. The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J Am Chem Soc. 1993;115:12593–12594. [Google Scholar]
- 40. Schleucher J, Schwendinger M, Sattler M, et al. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J Biomol NMR. 1994;4:301–306. [DOI] [PubMed] [Google Scholar]
- 41. Wishart DS, Sykes BD. The 13C chemical‐shift index: a simple method for the identification of protein secondary structure using 13C chemical‐shift data. J Biomol NMR. 1994;4:171–180. [DOI] [PubMed] [Google Scholar]
- 42. Findeisen M, Brand T, Berger S. A 1H‐NMR thermometer suitable for cryoprobes. Magn Reson Chem. 2007;45:175–178. [DOI] [PubMed] [Google Scholar]
- 43. Shimizu A, Ikeguchi M, Sugai S. Appropriateness of DSS and TSP as internal references for 1H NMR studies of molten globule proteins in aqueous media. J Biomol NMR. 1994;4:859–862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 A histogram (blue) showing the distribution of amide proton chemical shift residuals in the example data (from all residues for which curvature is not detected, at all temperatures). A t distribution (black line) fit to these residuals is superimposed.
Appendix S1: Supporting information