

Abstract
Cholangiocarcinoma (CCA) is the second most common malignancy arising in the liver. It carries a poor prognosis, in part because its pathogenesis is not well understood. The type 3 inositol 1,4,5-trisphosphate receptor (ITPR3) is the principal intracellular Ca2+ release channel in cholangiocytes and its increased expression has been related to the pathogenesis of malignancies in other types of tissues, so we investigated its role in CCA. ITPR3 expression was increased in both hilar and intrahepatic CCA samples as well as in CCA cell lines. Deletion of ITPR3 from CCA cells impaired proliferation and cell migration. A bioinformatic analysis suggested that over-expression of ITPR3 in CCA would have a mitochondrial phenotype, so this also was examined. ITPR3 normally is concentrated in a sub-apical region of endoplasmic reticulum (ER) in cholangiocytes, but both immunogold electron microscopy and super-resolution microscopy showed that ITPR3 in CCA cells also was in regions of ER in close association with mitochondria. Deletion of ITPR3 from these cells impaired mitochondrial Ca2+ signaling and led to cell death.
Conclusion:
ITPR3 expression in cholangiocytes becomes enhanced in cholangiocarcinoma. This contributes to malignant features, including cell proliferation and migration and enhanced mitochondrial Ca2+ signaling.
Keywords: Cholangiocyte, Apoptosis, Calcium signaling, Mitochondria, Cell proliferation
INTRODUCTION
Cholangiocarcinoma (CCA) is the second most common type of primary liver tumor and may arise along intrahepatic or extrahepatic bile ducts (1). The highest incidence of CCA is in northeastern Thailand but the incidence of this disease is increasing globally as well, while the 5-year survival rate has remained less than 5% (2). This poor survival rate in part may be because surgical resection is currently the only potentially curative treatment option, but most patients with CCA are diagnosed at a stage of disease that is too advanced for curative resection to be feasible. For non-resectable cases, current chemotherapy options typically are palliative. The limited efficacy of medical therapies for CCA may in part reflect that the molecular mechanism of biliary carcinogenesis remains incompletely understood.
Intracellular calcium (Ca2+) signaling pathways regulate a number of cellular processes that relate to tumor development and growth, including gene transcription (3), cell proliferation (4, 5), cell migration (6), apoptosis (7, 8), and necrosis (9). Ca2+ release into the cytosol through the inositol 1,4,5-trisphosphate receptor (ITPR) regulates these functions in most epithelia (10, 11), including cholangiocytes (12). The type 3 inositol 1,4,5-trisphosphate receptor (ITPR3) is the principal isoform in cholangiocytes (12) and several lines of evidence suggest this isoform may play a particularly important role in cancer. A mutation in the ITPR3 gene was identified in the genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma (13). ITPR3 is overexpressed in human gastric cancer (14), human glioblastoma (15), and colorectal cancer, where its expression level also correlates with tumor aggressiveness and patient survival (16). ITPR3 may be particularly important for regulating apoptosis (7), and that feature of this isoform has been related to the pathogenesis of several types of malignancies, including melanoma, mesothelioma, and prostate cancer (17, 18). The purpose of this study was to investigate whether and how ITPR3 is involved in the development of CCA.
EXPERIMENTAL PROCEDURES
Human ethics statement and study approval
Human liver tissue biopsies were obtained under the auspices of protocols approved by the Institutional Review Board on the Protection of the Rights of Human Subjects (Yale University). The Human Investigation Committee protocol number HIC-1208010665 was approved prior to the initiation of the study.
Liver histology and immunohistochemistry
Fourteen surgically resected specimens of normal livers and bile duct cancer tissues were obtained from patients admitted to Yale New Haven Hospital. Immunohistochemical analyses of ITPR3 expression were performed in liver biopsies from histologically normal livers and from patients with hilar cholangiocarcinoma and intrahepatic cholangiocarcinoma. Additional details are in Supporting Materials and Methods.
Cell culture
Two well-established normal human cholangiocyte cell lines: i) the H69 cells were kindly provided from Dr. Douglas M. Jefferson (Tufts University), and ii) the NHC cell line (19). Four cholangiocarcinoma cell lines: i) the MzCha1 cell line was kindly provided by Dr. Greg Fitz (University of Texas Southwestern), ii) the HuCCA1 cell line was kindly provided by Dr. Stitaya Sirisinha (Mahidol University), iii) the TFK-1 cell line was kindly provided by Dr. Mario Strazzabosco (Yale University), and iv) the HuCCT1 cell line was purchased from Japanese Collection of Research Bioresources (JCRB) Cell Bank (National Institutes of Biomedical Innovation, Health and Nutrition, Osaka, Japan). Additional details are in Supporting Materials and Methods.
Generation of CRISPR/Cas9-mediated ITPR3-knockout in cholangiocyte cell lines
The ITPR3 knockout in NHC, HuCCA1 and MzCha1 cells were established by using the CRISPR/Cas9 system. IP3R-III CRISPR/Cas9 KO plasmid and IP3R-III HDR plasmid were obtained from Santa Cruz Biotechnology. Additional details are in Supporting Materials and Methods.
Cell proliferation, Cell cycle, Wound healing and Transwell migration assay
Additional details are in Supporting Materials and Methods.
Three-dimensional cell culture and spheroid imaging
Cells were plated into ultra-low attachment spheroid plates and allowed spheroids to form. Live/dead staining of 3D spheroids was monitored by staining with calcein acetoxymethyl ester, ethidium homodimer-1, and Hoechst. Spheroids were imaged using Operetta High Content Imaging System. Additional details are in Supporting Materials and Methods.
Super-resolution fluorescence microscopy and ER-mitochondria contacts
Cells were co-transfected with GFP-ITPR3 (a gift from Dr. Colin W. Taylor, University of Cambridge) targeting to endoplasmic reticulum and TOMM20-mKO2-N1 targeting to mitochondria (a gift from Dr. Michael Davidson, Addgene plasmid # 54625). Additional details are in Supporting Materials and Methods.
Immunogold Electron microscopy
Sample preparation and imaging were performed at Center for Cellular and Molecular Imaging Electron Microscope Core Facility, Yale University. Additional details are in Supporting Materials and Methods.
Calcium imaging and mitochondrial labelling
Cells were transfected with mitochondrial Ca2+ sensor Inverse-Mito-Pericam, a gift from Dr. György Hajnóczky (MitoCare Center, Thomas Jefferson University). To visualize changes in mitochondrial Ca2+, cells were loaded with caged inositol triphosphate (ci-IP3/PM). Photo-releasing of ci-IP3/PM was activated by a flash of UV light. Inverse-Mito-Pericam fluorescence was monitored in real-time in a Bruker Opterra II swept-field confocal microscope. Additional details are in Supporting Materials and Methods.
Statistical analyses
All data are expressed as mean ± standard error of mean (SEM) of multiple independent experiments. Statistical analyses were performed using the two-tailed Student’s t-test. Differences with P < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 7 Software (GraphPad, La Jolla, CA).
FURTHER METHODOLOGICAL DETAILS
Detailed additional Materials and Methods are available in the Supporting Information.
RESULTS
ITPR3 expression is increased in cholangiocarcinoma.
To begin to examine the relationship between ITPR3 and cholangiocarcinoma (CCA), the expression of ITPR3 was examined in liver biopsies of patients with hilar (n=3) and intrahepatic (n=4) CCAs, using histologically normal liver biopsies as a control. Histological diagnoses were established based on examination of hematoxylin-eosin (H&E) stained specimens (Supporting Figure S1). Liver sections from controls showed normal hepatic parenchyma with normal appearing portal tracts. Liver sections from hilar cholangiocarcinomas showed atypical glandular structures with irregular profiles and marked cytologic atypia, and many glandular structures with round to irregular profiles, marked cytologic atypia and associated stromal desmoplastic response were seen in the intrahepatic cholangiocarcinomas (Supporting Figure S1). Immunohistochemistry of liver sections from both types of CCA specimens and histologically normal controls showed that ITPR3 expression was detected only in bile ducts (Figure 1A). In liver biopsies obtained from controls, quantitative confocal immunofluorescence showed that ITPR3 expression in cholangiocytes was low and was concentrated in the apical region, as has been described previously (12, 19), whereas labeling of ITPR3 in cholangiocytes was significantly more intense in patients with hilar or intrahepatic CCA (Figure 1A–C). ITPR3 protein expression also was compared between two normal cholangiocyte cell lines (H69 and NHC cells) and different types of biliary adenocarcinoma cell lines (MzCha1, HuCCA1, HuCCT1, and TFK-1 cells). Consistent with the histological and quantitative immunofluorescence findings in CCA patients, ITPR3 expression was higher in MzCha1 and HuCCA1 cells than in cells derived from normal cholangiocytes (Figures 1D and 1E), although ITPR3 expression was more variable in HuCCT1 and TFK-1 cells (Supporting Figure S2). Because MzCha1 and HuCCA1 cells more closely reflect the over-expression of ITPR3 observed in actual human CCA biopsy specimens, these two cell lines were used for functional experiments in this study.
Figure 1. ITPR3 staining is increased in cholangiocytes of patients with hilar and intrahepatic cholangiocarcinomas and in cholangiocarcinoma cell lines.
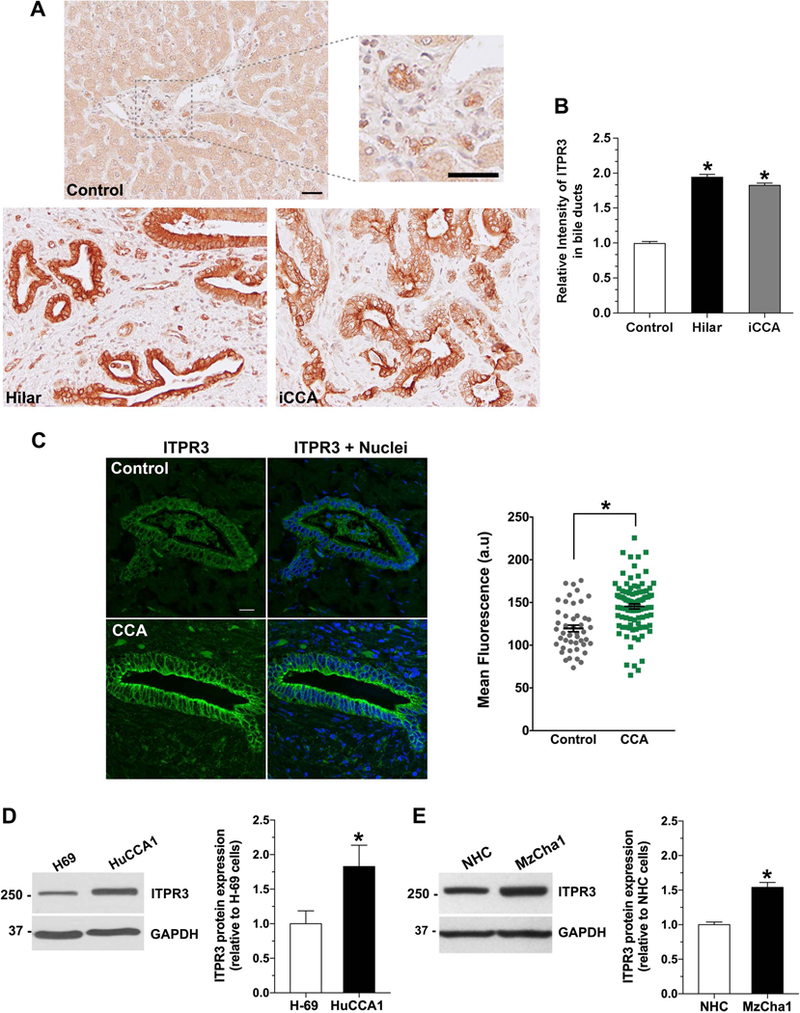
(A) Representative immunohistochemical (IHC) staining of ITPR3 in liver biopsy specimens from controls and patients with hilar cholangiocarcinoma and intrahepatic cholangiocarcinoma (iCCA) shows that ITPR3 staining is more intense in liver samples of CCA patients. Scale bars: 50 μm. (B) Quantitative analysis of the intensity of ITPR3 staining in the IHC images. Images are representative of what was observed in 3–7 patients, using 4–5 images per patient, *P < 0.0001. (C) Representative confocal immunofluorescence images of ITPR3 (left) and quantitative analysis of the intensity of ITPR3 staining in the images (right) of liver biopsy specimens from controls and CCA patients. ITPR3 expression (green) is detected only in bile ducts. Nuclei were labeled with a nuclear marker (blue). Labelling of ITPR3 in cholangiocytes in CCA patients (n=97 ducts from 7 patients) is greater than in controls (n=47 ducts from 7 patients; *P < 0.0001). Representative immunoblotting (top panel) and relative protein expression (bottom panel) of ITPR3 (D) in H69 and HuCCA1 cells and (E) in NHC and MzCha1 cells show that ITPR3 is increased in cholangiocarcinoma cell lines. *P < 0.05 (H69 versus HuCCA1) and *P < 0.001 (NHC versus MzCha1) (n=6).
ITPR3 contributes to cell proliferation and migration in cholangiocarcinoma cells.
Cell proliferation and migration are recognized characteristics of cancer cell survival (20). Calcium (Ca2+) signaling regulates both proliferation (4) and migration (6), including in cancer cells (21). To examine the role of ITPR3 in proliferation and migration of CCA cells, the CRISPR/Cas9 system was used to knockout ITPR3 in both MzCha1 and HuCCA1 cells (Figure 2A and Supporting Figure S3A). ITPR3 normally constitutes ~90% of the total ITPR pool and ITPR1 and ITPR2 together account for the remaining 10% (12). MzCha1 and HuCCA1 cells also predominantly express ITPR3 (Supporting Figure S4A and S4B), so we examined whether ITPR1 or ITPR2 expression changed to compensate for the loss of ITPR3 in knockout (KO) cells. ITPR1 expression was slightly increased in ITPR3-KO-MzCha1 cells but was slightly decreased in ITPR3-HuCCA1-KO cells, while ITPR2 was unchanged in either ITPR3-KO cell line (Supporting Figure S4C – S4F). The increase in the ITPR1 pool in ITPR3-KO-MzCha1 cells was not enough to compensate for the overall decrease in ITPR’s resulting from loss of ITPR3.
Figure 2. Loss of ITPR3 impairs cell proliferation and induces S phase arrest in cholangiocarcinoma cell lines.
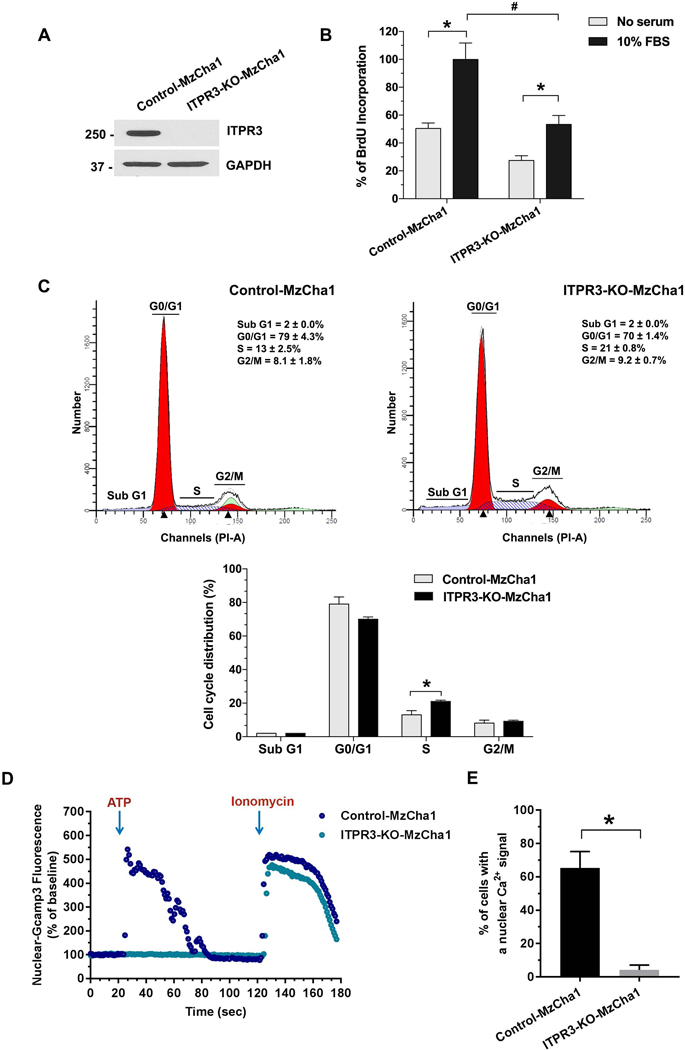
(A) Immunoblotting analysis of ITPR3 expression in control and ITPR3-KO-MzCha1 cells confirms loss of ITPR3 in the KO cells (n=4). (B) Cell proliferation is impaired by loss of ITPR3 in MzCha1 cells, as assessed by BrdU incorporation assay. The percentage of BrdU incorporation is enhanced by 10% fetal bovine serum (FBS) in both types of MzCha1 cells (*P < 0.001), but FBS-induced BrdU incorporation is half as great in ITPR3-KO-MzCha1 cells (#P < 0.0001, compared with control cells; n=4). (C) Representative flow cytometric histograms of propidium iodide staining (top panel) and quantitative analysis of cell cycle distribution (bottom panel) in control and ITPR3-KO MzCha1 cells shows that loss of ITPR3 arrests a significant fraction of cells in S phase. *P < 0.05, compared with control cells (n=3). (D) Ca2+ signaling in the nucleus of control and ITPR3-KO MzCha1 cells. Cells were transfected with the nuclear-localized Ca2+ indicator GCamp3 and then monitored by time lapse confocal microscopy while stimulated with ATP (20 μM). The amplitude of ATP-induced nuclear Ca2+ signals is much lower in ITPR3-KO-MzCha1 cells than in control cells, even though ionomycin-induced Ca2+ signals, reflecting the size of ER Ca2+ stores, are similar. Tracing is representative of what was observed in 9–10 coverslips, examining 4–12 cells per coverslip in three separate experiments. (E) The percentage of cells with a detectable nuclear Ca2+ signal in response to extracellular ATP is much greater in control than in ITPR3-KO-MzCha1 cells. *P < 0.0001, compared with control cells (4–12 cells per coverslip in 9–10 coverslips).
BrdU incorporation was significantly decreased in both ITPR3-KO-MzCha1 and ITPR3-KO-HuCCA1 cells, relative to their respective ITPR3 control cells (Figure 2B and Supporting Figure S3B), suggesting that ITPR3 is important for proliferation in CCA. Because ITPR-mediated Ca2+ signaling is also important in cell cycle progression (4, 22), flow cytometry was conducted to measure the effects of ITPR3 on the cell cycle. Loss of ITPR3 resulted in an increase in the percentage of cells in the S phase in both MzCha1 and HuCCA1 ITPR3-KO cells (Figure 2C and Supporting Figure S3C). Both cell proliferation and progression through the cell cycle depend on Ca2+ signals in the nucleus in particular (4), so the nuclear-targeted fluorescent calcium sensor GCaMP3 was used to specifically determine the role of ITPR3 in Ca2+ signaling in the nucleus. MzCha1 cells were stimulated with extracellular ATP (20 μM) because cholangiocytes express P2Y receptors that link to inositol trisphosphate (IP3)-mediated Ca2+ signaling (12, 19). ATP consistently increased Ca2+ in the nucleoplasm of control cells, but nuclear Ca2+ signals were minimal to absent in ITPR3-KO cells (Figure 2D and 2E). Subsequent stimulation with the calcium ionophore ionomycin (5 μM) elicited calcium signals with similar amplitudes in both types of cells (Figure 2D), suggesting that ER calcium stores were not diminished by lack of ITPR3. Collectively, these findings provide evidence that ITPR3 promotes proliferation in CCA by stimulating progression through the cell cycle and by enhancing calcium signals in the nucleus.
To examine the role of ITPR3 in cell motility, wound healing scratch assays and a transwell chamber migration analysis were performed. MzCha1 and HuCCA1 control cells each progressively filled the wound after 24 and then 48 hours, while approximately 70% of the wound remained unfilled after 48 hours in ITPR3-KO-MzCha1 cells (Figure 3) and >90% remained unfilled after 48 hours in ITPR3-KO-HuCCA1 cells (Supporting Figure S5A). Similarly, migration of ITPR3-KO-MzCha1 and ITPR3-KO-HuCCA1 cells each was markedly reduced compared to their respective control cells (Figure 3B and Supporting Figure S5B). These results suggest that ITPR3 participates not only in cell proliferation but also in cell motility in CCA.
Figure 3. Loss of ITPR3 impairs cell migration in cholangiocarcinoma cells.

(A) Representative images (top panel) and quantitative analysis of in vitro wound area coverage of control and ITPR3-KO MzCha1 cells (bottom panel). Photomicrographs were obtained at the indicated time points using a 10X objective. Loss of ITPR3 decreases cell migration in MzCha1 cells at 24 and 48 hours after wound scratching. Scale bar: 100 μm. *P < 0.01, compared with control cells (n=3). (B) Representative images of cell migration in the transwell migration assay (left panel) and quantitative analysis of number of cell migration in control MzCha1 and ITPR3-KO-MzCha1 cells (right panel). Loss of ITPR3 significantly decreases the number of migrating MzCha1 cells. Scale bar: 200 μm. *P < 0.001, compared with control cells (n=3).
Several mechanisms have been reported for downregulation of ITPR3 in cholangiocytes (19, 23), but factors that could increase ITPR3 are less clear. Increasing cholangiocyte proliferation has been attributed in part to activation of the sphingosine-1-phosphate receptor 2 (S1PR2) (24), so we investigated whether S1PR2 affects ITPR3 expression. The S1PR2 agonist taurocholate did not increase ITPR3, while the S1PR2 inhibitor JTE-013 did not decrease ITPR3 in MzCha1 cells (Supporting Figure S6A and 6B), suggesting that S1PR2 activation does not stimulate ITPR3 expression in CCA. Calcium signals may be increased not only as a result of increased ITPR expression but also because of increased channel activity, and O-linked glycosylation enhances calcium release from ITPR3 (25). Therefore, ITPR3 was immunoprecipitated from MzCha1, HuCCT1, and NHC cells, and the immunoprecipitated was probed for the O-GlcNAc modification. There was no significant difference in the fraction of ITPR3 that was O-glycosylated between NHC cells and either of the two CCA cell lines, suggesting that modulation of this modification does not contribute to enhanced calcium signaling in CCA (Supporting Figure S7).
Bioinformatic analysis of the role of ITPR3 in cholangiocarcinoma.
Analysis of the genetic landscape of CCA suggests a molecular classification system can be used to place most tumors into one of three subtypes (26). To understand whether and how this information can be used to understand the role of ITPR3, CRISPR/Cas9 was used to interrupt the ITPR3 gene in NHC cells and a microarray analysis of mRNA derived from NHC cells with and without an intact ITPR3 gene was performed using Affymetrix High Throughput Transcriptomics Array (HTA). Genes that were differentially expressed between wild-type and ITPR3-KO NHC cells were then compared with the RNA-seq expression data from CCA (26). Bioinformatic analysis identified six genes from the TCGA cholangiocarcinoma dataset that were significantly affected by loss of ITPR3 in NHC cells (Supporting Table 1). The microarray results for these genes were then subjected to verification by TaqMan RT-PCR, which resulted in identification of three genes. This included a significant upregulation of occludin (OCLN) and adaptor-related protein complex 1 sigma 3 (AP1S3), and downregulation of SH3-domain binding protein 5 (BTK-associated) (SH3BP5) (Supporting Figure S8A). These particular changes correspond to the IDH-mutant-enriched cluster of CCA cases (26), suggesting that ITPR3 affects CCA through a mitochondrial phenotype. To investigate the signaling pathways that link OCLN, AP1S3, and SH3BP5 to cell cycle progression, cellular migration and energy metabolism, a co-expression network analysis was performed using Exploratory Gene Association Networks (EGAN) software (Supporting Figure S8B). This analysis revealed that OCLN is co-expressed and interacts with genes related to tight junction biology, leukocyte trans-endothelial migration and cell adhesion, which suggests that the observed downregulation of OCLN in ITPR3-KO cells alters migratory properties of cholangiocarcinoma cells. SH3BP5 was found to have a significant interaction with the protein-coding gene of unknown function KIAA0040, so the functional relevance of SH3BP5 upregulation in cholangiocytes lacking ITPR3 is less clear. However, SH3BP5 is a known negative regulator of Bruton’s tyrosine kinase (BTK), which promotes development of malignancy by enhancing ITPR-mediated calcium release (27), and is a substrate of JNK (28), which suggests that this gene might work by modulating tyrosine kinase signaling in transformed cholangiocytes. No significant interactions were uncovered for AP1S3 by EGAN analysis.
ITPR3 accumulates in ER-mitochondrial junctions and affects mitochondrial calcium in MzCha1 cells.
ITPR3 normally is most concentrated in the sub-apical ER in cholangiocytes, where it regulates biliary bicarbonate secretion (12, 29). However, immunohistochemistry (Figure 1) suggests that the excess ITPR3 in CCA becomes localized in other parts of the cell as well. ITPRs that are localized to ER-mitochondrial junctions, or mitochondria-associated ER membranes (MAMs), could be of particular significance because such ITPRs have been linked to mitochondrial calcium signaling, metabolism, and regulation of various types of cell death (7, 9, 30). There is also evidence that ITPR3 may have a greater affinity than other ITPR isoforms for MAMs (7). Therefore, the possibility that some of the ITPR3 in CCA associate with mitochondria was investigated using three different approaches. First, super-resolution fluorescence microscopy was used to examine the association between a GFP-tagged ITPR3 and a red fluorescent protein (KO2-N1)-tagged mitochondria protein, TOMM20. These labeled proteins were transfected in MzCha1 cells, which then were examined with ~150 nm resolution (Figure 4A). Quantitative analysis of 19 cells showed that nearly all of the mitochondria were associated with ER containing GFP-ITPR3 (Mander’s coefficient, 0.91), while half of the ER co-localized with mitochondria (Mander’s coefficient, 0.53) (Figure 4B). Second, the subcellular distribution of native ITPR3 in MzCha1 cells also was examined by immunogold electron microscopy (Figure 4C). A frequency histogram showed that a higher fraction of ITPR3 are within 50 nm of mitochondria than any other sub-fraction (Figure 4D), representing 25.4% of total ITPR3. It has previously been established that ITPRs in such proximity to mitochondria are those within MAMs and are responsible for mitochondrial calcium signaling (31). Third, to determine the functional significance of ITPR3 in proximity to mitochondria, mitochondrial calcium signaling was measured in control, ITPR3-KO, and ITPR3-overexpressing MzCha1 cells. Cells were transfected with the mitochondrial-targeted, calcium-sensitive fluorescent protein inverse-mito-pericam, and overexpressing cells were co-transfected with an mCherry-tagged ITPR3 construct. All cells were loaded with the chemically ‘caged’ IP3 compound ci-IP3, then IP3 was photo-released by a controlled flash of UV light as the induced mitochondrial calcium signal was monitored by swept field confocal microscopy (Figure 5A). Photo-release of IP3 resulted in a 30% change in fluorescence in the control cells (Figure 5B), indicative of an IP3-induced mitochondrial calcium signal. In contrast, IP3 induced a significantly smaller increase in mitochondrial calcium in ITPR3-KO cells, which was not different from the nonspecific change in fluorescence seen in control cells subjected to the UV uncaging regimen that were not loaded with caged IP3 (Figure 5B). Conversely, IP3 induced a significantly larger increase in mitochondrial Ca2+ relative to controls in ITPR3-overexpressing MzCha1 cells (Supporting Figure S9A and S9B), and overexpressing cells also exhibited a more rapid increase in mitochondria Ca2+ (Supporting Figure S9B). These findings demonstrate that the level of ITPR3 expression directly relates to the speed and magnitude of mitochondrial Ca2+ signals in these cells. Mitochondria Ca2+ stimulates aerobic metabolism, so the effects of ITPR3 on mitochondrial metabolic state also were examined. There was no difference in mitochondrial membrane potential between control and ITPR3-KO-MzCha1 cells, as measured by the fluorescent indicator dye JC-1 (Supporting Figure S10A). However, there was a small but significant increase in basal respiratory rate in control cells relative to ITPR3-KO-MzCha1 cells (Supporting Figure S10B), which likely is driven by the increased mitochondrial calcium transmitted by ITPR3. Collectively, these findings provide both structural and functional evidence that a sub-fraction of ITPR3 in MzCha1 cells is in close proximity to mitochondria and is responsible for mitochondrial calcium signaling.
Figure 4. ITPR3 is in close proximity to mitochondria in MzCha1 cells.
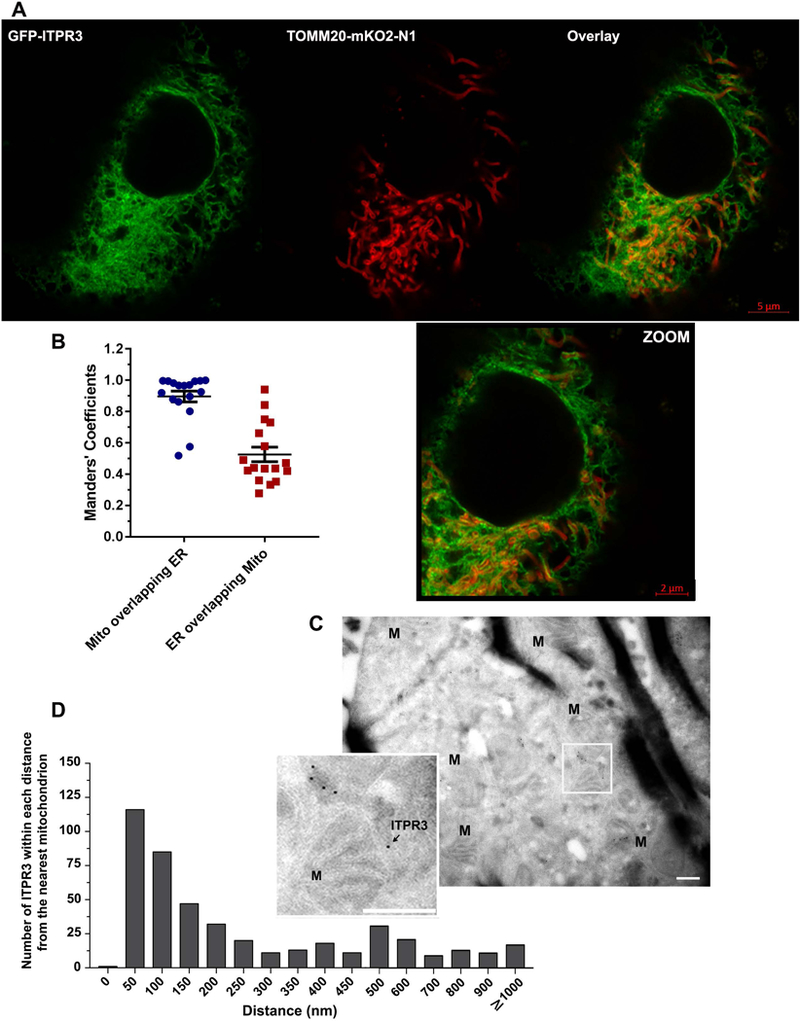
(A) Representative super-resolution image of GFP-ITPR3, which localizes to ER, and TOMM20-mKO2-N1, which localizes to the outer mitochondrial membrane, in a live MzCha1 cell. Green indicates GFP, red indicates mitochondrial-targeted KO2-N1, and yellow indicates colocalization of TOMM20-mKO2-N1 and GFP-ITPR3 labeling. Scale bars: 5 μm (top) and 2 μm (in the lower, zoomed image). (B) Manders’ overlap coefficient demonstrates the degree of colocalization of mitochondria (TOMM20-mKO2-N1) and ER (ITPR3) labeling. Most of the mitochondrial label overlaps with ER, and half of ER labeling overlaps with mitochondria. Values reflect individual data points plus mean ± SEM from n=19 images. (C) Representative immunogold electron micrograph of MzCha1 cells probed with ITPR3 antibody-linked gold particles. The zoomed region (inset) illustrates an example of ITPR3 in close proximity to a mitochondrion. Arrows indicate ITPR3-linked gold particles. Scale bars: 600 nm. Electron micrographs were obtained from 42 EM samples. (D) Frequency histogram of ITPR3 at each distance from their nearest mitochondria in MzCha1 cells. Distances ranging from 0 to ≥ 1000 nm (n=456 counted cells).
Figure 5. Loss of ITPR3 decreases mitochondrial Ca2+ signaling in MzCha1 cells.
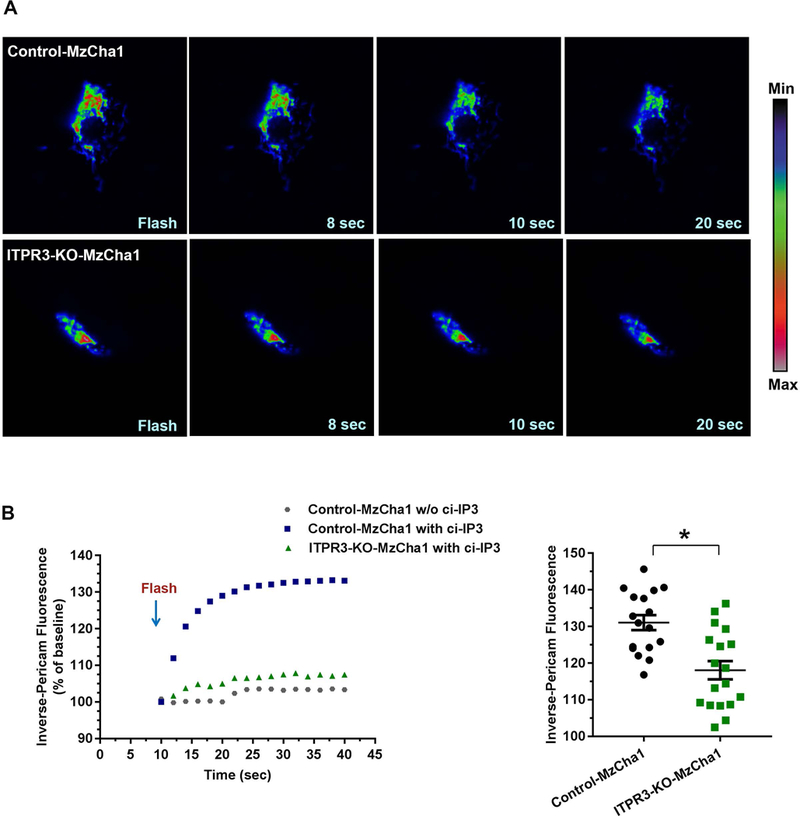
(A) Serial confocal images of mitochondrial Ca2+ signals in representative control and ITPR3-KO MzCha1 cells expressing inverse-mito-pericam. Caged IP3 was photo-released by a controlled near-UV (405 nm) flash as the induced mitochondrial Ca2+ signal was monitored by swept field confocal microscopy. Pseudo-color Ca2+ scale is shown on the right. (B) Quantitation of fluorescence shows that IP3-induced Ca2+ signals are decreased in ITPR3-KO cells. Left panel: representative tracings from single cells show an IP3-induced increase in mitochondrial Ca2+ in a control cell, with a much smaller increase in an ITPR3-KO cell, which is similar in magnitude to the small, non-specific increase seem in a control cell subjected to the same near-UV flash but not loaded with caged IP3. Right panel: summary from all experiments shows that IP3-induced mitochondrial Ca2+ signal is significantly greater in control cells than in ITPR3-KO cells. Values reflect individual data points plus mean ± SEM from 16–18 cells in 9–10 coverslips from each condition (*P < 0.0001).
Although our findings indicate that ITPR3 affects calcium signals in the nucleus (Figure 2D and 2E) and mitochondria (Figure 5) of MzCha1 cells, signaling in these microdomains may not always reflect what is occurring in the cytosol (31, 32). Cytosolic calcium signals were detected in the majority of MzCha1 cells stimulated with ATP, but this was blocked in nearly all cells pre-treated with the ITPR inhibitor Xestospongin C (P < 0.05; Supporting Figure S11A), confirming that ATP-induced calcium signals are mediated by IP3/ITPR. MzCha1 control cells were slightly more sensitive than ITPR3-KO cells to ATP, but there was no difference in the amplitude of calcium signals among responding cells (Supporting Figure S11B). There also was no difference in expression levels of other calcium-dependent proteins (NFATc1–4, calcineurins A-C, SERCA2, and calmodulin 1) between control and ITPR3-KO cells (Supporting Figure S11C). These findings are consistent with the importance of ITPR3’s effects on calcium signals in the nucleus and mitochondria even though effects on cytosolic calcium are minimal.
ITPR3 is required for cholangiocarcinoma cell survival.
Cancer cells become metabolically ‘addicted’ to calcium transferred from ER to mitochondria, with enhanced susceptibility to cell death when this is interrupted (9). Because our findings suggest that increased expression of ITPR3 leads to a mitochondrial phenotype that includes increased mitochondrial calcium signaling in CCA, the dependence of CCA cells on ITPR3 was examined. Flow cytometry with fluorescein isothiocyanate (FITC)-Annexin V and propidium iodide (PI) labeling was used to compare viability in ITPR3-KO and control cells (Figure 6). A significant fraction of ITPR3-KO-MzCha1 cells and ITPR3-KO-HuCCA1 cells were necrotic, whereas essentially no control cells were necrotic (Figure 6). The percentage of healthy cells was significantly decreased in ITPR3-KO-MzCha1 cells compared to controls (86.0 ± 2.3% in control MzCha1 cells versus 25.0 ± 1.3% in ITPR3-KO-MzCha1 cells, P < 0.0001). The decrease in healthy cells was largely due to necrosis (0.3 ± 0.01% in control MzCha1 cells versus 60.0 ± 0.9% in ITPR3-KO-MzCha1 cells, P < 0.0001) (Figures 6A and 6B). Similarly, loss of ITPR3 decreased the percentage of healthy HuCCA1 cells from 92.0 ± 1.0% in controls to 68.0 ± 1.6% in ITPR3-KO cells (P < 0.0001). In these cells, deletion of ITPR3 increased the percentage in both late apoptosis (6.1 ± 0.5% in control HuCCA1 cells versus 9.9 ± 1.4% in ITPR3-KO-HuCCA1, P < 0.05) and necrosis (0.6 ± 0.1% in control HuCCA1 versus 19.0 ± 1.1% in ITPR3-KO-HuCCA1, P < 0.0001) (Figures 6C and 6D). These changes in viability also were evaluated in an in vitro microtumor model. Three-dimensional (3D) spheroids of MzCha1 or HuCCA1 cells with or without ITPR3 were generated, and these spheroids were examined over a 9-day period by live/dead cell staining with calcein acetoxymethyl ester (calcein-AM) to label live cells and ethidium homodimer-1 (EthD-1) to label dead cells (Figure 7 and Supporting Figure S12). MzCha1 and HuCCA1 cells each formed 3D spheroids within 24 hours, and the size of the spheroids increased in a time-dependent manner. ITPR3-KO-MzCha1 and ITPR3-KO-HuCCA1 spheroids were similar in size compared to their respective controls, but the morphology and cell content were different. MzCha1 and HuCCA1 control spheroids exhibited intense live-cell calcein-AM staining throughout their interiors with a small amount of EthD-1 staining in the periphery. However, the ITPR3-KO-MzCha1 and ITPR3-KO-HuCCA1 spheroids appeared looser and less cohesive, with a decrease in the intensity of the calcein-AM signal and a progressive increase in EthD-1 signal in the central core, and this pattern progressed over time. These results indicate that control-spheroids remain viable and suggest that the capability of migration and repopulation are retained, whereas more necrotic and dead cells are found in the ITPR3-KO spheroids (Figure 7 and Supporting Figure S12). ITPR activity has also been related to autophagy and senescence, so these were examined in control and ITPR3-KO-MzCha1 cells. The autophagy marker LC3B-II was decreased in both ITPR3-KO-MzCha1 and ITPR3-KO-HuCCA1 cells relative to their controls, and LC3B-I also was decreased in ITPR3-KO-HuCCA1 cells, although Beclin 1 was not altered in either type of ITPR3-KO cell (Supporting Figure S13). In addition, senescence-associated beta-galactosidase (SA-β-Gal) activity was decreased in ITPR3-KO-MzCha1 cells (Supporting Figure S14), suggesting senescence is repressed in these cells. Finally, over-expression of ITPR3 led to cell death in ~75% of transiently transfected cells (Supporting Figure S15). This is consistent with the idea that not all cells can metabolically adapt to the increased mitochondrial calcium load that occurs when ITPR3 becomes over-expressed, but the cells that are able to adapt become dependent on this increased calcium load. Collectively, these findings provide evidence that CCA cells, like other types of cancer cells, become ‘addicted’ to enhanced calcium signaling and that this is mediated by increased ER-to-mitochondrial transfer of calcium via ITPR3. Loss of ITPR3 then leads to cell death largely due to necrosis, with lesser contributions from autophagy, senescence and apoptosis.
Figure 6. Loss of ITPR3 causes necrosis of cholangiocarcinoma cells.
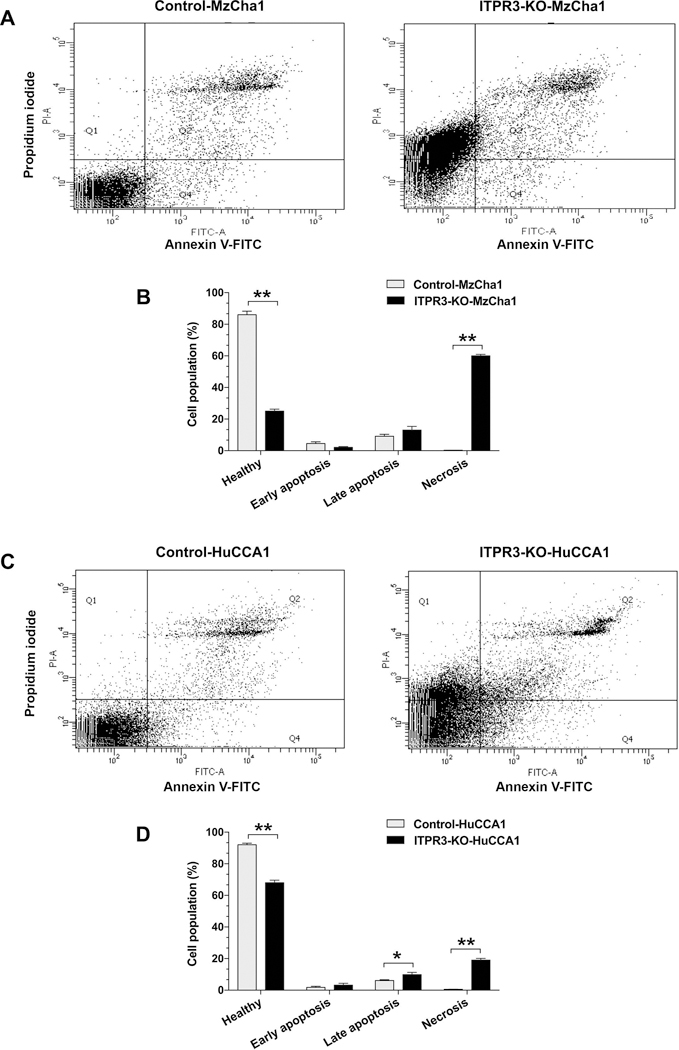
Cell viability and cell death were detected by dual labeling for annexin V-FITC and propidium iodide. (A) Flow cytometry analysis of labeling for annexin V-FITC and propidium iodide (PI) in control and ITPR3-KO MzCha1 cells. (B) Quantitation of the distribution of control and ITPR3-KO MzCha1 cells that are healthy, apoptotic or necrotic. The percentage of healthy cells was significantly lower, while the percentage that were necrotic was significantly higher, in ITPR3-KO-MzCha1 cells relative to controls (**P < 0.0001; n=3). (C) Flow cytometry analysis of labeling for annexin V-FITC and propidium iodide (PI) in control and ITPR3-KO HuCCA1 cells. (D) Quantitation of the distribution of control and ITPR3-KO HuCCA1 cells that are healthy, apoptotic or necrotic. The percentage of healthy cells was significantly lower, while the percentage that were in late apoptosis or necrotic was significantly higher, in ITPR3-KO-HuCCA1 cells relative to controls (*P < 0.05 and **P < 0.0001, compared with control cells; n=3).
Figure 7. Loss of ITPR3 results in increased death of MzCha1 cells in a 3D-spheroid model.
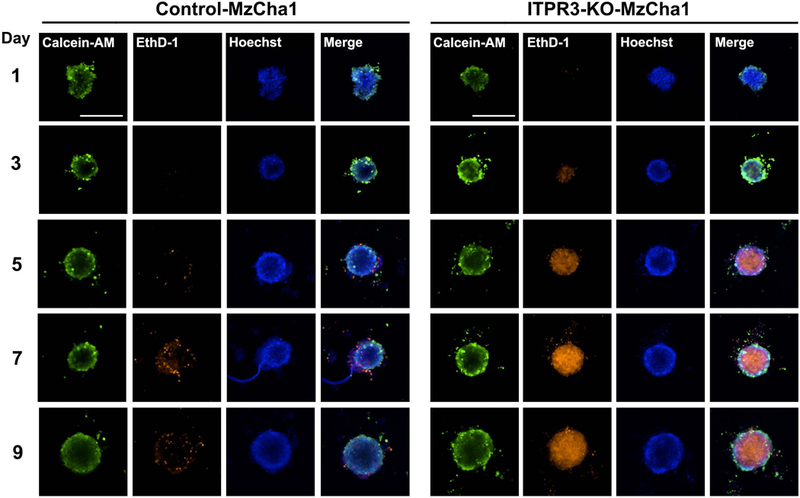
Representative fluorescence images of spheroids cultured from control (left panel) and ITPR3-KO (right panel) MzCha1 cells. Spheroids were co-labeled with calcein acetoxymethyl ester (calcein-AM) and ethidium homodimer-1 (EthD-1) to detect changes in growth and viability of the spheroids over 9 days. Composite images of representative spheroids stained with calcein-AM (green) for live cells, EthD-1 (orange) for dead cells, and Hoechst (blue) for nuclei, are shown. Scale bar: 500 μm. (n= 3 spheroids from 6 separate preparations for each time point and for each cell type, for a total of 180 spheroids that were examined).
DISCUSSION
The current work provides evidence that ITPR3, which is the principal intracellular calcium release channel in cholangiocytes, becomes over-expressed in both intrahepatic and hilar CCA. It is furthermore shown that this contributes to features that characterize the cancer biology of CCA, including enhanced cell proliferation and migration (6), plus the mitochondrial ‘addiction’ to calcium that can occur in malignancy (9). ITPR3 plays an important role in mediating cholangiocyte bicarbonate secretion (29, 33), and several factors have been identified that can decrease ITPR3 expression in cholangiocytes. These include the transcription factors Nrf2 (23) and NF-kB (19) and the micro-RNA miR506 (34). However, each of these inhibits rather than increases expression of ITPR3, which in turn contributes to various cholangiopathies and cholestasis. Factors that may instead increase ITPR3 expression are not yet known, but several possibilities are plausible. First, it is possible that certain transcription factors may increase rather than decrease ITPR3. For example, cAMP response element binding protein (CREB), via cAMP, increases transcription and thus expression of ITPR2 in hepatocytes (35), although no transcription factors that stimulate ITPR3 expression have been identified yet. Similarly, the oncoprotein MYC binds to each of the three ITPR isoforms (36) and this has been related to enhanced calcium-dependent activity of NUAK1, which promotes a malignant phenotype (37), but this has not yet been directly related to ITPR3 expression or development of CCA. Second, epigenetic factors such as demethylation play a well-known role in expression of a variety of oncogenes (38), but it is not yet known if this contributes to increased expression of ITPR3. Third, the effects of ITPR3 on mitochondrial Ca2+ in CCA likely depend on its aberrant localization to the MAM, and several factors have recently been identified that can contribute to this. Polycystin 2, which is expressed in cholangiocytes (39), modulates ITPR3 activity in the MAM (40), while IRE1α helps ITPR3 to localize there (41). Finally, ITPR3 is degraded in part by ubiquitination (42), so modulation of this pathway could also contribute to altered cellular ITPR3 expression. Indeed, abnormal expression of tumor suppressors that modify this degradation pathway, including BAP1 and PTEN, has been linked to altered ITPR3 and resulting tumor development in several types of malignancies (17, 18). Alternatively, the anti-apoptotic protein Bcl-2 promotes cancer by binding to ITPR to inhibit calcium release into mitochondria (27), although Mcl-1, which also inhibits mitochondrial calcium (43), is thought to be a more important anti-apoptotic protein in CCA. In each of these cases, however, a decrease rather than an increase in ITPR3 or its activity is thought to contribute to neoplasia, because loss of ITPR3 from MAMs can sometimes decrease apoptosis of cancer cells. The current findings suggest that CCA, like other digestive malignancies such as gastric (14) and colon (16) cancer, instead is enhanced by increased ITPR3 expression. This general effect has been explained by the observation that certain types of cancer cells become metabolically dependent on increased transmission of calcium into mitochondria (9). The current findings that ITPR3 localizes to MAMs and is responsible for mitochondrial calcium signals, and loss of ITPR3 results in necrotic cell death, suggests that this mechanism may occur in CCA as well.
The subcellular location of ITPRs plays an important role in determining their physiological effects. For example, ITPR2 is the principle isoform in hepatocytes and is concentrated in their sub-apical region, where it regulates secretion by targeting and activity of MRP2 (44) and BSEP (45). Although loss of ITPR2 contributes to canalicular cholestasis, mis-localization of ITPR2 can contribute as well, and occurs in clinical conditions such as estrogen- or endotoxin-induced cholestasis (45), or in situations in which lipid rafts in the apical membrane are disrupted (46). Similarly, the sub-apical localization of ITPR3 in cholangiocytes may be important for ductular bicarbonate secretion, because this ITPR pool is tightly linked to activation of the calcium-dependent apical chloride channel TMEM16a, which then links to chloride-bicarbonate exchange via AE2 (29). The current work provides evidence that ITPR3 in CCA also contributes to calcium signaling in two additional subcellular compartments, mitochondria and the nucleus, at least in part because the excess ITPR3 accumulates in these regions. Mitochondrial calcium signals have a variety of effects (31, 47), although cell metabolism (48) and ATP formation in particular (49) may be the most important ones in CCA. Calcium signals in the nucleus also have a variety of effects, including regulation of gene transcription (3), progression through the cell cycle (4), and cell proliferation (4, 5), and each of these may play a role in development of CCA. Calcium signals in the cell nucleus can be selectively induced by a variety of growth factors, whose cognate receptor tyrosine kinases (RTKs) can translocate to the nucleus to directly activate PLC and form IP3 there (5). This pathway is important in hepatocytes, where it participates in liver regeneration (5) as well as development of hepatocellular carcinoma (50), and the current work provides evidence that this pathway may be important in CCA as well. Further work will be required to understand the factors that draw ITPR3 to the sub-apical region in normal cholangiocytes and that allow its pathological accumulation elsewhere in CCA, as well as to determine whether these factors can be exploited for therapeutic benefit.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr. Xinran Liu and Kimberly Zichichi (Center for Cellular and Molecular Imaging Electron Microscopy Core Facility, Yale University) for their advice and assistance with immunogold electron microscopy. We thank Dr. Suparerk Borwornpinyo, Phichaya Suthivanich, and Kedchin Jearawuttanakul (Excellent Center for Drug Discovery, Mahidol University) for technical assistance, and Dr. Shuangge Steven Ma (Department of Biostatistics, Yale School of Public Health). We also thank Dr. David Yule (University of Rochester Medical center) for providing the mCherry-ITPR3 full-length plasmid, and the Yale Diabetes Research Center (DRC) IOMIC Flux Core for assistance with oxygen consumption rate measurements.
Financial Support: This study was funded by the Gladys Phillips Crofoot Professorship, Arnie Charbonneau Cancer Research Postdoctoral Fellowship, the National Institutes of Health (P01-DK57751, P30-DK34989, R56-DK99470, R01-DK114041, R01-DK112797, S10-OD023598 and P30-CA15083).
List of Abbreviations:
- CCA
cholangiocarcinoma
- IP3
inositol trisphosphate
- ITPR3
type 3 inositol trisphosphate receptor
- Ca2+
calcium ion
- iCCA
intrahepatic CCA
- NHC
normal human cholangiocytes
- ER
endoplasmic reticulum
- MAM
mitochondria-associated ER membranes
- Mcl-1
myeloid cell leukemia-1
Footnotes
Author names in bold designate shared co-first authorship.
REFERENCES
- 1.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013;145:1215–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet 2014;383:2168–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pusl T, Wu JJ, Zimmerman TL, Zhang L, Ehrlich BE, Berchtold MW, Hoek JB, et al. Epidermal growth factor-mediated activation of the ETS domain transcription factor Elk-1 requires nuclear calcium. J Biol Chem 2002;277:27517–27527. [DOI] [PubMed] [Google Scholar]
- 4.Rodrigues MA, Gomes DA, Leite MF, Grant W, Zhang L, Lam W, Cheng YC, et al. Nucleoplasmic calcium is required for cell proliferation. J Biol Chem 2007;282:17061–17068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amaya MJ, Oliveira AG, Guimaraes ES, Casteluber MC, Carvalho SM, Andrade LM, Pinto MC, et al. The insulin receptor translocates to the nucleus to regulate cell proliferation in liver. Hepatology 2014;59:274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature 2009;457:901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, et al. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. J Biol Chem 2005;280:40892–40900. [DOI] [PubMed] [Google Scholar]
- 8.Guerra MT, Fonseca EA, Melo FM, Andrade VA, Aguiar CJ, Andrade LM, Pinheiro AC, et al. Mitochondrial calcium regulates rat liver regeneration through the modulation of apoptosis. Hepatology 2011;54:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep 2016;15:219–220. [DOI] [PubMed] [Google Scholar]
- 10.Hirata K, Nathanson MH, Sears ML. Novel paracrine signaling mechanism in the ocular ciliary epithelium. Proc Natl Acad Sci U S A 1998;95:8381–8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amaya MJ, Nathanson MH. Calcium signaling in the liver. Compr Physiol 2013;3:515–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirata K, Dufour JF, Shibao K, Knickelbein R, O’Neill AF, Bode HP, Cassio D, et al. Regulation of Ca(2+) signaling in rat bile duct epithelia by inositol 1,4,5-trisphosphate receptor isoforms. Hepatology 2002;36:284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hedberg ML, Goh G, Chiosea SI, Bauman JE, Freilino ML, Zeng Y, Wang L, et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J Clin Invest 2016;126:1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakakura C, Hagiwara A, Nakanishi M, Shimomura K, Takagi T, Yasuoka R, Fujita Y, et al. Differential gene expression profiles of gastric cancer cells established from primary tumour and malignant ascites. Br J Cancer 2002;87:1153–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang SS, Han KS, Ku BM, Lee YK, Hong J, Shin HY, Almonte AG, et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res 2010;70:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibao K, Fiedler MJ, Nagata J, Minagawa N, Hirata K, Nakayama Y, Iwakiri Y, et al. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010;48:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bononi A, Giorgi C, Patergnani S, Larson D, Verbruggen K, Tanji M, Pellegrini L, et al. BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 2017;546:549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuchay S, Giorgi C, Simoneschi D, Pagan J, Missiroli S, Saraf A, Florens L, et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca(2+)-mediated apoptosis limiting tumour growth. Nature 2017;546:554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franca A, Filho A, Guerra MT, Weerachayaphorn J, Santos MLD, Njei B, Robert M, et al. Effects of endotoxin on type 3 inositol 1,4,5-trisphosphate receptor in human cholangiocytes. Hepatology 2019;69:817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 21.Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer 2011;11:609–618. [DOI] [PubMed] [Google Scholar]
- 22.Ciapa B, Pesando D, Wilding M, Whitaker M. Cell-cycle calcium transients driven by cyclic changes in inositol trisphosphate levels. Nature 1994;368:875–878. [DOI] [PubMed] [Google Scholar]
- 23.Weerachayaphorn J, Amaya MJ, Spirli C, Chansela P, Mitchell-Richards KA, Ananthanarayanan M, Nathanson MH. Nuclear Factor, Erythroid 2-Like 2 Regulates Expression of Type 3 Inositol 1,4,5-Trisphosphate Receptor and Calcium Signaling in Cholangiocytes. Gastroenterology 2015;149:211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Aoki H, Yang J, Peng K, Liu R, Li X, Qiang X, et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017;65:2005–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bimboese P, Gibson CJ, Schmidt S, Xiang W, Ehrlich BE. Isoform-specific regulation of the inositol 1,4,5-trisphosphate receptor by O-linked glycosylation. J Biol Chem 2011;286:15688–15697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farshidfar F, Zheng S, Gingras MC, Newton Y, Shih J, Robertson AG, Hinoue T, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep 2017;19:2878–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Distelhorst CW. Targeting Bcl-2-IP3 receptor interaction to treat cancer: A novel approach inspired by nearly a century treating cancer with adrenal corticosteroid hormones. Biochim Biophys Acta Mol Cell Res 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wiltshire C, Matsushita M, Tsukada S, Gillespie DA, May GH. A new c-Jun N-terminal kinase (JNK)-interacting protein, Sab (SH3BP5), associates with mitochondria. Biochem J 2002;367:577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minagawa N, Nagata J, Shibao K, Masyuk AI, Gomes DA, Rodrigues MA, Lesage G, et al. Cyclic AMP regulates bicarbonate secretion in cholangiocytes through release of ATP into bile. Gastroenterology 2007;133:1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, Ferrand M, et al. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 2014;5:3792. [DOI] [PubMed] [Google Scholar]
- 31.Feriod CN, Oliveira AG, Guerra MT, Nguyen L, Richards KM, Jurczak MJ, Ruan HB, et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol Commun 2017;1:23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leite MF, Thrower EC, Echevarria W, Koulen P, Hirata K, Bennett AM, Ehrlich BE, et al. Nuclear and cytosolic calcium are regulated independently. Proc Natl Acad Sci U S A 2003;100:2975–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shibao K, Hirata K, Robert ME, Nathanson MH. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology 2003;125:1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ananthanarayanan M, Banales JM, Guerra MT, Spirli C, Munoz-Garrido P, Mitchell-Richards K, Tafur D, et al. Post-translational regulation of the type III inositol 1,4,5-trisphosphate receptor by miRNA-506. J Biol Chem 2015;290:184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kruglov E, Ananthanarayanan M, Sousa P, Weerachayaphorn J, Guerra MT, Nathanson MH. Type 2 inositol trisphosphate receptor gene expression in hepatocytes is regulated by cyclic AMP. Biochem Biophys Res Commun 2017;486:659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014;511:483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Monteverde T, Tait-Mulder J, Hedley A, Knight JR, Sansom OJ, Murphy DJ. Calcium signalling links MYC to NUAK1. Oncogene 2018;37:982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehrlich M DNA methylation in cancer: too much, but also too little. Oncogene 2002;21:5400–5413. [DOI] [PubMed] [Google Scholar]
- 39.Fabris L, Fiorotto R, Spirli C, Cadamuro M, Mariotti V, Perugorria MJ, Banales JM, et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuo IY, Brill AL, Lemos FO, Jiang JY, Falcone JL, Kimmerling EP, Cai Y, et al. Polycystin 2 regulates mitochondrial Ca(2+) signaling, bioenergetics, and dynamics through mitofusin 2. Sci Signal 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carreras-Sureda A, Jana F, Urra H, Durand S, Mortenson DE, Sagredo A, Bustos G, et al. Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol 2019;21:755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wojcikiewicz RJ, Ernst SA, Yule DI. Secretagogues cause ubiquitination and down-regulation of inositol 1, 4,5-trisphosphate receptors in rat pancreatic acinar cells. Gastroenterology 1999;116:1194–1201. [DOI] [PubMed] [Google Scholar]
- 43.Minagawa N, Kruglov EA, Dranoff JA, Robert ME, Gores GJ, Nathanson MH. The anti-apoptotic protein Mcl-1 inhibits mitochondrial Ca2+ signals. J Biol Chem 2005;280:33637–33644. [DOI] [PubMed] [Google Scholar]
- 44.Cruz LN, Guerra MT, Kruglov E, Mennone A, Garcia CR, Chen J, Nathanson MH. Regulation of multidrug resistance-associated protein 2 by calcium signaling in mouse liver. Hepatology 2010;52:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kruglov EA, Gautam S, Guerra MT, Nathanson MH. Type 2 inositol 1,4,5-trisphosphate receptor modulates bile salt export pump activity in rat hepatocytes. Hepatology 2011;54:1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagata J, Guerra MT, Shugrue CA, Gomes DA, Nagata N, Nathanson MH. Lipid rafts establish calcium waves in hepatocytes. Gastroenterology 2007;133:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol 2000;529:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol 2015;17:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010;142:270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011;54:879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.