

Significance Statement
The specialized vessels comprising the renal vasculature are characterized by highly differentiated renal endothelial cell types, but this heterogeneity has been poorly inventoried. Using single-cell RNA sequencing, the authors developed a high-resolution atlas of mouse renal endothelial cells. They also investigated how medullary renal endothelial cells adapt to a switch from diuresis to antidiuresis. This study describes the molecular and metabolic adaptation of medullary renal endothelial cells to dehydration, and uncovers a role for mitochondrial oxidative phosphorylation in hyperosmolarity conditions to allow for urine concentration. The authors’ atlas of mouse renal endothelial cells provides a resource for future studies, and their findings may provide insights into cardiometabolic or kidney diseases involving hyperosmolarity and dehydration, in which urine concentration capacity is perturbed.
Keywords: renal endothelial cells, scRNA-sequencing, heterogeneity, dehydration, urine concentration, oxidative phosphorylation
Visual Abstract

Abstract
Background
Renal endothelial cells from glomerular, cortical, and medullary kidney compartments are exposed to different microenvironmental conditions and support specific kidney processes. However, the heterogeneous phenotypes of these cells remain incompletely inventoried. Osmotic homeostasis is vitally important for regulating cell volume and function, and in mammals, osmotic equilibrium is regulated through the countercurrent system in the renal medulla, where water exchange through endothelium occurs against an osmotic pressure gradient. Dehydration exposes medullary renal endothelial cells to extreme hyperosmolarity, and how these cells adapt to and survive in this hypertonic milieu is unknown.
Methods
We inventoried renal endothelial cell heterogeneity by single-cell RNA sequencing >40,000 mouse renal endothelial cells, and studied transcriptome changes during osmotic adaptation upon water deprivation. We validated our findings by immunostaining and functionally by targeting oxidative phosphorylation in a hyperosmolarity model in vitro and in dehydrated mice in vivo.
Results
We identified 24 renal endothelial cell phenotypes (of which eight were novel), highlighting extensive heterogeneity of these cells between and within the cortex, glomeruli, and medulla. In response to dehydration and hypertonicity, medullary renal endothelial cells upregulated the expression of genes involved in the hypoxia response, glycolysis, and—surprisingly—oxidative phosphorylation. Endothelial cells increased oxygen consumption when exposed to hyperosmolarity, whereas blocking oxidative phosphorylation compromised endothelial cell viability during hyperosmotic stress and impaired urine concentration during dehydration.
Conclusions
This study provides a high-resolution atlas of the renal endothelium and highlights extensive renal endothelial cell phenotypic heterogeneity, as well as a previously unrecognized role of oxidative phosphorylation in the metabolic adaptation of medullary renal endothelial cells to water deprivation.
The kidney is vital for organismal homeostasis, and has a complex vasculature to achieve this task. Within the kidney, renal endothelial cells (RECs) differ according to their origin and vascular bed.1,2 For instance, renal endothelial cells within glomeruli (gRECs) are fenestrated (but without diaphragm) to facilitate water passage but also to restrict filtration of high-molecular-weight substances.3 By contrast, cortical renal endothelial cells (cRECs) in larger vessels (arteries, arterioles) and medullary renal endothelial cells (mRECs) in the descending vasa recta (DVR) have a continuous lining, and are surrounded by pericyte/smooth muscle cells, which can regulate GFR in the cortex and osmolarity gradient preservation in the medulla. In addition, the parallel arrangement of the DVR and the ascending vasa recta (AVR) in the medulla causes an oxygen shunt, resulting in hypoxia in the renal papilla.4,5 The molecular heterogeneity of RECs from the cortex, glomeruli, and medulla has been investigated only using microarrays on bulk gREC, cREC, and mREC populations.6 Single-cell RNA sequencing (scRNA-seq) studies that sequenced enriched, freshly isolated RECs to construct a comprehensive taxonomy of RECs at high resolution are not available.
The heterogeneity of RECs may be of particular relevance for the medullary circulation. For example, dehydration exposes medullary cells to extreme hyperosmolarity.7–9 How the renal medullary endothelium adapts to such conditions is unknown, but could be critical for the function of the countercurrent system and hence capacity to concentrate urine. Because the osmolarity gradient is maximal in the medullary papilla, we utilized scRNA-seq to dissect the distinct responses of mRECs, gRECs, and cRECs to dehydration, and used the metabolic gene expression signature as a guidance to functionally confirm novel metabolic changes. To augment the resource value of our REC database, we provide access to http://www.vibcancer.be/software-tools/rec-dehydration to explore all genes that are shown in t-distributed stochastic neighbor embedding (t-SNE) plots or are listed in gene expression heatmaps.
Methods
Animals
Animal experiments have been approved by the Institutional Animal Ethics Committee of Katholieke Universiteit Leuven (KU Leuven) (approved project no. P144/2018, “Renal endothelial cells in health and disease: from full characterization to therapeutic intervention,” animal experiments 10, 11, and 15). All animals were maintained in a room with controlled temperature and humidity under a 12-hour light/dark cycle. No statistical method was used to predetermine sample size. For the scRNA-seq experiment, 12-week-old male mice (C57Bl6/J strain; Charles River Laboratories) were exposed to food and water ad libitum, or were water-deprived, for 12, 24, 36, or 48 hours (n=6 per group). Rodents are physiologically equipped to tolerate acute dehydration for periods of as long as 24 hours without overt signs of physiologic distress or behavioral abnormalities.10 On the basis of previous recommendations, longer periods of acute water deprivation are acceptable procedures, but only with suitable monitoring protocols.10 Following these recommendations,10 mice were tightly monitored assessing body weight loss (twice a day), behavior (physical vigor, activity), and any sign of major distress or suffering (hunched posture, fuzzy facial fur, sunken/recessed eyes) (three times a day), up to 48 hours of water deprivation. Humane end points were defined according to these parameters. None of the dehydrated mice included in the experiments showed signs of major distress or suffering as well as behavioral abnormalities. Mice were anesthetized with an intraperitoneal injection of ketamine/xylazine (Dechra, Innovet) and blood was collected from the right ventricle. Mice were then submitted to left heart perfusion with 3 ml PBS (1 ml/min) and perfused kidneys were collected in ice-cold HBSS. All 60 kidneys were collected the same day to avoid batch effects.
REC Isolation
Kidney capsules were removed and kidneys were further dissected under a microscope to separate cortex from medulla. Kidney cortices and medullae from each condition were then cut into small pieces in a collagenase digestion buffer (2 mg/ml collagenase I [C9891; Sigma-Aldrich], 27 IU/ml DNAse I [D4527; Sigma-Aldrich], and HBSS [14025100; Thermo Fisher Scientific]); 15 ml were used for cortex samples and 5 ml for medulla samples. The samples were then incubated for 40 minutes at 37°C, shaking every 10 minutes, and digestion was stopped by adding 20 ml ice-cold HBSS. Digested cortex and medulla samples were sequentially filtered through 70 and 40 µm filters and glomeruli were collected from the 40 µm filter used to filter the cortex samples, and were resuspended in HBSS. RECs from cortex and medulla were enriched using mouse CD31 magnetic microbeads (130–097–418; Miltenyi Biotec) and QuadroMACS separators with LS columns (Miltenyi Biotec). Collected glomeruli were further dissociated in a trypsin digestion buffer (0.2% trypsin [25200056; Thermo Fisher Scientific], 100 IU/ml DNAse I, 100 µg/ml heparin [Heparine LEO; LeoPharma]) for 23 minutes at 37°C. Trypsin was inactivated by adding 10% FBS in ice-cold HBSS. Glomerular cells were centrifuged for 6 minutes at 400×g, and resuspended in wash buffer (0.1% BSA in PBS). Cortical and medullary endothelial-enriched cells as well as glomerular cells were incubated with CD102-Alexa647 antibody (1/300, A15452; Thermo Fisher Scientific), CD45-PeCy7 antibody (1/500, 130–097–418; Thermo Fisher Scientific), CD73-PerCP-Cy5.5 antibody (1/100, 127213; BioLegend, for glomerular cells only), and viability dye eFluor450 (1/1000, 65–0863–14; Thermo Fisher Scientific) in FACS buffer (0.1% BSA, 2 mM EDTA in PBS) for 30 minutes at 4°C. OneComp ebeads (01–1111–42; Thermo Fisher Scientific) and cooked cells (20 minutes at 60°C) were used for compensation. A total of 100,000 viable endothelial cells (ECs) were FACS-sorted using a FACS Aria III (BD Biosciences), and collected in wash buffer (0.1% BSA in PBS). Cells were centrifuged for 6 minutes at 400×g, and resuspended in 40 µl wash buffer. Cell viability and number were assessed by staining cells with acridine orange and propidium iodide (F23001; Logos Biosystems) and using the automatic dual-fluorescence cell counter system (Luna-Fl; Logos Biosystems).
Single-Cell Droplet-Based RNA Sequencing and Data Preprocessing
The single-cell suspensions of FACS-sorted gRECs, cRECs, and mRECs were converted to barcoded scRNA-seq libraries using the Chromium Single Cell 3′ Library, Gel Bead & Multiplex Kit and Chip Kit (10× Genomics; PN-120237, PN-120236), aiming for approximately 4000 cells per library of control gRECs, cRECs, and mRECs, and 3000 cells of RECs from each compartment and for each dehydration time point. Samples were processed with 10× Genomics’ V2 barcoding chemistry. Cells from each sample were treated with the same master mix and in the same reaction vessel, by processing every single sample in a single well of a PCR plate. All samples were processed in parallel in the same thermal cycler. Libraries were sequenced on an Illumina HiSeq4000 and mapped to the mouse genome (build mm10) using CellRanger (10× Genomics, version 2.2.0), resulting in a sequencing depth of 63,692 reads per cell and a saturation rate of 92% on average. Data from the raw unfiltered matrix was further processed using R (version 3.4.4). After quality filtering on the basis of the number of detected genes, doublet exclusion, and mitochondrial read counts (see details below), a total of 40,662 high-quality RECs were obtained (15,419 gRECs; 11,762 cRECs; 13,481 mRECs for all conditions). One of the cREC samples (cREC4: cRECs isolated after 36 hours of water deprivation) did not pass the quality control because of a low number of unique molecular identifiers (UMIs) per cell (Supplemental Table 1) and was therefore excluded from downstream analyses. Among all high-quality cells, the total number of genes detected was 15,977, ranging between 1029 and 1309 (average of 1141) genes per cell. Data were first normalized for sequencing depth by dividing by the total number of UMIs in each cell, and then transformed to a log scale for each cell using the Seurat (http://satijalab.org/seurat/) NormalizeData function.11
For all of the samples, the following quality control steps were applied: (1) genes expressed by <50 cells or with a row mean of <0.001 were not considered; and (2) cells that had either <300 (low-quality cells) or >3000 expressed genes (possible doublets), or >10% of UMIs derived from the mitochondrial genome were removed. Dataset-specific cut-off values and parameter settings are listed in Supplemental Table 1.
scRNA-seq Bioinformatic Analysis
In Silico EC Selection
After autoscaling, the normalized data were first summarized by principal component analysis (PCA) using the flash PCA package, and the first 15 PCAs were visualized using t-SNE (Rtsne package) with a perplexity value of 200 and a learning rate of 100. Graph-based clustering was performed to group cells according to their gene expression profiles as implemented in Seurat (see Supplemental Table 1 for parameter settings). Cell clusters were annotated on the basis of canonical cell type markers, including Pecam-1, Icam2, and Cdh5 (ECs); and Hbb-a1, Hbb-a2, and Hbb-bs (red blood cells) to discriminate ECs from contaminating cells. No lymphatic ECs (Lyve1, Prox1), immune cells (Ptprc), or other renal cell types (tubular epithelial cells [Slc27a2, Lrp2, Slc12a1, Slc12a3, Pvalb, Aqp2, Hsd11b2, Atp6v1g3, Atp6v0d2, Insrr, Rhbg], podocytes [Nphs1, Nphs2], and mesangial and granular cells [Myl9, Ren1]) were detected. We did not identify a separate cluster, highly expressing a stress-response signature (artifactually resulting from the dissociation procedure).12 All downstream analyses were performed on ECs only.
EC Cluster Identification
We used the Seurat FindVariableGenes function to identify genes with high variability (see Supplemental Table 1 for parameter settings for each analysis). The normalized data were autoscaled and PCA was performed on variable genes or all genes (see Supplemental Table 1 for parameter settings for each analysis), followed by t-SNE to construct a two-dimensional representation of the data. To unbiasedly group control gRECs, cRECs, and mRECs, we performed PCA on highly variable genes, and used graph-based clustering as implemented in the FindClusters function of the Seurat package.11 In addition, to identify clusters of cells with discriminating gene expression patterns in all datasets, we color-coded t-SNE plots for each of the 15,977 detected genes using an in-house developed R/Shiny-based web tool. Cluster results were visualized using t-SNE to verify that all visually identified clusters were captured and not underpartitioned. Underpartitioned clusters that represented two distinct biologic phenotypes were subclustered. Overpartitioned clusters that represent the same biologic phenotype were merged into a single cluster. Clusters were considered only when containing at least 1% of the total number of cells per sample. We did not identify a separate cluster, highly expressing a stress-response signature (artifactually resulting from the dissociation procedure).12 Details of clustering parameters are listed in Supplemental Table 1.
To obtain ranked marker gene lists for each REC cluster, we performed pairwise differential gene expression analysis for each cluster against all other clusters independently, using the limma package (version 3.34.9). The results of each differential analysis were ordered on the basis of log2 fold change (genes with the highest fold change receiving the lowest rank number). We obtained a final ranked marker gene list for each cluster by calculating the rank product for all genes in all pairwise comparisons. This analysis was performed on gREC, cREC, and mREC samples taken separately, by using gene expression in control samples only, to avoid dehydration-induced effects.
To annotate clusters, we used canonical marker genes of artery, capillary, and vein ECs. In addition, we searched for a coherent set of genes involved in similar biologic processes within the top 50 ranking list of markers to further identify the associated REC phenotype. We also used gene set variation analysis (GSVA) to confirm upregulation of the identified biologic processes in the respective REC phenotypes (see below). Cells that could not be unambiguously assigned to a biologically meaningful phenotype might represent low-quality cells and were excluded from the analysis. Because of the manual microdissection of medulla from the cortex and of the REC isolation procedure, mREC and cREC samples contained a small cluster annotated as gRECs. This “contaminating” cluster was removed from the analyses for these two compartments.
GSVA
We used GSVA as implemented in the GSVA R-package (version 1.26.0) to convert the gene-by-cell matrix into a gene-set-by-cell matrix. Gene set analysis was performed using a set of 415 vascular related gene sets selected from the Molecular Signatures Database (MSigDB version 5.2 downloaded from http://bioinf.wehi.edu.au/software/MSigDB/). GSVA scores were only calculated for gene sets with a minimum of five detected genes. All other parameters were default.
Heatmap Analysis
All heatmaps are on the basis of cluster-averaged gene expression to account for cell-to-cell transcriptomic stochastics. Data were autoscaled for visualization. Heatmaps were produced using the Heatmaply R package (version 0.15.2). The data matrix for each heatmap can be downloaded from the accompanying web tool (see Data Availability below).
Single-Cell Regulatory Network Inference and Clustering Analysis
Single-cell regulatory network inference and clustering (SCENIC) scans differentially expressed genes for overrepresented transcription factor binding sites and analyzes coexpression of transcription factors and their putative target genes. Data were subsampled by randomly selecting 575 gRECs, cRECs, and mRECs for each control and dehydration time point and analyses were performed as described,13 using the SCENIC R package (version 1.1.0, which corresponds to RcisTarget 1.2.0, GENIE3 1.5.4, and AUCell 1.4.1) with RcisTarget motif databases (RcisTargets.mm9.motifDabases.20 k).
Venn Diagram Dehydration Analysis
To evaluate the dehydration effect on the gene signature between gREC, cREC, and mREC, we calculated the intersection of genes between the three compartments from the top 50 up- and downregulated genes for each dehydration time point, using Venn diagram calculation and visualization.
Gene Set Enrichment Analysis
We used gene set enrichment analysis (GSEA) as implemented in the clusterProfiler package (version 3.6.0) to compare gene expression signatures between mRECs at 48-hour dehydration and control mRECs.14 Gene set analysis was performed using a set of 415 vascular-related gene sets and 857 metabolic gene sets selected from the Molecular Signatures Database (MSigDB version 5.2 downloaded from http://bioinf.wehi.edu.au/software/MSigDB/). GSEA scores were calculated for sets with a minimum of ten detected genes, all other parameters were default.
Trajectory Inference
We used the SCORPIUS package (version 1.0.2) to place cells onto pseudotime trajectories.15 Analysis was performed on highly variable genes. Dimensionality reduction was performed using the reduce dimensionality function (we used 15 dimensions, all other parameters were default). Individual RECs from each cluster were subsequently placed onto linear pseudotime using the infer trajectory function of the SCORPIUS package with default settings. Activated RECs (angiogenic and response-to-IFN clusters) expressed a highly distinct signature and could therefore not be placed on the differentiation trajectory.
Jaccard Similarity Analysis
To measure the similarity of mRECs subclusters from dehydrated mice compared with those from control mice, we calculated the similarity of the top 50 marker gene sets using pairwise Jaccard similarity coefficients for all subclusters against each other. The Jaccard similarity coefficient is defined as the size of the intersection divided by the size of the union of sets:
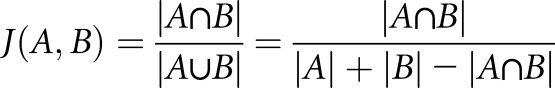 |
where J is the Jaccard index and A and B are two sets of marker genes. PCA was used for visualization.
Crossdataset Validation of the mRECs in Dehydration
Subclusters from the control mREC samples were projected on all samples (respective control and all dehydration time points), using the scmapCluster algorithm as implemented in the scmap package (version 1.1.5).16 We used the top 50 marker genes for each mREC subcluster in the control mREC taxonomy and a similarity threshold of 0.5; all other parameters were default. Below this threshold, cells were annotated as unassigned. The projection was visualized using a Sankey plot, similarity scores for each subcluster were visualized using a boxplot.
Data Visualization
The R implementation of the Plotly software was used for t-SNE, violin plot, and bar graph visualization.
Human Umbilical Vein Endothelial Cells
Human umbilical vein endothelial cells (HUVECs) were freshly isolated from different donors after obtaining their informed consent and under the ethical approval protocol S57123 (Ethics Committee Research UZ/KU Leuven), as previously described.17 The cells were routinely cultured in the appropriate gelatin-precoated (0.1% in PBS) cell culture plates in M199 medium (Thermo Fisher Scientific) containing 20% FBS, 2 mM L-glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin and endothelial cell growth factor supplements (ECGS/heparin; Promocell) or in endothelial cell growth medium (EGM2; Promocell) supplemented with endothelial cell growth medium supplement mix (Promocell), and were regularly tested for mycoplasma. For experiments with galactose medium, glucose-free EGM2 medium was reconstituted with 10 mM galactose in combination with dialyzed FBS (specifically requested customized medium; Promocell). The cells were used between passages 1 and 5 and all experiments were performed with HUVECs from at least three different donors.
HUVEC Model of Dehydration
Confluent HUVECs were adapted to hyperosmolarity by progressively increasing the medium osmolality from 300 to 900 mOsm/kg as follows: medium osmolality was changed to 380, 600, and 650 mOsm/kg every 12 hours, and up to 900 mOsm/kg by augmenting of 50 mOsm/kg every 2.5 hours. For seahorse experiments, the medium osmolality was increased up to 1200 mOsm/kg (50 mOsm/kg increase every 2.5 hours). To achieve this higher osmolality, EGM2 was supplemented with urea and NaCl (Sigma-Aldrich). For the 380 mOsm/kg osmolality, urea was supplemented at 40 mM and NaCl at 20 mM, and for the 600 mOsm/kg, urea was supplemented at 150 mM and NaCl at 75 mM. For adapting the medium from 650 to 1200 mOsm/kg, the urea molality was kept constant at 150 mM, whereas NaCl was increased progressively to achieve the desired osmolality. Cells were kept in hyperosmolar medium for at least 12 hours before performing any experiments.
Metabolic Assays
Seahorse Extracellular Flux Measurements
Freshly isolated CD31+MACS-enriched cRECs and mRECs from control and 48-hour dehydrated mice (n=3 mice per replicate per group) were seeded at a density of 500,000 cells per well on Seahorse XF24 tissue culture plates (Agilent) in complete mouse EC medium (M1168; Cell Biologics). Similarly, cultured HUVECs in normal or hyperosmolar medium (1200 mOsm/kg) were seeded at a density of 100,000 cells per well on Seahorse XF24 tissue culture plates in normal or hyperosmolar (1200 mOsm/kg) EGM2 medium. For the metformin experiments, cultured HUVECs in normal medium were pretreated with metformin (1, 10, or 20 mM) or control vehicle for 1 hour. Oxygen consumption rate (OCR) measurements were performed at 6-minute intervals (2 minutes mixing, 2 minutes recovery, 2 minutes measuring) in a Seahorse XF24 device. The addition of oligomycin (1.2 μM final; Sigma-Aldrich) allowed for the determination of ATP-coupled oxygen consumption rate (OCRATP). The addition of antimycine A (1 μM final; Sigma-Aldrich) allowed for the determination of basal OCR. HUVEC cultures were supplemented with ouabain at 0.5 µM immediately before OCR analyses to determine OCRATP related to Na+/K+ ATPase activity. The data were normalized to protein content.
Detection of Organic Osmolytes
Details are provided in Supplemental Appendix 1.
Biochemical Assays
Plasma and Urine Sample Analyses
Blood was collected from the right ventricle in anesthetized mice (see above) using potassium EDTA–coated syringes and centrifuged at 3000×g for 10 minutes. Plasma was collected and stored at −20°C until biochemical measurement. Urine was collected directly from the bladder. Plasma creatinine, urea, sodium, total protein levels, osmolality, and aspartate aminotransferase and lactate dehydrogenase (LDH) activity, as well as urine osmolality, were measured in the Central Medical Laboratory of the University Hospital of Leuven.
Bicinchoninic Acid Assay
Bicinchoninic acid assay (Pierce) was used to determine protein content following metabolic assays and for Western blot experiments. Cells were lysed using only RIPA buffer (Thermo Fisher Scientific) (for seahorse extracellular flux measurements) or in the presence of protease and phosphatase inhibitors (Roche, Vilvoorde, Belgium) (for Western blot analyses). For organic osmolyte detection, protein pellets were dissolved in 200 mM sodium hydroxide. Bicinchoninic acid assay was performed following the manufacturer’s guidelines.
Western Blot
Details are provided in Supplemental Appendix 1.
RNA Isolation and Quantitative RT-PCR
RNA was collected from HUVECs cultured in normal or hyperosmolar EGM2 (900 mOsm/kg), purified using the PureLink RNA Mini Kit (Invitrogen, Life Technologies) and converted to complementary DNA using the iScript cDNA synthesis kit (Bio-Rad). RNA expression analysis was performed by Taqman quantitative RT-PCR as described,18 using premade primer sets (Integrated DNA Technologies, identification numbers: Hs.PT.58.25051081, Hs.PT.58.4615682, Hs.PT.58.2145446, Hs.PT.58.24360893, Hs.PT.58.39555113, Hs.PT.58.45350074). For comparison of gene expression between conditions, expression (normalized to HPRT as endogenous control) is expressed relative to control condition.
LDH Release Viability Assay
HUVECs in normal or hyperosmolar EGM2 (900 mOsm/kg) were trypsinated and seeded at a density of 100,000 cells per well in 96-well plates. The following day, cells were treated with rotenone (2 μM) (Sigma-Aldrich) or oligomycin (1.2 μM; Sigma-Aldrich) in normal or hyperosmolar EGM2 (900 mOsm/kg) and in normal or hyperosmolar galactose medium (900 mOsm/kg) for 24 hours. LDH assay kit (Cytotoxicity Detection Kit; Roche) was used to quantify cellular LDH release following the manufacturer’s guidelines.
Flow Cytometry
Mitochondrial Content
HUVECs cultured in normal or hyperosmolar medium were trypsinated and incubated with MitoTracker Green FM (1/5000, M7514; Thermo Fisher Scientific) for 30 minutes at 37°C in phenol red–free EBM2 medium (Promocell) supplemented with endothelial cell growth medium supplement mix. Intensity of incorporated MitoTracker green was determined by fluorophore excitation with a 490 nm yellow laser and emission was recorded at 516 nm. Data were recorded by flow cytometry, and analyzed with the FlowJo 10.4.2 software (https://www.flowjo.com). MitoTracker staining was quantified by measuring median fluorescence intensity.
Superoxide Content Measurement
HUVECs cultured in normal or hyperosmolar medium were trypsinated and incubated with dihydroethidium (DHE; 1/250, D11347; Thermo Fisher Scientific) for 30 minutes at 37°C in phenol red–free EBM2 medium supplemented with endothelial cell growth medium supplement mix. Intensity of incorporated DHE was determined by fluorophore excitation with a 518 nm laser and emission was recorded at 606 nm. Data were recorded by flow cytometry, and analyzed with the FlowJo 10.4.2 software (https://www.flowjo.com). DHE staining was quantified by measuring median fluorescence intensity.
Histology and Immunohistochemistry
All methods for histology and immunostainings have been previously described.17,19 Mouse kidney samples were immediately fixed after collection in 4% paraformaldehyde overnight at 4°C, then dehydrated and embedded in paraffin. Briefly, kidneys were sectioned at 7 µm thickness and were stained using the following primary antibodies: CD105 (1/50, AF1320; R&D Systems), endomucin (1/50, 14–5851–82; Thermo Fisher Scientific), carbonic anhydrase–related protein (CA-VIII; 1/400, 12391–1-AP; Proteintech), S100A4 (1/50, A5114; Dako), PLVAP (1/40, Meca32-c; Developmental Studies Hybridoma Bank), FXYD6 (1/50, 15805–1-AP; Proteintech), and CRYAB (1/200, AB13497; Abcam). Sections were then incubated with the appropriate fluorescence-conjugated secondary antibodies (Thermo Fisher Scientific) or followed by amplification with the proper tyramide signal amplification systems (NEL704A001KT, NEL705A001KT, NEL701A001KT; Perkin Elmer). Tyramide signal amplification was used for CA-VIII, S100A4, PLVAP, and FXYD6 antibodies. Nuclei were counterstained with Hoechst (1 µg/ml, H3570; Thermo Fisher Scientific). Negative controls were performed omitting incubation with primary antibodies. Sections were mounted on glass slides in ProLong mounting medium. Images were acquired with a Zeiss LSM780 confocal microscope, coupled to ZEN software (Zeiss). Images were processed with ImageJ software.
Renal Hypoxia
Details are provided in Supplemental Appendix 1.
Total Mitochondrial Volume
HUVECs cultured in normal or hyperosmolar EGM2 (900 mOsm/kg) were fixed in 4% paraformaldehyde for 30 minutes and then immunostained with TOMM20 (1/200; Abcam) for mitochondria and Phalloidin-Alexa 488 (1/50; Life Technologies) for actin. Images were acquired with a Zeiss LSM780 confocal microscope, coupled to ZEN software. Mitochondrial volumes were defined using Leica MetaMorph AF 2.1 morphometry software package. A three-dimensional volume mask was created for each cell in which the mitochondrial volume was calculated on the basis of the thresholded TOMM20 signal.
In Vivo Metformin Treatment
For metformin treatment, 8- to 12-week-old male mice (C57Bl6/J strain) were exposed to food and water ad libitum, or were water-deprived and treated with 50 µl of either vehicle (water) or metformin (600 mg/kg; Sigma-Aldrich; a dose previously reported to influence urine concentration20) twice a day by gavage for 48 hours (n=5–17 per group). Dehydrated mice were closely followed up, as described in the Animals section. Mice were anesthetized with an intraperitoneal injection of ketamine/xylazine, blood was collected from the right ventricle and urine was collected from the bladder, and then analyzed as described above.
Statistical Analyses
Data are represented as mean±SEM. The presence of outliers was assessed using the ROUT test and eventual identified outliers were excluded from downstream analyses. Two-tailed statistical test was used for all analyses. Statistical significance was assessed with one sample t test or unpaired t test when comparing two conditions, and one- or two-way ANOVA with Bonferroni correction after the evaluation of variance homogeneity in Brown–Forsythe tests, or Kruskal–Wallis tests followed by Dunn tests, as appropriate, for multiple comparisons, using Prism 7 (GraphPad Prism). P<0.05 was considered statistically significant.
Data Availability
All raw sequencing data are available in ArrayExpress under accession number E-MTAB-8145. Normalized data can be downloaded from the accompanying web resource at http://www.vibcancer.be/software-tools/rec-dehydration. Custom code is available upon request.
Results
Isolation and scRNA-seq of RECs from Control and Dehydrated Mice
The kidney vasculature displays a hierarchically organized architecture1,2 (Figure 1A). To characterize the molecular heterogeneity of RECs and their adaptation to dehydration, we collected kidneys from mice under euhydration (controls) and mice deprived from drinking water for 12, 24, 36, and 48 hours (Figure 1B). RECs from glomeruli, cortex, and medulla were processed separately to obtain a similar resolution for further characterization (Figure 1B, Supplemental Figure 1A). All gREC, cREC, and mREC single-cell samples were subjected to scRNA-seq, resulting in 40,662 high-quality RECs (Figure 1B, Supplemental Figure 1B, Supplemental Table 1).
Figure 1.

Control RECs show kidney compartment-specific gene signatures. All data in (C–F) are generated using RECs from kidneys in control condition. (A) Simplified schematic overview of the nephron and glomerular vasculature. The renal artery supplies blood to the entire kidney, and branches into afferent arterioles, which enter glomeruli to form fenestrated glomerular capillaries where blood ultrafiltration takes place. Unlike most capillary beds, glomerular capillaries exit into efferent arterioles, which branch into cortical peritubular capillaries vascularizing the proximal and distal convoluted tubules. DVR branch off from the efferent arterioles of juxtamedullary nephrons, enter the medulla and branch into a capillary plexus that drains blood into AVR. Vasa recta surround the loop of Henle and are essential for concentrating the urine. Peritubular capillaries and AVR connect to renal veins in the cortex, which drain the blood from the kidney. (B) Schematic overview of the study design. Kidney cortex, medulla, and glomeruli were enzymatically digested until obtaining single-cell suspensions. After a pre-enrichment of CD31+ cRECs and mRECs via MACS, all RECs were purified by FACS. Single-cell RNA sequencing of these RECs resulted in 15,419 high-quality gRECs; 11,762 high-quality cRECs; and 13,481 high-quality mRECs for all conditions. (C) Expression-level scaled heatmap of all genes from RECs isolated from the three kidney compartments. Scale: light blue is low expression, red is high expression. (D) t-SNE plot showing REC clusters from the three kidney compartments. (E) Expression-level scaled heatmap of the top 50 marker genes of REC clusters isolated from the three kidney compartments. (F) t-SNE plots of RECs isolated from the three kidney compartments, color-coded for the expression of the indicated markers. Red arrowheads indicate cells with high expression of the indicated marker on the t-SNE plots. Scale: light blue is low expression, red is high expression. Asterisks indicate novel marker genes. (G) Expression-level scaled heatmap of the top 10 inferred transcription factor gene regulatory networks (as analyzed using SCENIC) in RECs from the three kidney compartments. Numbers between parentheses indicate the number of genes (g) that are part of the regulons for the respective transcription factors. Red corresponds to high transcription factor activity, blue corresponds to low transcription factor activity. See also Supplemental Figure 1 and Supplemental Tables 1 and 2.
Intercompartment REC Heterogeneity
We first constructed a REC taxonomy in control conditions. A heatmap representing all genes expressed by gRECs, cRECs, and mRECs showed that different REC types have unique transcriptome profiles (Figure 1, C and D). Using a rank product–based method, we identified the top 50 marker genes for each REC cluster (Supplemental Table 2). As reported, gRECs highly expressed Ehd3, Cyp4b1, and Tspan7,6,21,22 whereas mRECs showed higher expression of Igf1 and Cd366 (Figure 1, E and F, Supplemental Table 2). Specific markers for cRECs have not yet been described. Here, we found that cRECs highly expressed Igfbp3, Npr3, and other novel markers (Figure 1, E and F, Supplemental Table 2). In addition, Plvap, a marker of fenestrated ECs with diaphragm, was highly expressed in cRECs and mRECs, but not in gRECs, consistent with previous reports1 (Figure 1F). We identified markers for each REC type previously not highlighted in the literature (Figure 1, E and F, Supplemental Table 2). Some markers were not uniformly expressed throughout the clusters, but were identified only in a subset of cells within these clusters (Npr3 in cRECs; Aqp1, Cryab, and Ifi27l2a in mRECs) (Figure 1F, Supplemental Table 2), raising the question of whether these clusters contained additional REC subclusters.
To study the underlying molecular mechanisms driving the differentiation of the different REC phenotypes, we used SCENIC.13 We identified the top ten enriched transcription factor networks in gRECs, cRECs, or mRECs, including Gata5, Gata2, and Nuak2 regulons in gRECs; Irf1, Foxp1, and Sp1 networks in cRECs; and Pparg, Lef1, Nfe2l2, and Irf7 regulons in mRECs (Figure 1G). In agreement with our findings on gRECs, EC loss of gREC marker Gata5 causes glomerular lesions.23
Intracompartment REC Heterogeneity
To further characterize the heterogeneity of gRECs, cRECs, and mRECs, we used a shared nearest neighbor modularity optimization–based clustering algorithm to subcluster RECs on the basis of their gene expression signatures, resulting in 24 subclusters (Supplemental Figure 1, C–E). We used canonical marker genes of arterial (Sox17, Fbln5), fenestrated capillary (Plvap, Kdr), and venous (Plvap, Nr2f2) ECs, as well as the top 50 marker genes of each subcluster to identify the different REC phenotypes. The number of RECs per subcluster as well as the numbers of UMIs and genes per kidney compartment are displayed in Supplemental Figure 1, C–E. A detailed description of the marker genes (including novel markers, not previously highlighted in the literature), with their inferred putative biologic role used to identify each subcluster, is reported in Supplemental Table 3.
gRECs exhibited five subclusters (Figure 2, A and B, Supplemental Figure 2A, Supplemental Tables 3 and 4): afferent arteriolar ECs (G1), ECs from the terminal portion of the afferent arterioles associated with the juxtaglomerular apparatus (JGA) with enriched expression of cell-cell interaction genes (G2) (Supplemental Figure 2B), fenestrated capillary gRECs (G3), ECs expressing a mix of capillary and arterial EC markers (G4), and efferent arteriolar gRECs (G5). When taking advantage of a pseudotime algorithm to reconstruct the trajectory of the glomerular vascular bed, we observed that G4 positioned as an intermediate subcluster between G3 and G5, suggesting that it represents a gREC population between the glomerular capillaries and efferent arteriole, as observed also for G2 with the afferent arteriole (Figure 2C). Thus, in analogy with G2, G4 likely represents efferent arteriolar gRECs associated with the JGA (Figure 2, A–C).
Figure 2.

Control gRECs and cRECs exhibit intracompartment heterogeneity. All data shown were generated using gRECs and cRECs from kidneys in control condition. (A) gREC subclustering visualized by t-SNE plot. Asterisk indicates previously unrecognized subcluster. (B) Expression-level scaled heatmap of the top 20 marker genes in gREC subcluster phenotypes. (C) Pseudotime analysis of gREC subclusters and loess smoothed expression of the indicated gene markers in pseudotime in the indicated gREC subclusters. (D) cREC subclusters visualized by t-SNE plot. Asterisks indicate newly recognized subclusters. (E) Expression-level scaled heatmap of the top 20 marker genes in cREC subclusters. (F) Pseudotime analysis of cREC subclusters and loess smoothed gene expression of the indicated markers in pseudotime in the indicated cREC subclusters. Angiogenic and response-to-IFN cREC subclusters were not included in the pseudotime analysis. (G) Simplified schematic overview of the renal cortical vasculature. Inset shows larger magnification of the boxed area. Subclusters C4 and C5 are novel; the other vascular beds have been previously documented at the anatomic level. (H) Representative micrographs of mouse kidney sections immunostained for the endothelial cell marker CD105 (red), the vein/capillary marker endomucin (gray), and the novel marker CA-VIII (green) for the vein cluster (C7). Nuclei are counterstained with Hoechst (blue). Bottom images are magnifications of the respective boxed areas. Arrrowheads indicate endothelial cells from an artery and a vein. A t-SNE plot of cRECs, color-coded for the expression of Car8 is shown below. Scale: light blue is low expression, red is high. Note: CA-VIII denotes the protein, encoded by the Car8 gene. Scale bar, 200 µm (top panels) and 50 µm (bottom panels). See also Supplemental Figures 2 and 4, and Supplemental Tables 1, 3–5. Art, artery.
A total of nine cREC subclusters were identified (Figure 2, D and E, Supplemental Figure 2C, Supplemental Tables 3 and 4): C1, C2, and C3 represent ECs from arteries, arterioles, and efferent arterioles, respectively; C4 and C5 presented highly expressed capillary markers and were characterized by high (C4) or low (C5) ApoE expression; C6 showed an intermediate capillary-vein gene signature associated with postcapillary venules; and C7 included cRECs from veins. Pseudotime-based predictions of the renal cortical vascular trajectory confirmed this analysis, positioning the identified arterial, capillary, and vein subclusters in this order (Figure 2F). Validation at the protein level by immunostaining for CA-VIII confirmed expression of this novel venous cREC marker in endomucin-positive venous cRECs, but not in endomucin-negative arterial cRECs24 (Figure 2, G and H, Supplemental Figure 2D). We also discovered two novel capillary cREC subclusters: C8 showing an angiogenic phenotype, and C9 exhibiting an IFN-response phenotype (Figure 2, D and E, Supplemental Figure 2C, Supplemental Tables 3 and 4).
mRECs exhibited ten subclusters (Figure 3, A and B, Supplemental Figure 3A, Supplemental Tables 3 and 4): M1 represented mRECs from arterioles; M2 exhibited markers of the DVR; and M3 likely represented mRECs from the papillary portion of the descending vasa recta (papillary DVR). Although no known markers of the papillary DVR exist, these cells highly expressed hyperosmolarity responsive genes, consistent with the high osmolarity in the papilla.25 Immunostaining for S100A4 confirmed expression of this novel papillary DVR marker in mRECs in the papilla, but not in the inner and outer medulla (Figure 3, C and D, Supplemental Figure 3B). Fenestrated capillary subcluster M4 included mRECs from the medullary capillary plexuses. M5 represented postcapillary venous ECs, and M6 included vein-like mRECs from the AVR. Immunostaining for FXYD6 confirmed expression of this novel AVR marker in mRECs in the inner and outer medulla, but not the papilla (Figure 3, C and E, Supplemental Figure 3B). We also uncovered vein-like mRECs, which likely represented mRECs from the papillary portion of the ascending vasa recta (papillary AVR) (M7), as they showed enriched expression of hyperosmolarity responsive genes, similar to papillary DVR RECs. This was confirmed by immunostaining of the novel papillary AVR marker, CRYAB, expressed in the fenestrated PLVAP+ mRECs from AVR in the papilla (but not in the inner and outer medulla) (Figure 3, C and F, Supplemental Figure 3B). Although mRECs from both papillary AVR and papillary DVR exhibited distinct transcriptome profiles, they shared upregulated expression of hyperosmolarity-inducible genes and other novel marker genes (Supplemental Figure 3A). Pseudotime-based predictions of the renal medullary vascular trajectory confirmed this analysis, positioning the arterial, capillary, and vein subclusters in this order (Figure 3G). We also identified three new mREC subpopulations, including capillary mRECs representing an angiogenic phenotype (M8), as well as capillary (M9) and venous (M10) mRECs displaying a response-to-IFN phenotype (Figure 3, A and B, Supplemental Figure 3A).
Figure 3.

Control mRECs exhibit intracompartment heterogeneity. All data shown were generated using mRECs from kidneys in control condition. (A) mREC subclustering visualized by t-SNE plot. (B) Expression-level scaled heatmap of the top 20 marker genes in mREC subclusters. Asterisks indicate newly recognized subclusters. (C) Schematic overview of the renal papillary vasculature. Insets show larger magnifications of the respective boxed areas. (D) Representative micrographs of mouse kidney sections immunostained for the endothelial cell marker CD105 (red) and the novel marker S100A4 (green) for the DVR/papilla subcluster (M3). Nuclei are counterstained with Hoechst (blue). Top and middle panels are images of the papilla; middle images are magnifications of the respective boxed areas, with arrowheads indicating CD105+S100A4+ ECs. Bottom panels are images of outer/inner medulla. A t-SNE plot of mRECs color-coded for the expression of S100a4 is shown beneath the micrographs. Scale: light blue is low expression, red is high expression. Scale bar, 200 µm (top and bottom panels) and 50 µm (middle panels). (E) Representative micrographs of mouse kidney sections immunostained for the endothelial cell marker CD105 (red), the fenestrated endothelial marker PLVAP (gray), and the novel marker FXYD6 (green) for the AVR cluster (M6). Nuclei are counterstained with Hoechst (blue). Top panels are images of the papilla; middle and bottom panels are images of outer/inner medulla; bottom images are magnifications of the respective boxed areas, with arrowheads indicating CD105+PLVAP+ FXYD6+ ECs. A t-SNE plot of mRECs color-coded for the expression of Fxyd6 is shown beneath the micrographs. Scale: light blue is low expression, red is high expression. Scale bar, 200 µm (top and middle panels) and 50 µm (bottom panels). (F) Representative micrographs of mouse kidney sections immunostained for the endothelial cell marker CD105 (red), the fenestrated endothelial marker PLVAP (gray), and the novel marker CRYAB (green) for the AVR/papilla cluster (M7). Nuclei are counterstained with Hoechst (blue). Top and middle panels are images of the papilla of the mouse kidney; middle images are magnifications of the respective boxed areas. Higher magnification of the respective boxed areas is shown in insets, with arrowheads indicating CD105+PLVAP+CRYAB+ ECs. Bottom panels are images of outer/inner medulla of the mouse kidney. A t-SNE plot of mRECs color-coded for the expression of Cryab is shown on the right of the micrographs. Scale: light blue is low expression, red is high expression. Scale bar, 200 µm (top and bottom panels), 50 µm (middle panels), and 10 µm (insets in middle panels). (G) Pseudotime analysis of mREC subclusters and loess smoothed gene expression of the indicated markers in pseudotime in the indicated mREC subclusters. Angiogenic and response-to-IFN mREC subclusters were not included in the analysis. The t-SNE plots shown in (D–F) are also represented in Supplemental Figure 4A at a larger size for reasons of clarity. See also Supplemental Figures 3 and 4 and Supplemental Tables 1, 3–5.
GSVA showed high expression of genes encoding MHC class II protein complexes in response-to-IFN ECs (Supplemental Figure 4A, Supplemental Table 5). However, these RECs did not express detectable levels of the coactivators Cd80 and Cd86. Interestingly, cRECs from veins also highly expressed MHC II genes (Supplemental Figure 4A). GSVA revealed enrichment of transcripts involved in angiogenesis in angiogenic clusters from the cortex and medulla (Supplemental Figure 4B, Supplemental Table 5). Unbiased correlation heatmap analysis and hierarchical clustering revealed that gRECs (afferent and efferent arterioles, capillaries) represented a separate entity, resembling each other more than RECs from other kidney compartments, illustrating their high specification (Supplemental Figure 4C). Overall, this taxonomy captures the complex heterogeneity of RECs between and within kidney compartments. We used this atlas to study the transcriptome response of each REC compartment to dehydration.
Renal Endothelial Cells Differentially Respond to Dehydration
In response to acute dehydration, the kidney concentrates the urine, increases water reabsorption, and reduces glomerular filtration.7–9 To characterize the transcriptional adaptation of RECs to dehydration, mice were deprived of water for up to 48 hours, using a previously reported model, documented to induce weight loss, increased plasma urea, sodium, total protein and osmolality levels, urine osmolality, and decreased urine volume26–30 (Figure 1B). Water-deprived mice lost body weight, whereas their urine and plasma osmolality, plasma sodium and total protein levels, and plasma LDH and aspartate transferase activities were increased (Supplemental Figure 5, A–G). Plasma urea levels were slightly elevated at certain time points (Supplemental Figure 5H), whereas plasma creatinine levels remained unchanged (<0.15 mg/dl), indicating that the kidneys were still functional.
We performed a differential analysis of transcriptome profiles of gRECs, cRECs and mRECs at different time points after dehydration compared with their respective controls (Supplemental Table 6). Analysis of the top 50 up- and downregulated genes in RECs from the different compartments for each dehydration time point revealed kidney compartment-specific REC gene expression changes, with only a few genes in common (Figure 4A, Supplemental Figure 5I, Supplemental Table 6). Thus, RECs from different compartments respond distinctly to dehydration, illustrating the heterogeneity of RECs not only in baseline but also upon water deprivation. Quantification of the upregulated [log2(fold change) >0.2] or downregulated [log2(fold change) <0.2] genes in RECs from each compartment suggested that mRECs were the most affected by dehydration (Figure 4B). We therefore focused primarily on mRECs.
Figure 4.

mRECs are the most affected by dehydration. (A) Venn diagram of the top 50 up- and downregulated genes in RECs from the three kidney compartments at 48 hours of dehydration. (B) Number of dysregulated genes [(log2(fold change) >0.2 or <0.2] in gRECs, cRECs, and mRECs at the different dehydration time points versus control. Note for the 36-hour time point: the cREC sample did not meet sequencing quality standards and was therefore not included in downstream analyses. See also Supplemental Figure 5 and Supplemental Table 6.
Adaptation of mRECs to Hyperosmolarity during Dehydration
The medullary countercurrent system enables production of urine of widely varying osmolalities, and thereby exposes medullary renal cells to harsh conditions (elevated levels of solutes such as sodium chloride and urea) upon dehydration. Although the adaptation of medullary epithelial cells to survive in such hostile milieu are described,25,31,32 little is known about the mechanisms via which mRECs cope with and adapt to a hyperosmolarity challenge. We observed a shift in gene expression as early as 12 hours of water deprivation in mRECs (Figure 5, A and B). Their response to dehydration occurred in two phases: an early adaptation phase (12–24 hours) and a late response (36–48 hours) (Figure 5, A and B). Pseudotime analysis of mRECs showed that the arterial-capillary-venous trajectory was affected the most at 36–48 hours of water deprivation and that dehydration affected medullary capillaries the most (Figure 5C). Differential analysis of mREC transcriptome profiles in control and at 12, 24, 36, and 48 hours of water deprivation largely confirmed this result (for the upregulated genes, the 36-hour time point seemed to be a transition phase) (Figure 5, D and E).
Figure 5.

mRECs show a typical transcriptomic response to hyperosmolarity in dehydration. (A) Correlation heatmap of mRECs from the control condition and at different dehydration time points. Scale: red indicates a high transcriptome similarity, blue indicates a low transcriptome similarity. (B) t-SNE plot color-coded for mRECs from the control condition and at different dehydration time points. (C) Pseudotime analysis of mRECs from control conditions, and at 12–24 and 36–48 hours of dehydration. (D and E) Expression-level scaled heatmap of the top 50 upregulated (D) and downregulated (E) genes in mRECs at different dehydration time points. Red corresponds to high gene expression levels, blue corresponds to low gene expression levels. (F) Expression-level scaled heatmap of genes encoding heat shock proteins, or genes involved in cytoskeleton remodeling, DNA damage, and growth arrest, and immediate early genes in mRECs from control condition and at the different dehydration time points. Red corresponds to high expression levels, blue corresponds to low expression levels. (G) Expression-level scaled heatmap of cell volume regulation–related genes in mRECs from control condition and at the different dehydration time points. Red corresponds to high expression levels, blue corresponds to low expression levels. (H) Pathway map showing changes in transcript levels of genes belonging to myo-inositol and polyol pathways, and genes encoding transporters related to cell volume regulation in mRECs after 48 hours of dehydration compared with control. Scale bar: red corresponds to gene upregulation after dehydration, gray indicates that the change in gene expression did not reach the fold change threshold to be color-coded. See also Supplemental Figure 7 and Supplemental Table 1.
In the early phase, we observed a transient increase in expression of genes encoding heat shock proteins and immediate early genes, and initial changes in expression of genes involved in cytoskeleton remodeling, DNA damage, and growth arrest, as reported for renal epithelial cells upon dehydration or hyperosmolarity33 (Figure 5F). In the late phase, the changes in expression of genes involved in cytoskeleton remodeling, DNA damage, and growth arrest became more prominent (Figure 5F). Other transcriptome changes related to genes involved in cell volume regulation, especially encoding transporters and biosynthetic enzymes necessary to increase the intracellular concentration of inert osmolytes and sodium as well as water transport to equilibrate intra- and extracellular osmolarity.25,31,32 In agreement, mRECs upregulated the expression of genes involved in taurine transport (Slc6a6), myo-inositol transport (Slc5a3, transiently), sodium-coupled neutral amino acid transport (Slc38a2), sorbitol synthesis (Akr1b3), Na+/K+ ATPase (Atp1a1, Atp1b3, Fxyd2, Fxyd5), and water transport (Aqp1) (Figure 5, G and H). Not previously reported as a response mechanism to hyperosmolarity, we observed a transcriptome switch from myo-inositol transport (Slc5a3) to synthesis (Isyna1) (Figure 5, G and H). Contrary to what is described in epithelial cells,34 transcript levels of the Na+/K+/Cl− (NKCC) cotransporter (Slc12a2) were decreased in mRECs (Figure 5G).
mRECs from dehydration samples contained the same number of similar mREC subclusters as control mRECs (Supplemental Figure 6A). In addition, by using scMap16 and the top 50 marker genes of each mREC subcluster (M1–M10), mRECs from dehydration samples were confidently assigned to mREC subclusters in the control mREC taxonomy (similarity threshold >0.5; Supplemental Figure 6B). Thus, no novel dehydration-evoked cluster was identified in mRECs.
mRECs Adapt Their Metabolic Gene Expression Signature upon Dehydration
GSEA of mRECs from control versus 48-hour dehydrated mice, in combination with expression heatmap analysis, revealed upregulation upon dehydration of gene sets involved in hypoxia/hypoxic response, but surprisingly, also in inorganic ion transmembrane transport (genes encoding complex subunits of OXPHOS) (Figure 6, A and B, Supplemental Table 7), suggesting that mRECs might be exposed to deeper hypoxia, yet upregulated OXPHOS (see below). Upregulation of OXPHOS genes has never been reported in medullary epithelial cells exposed to hyperosmolarity. Consistent with a report highlighting an increase in hypoxia inducible factor 1α–positive cells in the renal medulla upon dehydration,35 we observed an increase in pimonidazole-stained hypoxic areas in the inner kidney medulla from dehydrated mice (48 hours) (Supplemental Figure 5J), validating the GSEA results. Also, SCENIC revealed that the Epas1 regulon (encoding hypoxia-inducible factor 2α) was upregulated in mRECs after dehydration (Figure 6C). We also identified upregulation of Rad21 regulon, involved in DNA repair,36,37 at all dehydration time points, whereas the Pparg regulon was downregulated (Figure 6C).
Figure 6.

mRECs metabolically adapt to dehydration. (A) GSEA of mRECs after 48 hours of dehydration compared with controls. (B) Expression-level scaled heatmap of oxidative phosphorylation-related genes for mRECs from the control condition and at different dehydration time points. (C) Expression-level scaled heatmap of the top up- and downregulated inferred transcription factor gene regulatory networks (as analyzed using SCENIC) in mRECs in control condition and at different dehydration time points. Numbers between parentheses indicate the number of genes (g) that are part of the regulons for the respective transcription factors. Red corresponds to high transcription factor activity, blue corresponds to low transcription factor activity. (D) GSEA using only metabolic gene sets of mRECs after 48 hours of dehydration compared with controls. (E–G) Expression-level scaled heatmap of ribosomal genes (E), proteasome genes (F), and glycolysis-related genes (G) for mRECs from the control condition and at different dehydration time points. (H) Pathway map showing changes in transcript levels of metabolic genes of glycolysis, TCA cycle, and oxidative phosphorylation in mRECs after 48-hour dehydration compared with controls. Scale bar: red corresponds to gene upregulation after dehydration, gray indicates that the change in gene expression did not reach the fold change threshold to be color-coded. (I) OCRATP of cRECs and mRECs isolated from control mice and from mice subjected to 48 hours of dehydration (DH). Data are from n=3 independent experiments, for which three mice were pooled each time. (J) Expression-level scaled heatmap of oxidative phosphorylation-related genes for mRECs from papillary subclusters or other medullary subclusters in the control condition and after 48 hours dehydration. See also Supplemental Figure 5 and Supplemental Table 7. Statistical test: unpaired t test, *P<0.05.
Considering that the unbiased GSEA yielded an upregulation of mainly metabolic pathway genes, together with hypoxia, we performed a GSEA focusing on metabolic pathways only (Figure 6D, Supplemental Table 7). This analysis revealed an upregulation of ribosome- and proteasome-related genes, suggesting high protein turnover (Figure 6, E and F). Also, genes involved in OXPHOS and glycolysis were upregulated in mRECs after 36–48 hours of dehydration (Figure 6, B and G), whereas expression of TCA cycle–related genes did not show prominent changes, as visualized by metabolic pathway mapping (Figure 6H). Consistently, freshly isolated mRECs from mice exposed to 48 hours of water deprivation showed a higher level of oxygen consumption coupled to ATP synthesis, whereas oxygen consumption was not affected in cRECs from the same animals (Figure 6I). Taking advantage of the mREC taxonomy, we observed that OXPHOS genes were enriched in papillary mRECs compared with other mRECs in both control and 48 hours dehydration conditions (Figure 6J).
In contrast to mRECs, which launched a response in two clearly distinct phases, each time point after dehydration largely evoked a different response in gRECs (Supplemental Figure 7, A and B). In addition, changes in expression of the aforementioned genes involved in cell volume regulation, metabolism, protein turnover, and stress response were different in gRECs (Supplemental Figure 7, C–H). Overall, the adaptive response of gRECs to dehydration was qualitatively and quantitatively different, likely because these cells are not exposed to such high hyperosmolarity and are influenced by their local microenvironment.
OXPHOS Upregulation Promotes EC Survival and Urine Concentration
To investigate whether the upregulation of the OXPHOS gene signature and oxygen consumption in mRECs after dehydration was triggered by hyperosmolarity, we developed an in vitro model using HUVECs, by exposing them to a progressive increase in urea and NaCl concentration in the culture medium, the main osmolytes in the kidney medullary interstitium,38 until reaching an osmolality of 900–1200 mOsm/kg in the medium corresponding to osmolality values close to the maximal urine concentration capacity in humans.38 Consistent with the mREC data in dehydration, HUVECs exposed to hyperosmolarity upregulated several osmolytes necessary to maintain cell volume, such as sorbitol and glutamine, among others (Supplemental Figure 8A). They also upregulated genes encoding the different complex subunits of OXPHOS (Figure 7A), and augmented OCRATP (Figure 7B). The Na+/K+ ATPase pump plays a critical role in maintaining physiologic concentrations of intracellular sodium, especially in hyperosmolarity conditions, a process requiring high amounts of ATP.25,39,40 As in mRECs during dehydration (see above), protein levels of this pump were upregulated in HUVECs exposed to hyperosmolarity (Supplemental Figure 8B). In agreement, treatment of hyperosmolarity-exposed HUVECs with ouabain, a Na+/K+ ATPase inhibitor, inhibited the increase in OCRATP (Figure 7B), suggesting that the increase in OXPHOS is necessary to sustain ATP production for sufficient Na+/K+ ATPase activity.
Figure 7.

ECs survive hyperosmolarity during dehydration by upregulating oxidative phosphorylation. (A) Quantitative RT-PCR analysis of OXPHOS complex subunit-encoding transcripts (NDUFA7, SDHB, UQCR10, COX4I1, ATP5H) in control and hyperosmolarity-exposed HUVECs (n=4). (B) OCRATP of control and hyperosmolarity-exposed HUVECs, in the absence or presence of ouabain (0.5 µM) (n=3). (C) MitoTracker green median fluorescence intensity of control and hyperosmolarity-exposed HUVECs (n=10). (D) Left: representative images of control and hyperosmolarity-exposed HUVECs stained for the mitochondrial marker TOMM20 (red), the cytoskeleton phalloidin (green), and the nucleus (Hoechst, blue). Scale bar: 20 µM. Right: total mitochondrial volume per cell of TOMM20-stained control and hyperosmolarity-exposed HUVECs (n=4). (E) DHE median fluorescence intensity of control and hyperosmolarity-exposed HUVECs (n=8). (F) Cell death quantification of control and hyperosmolarity-exposed HUVECs in the presence or absence of OXPHOS inhibitors (rotenone 2 µM; oligomycin 1.2 µM) in control medium or medium in which glucose was replaced by galactose (n=3–6). (G and H) Plasma urea levels (G) and urine osmolality (H) of mice treated with vehicle or metformin (600 mg/kg, twice a day) and submitted to 48 hours of dehydration (DH) (n=5–17). Data are mean±SEM. See also Supplemental Figure 8. Statistical tests: one-sample t test, unpaired t test, one- and two-way ANOVA/Bonferroni, *P<0.05 compared with control; #P<0.05 compared with hyperosmolarity or dehydration.
Hyperosmolarity-induced upregulation of OXPHOS was associated with an increase in mitochondrial content and reactive oxygen species levels (Figure 7, C–E). To study the functional relevance of the increased OXPHOS gene expression, we treated hyperosmolarity-exposed HUVECs with OXPHOS inhibitors (complex I inhibitor rotenone; complex V inhibitor oligomycin). OXPHOS inhibition compromised HUVEC survival when cells were cultured in a hyperosmolar medium, in which glucose was replaced by galactose to increase aerobic over anaerobic metabolism41–43 (Figure 7F). By contrast, control ECs exposed to the same medium were not significantly affected by OXPHOS inhibitors, highlighting a key role for OXPHOS in EC survival in hyperosmolarity conditions (Figure 7F).
Furthermore, treatment of mice with metformin, a multifunctional compound that inhibits complex I of OXPHOS and oxygen consumption in ECs (Supplemental Figure 8C; consistent with previous reports44,45), during dehydration elevated plasma urea levels, without altering plasma creatinine levels (<0.12 mg/dl), consistent with more severe dehydration (Figure 7G).26 Notably, metformin treatment largely prevented the increase in urine osmolality during dehydration, reflecting an impaired capability in urine concentration (Figure 7H). Overall, mRECs adapted to hyperosmolarity upon dehydration by upregulating an OXPHOS gene signature and oxygen consumption, whereas treatment with a multifunctional OXPHOS blocker (see Limitations of the Study) impaired the physiologic ability of mice to concentrate urine to spare body fluid.
Discussion
Renal Endothelial Cell Heterogeneity in Health
Using scRNA-seq, we revealed a total of 24 different REC subpopulations, of which eight were previously not recognized. In the glomerular vasculature, we identified a new gREC subpopulation (G4) presumably lining the juxtaglomerular section of the efferent arteriole. Interestingly, gRECs in afferent arterioles associated with the JGA (G2) highly expressed genes encoding cell-cell interaction molecules, possibly to control processes (renin production, tubulo-glomerular feedback) that require close interactions between gRECs, renin-producing cells, and other cells in the JGA.46 For instance, enriched Dll4 expression in this gREC cluster might signal to Notch-expressing granular cells to maintain renin production.6,47 Consistent with their role in fine-tuning GFR,48 gRECs from afferent and efferent arterioles highly expressed marker genes encoding vasoregulatory molecules. In the cortical vasculature, we deconvoluted cortical capillaries in two subpopulations, characterized by high (C4) or low (C5) ApoE expression and other transcriptome signatures. In the medullary vasculature, we identified novel markers of DVR mRECs, including, for instance, Scin encoding Scinderin (binding the water channel aquaporin-2 in collecting duct epithelial cells49). The high expression of Scin and Aqp1 in DVR mRECs raises the question whether a similar interaction of Scinderin with AQP1 may be operational in mRECs. Other novel markers of DVR mRECs included genes involved in vasodilation (Adipor2, Hpgd), consistent with the role of DVR mRECs in regulating medullary blood flow and osmolarity gradient formation.50 We also identified new markers of mRECs of the DVR and AVR in the medullary papilla, which were poorly characterized in the past. For instance, papillary AVR mRECs highly expressed Cryab involved in the hyperosmolarity response51 and Car2, the loss of which causes polyuria and a failure to augment the medullary osmolarity and thereby to concentrate the urine.52
We also uncovered capillary and vein mREC and cREC populations showing a response-to-IFN phenotype as well as antigen processing and presentation-related genes, but not Cd80 and Cd86, suggesting participation in immune surveillance as semiprofessional antigen presenting cells, capable of activating only antigen-experienced immune cells.53 Angiogenic capillary mRECs and cRECs were also identified, although whether they are involved in repair/regeneration of damaged RECs requires further study.
Renal Endothelial Cell Response to Dehydration
The fundamental question of how mRECs adapt to dehydration and survive in hyperosmolarity conditions has not received much attention. This is highly relevant because ECs have long half-lives, and mRECs are exposed to inhospitable conditions during dehydration, which would be lethal to other cell types in the body. Dehydration-exposed mRECs upregulated expression of genes involved in heat shock proteins, cytoskeleton rearrangement, and DNA damage/growth arrest, consistent with a response to the hypertonic effect of high NaCl and to the denaturating effect of urea on cellular macromolecules,32,38 as described in renal inner medulla epithelial cells.32 mRECs from dehydrated mice also highly expressed genes, involved in proteasome degradation, possibly of denatured proteins, and increased the expression of ribosomal genes, possibly to compensate for the enhanced protein turnover.54,55
Medullary epithelial cells accommodate hyperosmolarity by accumulating organic metabolites as inert osmolytes.25,32,56–58 In agreement, mRECs upregulated the expression of genes involved in water transport, taurine/sodium and amino acid/sodium cotransport, and myo-inositol and sorbitol synthesis, as well as genes involved in Na+/K+-ATPase–mediated sodium export during dehydration, presumably to equilibrate osmotically with their hypertonic interstitium while preventing toxic intracellular Na+ accumulation. The low medullary oxygen, resulting in part from the low blood flow rate in the vasa recta, is an inevitable accompaniment of urinary concentration, especially in antidiuretic conditions.59 Deeper hypoxia in the medulla during dehydration may contribute to the upregulated expression of glycolytic genes in mRECs, as also occurs in medullary renal epithelial cells, which primarily rely on glycolysis for ATP production.25,60
Interestingly, although ECs are glycolysis-addicted and produce most of their ATP glycolytically,61 mRECs upregulated OXPHOS gene expression and oxygen consumption upon dehydration, while being exposed to deeper hypoxia, possibly to use the available oxygen more efficiently. Furthermore, OXPHOS inhibition compromised EC survival in hyperosmolar conditions in vitro and largely prevented urine concentration in dehydrated mice in vivo. Such a role for OXPHOS in the response to hyperosmolarity has not been documented previously, also not in medullary epithelial cells. We speculate that hyperosmolarity-exposed mRECs upregulate OXPHOS to provide ATP for energy-demanding processes such as protein synthesis, volume and osmolyte content regulation, and maintenance of intracellular sodium concentration requiring sodium export driven by the Na+/K+ ATPase,25,39,40 especially because ATP production from glycolysis can be compromised if a fraction of the glycolytic intermediates is shunted for osmolyte (sorbitol; myo-inositol) synthesis in hyperosmolarity, and OXPHOS is a more productive pathway to generate ATP per mole of glucose than glycolysis. Hyperosmolarity-exposed mRECs might also upregulate OXPHOS to produce “metabolic water” during mitochondrial respiration, resulting from the oxidation of organic materials.62–65 Whether and to which extent metabolic water production by hyperosmolarity-exposed mRECs contributes to cell volume regulation in dehydration remains to be determined.
In addition to providing a detailed REC taxonomy in baseline condition, this study also offers insight in the transcriptome adaptation of RECs to dehydration at the single-cell level, highlighting a previously unrecognized role of OXPHOS in the mREC response to hyperosmolarity and the ability to concentrate urine in dehydration. As such, this study may constitute grounds for examining the response of medullary epithelial cells upon hyperosmolarity challenge in the future. Furthermore, the publicly available extensive dataset is a rich resource for future data mining.
Limitations of the Study
There were several limitations of our study. First, we used metformin as a non-toxic compound to inhibit OXPHOS in RECs in dehydrated mice in vivo, and acknowledge that this compound may have additional activities66,67 and can act indirectly, and hence more direct proof is required in the future. Second, although we did not identify REC subclusters expressing a tissue dissociation artifact gene signature, we cannot exclude the possibility that the REC isolation procedure might have influenced REC transcriptomes. To minimize such effect, we used similar dissociation protocols. Third, unfortunately, the insufficient number of cRECs at 24 hours of dehydration precluded us from confidently characterizing their possible different transcriptome response to dehydration. Fourth, although we demonstrate that freshly isolated mRECs from dehydrated mice have elevated oxygen consumption, additional studies are required to further characterize possible elevations of OXPHOS and oxidative stress in vivo by complementary quantitative methods. Finally, we also acknowledge that water deprivation may evoke additional systemic and local effects, such as reducing caloric intake, BP, and urine volume, and altered mechanostimulatory stimuli (lower blood and urine flow),10,26,28,30 which may contribute to the observed findings.
Disclosures
None.
Funding
Dr. Dumas and Dr. Falkenberg are supported by Marie Curie-individual fellowships. Ms. Conchinha, Dr. Goveia, Dr. Kalucka, and Dr. Rohlenova are supported by the “Fonds voor Wetenschappelijk Onderzoek” (FWO), Dr. Teuwen by University of Antwerp. Dr. Khan is supported by a grant from Kom op Tegen Kanger (Stand up to Cancer, Flemish Cancer Society). Prof. Bolund and Prof. Luo are supported by the Sanming Project of Medicine in Shenzhen (SZSM20162074). Dr. Lin is supported by the Lundbeck Foundation (R219-2016-1375) and a Sapere Aude starting grant from the Danmarks Frie Forskningsfond (8048-00072A). The work of Prof. Luo is supported by Beijing Genomics Institute-Research, the Danish Research Council for Independent Research (DFF-1337-00128), the Sapere Aude Young Research Talent Prize (DFF-1335-00763A), and Aarhus University strategic grant (AU-iCRISPR). Prof. Li is supported by the State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center at the Sun Yat-Sen University and the National Natural Science Foundation of China (81670855), a Guandgong Province Leading Expert Program grant and the Key Program of Guangzhou Scientific Research Plan (3030901006074). The work of Prof. Carmeliet is supported by the Vlaams Instituut voor Biotechnologie TechWatch program, long-term structural Methusalem funding by the Flemish Government, grants from the Research Foundation Flanders (FWO-Vlaanderen), Foundation against Cancer (2016-078), Kom op Tegen Kanker (Stand up to Cancer, Flemish Cancer Society), European Research Council Proof of Concept (ERC-713758), and ERC advanced research grant (EU-ERC743074). This study was sponsored by Regenerative Medicine Crossing Borders.
Supplementary Material
Acknowledgments
Dr. Dumas, Dr. Meta, Ms. Borri, Dr. Goveia, and Dr. Rohlenova designed all experiments. Dr. Dumas, Dr. Meta, Ms. Borri, Dr. Chen, and Dr. Lu setup renal endothelial cell isolation protocol and performed in vivo and cell culture experiments, as well as biochemical and molecular assays. Dr. Dumas, Dr. Meta, Ms. Borri, Ms. Conchinha, Dr. Falkenberg, and Dr. Teuwen prepared scRNA-seq samples. Dr. de Rooij and Ms. Parys performed FACS sorting of RECs. Dr. Lin and Prof. Luo performed 10X single-cell sequencing. Dr. Goveia, Dr. Khan, and Dr. Karakach preprocessed scRNA-seq data. Dr. Dumas, Dr. Meta, Dr. Goveia, and Dr. Rohlenova performed the bioinformatics analysis. Dr. Dumas and Dr. Vinckier performed immunostaining and confocal imaging. Dr. Dumas, Dr. Meta, Ms. Borri, Dr. Kalucka, and Ms. De Legher performed metabolic experiments. Mr. Taverna setup the online database. Dr. Schoonjans, Dr. Lin, Prof. Bolund, Prof. Dewerchin, Dr. Eelen, Prof. Rabelink, Prof. Li, and Prof. Luo provided advice and discussed results. Dr. Dumas, Dr. Meta, and Prof. Carmeliet wrote the manuscript. Prof. Carmeliet conceptualized the study. All authors discussed results and commented on the manuscript.
We thank Ms. A. Manderveld and Ms. A. Carton for their assistance with immunostaining, Dr. B. Ghesquière for the mass spectrometry experiments, and laboratory members and Prof. N. Chandel (Chicago, IL) for their feedback and discussion.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Endothelial Cell Identity, Heterogeneity and Plasticity in the Kidney,” on pages 1–2.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2019080832/-/DCSupplemental.
Supplemental Figure 1. EC selection and data metrics for gRECs, cRECs, and mRECs.
Supplemental Figure 2. Identification of gREC and cREC subclusters.
Supplemental Figure 3. Identification of mREC subclusters.
Supplemental Figure 4. gREC, cREC, and mREC subcluster analysis.
Supplemental Figure 5. REC molecular adaptation to dehydration.
Supplemental Figure 6. Response of mREC subpopulations to dehydration.
Supplemental Figure 7. Molecular and metabolic adaptation of gRECs to dehydration.
Supplemental Figure 8. In vitro dehydration model characterization.
Supplemental Appendix 1. Supplemental methods, metabolic assays (detection of organic osmolytes), Western blot, and histology and immunohistochemistry (renal hypoxia).
Supplemental Table 1. scRNA-seq data processing and visualization.
Supplemental Table 2. Top 50 marker genes for gRECs, cRECs, and mRECs in control.
Supplemental Table 3. Molecular taxonomy of phenotypes of freshly isolated mouse RECs in control.
Supplemental Table 4. Top 50 marker genes for gREC, cREC, and mREC phenotypes in control.
Supplemental Table 5. GSVA of cREC and mREC phenotypes in control.
Supplemental Table 6. Differential analyses of cRECs, mRECs, and gRECs in control versus different time points of dehydration.
Supplemental Table 7. GSEA of mRECs in control versus 48-hour dehydration.
References
- 1.Molema G, Aird WC: Vascular heterogeneity in the kidney. Semin Nephrol 32: 145–155, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, et al.: Endothelium structure and function in kidney health and disease. Nat Rev Nephrol 15: 87–108, 2019 [DOI] [PubMed] [Google Scholar]
- 3.Kriz W: Fenestrated glomerular capillaries are unique. J Am Soc Nephrol 19: 1439–1440, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Eckardt KU, Bernhardt WM, Weidemann A, Warnecke C, Rosenberger C, Wiesener MS, et al.: Role of hypoxia in the pathogenesis of renal disease. Kidney Int Suppl S46–S51, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Pallone TL, Edwards A, Mattson DL: Renal medullary circulation. Compr Physiol 2: 97–140, 2012 [DOI] [PubMed] [Google Scholar]
- 6.Brunskill EW, Sequeira-Lopez ML, Pentz ES, Lin E, Yu J, Aronow BJ, et al.: Genes that confer the identity of the renin cell. J Am Soc Nephrol 22: 2213–2225, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otani H, Kaya M, Tsujita J: Effect of the volume of fluid ingested on urine concentrating ability during prolonged heavy exercise in a hot environment. J Sports Sci Med 12: 197–204, 2013 [PMC free article] [PubMed] [Google Scholar]
- 8.Popkin BM, D’Anci KE, Rosenberg IH: Water, hydration, and health. Nutr Rev 68: 439–458, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bongers CCWG, Alsady M, Nijenhuis T, Tulp ADM, Eijsvogels TMH, Deen PMT, et al.: Impact of acute versus prolonged exercise and dehydration on kidney function and injury. Physiol Rep 6: e13734, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rowland NE: Food or fluid restriction in common laboratory animals: Balancing welfare considerations with scientific inquiry. Comp Med 57: 149–160, 2007 [PubMed] [Google Scholar]
- 11.Satija R, Farrell JA, Gennert D, Schier AF, Regev A: Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33: 495–502, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Brink SC, Sage F, Vértesy Á, Spanjaard B, Peterson-Maduro J, Baron CS, et al.: Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat Methods 14: 935–936, 2017 [DOI] [PubMed] [Google Scholar]
- 13.Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al.: SCENIC: Single-cell regulatory network inference and clustering. Nat Methods 14: 1083–1086, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu G, Wang LG, Han Y, He QY: clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16: 284–287, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cannoodt R, Saelens W, Saeys Y: Computational methods for trajectory inference from single-cell transcriptomics. Eur J Immunol 46: 2496–2506, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Kiselev VY, Yiu A, Hemberg M: Scmap: Projection of single-cell RNA-seq data across data sets. Nat Methods 15: 359–362, 2018 [DOI] [PubMed] [Google Scholar]
- 17.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, et al.: Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154: 651–663, 2013 [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, et al.: Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 7: 575–583, 2001 [DOI] [PubMed] [Google Scholar]
- 19.Cantelmo AR, Conradi LC, Brajic A, Goveia J, Kalucka J, Pircher A, et al.: Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30: 968–985, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efe O, Klein JD, LaRocque LM, Ren H, Sands JM: Metformin improves urine concentration in rodents with nephrogenic diabetes insipidus. JCI Insight 1: 88409, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karaiskos N, Rahmatollahi M, Boltengagen A, Liu H, Hoehne M, Rinschen M, et al.: A single-cell transcriptome atlas of the mouse glomerulus. J Am Soc Nephrol 29: 2060–2068, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takemoto M, He L, Norlin J, Patrakka J, Xiao Z, Petrova T, et al.: Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J 25: 1160–1174, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Messaoudi S, He Y, Gutsol A, Wight A, Hébert RL, Vilmundarson RO, et al.: Endothelial Gata5 transcription factor regulates blood pressure. Nat Commun 6: 8835, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.dela Paz NG, D’Amore PA: Arterial versus venous endothelial cells. Cell Tissue Res 335: 5–16, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neuhofer W, Beck FX: Cell survival in the hostile environment of the renal medulla. Annu Rev Physiol 67: 531–555, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Bekkevold CM, Robertson KL, Reinhard MK, Battles AH, Rowland NE: Dehydration parameters and standards for laboratory mice. J Am Assoc Lab Anim Sci 52: 233–239, 2013 [PMC free article] [PubMed] [Google Scholar]
- 27.Cui FX, Liu HQ, Zou ZM, Li H: Metabolic responses to water deprivation in C57BL/6J mice using a proton nuclear magnetic resonance-based metabonomics approach. Rsc Adv 5: 80142–80149, 2015 [Google Scholar]
- 28.Faraco G, Wijasa TS, Park L, Moore J, Anrather J, Iadecola C: Water deprivation induces neurovascular and cognitive dysfunction through vasopressin-induced oxidative stress. J Cereb Blood Flow Metab 34: 852–860, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris M, Li P, Callahan MF, Oliverio MI, Coffman TM, Bosch SM, et al.: Neuroendocrine effects of dehydration in mice lacking the angiotensin AT1a receptor. Hypertension 33: 482–486, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Nagakura A, Hiyama TY, Noda M: Na(x)-deficient mice show normal vasopressin response to dehydration. Neurosci Lett 472: 161–165, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Berl T: How do kidney cells adapt to survive in hypertonic inner medulla? Trans Am Clin Climatol Assoc 120: 389–401, 2009 [PMC free article] [PubMed] [Google Scholar]
- 32.Burg MB, Ferraris JD, Dmitrieva NI: Cellular response to hyperosmotic stresses. Physiol Rev 87: 1441–1474, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Hall A: Rho GTPases and the actin cytoskeleton. Science 279: 509–514, 1998 [DOI] [PubMed] [Google Scholar]
- 34.Ikebe M, Nonoguchi H, Nakayama Y, Tashima Y, Tomita K: Upregulation of the secretory-type Na(+)/K(+)/2Cl(-)-cotransporter in the kidney by metabolic acidosis and dehydration in rats. J Am Soc Nephrol 12: 423–430, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Manotham K, Tanaka T, Ohse T, Kojima I, Miyata T, Inagi R, et al.: A biologic role of HIF-1 in the renal medulla. Kidney Int 67: 1428–1439, 2005 [DOI] [PubMed] [Google Scholar]
- 36.Bauerschmidt C, Arrichiello C, Burdak-Rothkamm S, Woodcock M, Hill MA, Stevens DL, et al.: Cohesin promotes the repair of ionizing radiation-induced DNA double-strand breaks in replicated chromatin. Nucleic Acids Res 38: 477–487, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu H, Balakrishnan K, Malaterre J, Beasley M, Yan Y, Essers J, et al.: Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS One 5: e12112, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sands JM, Layton HE: The physiology of urinary concentration: An update. Semin Nephrol 29: 178–195, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weigand KM, Swarts HG, Fedosova NU, Russel FG, Koenderink JB: Na,K-ATPase activity modulates Src activation: A role for ATP/ADP ratio. Biochim Biophys Acta 1818: 1269–1273, 2012 [DOI] [PubMed] [Google Scholar]
- 40.Whittam R, Willis JS: Ion movements and oxygen consumption in kidney cortex slices. J Physiol 168: 158–177, 1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, et al.: Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS One 6: e28536, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y: Circumventing the Crabtree effect: Replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci 97: 539–547, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Warburg O, Geissler AW, Lorenz S: [On growth of cancer cells in media in which glucose is replaced by galactose]. Hoppe Seylers Z Physiol Chem 348: 1686–1687, 1967 [PubMed] [Google Scholar]
- 44.Detaille D, Guigas B, Chauvin C, Batandier C, Fontaine E, Wiernsperger N, et al.: Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 54: 2179–2187, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Detaille D, Vial G, Borel AL, Cottet-Rouselle C, Hallakou-Bozec S, Bolze S, et al.: Imeglimin prevents human endothelial cell death by inhibiting mitochondrial permeability transition without inhibiting mitochondrial respiration. Cell Death Discov 2: 15072, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurtz L, Madsen K, Kurt B, Jensen BL, Walter S, Banas B, et al.: High-level connexin expression in the human juxtaglomerular apparatus. Nephron, Physiol 116: 1–8, 2010 [DOI] [PubMed] [Google Scholar]
- 47.Castellanos Rivera RM, Monteagudo MC, Pentz ES, Glenn ST, Gross KW, Carretero O, et al.: Transcriptional regulator RBP-J regulates the number and plasticity of renin cells. Physiol Genomics 43: 1021–1028, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ito S, Abe K: Contractile properties of afferent and efferent arterioles. Clin Exp Pharmacol Physiol 24: 532–535, 1997 [DOI] [PubMed] [Google Scholar]
- 49.Noda Y, Horikawa S, Katayama Y, Sasaki S: Identification of a multiprotein “motor” complex binding to water channel aquaporin-2. Biochem Biophys Res Commun 330: 1041–1047, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Pallone TL, Turner MR, Edwards A, Jamison RL: Countercurrent exchange in the renal medulla. Am J Physiol Regul Integr Comp Physiol 284: R1153–R1175, 2003 [DOI] [PubMed] [Google Scholar]
- 51.Lee SD, Choi SY, Lim SW, Lamitina ST, Ho SN, Go WY, et al.: TonEBP stimulates multiple cellular pathways for adaptation to hypertonic stress: Organic osmolyte-dependent and -independent pathways. Am J Physiol Renal Physiol 300: F707–F715, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krishnan D, Pan W, Beggs MR, Trepiccione F, Chambrey R, Eladari D, et al.: Deficiency of carbonic anhydrase II results in a urinary concentrating defect. Front Physiol 8: 1108, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blum JS, Wearsch PA, Cresswell P: Pathways of antigen processing. Annu Rev Immunol 31: 443–473, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dall’Asta V, Bussolati O, Sala R, Parolari A, Alamanni F, Biglioli P, et al.: Amino acids are compatible osmolytes for volume recovery after hypertonic shrinkage in vascular endothelial cells. Am J Physiol 276: C865–C872, 1999 [DOI] [PubMed] [Google Scholar]
- 55.Law RO: Amino acids as volume-regulatory osmolytes in mammalian cells. Comp Biochem Physiol A Comp Physiol 99: 263–277, 1991 [DOI] [PubMed] [Google Scholar]
- 56.Lang F, Busch GL, Ritter M, Völkl H, Waldegger S, Gulbins E, et al.: Functional significance of cell volume regulatory mechanisms. Physiol Rev 78: 247–306, 1998 [DOI] [PubMed] [Google Scholar]
- 57.Handler JS, Kwon HM: Regulation of renal cell organic osmolyte transport by tonicity. Am J Physiol 265: C1449–C1455, 1993 [DOI] [PubMed] [Google Scholar]
- 58.Kinne RK: The role of organic osmolytes in osmoregulation: From bacteria to mammals. J Exp Zool 265: 346–355, 1993 [DOI] [PubMed] [Google Scholar]
- 59.Brezis M, Rosen S: Hypoxia of the renal medulla--its implications for disease. N Engl J Med 332: 647–655, 1995 [DOI] [PubMed] [Google Scholar]
- 60.Bagnasco S, Good D, Balaban R, Burg M: Lactate production in isolated segments of the rat nephron. Am J Physiol 248: F522–F526, 1985 [DOI] [PubMed] [Google Scholar]
- 61.Eelen G, de Zeeuw P, Simons M, Carmeliet P: Endothelial cell metabolism in normal and diseased vasculature. Circ Res 116: 1231–1244, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mellanby K: Metabolic water and desiccation. Nature 150: 21, 1942 [Google Scholar]
- 63.Ziegler EE, Filer LJ, International Life Sciences Institute-Nutrition Foundation : Present Knowledge in Nutrition, Washington, D.C., ILSI Press, International Life Sciences Institute, 1996 [Google Scholar]
- 64.Takei Y, Bartolo RC, Fujihara H, Ueta Y, Donald JA: Water deprivation induces appetite and alters metabolic strategy in Notomys alexis: Unique mechanisms for water production in the desert. Proc Biol Sci 279: 2599–2608, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rutkowska J, Sadowska ET, Cichoń M, Bauchinger U: Increased fat catabolism sustains water balance during fasting in zebra finches. J Exp Biol 219: 2623–2628, 2016 [DOI] [PubMed] [Google Scholar]
- 66.Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B: Metformin: From mechanisms of action to therapies. Cell Metab 20: 953–966, 2014 [DOI] [PubMed] [Google Scholar]
- 67.Rena G, Hardie DG, Pearson ER: The mechanisms of action of metformin. Diabetologia 60: 1577–1585, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data are available in ArrayExpress under accession number E-MTAB-8145. Normalized data can be downloaded from the accompanying web resource at http://www.vibcancer.be/software-tools/rec-dehydration. Custom code is available upon request.