

Abstract
Despite untiring efforts to develop therapies for pancreatic ductal adenocarcinoma (PDAC), survival statistics remain dismal, necessitating distinct approaches. Photodynamic priming (PDP), which improves drug delivery and combination regimens, as well as tumor photodestruction are key attributes of photodynamic therapy (PDT), making it a distinctive clinical option for PDAC. Localized, high-payload nanomedicine-assisted delivery of photosensitizers (PSs), with molecular specificity and controlled photoactivation, thus becomes critical in order to reduce collateral toxicity during more expansive photodynamic activation procedures with curative intent. As such, targeted photoactivable lipid-based nanomedicines are an ideal candidate but have failed to provide greater than two-fold cancer cell selectivity, if at all, due to their extensive multivariant physical, optical, and chemical complexity. Here, we report (1) a systematic multivariant tuning approach to engineer (Cet, anti-EGFR mAb) photoimmunonanoconjugates (PINs), and (2) stroma-rich heterotypic PDAC in vitro and in vivo models incorporating patient-derived pancreatic cancer-associated fibroblasts (PCAFs) that recapitulate the desmoplasia observed in the clinic. These offer a comprehensive, disease-specific framework for the development of Cet-PINs. Specificity-tuning of the PINs, in terms of PS lipid anchoring, electrostatic modulation, Cet orientation, and Cet surface densities, achieved ~16-fold binding specificities and rapid penetration of the heterotypic organoids within 1 h, thereby providing a ~16-fold enhancement in molecular targeted NIR photodestruction. As a demonstration of their inherent amenability for multifunctionality, encapsulation of high payloads of gemcitabine hydrochloride, 5-fluorouracil, and oxaliplatin within the Cet-PINs further improved their antitumor efficacy in the heterotypic organoids. In heterotypic desmoplastic tumors, the Cet-PINs efficiently penetrated up to 470 μm away from blood vessels, and photodynamic activation resulted in substantial tumor necrosis, which was not elicited in T47D tumors (low EGFR) or when using untargeted constructs in both tumor types. Photodynamic activation of the Cet-PINs in the heterotypic desmoplastic tumors resulted in collagen photomodulation, with a 1.5-fold reduction in collagen density, suggesting that PDP may also hold potential for conquering desmoplasia. The in vivo safety profile of photodynamic activation of the Cet-PINs was also substantially improved, as compared to the untargeted constructs. While treatment using the Cet-PINs did not cause any detriment to the mice’s health or to healthy proximal tissue, photodynamic activation of untargeted constructs induced severe acute cachexia and weight loss in all treated mice, with substantial peripheral skin necrosis, muscle necrosis, and bowel perforation. This study is the first report demonstrating the true value of molecular targeting for NIR-activable PINs. These constructs integrate high payload delivery, efficient photodestruction, molecular precision, and collagen photomodulation in desmoplastic PDAC tumors in a single treatment using a single construct. Such combined PIN platforms and heterocellular models open up an array of further multiplexed combination therapies to synergistically control desmoplastic tumor progression and extend PDAC patient survival.
Keywords: Nanoengineering, photoimmunonanoconjugates, pancreatic ductal adenocarcinoma, NIR photodynamic activation
Graphical Abstract
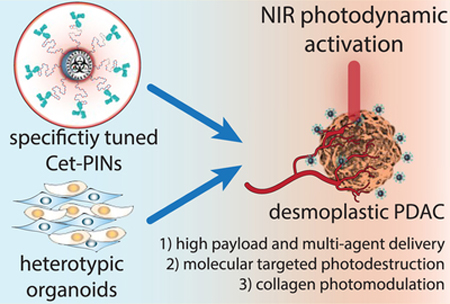
Pancreatic ductal adenocarcinoma (PDAC) is a particularly stubborn malignancy with dismal prognoses. Five-year patient survival rates are less than 5%, and even with the most aggressive and toxic treatment regimens, such as FOLFIRINOX, median survival does not exceed 11.1 months.1,2 PDAC is characterized by desmoplasia, a reaction leading to dense stromal deposition of collagen-rich extracellular matrix, predominantly by activated pancreatic cancer-associated fibroblasts (PCAFs).3–5 Desmoplasia limits the delivery of therapeutic agents, including nanomedicines, contributing to treatment resistance.6 Ideally, the preclinical models used to evaluate novel therapeutics for PDAC must capture and present features of desmoplasia. We demonstrate in this study that this is made feasible by heterotypic PDAC models that incorporate patient-derived PCAFs.
Photodynamic activation of photosensitizers (PSs) is capable of inducing significant and light dose-dependent tumor necrosis in PDAC patients using current clinical regimens for photodynamic therapy (PDT).7 In addition, a cooperative mechanistic photodynamic priming (PDP) of the tumor microenvironment improves the outcomes of subsequent combination therapies.7–11 The priming role of PDT has not been studied in depth, although it holds considerable promise for microenvironmental modulation such as increased drug permeability.11–13 This becomes critical as nanomedicines evolve, with prior evidence that PDP improves the delivery of Doxil and liposomal irinotecan to tumors.11,12 The ideal photoactivable nanomedicine would therefore combine features of PDAC tumor destruction with photodynamic priming using a single construct and a single near-infrared (NIR) photodynamic activation procedure.
PDT regimens in current early phase clinical trials achieve controllable zones of tumor necrosis but exhibit some adverse toxicities including prolonged skin phototoxicity, nausea, photosensitivity, skin hyperpigmentation, and higher healthy tissue accumulation of the PS that increases the risk of off-target photodamage. However, as clinical procedures evolve towards curative PDT regimens, higher PS doses and expansive regions of irradiation are required. Administration of higher PS doses has resulted in cases of hepatotoxicity resulting from hemolysis, dysregulated blood pressure and heart rate, and cardiorespiratory events leading to fatalities.15,16 Furthermore, expansive illumination protocols around critical anatomical sites have led to collateral phototoxicity. For example, PDT of PDAC tumors involving the gastroduodenal artery resulted in cases of gastrointestinal bleeds. If expansive and complete PDAC treatment is to be achieved safely, higher PS payload delivery to tumors is critically needed in addition to molecular precision of photodamage. Molecular targeted photodynamic activation can enhance the tolerability to higher light doses during PDT,17 thereby maximizing the extent of PDAC tumor damage while sparing critical healthy vicinal tissue.
Nanomedicines are an attractive and clinically proven strategy to reduce the systemic toxicity of chemotherapeutics while increasing localized tumor delivery.18 Photonanomedicines, light-activable nanotherapeutics, combine the attractive tumor-delivery and anticancer agent tolerability properties of nanomedicines with the potent antitumor potential of PDT.7,19 Considering the toxicities associated with high PS administration, localized high-payload delivery within desmoplastic PDAC tumors using photonanomedicines thus becomes critical, as well as spatiotemporally controlled photoactivation, to reduce collateral toxicity. Ideally, the highest-payload nanoparticle PS carrier would be constituted of the photodynamic agent itself. This simplified nanoparticle approach is also advantageous in that it circumvents issues related to complex nanosystems and thus simplicity has been correlated with impact.20 An elegant example of such high-payload photonanomedicines includes metal–organic frameworks, which are ordered in a conformation that preserves the photochemistry of the photosensitizer.21 However, introducing functionality to photonanomedicines naturally increases the complexity of the nanosystem, and a balance between rigorous and reproducible fabrication, functionality, and simplicity becomes critical.22 Furthermore, the clinical relevance of multifunctional photonanomedicines is of paramount importance for expediting the translation of critically needed PDAC treatments. Visudyne is the first clinically approved lipid-based photonanomedicine formulation of benzoporphyrin derivative (BPD) that has already shown superior performance in PDAC patients.7 Given that 80% of pancreatic cancers are unresectable,23 Visudyne-PDT has been shown to be particularly powerful in PDAC clinical trials, with one instance of converting an unresectable PDAC tumor into one that underwent R0 Whipple’s pancreaticoduodenectomy.7
The ultimate goal and challenge is to thus strike a balance between efficacy and toxicity. This study is a significant stride toward that goal by presenting photoimmunonanoconjugates (PINs) that can efficiently destroy desmoplastic PDAC tumors with the molecular precision of antibody therapeutics while also modulating the tumor stroma by reducing collagen density (Scheme 1). A number of agents that modulate the stroma in PDAC are currently being tested in clinical trials, such as the angiotensin inhibitor losartan, the micropinocytosis inhibitor hydroxychloroquinine, enzymes that digest hyaluronic acid and agonists of the retinoic acid receptor and the vitamin D receptor.5,24 In this paper, we present a single therapeutic NIR-activable nanoconjugate with collagen-modulating characteristics, high-payload drug delivery, and molecular targeted tumor killing potential that promises to enhance the safety of curative clinical PDT procedures.
Scheme 1.

Molecular Targeted NIR Photodynamic Activation in Heterotypic Desmoplastic PDAC using Specificity-Tuned Photoimmunonanoconjugatesa,b
a(a) Conceptual representation of EGFR-specific, molecular targeted NIR photodynamic activation in heterotypic desmoplastic PDAC that results in nanoconjugate disintegration, targeted necrosis of desmoplastic tumor tissue, and photomodulation of collagen; (b) Effective targeted photodestruction of desmoplastic PDAC tissue is achieved by high-payload, cetuximab directed photoimmunonanoconjugates (PINs), which have been tuned to achieve the highest degree of molecular specificity. b Three-dimensional (3D) representations of Cetuximab (IgG2a, Protein Data Bank (PDB) ID: 1IGT)25 and Protein Z (Z domain, Protein A from Staph. Aureus, PDB ID: 2SPZ)26 were projected using Jmol: an open-source Java viewer for chemical structures in 3D.27
Particularly in the case of problematic diseases such as desmoplastic PDAC, the high-throughput in vitro testing routines used to guide the development of these specify-tuned PINs must be able to recapitulate the physical and biochemical barriers presented by clinical manifestations of the disease. Not only must there be intelligence in the design of such complex nanomedicines but there must also be creativity in the design of disease-recapitulating, high-throughput testing platforms to provide a meaningful framework for comprehensive evaluation. In this study, we develop heterotypic PDAC organoids and in vivo tumors using MIA PaCa-2 cancer cells and patient-derived PCAFs as an intelligent, disease-specific model to guide the engineering and evaluation of molecular targeted, NIR activable PINs.
Such complex molecular targeted, NIR-activable PINs require diligence in their fabrication and strategic modulation of their codependent constituents to simultaneously capture and retain the following salient features of optimal PINs: molecular specificity, high-payload delivery, and potent photochemical activity. Although previous attempts to provide molecular specificity for lipidic nanoconstructs carrying PSs have shown up to two-fold improvements in binding specificities and phototoxicity in cell cultures, many have failed, and little evidence of in vivo efficacy in solid tumors has been demonstrated to date.28–36 This is a direct result of the complexity of preparing functionalized nanoconjugates that incorporate light-activable features. To encompass the intricacies of fabricating molecular targeted PINs that retain the motivating features described above, we adopt a rational multivariant engineering approach to engineer specificity-tuned, Cetuximab (Cet; anti-EGFR mAb) targeted nanolipid formulations of BPD. These PINs are inspired by the promising high-payload clinical formulation of BPD, Visudyne, and the molecular targeted clinical antibody Cet. With EGFR expression observed in up to 85% of patients with PDAC,37 and anti-EGFR antibodies currently being used for image-guided surgical resection of PDAC in the clinic,38 Cet becomes an ideal and opportune candidate for molecular targeted NIR photodynamic activation.
The interdependent factors of the optimal Cet-PIN that we show to be critical, and have modulated for maximal specificity, include the lipidation and stable membrane-anchoring of BPD, tuning the surface electrostatics to minimize heterogeneity in cellular uptake, and controlling Cet surface orientations and densities using either site-specific Protein Z-mediated, or stochastic copper-free click chemistry. We show in this study that the specificity-tuned Cet-PINs that are capable of destroying desmoplastic heterotypic PDAC organoids and xenografted tumors in vivo combine a lysophospholipidanchored variant of BPD, an anionic ζ-potential of ‒20.7 ± 1.6 mV, and a surface density of 517.5 ± 138.6 Cet per μm2 of stochastically oriented Cet. As compared to direct BPD-Cet photoimmunoconjugates (PICs) with a maximum payload of 10 BPD molecules per Cet,39 the specificity-tuned Cet-PINs engineered in this study retained a striking 60% of the hydrophobic PS’s photodynamic activity and remained tumor-specific and colloidally stable at a high payload of ~600 BPD molecules per construct. Although recent elegant efforts to maximize the PS payloads of PICs have achieved up to 15 PS molecules per antibody,40 the Cet-PINs offer higher PS payloads and offer the potential of multiagent delivery. Our specificity-tuned Cet-PINs demonstrated the highest ever reported cancer cell binding specificities of all targeted photoactivable nanoconstructs, with up to 100-fold preferential binding, and up to 30-fold improvements in EGFR-specific photokilling of MIA PaCa-2 cells in monolayer. More importantly, the specificity-tuned Cet-PIN demonstrated up to 16.9-fold binding specificities in the heterotypic PDAC organoids containing PCAFs and rapidly permeated the organoids within 1 h of incubation. This translated to a remarkable ~16-fold enhancement in molecular targeted photodynamic destruction that was agnostic to PCAF-induced treatment resistance in the heterotypic organoids.
Using non-invasive in vivo photoacoustic imaging, we further show that the perivascular tumor penetration of the optimized Cet-PINs ranged from 174–473 μm within heterotypic desmoplastic MIA PaCa-2 + PCAF tumors. In this first report of a specificity-tuned Cet-PIN for molecular targeted NIR photodynamic activation in desmoplastic heterotypic PDAC tumors, we show that treatment induces substantial tumor necrosis throughout the entirety of the tumor cross-section at an administered BPD equivalent dose that is 10-fold lower than the human equivalent dose required.7 Interestingly, molecular targeted NIR photodynamic activation also resulted in the photomodulation of tumor collagen, reducing the fractional collagen area by 1.5-fold.
The significance of this desmoplasia-modulating, molecular targeted NIR photodynamic activation approach lies in that it accelerates the conceptual shift in light-activation for drug delivery and therapeutic combinations. Recent work by our group9,11 and others41–43 demonstrated that combination therapies using light-activated nanoconstructs synergistically enhance efficacy. These, along with ongoing clinical efforts to introduce molecular specificity to chemotherapy-encapsulating nanomedicines for PDAC and other cancers (MM-310, Ephrin A2 targeted liposomal docetaxel),44 accentuate the high translational potential of our Cet-PINs. As a proof of concept, we also show that encapsulation of the front-line PDAC chemotherapeutics gemcitabine hydrochloride, 5-fluorouracil, and oxaliplatin, within the Cet-PINs, yields chemo-per-Cet payloads that are not achievable with direct antibody conjugates. We show that the Cet-PIN-Gemcitabine (41 chemo per Cet payload), the Cet-PIN-5-fluorouracil (1200 chemo per Cet payload), and the Cet-PIN-Oxaliplatin (20 chemo per Cet payload) all improve treatment response in heterotypic MIA PaCa-2 + PCAF organoids following photodynamic activation. They do so by combining photodynamic therapy with chemotherapy in a single construct at payloads that are not achievable in direct antibody conjugates without impairing binding specificity. The release of the entrapped, water-soluble agents is also regulated by NIR photochemical activation of the BPD-PC, as the presence of sodium azide can inhibit the release of calcein disodium salt, a fluorescent surrogate for entrapped chemotherapeutics (Figure S5f).
We here present a uniquely powerful modality with spatiotemporally controlled light-activable features, molecular specificity, and desmoplasia-modulating characteristics. Being a key critical barrier to drug delivery in PDAC, we envisage that the photomodulation of collagen by a single targeted nanoconjugate will also have a marked impact on subsequent Cet-PIN delivery for repeated cycles of NIR photodynamic activation.
With existing treatments currently falling short of providing effective control of PDAC progression, in addition to inducing significant dose-limiting comorbidities, there is a critical unmet need for a paradigm shift in therapeutic strategies. With the capacity to provide safer photodynamic illumination protocols and multiagent delivery, the integrated molecular targeted NIR photodynamic activation modality using specificity-tuned Cet-PINs that we present here is a significant stride forward in the pursuit for rationally designed treatments with improved patient outcomes.
Results and Discussion.
Although a few prior examples of targeted photoactivable lipid-based nanomedicines have shown some promise in vitro, most have often failed to demonstrate more than two-fold selectivity of binding and phototoxicity, if at all.28–36 In vivo, little evidence exists in the literature that solid tumors can be effectively destroyed using such molecular targeted lipid-based constructs.36 These failures underscore a fundamental, multiparametric complexity in developing and optimizing targeted photoactivable lipid-based nanomedicines, which have been generally overlooked and unaddressed in the literature. These complexities are either PDT-related, nanoconstruct-related, or combinations of both. Using the following systematic specificity-tuning process to engineer optimal Cet-PINs, we identify and modulate key nanoconstruct parameters to deliver the highest degrees of molecular specificity, tumor tissue penetration, and targeted photokilling in monotypic and heterotypic PDAC organoids and in vivo xenograft tumors.
Chemical Modulation of Benzoporphyrin Derivate Membrane Stability by Lipidation.
In addition to the existing challenges facing the fabrication of ligand targeted nanoconstructs,45 the NIR light-activable component of PINs introduces significant variability in the physicochemical properties of the plethora of promising clinical PSs available and variability in the stability within their nanoconstruct carriers. Here, we show that PS-nanoconstruct stability is non-trivial, yet it is of paramount importance for their molecular targeting specificity.28–34 One example of this variability is reported in our previous study, which shows that non-covalent antibody-associated liposomal BPD constructs exhibit some degree of binding specificity but suffer from substantial PS leakage as the extent of antibody adsorption increases.46 Similarly, another study found that transferrin targeted liposomal hypericin was ineffective in vivo due to substantial leakage of the PS.36 This relationship between PS hydrophobicity and liposomal association has also been a focal point of interest in the literature.47 As such, we have adopted a lipid anchoring strategy for BPD to modulate its membrane stability and promote nanoconstruct integrity.48 In this study, we found that BPD hydrophobically entrapped in a liposomal membrane rapidly and non-specifically leeches into OVCAR-5 cells that were used as a model high-EGFR expressing cell line, unlike a lipidated fluorophore (DSPE-Liss Rhod B) membrane anchored to the same construct (Figure S1a,b). Without the stable membrane insertion of BPD into the nanoconstruct, attempts at antibody targeted delivery of BPD become futile. Lipidic bilayered nanovesicles formed of lipidated porphyrins, first reported in 2002,49 have been previously prepared by the conjugation of porphyrins to phospholipid acyl chains,49 glycerol moieties,48,50,51 phosphate head groups,52,53 and PEG chains.48,54 Recent studies by Zheng and Lovell have demonstrated the unique properties of lipidated porphyrin constituents of nanomedicines and nanotheranostics.43,50 We thus synthesized a panel of liposomal nanoformulations of BPD and its lipidated variants conjugated to 16:0 lyso PC, 20:0 lyso PC, and cholesterol through Steglich esterification or to DSPE-PEG2000-NH2 by EDC-amide coupling.48 Cholesterol was selected as a classical membrane anchor,55 16:0 lyso PC and 20 lyso PC were compared for their varying acyl chain length, and DSPE-PEG2000-NH2 was selected for its double acyl chain membrane anchors. The chemical structures of BPD and its lipidated variants used in this study are shown in Figure 1a. Lipidation of BPD had no impact on its absorption properties, as determined by the lack of spectral shifts in the Soret or Q-band maxima of BPD (Figure S1c) or the Q-band full-width half maxima.48 Our prior work has shown that the photochemistry of BPD is also not impaired following conjugation to lipids, making them suitable alternative NIR-photoactivable agents for the specificity-tuned constructs engineered here.48 Conjugation of BPD to cholesterol resulted in the loss of its membrane-inserting properties and was not used further. DSPE-PEG2000-BPD nanoconstructs were unstable, exhibiting a polydispersity index (P.D.I.) greater than 0.2, possibly due to the terminal extension of the hydrophobic BPD molecules into the external aqueous phase. However, the 16:0 BPD-PC and 20:0 BPD-PC nanoconstructs remained colloidally stable with the BPD inserted into the hydrophobic bilayer (Figure S1d). When incubated with OVCAR-5 cells for 30 min, 16:0 and 20:0 BPD-PC nanoconstructs demonstrated no PS leaching at up to 500 μM BPD equivalent and reduced non-specific uptake over 24 h (Figure 1b; Figure S1e). The 16:0 BPD-PC variant, referred to as BPD-PC from hereon, was selected for the remaining studies as its membrane stability was equal to that of the 20:0 PC variant and as its acyl chain length matched that of DPPC, the main phospholipid constituent used for the Cet-PINs. Nanoformulation of BPD-PC preserved 60% of the fluorescence and photodynamic activity of the hydrophobic PS in PBS, which was 21% greater than that of the BPD nanoconstructs (Figure S1f). This favorable opto-chemical superiority of the high-payload BPD-PC nanoconstructs is attributed to the steric prevention of BPD aggregates in the bilayer when lipidated and further motivated its selection as the PS variant for the specificity-tuned Cet-PINs.
Figure 1.

Varying lipid anchors for BPD stabilization: (a) chemical structures of BPD and its lipidated variants conjugated to cholesterol, 20:0 lyso PC, 16:0 lyso PC, and DSPE-PEG2000-NH2 as anchors to promote membrane stability and eventually specificity when targeted with Cet. (b) Flow cytometry analysis reveals that the nanoconstructs formulated with 16:0 and 20:0 BPD-PC variants prevent non-specific PS leeching into OVCAR-5 cells, as compared to non-lipidated BPD nanoconstructs. Tuning nanoconstruct ζ-potential: (c) BPD-PC nanoconstructs doped with 0.5 mol % DSPE- PEG2000-DBCO for copper-free click conjugation to Cet aggregate in the absence of cationic DOTAP or anionic DOPG lipid dopants. (d) ζ-Potential of DOTAP and DOPG-containing BPD-PC nanoconstructs. (e) Min-max box plots of pooled BPD-PC nanoconstruct uptake rates in cancer cells expressing varying degrees of EGFR reveal that the anionic nanoconstructs have a favorable, lower variance in uptake rates before Cet conjugation (two-tailed t test, F test of variance). Tuning surface Cet grafting: (f) Degree of conjugation of site-specific Cet-Pz is more efficient than the conjugation of stochastic Cet at all the lipid:Cet mass ratios tested (mean ± S.E., one-way ANOVA with a Tukey post-test; n = 3 for b, c, d, and f; n = 6 for each cell line x rates from three cell lines pooled for each construct).
Steric and Electrostatic Tuning To Regulate Specificity.
To conjugate Cet site-specifically or stochastically to the PINs using bioorthogonal and chemoorthogonal copper-free click chemistry, a strained dibenzocyclooctyl (DBCO) moiety was needed at the BPD-PC nanoconstruct surface. However, DBCO functional groups are hydrophobic and promote nanoconstruct aggregation. The stability of BPD-PC nanoconstructs doped with a total of 5 mol % DSPE-PEG2000 was increasingly compromised by incorporating small amounts of DSPE- PEG2000-DBCO. BPD-PC nanoconstructs doped with 2 mol % DSPE-PEG2000-DBCO and 3 mol % DSPE-mPEG2000 were immediately unstable following extrusion (Figure S1h). Although the nanoconstructs with 1% DSPE-PEG2000-DBCO and 4% DSPE-mPEG2000 were stable following preparation, they precipitated overnight. Colloidal stability was only achieved at 0.5 mol % DSPE-PEG2000-DBCO doping when counter-stabilized with 4.5 mol % DSPE-mPEG2000 (Figure S1h). Not only was the modulation of DSPE-PEG2000-DBCO doping critical for stability, electrostatic repulsion of nanoconstructs was also needed and was achieved by the incorporation of 7.9 mol % of either DOTAP (cationic lipid) or DOPG (anionic lipid) (Figure 1c,d).
Electrostatic charge is a prime example of a biochemical PIN variable that has not been investigated in the context of antibody targeting of nanoconstructs in general, let alone for NIR activable PINs. Electrostatics have long been assumed to play a binary role in nanoparticle delivery: cationic nanoparticles promote cell membrane association,56 while anionic nanoparticles exhibit more favorable pharmacokinetics in vivo.57 Here, we show that the tuning of electrostatic charge to an anionic ζ-potential plays a critical role in achieving consistent cellular interactions within a heterogeneous EGFR-expressing population. We found that the electrostatic charge of the constructs influenced the intercellular variability of uptake rates before targeting between A431, OVCAR-5, and T47D cells that have varying degrees of EGFR expression (Figure S1i). Variability in intercellular uptake (prior to Cet conjugation) is a critical barrier for consistent cellular binding and uptake after targeting. This has negative consequences when attempting to maintain a consistent degree of specificity in heterogeneous tumor populations. The mean pooled cellular uptake rates of cationic and anionic BPD-PC nanoconstructs before targeting were similar; however, we found that the intercellular variance was significantly lower with the anionic DOPG-doped nanoconstructs (two-tailed unpaired t test, F test P < 0.0001, Figure 1e). Considering that the degree of variability in uptake rates of the anionic BPD nanoconstructs (prior to Cet conjugation) between the different cell lines tested was lower than that of the cationic nanoconstructs, the anionic constructs were selected as the platform for antibody conjugation and further PIN tuning and evaluation.
Modulating Antibody Orientation and Density of Surface Grafting Using Copper-Free Click Chemistry.
Antibody targeting of nanoconstructs in general often suffers from complexities in conjugation that are centered around perturbative, inconsistent chemical coupling, and antibody disorientation.58 Recent work has demonstrated the critical need for fine-tuning antibody targeted nanotherapeutics, as the molecular specificity can be markedly impaired when an one-size-fits all approach to nanoengineering is adopted.45 To circumvent issues pertaining to structurally perturbative anti-body conjugation techniques that are common to PINs, such as amine-sulfhydryl cross-linkers29,30,34 and thiol-maleimide chemistry,32,59,60 we adopted chemoorthogonal and bioorthogonal copper-free click chemistry61–66 to couple Cet to BPD-PC nanoconstructs. Following promising findings of an Fc-specific photo-cross-linked Protein Z (Pz) intermediary,67,68 we site-specifically conjugated Cet to BPD-PC nanoconstructs and compared their efficiencies with stochastically conjugated Cet constructs to determine the optimal antibody orientation for maximal PIN specificity.32,69–72
Cet was site-specifically modified with an azido-Protein Z intermediary, photo-cross-linked to the Fc fragment (Cet-Pz), or stochastically modified with a single azido-PEG moiety (validated by Cy5-DBCO labeling; Figure S2a–c). At all lipid/Cet mass ratios greater than 1:0.05, site-specific conjugation of Cet-Pz was more efficient than stochastic conjugation of Cet, suggesting that the constant site-specific availability of the azido linker offers superior control over conjugation (Figure 1f).
As with other bioconjugate systems, the tendency for adsorption and non-covalent interactions compromises the integrity of the nanoconstructs in biological milieu and can impair molecular specificity. Thus, we validated the chemical conjugation of site-specific Cet-Pz and stochastic Cet to the nanoconstructs using two methods. The first was by preinactivating the nanoconstruct surface DBCO by incubation with 0.1 M sodium azide for 24 h (Figure S2d). Both stochastic and site-specific conjugation of Cet to DBCO-inactivated PINs were significantly decreased (Figure S2e,f). Second, the degree of non-covalent Cet adsorption was also assessed by incubating site-specific Cet-Pz and stochastic Cet with nanoconstructs containing non-reactive DSPE-PEG-NH2 in place of the DBCO lipid. Cet adsorption was found to be negligible in both Cet preparations (Figure S2e,f).
Table S1 provides details of the full physical characterization of the site-specific Cet-Pz-PINs and stochastic Cet-PINs prepared, where the polydispersity indices of the Cet-Pz-PINs are greater than those of the Cet-PINs, suggesting a greater disparity in size distribution. However, the anionic charge is maintained in all PINs prepared, which is crucial to minimize variations in nanoconstruct–cell interactions at the various antibody surface densities tested.
Tuned Cellular Binding Specificity and Targeted Phototoxicity in 2D Monolayer Cultures.
To tune the surface grafting of site-specific Cet-Pz (Figure 2a) and stochastic Cet (Figure 2b) to PINs, the binding specificity at different surface densities was measured in A431 ((2–4) × 106 EGFR/cell),73 OVCAR-5 (4 × 105 EGFR/cell),74 T47D (7 × 103 EGFR/cell),75 and CHO-WT (EGFR null)76 cells using flow cytometry following a 30 min incubation period in serum-containing media at 37 °C. The most efficient Cet-Pz surface density was found to be 1129.1 ± 66.6 Cet-Pz per μm2, which equates to ~100 Cet-Pz molecules per PIN. This site-specific Cet-Pz-PIN exhibited 36.2-fold preferential binding to A431 cells over CHO-WT cells (Figures 2a and S3a). The most efficient stochastic Cet density was found to be only 517.5 ± 138.6 Cet per μm2, which equates to ~30 Cet molecules per PIN. Surprisingly, the stochastic Cet-PIN was found to exhibit unprecedented preferential binding specificities to A431 cells (97.3-fold), OVCAR-5 cells (10.5- fold), and T47D cells (2.2-fold), as compared to CHO-WT cells (Figures 2b and S3a). The site-specific or stochastic nanoconjugate demonstrating the highest specificity, ~100 Cet-Pz per PIN (referred to as Cet-Pz-PIN from hereon) or ~30 Cet per PIN (referred to as Cet-PIN from hereon), respectively, was used for the remainder of the studies. Although optimal specificity would ultimately confer the greatest degree of preference of PINs for high-EGFR-expressing cancer tissue over low-EGFR-expressing healthy tissue, the specificity of PINs can also be represented as the binding efficiencies to EGFR-expressing cells, with respect to untargeted constructs. As such, we have also compared the binding specificity of the Cet-Pz-PINs and the Cet-PINs (with respect to untargeted constructs) in A431 cells, OVCAR-5 cells, T47D cells, and CHO-WT cells. The highest degree of specificity of 26-fold with respect to untargeted constructs was found to be for the A431 cells when using the Cet-PIN formulation. Overall, we found that the binding of the Cet-Pz-PINs and the Cet-PINs correlated positively with the EGFR expression levels in all the cell lines tested (Figure 2d; Pearson’s r = 0.9918 and P = 0.0082 for the Cet-Pz-PINs; r = 0.9992 and P = 0.0008 for the Cet-PINs). However, upon quantifying the area-under-the-curve of the correlation, the Cet-PINs were found to be 2.72-fold more efficient at EGFR binding than the Cet-Pz-PINs (1.50 × 1010 vs 4.74 × 109, respectively). Furthermore, we found that targeting using the stochastic Cet-PIN increased the rates of cancer-cell specific (OVCAR-5) internalization in a manner that was dependent on the stochastic Cet surface density (Figure S6e). In addition to providing the highest degree of molecular specificity, the ~30 Cet surface density per PIN provided the highest rate of cellular internalization, expediting the uptake of the therapeutic photosensitizing entities. This optimal stochastic Cet-PIN was therefore used for all further molecular targeted NIR photodynamic activation studies.
Figure 2.

Binding of PINs conjugated to various densities of (a) site-specific Cet-Pz and (b) stochastic Cet to A431 (2–4 × 106 EGFR/cell), OVCAR-5(4 × 105 EGFR/cell), T47D (7 × 103 EGFR/cell), and CHO-WT (0 EGFR/cell) cell lines. (c) Binding specificity of the most efficient site-specific 100 Cet-Pz PINs and the stochastic 30 Cet-PINs is also presented with respect to the untargeted nanoconstructs (0 Cet per PIN molecule) for each cell line. (d) Pearson’s r correlation between Cet-Pz-PIN and Cet-PIN binding, and cellular expression levels of EGFR revealed that the binding of both constructs positively correlates with EGFR expression, but the Cet-PIN binding is more efficient (mean ± S.E., two-tailed t test). Representative phototoxicity dose-response curves of the untargeted construct and the specificity-tuned Cet-PIN in (e) MIA PaCa-2, (f) healthy CHO-WT, and (g) healthy CHO-EGFR cells demonstrate the EGFR-dependence of phototoxicity. (h) Molecular targeted phototoxicity was evaluated in a panel of cell lines expressing varying degrees of EGFR, and the differences in IC50 values between NIR photodestruction using untargeted constructs and the optimal specificity-tuned Cet-PINs are presented. The NIR photodynamic activation regimen used was 690 nm light irradiation with 20 J/cm2 at 150 mW/cm2 (mean ± S.E.; n = 3 for a–d and n = 8 for e–h; one-way ANOVA with a Tukey post-test; *** = P ≤ 0.001; ** = P ≤ 0.01; * = P ≤ 0.05).
To further authenticate the binding specificity of the optimal specificity-tuned Cet-PIN, binding to OVCAR-5 cells was competitively inhibited by the presence of free Cet at a 100-fold molar excess to the concentration of Cet conjugated to the PINs. Free 100× Cet resulted in greater than 80% inhibition in cellular binding, whereas the presence of 100× molar excess of free Trastuzumab or human IgG sham had no inhibitory effect, thereby further confirming the true molecular specificity of the optimal specificity-tuned Cet-PINs (Figure S3c).
It was interesting to find that at all Cet densities, cancer cell binding of the optimal stochastic Cet-PIN was superior to the site-specific Cet-Pz-PIN, suggesting that the stochastic Cet orientation at the PIN surface may offer the conformational freedom with the potential for optimal EGFR epitope binding. The elegant Pz approach was leveraged here to achieve a tightly regulated conjugation with high yields (Figure 1f).67 Despite its inferior conjugation efficiency, the stochastic conjugation, which is simpler and less expensive than the site-specific conjugation, was 2.72-fold more efficient. This further points to the complexity of conjugation of targeting moieties to nanoconstructs and the importance of the robust specificity tuning we perform here.
The efficacy of molecular targeted NIR photodynamic activation using the specificity-tuned Cet-PIN was then compared with an untargeted BPD-PC nanoconstruct in a panel of cell lines with varying degrees of EGFR expression using 690 nm NIR light irradiation at irradiance of 150 mW/cm2 and a fluence of 20 J/cm2. In the MIA PaCa-2 cells, targeting improved the efficacy of photodestruction by 32.7-fold, reducing the IC50 from 271.4 nM to 9.3 nM (Figure 2e,f,g; Table S2). As an ultimate control, molecular targeted NIR photodynamic activation was tested in CHO-WT (EGFR-null) and CHO-EGFR cells stably transfected with full-length EGFR.77 Cet-PINs did not reduce the IC50 of photodestruction in CHO-WT cells, as compared to untargeted constructs, whereas a remarkable 688.5-fold reduction in IC50 was observed with Cet-PINs in CHO-EGFR cells, reducing the IC50 from 3717.7 nM to 5.4 nM (Figure 2e–h, Table S2). Similar EGFR- dependent trends of photodestruction were observed for A431, OVCAR-5, and T47D cells (Figure 2h; Figure S3; Table S2). Molecular targeted phototoxicity of the Cet-PINs was found to be significantly higher than that of Cet-Pz-PINs following NIR photodynamic activation, which is consistent with its superior binding efficiencies (Figure S 3d).
The findings of the 2D in vitro testing rigorously confirm that the specificity-tuned Cet-PINs mediate an EGFR-dependent, molecular targeted NIR photodynamic destruction of cancer cells. As is consistent with other antibody-targeted therapies, Cet-PINs and Cet-Pz-PINs were found to be internalized through the endolysosomal pathway and remained in lysosomes up to 24 h of incubation (Figure S4).29,78–81 It is worth noting that for all the PIN concentrations tested, no dark toxicity was observed (Figure S5), as is ideal for an NIR activable therapeutic nanosystem.
Heterotypic Binding Specificity and Penetration.
In light of the evident complexity of optimal PIN preparation, a critical need also exists for disease-recapitulating in vitro high-throughput screening platforms to rapidly validate their specificity and ability to penetrate desmoplastic PDAC tumor tissue. 3D organoid tumor models have been previously used to evaluate liposome82 and nanoparticle penetration,83,84 PIC selectivity,85 and evaluation of PDT-based treatment regimens. 86–88
This is the first report of the binding specificity, penetration, and targeted phototoxicity of specificity-tuned Cet-PINs in both monotypic and heterotypic MIA PaCa-2 PDAC organoids cultured in the presence of PCAFs.89 The PCAFs are interspersed throughout the heterotypic organoids 48 h after seeding and represent 52.8% of the organoid cell population (Figure 3c; 6.8% intensity thresholding and 3-infinity pixel inclusion). PCAFs are critical stromal partners that are responsible for desmoplasia, in addition to paracrine signaling that is involved in treatment resistance in PDAC. The desmoplastic reaction results in stromal matrix deposition that constitutes up to 90% of the total PDAC tumor volumes.3,4 Desmoplasia, as discussed earlier, is a critical barrier that often prevents the penetration of therapeutics through PDAC tumor tissue. Given that PCAFs are supportive of PDAC progression and treatment resistance, it is also important that the PCAF component of tumors is also simultaneously destroyed following molecular targeted NIR photodynamic activation. However, depletion of the stroma alone must be avoided, as it can promote tumor metastasis. Recent work has shown that cancer-associated fibroblasts significantly overexpress and upregulate EGFR upon activation by vicinal cancer cells, as compared to normal fibroblasts residing in healthy tissue.90,91 This makes the PCAFs viable targets for the Cet-PINs in addition to the EGFR-overexpressing PDAC cells. Considering that MIA-PaCa-2 cells express 1.7 × 105 EGFR/cell,92 we calculated the approximate EGFR expression of PCAF cells to be 3.5 × 104 EGFR/cell based on the relative receptor expression levels of the two cell lines (Figure S6a). This is consistent with the relative Cet-PIN binding efficiencies to the two cell lines (Figure S6b–d). Importantly, no statistically significant difference was observed between the Cet-PIN binding patterns and the respective EGFR expression patterns in the two cell lines (Figure S6d; one-way ANOVA with Tukey post-test), further validating the molecular specificity of the Cet-PINs.
Figure 3.

(a) Representative white light and sum-of all-slices two photon fluorescent image composites of heterotypic PDAC organoids at 1 h, 6 h, and 24 h incubation with untargeted constructs or Cet-PINs both labeled with Alexa Fluor 680. The organoids with the highest signals for each time point were used to establish the image acquisition parameters. (b) Organoid images were used to quantify the Cet-PIN binding specificity toward the monotypic and heterotypic PDAC organoids at 1 h, 6 h, and 24 h incubation, with respect to the untargeted construct. The highest specificity of Cet-PINs in both types of organoids was found to be at 6 h of incubation. (c) Characterization of spatial distribution of MIA PaCa-2 cells (blue, DiD labeled) in monotypic and heterotypic PDAC organoids containing PCAFs (orange; DiO labeled). (d) Two-photon images of Cet-PIN distribution throughout the monotypic and heterotypic PDAC organoids at 1 h, 6 h, and 24 h incubation. 3D renders of the PINs bound to the organoids are in orange and penetration in 2D cross-sections through 50% of the organoid’s z-plane are shown in cyan, revealing efficient penetration through the organoids as early as 1 h incubation (scale bars in all images are 200 μm, specific 200 μm dimensions in (d) are highlighted, mean ± S.E., one-way ANOVA with a Tukey post-test; ∗∗∗ = P ≤ 0.001; ∗∗ = P ≤ 0.01; significance with regards to respective organoids at all time points; n = 3 organoids per condition).
Using two-photon microscopy, we assessed the penetration of the specificity-tuned Cet-PINs through monotypic and heterotypic MIA PaCa-2 organoids that include PCAFs, which are usually responsible for desmoplasia-limited drug penetration in PDAC. Cet-PINs tagged with Alexa Fluor 680 were incubated with the monotypic and heterotypic organoids for 1, 6, and 24 h. Representative images of the heterotypic heterotypic MIA PaCa-2 + PCAF organoids reveal that the Cet-PINs tagged with Alexa Fluor 680 exhibit a considerably higher binding affinity for the organoids at all time points than the untargeted constructs (Figure 3a). Image analysis of the optical z-stacks using a brightfield mask revealed that the highest specificity of 12.5-fold and 16.9-fold was observed for the monotypic MIA PaCa-2 and heterotypic MIA PaCa-2 + PCAF organoids at 6 h of incubation, resepectively (Figure 3b). At 50% optical sections through the organoid z-planes, we show that the Cet-PINs efficiently penetrated the organoids even at only 1 h of incubation. We then quantified the binding specificity of the Cet-PINs to the monotypic MIA PaCa-2 and heterotypic MIA PaCa-2 + PCAF organoids at 1 h, 6 h, and 24 h of incubation, with respect to untargeted construct controls. The three-dimensional distribution of Alexa Fluor 680 tagged Cet-PINs within the organoids was imaged using two photon microscopy, and 3D reconstructions of the Cet-PIN-bound organoids were generated (Figure 3d).
Molecular Targeted NIR Photodynamic Activation in Monotypic and Heterotypic PDAC Organoids.
After 48 h of seeding, targeted NIR photodynamic activation experiments were performed using a 6 h incubation time, which was determined to result in maximal binding specificity and efficient Cet-PIN penetration in the monotypic and heterotypic PDAC organoids (Figure 4a). Considering that one of the motivating factors for developing such complex molecular targeted PINs for desmoplastic PDAC is to increase the payload of PS delivery in PDAC tissue, we compared the efficacy of high-payload delivery of the specificity-tuned Cet-PINs with lower-payload direct Cet-BPD photoimmunoconjugates (PICs)39 and treated the organoids with them at equivalent Cet concentrations. The specificity-tuned Cet-PINs, untargeted constructs, and PICs were incubated for 6 h with increasing concentrations of BPD equivalent that were normalized to the concentration of Cet equivalent. The monotypic and heterotypic PDAC organoids were then irradiated with 40 J/cm2 of 690 nm light at 150 mW/ cm2. Seventy-two hours following treatment, the LIVE/DEAD viability assay was performed on the organoids. The Comprehensive Image Analysis Procedure for Structurally complex Organotypic cultures (CALYPSO)86 was used to generate viability heatmap images (Figure 4b; Figure S6g) and to quantify the fractional viability (Figure 4c) of the PDAC organoids. The IC20 of NIR photodestruction using the Cet-PINs in the heterotypic MIA PaCa-2 + PCAF organoids was 105.4 nM BPD equivalent, which was almost identical to that of monotypic MIA PaCa-2 organoids (103.4 nM BPD equivalent). Furthermore, we found that the Cet-PINs improved the NIR photodestruction efficacy by reducing IC20 values 16.8-fold in the monotypic MIA PaCa-2 organoids and 16.3-fold in the heterotypic MIA PaCa-2 + PCAF organoids, as compared to the untargeted construct (Figure 4c). The importance of these results lies in the fact that the specificity-tuned Cet-PINs were capable of circumventing any treatment resistance that is typically conferred by the presence of cancer-associated fibroblasts. It must be noted, ss however, that at the highest concentration of 2000 nM BPD equivalent, the mean heterotypic organoid viability was found to be 15.19% greater than the mean monotypic organoid viability. This is consistent with the residual viable cells that are observed following treatment with the highest concentration of 2000 nM BPD equivalent of Cet-PINs, which are not observed to the same extent in the monotypic organoids. Resistance to NIR photodynamic activation is a complex and multifaceted phenomenon. Factors that influence resistance in heterotypic three-dimensional organoids include microscale differences in light distribution and oxygen saturation, mechanistic cellular resistance to NIR photodynamic activation, PCAF activation status following treatment, and dynamic changes to EGFR expression in response to therapy. Understanding the mechanisms for the marginal residual disease following treatment in the heterotypic organoids is critical for designing appropriate dose-parameters for photodynamic activation and serve as the focus of future studies.
Figure 4.

(a) Schematic representation of the culturing procedures and treatment regimen of monotypic and heterotypic PDAC organoids followed by imaging-based CALYPSO analysis of treatment response. (b) Viability heatmap images of heterotypic PDAC organoids following molecular targeted NIR photodynamic activation with increasing concentrations of PIC, untargeted construct, and the specificity-tuned Cet-PIN. (c) CALYPSO image analysis framework was used for the quantitation of monotypic and heterotypic PDAC organoid fractional viability following molecular targeted NIR photodynamic activation, comparing both monotypic and heterotypic PDAC organoids treated using the PIC, an untargeted construct and the specificity-tuned Cet-PIN. (d) Heterotypic PDAC organoids treated using Cet-PINs, Cet-PIN-OxPt, Cet-PIN-Gem, and Cet-PIN-5-FU with NIR photodynamic activation. The NIR photodynamic activation regimen used was 690 nm light irradiation with 40 J/cm2 at 150 mW/cm2 (scale bars are 1 mm, box plots with min–max data presentation, one-way ANOVA with a Tukey post-test for PIC and Cet-PIN, * = P ≤ 0.05, *** = P ≤ 0.001, **** = P ≤ 0.0001; mean ± S.E.M., n = 4–8 organoids per condition).
Importantly, we found that the PIC exerted no phototoxic effects on the monotypic and heterotypic PDAC organoids at the Cet concentration equivalents used in this study, and as such, no IC20 values were derived. The PIC represents the current state of the art for molecular targeted photodynamic activation in the clinic,93 and due to its limited payload efficiency, the treatment will remain mostly as a monotherapy. To exemplify the multifunctionality of our Cet-PIN platforms, the PDAC front line chemotherapeutics oxaliplatin, gemcitabine hydrochloride, and 5-fluorouracil were entrapped within the specificity tuned Cet-PINs (Cet-PIN-OxPt, Cet-PIN-Gem, and Cet-PIN-5-FU, respectively). The chemotherapy loaded Cet-PINs all provide Cet payloads that are not achievable with direct antibody conjugates, especially with regard to PICs that are already modified with up to eight PS molecules (Cet-PIN-OxPt, 20 molecules per Cet payload; Cet-PIN-Gem, 41 molecules per Cet payload; Cet-PIN-5-FU, 1200 molecules per Cet payload). Following incubation and NIR photodynamic activation within the heterotypic PDAC organoids, the Cet-PIN-OxPt, Cet-PIN-Gem, and Cet-PIN-5-FU all significantly increased the antitumor efficacy of the treatment, with respect to the chemotherapy-free Cet-PINs at both 1 μM and 2 μM BPD-PC equivalent of nanoconstructs (Figure 4d).
Our findings underscore the value of using such high-payload PINs with the amenability for multimodal therapy, given that their biochemical tuning is diligent and rigorous to confer both sufficient tumor organoid tissue penetration and optimal binding specificity.
In Vivo Molecular Targeted NIR Photodynamic Activation of Desmoplastic Heterotypic PDAC Tumors.
As the ultimate utility of our tuned Cet-PINs is for in vivo NIR photodynamic activation regimens, we performed acute in vivo evaluation of the perivascular penetration and molecular targeted NIR photodestruction of desmoplastic PDAC using the specificity-tuned Cet-PINs, as outlined in Figure 5a. In a subcutaneous heterotypic murine xenograft model of pancreatic cancer consisting of MIA PaCa-2 and PCAF cells, we first quantified the degree of Cet-PIN tumor penetration using photoacoustic imaging. The tumors were imaged prior to the administration of the Cet-PIN labeled with IRDye800CW (Cet-PIN-IRDye800) to generate 2D and 3D baseline mapping of tumor blood vessels and imaged again 12 h after intravenous administration of the Cet-PIN-IRDye800 to generate 2D and 3D mapping of the tumoral distribution of the construct (Figure 5b). The validity of the photoacoustic signals arising from the Cet-PIN-IRDye800 was confirmed by quantitation of the photoacoustic tumor images before and after administration (Figure 5c). Using 15 randomly selected vessels from 2D cross-sectional images from three different mice, the perivascular distance of Cet-PIN penetration was quantified and found to range from 174–473 μm(Figure 5d).
Figure 5.
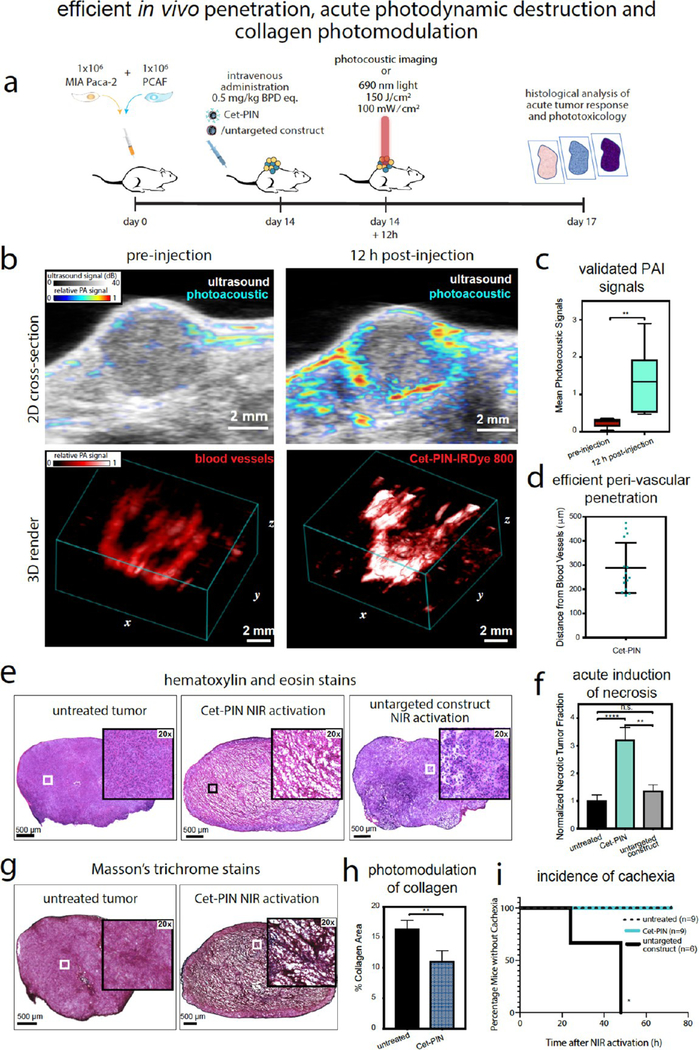
(a) Schematic representation of the time-scale of in vivo heterotypic tumor implantation, Cet-PIN administration, photoacoustic imaging, or molecular targeted NIR photodynamic activation and histological analyses of tumor responses to therapy. (b) Two-dimensional cross-sections and three-dimensional renders of heterotypic MIA PaCa-2 + PCAF tumors in vivo before and after intravenous administration of the Cet-PIN tagged with the photoacoustic contrast agent IRDye800CW (Cet-PIN-IRDye800). (c) Quantitation of photoacoustic signals before and after intravenous administration of the Cet-PIN-IRDye800 validates that the detected photoacoustic signal is arising from the administered construct. (d) Quantitation of perivascular tumor penetration of the Cet-PIN-IRDye800 shows that the constructs exhibit efficient tumor penetration in vivo. (e) HE stains of untreated tumors and tumors treated with NIRphotodynamic activation of the Cet-PIN or the untargeted constructs. Significant necrosis throughout the tumor cross-section is only observed in the tumors treated with the Cet-PIN at72h following therapy. (f) Quantitation of HE images in (e) reveals a significant increase in necrotic area following NIR photodynamic activation using the Cet-PIN, which is not the case following NIR photodynamic activation of tumors in mice treated with the untargeted construct. (g) Masson’s trichrome stains of tumor sections with and without NIR photodynamic activation using the Cet-PIN reveal a significant reduction in collagen content (blue), following therapy. This is quantified in (h). (i) Incidence of acute mouse cachexia during the 72 h following treatment. Cachexia was observed in 100% of mice 72 h following NIRphotodynamic activation of untargeted constructs, whereas untreated mice and mice treated with NIR photodynamic activation of Cet-PINs remained healthy (means ± S.E., two-tailed t test, ** = P ≤ 0.01; n = 3 mice × 3 z-planes for b and c; n = 6 mice × 6 slices per tumor for e and f, n = 3 mice (9 total untreated tumor slices) in g and h; n =4 mice (8 total PDT-treated tumor slices) in g and h.; Logrank test, *= P ≤ 0.05, n = 6–9 in d).
To evaluate the efficacy of the specificity-tuned Cet-PINs in desmoplastic PDAC tumors in vivo, we administered 0.5 mg/kg BPD equivalent of the constructs into mice bearing MIA PaCa-2 + PCAF xenograft tumors. Notably, this administered dose is ~10-fold lower than the Visudyne dose administered to patients undergoing PDT for PDAC, underscoring the efficiency of molecular targeted PS delivery with our specificity-tuned Cet-PIN formulation.7 This further highlights the power of intelligently engineering high-payload photonanomedicine systems to reduce the adverse effects associated with high PS administrations. NIR photodynamic activation was performed 12 h following intravenous administration of the Cet-PINs. Seventy-two hours following in vivo molecular targeted NIR photodynamic activation, the heterotypic PDAC tumors were harvested, sectioned, and stained using hematoxylin and eosin (HE) to evaluate tissue necrosis and Masson’s Trichrome stain to evaluate the collagen content in the tumors with and without therapy. It was found that only molecular targeted NIR photodynamic activation induced substantial necrosis throughout the entire cross-section of the tumors, as treatment using the untargeted constructs failed to induce significant tumor necrosis (Figure 5e). This was also quantified using image analysis, showing a statistically significant ~3-fold increase in fractional necrotic area 72 h following molecular targeted NIR photodynamic activation, with a neglible and non-statistically significant increase in necrosis following treatment using the untargeted constructs (Figure 5f).
Of considerable significance, we show that molecular targeted NIR photodynamic activation using the specificity-tuned Cet-PINs directly reduced the density of tumor collagen by 1.5-fold (Figure 5g,h; blue). The photomodulation of tumor collagen, which is produced by the activated PCAF cells within the tumor, is of paramount importance, as it demonstrates the potential for using our optimized Cet-PIN and NIR photodynamic activation to improve the tumor delivery and permeability of Cet-PINs during repeated treatment cycles that are known to be significantly limited by desmoplastic tumor stroma.
Molecular targeting of nanomedicines has also been reported to increase the antitumor efficacy of anticancer treatments, and thus, we have shown this to be the case for the Cet-PINs here. However, the ultimate goal of molecular targeting of such high-payload systems such as the Cet-PINs we present here is to allow for expansive and unrestricted illumination protocols during photodynamic treatment of in vivo pancreatic tumors. This is achieved by restricting phototoxicity to the receptor-overexpressing tumor tissue, thereby maximizing the safety of the illumination protocol. With regards to the molecular specificity of photodamage induced by NIR photodynamic activation of the Cet-PINs, we show that treatment of low EGFR-expressing T47D tumors using Cet-PINs did not significantly increase tumor necrosis (Figure S6d). In addition, there was no significant difference in fractional tumor necrosis between treatment using the Cet-PINs and the untargeted constructs. As improving the safety of photodynamic therapy protocols using molecular targeting is the priority, we evaluated the health of the mice following NIR photodynamic activation with either the Cet-PINs or untargeted constructs.
Within 72 h following NIR photodynamic activation in mice administered with untargeted constructs, all mice had developed severe cachexia and moribundity (Figure S7a), whereas all mice treated with Cet-PINs remained healthy following NIR photodynamic activation (Figure 5i). Mice treated with untargeted constructs also exhibited significant acute weight loss from as early as 48 h following NIR photodynamic activation, whereas mice treated with Cet-PINs experienced no change in body mass following NIR photodynamic activation (Figure S7b). The most common cause of toxicity in patients undergoing PDT within the vicinity of the peritoneum is bowel perforation.94 As is consistent with the clinical observations, we found that the mice with cachexia following treatment with untargeted constructs had visible signs of bowel photodamage and ulceration, in addition to bowel perforation that is visible in histological tissue sections (Figure S7c). Unlike treatment with the Cet-PINs, in the mice treated with untargeted constructs, there was substantial blistering in the skin covering the irradiated tumor, which was corroborated by necrosis in the epidermis that was visualized using HE staining (Figure S8). Furthermore, the muscle tissue surrounding the tumor that was also exposed to the NIR irradiation following administration of untargeted constructs exhibited visible signs of bruising, with one instance of a mouse losing function of the respective limb. HE staining of sections of vicinal muscle surrounding the irradiated tumors also showed significant necrosis and tissue damage following treatment using untargeted constructs. Liver segments beneath the irradiated tumors, however, appeared mostly unaffected. Unlike treatment using the untargeted constructs in this study in addition to untargeted BPD nanoformulations previously reported,95 only very mild erythema in the skin directly illuminated with NIR light was observed, which was resolved within 3 days. As such, our study concludes that molecular targeting using the specificity tuned Cet-PINs preserved the health of the mice, protected them from any signs of toxicity, morbidity, or moribundity, and spared all off-target tissue from non-specific phototoxicity, all while concomitantly improving anti-tumor efficacy.
Conclusions.
The dismal nature of PDAC therapy is attributed to a number of limiting factors including drug delivery and microenvironmental effects. While there is no shortage of highly potent, and likewise toxic, therapeutic agents to treat PDAC, systemic and dose-limiting toxicities in up to 40% of patients frequently result in termination of treatment.96 As such, there is a critical need for a distinct treatment modality that addresses these limitations, in addition to providing direct, controlled damage to the tumor. PDT is an example of such a distinctive modality, whereby photodynamic activation results in photodynamic priming of the tumor, improving delivery and sensitizing the tumor to subsequent combination therapies.
Our study presents a unique demonstration of how a single, specificity-tuned photoimmunonanoconjugate (PIN) offers a distinct NIR-activable therapeutic regimen for desmoplastic PDAC with unprecedented molecular specificities of binding and targeted phototoxicities. Using a single clinically inspired nanoconjugate rigorously tuned for optimal specificity, we present the simultaneous EGFR molecular targeted photodynamic tumor destruction and photomodulation of tumor collagen. Considering that there is no shortage of toxic PDAC agents, the power of the PIN approach does not lie solely in its capacity to destroy tumor tissue. Improving the efficacy of targeted photosensitizer conjugates, such as low-payload photoimmunoconjugates (PICs), can be achieved by tethering to charged polymers and polymeric nanoparticles.97–100 However, the value of the PIN approach we present here lies in its capacity to integrate the following important facets in a single construct and a single treatment application: high-payload photosensitizer delivery, multiagent entrapment for multimodal therapy, molecular specificity, spatiotemporal control over photoinduced tumor necrosis, and photomodulatory effects of the desmoplastic tumor stroma. Furthermore, it is evident that NIR photodynamic activation of the specificity-tuned PINs in vivo results in previously unreported photomodulatory effects in desmoplastic PDAC tumors, which has implications in alleviating stroma-induced treatment resistance.
Given that PDAC is particularly challenging to treat due to the dense stromal deposition arising from activated PCAFs, the high throughput in vitro screening models used must be appropriate to recapitulate elements of these stromal barriers. Motivated by this necessity, we developed heterotypic PDAC organoids that contain MIA PaCa-2 cancer cells and patient-derived PCAF cells in this study. Using this platform, we evaluated our high payload BPD-entrapping, specificity-tuned Cet-PINs. We show that the Cet-PINs rapidly penetrate the heterotypic PDAC organoids in under 1 h, exhibit binding specificities of up to ~16.9-fold, and improve molecular targeted NIR photodynamic killing efficacy by up to ~16-fold. Cet-PINs, encapsulated with the front-line PDAC chemotherapeutics oxaliplatin, gemcitabine hydrochloride, and 5-fluorouracil, further improved the antitumor efficacy in the heterotypic PDAC organoids, demonstrating a clear technical advance as compared to PICs.
For the first time, we here show that specificity-tuned Cet-PINs provide molecular targeted NIR photodynamic activation with excellent efficiencies of photodestruction in desmoplastic heterotypic PDAC tumors in vivo using a photosensitizer dose-equivalent that is 10-fold lower than the human equivalent dose needed for inducing effective PDAC necrosis in the clinic. At Cet-PIN doses and NIR light doses capable of inducing substantial PDAC tumor necrosis, vicinal healthy muscle tissue and skin covering the tumor that was exposed to the NIR beam were entirely unaffected. Conversely, equivalent doses of untargeted construct and NIR photodynamic activation were not capable of inducing similar tumor photodamage but did induce severe necrosis of the overlying skin, proximal muscle, and underlying bowel tissue that became perforated. Unlike the Cet-PINs, the untargeted constructs induced extreme weight loss, moribundity and cachexia in all treated mice. The findings were present here are the first to emphasize the criticality of imparting molecular specificity for light-activable nanomedicines. This ultimately allows for increased tolerability to photodynamic activation, allowing the field to move forward from the current accomplishment of controlled zones of tumor necrosis in patients, toward expansive curative illumination protocols that also spare healthy pancreatic tissue and vasculature. Furthermore, molecular targeted NIR photodynamic activation resulted in photomodulation of the desmoplastic tumor stroma, significantly reducing tumor collagen by 1.5-fold. The benefits of the treatment regimen provided by a single nanoconstruct with a single NIRphotodynamic activation process have significant implications in targeted drug delivery, whereby photomodulation of collagen can improve PDAC response to subsequent rounds of treatment. By modulating the tumor biology to provide favorable microenvironments, photodynamic priming using specificity-tuned PINs paves the way for critically needed multimodal therapies in the pursuit of paradigm-shifting PDAC treatment regimens. The findings we present here warrant critical further investigation into the molecular basis for the photomodulation of desmoplastic collagen within the heterotypic PDAC tumors provided by NIR photodynamic activation of the specificity-tuned Cet-PIN. Future avenues of investigation include elucidating the effect of NIR photodynamic activation on the photooxidation of collagen, collagen order and tortuosity, desmoplastic tumor elasticity, autocrine and paracrine signaling pathways in PDAC and PCAF cells, and the PCAF activation status following treatment.
PDACs are also characterized by high rates of KRAS mutations (70–90%), rendering them resistant to Cet (Erbitux) therapy for EGFR blockade,101 irrespective of the high rates of EGFR expression in patients with PDAC (85%).37 This has led to the failure of Cet (Erbitux) as part of a combination treatment for PDAC. As such, Cet is currently used for clinical tumor imaging only.38 Our prior work has shown that photodynamic priming of KRAS mutant ovarian cancers resensitizes the tumor cells to EGFR blockade using Cet, making the synergistic outcome superior to single treatments, which are both minimal and modest.102 With the capacity to also prime KRAS mutant PDAC cells for EGFR blockade using Cet, photodynamic priming by the NIR photoactivation of the specificity-tuned Cet-PIN is also expected to exert molecular synergy in the most resistant of PDAC subtypes, further enhancing outcomes.
This study forms the basis of integrating multimodal mechanistically inspired combination therapies within the Cet-PIN framework to reduce the toxicities associated with current PDAC treatments and high PS doses, to enhance their delivery to desmoplastic tumors, and to promote synergistic, spatiotemporally controlled therapies. These strategies that are designed to simultaneously mitigate multiple hurdles to effective PDAC management promise to push current clinical PDT regimens toward more complete, curative NIR-activated photodynamic treatment protocols while sparing healthy vicinal tissue and organ function to extend patient survival and maximize patient quality of life.
Supplementary Material
ACKNOWLEDGMENTS
We thank Drs. Danian Cao, Jie Zhao, Brijesh Bhayana, James Hui, and Ahmed Alkhateeb for their technical assistance and Drs. Bryan Spring, Sriram Anbil, Imran Rizvi, Akilan Palanisami, and Huang-Chiao Huang for insightful discussions. We also thank Dr. Andrew Tsourkas for providing the bacterial Protein Z expression cultures and DNA plasmid vectors.
Funding
Financial support was provided by the National Institutes of Health Grant Nos. P01CA084203, R01CA156177, R01CA160998, R21CA220143, and S100D012326 to T.H. and K99CA215301 to G.O. We also gratefully acknowledge the Bullock-Wellman Fellowship to G.O. and the IIRSIP Fellowship from the Higher Education Commission (HEC) of Pakistan to S.B.
ABBREVIATIONS
- PINs
photoimmunonanoconjugates
- BPD
benzoporphyrin derivative
- BPD-PC
16:0 lysophosphocholine BPD conjugate
- TLC
thin layer chromatography
- NIR
near-infrared
- Cet
cetuximab
- PCAF
pancreatic cancer-associated fibroblasts
- Pz
protein Z
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.9b00859.
Materials and methods with supplementary results (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Conroy T; Desseigne F; Ychou M; Bouche O; Guimbaud R; Becouarn Y; Adenis A; Raoul JL; Gourgou-Bourgade S; de la Fouchardiere C; Bennouna J; Bachet JB; Khemissa-Akouz F; Pere-Verge D; Delbaldo C; Assenat E; Chauffert B; Michel P; Montoto-Grillot C; Ducreux M. Groupe Tumeurs Digestives of, U.; Intergroup, P. N. Engl.J. Med 2011, 364 (19), 1817–25. [DOI] [PubMed] [Google Scholar]
- (2).Ryan DP; Hong TS; Bardeesy NN Engl. J. Med 2014, 371 (22), 2140–1. [DOI] [PubMed] [Google Scholar]
- (3).Bolm L; Cigolla S; Wittel UA; Hopt UT; Keck T; Rades D; Bronsert P; Wellner UF Transl. Oncol 2017, 10 (4), 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Waghray M; Yalamanchili M; di Magliano MP; Simeone DM Curr. Opin. Gastroenterol 2013, 29 (5), 537–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Weniger M; Honselmann KC; Liss AS The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10 (9), 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Neesse A; Michl P; Frese KK; Feig C; Cook N; Jacobetz MA; Lolkema MP; Buchholz M; Olive KP; Gress TM; Tuveson DA Gut 2011, 60 (6), 861–8. [DOI] [PubMed] [Google Scholar]
- (7).Huggett MT; Jermyn M; Gillams A; Illing R; Mosse S; Novelli M; Kent E; Bown SG; Hasan T; Pogue BW; Pereira SP Br. J. Cancer 2014, 110 (7), 1698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).DeWitt JM; Sandrasegaran K; O’Neil B; House MG; Zyromski NJ; Sehdev A; Perkins SM; Flynn J; McCranor L; Shahda S. Gastrointest. Endosc 2019, 89 (2), 390–398. [DOI] [PubMed] [Google Scholar]
- (9).Spring BQ; Bryan Sears R; Zheng LZ; Mai Z; Watanabe R; Sherwood ME; Schoenfeld DA; Pogue BW; Pereira SP; Villa E,; Hasan T. Nat. Nanotechnol 2016, 11 (4), 378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Celli JP; Solban N; Liang A; Pereira SP; Hasan T. Lasers Surg. Med 2011, 43 (7), 565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Huang HC; Rizvi I; Liu J; Anbil S; Kalra A; Lee H; Baglo Y; Paz N; Hayden D; Pereira S; Pogue BW; Fitzgerald J; Hasan T. CancerRes. 2018, 78 (2), 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Snyder JW; Greco WR; Bellnier DA; Vaughan L; Henderson BW Cancer Res. 2003, 63 (23), 8126–31. [PubMed] [Google Scholar]
- (13).Kobayashi H; Choyke PL Nanoscale 2016, 8 (25), 12504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bown SG; Rogowska AZ; Whitelaw DE; Lees WR; Lovat LB; Ripley P; Jones L; Wyld P; Gillams A; Hatfield AW Gut 2002, 50 (4), 549–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Visudyne. Control number: 189192 ProductMonograph; Novartis Pharmaceuticals Canada Inc. [Google Scholar]
- (16).NX Development Corp. (NXDC) Launches FDA-Approved Gleolan (aminolevulinic acid HCl) for Enhanced Visualization of High-Grade Gliomas (including Glioblastomas). NX Development Corp Briefing Package; NXDC, 2018 [Google Scholar]
- (17).Spring BQ; Abu-Yousif AO; Palanisami A; Rizvi I; Zheng X; Mai Z; Anbil S; Sears RB; Mensah LB; Goldschmidt R; Erdem SS; Oliva E; Hasan T. Proc. Natl. Acad. Sci. U. S.A 2014,111 (10), E933–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Min Y; Caster JM; Eblan MJ; Wang AZ Chem. Rev 2015, 115 (19), 11147–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Obaid G; Broekgaarden M; Bulin AL; Huang HC; Kuriakose J; Liu J; Hasan T. Nanoscale 2016, 8 (25), 12471–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tian X; Lind KR; Yuan B; Shaw S; Siemianowski O; Cademartiri L. Adv. Mater 2017, 29 (17), 1604681. [DOI] [PubMed] [Google Scholar]
- (21).Lismont M; Dreesen L; Wuttke S. Adv. Funct. Mater 2017, 27 (14), 1606314. [Google Scholar]
- (22).Freund R; Lachelt U; Gruber T; Ruhle B; Wuttke S. ACS Nano 2018, 12 (3), 2094–2105. [DOI] [PubMed] [Google Scholar]
- (23).Oliveira-Cunha M; Newman WG; Siriwardena AK Cancers 2011, 3 (2), 1513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).(a) ClinicalTrials.gov Identifier: NCT03563248, Losartan and Nivolumab in Combination With FOLFIRINOX and SBRT in Localized Pancreatic Cancer. Clinicaltrails.gov; U.S. National Library of Medicine, 2019. http://www.clinicaltrials.gov (accessed September 19, 2019). [Google Scholar]; (b) ClinicalTrials.gov Identifier: NCT03825289, Trametinib and Hydroxychloroquine in Treating Patients With Pancreatic Cancer (THREAD). Clinicaltrails.gov; U.S. National Library of Medicine, 2019. http://www.clinicaltrials.gov (accessed September 19, 2019). [Google Scholar]; (c) ClinicalTrials.gov Identifier: NCT03634332, Second-line Study of PEGPH20 and Pembro for HA High Metastatic PDAC. Clinicaltrails.gov; U.S. National Library of Medicine, 2019. http://www.clinicaltrials.gov (accessed September 19, 2019). [Google Scholar]
- (25).Harris LJ; Larson SB; Hasel KW; McPherson A. Biochemistry 1997, 36 (7), 1581–97. [DOI] [PubMed] [Google Scholar]
- (26).Tashiro M; Tejero R; Zimmerman DE; Celda B; Nilsson B,; Montelione GT J. Mol. Biol 1997, 272 (4), 573–90. [DOI] [PubMed] [Google Scholar]
- (27).Jmol, 2019. http://www.jmol.org/ (accessed April 10, 2019).
- (28).Paszko E; Vaz GM; Ehrhardt C; Senge MO Eur. J. Pharm. Sci 2013, 48 (1), 202–210. [DOI] [PubMed] [Google Scholar]
- (29).Bergstrom LC; Vucenik I; Hagen IK; Chernomorsky SA; Poretz RD J. Photochem. Photobiol., B 1994, 24 (1), 17–23. [DOI] [PubMed] [Google Scholar]
- (30).Yemul S; Berger C; Estabrook A; Suarez S; Edelson R; Bayley H. Proc. Natl. Acad. Sci. U. S. A 1987, 84 (1), 246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).García-Díaz M; Nonell S; Villanueva A; Stockert JC; Cañete M; Casadό A; Mora M; Sagrista ML Biochim. Biophys. Acta, Biomembr 2011, 1808 (4), 1063–1071. [DOI] [PubMed] [Google Scholar]
- (32).Broekgaarden M; van Vught R; Oliveira S; Roovers RC; van Bergen en Henegouwen PM; Pieters RJ; Van Gulik TM; Breukink E; Heger M. Nanoscale 2016, 8 (12), 6490–4. [DOI] [PubMed] [Google Scholar]
- (33).Morgan J; Gray A; Huehns E. Br.J. Cancer 1989, 59 (3), 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morgan J; MacRobert A; Gray A; Huehns E. Br. J. Cancer 1992, 65 (1), 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cui S; Yin D; Chen Y; Di Y; Chen H; Ma Y; Achilefu S; Gu Y. ACS Nano 2013, 7 (1), 676–88. [DOI] [PubMed] [Google Scholar]
- (36).Derycke AS; DeWitte PA Int. J. Oncol 2002, 20 (1), 181–7. [PubMed] [Google Scholar]
- (37).Barton CM; Hall PA; Hughes CM; Gullick WJ; Lemoine NR J. Pathol 1991, 163 (2), 111–6. [DOI] [PubMed] [Google Scholar]
- (38).ClinicalTrials.gov Identifier: NCT03384238, Panitumumab-IRDye800 in Patients With Pancreatic Cancer Undergoing Surgery. ClinicalTrials.gov; U.S. National Library of Medicine, 2019. ( accessed April 10,2019). [Google Scholar]
- (39).Savellano MD; Hasan T. Photochem. Photobiol 2003, 77 (4), 431–439. [DOI] [PubMed] [Google Scholar]
- (40).Bryden F; Maruani A; Rodrigues JMM; Cheng MHY; Savoie H; Beeby A; Chudasama V; Boyle RW Bioconjugate Chem. 2018, 29 (1), 176–181. [DOI] [PubMed] [Google Scholar]
- (41).He C; Liu D; Lin W ACS Nano 2015, 9 (1), 991–1003. [DOI] [PubMed] [Google Scholar]
- (42).He C; Duan X; Guo N; Chan C; Poon C; Weichselbaum RR; Lin W. Nat. Commun 2016, 7, 12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Carter KA; Luo D; Razi A; Geng J; Shao S; Ortega J; Lovell JF Theranostics 2016, 6 (13), 2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kamoun WS; Oyama S; Kornaga T; Feng T; Luus L; Pham MT; Kirpotin DB; Marks JD; Geddie M; Xu L. Nanoliposomal targeting of Ephrin receptor A2 (EphA2): Clinical translation; AACR, 2016. [Google Scholar]
- (45).Dai Q; Wilhelm S; Ding D; Syed AM; Sindhwani S; Zhang Y; Chen YY; MacMillan P; Chan WCW ACS Nano 2018,12 (8), 8423–8435. [DOI] [PubMed] [Google Scholar]
- (46).Mir Y; Elrington SA; Hasan T. Nanomedicine 2013, 9 (7), 1114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ben-Dror S; Bronshtein I; Wiehe A; Roder B; Senge MO; Ehrenberg B. Photochem. Photobiol 2006, 82 (3), 695–701. [DOI] [PubMed] [Google Scholar]
- (48).Obaid G; Jin W; Bano S; Kessel D; Hasan T. Photochem. Photobiol 2019, 95 (1), 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Komatsu T; Moritake M; Nakagawa A; Tsuchida E. Chem. - Eur. J 2002, 8 (23), 5469–80. [DOI] [PubMed] [Google Scholar]
- (50).Lovell JF; Jin CS; Huynh E; Jin H; Kim C; Rubinstein JL; Chan WC; Cao W; Wang LV; Zheng G. Nat. Mater 2011,10 (4)324. [DOI] [PubMed] [Google Scholar]
- (51).Rizvi I; Obaid G; Bano S; Hasan T; Kessel D. Lasers Surg. Med 2018, 50 (5), 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Riske KA; Sudbrack TP; Archilha NL; Uchoa AF; Schroder AP; Marques CM; Baptista MS; Itri R. Biophys. J 2009, 97 (5), 1362–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Nathan E; Vijayashree K; Harikrishna A; TakafUji M; Jintoku H; Ihara H; Rao NM Photochem. Photobiol Sci 2016, 15 (12) 1476–1483. [DOI] [PubMed] [Google Scholar]
- (54).Park H; Lee J; Jeong S; Im BN; Kim MK; Yang SG; Na K. Adv. Healthcare Mater 2016, 5 (24), 3139–3147. [DOI] [PubMed] [Google Scholar]
- (55).Achalkumar AS; Bushby RJ; Evans SD Soft Matter 2010, 6(24), 6036–6051. [Google Scholar]
- (56).Zelphati O; Uyechi LS; Barron LG; Szoka FC Jr. Biochim. Biophys. Acta, Lipids Lipid Metab 1998, 1390 (2), 119–33. [DOI] [PubMed] [Google Scholar]
- (57).Cullis PR; Chonn A; Semple SC Adv. Drug Delivery Rev 1998, 32 (1–2), 3–17. [DOI] [PubMed] [Google Scholar]
- (58).Richards DA; Maruani A; Chudasama V. Chemical Science 2017, 8 (1), 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kirchhof S; Strasser A; Wittmann H-J; Messmann V; Hammer N; Goepferich AM; Brandl FP J. Mater. Chem. B 2015, 3(3), 449–457. [DOI] [PubMed] [Google Scholar]
- (60).Staros JV; Wright RW; Swingle DM Anal. Biochem 1986, 156 (1), 220–2. [DOI] [PubMed] [Google Scholar]
- (61).Oude Blenke E; Klaasse G; Merten H; Pluckthun A; Mastrobattista E; Martin NI J. Controlled Release 2015, 202, 14–20. [DOI] [PubMed] [Google Scholar]
- (62).Koo H; Lee S; Na JH; Kim SH; Hahn SK; Choi K; Kwon IC; Jeong SY; Kim K. Angew. Chem. Int. Ed 2012, 51 (47), 11836–11840. [DOI] [PubMed] [Google Scholar]
- (63).Baskin JM; Prescher JA; Laughlin ST; Agard NJ; Chang PV; Miller IA; Lo A; Codelli JA; Bertozzi CR Proc. NaÜ.Acad. Sci. U. S. A 2007, 104 (43), 16793–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Elias DR; Cheng Z; Tsourkas A. Small 2010, 6 (21), 2460–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Alam S; Alves DS; Whitehead SA; Bayer AM; McNitt CD; Popik VV; Barrera FN; Best MD Bioconjugate Chem. 2015,26 (6), 1021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Zhang H; Weingart J; Jiang R; Peng J; Wu Q; Sun X-L Bioconjugate Chem. 2013, 24 (4), 550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hui JZ; Tsourkas A. Bioconjugate Chem. 2014, 25 (9), 1709–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Hui JZ; Tamsen S; Song Y; Tsourkas A. Bioconjugate Chem. 2015, 26 (8), 1456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Cheng WW; Allen TM J. Controlled Release 2008, 126 (1), 50–8. [DOI] [PubMed] [Google Scholar]
- (70).Cheng W; Allen T. Expert Opin. Drug Delivery 2010, 7 (4), 461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Park JW; Hong K; Kirpotin DB; Colbern G; Shalaby R; Baselga J; Shao Y; Nielsen UB; Marks JD; Moore D; Papahadjopoulos D; Benz CC Clin. Cancer Res 2002, 8 (4), 1172–81. [PubMed] [Google Scholar]
- (72).Iden DL; Allen TM Biochim. Biophys. Actaj Biomembr 2001, 1513 (2), 207–16. [DOI] [PubMed] [Google Scholar]
- (73).Benveniste M; Livneh E; Schlessinger J; Kam ZJ Cell Biol. 1988, 106 (6), 1903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Savellano MD; Hasan T. Clin. CancerRes 2005,11 (4), 1658–68. [DOI] [PubMed] [Google Scholar]
- (75).Beerli RR; Graus-Porta D; Woods-Cook K; Chen X; Yarden Y; Hynes NE Mol. Cell. Biol 1995, 15 (12), 6496–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Abu-Yousif AO; Moor AC; Zheng X; Savellano MD; Yu W; Selbo PK; Hasan T. CancerLett. 2012, 321 (2), 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Heitner T; Moor A; Garrison JL; Marks C; Hasan T; Marks JD J. Immunol. Methods 2001, 248 (1–2), 17–30. [DOI] [PubMed] [Google Scholar]
- (78).Wileman T; Harding C; Stahl P. Biochem.J 1985,232 (1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Ritchie M; Tchistiakova L; Scott N. MAbs 2013,5 (1), 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Rejman J .; Oberle V; Zuhorn IS; Hoekstra D. Biochem. J 2004, 377 (1), 159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Bareford LM; Swaan PW Adv. Drug Delivery Rev 2007, 59 (8), 748–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Takechi-Haraya Y; Goda Y; Sakai-Kato K. Mol. Pharmaceutics 2017, 14, 2158. [DOI] [PubMed] [Google Scholar]
- (83).Ma HL; Jiang Q; Han S; Wu Y; Cui Tomshine J; Wang D; Gan Y; Zou G; Liang XJ Mol. Imaging 2012, 11 (6), 487–98. [PubMed] [Google Scholar]
- (84).Zanoni M; Piccinini F; Arienti C; Zamagni A; Santi S; Polico R; Bevilacqua A; Tesei A. Sci. Rep 2016, 6, 19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Rahmanzadeh R; Rai P; Celli JP; Rizvi I; Baron-Lühr B; Gerdes J; Hasan T. Cancer Res. 2010, 70 (22), 9234–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Bulin AL; Broekgaarden M; Hasan T. Sci. Rep 2017, 7 (1), 16645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Rizvi I; Anbil S; Alagic N; Celli J; Zheng LZ; Palanisami A; Glidden MD; Pogue BW; Hasan T. Photochem. Photobiol 2013, 89 (4), 942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Celli JP; Rizvi I; Blanden AR; Massodi I; Glidden MD; Pogue BW; Hasan T. Sci. Rep 2015, 4, 3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Waghray M; Yalamanchili M; Dziubinski M; Zeinali M; Erkkinen M; Yang H; Schradle KA; Urs S; Pasca Di Magliano M; Welling TH; Palmbos PL; Abel EV; Sahai V; Nagrath S; Wang L; Simeone DM Cancer Discovery 2016, 6 (8), 886–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Epstein Shochet G; Brook E; Eyal O; Edelstein E; Shitrit D. Am. J. Physiol.: Lung Cell. Mol. Physiol 2019, 316, L1025. [DOI] [PubMed] [Google Scholar]
- (91).LeBleu VS; Kalluri R Dis. Models & Mech 2018, 11 (4), No. dmm029447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Smith JJ; Derynck R; Korc M. Proc. Natl. Acad. Sci. U. S. A 1987, 84 (21), 7567–7570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).ClinicalTrials.gov Identifier: NCT03769506, Photoimmuno-therapy (PIT) Study in Recurrent Head/Neck Cancer for Patients Who Have Failed at Least Two Lines of Therapy. ClinicalTrials.gov; U.S. National Library of Medicine, 2019. (accessed April 10, 2019). [Google Scholar]
- (94).DeLaney TF; Sindelar WF; Tochner Z; Smith PD; Friauf WS; Thomas G; Dachowski L; Cole JW; Steinberg SM; Glatstein E. Int. J. Radiat. Oncol. Biol. Phys 1993, 25 (3), 445–57. [DOI] [PubMed] [Google Scholar]
- (95).Mallidi S; Watanabe K; Timerman D; Schoenfeld D; Hasan T. Theranostics 2015, 5 (3), 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Boone BA; Steve J; Krasinskas AM; Zureikat AH; Lembersky BC; Gibson MK; Stoller RG; Zeh HJ; Bahary NJ Surg. Oncol 2013, 108 (4), 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Soukos NS; Hamblin MR; Hasan T. Photochem. Photobiol 1997, 65 (4), 723–9. [DOI] [PubMed] [Google Scholar]
- (98).Hamblin MR; Miller JL; Hasan T. CancerRes. 1996,56 (22), 5205–5210. [PubMed] [Google Scholar]
- (99).Molpus KL; Hamblin MR; Rizvi I; Hasan T. Gynecol. Oncol 2000, 76 (3), 397–404. [DOI] [PubMed] [Google Scholar]
- (100).Huang HC; Pigula M; Fang Y; Hasan T. Small 2018, 14, No. e1800236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Kullmann F; Hartmann A; Stohr R; Messmann H; Dollinger MM; Trojan J; Fuchs M; Hollerbach S; Harder J; Troppmann M; Kutscheidt A; Endlicher E. Oncology 2011, 81 (1), 3–8. [DOI] [PubMed] [Google Scholar]
- (102).del Carmen MG; Rizvi I; Chang Y; Moor AC; Oliva E; Sherwood M; Pogue B; Hasan TJ Natl. Cancer Inst 2005, 97 (20), 1516–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.