

Abstract
Immunotherapy targeting programmed cell death-1 (PD-1) induces durable antitumor efficacy in many types of cancer. However, such clinical benefit is limited because of the insufficient reinvigoration of antitumor immunity with the drug alone; therefore, rational therapeutic combinations are required to improve its efficacy. In our preclinical study, we evaluated the antitumor effect of U3-1402, a human epidermal growth factor receptor 3–targeting (HER3–targeting) antibody-drug conjugate, and its potential synergism with PD-1 inhibition. Using a syngeneic mouse tumor model that is refractory to anti–PD-1 therapy, we found that treatment with U3-1402 exhibited an obvious antitumor effect via direct lysis of tumor cells. Disruption of tumor cells by U3-1402 enhanced the infiltration of innate and adaptive immune cells. Chemotherapy with exatecan derivative (Dxd, the drug payload of U3-1402) revealed that the enhanced antitumor immunity produced by U3-1402 was associated with the induction of alarmins, including high-mobility group box-1 (HMGB-1), via tumor-specific cytotoxicity. Notably, U3-1402 significantly sensitized the tumor to PD-1 blockade, as a combination of U3-1402 and the PD-1 inhibitor significantly enhanced antitumor immunity. Further, clinical analyses indicated that tumor-specific HER3 expression was frequently observed in patients with PD-1 inhibitor–resistant solid tumors. Overall, U3-1402 is a promising candidate as a partner of immunotherapy for such patients.
Keywords: Oncology
Keywords: Cancer immunotherapy

Introduction
Immune checkpoint inhibitor (ICI) therapy has emerged as a standard of care treatment for many types of cancer. The mainstay of such immunotherapy is the programmed cell death-1 (PD-1)/programmed cell death-ligand 1 (PD-L1) axis blockade, and this includes nivolumab, pembrolizumab, atezolizumab, durvalumab, and avelumab, all of which have been recently approved as therapeutic agents for cancer treatment (1). Previous clinical trials have shown that these drugs confer longer survival benefit than conventional antitumor chemotherapies for solid cancers, including malignant melanoma (MM), non–small cell lung cancer (NSCLC), head and neck cancer (HNC), gastric cancer (GC), and many other types of malignancies. Importantly, however, only a limited fraction of patients (approximately 10%–30%) with these cancers receive such clinical benefit (2–6). The insufficient therapeutic outcome is caused by several undesirable pretreatment immune states: (a) the absence of preexisting antitumor immunity because of insufficient innate or adaptive immune recognition of cancer cells as foreign (immunological ignorance), (b) the incapability of antitumor immune cells to be reinvigorated by immunotherapy (terminal exhaustion/hyperexhaustion), and (c) the presence of immune suppressors such as myeloid-derived suppressor cells (MDSCs), Tregs, cancer-associated fibroblasts, and aberrant angiogenesis (7).
While many combinatorial treatment strategies are currently expected to overcome this immunological resistance to PD-1/PD-L1 inhibition alone, the most promising partner of the PD-1/PD-L1 inhibitor is cytotoxic chemotherapy (1, 8, 9). This concept was supported by recently published clinical trials targeting advanced NSCLC, which showed that the combination of cytotoxic chemotherapy with PD-1 inhibition significantly improved survival outcomes compared with the standard of care treatment (10–13). However, substantial immunological downsides of conventional cytotoxic chemotherapy as a partner of immunotherapy are also reported. For example, myelosuppression, lymphocyte depletion, and insufficient antitumor cytotoxicity (objective response rates [ORR] of conventional cytotoxic chemotherapies are, at best, 20%–30% in most types of solid cancers) due to nonspecific mode of action all result in less potential to activate antitumor immunity (14). An antibody-drug conjugate (ADC) is advantageous in this regard because such agents specifically deliver their payload (a cytotoxic drug) to the tumor cells via their cancer cell–targeting carrier (an antibody) (14). Indeed, previous landmark clinical trials reported that ado-trastuzumab emtansine (T-DM1), an anti–human epidermal growth factor receptor 2 (anti-HER2) monoclonal antibody linked to the cytotoxic antimicrotubule agent DM1, was clearly superior to conventional cytotoxic chemotherapies in HER2-expressing breast cancer (15, 16). Furthermore, a recent early clinical trial of DS-8201a, an anti-HER2 ADC carrying the highly potent topoisomerase I inhibitor DXd, demonstrated a promising result for HER2-positive solid cancers (17–19). With these results, ADC may be considered as a replacement for conventional cytotoxic chemotherapies, at least in advanced HER2-positive cancers.
HER3, also known as ERBB3, is broadly expressed in various types of human cancer and is associated with poor prognosis (20, 21). HER3 is expressed on cancer cell surfaces and transduces signals through the downstream PI3K/AKT pathway upon coupling with other HER family members, prevents cells from undergoing apoptosis, and causes drug resistance, including resistance to anti-HER1 or anti-HER2 inhibitors (20). Patritumab, one of several HER3-targeting antibodies investigated in clinical trials, produces its antitumor efficacy presumably by inhibiting HER3 ligand binding. However, patritumab did not show any significant survival benefit in previous clinical trials in NSCLC and HNC, requiring the development of other strategies to fight against HER3-expressing cancers (22, 23). U3-1402, a potential first-in-class anti-HER3 ADC (with patritumab as the carrier and DXd as the payload), is currently under development to act on these targets (24). Indeed, an early report of a clinical trial suggested that U3-1402 could be safely administered and demonstrated promising antitumor efficacy in heavily treated HER3-expressing (immunohistochemical HER3 score of tumor cells was 2+/3+) metastatic breast cancer (the ORR was 47%, and the disease control rate was 94%, both of which were far superior to that of the historical control) (25). This satisfactory result has led to the investigation here of U3-1402 in combination with cancer immunotherapy.
Considering the above findings, a logical next step is to explore the therapeutic potential of U3-1402 as a partner to PD-1/PD-L1 inhibition to provide improved treatment of HER3-expressing cancers. Our study evaluated how U3-1402 stimulates the immune system and cooperates with PD-1/PD-L1 axis blockade in preclinical and translational experiments using mouse models and clinical specimens.
Results
U3-1402 improves the functional activity of tumor-infiltrating CD8+ T lymphocytes in HER3-expressing tumors.
First, we evaluated the antitumor efficacy of U3-1402 in HER3-expressing tumors. We established a syngeneic mouse tumor model carrying HER3-expressing cancer cells by subcutaneous inoculation of clone M-3 (CM-3), a previously established mouse melanoma cell line also known as Cloudman S91, to the right-side flank of DBA/2NCrl mice (26, 27). CM-3 tumors were harvested from the mouse model, and the cancer cell expression of HER3 was demonstrated by immunohistochemical analyses using the known HER3-expressing human breast cancer cell line MDA-MB-453 as a positive control (28), whereas CT26 mouse colon cancer cells did not express HER3 (Figure 1A). Additionally, flow cytometry analyses using patritumab as a primary antibody to recognize HER3 confirmed cell-membrane expression of HER3 on CM-3 tumor cells, which was equivalent or somewhat superior to that on the known HER3-expressing human lung cancer cells HCC827 (29), indicating the adequacy of CM-3 to evaluate the antitumor effects of U3-1402 (Figure 1B). Indeed, in in vitro growth inhibition assays, wherein the drug concentration corresponded to that achievable in ongoing human clinical trials (U3-1402: <100 μg/ml) (25), CM-3 cells were sensitive to U3-1402, while HER3-negative CT26 cells were not (Figure 1C). The in vitro growth inhibition assay also showed that patritumab did not affect the growth of CM-3 cells, and this verified that the observed cytotoxic activity of U3-1402 was caused by its payload DXd and not by HER3 signal blockade by patritumab (Figure 1C). In addition, the low concentration of DXd (<10 nM), which corresponded to that of free DXd in plasma released from U3-1402 (25), did not affect cell viability in either HER3-positive or HER-3 negative cells, again indicating that the cytotoxicity of U3-1402 was not caused by an HER3-independent mechanism (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI126598DS1). These findings collectively explain that U3-1402 exhibited HER3-expression–dependent cytotoxic activity in CM-3 cells.
Figure 1. U3-1402 exhibits HER3-dependent cytotoxicity and improves CD8+ TILs function.

(A) Images of membranous HER3 immunostaining of CM-3 (left), MDA-MB-453 (middle), and CT26 cells (right). Scale bars: 100 mm. (B) Flow cytometry analysis of membranous HER3 expression. Data are representative of 3 independent experiments. (C) Left: in vitro growth inhibition assay for U3-1402 with CM-3 or CT26 cells. Right: in vitro growth inhibition assay for U3-1402 or patritumab with CM-3 cells. Data represent mean ± SEM of 6 replicate and 2 independent experiments. (D) Schematic of in vivo experiments. (E) Left: tumor volume curve of subcutaneous CM-3 tumors. Right: tumor volume 11 days after treatment initiation. n = 4–6 for each arm, pooled from 2 independent experiments. (F and G) Flow cytometry analysis of CD8+ TILs. n = 9–10, pooled from 2 independent experiments (F) or 4–5 (G) for each arm. (H) Left: flow cytometry analysis of IFN-γ– and TNF-α–producing CD8+ TILs. n = 6–7 for each arm. Right: representative flow cytometric plots of IFN-γ– and TNF-α–producing CD8+ TILs. Values in the figures indicate the frequency of IFN-γ– and TNF-α–producing CD8+ TILs. (I) Left: tumor volume curve of subcutaneous CM-3 tumors treated as indicated. Right: tumor volume 14 days after treatment initiation. n = 12 for each arm, pooled from 4 independent experiments. P values in E–I are shown on the horizontal lines. Each dot in E–I represents 1 tumor. Data were assessed by unpaired t tests.
Next, we performed in vivo experiments to evaluate the antitumor effects of U3-1402 using the syngeneic mouse HER3-expressing tumor model. A schematic of our in vivo experimental study is depicted in Figure 1D. Treatment was initiated when tumor volume was 80–250 mm3. As expected, U3-1402 significantly inhibited tumor growth compared with vehicle treatment (Figure 1E). Although we assumed an increase in the number of tumor-infiltrating CD8+ T cells (CD8+ TILs) following U3-1402 treatment, flow cytometry analysis demonstrated that there was no significant difference in CD8+ TIL density between the vehicle and U3-1402 treatment arms at this time point (Figure 1F). However, we noticed that the expressions of inhibitory molecules, such as PD-1, lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin and mucin-domain containing protein-3 (TIM-3), on CD8+ TILs were downregulated after U3-1402 treatment. Since cells that highly express multiple inhibitory molecules represent hyperexhausted or unrecoverable T cells (30), our findings suggest that U3-1402 treatment rescues CD8+ TILs from extreme exhaustion (Figure 1G). Indeed, CD8+ TILs (CD45+CD11b–CD4–CD8+) from the U3-1402 group produced more IFN-γ and TNF-α than CD8+ TILs from the control group upon ex vivo stimulation with tumor cells (Figure 1H and Supplemental Figure 2A). Moreover, CD4+ TILs (CD45+CD11b–CD4+CD8–) from the U3-1402–treated tumors also produced more multiple cytokines, including IFN-γ, TNF-α, and IL-2, than those from the control tumors, and the levels of the inflammatory cytokines produced by NK cells (CD45+CD11blo-positiveFSCloSSCloCD4–CD8–) were greater in the U3-1402 arm than in the control arm (Supplemental Figure 2, B and C). Furthermore, in vivo CD8+ cell depletion weakened U3-1402–induced antitumor efficacy and decreased survival (Figure 1I and Supplemental Figure 3). To further clarify whether these positive effects of U3-1402 on antitumor immunity in HER3-expressing tumors require anti-HER3 antibody–dependent DXd delivery to tumor cells, we also performed additional in vivo experiments to treat mice harboring the CM-3 tumor (80–250 mm3) with free payload DXd, the dose of which was equivalent to that of DXd loaded on U3-1402 (1.5 μmol/kg body weight). This nonspecific treatment did not inhibit tumor growth or improve cytokine production of tumor-infiltrating immune cells, implying that the induction of antitumor immunity by U3-1402 requires an anti-HER3 antibody as a potent carrier of DXd (Supplemental Figure 4). Together, these results show that, in addition to its direct cytotoxicity in tumor cells, U3-1402 improves CD8+ TIL function and that of other antitumor immune cells, thus accelerating the control of tumor growth.
U3-1402 sensitizes HER3-expressing tumors to PD-1 inhibitor therapy.
The data thus far suggest that U3-1402 can be a rational chemotherapeutic agent for ICI combination therapy to improve antitumor immunity; therefore, we next examined its efficacy along with PD-1 inhibitor treatment. When treatment was initiated at a low tumor burden (tumor volumes of 40–80 mm3), either anti–PD-1 or U3-1402 alone significantly inhibited the tumor growth as compared with vehicle treatment, and the combination (combo) treatment of U3-1402 with anti–PD-1 was more effective than each drug alone (Figure 2A and Supplemental Figure 5A). In contrast, anti–PD-1 alone was no longer effective for animals carrying high tumor burdens (tumor volumes of 80–250 mm3) (Figure 2, B and C). This difference in the antitumor efficacy of anti–PD-1 alone could be at least partially explained on the basis of the difference in the intratumoral T cell status based on the tumor burdens. In particular, effector T cell marker T-bet or proliferation marker Ki-67 was downregulated in the TILs in the high–tumor burden group compared with those in the low–tumor-burden group, hinting that the TILs had low capacity to be reinvigorated by anti–PD-1 alone when the tumor burden was high (Supplemental Figure 5, B and C). Importantly, however, treatment with U3-1402 alone was still effective even in this experimental setting, wherein the tumor burden was high (Figure 2, B and C).
Figure 2. U3-1402 sensitizes HER3-expressing tumors to PD-1–blockade therapy.

(A) Left: tumor volume curve of subcutaneous CM-3 tumors treated as indicated. Right: tumor volume 14 days after treatment initiation. n = 11 for each arm, pooled from 4 independent experiments. Treatment was initiated when tumor sizes reached 40–80 mm3 (low tumor burden) (B) Left: tumor volume curve of subcutaneous CM-3 tumors treated as indicated. Middle and right: tumor volume at 14 days (middle) or 18 days (right) after treatment initiation. Treatment was initiated when tumor sizes reached 80–250 mm3 (high tumor burden). (C) Survival curve of CM-3 tumor–carrying mice shown in B. * P < 0.05; **P < 0.01, respectively. (D) Left: tumor volume curve of subcutaneous CM-3 tumors treated as indicated. Middle: tumor volume at 11 days after treatment initiation. P values are shown on horizontal lines. Right: survival curve for mice treated as indicated. n = 10 for each arm. Treatment was initiated when tumor size reached 80–250 mm3 (high tumor burden). P values in A and B are shown on horizontal lines only when they were less than 0.05 in multiple comparisons. Each dot in A, B, and D represents 1 tumor. Data were assessed by unpaired t test (B and D) or 1-way ANOVA with Tukey’s correction for multiple comparisons (A and B). Differences in survival curves were assessed using a log-rank test (C and D).
Additionally, the antitumor effect of U3-1402 was further enhanced in the presence of anti–PD-1 therapy (Figure 2, B and C), suggesting that the potent cytotoxic effects of U3-1402 potentiate the antitumor efficacy of PD-1–blockade, even for the tumors insensitive to anti–PD-1 alone. Body weight loss was not observed during the treatment course, implying that there was no significant health-related toxicity (Supplemental Figure 6A). The combination of anti–PD-1 and patritumab (the anti-HER3 antibody) did not show synergistic antitumor effects (Figure 2D). This lack of synergism confirmed that the better antitumor effect achieved by the combination of U3-1402 with PD-1 inhibition was not caused solely by either HER3 signal blockade or antibody-dependent cell-mediated cytotoxicity (ADCC) by patritumab.
Furthermore, we performed additional in vivo treatment studies using another HER3-expressing B16-F10 cancer cell line to validate the combinatorial effect of U3-1402 and PD-1 inhibition on tumor growth. We identified that membranous HER3 expression of B16-F10 was not as high as that of CM-3 (Supplemental Figure 6B), while flow cytometry analyses performed after in vitro IFN-γ exposure revealed a stronger membranous PD-L1 expression on B16-F10 cells compared with CM-3 cells (Supplemental Figure 6C). Whole-exome sequencing analysis revealed that the nonsynonymous tumor mutation burden (TMB), which is a potential predictive factor for ICI treatment efficacy in human studies (1), was lower in CM-3 cells than in B16-F10 cells in our laboratory (Supplemental Figure 6C); however, B16-F10 and CM-3 cells can be generally categorized as poorly immunogenic and moderately immunogenic, respectively, according to previous studies (31–33). In this HER3loPD-L1hiTMBhi poorly immunogenic B16-F10 tumor model (in contrast to the CM-3 tumor model: HER3highPD-L1loTMBlo moderately immunogenic model), anti–PD-1 therapy did not delay the tumor growth even at a markedly low tumor burden (20–80 mm3). However, U3-1402 was partially effective in this setting, although the antitumor effect was not typical presumably because of the low extent of HER3 expression. Further, addition of PD-1 inhibitor to U3-1402 significantly delayed the tumor growth and prolonged the survival of the treated mice, suggesting a promising effect of U3-1402 when synergized with PD-1 inhibition in such poorly immunogenic tumors (Supplemental Figure 6D). Overall, these results suggested that U3-1402 sensitized the insensitive tumors to PD-1 blockade therapy that presumably acted via immune activation induced by HER3-expressing tumor–specific cytotoxicity.
Combination of U3-1402 and PD-1 blockade induces intratumoral infiltration of multiple innate and adaptive immune cells with less suppressive signatures.
To clarify the mechanisms by which U3-1402 improves antitumor immunity, we performed flow cytometry analyses of tumor-infiltrating immune cells 8 days after treatment in the CM-3 high–tumor burden model. We noticed a relative increase in the number of CD45+ cells in U3-1402–treated, but not in anti–PD-1–treated, animals. Notably, the combination of U3-1402 and anti–PD-1 significantly increased the number of CD45+ cells (Figure 3A). These included a variety of immune cells, such as DCs (CD11b+CD11c+), tumor-associated macrophages (TAMs) (CD11b+CD11c–Gr1–F4/80+), NK cells (CD11b+CD11c–F4/80–NKp46+), and CD4+ TILs (CD11b–CD4+CD8–) (Figure 3A). Consistent with our previous results (Figure 1F), CD8+ TILs (CD11b–CD4–CD8+) were not increased by U3-1402 irrespective of the presence or absence of PD-1 inhibition (Figure 3A). Importantly, there were no significant changes in the density of Tregs (CD11b–CD4+CD25+FOXP3+) and MDSCs (CD11b+Gr1+) (Supplemental Figure 7), suggesting that combination therapy using U3-1402 and anti–PD-1 did not exacerbate the immune-suppressive environment in the tumor. Rather, not only CD8+ TILs, but also CD4+ TILs, exhibited fewer exhausted phenotypes, as determined by the downregulation of several inhibitory molecules in animals that received combination therapy (Figure 3B). Intratumoral myeloid-derived cells (CD45+CD11b+) were also found to retain their ability to express MHC as well as costimulatory molecules, particularly in animals that received combination therapy (Figure 3C). Notably, the loss of the immunosuppressive M2-like phenotype in TAMs in the combination therapy group was implied by the significant decrease of arginase expression (Figure 3D). These analyses of tumor-infiltrating T cells and myeloid cells are suggestive of collective improvement of antitumor immunity through U3-1402 and convinced us of its potential concurrent use with immunotherapy.
Figure 3. U3-1402 induces innate and adaptive immune cell infiltration and activation, which is enhanced by PD-1 inhibitor therapy.

(A) Flow cytometry analysis of the indicated cell types. Each dot represents 1 tumor. n = 16–22 for each arm, pooled from 5 independent experiments (for NK [middle of bottom], n = 7–9 for each arm, pooled from 2 independent experiments). P values are shown on horizontal lines only when they were less than 0.05 in multiple comparisons. (B and C) Flow cytometry analysis of the indicated cell types. Each dot represents 1 tumor. n = 4–6 for each arm. P values are shown on horizontal lines only when they were less than 0.05 in multiple comparisons. (D) Left: Representative flow cytometric plots of arginase-producing TAMs. Each dot represents 1 tumor. n = 9–10 for each arm, pooled from 2 independent experiments. P values are shown on horizontal lines. Right: flow cytometry representing arginase-producing TAMs. Each value in the figures indicates the frequency of arginase-producing TAMs. Data were assessed by unpaired t test (D) or 1-way ANOVA with Tukey’s correction for multiple comparisons (A–C).
Multiple innate and adaptive immune cells contribute to U3-1402–induced antitumor immunity.
We next investigated whether the infiltrated intratumoral immune cells directly affect the antitumor efficacy of U3-1402 with or without PD-1 inhibition. Ex vivo T cell restimulation analyses in the CM-3 high–tumor burden model showed that IFN-γ production of both CD4+ TILs and CD8+ TILs was upregulated in the U3-1402 arm compared with that in the vehicle and anti–PD-1 alone arms and was even more pronounced in the combination arm (Figure 4, A and B). In addition, there was a relative increase in granzyme-B expression by NK cells in the U3-1402 arm and a significant increase in the combination arm (Figure 4C). Furthermore, these antitumor immune cells gained the capacity to produce multiple cytokines, including TNF-α or IL-2 (Supplemental Figure 8). These results suggest that combination therapy using U3-1402 and anti–PD-1 is capable of reinvigorating the functions of multiple antitumor immune cells that have infiltrated in the tumor.
Figure 4. U3-1402 potentiates immune cell antitumor activity.

(A–C) Left: flow cytometry analysis of the indicated cell types. Each dot represents 1 tumor. n = 9 for each arm, pooled from 2 independent experiments. P values are shown on horizontal lines only when they were left 0.05 in multiple comparisons. Right: representative flow cytometric plots of IFN-γ– (A and B) or granzyme-B–producing (C) CD8+ TILs (A), CD4+ TILs (B), and NK cells (C). Each value in the figures indicates the frequency of each cell type. Data were assessed by 1-way ANOVA with Tukey’s correction for multiple comparisons.
In vivo depletion of CD8+ cells confirmed that the CD8+ TILs contributed to the antitumor efficacy of U3-1402 with PD-1 inhibition (Figure 5A). Additional CD4+ cell depletion revealed that the antitumor efficacy of CD8+ TILs was further enhanced by CD4+ TILs in U3-1402 and anti–PD-1–treated animals (Figure 5B). In vivo depletion of not only adaptive immune cells, but also of NK cells by the anti–Asialo-GM1 antibody significantly exacerbated tumor burden, which is also indicative of the contribution of NK cells to the antitumor immunity induced by U3-1402 (Figure 5C). These findings confirmed that the antitumor immunity induced by U3-1402 is mediated by the cooperation of multiple antitumor immune cells, including CD8+ TILs, CD4+ TILs, and NK cells. Note that HER3 expression was not observed on these antitumor immune cells (Supplemental Figure 9), which excludes the undesirable destruction of antitumor immune cells by U3-1402.
Figure 5. U3-1402 requires activated antitumor immune cells to cooperate with PD-1 inhibition.

(A and B) Left: tumor volume curve of subcutaneous CM-3 tumors treated as indicated. Middle: tumor volume 14 days after treatment initiation. Each dot represents 1 tumor. n = 12 for each arm, pooled from 4 independent experiments. P values are shown on the horizontal line. Right: survival curve of mice treated as indicated. (C) Left: tumor volume curve of subcutaneous CM-3 tumors treated as indicated. Middle and right: tumor volume 14 days after treatment initiation, as indicated. Each dot represents 1 tumor. n = 10 for each arm, pooled from 4 independent experiments. P values are shown on the horizontal line. Data were assessed by unpaired t test (A–C). Differences in survival curves were assessed using a log-rank test (A and B).
DXd induces immunogenic cell death characterized by HMGB-1 release.
Since several types of cytotoxic chemotherapies are known to induce immunogenic cell death (ICD), which accelerates antitumor immune response (8), we investigated whether this could be the case for U3-1402 treatment. Indeed, vaccination with DXd-treated CM-3 cells on the left flanks of mice inhibited the growth of untreated CM-3 cells administered contralaterally, suggesting that CM-3–specific immune responses were elicited by administration of DXd-treated cells (Figure 6A). We next investigated whether high-mobility group box-1 (HMGB-1), a representative of damage-associated molecular pattern (DAMP), was released from DXd-treated cancer cells. ELISA with supernatants from a DXd-treated cell culture medium indicated significant HMGB-1 release from those cells, comparable to that of the traditional ICD-inducer mitoxantrone (MTx) (Figure 6B). It has been reported that HMGB-1 released from tumor cells can attract NK cells and myeloid cells into the tumor site and maturate their functions, including antigen presentation capacity, ultimately leading to antitumor cytotoxic immune cell activation (8, 34). Indeed, our flow cytometry analysis of intratumoral cells indicated increased infiltration of these kinds of cells in U3-1402–treated tumors (Supplemental Figure 10A). Moreover, the analyses revealed distinct expression of TLR4, one of the major HMGB-1 receptors, on the tumor-infiltrating myeloid-derived cells (CD45+CD11bhiNKp46–) including DCs (CD45+CD11bhiNKp46–CD11c+) and TAMs (CD45+CD11bhiNKp46–CD11c–), and modest expression of TLR4 on the tumor-infiltrating NK cells (CD45+CD11bloNKp46+), but not on other non–myeloid-derived immune cells, including T cells (CD45+CD11b–) (Supplemental Figure 10B). Of note, there was a significant increase of TLR4+ myeloid-derived cell infiltration in tumors with U3-1402 treatment (Figure 6C). Interestingly, U3-1402 treatment significantly decreased the frequency of scavenger receptor class A member 5–expressing (SCARA5-expressing) cells in tumor-infiltrating TLR4+ myeloid-derived cells (Figure 6D). SCARA5 is a negative regulatory receptor for HMGB-1 that inhibits the TLR4 pathway, thus resulting in immunosuppression, and is dominantly expressed on immunosuppressive M2-type macrophages (35, 36). Therefore, SCARA5 downregulation in TLR4+ tumor-infiltrating myeloid-derived cells further supported the positive immune effect of U3-1402 on myeloid lineage. Indeed, intratumoral myeloid-derived cells produced more TNF-α on treatment with rather than without U3-1402 (Figure 6E). These findings collectively suggest that HMGB-1 release from tumor cells upon U3-1402 treatment induced intratumoral infiltration of myeloid-derived cells and NK cells and stimulated their antitumor activity. These assumptions were also supported by our previous experimental findings for NK cells and myeloid-derived cells (Figure 3, C and D, Figure 4C, Figure 5C, Supplemental Figure 2D, and Supplemental Figure 8C). Thus, our data suggest that the specific toxicity to HER3-expressing tumor cells induced by U3-1402 is mediated by stimulated innate and adaptive antitumor immunity, which subsequently sensitizes the tumor to immunotherapy.
Figure 6. DXd causes HMGB-1 release and immune activation with damaged cancer cells.

(A) Left: tumor volume curve of subcutaneous CM-3 tumors. Mice were vaccinated as indicated. Right: tumor volume 21 days after treatment initiation. Each dot represents 1 tumor. n = 10 for each arm. Data are representative of 2 independent experiments. (B) ELISA of extracellular HMGB-1. Data represent mean ± SEM of 3 replicates and are representative of 2 independent experiments. (C) Flow cytometry analysis of TLR4-expressing myeloid cells in tumors. Each dot represents 1 tumor. n = 6–7 for each arm. (D) Left: flow cytometry analysis of the indicated cell types. Each dot represents 1 tumor. n = 6–7 for each arm. Right: representative flow cytometric plots of SCARA5-expressing TLR4+ myeloid cells in tumors. A negative control using isotype control antibody is shown as a reference on the left side. Each value in the figures indicates the frequency of each cell type. (E) Left: flow cytometry analysis of the indicated cell types. Each dot represents 1 tumor. n = 6–7 for each arm. Right: Representative flow cytometric plots of TNF-α–producing myeloid cells in tumors. Each value in the figures indicates the frequency of each cell type. P values in A, C, D, and E are shown on the horizontal lines. Data were assessed by unpaired t test.
Marked HER3 expression in cancer cells resistant to PD-1 inhibitor treatment in humans.
The above preclinical evaluation revealed that U3-1402 is a promising drug for treating HER3-expressing cancer when partnered with PD-1/PD-L1 blockade. Thus, we required further clinical investigation to identify patients who had the potential to receive clinical benefit through such a combination strategy. From a clinical database in our institute, 83 cases with PD-1 inhibitor–treated advanced solid cancer whose pretreatment tumor tissues were available for immunohistochemical analyses were recruited. Table 1 depicts their clinical characteristics. Patients with HNC, MM, GC, and NSCLC were included. All tumor tissues were biopsied before PD-1 inhibitor treatment, except for one patient who was biopsied after treatment. The overall survival (OS) was 10.8 months (Figure 7A). There was no significant difference in OS based on cancer types (Figure 7A). These results were consistent with those of published clinical trials (2–5). As expected, the OS was significantly longer in PD-1 inhibitor responders than it was in nonresponders (Figure 7B). We interpreted these findings as again underscoring the urgent need for a treatment strategy to improve survival outcomes of the nonresponders.
Table 1. Clinical characteristics of 83 patients treated with PD-1 inhibitors.
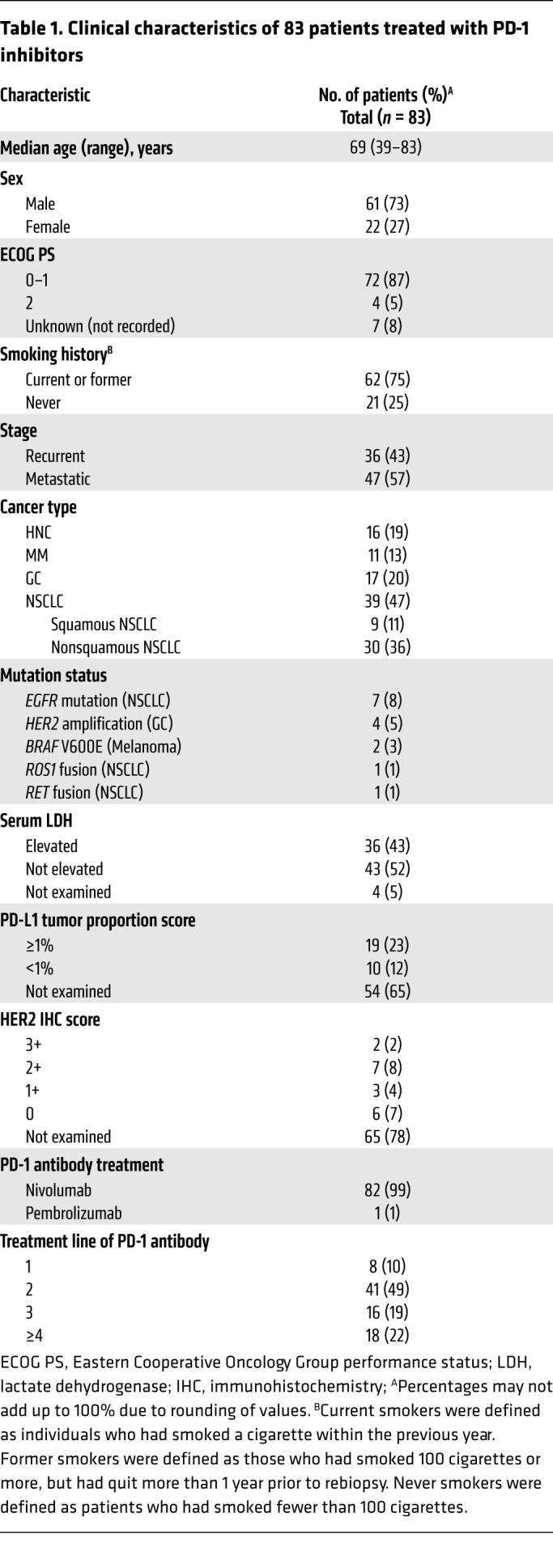
Figure 7. HER3 expression is frequently observed in PD-1 inhibitor–resistant patients with solid cancer.

(A) Kaplan-Meier curves for OS in patients treated with PD-1 inhibitors (left: all patients; right: each cancer type). (B) Kaplan-Meier curves for OS in responders and nonresponders to PD-1 inhibitor treatment. Vertical bars denote censoring. (C and D) Immunohistochemical HER3 scores of FFPE-derived tumor tissues obtained from all patients treated with PD-1 inhibitors (C) and nonresponders to PD-1 inhibitor treatment (D). (E) HER2 and HER3 immunohistochemical scores of 18 individual patients. No correlation was shown between scores by nonparametric Spearman’s rank correlation tests (E). Differences in survival curves were assessed using a log-rank test (A and B).
We next performed immunohistochemical analysis of HER3 expression to determine candidates who would obtain clinical benefit from U3-1402 among these patients. HER3 expression levels were categorized by HER3 score using an algorithm based on the guidelines of HER2 Testing in Breast Cancer (Supplemental Figure 11 and refs. 25, 37). For example, our experimental model CM-3 tumor was scored as 3+ and the B16-F10 tumor was scored as 2+, while the CT26 cell block and mouse spleen were scored as 0. The ongoing clinical trial of U3-1402 included cases with HER3 scores of 2+/3+ (25). Thus, we defined 2+/3+ as HER3-positive in our clinical study. The immunohistochemical analysis of our clinical specimens showed that there was a specific subset with significant HER3 expression (63%). Detailed results are shown in Figure 7C. There was universal HER3 expression among the 4 types of cancer, although the expression was more often observed in MM and GC in our cohort. We detected no significant difference in treatment outcome between HER3-positive and HER3-negative patients (Supplemental Figure 12A). Meanwhile, a substantial proportion of the patients were HER3-positive among the PD-1 inhibitor nonresponders (Figure 7D). Furthermore, some patients were HER3 positive among the PD-1 inhibitor responders (Supplemental Figure 12B). Overall, the combination treatment strategy of U3-1402 with PD-1/PD-L1 blockade is promising for these HER3-positive patients, and in particular, we expect that the survival outcome of the HER3-positive nonresponders would be improved by such combination immunotherapy.
In addition, our clinical interest was whether HER3-targeting U3-1402 treatment overlaps with HER2-targeting cytotoxic chemotherapy. Thus, we analyzed the concordance between HER2 and HER3 expression using immunohistochemical analysis. HER2 expression data were available from 18 patients evaluated for HER2 expression in their tumor tissue by their clinics (17 cases with GC, 1 case with NS). These limited data suggested that HER3 expression was independent of HER2 expression (Figure 7E).
Discussion
In this study, we show that HER3-targeting U3-1402 therapy can elicit potent antitumor immunity even for animals with tumors insensitive to anti–PD-1 therapy and that the efficacy is more pronounced in the presence of PD-1 inhibition, suggesting that U3-1402 sensitizes the insensitive tumors to PD-1 blockade. This effect was mediated by both the massive infiltration of various innate and adaptive immune cells within the tumor without acceleration of the immune suppressive environment and the reinvigoration of TILs from functional exhaustion. In vivo depletion experiments demonstrated the contribution of multiple antitumor immune cells, including CD8+ TILs, CD4+ TILs, and NK cells. This immune modulation of U3-1402 therapy was associated with the ICD of HER3-expressing cancer cells and the resultant release of the alarmin HMGB-1. Since high levels of HER3 expression on tumor cells are evident in a large proportion of patients, including nonresponders to anti–PD-1 therapy, we propose HER3-targeting U3-1402 therapy as a candidate for combinational therapy with PD-1/PD-L1 axis blockade.
Although HER3 expression is not commonly observed in mouse cancer cells, a potential of DXd to stimulate antitumor immunity was reproduced by another study using a murine colon cancer model (21). Here, DS-8201a carrying DXd was shown to enhance antitumor immunity, and the effect was possibly synergistic with PD-1 inhibition in HER2-expressing mouse colon cancer tumors (17). Consistent with the current HER3-targeting U3-1402 therapy, DS-8201a therapy also led to the upregulation of CD86 on antigen-presenting cells. Induced adaptive and memory T cell formation were also verified by the rejection of rechallenged tumors in DS-8201a–pretreated mice. Additionally, expressions of MHC class I and PD-L1 on cancer cells of DS-8201a–treated mice were upregulated compared with those of untreated mice, suggesting that the IFN-γ production of immune cells was increased by DXd exposure to cancer cells. These previous findings regarding DXd support the results of our U3-1402 study, and our study provided further detailed and comprehensive observations with thorough examinations using a PD-1 inhibitor.
The mechanism of U3-1402–induced antitumor immunity was not fully elucidated in this study. However, HMGB-1 was implicated as a contributor. DXd, a cytotoxic drug payload of U3-1402, was previously found to induce apoptosis of cancer cells (24, 38), and such apoptotic cancer cells can be immunogenic through DAMPs released from them (8). Indeed, our experiments showed a considerable release of HMGB-1 from DXd-treated cancer cells, which potentially work as an adjuvant to improve multiple antitumor immunities. This characteristic of DXd is notable, as not all chemotherapeutics induce HMGB-1 (9, 39). Furthermore, analyses with the anti-HER3 antibody patritumab indicated that the antitumor immune effect of U3-1402 was not induced by HER3 signal blockade or ADCC, convincing us of the contribution of the direct cytotoxic effects of DXd to the enhanced immunogenicity of the treated cancer cells.
Another possible mechanism could be the reduction of a suppressive factor and levels of antigenic stimulation by direct antitumor cytotoxicity induced by U3-1402. Previous findings may explain this, in that high tumor burdens were associated with poor therapeutic efficacy of PD-1/PD-L1 blockade (40, 41). Huang et al. indicated that a high tumor burden leads to the induction of severe exhaustion in antitumor T cells, which is characterized by the aberrant expression of several inhibitory immune checkpoint molecules, including PD-1, LAG-3, and TIM-3 (40). Indeed, a recent clinical observational study confirmed the association of high tumor burdens with poor treatment outcomes following PD-1 inhibitor therapy in MM (41). This detrimental effect of tumor burden on anti–PD-1 therapy was posited to be caused by excessive and rigorous antigen exposure to T cell receptors, resulting in hyperexhaustion characterized by the upregulation of key immune inhibitory molecules (7, 30, 40). This theory was also consistently supported by other studies using a viral infection model (42, 43). In our study, animals carrying high tumor burdens did not respond to PD-1 inhibition alone, although animals carrying low tumor burdens responded well. This observation also implied that T cells became unrecoverable from PD-1 inhibition when the tumor antigen load was excessive. Thus, decreasing tumor burden through the potent antitumor cytotoxic effect of U3-1402 possibly rescued T cells from extreme exhaustion by reducing the tumor antigen load. This assumption was also supported by the downregulation of immune checkpoint molecules on CD8+ and CD4+ TILs in animals treated with a combination of U3-1402 and anti–PD-1. From this point of view, U3-1402 is an ideal partner for PD-1/PD-L1 axis blockade therapy because it is more potent in decreasing tumor burden compared with conventional cytotoxic chemotherapies, due to its antitumor-specific characteristics (14).
It should be noted that conventional cytotoxic chemotherapies usually interfere with immune responses by exerting direct damage to immune cells (14). However, HER3 expression was observed exclusively on tumor cells, but not on immune cells in our mouse model, suggesting that U3-1402 does not exert any direct effects on immune cells. Conversely, there is a concern that DXd is possibly released from dying cancer cells. Released DXd has the potential to kill surrounding cells, and this is called a bystander effect (44). This bystander effect is advantageous, as it may result in more potent antitumor cytotoxicity, especially when the HER3 expression of cancer cells is heterogeneous and partly includes HER3-negative tumor cells, which cannot respond to U3-1402. In contrast, a possible negative impact of the bystander effect on antitumor immunity should also be of concern, namely, the released DXd can impair the surrounding immune cells in the tumor microenvironment. However, a previous in vitro examination indicated that DXd directly enhanced and stimulated DC activity to upregulate CD86 and MHC class II expression (17). The direct effects of DXd on other types of immune cells remains unknown, but any negative impact of U3-1402 on intratumoral immune cells was not suggested in our study.
The strong potential for ADCs to be ideal partners for ICI because of their favorable potencies of immune activators is supported by other preclinical and translational studies (14, 17, 45). Accordingly, a few early clinical trials using the combination of ADCs and PD-1/PD-L1 inhibitors have already been conducted thus far. In the KATE2 trial, a randomized phase II study of atezolizumab and T-DM1 in heavily treated HER2-expressing breast cancer, a significant antitumor effect was not observed in the intention-to-treat population, although a potential survival benefit of such combinatorial therapy was suggested in PD-L1–positive patients (46). Furthermore, an early clinical trial evaluating the safety and efficacy of the combination of DS-8201a and nivolumab is now in progress (ClinicalTrials.gov NCT03523572). However, these potentially emerging clinical benefits are currently limited to HER2-expressing cancers, motivating us to develop therapeutic strategies using ADCs targeting the other molecules broadly expressed on cancer cells. From this perspective, U3-1402 has a significant advantage regarding its targeting molecule, as HER3 is widely expressed on various types of cancer cells (20, 21). Previous studies showed that common types of cancers, including HNC, MM, GC, and NSCLC, express HER3, which was confirmed by our findings.
Furthermore, these cancer types are at least partially sensitive to PD-1/PD-L1 axis blockade therapy, and our clinical exams underscored the importance of U3-1402 as a sensitizer to such immunotherapy. In addition, HER3 expression is also known to be expressed on immune-tolerant cancers such as colon cancer, pancreatic cancer, and breast cancer (21). Thus, U3-1402 is expected to serve as a potent inducer of antitumor immunity, facilitating immunotherapy for patients with these aggressive cancers, as reported herein using the poorly immunogenic B16-F10 model. HER3 expression was reported to be a poor prognostic factor, although our study did not confirm this characteristic of HER3 expression in our limited cohort (20, 21). This discrepancy might be caused by insufficient power to detect the significant differences or by heterogeneous patient characteristics in our study. Only some patients receiving PD-1 inhibition therapy were included in our retrospective clinical sample study, and the number of patients was not perfectly sufficient to derive a clinically definitive conclusion. Therefore, further clinical evaluation of HER3 is needed to confirm its significance in this clinical setting. However, the clinical significance of HER3 expression should be noted because there were a considerable number of cases with HER3 expression among the PD-1 inhibition nonresponders who have notably shorter survival terms. Given the consistent and promising data from our preclinical study, we suggest U3-1402 as a potential partner of PD-1/PD-L1 axis blockade for these patients to improve their survival outcomes. HER3 signal blockade therapy alone has failed to exhibit survival benefits in previous clinical studies (22, 23). However, U3-1402 is a promising drug because it potently induces antitumor immunity through its tumor-specific immunogenic cytotoxicity with DXd, and its antitumor effect is independent of HER3 signal blockade.
Additionally, a preliminary result from our small analysis of HER2/HER3 expression showed that the candidate of U3-1402 could, apparently, be distinguished from that of HER2-targeting ADCs, including DS-8201a. Therefore, an HER3-targeting strategy using ADC will possibly rescue numerous cancer patients who do not receive therapeutic benefit from HER2-based therapy. To the best of our knowledge, the current study is the first to demonstrate the great potential of U3-1402 as a partner to PD-1/PD-L1 axis blockade therapy, thus prompting further evaluation of its clinical potential and subsequent clinical trials to evaluate the safety and efficacy of the combination treatment of U3-1402 and PD-1/PD-L1 inhibitors in HER3-expressing cancers.
Methods
Cells and reagents.
CM-3, CT-26, HCC827, and B16-F10 cells were obtained from ATCC. Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C, in Ham’s F-12K medium (Gibco; Thermo Fisher Scientific) supplemented with heat-inactivated 15% horse serum (Gibco, Thermo Fisher Scientific) and 2.5% FBS (Biowest) for CM-3, in RPMI-1640 medium (Sigma-Aldrich) supplemented with heat-inactivated 10% FBS for CT-26 and HCC827 cells, and in DMEM (Sigma-Aldrich) supplemented with heat-inactivated 10% FBS for B16-F10 cells. All media were also supplemented with 1% penicillin–streptomycin–amphotericin B (Wako Pure Chemical Industries). Cells were regularly tested for Mycoplasma contamination. Their growth behaviors were identical to their original characteristics. Acetate-buffered saline (ABS), U3-1402, patritumab, and DXd were provided by Daiichi-Sankyo under a material transfer agreement. The anti–PD-1 antibody (clone RMP1-14; catalog BE0146), anti-CD8 antibody (clone 53-6.7; catalog BE0004-1), and anti-CD4 antibody (clone GK1.5; catalog BE0003-1) were purchased from Bio X Cell. The anti–Asialo-GM1 antibody (clone Poly21460; catalog 146002) was purchased from BioLegend.
In vitro growth inhibition assay.
CM-3 and CT26 cells were plated in 96-well round-bottomed plates at a cell density of 5 × 103 and 2 × 103 cells/well, respectively. U3-1402, patritumab, and DXd were added at a range of concentrations. Following a 5- to 6-day incubation, cell viability was assessed with the use of the CellTiter-Glo 3D Luminescent Cell Viability Assay (Promega). Luminescence values were expressed as percentages of those observed for untreated cells.
In vivo tumor models and treatments.
All mice were housed under specific pathogen–free conditions. Five- to seven-week-old female DBA/2NCrl mice (Hamaguchi Laboratory, Osaka, Japan) were inoculated subcutaneously on their right flanks with 5 × 105 CM-3 cells. In B16-F10 models, 5- to 7-week-old female C57BL/6 mice (CLEA Japan) were inoculated subcutaneously on their right flanks with 5 × 105 B16-F10 cells. Once the tumors had reached the target volume (day 0), mice were randomly assigned to each of the treatment arms. Mice received intraperitoneal injections of ABS (200 μL, weekly; vehicle), U3-1402 (30 mg/kg body weight in 200 μL ABS, weekly), anti–PD-1 antibody (10 mg/kg body weight in 200 μL PBS, twice a week), or a combination of U3-1402 and anti–PD-1. Patritumab (30 mg/kg body weight in 200 μL ABS) and DXd (1.5 μmol/kg body weight in 200 μL PBS) were administered intraperitoneally once a week. These treatments were continued for 2 weeks. For in vivo depletion of CD8+ and CD4+ cells, anti-CD8– and anti-CD4–depleting antibodies were administered intraperitoneally at day −2 and day 0 and then weekly until the end of the experiments. For NK cell depletion, the anti–Asialo-GM1 antibody was administered intraperitoneally in the same way. Tumor volumes and mouse body weights were recorded twice a week until termination. Tumor volume was defined as 1/2 × length × width2. Mice were euthanized when tumors became necrotic or grew to a volume of 2000 mm3 or when tumors were harvested for analyses.
Flow cytometry analysis.
Harvested tumors and spleens from mice were mechanically dissociated and digested with 200 U/ml collagenase type IV (Sigma-Aldrich) and 100 μg/ml DNase I (Sigma-Aldrich) solutions. Single-cell suspensions were prepared using a 20-gauge syringe needle and a 70 μm cell strainer, washed 3 times with cold PBS, and then treated with Fc block (2.4G2; BD Biosciences — Pharmingen). In the case of the cultured cells, cells were dissociated using Accutase cell-detachment solution (BD Biosciences) and washed 3 times with 0.5% BSA in cold PBS. Thereafter, these cells were stained with a Zombie Fixable Viability Kit (BioLegend) to discriminate live and dead cells, then stained against the indicated markers and washed twice with Stain Buffer containing FBS (BD Biosciences) before flow cytometry analyses. Tumor weights were measured before tumor dissociation, and numbers of cells per mg were used as absolute numbers. The following antibodies recognizing the indicated antigens were used: CD45 (30-F11; catalog 103129; BioLegend), CD11b (M1/70; catalog 101245 and 101255; BioLegend), CD4 (RM4-5; catalog 563151; BD Horizon), CD8a (53-6.7; catalog 563786; BD Horizon), CD25 (PC61; catalog 102033; BioLegend), FOXP3 (FJK-16s; catalog 12-5773-82; eBiosciences), CD11c (HL3; catalog 561119 and 562782; BD Biosciences), F4/80 (BM8; catalog 123141; BioLegend), Gr-1 (RB6-8C5; catalog 108419; BioLegend), NKp46 (29A1.4; catalog 740627; BD Biosciences), PD-1 (J-43; catalog 562671; BD Biosciences), LAG-3 (C9B7W; catalog 564673; BD Biosciences), TIM-3 (RMT3-23; catalog 12-5870-82; eBiosciences), T-bet (4B10; catalog 25-5825-82; eBiosciences), Ki-67 (B56; catalog 562899; BD Horizon), IFN-γ (XMG1.2; catalog 12-7311-82; eBiosciences), TNF-α (MP6-XT22; catalog 17-7321-82; eBioscience), IL-2 (JES6-5H4; catalog 503826; BioLegend), granzyme-B (GB11; catalog GRB05; Invitrogen), CD86 (GL-1; catalog 105039; BioLegend), MHC class II (M5/114.15.2; catalog 107615; BioLegend), arginase (catalog IC5868A; R&D Systems), and TLR4 (SA15-21; catalog 145403; BioLegend). To detect membranous HER3 expression, cells were treated with patritumab (25 μg/ml) as primary antibodies and then stained with anti-human IgG-PE (M1310G05; catalog 410707; BioLegend) as a secondary antibody. To detect membrane SCARA5 expression, cells were treated with anti-SCARA5 IgG (catalog AF4754-SP; R&D Systems) as the primary antibody and then anti-sheep IgG-PE (catalog F0127; R&D Systems) as the secondary antibody. In in vitro experiments performed to detect membrane PD-L1 expression, cultured cells were seeded at a density of 1 × 106 cells in culture dishes (10 cm) 1 day before treatment with recombinant murine IFN-γ (10 ng/ml; PEPROTECH) for 24 hours at 37°C and then harvested for flow cytometry analyses with BV650-conjugated anti–PD-L1 antibody (MIH5; catalog 740614; BD Biosciences). All antibodies were diluted with stain buffer containing FBS. The Transcription Factor Buffer Set (BD Biosciences — Pharmingen) was used for intracellular marker staining. For the ex vivo restimulation assay, tumor-derived immune cells and cancer cells were treated together with brefeldin A (Sigma-Aldrich) and eBioscience Cell Stimulation Cocktail (PMA/ionomycin) for 6 hours at 37°C. All flow cytometry experiments were performed on LSRFortessa X-20 (BD Biosciences) and analyzed using FlowJo software. Fluorescence minus one (FMO) controls and corresponding isotype controls were used as negative controls.
Whole-exome sequencing and exome analysis pipeline.
Genomic DNA was extracted from fresh-frozen tumor tissues and normal tissues (tails) with the use of a QIAamp DNA Mini Kit (QIAGEN) with RNase A (QIAGEN). DNA quality control was performed by agarose gel electrophoresis, and the purity of the DNA was analyzed by NanoDrop 2000 (Thermo Fisher Scientific). The purified genomic DNA was quantified by Picogreen (Thermo Fisher Scientific), and a minimum of 1 μg of DNA was used for whole-exome library construction. Whole-exome capture libraries were constructed with the use of Sure-Select Mouse All Exon, version 6.0 (Agilent Technologies). The exome libraries were quantified by real-time PCR using a Kapa Library Quantification Kit (Kapa Biosystems), and library quality control was run on the Agilent Bioanalyzer 2100 (Agilent Technologies). The constructed libraries were sequenced using an Illumina NovaSeq 6000 sequencer, yielding an average of 59 million reads (8.8 gigabases). The mean depths of target regions were 65.6–101.5. Sequencing reads were aligned to the GRCm38 (mm10) mouse genome assembly using the Burrows-Wheeler Alignment (BWA) tool, version 0.7.12. Duplicate reads were then identified and removed by using the Picard MarkDuplicates tool,version 1.130, and single-nucleotide and indel variants were called using MuTect2 of Genome Analysis Toolkit (GATK), version 3.8. The called variants were annotated by SnpEff tool, version 4.1. Variants were filtered out if their total reads in tumor or normal samples were less than 10, their variant reads in tumor samples were less than 3, their variant allele frequencies in tumor samples were less than 10%, and their allele frequencies in normal tissues were greater than 3%. Accepted variants that were nonsynonymous were counted as nonsynonymous TMBs.
Extracellular HMGB-1.
CM-3 cells were seeded at a density of 4 × 105 cells in culture dishes (10 cm) 4 days before treatment with DXd (50 μM) or MTx (2 μM; Sigma-Aldrich) for 24 hours at 37°C. As a negative control, 1% DMSO was used. As a positive control, CT-26 cells were seeded at a density of 2 × 105 cells in culture dishes (10 cm) 4 days before treatment with MTx (2 μM) for 24 hours at 37°C. Thereafter, the culture media were centrifuged and supernatants were collected for analysis using an HMGB-1 ELISA Kit (Shinotest).
In vivo experiments with tumor cells pretreated with drugs in vitro.
CM-3 cells were seeded at a density of 8 × 105 cells in culture dishes (150 cm2) 4 days before treatment with DXd (50 μM) for 24 hours. Viable and dead pretreated cells were counted using trypan blue staining, and 5 × 105 pretreated cells were injected subcutaneously in the left flanks of DBA/2NCrl mice. The proportion of dead cells was higher than 50%. As a negative control, PBS was similarly injected subcutaneously in the left flanks of control mice. Seven days later, mice were subcutaneously inoculated with 5 × 105 live untreated CM-3 cells in their right flanks. Thereafter, tumor volume was measured twice a week.
Clinical data.
This retrospective clinical analysis aimed to evaluate HER3 positivity among nonresponders to PD-1 inhibitor therapy. We reviewed the medical records of patients with recurrent/metastatic HNC, MM, GC, and NSCLC who were 20 years old or older and treated with anti–PD-1 antibody at the Department of Medical Oncology, Kindai University Hospital, during the period between December 2015 and March 2018. Only patients with adequate available formalin-fixed paraffin-embedded (FFPE) tumor tissue samples were included. The data regarding clinicopathological features and treatment history were extracted. The data were updated as of July 31, 2018. Responses were assessed by investigators according to RECIST, volume 1.1. OS was measured from treatment initiation to death from any cause. Patients without documented clinical or radiographic disease progression or who were still alive were censored on the date of the last follow-up.
Immunohistochemical analysis.
Membranous tumor HER3 expression was evaluated by immunohistochemical analysis. FFPE-tumor tissue–derived 4 μm sections were immunostained with anti-HER3 rabbit monoclonal antibody (D22C5; catalog 12708S; Cell Signaling Technology) using an autostainer (Leica Bond III). Briefly, the sections were baked at 65°C for 30 minutes and dewaxed with Bond dewaxing solution. Antigen retrieval was performed with Bond Epitope Retrieval Solution 2 (EDTA based pH 9.0) at 100°C for 20 minutes. All of the following procedures were done at room temperature. Peroxidase blocking was then performed for 5 minutes, and the sections were washed 3 times. Thereafter, they were incubated with anti-HER3 antibody for 30 minutes, followed by washing 3 times. The HRP-labeled polymer was exposed to the sections for 8 minutes and then washed 3 times. The sections were treated with DAB for 10 minutes. Counterstaining was performed with hematoxylin or Giemsa. An IgG isotype control (DA1E; catalog 3900; Cell Signaling Technology) was used as a negative control. The immunostained sections were reviewed by board-certified pathologists who were blinded to the clinical data. They evaluated HER3 expression using a HER2-staining scoring algorithm. In detail, HER3 staining was categorized by intensity as 0, 1+, 2+, and 3+ (Supplemental Figure 11). Scores were defined as follows: 0, no staining or membrane staining in 10 % or less of the tumor cells; 1+, faint or barely perceptible incomplete membrane staining in more than 10 % of tumor cells; 2+, weak-to-moderate complete membrane staining observed in more than 10 % of tumor cells; and 3+, circumferential membrane staining that was complete, intense, and in more than 10 % of tumor cells. This categorization is based on a HER2 scoring algorithm as described in the guideline of HER2 Testing in Breast Cancer and is currently in use in the ongoing clinical trial of U3-1402 (25, 37). FFPE-cell block–derived 4 μm section of MDA-MB-453 cells as a positive control was obtained from Pathology Institute Corp.
Statistics.
Error bars indicate SEM. Independent experiments are presented individually or combined, as explained in the figure legends. Unpaired t tests were applied to compare continuous variables unless indicated otherwise. Analysis of experiments with more than 2 groups was performed using 1-way ANOVA with Tukey’s correction for multiple comparisons. No blinding was performed for the experimental studies. Differences in survival curves constructed by the Kaplan-Meier method were assessed using a log-rank test. Missing data were not imputed. All P values were based on a 2-sided hypothesis. Statistical analyses were performed using JMP software,version 14.0.0 (SAS Institute). Data were graphically depicted using JMP software and GraphPad Prism 7.0 (GraphPad Software).
Study approval.
All experimental animal procedures were performed in accordance with Recommendations for the Handling of Laboratory Animals for Biomedical Research compiled by the Committee on Safety and Ethical Handling Regulations of Laboratory Animal Experiments of Kindai University. The study was also reviewed and approved by the Animal Ethics Committee of Kindai University. The human studies were performed according to the Declaration of Helsinki and approved by the Institutional Review Board of Kindai University. Patients provided written informed consent, where applicable, or such informed consent was waived by the Institutional Review Board–approved protocols, as this was a retrospective study only using de-identified data and excess archival tissues. The authors followed Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines for animal studies and REporting recommendations for tumour MARKer prognostic studies (REMARK) guidelines and STROBE guidelines for human studies.
Author contributions
K Haratani developed the hypothesis, designed and performed the experiments, collected and analyzed the clinical and pathological data, wrote the manuscript, and supervised the project. KY developed the hypothesis, analyzed the data, wrote the manuscript, supervised the project, and acquired research funding. ST analyzed the data, wrote the manuscript, and supervised the project. OM performed the pathological analyses and wrote the manuscript. RK and NT collected the clinical data and performed the experiments. HK, KT, HH, and MT collected the clinical data. NM designed the pathological methodology. TK and K Hirotani interpreted the experimental data. JT, K Nishio, KD, MM, and K Nakagawa interpreted the data and supervised the project. All authors critically revised the manuscript for important intellectual content and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Haruka Yamaguchi, Yume Shinkai, Michiko Kitano, Mami Kitano, and Ai Yukumoto at the Department of Medical Oncology, Kindai University Faculty of Medicine, for their technical support during sample preparation. We also thank Shinji Kurashimo at the Central Research Facilities, Kindai University Faculty of Medicine, for his technical support with flow cytometry analyses. We thank Kenji Chamoto and Ryusuke Hatae at the Department of Immunology and Genomic Medicine, Graduate School of Medicine Kyoto University, for their kind help and technically meaningful advice. This research study was financially supported by Daiichi-Sankyo. ABS, U3-1402, patritumab, and DXd were provided by Daiichi-Sankyo under a material transfer agreement.
Version 1. 10/29/2019
In-Press Preview
Version 2. 12/03/2019
Electronic publication
Version 3. 01/02/2020
Print issue publication
Footnotes
Conflict of interest: K Haratani has received research funding from AstraZeneca. KY has received research funding from Daiichi-Sankyo and has a patent pending with Daiichi-Sankyo (WO2015048804A2, WO2018123999A1, and WO2018159582A1). HH has received honoraria from AstraZeneca, Bristol-Myers Squibb, and Ono Pharmaceutical Co. and has a patent pending with Ono Pharmaceutical Co. and Sysmex (Japanese Patent 2018-59834). NM, TK, and K Hirotani are employees of Daiichi-Sankyo. JT has received research funding from Eisai and Lilly. K Nishio has received honoraria from Eisai and research funding from Ignyta, Korea Otsuka Pharmaceutical Co. Ltd., and Boehringer Ingelheim. MM has received advisory fees from SNBL and Unmet Medical Needs Pharma Inc. K Nakagawa has received honoraria from Astellas, AstraZeneca, Lilly, MSD KK, and Pfizer Japan as well as research funding from AbbVie, Astellas, Daiichi-Sankyo, Eisai, MSD KK, Boehringer Ingelheim, and Takeda and has patents pending with Daiichi-Sankyo (WO2015048804A2, WO2018123999A1, and WO2018159582A1) and Ono Pharmaceutical Co. and Sysmex (Japanese Patent 2018-59834).
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(1):374–388.https://doi.org/10.1172/JCI126598.
Contributor Information
Koji Haratani, Email: haratani_k@med.kindai.ac.jp.
Kimio Yonesaka, Email: yonesaka@med.kindai.ac.jp.
Shiki Takamura, Email: takamura@med.kindai.ac.jp.
Osamu Maenishi, Email: m-roadster@iris.eonet.ne.jp.
Ryoji Kato, Email: kato.r@med.kindai.ac.jp.
Naoki Takegawa, Email: takegawa_n@med.kindai.ac.jp.
Hisato Kawakami, Email: kawakami_h@med.kindai.ac.jp.
Kaoru Tanaka, Email: katanaka@med.kindai.ac.jp.
Hidetoshi Hayashi, Email: hidet31@med.kindai.ac.jp.
Masayuki Takeda, Email: takeda_m@med.kindai.ac.jp.
Naoyuki Maeda, Email: maeda.naoyuki.ct@daiichisankyo.co.jp.
Takashi Kagari, Email: kagari.takashi.z4@daiichisankyo.co.jp.
Kenji Hirotani, Email: hirotani.kenji.br@daiichisankyo.co.jp.
Junji Tsurutani, Email: tsurutaj@med.showa-u.ac.jp.
Kazuto Nishio, Email: knishio@med.kindai.ac.jp.
Katsumi Doi, Email: kdoi@med.kindai.ac.jp.
Masaaki Miyazawa, Email: masaaki@med.kindai.ac.jp.
Kazuhiko Nakagawa, Email: nakagawa@med.kindai.ac.jp.
References
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weber JS, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16(4):375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 3.Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 4.Ferris RL, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–1867. doi: 10.1056/NEJMoa1602252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kang YK, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10111):2461–2471. doi: 10.1016/S0140-6736(17)31827-5. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. 2015;28(6):690–714. doi: 10.1016/j.ccell.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Bezu L, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol. 2015;6:187. doi: 10.3389/fimmu.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandhi L, et al. Pembrolizumab plus chemotherapy in metastatic non-small-cell lung cancer. N Engl J Med. 2018;378(22):2078–2092. doi: 10.1056/NEJMoa1801005. [DOI] [PubMed] [Google Scholar]
- 11.Socinski MA, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–2301. doi: 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 12.Jotte RM, et al. IMpower131: Primary PFS and safety analysis of a randomized phase III study of atezolizumab + carboplatin + paclitaxel or nab-paclitaxel vs carboplatin + nabpaclitaxel as 1L therapy in advanced squamous NSCLC. J Clin Oncol. 2018;36(18):2snPEzb [Google Scholar]
- 13.Paz-Ares L, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med. 2018;379(21):2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 14.Gerber HP, Sapra P, Loganzo F, May C. Combining antibody-drug conjugates and immune-mediated cancer therapy: What to expect? Biochem Pharmacol. 2016;102:1–6. doi: 10.1016/j.bcp.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 15.Verma S, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91. doi: 10.1056/NEJMoa1209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krop IE, et al. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017;18(6):743–754. doi: 10.1016/S1470-2045(17)30313-3. [DOI] [PubMed] [Google Scholar]
- 17.Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther. 2018;17(7):1494–1503. doi: 10.1158/1535-7163.MCT-17-0749. [DOI] [PubMed] [Google Scholar]
- 18.Tamura K, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive breast cancer previously treated with trastuzumab emtansine: a dose-expansion, phase 1 study. Lancet Oncol. 2019;20(6):816–826. doi: 10.1016/S1470-2045(19)30097-X. [DOI] [PubMed] [Google Scholar]
- 19.Shitara K, et al. Trastuzumab deruxtecan (DS-8201a) in patients with advanced HER2-positive gastric cancer: a dose-expansion, phase 1 study. Lancet Oncol. 2019;20(6):827–836. doi: 10.1016/S1470-2045(19)30088-9. [DOI] [PubMed] [Google Scholar]
- 20.Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010;16(5):1373–1383. doi: 10.1158/1078-0432.CCR-09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ocana A, Vera-Badillo F, Seruga B, Templeton A, Pandiella A, Amir E. HER3 overexpression and survival in solid tumors: a meta-analysis. J Natl Cancer Inst. 2013;105(4):266–273. doi: 10.1093/jnci/djs501. [DOI] [PubMed] [Google Scholar]
- 22.Yonesaka K, et al. Circulating heregulin level is associated with the efficacy of patritumab combined with erlotinib in patients with non-small cell lung cancer. Lung Cancer. 2017;105:1–6. doi: 10.1016/j.lungcan.2016.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Harrington KJ, et al. Randomized phase 2 trial of patritumab (P) or placebo (PBO) + cetuximab (C) + cisplatin (CIS) or carboplatin (CAR) for recurrent and/or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN) J Clin Oncol. 2018;36(15):6045 [Google Scholar]
- 24.Yonesaka K, et al. An HER3-targeting antibody-drug conjugate incorporating a DNA topoisomerase I inhibitor U3-1402 conquers EGFR tyrosine kinase inhibitor-resistant NSCLC. Oncogene. 2019;38(9):1398–1409. doi: 10.1038/s41388-018-0517-4. [DOI] [PubMed] [Google Scholar]
- 25.Kogawa T, et al. Single agent activity of U3-1402, a HER3-targeting antibody-drug conjugate, in breast cancer patients: Phase 1 dose escalation study. J Clin Oncol. 2018;36(15):2512 [Google Scholar]
- 26.Yasamura Y, Tashjian AH, Sato GH. Establishment of four functional, clonal strains of animal cells in culture. Science. 1966;154(3753):1186–1189. doi: 10.1126/science.154.3753.1186. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt W, et al. Cell-free tumor antigen peptide-based cancer vaccines. Proc Natl Acad Sci U S A. 1997;94(7):3262–3267. doi: 10.1073/pnas.94.7.3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanley A, Ashrafi GH, Seddon AM, Modjtahedi H. Synergistic effects of various Her inhibitors in combination with IGF-1R, C-MET and Src targeting agents in breast cancer cell lines. Sci Rep. 2017;7(1):3964. doi: 10.1038/s41598-017-04301-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelman JA, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 30.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peter I, Mezzacasa A, LeDonne P, Dummer R, Hemmi S. Comparative analysis of immunocritical melanoma markers in the mouse melanoma cell lines B16, K1735 and S91-M3. Melanoma Res. 2001;11(1):21–30. doi: 10.1097/00008390-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Lechner MG, et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36(9):477–489. doi: 10.1097/01.cji.0000436722.46675.4a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu JW, et al. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS One. 2018;13(11):e0206223. doi: 10.1371/journal.pone.0206223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parodi M, et al. Natural Killer (NK)/melanoma cell interaction induces NK-mediated release of chemotactic high mobility group box-1 (HMGB1) capable of amplifying NK cell recruitment. Oncoimmunology. 2015;4(12):e1052353. doi: 10.1080/2162402X.2015.1052353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo DY, Cao C, Zhang XY, Xiang LX, Shao JZ. Scavenger Receptor SCARA5 Acts as an HMGB1 Recognition Molecule Negatively Involved in HMGB1-Mediated Inflammation in Fish Models. J Immunol. 2016;197(8):3198–3213. doi: 10.4049/jimmunol.1600438. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, et al. Tumor hypoxia enhances non-small cell lung cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget. 2014;5(20):9664–9677. doi: 10.18632/oncotarget.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wolff AC, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists Clinical Practice Guideline Focused Update. J Clin Oncol. 2018;36(20):2105–2122. doi: 10.1200/JCO.2018.77.8738. [DOI] [PubMed] [Google Scholar]
- 38.Ogitani Y, et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res. 2016;22(20):5097–5108. doi: 10.1158/1078-0432.CCR-15-2822. [DOI] [PubMed] [Google Scholar]
- 39.Pfirschke C, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44(2):343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang AC, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joseph RW, et al. Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res. 2018;24(20):4960–4967. doi: 10.1158/1078-0432.CCR-17-2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105(39):15016–15021. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takamura S, et al. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J Immunol. 2010;184(9):4696–4707. doi: 10.4049/jimmunol.0903478. [DOI] [PubMed] [Google Scholar]
- 44.Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107(7):1039–1046. doi: 10.1111/cas.12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Müller P, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med. 2015;7(315):315ra188. doi: 10.1126/scitranslmed.aac4925. [DOI] [PubMed] [Google Scholar]
- 46. Emens L, et al. Results from KATE2, a randomized phase 2 study of atezolizumab (atezo)+trastuzumab emtansine (T-DM1) vs placebo (pbo)+T-DM1 in previously treated HER2+ advanced breast cancer (BC) [abstract]. Paper presented at: 2018 San Antonio Breast Cancer Symposium; December 4–8, 2018; San Antonio, Texas, USA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.